

Abstract
Malignant melanoma is a highly aggressive form of skin cancer responsible for the majority of skin cancer-related deaths. Recent insight into the heterogeneous nature of melanoma suggests more personalised treatments may be necessary to overcome drug resistance and improve patient care. To this end, reliable molecular signatures that can accurately predict treatment responsiveness need to be identified. In this study, we applied multiplex phosphoproteomic profiling across a panel of 24 melanoma cell lines with different disease-relevant mutations, to predict responsiveness to MEK inhibitor trametinib. Supported by multivariate statistical analysis and multidimensional pattern recognition algorithms, the responsiveness of individual cell lines to trametinib could be predicted with high accuracy (83% correct predictions), independent of mutation status. We also successfully employed this approach to case specifically predict whether individual melanoma cell lines could be sensitised to trametinib. Our predictions identified that combining MEK inhibition with selective targeting of c-JUN and/or FAK, using siRNA-based depletion or pharmacological inhibitors, sensitised resistant cell lines and significantly enhanced treatment efficacy. Our study indicates that multiplex proteomic analyses coupled with pattern recognition approaches could assist in personalising trametinib-based treatment decisions in the future.
Subject terms: Tumour heterogeneity, Tumour biomarkers, Proteomics
Introduction
Malignant melanoma is a highly aggressive form of skin cancer with the highest mortality rate among dermatological cancers. It is also a highly heterogeneous disease [1–3], which carries one of the highest mutational burdens [4, 5]. The mitogen-activated protein kinase (MAPK) signalling pathway [GTPase Ras (RAS)-RAF proto-oncogene serine/threonine-protein kinase (RAF)-Dual specificity mitogen-activated protein kinase kinase 1 (MEK)-extracellular signal-regulated kinase-1 (ERK)] is one of the most active and also most extensively studied pathways in melanoma [6]. Genetic alterations causing aberrant activation of members of the MAPK pathway are common in melanoma. The BRAF gene is mutated in >50% of melanomas, with the activating valine to glutamine (V600E) substitution in codon 600 being most prominent [7]. Activating mutations in RAS genes, most often in GTPase NRas (NRAS), are found in about 25% of melanomas [8]. Although mutations in MEK are less frequent, pathway activation caused by mutations in the RAF, RAS or receptor tyrosine kinases are mediated through the MEK (MAP/ERK kinase) axis, making inhibition of MEK an attractive therapeutic strategy [9].
The first generation of MEK inhibitors showed limited clinical benefits in unselected melanoma patients as a single agent [10] or when combined with chemotherapy [11]. However, clinical studies with second-generation MEK inhibitors were successful and led to the Food and Drug Administration approval of trametinib (GSK1120212), an allosteric and selective inhibitor of MEK1 and MEK2, with an IC50 of 0.7–14.9 nmol/l [12]. In a phase III clinical trial, trametinib demonstrated significant clinical benefit for melanoma patients with BRAF V600E/K mutations when compared with chemotherapy [13]. Furthermore, trametinib plus dabrafenib significantly improved overall survival in previously untreated metastatic melanoma patients harbouring BRAF V600E/K mutations, without increasing the overall toxicity [14]. Although trametinib has not been comprehensively tested in NRAS mutant melanoma, another MEK inhibitor, binimetinib, improved progression-free survival when compared with dacarbazine, highlighting this as a potential treatment strategy for NRAS mutant melanoma [15].
Various reports suggested that activating mutations in the BRAF gene represent the most important predictive biomarkers for sensitivity to MEK inhibitors [16–18]. However, clinical data indicate that although RAF mutant melanomas seem to be more sensitive to MEK inhibition, tumours with these mutations are not uniformly responsive [19]. Similarly, sensitivity varies strongly within NRAS-mutated melanomas, resulting in limited clinical benefits in these patient cohorts and highlighting the need for reliable molecular markers for treatment responsiveness [20, 21]. The lack of efficacy and durability of responses to MEK inhibitors might originate from several signalling pathways that alone or in combination cause or regulate intrinsic and adaptive resistance. This includes, for example, reactivation of the MAPK pathway through BRAF amplifications, MEK1/2 mutations or NRAS mutation, resulting in continued ERK activation in the presence of inhibitors [22–24]. Moreover, the activation of parallel signalling pathways such as the phosphoinositd 3-kinase (PI3K)/phosphatase and tensin homolog (PTEN)/RAC-alpha serine/threonine-protein kinase (AKT) [25], Src/Focal adhesion kinase (FAK)/signal transducer and activator of transcription 3 (STAT3) [26, 27] or mitogen-activated protein kinase 8 (JNK)/transcription factor AP-1 (c-JUN) [28] can stimulate upregulation of several growth factor receptors, thereby possibly contributing to relapse by increased tumour cell proliferation.
In this study, we examined a broad spectrum of kinase-dependant signalling pathways that have been shown to play important roles in cellular viability or are frequently overactive or suppressed in malignant neoplasms, including melanoma (Table 1). We hypothesised that the pre-treatment activities of these pathways and cascades, measured by phosphoprotein signatures, might carry information on whether trametinib treatment can induce growth arrest or cell death. As proof-of-concept, this was tested in a panel of 24 human melanoma cell lines, using an approach of data-driven multivariate statistical modelling and pattern recognition. Our study defines novel avenues to address the need for responsiveness biomarkers for trametinib treatments, as well as means to case specifically predict sensitisation strategies for cases where trametinib as a single-agent treatment remains ineffective.
Table 1.
Phosphoproteins used in multiplex analysis
| Protein | Full name | Gene names | Phosphorylation site | Function | UniProt entry |
|---|---|---|---|---|---|
| MEK | Dual specificity mitogen-activated protein kinase kinase-1 | MAP2K1, MEK1, PRKMK1 | S217/S221 | Cell growth, survival, differentiation | Q02750 |
| ERK | Mitogen-activated protein kinase-1 | MK01, MAPK1, ERK2, PRKM1, PRKM2 | T202/Y204 | Cell growth, adhesion, survival, differentiation | P28482 |
| AKT | RAC-alpha serine/threonine-protein kinase | AKT1, PKB, RAC | S473 | Metabolism, proliferation, cell survival, cell growth, angiogenesis | P31749 |
| FAK | Focal adhesion kinase-1 | PTK2, FAK, FAK1 | Y397 | Cell migration, adhesion, cell cycle, progression, proliferation, apoptosis | Q05397 |
| C-JUN | Transcription factor AP-1 | JUN | S63 | Cell cycle progression, proliferation angiogenesis, apoptosis | P05412 |
| STAT3 | Signal transducer and activator of transcription 3 | STAT3, APRF | Y705 | Cell growth, proliferation, migration, differentiation, survival, apoptosis | P40763 |
| P53 | Cellular tumour antigen p53 | TP53, P53 | S46 | Growth arrest, apoptosis, cell cycle regulation | P04637 |
| IκBa | NF-kappa-B inhibitor alpha | NFKBIA, IKBA, MAD3, NFKBI | S32/S36 | Proliferation, apoptosis | P25963 |
| WNK | Serine/threonine-protein kinase WNK1 | WNK1, HSN2, KDP | T60 | Electrolyte homeostasis, cell signalling, survival, proliferation | Q9H4A3 |
| CREB | Cyclic AMP-responsive element-binding protein 1 | CREB1 | S133 | Proliferation, migration, tumour suppression | P16220 |
| PTN11 | Tyrosine-protein phosphatase non-receptor type 11 | PTPN11, PTP2C, SHPTP2 | Y542 | Signal transducer | Q06124 |
Materials and methods
Materials
Trametinib was purchased from LC labs (MA, USA), FAK and c-JUN small interfering RNAs (siRNAs) were purchased from Qiagen (Hilden, Germany), JNK inhibitor V (CAS 345987-15-7) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA), and defactinib (VS-6063, PF-04554878) was purchased from Selleckchem (Houston, TX, US).
Melanoma cell lines
Fourteen metastatic melanoma cell lines SK-MEL5 (ATCC; HTB-70), MALME-3M (ATCC; HTB-64), SK-MEL2 (ATCC; HTB-68), WM3060 (Wistar; WC00126), WM1791c (Wistar; WC00086), IGR-37 (DSMZ; ACC 237), WM1205-LU (ATCC; CRL2812), WM451-LU (ATCC; CRL-2813), SK-MEL147, SK-MEL19, SK-MEL13, MeWo (ATCC; HTB-65), WM3734 (Rockland; WM3734-01-0001) and WM852 (Rockland; WM852-01-0001); eight primary cell lines A375 (ATCC; CRL-1619), WM1552c (ATCC; CRL-2808), SK-MEL28 (ATCC; HTB-72), WM793B (ATCC; CRL-2806), MEL-JUSO (DSMZ; ACC74), WM1366 (Wistar; WC00078), WM115 (ATCC; CRL-1675), WM35 (Wistar; WC00060) and two cell lines with unknown progression stage WM3248 (Rockland; WM3248-01-0001) and WM1819 (Rockland; WM1819-01-0001) were obtained from ATCC (Manassas, VA, USA), Wistar Institute (Philadelphia, PA, USA), Rockland (Limerick, PA, USA) or DSMZ (Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany). Cells were grown in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% foetal bovine serum (Life Technologies, MA, USA), 1% penicillin/streptomycin (Life Technologies, MA, USA) and were maintained in a humidified incubator at 37 oC and 5% CO2.
Cell viability measurements
Melanoma cells were seeded into 96-well plates at various cell densities (4000–10,000 cells/well) and allowed to adhere overnight. To investigate the responsiveness of the cell lines, cells were treated with trametinib, defactinib or JNK inhibitor V for up to 48 h. Following treatment, the medium was replaced, and cells were treated with 10 µg/ml of resazurin (Santa Cruz, TX, USA) for 2 h at 37 oC. After incubation, the fluorescence intensity of each sample was measured at 530/25 nm excitation and 590/35 emission wavelengths using a Varioskan LUX multi-well plate reader (Thermo Scientific, MA, USA).
Flow cytometry
For cell death measurements, flow cytometry was used to assess the number of PI+ and Annexin V+ cells following siRNA transfection and/or treatment with trametinib in the presence or absence of pan-Caspase inhibitor (fluoromethylketone); N-(2-Quinolyl)-L-valyl-L-aspartyl-(2,6-difluorophenoxy) methylketone (Q-VD-OPh) (30 μM). Cells were harvested with trypsin-EDTA and pelleted by centrifugation at 1200 rpm for 5 min at 4 oC. Cells were then incubated in 100 μl of binding buffer (BD Biosciences) containing Annexin V-green fluorescent protein (GFP) conjugate (3 μg/ml) and propidium iodide (PI) (2 μg/ml) for 10 min in the dark. Flow cytometry was performed on a MACSQuant Analyzer 10 (Miltenyi Biotech), using the blue 488 nm laser for excitation (emission from 655 to 730 nm) of PI and blue 488 nm laser for excitation (emission from 500-550 nm) of GFP. For each cell line, a total of 2 × 104 cells were gated.
siRNA transfection
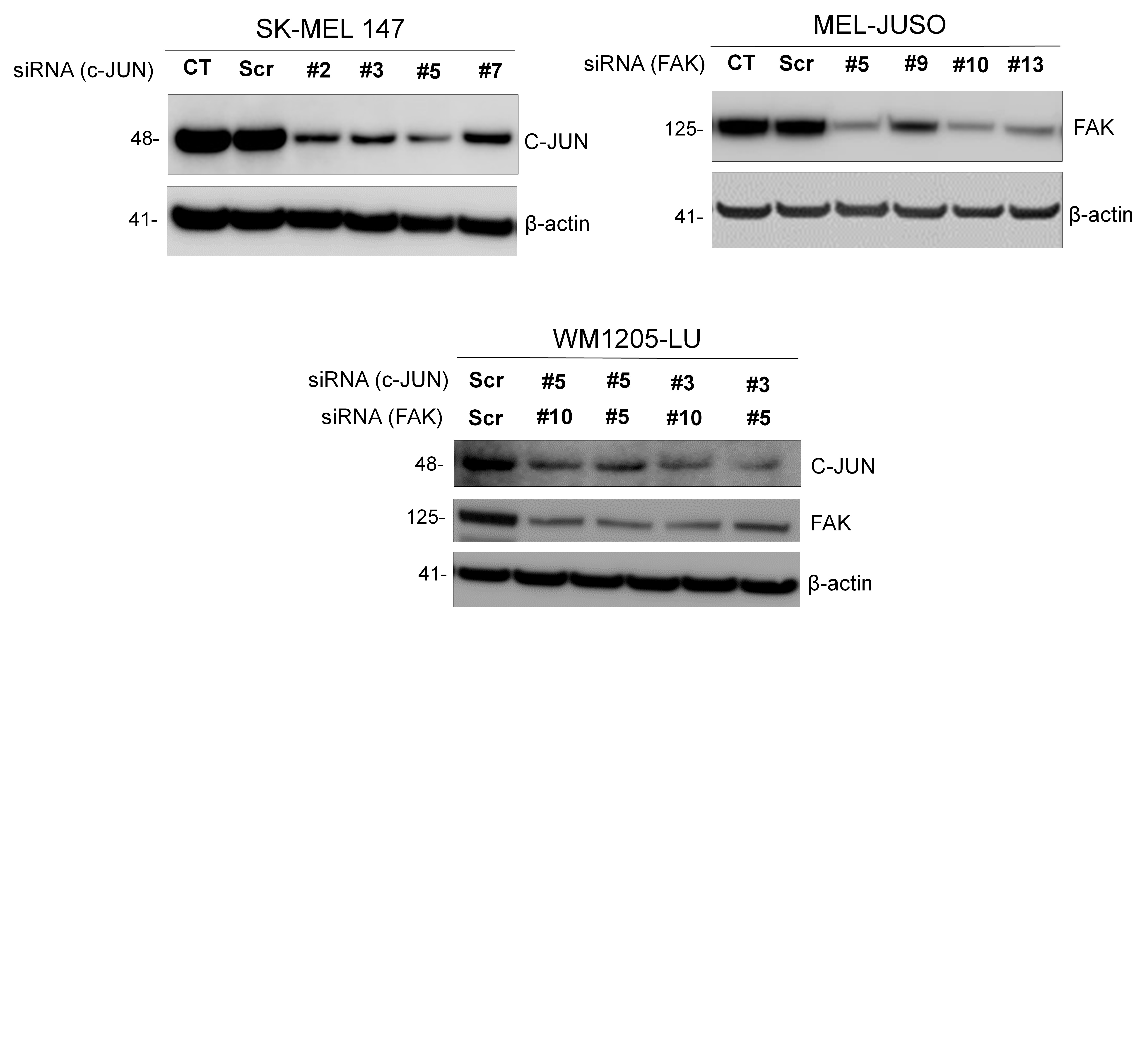
To downregulate c-JUN and FAK protein amounts, we used different sequences of siRNA (JUN siRNA FlexiTube GeneSolution—Cat# GS3725 and FAK siRNA FlexiTube GeneSolution—Cat# GS5747) and non-silencing control sequences (Qiagen). Cells were transfected with siRNA (100 nM) in antibiotic-free Opti-MEM (Gibco) medium with Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s instructions. Following 48 h of incubation, the transfection mixture was replaced with fresh medium. Protein depletion was validated by immunoblotting.
xMAP assays
Following the addition of ProtATonce lysis buffer (ProtATonce, Athens, Greece), cells were lysed by freezing/thawing and cellular debris was removed by centrifugation at 2700 g for 20 min. The protein concentration of the lysates was adjusted to 250 µg/µL using a BCA assay kit (PierceTM). xMAP assays were performed on a Luminex FLEXMAP 3D® platform (Luminex, Austin, TX), using a custom-developed phosphoprotein 11-plex panel (ProtATonce, Athens, Greece): dual specificity MAPK kinase-1 (MEK), ERK1, RAC-alpha serine/threonine-protein kinase (AKT), FAK-1, transcription factor AP-1 (c-JUN), STAT3, cellular tumour antigen p53 (P53), NF-kappa-B inhibitor alpha (IκBa), serine/threonine-protein kinase WNK1 (WNK), cyclic AMP-responsive element-binding protein 1 (CREB), tyrosine-protein phosphatase non-receptor type 11 (SHP2). Custom antibody-coupled beads were technically validated as described before [29].
Western blot analysis
Cells were homogenised in lysis buffer containing 150 mM NaCl, 1 mM EDTA, 20 mM TRIS, 1% Triton x-100 (pH 7.6), protease and phosphatase inhibitors (Roche). The protein concentration of lysates was determined with Bradford assay. Next, 20 µg total cellular lysates were mixed with 5 × Lämmli sample buffer, boiled at 95 oC for 10 min and separated on 4–12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels (Invitrogen, USA). Proteins were transferred to nitrocellulose membranes using an iBlot gel transfer device (Life Technologies, Invitrogen). The membranes were blocked in 5% bovine serum albumin in tris-buffered saline with 0.1% Tween 20 (TBST) for 1 h at room temperature. Primary antibodies were added after blocking and membranes were incubated overnight at 4 oC. The following primary antibodies were used: rabbit polyclonal caspase-3 (Cat# 9662, Cell Signaling); mouse monoclonal PARP (Cat# 551024, BD Pharmingen), rabbit monoclonal c-JUN (Cat# 9165, Cell Signaling) rabbit monoclonal FAK (Cat# ab40794, Abcam), mouse monoclonal MEK1/2 (Cat#4694 s, Cell Signaling), mouse monoclonal ERK1/2 (Cat#4696 s, Cell Signaling), mouse monoclonal β-tubulin (Cat#T6793, Sigma) and mouse monoclonal β-actin (Cat# 3700, Cell Signalling). After incubation, the membranes were washed three times with TBST for 10 min, prior to incubation with anti-mouse (Lot# 132504, Dianova), anti-rabbit (Lot# 132676, Dianova) or anti-mouse (Lot# 7076, Cell Signaling) secondary antibody for 1 h at room temperature. Protein bands (PARP, proCas3, β-actin) were visualised using horseradish peroxidase (HRP) substrate (SuperSignal West Dura Extended Duration Substrate, Thermo Scientific) and images were captured using Amersham 600 imager (GE Healthcare Life Sciences). For visualisation of total MEK and ERK, a Clarity Western ECL Substrate (Cat# 1705061) was used and images were captured using ChemidocTM MP system (Bio-Rad, Nazareth Eke, Belgium)
Data processing and analysis for data-driven modelling
All data processing and analysis were performed using a customised version of a previously published method [30]. The pipeline was developed for MATLAB 2017b (The Mathworks, UK), equipped with the statistical toolbox. Prior to statistical analysis, protein phosphorylation data were mean centred and scaled, dividing by the respective standard deviation. A principal component analysis (PCA) was performed on the standardised data set and the PCs with an eigenvalue > 1 were used for subsequent analyses. Linear discriminant analysis (LDA) was applied to objectively assess the accuracy of response class separation in the space defined by the first four principal components (PCs). Leave-one-out cross-validation (LOOCV) was applied iteratively to the cell line panel to assess predictive capacity. For each iteration, data from 23 cell lines were used as a training set to define the PC space, followed by LDA, and one test cell line was subsequently placed according to its protein expression profile. It was then determined if test cell lines positioned in the correct responsiveness subspace. To predict the sensitisation to trametinib after c-JUN and/or FAK knock down, the latter protein values were set to zero and new response classes were predicted from cell line repositioning in the PC space.
Data presentation and statistical analysis
Data were graphed and statistically analysed using the computer programs Matlab 2017b (The MathWorks), GraphPad Prism 6 (GraphPad Software) or R (R Core Team).
Results
PCA highlights that phosphoprotein expression patterns in melanoma are highly heterogeneous
To study heterogeneity in the basal phosphorylation status of kinase-dependent signalling pathways in melanoma, we quantified the phosphorylation of 11 kinases and kinase targets in a panel of 24 melanoma cell lines harbouring disease-relevant mutations (Fig. 1a). The selected kinases encompassed direct targets of trametinib (pMEK), upstream and downstream signalling pathways (pERK, pPTN11, pFAK), parallel signalling pathways (pAKT, pIKBa, pWNK) and transcription factors (pc-JUN, pSTAT3, pCREB, pP53). As expected, phosphorylation of MAPK pathway components (pMEK and pERK) was increased in BRAF mutant cell lines compared with BRAF wild-type cells. Furthermore, NRAS mutant cells showed increased phosphorylation levels of pSTAT3 when compared with other cell lines. Whereas the phosphorylation levels of some proteins like pWNK were rather homogeneous across the BRAF and NRAS-mutated cell lines and elevated when compared with BRAF/NRAS wild-type cells, the phosphorylation status of most other proteins varied dramatically (Fig. 1a).
Fig. 1.

Phosphoprotein expression patterns in melanoma are highly heterogeneous. a The phosphorylation status of 11 kinases and kinase targets was determined in 24 unstimulated melanoma cell lines using xMAP ELISA. Circles summarise 264 quantifications. Circle sizes are proportional to the protein phosphorylation level analysed from n = 3 independently performed experiments. b A principal component analysis (PCA) was applied to the phosphoprotein data. Results are shown as a scree plot where bar graphs indicate the contribution of each principal component (PC) toward explaining the data variance. c The coefficients for all proteins in the first four PCs are shown as bar graphs. d Graphical illustration of the distribution of melanoma cell lines within the space defined by the first three PCs
We used these protein data to analyse if protein phosphorylation patterns correlated between multiple cell lines. Applying a PCA, we found that at least four PCs are required to capture a minimum of 70% of the variance of the original data set (Fig. 1b). The weighting coefficients of the respective phosphoproteins for the first four PCs are shown in Fig. 1c. The first PC is strongly influenced by various proteins, including for example pERK1, pMEK1, pIKBa and pPTN11. In contrast, pc-JUN contributed strongly to PC2, pFAK to the third PC and pCREB to the fourth PC. To visualise the positioning of the 24 cell lines in the PC space, we used the first three PCs (Fig. 1d). Importantly, we did not observe any spatially separated cell clusters in the PC space, indicating that phosphoprotein patterns between the cell lines are highly heterogeneous.
Trametinib induces apoptosis-dependent loss of cell viability, but responsiveness is heterogeneous
We next studied the efficacy of trametinib treatment across the melanoma cell line panel. Cell viability measurements demonstrated that responsiveness was heterogeneous, with both treatment responders and cell lines largely resistant to trametinib observed (Fig. 2a). Responders and resistant cell lines, defined by a 50% viability cut-off, were detected in the groups of NRAS mutated, BRAF mutated, as well as wild-type cell lines, indicating that the cellular mutation status might carry only limited information on trametinib responsiveness. Furthermore, treatment responsiveness does not correlate with the tumour progression stage from which the cell lines were derived (Supplemental Table 1).
Fig. 2.

Responsiveness of melanoma cell lines to MEK inhibitor trametinib. a Cells were treated with 10 nM trametinib for 48 h and cell viability determined by resazurin reduction assay. Data show cell viability relative to untreated controls (means + S.D. from n = 3 independently performed experiments). b Three responsive and, c three resistant cell lines were treated with increasing concentrations of trametinib or with 10 nM trametinib and 30 µM Q-VD-OPh. The phosphorylation of MEK and ERK kinases were determined (24 h) using multiplex ELISA. Data show averaged median fluorescence intensity (MFI) for each protein (means ± S.D. from n = 3 independent experiments). Western blots show levels of total MEK and total ERK kinases (24 h). β-Tubulin served as loading control. d Three responsive and, e three resistant melanoma cell lines were treated with increasing concentrations of trametinib or with 10 nM trametinib and 30 µM Q-VD-OPh. After 48-h cell death was assessed by propidium iodide uptake and Annexin V-GFP staining. Bars represent mean values ± S.E.M. from three independently performed experiments. Western blots show processing of caspase-3 and PARP at 48 h. β-Actin served as loading control
As trametinib is a specific MAPK inhibitor, we next investigated its effects on phospho and total amounts of MEK and ERK kinases in three representative cell lines from sensitive or resistant subgroups, respectively (Fig. 2b, c). Although the amounts of total protein remained unchanged across all the cell lines tested (Fig. 2b, c), trametinib suppressed activity of the MAPK pathway in both responsive and resistant cells (Fig. 2b, c). In MEL-JUSO and SK-MEL147 cells, trametinib expectedly resulted in inhibition of ERK, but not MEK (Fig. 2c). We, furthermore, studied the type of cell death being induced by trametinib in both groups (Fig. 2d, e). Cell death, measured by propidium iodide uptake, was induced dose dependently in responsive cell lines, irrespective of the individual mutation status. In most cases, cell death was prevented effectively by pan-caspase inhibition with Q-VD-OPh, indicating that apoptosis was the primary cell death modality (Fig. 2d). Correspondingly, loss of pro-caspase-3 and poly [ADP-ribose] polymerase 1 (PARP) cleavage was detected in responsive cell lines (Fig. 2d). As expected, resistant cell lines did not show any indication for apoptotic cell death (Fig. 2e). Taken together, these findings indicate that trametinib can induce apoptosis independent of the mutation status of melanoma cells. Besides active compensatory responses by which reliance on or inhibition of the MAPK pathway can be overcome, also the pre-treatment activity of other kinases could co-determine trametinib responsiveness.
Phosphoprotein signatures allow predicting trametinib responsiveness
We next investigated if cellular phosphoprotein signatures prior to treatment might carry information on whether individual melanoma cell lines are likely to respond to treatment or not. For visualisation purposes, we therefore colour coded the cell lines in the PC space according to their responsiveness towards trametinib (Fig. 3a). Although it appeared that responsive and resistant cell lines tended to spatially separate in 3D space, we also applied LDA as a more objective cluster separation algorithm in the hyperspace defined by PCs with an eigenvalue above 1 (PCs 1–4). LDA correctly separated 21 out of 24 cell lines (87.5%; Fig. 3b), confirming that phosphoprotein signatures can separate melanoma cell lines into responders to trametinib and resistant cell lines.
Fig. 3.

Phosphoprotein signatures predict trametinib responsiveness. a Melanoma cell lines in the PC space were colour coded according to their responsiveness to trametinib (black, resistant; yellow, responsive). b Accuracy of response group separation in 4D PC space by LDA. c Schematic representation of leave-one-out cross-validation (LOOCV) workflow. d Accuracy of predicting drug responsiveness of test cell lines to trametinib
To test if these signatures are sufficiently strong to predict responsiveness on a case-by-case basis, we performed LOOCV. For this, we ran 24 PCAs, each using different sets of phosphoprotein signatures of 23 cell lines, and used LDA to define hyperspace regions of responsiveness and resistance. The individually omitted cell lines were then positioned into the respective PC spaces according to their phosphoprotein signatures. If the tested cell line positioned into the correct response region, the prediction was considered successful (see Fig. 3c for a visualisation of this principle and Supplemental Fig. 1 for a visualisation of the results, limited to the first two PCs). LOOCV accurately predicted trametinib responsiveness of 20 out of 24 cell lines (83.3%) (Fig. 3d), demonstrating that phosphoprotein signatures carry information from which trametinib responsiveness can be deduced with high accuracy.
Accurate prediction of sensitisation to trametinib by individual or combined knock down of c-JUN and FAK
Since our model was able to predict trametinib responsiveness based on pre-treatment protein phosphorylation amounts, we next tested whether this information could be used to create case-specific predictions on how to sensitise previously unresponsive cells to trametinib. We found that kinases such as pERK, pMEK, pIKBa, pPTN11 and pWNK in the first PC contributed equally to the positioning of melanoma cells in the multidimensional space. However, in the second PC, c-JUN had the highest weighting coefficient (compare with Fig. 1c). Similarly, FAK contributed strongly to the third PC. To test if targeting these kinases in individual cell lines would likely cause trametinib sensitisation, we visualised how cell line positions in the PC space would change upon elimination of these proteins. We used the PCA weighting coefficients to calculate the repositioning vectors resulting from eliminating c-JUN or FAK (Fig. 4a), and then determined the cell line-specific repositioning in the PC space. Applied to SK-MEL147 and MEL-JUSO cells, we predicted sensitisation to trametinib upon removal of c-JUN or FAK, respectively (Figs. 4b, d). In contrast, eliminating FAK in SK-MEL147 or c-JUN in MEL-JUSO were predicted not to cause trametinib sensitisation. To test these predictions, we targeted c-JUN or FAK by siRNAs in both cell lines and experimentally determined trametinib-induced cell death (Figs. 4c, e). As predicted, c-JUN depletion, but not FAK depletion, sensitised SK-MEL147 cells to trametinib (Fig. 4c, for knock down efficiency see Figure S2). Similarly, knock down of FAK but not c-JUN sensitised MEL-JUSO cells to apoptosis induced by trametinib (Fig. 4e).
Fig. 4.

Accurate prediction of sensitisation to trametinib by individual knock down of c-JUN and FAK. a Replacement vectors in 3D PC space. The movement vectors indicate the direction of cell line displacement in the PC space upon depleting or inhibiting c-JUN or FAK proteins, respectively. b Vectors indicate the movement for SK-MEL147 cells within the 3D PC space as a consequence of depleting basal c-JUN or FAK expression in this cell line. SK-MEL147 cells move towards regions populated by responsive cell lines only upon c-JUN but not FAK knock down. c Experimental validation of predictions. c-JUN or FAK were depleted by siRNA transfection (48 h), and cells were treated with trametinib and Q-VD-OPh for 48 h. Cell death was determined using PI/Annexin V-GFP staining (means + S.E.M. from n = 3 independent experiments). Statistical analysis was performed using Student's t-test. d Vectors indicate the movements for MEL-JUSO cells within the 3D PC space upon c-JUN or FAK depletion. MEL-JUSO cells move towards regions populated by responsive cell lines only upon pFAK but not pc-JUN knock down. e Experimental validation of predictions. c-JUN or FAK were depleted by siRNA transfection (48 h), and cells were treated with trametinib and Q-VD-OPh for 48 h. Cell death was determined using PI/Annexin V-GFP staining (means + S.E.M. from n = 3 independent experiments). Statistical analysis was performed using Student's t-test
In silico predictions for WM1205-LU cells demonstrated that the combined elimination of both, c-JUN and FAK, might effectively sensitise to trametinib (Fig. 5a). Indeed, we confirmed experimentally, with co-depletion of c-JUN/FAK significantly enhancing trametinib-induced apoptosis in WM1205-LU cells (Fig. 5b, see Supplemental Fig. 2 for knock down efficiency). Taken together, these results show that cellular phosphoprotein signatures can be employed to generate efficient and case-specific predictions for targeted interventions that significantly increase trametinib responsiveness.
Fig. 5.

Accurate prediction of sensitisation to trametinib by combined knock down of c-JUN and FAK. a Vectors indicate the movement for WM1205-LU cells within the 3D PC space, as predicted by co-depleting c-JUN and FAK. WM1205-LU cells move towards regions populated by responsive cell lines upon co-depleting c-JUN and FAK. b Experimental validation of prediction. c-JUN or FAK were co-depleted by siRNA transfection (48 h), and cells were treated with trametinib and Q-VD-OPh for 48 h. Cell death was determined using PI/Annexin V-GFP staining (means + S.E.M. from n = 3 independent experiments). Statistical analysis was performed using Student's t-test
Accurate prediction of cell line sensitisation to trametinib by individual or combined pharmacological targeting of c-JUN and FAK
As siRNA-based target depletion is of limited translational value, we next tested if case-specific predictions on trametinib sensitisation, as shown in Figs. 4 and 5, held true for settings in which c-JUN and FAK were pharmacologically targeted. In SK-MEL147 cells, we aimed to reduce pc-JUN via inhibition of its upstream activator JNK, using JNKi V as a commonly employed inhibitor. As shown in Fig. 6a, JNKi V resulted in partial dephosphorylation of pc-JUN in single and combination treatment with trametinib. Neither treatment affected phosphorylation of MEK, but, as expected, trametinib suppressed MAPK pathway activity at the level of pERK. As predicted, the combination treatment was superior in reducing cell viability than the single-drug treatment (Fig. 6b). In MEL-JUSO cells, trametinib expectedly resulted in inhibition of ERK, but not MEK (Fig. 6c). FAK inhibitor defactinib significantly reduced the amounts pFAK in both in single and combination treatment. The combination of trametinib with defactinib was significantly more potent than the single-agent treatments in reducing cell viability (Fig. 6d).
Fig. 6.

Accurate prediction of cell line sensitisation to trametinib by pharmacological inhibitors. a, b SK-MEL147 cells were pre-treated with JNK inhibitor V (5 µM); c, d MEL-JUSO cells were pre-treated with Defactinib (5 µM); e, f WM1205-LU cells were pre-treated with either inhibitor or a combination of both. Subsequently, cells were treated with trametinib (10 nM) and phosphorylation of key kinases, as well as cell viability were determined (48 h) using multiplex ELISA and resazurin reduction assay, respectively. Data show averaged median fluorescence intensity (MFI) for each protein (means ± S.D. from n = 3 independent experiments) and or cell viability relative to control values of 100% (means + S.E.M. from n = 3 independent experiments). Statistical analysis was performed using two-way ANOVA with post-hoc Tukey test
As predicted in Fig. 5, sensitisation of WM1205-LU cells was achieved specifically by the triple treatment combination of trametinib, JNKi V and defactinib (Fig. 6f), with pERK, pc-JUN and pFAK being strongly reduced (Fig. 6e), corresponding to on target activity of the inhibitors. Taken together, our findings confirm that accurate case-specific predictions can be made on which kinases might contribute to trametinib resistance and that pharmacological interventions tailored to the respective resistance scenarios can restore trametinib responsiveness in otherwise resistant cell lines.
Discussion and conclusion
With the exception of BRAF V600E inhibitors in accordingly stratified patients, responsiveness to MAPK family inhibitors in melanoma patients is highly heterogeneous [3, 31]. To improve such targeted therapies and to further personalise treatment, biomarkers or biomarker signatures are required that can more reliably predict drug responsiveness for individual patients. Here, we present the pre-clinical proof-of-concept for a new stratification method based on phosphoprotein profiling that can predict the therapeutic response to MEK inhibitor trametinib in melanoma cells and that, at the same time, offers solutions to case specifically overcome trametinib resistance. Our approach in the future might hold potential to complement the current state-of-the-art genetic profiling of BRAF and NRAS mutation status and could contribute to more precisely define which combination of kinase inhibitors to administer to individual patients. As such, the heterogeneity and complexity of phosphoprotein patterns, likely indicative of the activity of diverse signal transduction pathways, could add value to established genetic profiling. However, it needs to be noted that besides tumour-to-tumour differences also intra-tumour heterogeneity contributes to treatment success [2, 3]. Capturing the tumour-intrinsic variance in phosphoprotein patterns, as could arise, for example, from differences in the tumour microenvironment or from tumour-intrinsic heterogeneity in mutation loads, would provide scope for further improving prognostication of long-term treatment success.
The combination of phosphoprotein profiling, multivariate statistics and pattern recognition was able to identify trametinib non-responders within the group of BRAF mutant melanoma cell lines and concomitantly offers solutions to improve the response. This is noteworthy, since approximately 20% of patents with BRAF-mutated melanoma tumours are not responsive to the MAPK inhibitors treatment due to intrinsic resistance [32]. RAS driver mutations are often considered as undruggable and represent an unmet medical need in melanoma management [33, 34]. Currently, MEK inhibitors are under clinical investigation for NRAS-mutated melanoma, and the stratification method presented here could help identify those patient subpopulations that might benefit from trametinib treatment. If the approach presented here will prove successful in identifying tumours that are dependent on MAPK pathway activation but do not harbour BRAF or NRAS mutations, successfully predicting responsive to MEK inhibitors such as trametinib for these cohorts would further extend the number of patients that in the future could benefit from MEK inhibitor-based therapies.
c-JUN and FAK are known regulators of cell growth and proliferation [35, 36] and, among the kinases investigated, appeared as critical suppressors of trametinib responsiveness in a subset of melanoma cells. c-JUN has been implicated previously in melanoma tumour development [37–40] and recent studies indeed identified c-JUN as an important co-regulator of BRAF/MEK inhibitor responsiveness in melanoma [28, 41, 42]. By co-targeting the JNK/c-JUN pathway in either BRAF or NRAS-mutated melanoma, we could sensitise previously resistant cells to trametinib, further corroborating that c-JUN could play a major role as a future therapeutic target in melanoma. Similarly, we identified FAK as a regulator of trametinib resistance, and FAK has previously been implicated in promoting an aggressive phenotype in melanoma [43] and in co-determining chemotherapy resistance [44]. Various groups identified that FAK is upregulated in other advanced-stage solid tumours, such as ovarian and breast cancer, and likely implicated in promoting tumour progression, metastasis, invasion and migration [45–47]. Interestingly, Fallahi-Sichani and colleagues recently showed that depletion of both, c-JUN and FAK, significantly increased sensitivity to vemurafenib in melanoma cells [48], which further supports our findings. Although JNK inhibitors, as upstream suppressors of c-JUN activity, are still in pre-clinical development, FAK inhibitor co-treatments are already being explored in phase II clinical trials for advanced solid malignancies (NCT02758587, NCT02428270).
Different proteomic profiling technologies, such as reverse phase protein arrays and mass spectrometry, are being explored that feed into pipelines geared towards predicting responses to targeted therapies, and discovering drug resistance mechanism or identifying optimal combination therapies [49–52]. As a big advantage, high-throughput quantitative multiplex proteomic measurements require only low amounts of patient material, provide comprehensive data, can be intrinsically calibrated and are fast. As a methodological framework, multiplex-based measurements are therefore attractive in clinical scenarios where optimal treatment decisions need to be made in reasonably short times. In melanoma, this might be of particular value in rapidly progressing, aggressive metastatic disease, where ineffective treatments with targeted kinase inhibitors and lengthy periods of time for immunotherapies to become effective cannot be tolerated.
In conclusion, we here describe that quantitative multiplex phosphoprotein analysis can be merged with data-driven mathematical modelling to predict trametinib responsiveness of melanoma cells and how treatment efficacy can be optimised by further case-specific targeted interventions. In the future, it will be of interest to study if these approaches can be extended towards more clinical settings, as well as to malignant neoplasms other than melanoma.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
LGA, MR, DK, PA and TS receive funding through the EU Horizon 2020 MEL-PLEX program (grant agreement #642295). LGA also receives funding through the Horizon 2020 SyMBioSys program (grant agreement #675585). LGA and VP also receives funding from European Union and Greek National Funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call Research-Create-Innovate (project code:T1EDK-03532). MR also receives support from the German Research Foundation (FOR2036; MO 3226/1–1) and the Health Research Board Ireland (HRA POR 2013 245). DK also receives funding from Federal Ministry of Education and Research (BMBF: FKZ 031A423A, Melanoma Sensitivity). TS also receives funding from the Luxembourg National Research Fund (FNR: BMBF/BM/7643621, Melanoma Sensitivity). The authors wish to thank Prof Thomas Brunner (University of Konstanz, Germany) for helpful advice.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by G Melino
These authors contributed equally: Markus Rehm, Leonidas G. Alexopoulos
Electronic supplementary material
The online version of this article (10.1038/s41418-018-0210-8) contains supplementary material, which is available to authorised users.
References
- 1.Krepler C, Sproesser K, Brafford P, Beqiri M, Garman B, Xiao M, et al. A comprehensive patient-derived xenograft collection representing the heterogeneity of melanoma. Cell Rep. 2017;21:1953–67. doi: 10.1016/j.celrep.2017.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reuben A, Spencer CN, Prieto PA, Gopalakrishnan V, Reddy SM, Miller JP, et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom Med. 2017;2:10. doi: 10.1038/s41525-017-0013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menzies AM, Haydu LE, Carlino MS, Azer MWF, Carr PJA, Kefford RF, et al. Inter- and intra-patient heterogeneity of response and progression to targeted therapy in metastatic melanoma. PLoS ONE. 2014;9:1–9. doi: 10.1371/journal.pone.0085004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. doi: 10.1186/s13073-017-0424-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang AX, Qi XY. Targeting RAS/RAF/MEK/ERK signaling in metastatic melanoma. IUBMB Life. 2013;65:748–58. doi: 10.1002/iub.1193. [DOI] [PubMed] [Google Scholar]
- 7.Spagnolo F, Ghiorzo P, Orgiano L, Pastorino L, Picasso V, Tornari E, et al. BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. Onco Targets Ther. 2015;8:157–68. doi: 10.2147/OTT.S39096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kunz M. Oncogenes in melanoma: an update. Eur J Cell Biol. 2014;93:1–10. doi: 10.1016/j.ejcb.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Caunt Christopher J., Sale Matthew J., Smith Paul D., Cook Simon J. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nature Reviews Cancer. 2015;15(10):577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- 10.Kirkwood JM, Bastholt L, Robert C, Sosman J, Larkin J, Hersey P, et al. Phase II, open-label, randomized trial of the MEK1/2 inhibitor selumetinib as monotherapy versus temozolomide in patients with advanced melanoma. Clin Cancer Res. 2012;18:555–67. doi: 10.1158/1078-0432.CCR-11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta A, Love S, Schuh A, Shanyinde M, Larkin JM, Plummer R, et al. DOC-MEK: a double-blind randomized phase II trial of docetaxel with or without selumetinib in wild-type BRAF advanced melanoma. Ann Oncol. 2014;25:968–74. doi: 10.1093/annonc/mdu054. [DOI] [PubMed] [Google Scholar]
- 12.Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011;17:989–1000. doi: 10.1158/1078-0432.CCR-10-2200. [DOI] [PubMed] [Google Scholar]
- 13.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 14.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2014;372:141116004513004. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 15.Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017;18:435–45. doi: 10.1016/S1470-2045(17)30180-8. [DOI] [PubMed] [Google Scholar]
- 16.Garon EB, Finn RS, Hosmer W, Dering J, Ginther C, Adhami S, et al. Identification of common predictive markers of in vitro response to the Mek inhibitor selumetinib (AZD6244; ARRY-142886) in human breast cancer and non-small cell lung cancer cell lines. Mol Cancer Ther. 2010;9:1985–94. doi: 10.1158/1535-7163.MCT-10-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jing J, Greshock J, Holbrook JD, Gilmartin A, Zhang X, McNeil E, et al. Comprehensive predictive biomarker analysis for MEK inhibitor GSK1120212. Mol Cancer Ther. 2012;11:720–9. doi: 10.1158/1535-7163.MCT-11-0505. [DOI] [PubMed] [Google Scholar]
- 19.Sznol M. Molecular markers of response to treatment for melanoma. Cancer J. 2011;17:127–33. doi: 10.1097/PPO.0b013e318212dd5a. [DOI] [PubMed] [Google Scholar]
- 20.Thumar J, Shahbazian D, Aziz SA, Jilaveanu LB, Kluger HM. MEK targeting in N-RAS mutated metastatic melanoma. Mol Cancer. 2014;13:45. doi: 10.1186/1476-4598-13-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jakob JA, Bassett RL, Ng CS, Curry JL, Joseph RW, Alvarado GC, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014–23. doi: 10.1002/cncr.26724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bagrodia S, Smeal T, Abraham RT. Mechanisms of intrinsic and acquired resistance to kinase-targeted therapies. Pigment Cell Melanoma Res. 2012;25:819–31. doi: 10.1111/pcmr.12007. [DOI] [PubMed] [Google Scholar]
- 23.Lassen A, Atefi M, Robert L, Wong DJ, Cerniglia M, Comin-Anduix B, et al. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol Cancer. 2014;13:83. doi: 10.1186/1476-4598-13-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, et al. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J Biol Chem. 2012;287:28087–98. doi: 10.1074/jbc.M112.377218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perna D, Karreth FA, Rust AG, Perez-Mancera PA, Rashid M, Iorio F, et al. BRAF inhibitor resistance mediated by the AKT pathway in an oncogenic BRAF mouse melanoma model. Proc Natl Acad Sci USA. 2015;112:E536–45. doi: 10.1073/pnas.1418163112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastas- Rev. 2005;24:315–27. doi: 10.1007/s10555-005-1580-1. [DOI] [PubMed] [Google Scholar]
- 27.Vultur A, Villanueva J, Krepler C, Rajan G, Chen Q, Xiao M, et al. MEK inhibition affects STAT3 signaling and invasion in human melanoma cell lines. Oncogene. 2014;33:1850–61. doi: 10.1038/onc.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramsdale R, Jorissen RN, Li FZ, Al-Obaidi S, Ward T, Sheppard KE, et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal. 2015;8:ra82. doi: 10.1126/scisignal.aab1111. [DOI] [PubMed] [Google Scholar]
- 29.Poussin C, Mathis C, Alexopoulos LG, Messinis DE, Dulize RHJ, Belcastro V, et al. The species translation challenge—A systems biology perspective on human and rat bronchial epithelial cells. Sci Data. 2014;1:1–14. doi: 10.1038/sdata.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Passante E, Würstle ML, Hellwig CT, Leverkus M, Rehm M. Systems analysis of apoptosis protein expression allows the case-specific prediction of cell death responsiveness of melanoma cells. Cell Death Differ. 2013;20:1521–31. doi: 10.1038/cdd.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roesch A. Tumor heterogeneity and plasticity as elusive drivers for resistance to MAPK pathway inhibition in melanoma. Oncogene. 2015;34:2951–7. doi: 10.1038/onc.2014.249. [DOI] [PubMed] [Google Scholar]
- 32.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828–51. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandalà M, Merelli B, Massi D. Nras in melanoma: targeting the undruggable target. Crit Rev Oncol Hematol. 2014;92:107–22. doi: 10.1016/j.critrevonc.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zenz R, Wagner EF. Jun signalling in the epidermis: from developmental defects to psoriasis and skin tumors. Int J Biochem Cell Biol. 2006;38:1043–9. doi: 10.1016/j.biocel.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 37.Spangler B, Vardimon L, Bosserhoff AK, Kuphal S. Post-transcriptional regulation controlled by E-cadherin is important for c-Jun activity in melanoma. Pigment Cell Melanoma Res. 2011;24:148–64. doi: 10.1111/j.1755-148X.2010.00787.x. [DOI] [PubMed] [Google Scholar]
- 38.Spangler B, Kappelmann M, Schittek B, Meierjohann S, Vardimon L, Bosserhoff AK, et al. ETS-1/RhoC signaling regulates the transcription factor c-Jun in melanoma. Int J Cancer. 2012;130:2801–11. doi: 10.1002/ijc.26277. [DOI] [PubMed] [Google Scholar]
- 39.Lopez-Bergami P, Kim H, Dewing A, Goydos J, Aaronson S, Ronai Z. c-Jun regulates phosphoinositide-dependent kinase 1 transcription: implication for Akt and protein kinase C activities and melanoma tumorigenesis. J Biol Chem. 2010;285:903–13. doi: 10.1074/jbc.M109.075630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurzov EN, Bakiri L, Alfaro JM, Wagner EF, Izquierdo M. Targeting c-Jun and JunB proteins as potential anticancer cell therapy. Oncogene. 2008;27:641–52. doi: 10.1038/sj.onc.1210690. [DOI] [PubMed] [Google Scholar]
- 41.Fallahi-Sichani M, Moerke NJ, Niepel M, Zhang T, Gray NS, Sorger PK. Systematic analysis of BRAFV600E melanomas reveals a role for JNK/c-Jun pathway in adaptive resistance to drug-induced apoptosis. Mol Syst Biol. 2015;11:797. doi: 10.15252/msb.20145877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Titz B, Lomova A, Le A, Hugo W, Kong X, Ten Hoeve J, et al. JUN dependency in distinct early and late BRAF inhibition adaptation states of melanoma. Cell Discov. 2016;2:16028. doi: 10.1038/celldisc.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hess AR, Postovit LM, Margaryan NV, Seftor EA, Schneider GB, Seftor REB, et al. Focal adhesion kinase promotes the aggressive melanoma phenotype. Cancer Res. 2005;65:9851–60. doi: 10.1158/0008-5472.CAN-05-2172. [DOI] [PubMed] [Google Scholar]
- 44.Tavora B, Reynolds LE, Batista S, Demircioglu F, Fernandez I, Lechertier T, et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature. 2014;514:112–6. doi: 10.1038/nature13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roy-Luzarraga M, Hodivala-Dilke K. Molecular pathways: endothelial cell FAK-A target for cancer treatment. Clin Cancer Res. 2016;22:3718–24. doi: 10.1158/1078-0432.CCR-14-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kolli-Bouhafs K, Sick E, Noulet F, Gies JP, De Mey J, Rondé P. FAK competes for Src to promote migration against invasion in melanoma cells. Cell Death Dis. 2014;5:e1379. doi: 10.1038/cddis.2014.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pei G, Lan Y, Chen D, Ji L, Hua ZC. FAK regulates E-cadherin expression via p-SrcY416/p-ERK1/2/p-Stat3Y705 and PPARγ/miR-125b/Stat3 signaling pathway in B16F10 melanoma cells. Oncotarget. 2017;8:13898–908. doi: 10.18632/oncotarget.14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fallahi-Sichani M, Becker V, Izar B, Baker GJ, Lin JR, Boswell SA, et al. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol Syst Biol. 2017;13:1–24. doi: 10.15252/msb.20166796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rebecca VW, Wood E, Fedorenko IV, Paraiso KHT, Haarberg HE, Chen Y, et al. Evaluating melanoma drug response and therapeutic escape with quantitative proteomics. Mol Cell Proteom. 2014;13:1844–54. doi: 10.1074/mcp.M113.037424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, et al. Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2012;2:798–811. doi: 10.1158/2159-8290.CD-12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mueller C, Liotta LA, Espina V. Reverse phase protein microarrays advance to use in clinical trials. Mol Oncol. 2010;4:461–81. doi: 10.1016/j.molonc.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klammer M, Kaminski M, Zedler A, Oppermann F, Blencke S, Marx S, et al. Phosphosignature predicts dasatinib response in non-small cell lung cancer. Mol Cell Proteom. 2012;11:651–68. doi: 10.1074/mcp.M111.016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.