

Abstract
During neurogenesis, proliferating neural precursor cells (NPC) exit the cell cycle and differentiate into postmitotic neurons. The proteins that regulate cell survival through the stages of differentiation, however, are still poorly understood. Here, we examined the roles of the anti-apoptotic Bcl-2 proteins, Mcl-1 and Bcl-xL, in promoting survival as cells progress through the stages of neurogenesis in the mouse embryonic central nervous system. We used Nestin-mediated, nervous system-specific conditional deletion of mcl-1, bcl-x or both to identify their distinct and overlapping roles. Individual conditional deletion of mcl-1 (MKO) and bcl-x (BKO) suggested sequential roles in promoting cell survival during developmental neurogenesis. In the MKO embryo, apoptosis begins at embryonic day 10 (E10) in the proliferating NPC population throughout the entire developing nervous system. In the BKO embryo, apoptosis begins later at E11 within the postmitotic neuron populations. In the double (mcl-1 and bcl-x) conditional knockout (DKO), cell death extended throughout both proliferating and non-proliferating cell populations resulting in embryonic lethality at E12, earlier than in either the MKO or BKO. Apoptotic cell death of the entire central nervous system in the DKO demonstrates that both genes are necessary for cell survival during developmental neurogenesis. To determine whether Mcl-1 and Bcl-xL have overlapping anti-apoptotic roles during neurogenesis, we examined the impact of gene dosage. Loss of a single bcl-x allele in the MKO embryo exasperated apoptotic cell death within the NPC population revealing a novel anti-apoptotic role for Bcl-xL in proliferating NPCs. Cells were rescued from apoptosis in both the MKO and BKO embryos by breeding with the Bax null mouse line indicating that Mcl-1 and Bcl-xL have a common pro-apoptotic target during developmental neurogenesis. Taken together, these findings demonstrate that Mcl-1 and Bcl-xL are the two essential anti-apoptotic Bcl-2 proteins required for the survival of the developing mammalian nervous system.
Subject terms: Neuroscience, Development, Cell biology, Gene expression
Introduction
In the developing nervous system, neural stem cells and progenitor cells, collectively called neural precursor cells (NPC), divide to form the differentiated neurons and macroglia of the mature nervous system. In the mouse, the NPC pool expands from embryonic day 9–10 (E9–E10) and neurogenesis begins at E11 when NPCs exit the cell cycle and initiate differentiation. Apoptosis plays a key role in both patterning and regulating the total number of cells in the nervous system [1]. The B-cell lymphoma 2 (Bcl-2) family of pro-apoptotic and anti-apoptotic proteins controls initiation of the apoptotic pathway [2]. The anti-apoptotic members of the Bcl-2 family, myeloid cell leukemia-1 (Mcl-1) and Bcl-2-related gene long isoform (Bcl-xL), have crucial roles in nervous system development. Each protein appears to function at a specific stage in neurodevelopment; however, it is unclear whether their roles are distinct or overlap.
Mcl-1 was first identified as a gene that is upregulated during the differentiation of myeloid cells [3]. Conditional Mcl-1 knockout mice have since demonstrated that Mcl-1 is key for the survival of precursor populations within the hematopoietic, hepatocytic, and dermal tissues [4–7]. In the nervous system-specific Mcl-1 conditional knockout mice (MKO), extensive apoptosis of NPCs occurs in the developing forebrain [8]. Mcl-1 is the only known anti-apoptotic Bcl-2 protein required for the survival of both embryonic and adult NPCs [8, 9]. Mice with germline knockout of anti-apoptotic A1, Bcl-w or Bcl-2 have no apparent developmental defects in the CNS [10–14]. Knockdown of Mcl-1 in postmitotic neurons, however, does not result in apoptosis, but autophagic cell death [15]. This suggests that Mcl-1’s anti-apoptotic role has a defined temporal requirement during neurogenesis.
The anti-apoptotic bcl-x long (bcl-xL) isoform is one of five isoforms on the bcl-x gene, and is the predominant isoform in the nervous system [16–18]. Bcl-xL expression coincides with the start of neurogenesis at E11 and is expressed in postmitotic neurons throughout the nervous system by E13.5 [19, 20]. Germline bcl-x knockouts are embryonic lethal at E13 with apoptosis of immature neurons and hematopoietic cells [21]. Neuron-specific bcl-x conditional knockouts demonstrated that bcl-x is required for the survival of catecholaminergic neurons, upper layer cortical neurons, and CA1–CA3 hippocampal neurons [22, 23]. Immature neurons have a greater dependency on Bcl-xL than mature neurons as knocking out bcl-x in immature retinal ganglion neurons results in apoptosis, whereas adult neurons survive [24]. Furthermore, conditional bcl-x knockout in NPCs revealed that Bcl-xL dependency begins within 24 h of exiting the cell cycle [25]. These studies demonstrate that similar to Mcl-1, the anti-apoptotic role of Bcl-xL in neurogenesis is temporal.
The distinct temporal requirements of Mcl-1 and Bcl-xL during neurogenesis suggests a transition in anti-apoptotic protein dependency from Mcl-1 to Bcl-xL. To determine whether their roles are functionally distinct or overlap, we generated a double (mcl-1 and bcl-x) conditional knockout (DKO) mouse. We show that survival of the entire nervous system is dependent on the combined expression of Mcl-1 and Bcl-xL during neurogenesis.
Results
Mcl-1 and Bcl-xL have distinct anti-apoptotic roles in the developing spinal cord
To investigate the individual anti-apoptotic roles of Mcl-1 and Bcl-xL in developmental neurogenesis, we first examined whether both were expressed in the early embryonic spinal cord. qRT-PCR revealed that Mcl-1 and Bcl-xL mRNA are expressed in the spinal cord at E10 and appear to increase with the onset of neurogenesis (Suppl. Figure 1). To determine whether Mcl-1 and Bcl-xL are required for cell survival during early spinal cord neurogenesis, we used nervous system-specific conditional knockout mice for mcl-1 (MKO) and bcl-x (BKO) [8, 25]. Apoptotic cells were identified by two key markers of apoptosis—a completely condensed nucleus as demonstrated with Hoechst nuclear stain combined with immunopositivity for active Caspase-3 (AC3). A few apoptotic cells were first observed at E9 in the ventral spinal cord of MKO embryos (data not shown) and by E10, apoptosis had spread across the ventral spinal cord (Fig. 1a). Few to no apoptotic cells were observed in comparable sections through the E10 spinal cords of control (CTL), heterozygotes (not shown), and BKO embryos. By E11, the majority of apoptotic cells in the MKO spinal cord were located dorsally, whereas in the E11 BKO embryo, apoptosis was just beginning in the ventrolateral horns (Fig. 1b). As development proceeded, apoptosis was still observed in the E13 dorsal spinal cord of MKO embryos while in E13 BKO embryos, apoptosis was spreading into the dorsal spinal cord (Fig. 1c). In both MKO and BKO embryos, apoptosis began in the ventral spinal cord and spread into the dorsal spinal cord over 1–2 days.
Fig. 1.

The onset of apoptosis occurs earlier in the MKO embryo than the BKO embryo. Representative photomicrographs of active Caspase-3 (AC3) immunohistochemistry labeling of apoptotic cell death in horizontal spinal cord sections at (a) E10, (b) E11, and (c) E13 from CTL, MKO, and BKO embryos. a In MKO embryos, apoptosis appears first in the ventral spinal cord at E10 whereas no apoptosis is observed in the BKO spinal cord. b By E11, apoptosis is predominantly in the dorsal spinal cord of the MKO embryo. Apoptosis begins at E11 in BKO embryos and similar to the MKO embryo, it is first observed within the ventral spinal cord. c By E13, apoptosis is only located in the dorsal spinal cord in the MKO embryo and in the BKO embryo, apoptosis has spread across the ventral and into dorsal spinal cord. D dorsal, V ventral. Scale bar = 200 μm (a–c)
Apoptosis in the MKO and BKO spinal cord appears spatially and temporally distinct. To examine the proliferative state of apoptotic cells in both MKO and BKO spinal cords, pregnant dams were given a BrdU pulse prior to euthanasia at E11. Dual immunohistochemistry for AC3 and BrdU on spinal cord sections revealed that apoptotic cells in the MKO embryo were BrdU+ proliferating cells, but in the BKO embryo apoptotic cells were BrdU− (Fig. 2a, b). Mcl-1 and Bcl-xL therefore appear to be required for the survival of proliferating and differentiating cells within the developing spinal cord, respectively, suggesting they have distinct roles.
Fig. 2.

Apoptosis appears in spatially distinct cell populations in MKO and BKO embryos. a Representative photomicrographs of horizontal spinal cord sections from E11 CTL, MKO, and E13 BKO embryos labeled for active Caspase-3 (AC3) (green) and BrdU (pink). A BrdU pulse was given 2 h prior to euthanasia to label proliferating cells. b Higher magnification photomicrographs of boxed regions in a revealed co-labeling of AC3 and BrdU in MKO tissue, but not in CTL or BKO tissue. Arrows point to double labeled cells
Conditional deletion of Mcl-1 and Bcl-xL leads to a complete loss of the central nervous system
To examine the role of both Mcl-1 and Bcl-xL on nervous system development, we generated a nervous system-specific, conditional double mcl-1 and bcl-x knockout mouse (DKO). The DKO embryo was lethal at E12 (Table 1), preceeding the lethality of the MKO at E16 and the BKO at E19 [8, 25]. Mice heterozygous for mcl-1 and bcl-x (Mcl-1 Het:Bcl-x Het) were born at normal Mendelian ratios and were fertile. A single DKO embryo was discovered at E14, however tissue histology revealed complete loss of the entire central nervous system (Fig. 3a). Numerous red blood cells were observed within the head region caused by extensive bleeding likely due to a lack of support for the newly forming vasculature.
Table 1.
Survival of DKO embryos
| Embryonic time-point | |||||
|---|---|---|---|---|---|
| E10 | E11 | E12 | E13 | E14 | |
|
Number of DKOs (%) |
5 (11.4%) |
5 (11.9%) |
2 (5.6%) |
1 (2.9%) |
1 (1.4%) |
|
Total # of embryos (# of litters) |
44 (5) |
42 (5) |
36 (4) |
35 (4) |
71 (9) |
|
Expected number of DKOs Mendelian ratio (%) |
3 (6.8%) |
3 (7.1%) |
3 (8.3%) |
3 (8.6%) |
6 (8.5%) |
Parental crosses were either (i) Mcl-1+/flox:Bcl-xflox/flox x Nestin+/Cre: Mcl-1+/flox Bcl-x+/flox to provide an expected 6.25% DKO embryos or (ii) Mcl-1+/flox:Bcl-x+/flox x Nestin+/Cre: Mcl-1+/flox Bcl-x+/flox to provide an expected 3.125% DKO embryos. Mendelian ratios were calculated by dividing the expected number of DKO embryos versus total embryos. + = wild type allele, flox = floxed allele, Cre = Cre allele
Fig. 3.

Conditional deletion of both mcl-1 and bcl-x—a double conditional knockout (DKO) results in complete loss of the CNS by E14. a Photomicrographs of representative cresyl violet-stained sagittal sections of E14 CTL, MKO, BKO, and DKO embryos. Representative higher magnification photomicrographs of cresyl violet stained sections of (b) cortex and (c) spinal cords for each genotype. Scale bars = 2000 μm (a), 100 μm (b, c)
To compare brain and spinal cord histology, we performed cresyl violet staining on midline sagittal sections from E14 CTL, MKO, BKO, and DKO embryos. The thickness of the developing cortical plate appeared similar in CTL and BKO embryos, but appeared much thinner in the MKO consistent with the extensive apoptosis that occurs in the forebrain at this time [8] (Fig. 3b). In contrast, only a remnant of tissue was observed around the periphery of the brain cavity in the DKO embryo. A comparison of the sagittal spinal cord at the level of the heart revealed that both MKO and BKO spinal cords appeared thinner than CTL spinal cords (Fig. 3c). The DKO spinal cord had the most extensive loss of neural tissue compared to all other genotypes demonstrating that Mcl-1 and Bcl-xL are the two key anti-apoptotic proteins required for cell survival in the developing nervous system.
Manipulating gene dosage reveals overlapping roles for Mcl-1 and Bcl-xL in nervous system development
To determine whether Mcl-1 and Bcl-xL have overlapping anti-apoptotic roles in nervous system development, we examined the impact of gene dosage. We focussed on embryonic age E11, when apoptosis is observed in the MKO and BKO embryos in distinct dorsal and ventral regions of the spinal cord, respectively (Fig.1). Histology and AC3 immunohistochemistry was performed on sagittal sections of the spinal cord (Fig. 4), brainstem (Suppl. Figure 2) and forebrain (Suppl. Figure 3). Few to no apoptotic cells were observed in CTL or double heterozygous (Mcl-1 Het:Bcl-x Het) embryos in all three regions investigated. In contrast, apoptosis was extensive throughout the developing nervous system in the MKO embryo. Cell death extended throughout the dorsal spinal cord and the ventricular zone of the brainstem and forebrain. Loss of a single bcl-x allele on the MKO background (MKO:Bcl-x Het) resulted in an increase in apoptosis within each region impacted in the MKO embryo alone (Fig. 4 and Suppl. Figures 2, 3). In the BKO embryo, apoptosis was observed in the ventral spinal cord and apical region of the brainstem, but not in the dorsal forebrain (Fig. 4 and Suppl. Figures 1 and 3). Interestingly, loss of a single mcl-1 allele on the BKO background (Mcl-1 Het:BKO) also caused an increase in apoptosis, yet within the region impacted in the MKO embryo (Fig. 4). An examination of apoptosis in the brainstem of the Mcl-1 Het:BKO embryo similarly showed an increase in apoptosis within the ventricular zone (Suppl. Figure 2b). Within the developing forebrain of the Mcl-1 Het:BKO embryo, a few apoptotic cells were observed within the pallium and subpallium (Suppl. Figure 3b arrows). In the DKO embryo, apoptotic cell death was extensive throughout the entire developing nervous system (Fig. 4 and Suppl. Figures 2 and 3).
Fig. 4.

Gene dosage affects the onset of apoptosis in the spinal cord. a Photomicrograph of a cresyl violet stained sagittal section through an E11 mouse embryo. Black box indicates region of spinal cord examined. b Representative photomicrographs of sagittal spinal cord sections from E11 CTL, Mcl-1 HET:Bcl-x HET, MKO, MKO:Bcl-x HET, BKO, Mcl-1 HET:BKO, and DKO animals (n = a minimum of 3/genotype). Apoptotic cells are labeled with active caspase-3 (AC3) immunohistochemistry, while cresyl violet staining shows tissue histology. Larger boxed area in b indicates region of higher magnification photomicrographs for active caspase-3 stained sections, while smaller inset indicates region shown in cresyl violet photomicrographs. R rostral, C caudal, D dorsal, V ventral
We next examined whether proliferating or differentiating cell populations were affected with the increased cell death observed in the MKO:Bcl-x Het and Mcl-1 Het:BKO embryos. Antibodies to Ki67 were used to label proliferating cells and antibodies to βIII-tubulin (Tuj1) were used to label immature neurons, the differentiating population (Fig. 5a, b). During apoptosis, once nuclear condensation has occurred, cell-specific markers are often already degraded making it difficult to identify the cell type. Apoptotic cell counts were therefore performed in distinct locations in Ki67+ proliferating cells at the center of the spinal cord and in Tuj1+ differentiating neurons in the ventrolateral horns (Fig. 5a, b). In MKO embryos, apoptosis was primarily observed within the Ki67+ proliferating population (26.3 ± 7.2%, n = 5) with fewer apoptotic cells (4.0 ± 3.0%, n = 5) in the Tuj1+ differentiating population. In contrast to MKO embryos, a two-fold increase in apoptosis was observed in MKO:Bcl-x Het embryos within both the Ki67+ population (54.1 ± 17.3%, n = 4) and the Tuj1+ population (8.1 ± 6.3%, n = 4) (Fig. 5c, d). In BKO embryos, apoptosis is just beginning at E11 and is confined to the Tuj1+ cells in the ventrolateral horns (4.7 ± 0.4%, n = 3). In comparison to BKO embryos, a two-fold increase in apoptosis was observed in the Mcl-1 Het:BKO embryo within the differentiating Tuj1+ population of the ventral horn (11 ± 3.8%, n = 3) but no significant increase in apoptosis was observed within the proliferating Ki67+ population.
Fig. 5.

An anti-apoptotic role for Bcl-xL in neural precursor cell populations. a Representative coronal spinal cord sections from E11 Mcl-1 HET:Bcl-x HET, MKO, MKO:Bcl-x HET, BKO, and Mcl-1 HET:BKO embryos immunostained with antibodies to AC3 and Ki67. Higher magnification of boxed areas in a showing double labeled cells. b Representative coronal spinal cord sections from E11 Mcl-1 HET:Bcl-x HET (TRH triple heterozygous), MKO, MKO:Bcl-x HET, BKO, and Mcl-1 HET:BKO embryos immunostained with antibodies to AC3 and βIII-tubulin (Tuj1). Higher magnification of boxed areas in b showing double-labeled cells. c Counts of apoptotic cells within the proliferating population of the spinal cord as shown in a. There was no significant differences in the number of apoptotic cells between the TRH, BKO, and BKO:Mcl-1 HET. In contrast, the MKO:Bcl-x HET had significantly more apoptotic cells than the MKO (p < 0.01) and the TRH, BKO, and BKO:Mcl-1 HET (p < 0.001) and the MKO had significantly more apoptotic cells than the BKO:Mcl-1 HET (p < 0.05) and the TRH, BKO, MKO:Bcl-x HET (p < 0.01). d Counts of apoptotic cells within the ventral lateral horns of the spinal cord as shown in b. Overall fewer apoptotic cells were observed in the ventral horns versus in the proliferating NPCs in the center of the spinal cord. A significant increase in the number of apoptotic cells was observed in the BKO:Mcl-1 HET versus the TRH (p < 0.05). Individual means ± SD are shown for each embryo. *p < 0.05, **p < 0.01, ***p < 0.001
To determine whether a change in protein expression could account for the compensatory effects we observed in the double mutant embryos, we quantified Mcl-1 and Bcl-xL protein expression in the single conditional knockouts. Spinal cord, brainstem and forebrain tissue was collected from CTL, Mcl-1 Het, and MKO embryos and Western analysis performed (Suppl. Figure 4a–f). Mcl-1 protein is efficiently excised in the MKO nervous system. The remnant of Mcl-1 protein observed in the MKO spinal cord can be attributed to contamination by non-neural tissue during dissection. No compensatory increase in Bcl-xL protein expression was observed (Suppl. Figure 4a–f). Similarly, in CTL, Bcl-x Het and BKO embryos, Mcl-1 and Bcl-xL protein levels were assessed (Suppl. Figure 4g–l). Bcl-xL protein levels were reduced to 25% of control in BKO embryos. Mcl-1 protein expression was not significantly different across the three genotypes, indicating there is no compensatory increase in Mcl-1 protein in the BKO (Suppl. Figure 4g–l). The overlapping and compensatory role of the anti-apoptotic proteins therefore is not due to an increase in protein expression in the mutant embryos.
Bax is a common pro-apoptotic target for Mcl-1 and Bcl-xL during nervous system development
We next questioned whether this compensation by Mcl-1 and Bcl-xL could be explained by them targeting a common pro-apoptotic protein. The two key pro-apoptotic Bcl-2 proteins, Bax and Bak act at the mitochondria to initiate apoptosis [26]. Previous studies have demonstrated that germline deletion of Bak does not impact nervous system development [27]. In contrast, germline deletion of Bax (Bax Null) affects the development of several neuronal populations including motor neurons resulting in supernumerary cells [28]. Furthermore, Bax protein expression has been shown to increase in differentiating neurons [20]. Although both Bax and Bak are expressed in the developing spinal cord and appear to increase with the onset of neurogenesis (Suppl. Figure 1), we focussed on Bax because of its known role in nervous system development and its affinity for both Mcl-1 and Bcl-xL [29]. If Bax is a common target for Mcl-1 and Bcl-xL, we would expect that deletion of Bax would rescue apoptotic cell death in the MKO and BKO embryos, respectively.
MKO mice were bred with the Bax null mouse line and embryos were collected at E11. Immunohistochemistry for AC3 was performed on horizontal spinal cord sections through the lumbar spinal cord of double heterozygotes (Mcl-1 Het:Bax Het), MKO, and MKO:Bax Null embryos. Apoptotic cells were counted in the dorsal region of the lumbar spinal and compared across genotypes. Cell counts revealed that Bax deletion significantly rescued the majority of apoptosis in the MKO embryo (Table 2 and Fig. 6a, b). A similar result was observed when BKO mice were bred with the Bax Null mice. Apoptotic cell death was examined at E13 when apoptosis is observed throughout the lumbar ventral spinal cord [25] (Fig. 6c, d). Cell counts revealed that Bax deletion rescued apoptosis in the BKO embryo (Table 3). Taken together, these results demonstrate that Bax is the main target for Mcl-1 and Bcl-xL in the developing nervous system.
Table 2.
Apoptotic cell counts in the dorsal spinal cords of E11 double heterozygous (Mcl-1 HET:Bax HET), MKO, and MKO:Bax Null Embryos
| Mcl-1 HET:Bax HET | MKO | MKO:Bax Null | |
|---|---|---|---|
| Apoptotic cells | 0.3 ± 0.4 | 78.4 ± 22.0 | 4.1 ± 2.2 |
| Total nuclei | 301.4 ± 27.2 | 286.0 ± 18.5 | 295.1 ± 6.7 |
| % Apoptotic cells | 0.06 ± 0.14 | 27.76 ± 9.55* | 1.38 ± 0.73 |
| # of Embryos | 5 | 4 | 3 |
Apoptotic cells were identified as immunopositive for active Caspase-3 with completely condensed nuclei. Cells were counted within a 125 µm × 125 µm box placed midway in the dorsal spinal cord. Means ± SD are indicated
*p < 0.0001
Fig. 6.

Deletion of Bax rescues cell death in the MKO and BKO embryo. a Representative horizontal sections through the E11 lumbar spinal cord of Mcl-1 HET:Bax HET, MKO, and MKO:Bax Null embryos immunostained with antibodies to AC3 showed that deletion of Bax rescues the majority of apoptotic cells in the MKO spinal cord. b Higher magnification of the area for apoptotic cell counts (AC3) and total nuclei counts (Hoechst) in each of the genotypes. c Representative coronal sections through the spinal cord of E13 Bcl-x HET:Bax Het, BKO, and BKO:Bax Null embryos. d Higher magnification of the area for apoptotic cell counts (AC3) and total nuclei counts (Hoechst) in each of the genotypes
Table 3.
Apoptotic cell counts in the ventral spinal cords of E13 double heterozygous (Bcl-x HET:Bax HET), BKO, and BKO:Bax Null Embryos
| Bcl-x HET:Bax HET | BKO | BKO:Bax Null | |
|---|---|---|---|
| Apoptotic cells | 0.0 ± 0.0 | 30.2 ± 8.1 | 0.0 ± 0.0 |
| Total nuclei | 151.5 ± 19.8 | 153.5 ± 8.7 | 145.5 ± 16.8 |
| % Apoptotic cells | 0.0 ± 0.0 | 19.6 ± 4.7* | 0.0 ± 0.0 |
| # of Embryos | 4 | 4 | 4 |
Apoptotic cells were identified as immunopositive for active Caspase-3 with completely condensed nuclei. Cells were counted within a 150 µm × 150 µm box placed in the ventrolateral horn of the spinal cord. Means ± SD are indicated
*p < 0.0001
Discussion
In this study, we examined the anti-apoptotic roles of Mcl-1 and Bcl-xL in mammalian developmental neurogenesis. We have three main findings (i) Mcl-1 and Bcl-xL are essential for cell survival during developmental neurogenesis; (ii) Bcl-xL’s anti-apoptotic role can partially compensate for Mcl-1 in differentiating NPCs; and (iii) Bax is a common pro-apoptotic target for both Mcl-1 and Bcl-xL. Overall, this study demonstrates that Mcl-1 and Bcl-xL are the crucial anti-apoptotic Bcl-2 members required for nervous system development.
In the mouse embryo, neurogenesis begins once neural tube formation is complete at E10 [30]. Previous studies have shown that both Mcl-1 and Bcl-xL are expressed in the developing nervous system at the onset of neurogenesis [8, 20]. In our conditional knockouts, apoptosis is first observed at E10 in the MKO and E11 in the BKO embryo coinciding with the onset of neurogenesis. Both the MKO and BKO mouse lines were generated by the Nestin-Cre transgenic mouse line with the Nestin promoter becoming active in primitive neuroepithelial cells at E7.5 [31]. Despite the loss of mcl-1, bcl-x, or both genes, the neural tube still formed. This suggests that Mcl-1 and Bcl-xL are not required for the survival of the primitive neuroepithelial cells that form the neural tube. Similarly, loss of the pro-apoptotic Bcl-2 proteins in the Bax null and the Bax:Bak double knockout mouse does not affect the survival of the primitive neuroepithelial cells because although the brains are larger and contain more neurons, the overall brain morphology is normal [27, 32, 33]. In contrast, exencephaly and significant morphological changes are observed in the developing brains of knockout mice for the pro-apoptotic genes, Apaf-1, Caspase-3, and Caspase-9 and for the pro-autophagy gene, Ambra1, demonstrating that each of these genes affect the survival of the primitive neuroepithelial cells [32, 34–39]. The Bcl-2 family, therefore, is required for the survival of the definitive neural precursor population but not the primitive neuroepithelial cells that are the founder population of the developing nervous system [32]. Our findings further demonstrate that the role of the Bcl-2 family begins at the onset of neurogenesis as NPCs exit the cell cycle and initiate neural differentiation.
The pattern of apoptosis in the individual conditional knockouts, MKO and BKO, suggests that Mcl-1 and Bcl-xL have distinct, sequential roles in developmental neurogenesis [8, 21, 25]. However, our gene dosage experiments revealed that loss of a single bcl-x allele in the MKO:Bcl-x HET resulted in a two-fold increase in apoptosis of the Ki67+ proliferating population. As apoptosis is not observed in the proliferating population in the BKO embryo, this shows that Bcl-xL can partially compensate for the role of Mcl-1 in NPCs. In the Tuj1+ differentiating population, both Mcl-1 and Bcl-xL promote cell survival as loss of either mcl-1, bcl-x or a combination of both increases apoptosis in this population. This is consistent with previous studies examining the individual gene knockouts [8, 21, 25]. NPCs, therefore, are dependent on Mcl-1 at the start of neurogenesis and become dependent on both genes as neurogenesis proceeds (Fig. 7). This dependence on Mcl-1 and Bcl-xL to prevent apoptosis, however, is short-lived. Conditional deletion of either gene in mature neurons does not result in immediate apoptotic cell death, rather neurons die by autophagy with the loss of Mcl-1, or survive with a heightened sensitivity to apoptotic stimuli in the loss of Bcl-xL [15, 24]. Neurogenesis, therefore, highlights a sensitive period in which NPCs exit the cell cycle to begin differentiation and depend on the expression of anti-apoptotic proteins Mcl-1 and Bcl-xL to prevent activation of the apoptotic pathway.
Fig. 7.
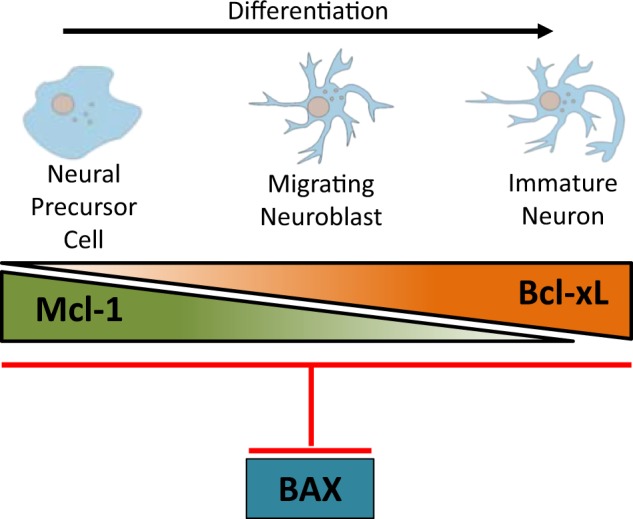
The distinct and overlapping roles of anti-apoptotic Mcl-1 and Bcl-xL in mammalian neurogenesis. Mcl-1 is required at the earlier stages of neurogenesis specifically for the survival of NPCs as they exit the cell cycle and start to differentiate. Our gene dosage data shows that Bcl-xL partially compensates for Mcl-1 in NPCs. As cells become immature neurons, Bcl-xL takes on a more prominent role, becoming the main anti-apoptotic Bcl-2 family member
The finding that Bcl-xL can functionally overlap with Mcl-1 in NPCs suggested that both anti-apoptotic proteins may inhibit a common pro-apoptotic protein. Although it has previously been shown that Bax deletion rescues apoptosis in the nervous system of bcl-x null mice [40], it was not clear whether Mcl-1 also inhibited Bax in NPCs. Our rescue experiments confirmed that both Mcl-1 and Bcl-xL promote survival by blocking Bax activation in NPCs and immature neurons. The transition from Mcl-1 dependency to Bcl-xL dependency therefore is not due to a difference in Bax inhibition. Differences in the overall expression of Mcl-1 and Bcl-xL within cells likely do not affect the transition as Mcl-1 mRNA is highly expressed in both NPCs and post mitotic neurons [8]. Bcl-xL protein is expressed at low levels in NPCs and at much higher levels in immature neurons [20]. However, a change in BH3-only protein expression may account for the transition from Mcl-1 to Bcl-xL dependency. Mcl-1 and Bcl-xL have both distinct and common BH3-only binding partners and their binding affinities for the common BH3-only proteins also differ (reviewed in ref. [2]). Knockout mice for the individual BH3-only proteins have failed to show a nervous system defect (Bad [41], Bid [42], Bim [43], Noxa [44, 45], and PUMA [45]). Deletion of Bim in the germline bcl-x null mouse rescued apoptosis of hematopoietic cells but not neurons, and also failed to rescue the embryonic lethality [46]. In contrast, deletion of both Bim and PUMA failed to rescue neutrophil apoptosis in a myeloid cell conditional Mcl-1 knockout demonstrating cell-specific differences in BH3-only functions [47]. Combinations of multiple BH3-only knockouts have resulted in nervous system defects. For instance, the triple knockout of Bid, Bim and PUMA results in a similar phenotype as the Bax:Bak double knockout with developmental defects including supernumerary cells throughout the developing nervous system [48]. The individual deletion of BBC3/PUMA alone reduced developmental cell death in the retina comparable to that observed in the Bax-/- embryo, but a similar effect on the central nervous system has not been observed [49]. However, in the adult mouse brain individual deletion of either Bim or PUMA prevents apoptosis in the hippocampus resulting in the increased survival of newly generated neurons [50]. Further study, therefore, is necessary to determine whether the change from Mcl-1 dependency to Bcl-xL dependency can be attributed to a change in the expression profile of the BH3-only proteins during developmental neurogenesis.
Tissue-specific knockouts of Mcl-1 and Bcl-xL have revealed similarities and differences with their roles in the developing nervous system. A role for the Bcl-2 family in cell survival is first observed at E10.5 in the developing nervous system with the onset of neurogenesis and in the hematopoietic system with the appearance of the definitive hematopoietic stem cells [51]. Cell-specific knockouts have shown that Mcl-1 is required for the survival of most precursor cells within the hematopoietic system including hematopoietic stem cells, the common lymphocyte progenitor and common myeloid cell progenitor [4], B and T lymphocytes [5], and plasma cells [52]. Tamoxifen-inducible deletion of Mcl-1 revealed that it is required for B-cell survival throughout their lifespan and a reduction in Mcl-1 expression greatly enhances B-cell sensitivity to apoptotic stimuli [53]. Similarly, we have previously demonstrated that Mcl-1 is required for the survival of NPC in development and in the adult brain [8, 9]. In most hematopoietic precursor populations, Mcl-1 is the primary anti-apoptotic protein and Bcl-xL functions in a secondary role. However, double knockout of Mcl-1 and Bcl-xL revealed that Bcl-xL was required for the survival of megakaryocytes and loss of Mcl-1 sensitized cells to apoptotic stimuli but did not cause their death [54]. Interestingly, in the developing liver, Mcl-1, and Bcl-xL appear to have a cooperative role in maintaining the survival of embryonic and adult hepatocytes. The loss of a single allele of each gene results in spontaneous hepatocyte apoptosis [55]. These studies demonstrate that Mcl-1 and Bcl-xL have distinct roles depending on the cell type.
In conclusion, we show that conditional deletion of both mcl-1 and bcl-x in our DKO results in death of the entire central nervous system by E14 demonstrating that Mcl-1 and Bcl-xL are required for nervous system development. The gene dosage experiments uncovered a novel role for Bcl-xL showing that Bcl-xL has an anti-apoptotic supportive function in NPCs during neurogenesis. Finally, with rescue experiments, we found that both Mcl-1 and Bcl-xL promote cell survival by blocking Bax activation in neurogenesis. Altogether this study demonstrates the distinct and overlapping roles of Mcl-1 and Bcl-xL in developmental neurogenesis.
Materials and methods
Mice
Mice were housed on a 12 h light/dark cycle with access to food and water ad libitum. All experiments were approved by Memorial University’s Animal Care Ethics committee, adhering to the Guidelines of the Canadian Council on Animal Care.
Nestin cre (cre+/−) mice were generated in the laboratory of Dr. R. Slack [8], mcl-1 floxed mice were generated in the laboratory of Dr. S. Korsmeyer [4], and bcl-x floxed mice were generated in the laboratory of Dr. L. Hennighausen [56]. The bcl-x floxed mice were transferred from a FVBN background to a C57Bl/6 background by backcrossing to C57Bl/6 for six generations. Bax null mice were obtained from Jackson Labs [57]. All strains were maintained on a C57BL/6 background. Tail samples were collected for genotyping by PCR with previously published primers for cre recombinase, mcl-1 [8, 9], bcl-x [25, 56], and bax [57].
Nestin cre transgenic mice were bred with floxed mice to generate conditional knockout mice (KO) of mcl-1 (MKO), bcl-x (BKO), as well as mcl-1 and bcl-x double knockouts (DKO). Knockout mice were compared to littermate control (CTL) embryos that were wild type or mcl-1+/f or bcl-x+/f (+ = wild type allele, f = floxed allele) and to littermate heterozyous embryos, such as nes+/cre:mcl-1+/f (Mcl-1 Het) and nes+/cre:bcl-x+/f (Bcl-x Het) and double heterozygous embryos nes+/cre:mcl-1+/f:bcl-x+/f (Mcl-1 Het:Bcl-x Het). The bax null mouse line was bred with the MKO and BKO mouse lines to generate the MKO:Bax null and BKO:Bax null lines, respectively. Breeding pairs were placed in the same cage until the formation of a vaginal plug, up to a maximum period of 3 days. Female mice were checked twice daily for a vaginal plug, which at the point of discovery was designated as embryonic day 0.5 (E0.5).
Tissue fixation and immunohistochemistry
At the time of sacrifice, pregnant dams were euthanized with an intraperitoneal injection of Euthanyl (250 mg/mL sodium pentobarbital-Vètoquinol, IEUS001, Lavaltrie, Quebec, Canada) followed by cervical dislocation. Embryos were fixed overnight in 4% paraformaldehyde in 1× PBS (pH 7.4). Following fixation, embryos were cryoprotected in 30% sucrose (w/v) in 1× PBS and embedded and frozen in OCT compound.
All tissues used for immunohistochemical staining were sectioned on a Microm cryostat, and 14 μm-thick sections were collected and processed according to previously published protocols [25]. Primary antibodies used include: AC3 (BD BioSciences, Cat# 559565, RRID:AB_397274), β-tubulin isoform III (Millipore, Cat# MAB1637, RRID:AB_2210524), Bromodeoxyuridine (BrdU) (BD BioSciences, Cat# 347580, RRID:AB_400326), and Ki67 (BD Biosciences, Cat# 550609, RRID:AB_393778). Primary antibodies were detected with fluorescent secondary antibodies anti-rabbit IgG tagged with Alexa Fluor 488 or 594 (Thermo Fisher Scientific, Cat# A21206, RRID:AB_2535792 and Cat# A21207, RRID:AB_141637) and anti-mouse IgG tagged with Alexa Fluor 488 or 594 (Thermo Fisher Scientific, Cat# A21202, RRID:AB_2535788 and Cat# A21203, RRID:AB_2535789). Hoechst staining was performed to label cell nuclei (Sigma Chemical Company, H33258 Cat# B1155). For BrdU immunohistochemical staining, slides were pretreated in 2 N HCl at 37 °C for 30 min followed by 1 M NaB4O7 pH 8.0 for 10 min [58].
Western analysis and quantification of protein blots
Spinal cord, brainstem, and forebrain tissue were dissected from E12 embryos and digested in lysis buffer. Protein extraction and Western blotting were completed as previously described [59]. Primary antibodies Mcl-1 (Rockland, 600-401-394, RRID:AB_2266446), Bcl-xL (Cell Signaling Technology, Cat# 2764, RRID:AB_2228008), and β-actin (Sigma-Aldrich, Cat# A5316, RRID:AB_476743) were applied to blots overnight, followed by the application of a horse-radish peroxidase-tagged secondary antibody (Bio-Rad, 1706515/1706516, Mississauga, Ontario, Canada). Blots were developed using a chemiluminescence reaction kit (Perkin Elmer Labs Inc., 02118-2512, Waltham, MA, USA) according to the manufacturer’s instructions. Densitometry analysis of Mcl-1, Bcl-xL, and β-actin bands were completed using ImageJ according to the protocol outlined in the user guide (http://imagej.nih.gov/ij/). Experiments were performed in triplicate and relative protein expression was compared by one-way ANOVA followed by Tukey’s post hoc analysis with significance at p < 0.05.
qPCR
RNA was extracted from dissected spinal cord tissue from E10 and E12 wild type embryos using TRIzol Reagent (Life Technologies, 15596-026, Burlington, Ontario, Canada) according to the manufacturer’s instructions. E10 samples were pooled from three embryos, while E12 samples were from individual embryos. RNA samples were DNase treated followed by cDNA synthesis using the RevertAid H Minus first strand cDNA Synthesis kit (Thermo Scientific, K1631, Waltham, Massachusetts, USA). Quantitative PCR (q-PCR) was performed using StepOnePlus Real-Time PCR System (Life Technologies, Burlington, Ontario, Canada). cDNA was mixed with Power SYBR Green PCR master mix (Life Technologies, 4367659, Burlington, Ontario, Canada) with primers for bax (NM_007527.3, forward: 5′-TGAAGACAGGGGCCTTTTTG-3′, reverse: 5′-AATTCGCCGGAGACACTCG-3′, PrimerBank 6680770a1); bak (NM_007523.2, forward: 5′-CAACCCCGAGATGGACAACTT-3′, reverse: 5′-CGTAGCGCCGGTTAATATCAT-3′, PrimerBank 15553445a1); bcl-x (NM_009743.4, Qiagen, QT00149254); gapdh (NM_008084, Qiagen, QT01658692) and mcl-1 (Qiagen, QT00107436). Quantification of genes for each sample was done in triplicate. GAPDH was amplified as an internal control, negative control wells with the absence of cDNA template, as well as RNA only wells were used to verify lack of genomic contamination.
Microscopy
All immunostained slides were viewed using a Zeiss AxioImager Z.1 microscope (Carl Zeiss Microscopy, Jenna, Germany) and fluorescence produced using a Colibri LED light source (Carl Zeiss Microscopy, Jenna, Germany). Photomicrographs were taken using a Zeiss AxioCam MRm camera (Carl Zeiss Microscopy, Jenna, Germany) using Zeiss AxioVision v4.8 software. Low magnification images were taken using a Zeiss Stemi-2000 dissecting microscope (Carl Zeiss Microscopy, Jenna, Germany) and Zeiss AxioCam MRm camera (Carl Zeiss Microscopy, Jenna, Germany).
Cell counts and statistics
Apoptotic cells were counted on horizontal spinal cord sections from E11 and E13 embryos. Apoptotic cells were identified as being positive for AC3 and having completely condensed nuclei as determined by Hoechst staining as previously demonstrated [58]. Apoptotic cells were counted within a 100 μm × 100 μm boxed area within the Tuj1+ cells of the ventrolateral horn and at the midway point of the spinal cord in the Ki67+ cells near the midline in E11 Ctl, MKO, MKO:Bclx HET, BKO, and BKO:Mcl-1 HET embryos. Two boxed areas were counted within the Tuj1+ cells and Ki67+ cells per embryo and displayed as percent apoptotic cells from the total number of Hoechst+ nuclei. The means were compared with one-way ANOVA followed by Tukey’s post hoc analysis with significant differences evaluated at p < 0.05. For the Bax rescue experiments in the MKO embryo, apoptotic cells were counted within a boxed area of 125 μm × 125 μm placed midway in the dorsal spinal cord (Fig. 6A). Bax rescue experiments in the BKO embryo were performed at E13 and apoptotic cells were counted within a boxed area of 150 μm × 150 μm placed midway in the ventral spinal cord (Fig. 6C). Counts were performed on three spinal cord sections per embryo with a 140 μm interval between sections. Mean counts were compared across genotypes with one-way ANOVA followed by Tukey’s post hoc analysis with significant differences evaluated at p < 0.05.
Electronic supplementary material
Acknowledgements
This work was supported by grants from the Canadian Institute of Health Research (CIHR), the Research and Development Corporation of Newfoundland and Labrador (RDC-NL) and a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) to J.L.V. L.C.F. was supported by an NSERC studentship, A.K. was supported by an NSERC-USRA. We thank Jieying Xiong, S.M. Mahmudul Hasan, and Yegappan Suppiah for technical assistance.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41418-018-0225-1) contains supplementary material, which is available to authorized users.
References
- 1.Buss RR, Sun W, Oppenheim RW. Adaptive roles of programmed cell death during nervous system development. Annu Rev Neurosci. 2006;29:1–35. doi: 10.1146/annurev.neuro.29.051605.112800. [DOI] [PubMed] [Google Scholar]
- 2.Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80. doi: 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA. 1993;90:3516–20. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307:1101–4. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 5.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–6. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 6.Sitailo LA, Jerome-Morais A, Denning MF. Mcl-1 functions as major epidermal survival protein required for proper keratinocyte differentiation. J Invest Dermatol. 2009;129:1351–60. doi: 10.1038/jid.2008.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vick B, Weber A, Urbanik T, Maass T, Teufel A, Krammer PH, et al. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology. 2009;49:627–36. doi: 10.1002/hep.22664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arbour N, Vanderluit JL, Le Grand JN, Jahani-Asl A, Ruzhynsky VA, Cheung EC, et al. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J Neurosci. 2008;28:6068–78. doi: 10.1523/JNEUROSCI.4940-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malone CD, Hasan SM, Roome RB, Xiong J, Furlong M, Opferman JT, et al. Mcl-1 regulates the survival of adult neural precursor cells. Mol Cell Neurosci. 2012;49:439–47. doi: 10.1016/j.mcn.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Michaelidis TM, Sendtner M, Cooper JD, Airaksinen MS, Holtmann B, Meyer M, et al. Inactivation of bcl-2 results in progressive degeneration of motoneurons, sympathetic and sensory neurons during early postnatal development. Neuron. 1996;17:75–89. doi: 10.1016/S0896-6273(00)80282-2. [DOI] [PubMed] [Google Scholar]
- 11.Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N, et al. Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc Natl Acad Sci USA. 1998;95:12424–31. doi: 10.1073/pnas.95.21.12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ross AJ, Waymire KG, Moss JE, Parlow AF, Skinner MK, Russell LD, et al. Testicular degeneration in Bclw-deficient mice. Nat Genet. 1998;18:251–6. doi: 10.1038/ng0398-251. [DOI] [PubMed] [Google Scholar]
- 13.Schenk RL, Tuzlak S, Carrington EM, Zhan Y, Heinzel S, Teh CE, et al. Characterisation of mice lacking all functional isoforms of the pro-survival BCL-2 family member A1 reveals minor defects in the haematopoietic compartment. Cell Death Differ. 2017;24:534–45. doi: 10.1038/cdd.2016.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tuzlak S, Schenk RL, Vasanthakumar A, Preston SP, Haschka MD, Zotos D, et al. The BCL-2 pro-survival protein A1 is dispensable for T cell homeostasis on viral infection. Cell Death Differ. 2017;24:523–33. doi: 10.1038/cdd.2016.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS, et al. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011;30:395–407. doi: 10.1038/emboj.2010.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-N. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez-Garcia M, Perez-Ballestero R, Ding L, Duan L, Boise LH, Thompson CB, et al. bcl-XL is the major bcl-x mRNA form expressed during murine development and its product localizes to mitochondria. Development. 1994;120:3033–42. doi: 10.1242/dev.120.10.3033. [DOI] [PubMed] [Google Scholar]
- 18.Yang XF, Ye Q, Press B, Han RZ, Bassing CH, Sleckman BP, et al. Analysis of the complex genomic structure of Bcl-x and its relationship to Bcl-x(gamma) expression after CD28-dependent costimulation. Mol Immunol. 2002;39:45–55. doi: 10.1016/S0161-5890(02)00049-4. [DOI] [PubMed] [Google Scholar]
- 19.Frankowski H, Missotten M, Fernandez PA, Martinou I, Michel P, Sadoul R, et al. Function and expression of the Bcl-x gene in the developing and adult nervous system. Neuroreport. 1995;6:1917–21. doi: 10.1097/00001756-199510020-00023. [DOI] [PubMed] [Google Scholar]
- 20.Krajewska M, Mai JK, Zapata JM, Ashwell KW, Schendel SL, Reed JC, et al. Dynamics of expression of apoptosis-regulatory proteins Bid, Bcl-2, Bcl-X, Bax and Bak during development of murine nervous system. Cell Death Differ. 2002;9:145–57. doi: 10.1038/sj.cdd.4400934. [DOI] [PubMed] [Google Scholar]
- 21.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, et al. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–10. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura A, Swahari V, Plestant C, Smith I, McCoy E, Smith S, et al. Bcl-xL is essential for the survival and function of differentiated neurons in the cortex that control complex behaviors. J Neurosci. 2016;36:5448–61. doi: 10.1523/JNEUROSCI.4247-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savitt JM, Jang SS, Mu W, Dawson VL, Dawson TM. Bcl-x is required for proper development of the mouse substantia nigra. J Neurosci. 2005;25:6721–8. doi: 10.1523/JNEUROSCI.0760-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harder JM, Ding Q, Fernandes KA, Cherry JD, Gan L, Libby RT. BCL2L1 (BCL-X) promotes survival of adult and developing retinal ganglion cells. Mol Cell Neurosci. 2012;51:53–59. doi: 10.1016/j.mcn.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fogarty LC, Song B, Suppiah Y, Hasan SM, Martin HC, Hogan SE, et al. Bcl-xL dependency coincides with the onset of neurogenesis in the developing mammalian spinal cord. Mol Cell Neurosci. 2016;77:34–46. doi: 10.1016/j.mcn.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 26.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 27.Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–99. doi: 10.1016/S1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–11. doi: 10.1016/S0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 29.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 30.Bielen H, Pal S, Tole S, Houart C. Temporal variations in early developmental decisions: an engine of forebrain evolution. Curr Opin Neurobiol. 2017;42:152–9. doi: 10.1016/j.conb.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–95. doi: 10.1016/0092-8674(90)90662-X. [DOI] [PubMed] [Google Scholar]
- 32.Roth KA, Kuan C, Haydar TF, D’Sa-Eipper C, Shindler KS, Zheng TS, et al. Epistatic and independent functions of caspase-3 and Bcl-X(L) in developmental programmed cell death. Proc Natl Acad Sci USA. 2000;97:466–71. doi: 10.1073/pnas.97.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci. 1998;18:1428–39. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–37. doi: 10.1016/S0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- 35.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–5. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 36.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–52. doi: 10.1016/S0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 37.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–37. doi: 10.1016/S0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 38.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, et al. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–72. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–50. doi: 10.1016/S0092-8674(00)81733-X. [DOI] [PubMed] [Google Scholar]
- 40.Shindler KS, Latham CB, Roth KA. Bax deficiency prevents the increased cell death of immature neurons in bcl-x-deficient mice. J Neurosci. 1997;17:3112–9. doi: 10.1523/JNEUROSCI.17-09-03112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, et al. Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100:9324–9. doi: 10.1073/pnas.1533446100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leonard JR, D’Sa C, Cahn BR, Korsmeyer SJ, Roth KA. Bid regulation of neuronal apoptosis. Brain Res Dev Brain Res. 2001;128:187–90. doi: 10.1016/S0165-3806(01)00174-2. [DOI] [PubMed] [Google Scholar]
- 43.Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–8. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 44.Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, et al. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003;17:2233–8. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–8. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 46.Akhtar RS, Klocke BJ, Strasser A, Roth KA. Loss of BH3-only protein Bim inhibits apoptosis of hemopoietic cells in the fetal liver and male germ cells but not neuronal cells in bcl-x-deficient mice. J Histochem Cytochem. 2008;56:921–7. doi: 10.1369/jhc.2008.951749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steimer DA, Boyd K, Takeuchi O, Fisher JK, Zambetti GP, Opferman JT. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood. 2009;113:2805–15. doi: 10.1182/blood-2008-05-159145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–3. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harder JM, Libby RT. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol Neurodegener. 2011;6:50. doi: 10.1186/1750-1326-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bunk EC, Konig HG, Bernas T, Engel T, Henshall DC, Kirby BP, et al. BH3-only proteins BIM and PUMA in the regulation of survival and neuronal differentiation of newly generated cells in the adult mouse hippocampus. Cell Death Dis. 2010;1:e15. doi: 10.1038/cddis.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muller AM, Medvinsky A, Strouboulis J, Grosveld F, Dzierzak E. Development of hematopoietic stem cell activity in the mouse embryo. Immunity. 1994;1:291–301. doi: 10.1016/1074-7613(94)90081-7. [DOI] [PubMed] [Google Scholar]
- 52.Peperzak V, Vikstrom I, Walker J, Glaser SP, LePage M, Coquery CM, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol. 2013;14:290–7. doi: 10.1038/ni.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vikstrom IB, Slomp A, Carrington EM, Moesbergen LM, Chang C, Kelly GL, et al. MCL-1 is required throughout B-cell development and its loss sensitizes specific B-cell subsets to inhibition of BCL-2 or BCL-XL. Cell Death Dis. 2016;7:e2345. doi: 10.1038/cddis.2016.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Debrincat MA, Josefsson EC, James C, Henley KJ, Ellis S, Lebois M, et al. Mcl-1 and Bcl-x(L) coordinately regulate megakaryocyte survival. Blood. 2012;119:5850–8. doi: 10.1182/blood-2011-12-398834. [DOI] [PubMed] [Google Scholar]
- 55.Hikita H, Takehara T, Shimizu S, Kodama T, Li W, Miyagi T, et al. Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology. 2009;50:1217–26. doi: 10.1002/hep.23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rucker EB, 3rd, Dierisseau P, Wagner KU, Garrett L, Wynshaw-Boris A, Flaws JA, et al. Bcl-x and Bax regulate mouse primordial germ cell survival and apoptosis during embryogenesis. Mol Endocrinol. 2000;14:1038–52. doi: 10.1210/mend.14.7.0465. [DOI] [PubMed] [Google Scholar]
- 57.Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 58.Hasan SM, Sheen AD, Power AM, Langevin LM, Xiong J, Furlong M, et al. Mcl1 regulates the terminal mitosis of neural precursor cells in the mammalian brain through p27Kip1. Development. 2013;140:3118–27. doi: 10.1242/dev.090910. [DOI] [PubMed] [Google Scholar]
- 59.Vanderluit JL, Ferguson KL, Nikoletopoulou V, Parker M, Ruzhynsky V, Alexson T, et al. p107 regulates neural precursor cells in the mammalian brain. J Cell Biol. 2004;166:853–63. doi: 10.1083/jcb.200403156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.