

Abstract
Cancer cells are known to upregulate aerobic glycolysis to promote growth, proliferation, and survival. However, the role of mitochondrial respiration in tumorigenesis remains elusive. Here we report that inhibition of mitochondrial function by silencing TFAM, a key transcription factor essential for mitochondrial DNA (mtDNA) replication and the transcription of mtDNA-encoded genes, markedly reduced tumor-initiating potential of colon cancer cells. Knockdown of TFAM significantly decreased mitochondrial respiration in colon cancer cells; however, the cellular levels of ATP remained largely unchanged as a result of increased glycolysis. This metabolic alteration rendered cancer cells highly susceptible to glucose deprivation. Interestingly, upregulation of glycolysis was independent of hypoxia-inducible factor-1 (HIF1) as TFAM knockdown cells fail to stabilize HIF1α under hypoxic conditions. Moreover, knockdown of TFAM results in decreased expression of genes-associated cancer stem cells downstream of Wnt/β-catenin signaling. Metabolic analysis reveals that the level of α-ketoglutarate (α-KG) was significantly upregulated in TFAM knockout cells. Silencing of prolyl hydroxylase domain-containing protein 2 (PHD2), a α-KG-dependent dioxyenase, rescued the expression of target genes of both HIF1α and Wnt/β-catenin. Furthermore, intestinal-specific knockout of TFAM prevents tumor formation in Apc-mutant mouse models of colon cancer. Taken together, our findings identify a novel role of mitochondria-mediated retrograde signaling in regulating Wnt signaling and tumor initiation in colon cancer.
Subject terms: Cancer metabolism, Experimental models of disease
Introduction
Altered metabolism is considered a hallmark of cancer. Since the first report of aerobic glycolysis by Warburg, the most prevalent metabolic remodeling described in cancer cells is that cancer cells preferentially use glycolysis even in the presence of oxygen [1]. However, in contrast to the initial misconception that cancer cells adapt aerobic glycolysis as a result of impaired mitochondrial function, emerging evidence indicates that a majority of tumor cells maintain the capacity to produce energy through mitochondrial oxidative phosphorylation (OXPHOS), and by controlling the production of a large numbers of metabolites, mitochondria may serve as signaling organelles that regulate cell proliferation and tumorigenesis [2–4].
Mammalian mitochondria contain multiple copies of double-stranded, circular mitochondrial DNA (mtDNA). Human mtDNA encodes 37 genes, within them 13 encode protein components of the electron transport chain in complex I, III, IV, and V [5]. The subunits of mitochondrial complex II, succinate dehydrogenase (SDH), are encoded entirely by nuclear DNAs. Mitochondrial transcription factor A (TFAM), a nuclear-encoded factor present in mitochondria, is a key regulator of mtDNA replication and mtDNA transcription [6–8]. Germ line disruption of mouse TFAM leads to severe mtDNA depletion, OXPHOS deficiency, and embryonic lethality [9]. Knockout of TFAM in lung tissues largely abolished the formation of lung adenocarcinoma driven by KrasG12D in vivo [10]. In addition, mitochondrial OXPHOS has been implicated in the maintenance of cancer stem cells in different cancers, including glioblastoma and pancreatic cancer [11–14]. However, the molecular mechanism underlying mitochondria-dependent regulation of colon cancer remains largely unknown.
In this study, we investigated the functional importance of mitochondrial metabolism in regulating tumor growth and initiation in colon cancer. Silencing TFAM altered cellular metabolism and reduced the expression of genes associated with cancer stem cells. Importantly, intestinal-specific deletion of TFAM inhibited tumorigenesis in Apc-driven mouse models of colon cancer.
Results
Knockdown of TFAM decreases mitochondrial activity and increases glycolysis in colon cancer cells
To begin elucidating the role of mitochondria in colon cancer, we silenced TFAM in DLD1 and HCT116 cells using two different lentiviral shRNA targeting sequences (Fig. 1a), and showed that TFAM depletion resulted in a significant decrease in the expression of genes in mitochondrial complex I, III, IV, and V (mt-ND1, mt-CYB, mt-CO1, and mt-ATP6, respectively) and mtDNA copy number (Supplementary Fig. S1). Interestingly, SDHB, a nuclear encoded subunit of succinate dehydrogenase in complex II, was also downregulated in TFAM knockdown cells (Supplementary Fig. S1). Collectively, these data confirmed that silencing TFAM effectively decreases mitochondrial gene expression and mtDNA replication. No compensatory increase in mitochondrial complex II was observed in TFAM knockdown colon cancer cells.
Fig. 1.

Downregulation of TFAM inhibits mitochondrial respiration and promotes glycolysis in colon cancer cells. a The expression of TFAM was downregulated in stable TFAM knockdown DLD1 and HCT116 cells. Protein lysates from control and knockdown cells were analyzed by western blot. Two different shRNA targeting sequences were used for knocking down TFAM. b, c Control and TFAM knockdown DLD1 (b) and HCT116 (c) cells were subjected to the Mito stress test to obtain OCR measurements using the Seahorse XF96 Extracellular Flux analyzer. Results were quantified and the relative levels of OCR associated with basal and maximal respiration, ATP production and reserved capacity were calculated. Data represent the mean ± SD (n = 3, ¶p < 0.0001 and §p < 0.001). d Cellular ATP levels were measured in control and TFAM knockdown DLD1 and HCT116 cells. Data represent the mean ± SD (n = 4, #p < 0.05). e, f Control and TFAM knockdown DLD1 (e) and HCT116 (f) cells were subjected to the glycolysis stress test to obtain ECAR measurements using the Seahorse XF96 Extracellular Flux analyzer. Results were quantified and levels of glycolysis, glycolytic capacity and glycolytic reserve were calculated. Data represent the mean ± SD (n = 3, ¶p < 0.0001 and §p < 0.001). g, h Control and TFAM knockdown DLD1 (g) and HCT116 (h) cells were cultured in the control medium (high-glucose), glucose-free medium or treated with 2-DG for 48 h. The relative cell survival was determined by comparing to the control condition. Data represent the mean ± SD (n = 4, ¶p < 0.0001, §p < 0.001, and *p < 0.01 compared to sh-control within individual culture conditions)
We next determined the mitochondrial activity in control and TFAM knockdown cells using Seahorse Extracellular Flux Analyzer. Results from Mito Stress Tests showed that oxygen consumption rates (OCR) associated with both basal and maximal respiration, as well as respiration used for ATP production and reserved mitochondrial capacities, were significantly decreased in TFAM knockdown cells (Fig. 1b, c). However, the cellular ATP levels were either slightly decreased or remained largely unchanged in TFAM knockdown cells despite significant reductions in mitochondrial activity (Fig. 1d), suggesting that other metabolic pathways may compensate for the loss of mitochondrial respiration to meet the energetic need of cancer cells.
Since cancer cells are known to upregulate glycolysis, we investigated if glycolysis is activated in cells with impaired mitochondrial function. Results from Seahorse Glycolysis Stress Tests indicated that TFAM depletion significantly increased the extracellular acidification rate (ECAR) associated with glycolysis (Fig. 1e, f), whereas the ECAR levels associated with glycolytic capacity and glycolysis reserve were decreased (Fig. 1e, f). Thus, colon cancer cells are capable of exploiting cellular-reserved glycolytic capacities to compensate for decreased mitochondrial respiration. Consistent with increased ECAR derived from glycolysis, knockdown of TFAM increased both glucose consumption and lactate production (Supplementary Fig. S2). To determine the functional implication of increased glycolysis, we blocked glycolysis by either culturing cells under glucose starvation condition or by treating cells with 2-deoxy-D-glucose (2-DG), an inhibitor of glycolysis. Whereas the growth of TFAM knockdown cells was reduced by only ~10–20% in glucose-containing media, limiting glucose availability or blocking glycolysis preferentially decreased the survival of TFAM knockdown cells (Fig. 1g, h), thus suggesting that inhibition of mitochondrial respiration renders cancer cells hypersensitive to glucose deprivation.
Impaired mitochondrial respiration prevents stabilization of HIF1α under hypoxia
Since HIF1α has been implicated in promoting the glycolytic phenotype in solid tumors [15], we determined the expression of HIF1α under both normoxia and hypoxia conditions in TFAM knockdown cells. Unexpectedly, we found that while HIF1α protein was readily stabilized in control cells under hypoxia, silencing TFAM markedly limited the extent of HIF1α stabilization (Fig. 2a). The expression of HIF1α under normoxia was also lower in TFAM knockdown cells. Additionally, TFAM depletion inhibited basal and hypoxia-induced upregulation of HIF1α target genes associated with glycolysis, including LDHA, PDK1, HK2, and SLC2A1 (Glut1) (Fig. 2b). Taken together, our results suggest that HIF1α-mediated transcriptional activation of glycolysis genes is not involved in promoting the glycolytic phenotype in TFAM knockdown cells. Instead, cancer cells with impaired mitochondrial respiration can upregulate glycolysis by mobilizing reserved glycolytic capacities.
Fig. 2.
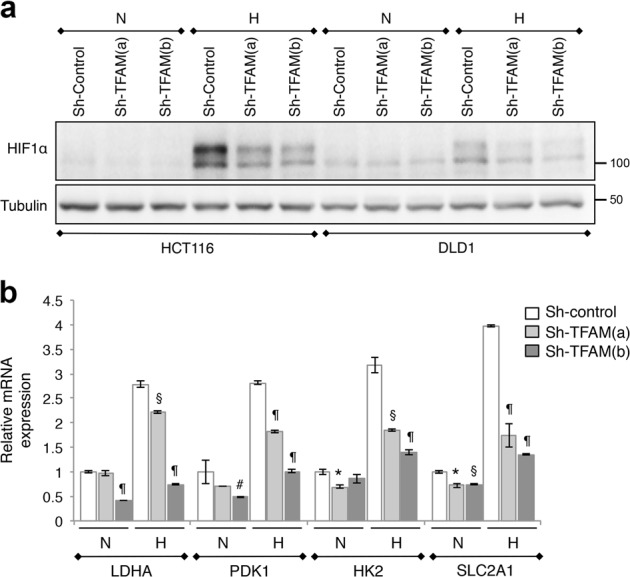
Mitochondrial respiration is required for HIF1α stabilization under hypoxia. a Control and TFAM knockdown HCT116 and DLD1 cells were cultured under normoxia (N, normoxia) or hypoxia (H, 1% O2) condition for 24 h. Cell lysates were analyzed for HIF1α expression using western blot. b The expression levels of HIF1α target genes, including LDHA, HK2, PDK1, and SLC2A1 (Glut1), were analyzed using qRT-PCR in control and TFAM knockdown DLD1 cells. Data represent the mean ± SD (n = 3, ¶p < 0.0001, §p < 0.001, *p < 0.01, and #p < 0.05 compared to sh-control within individual culture conditions)
Downregulation of mitochondrial respiration decreases Wnt signaling
Since the activation of Wnt/β-catenin signaling plays an important role in colon cancer, we next investigated the functional interaction between mitochondrial respiration and Wnt signaling. Despite having activating mutations in the Wnt pathway, colon cancer cells with the highest levels of Wnt pathway activation are shown to display cancer stem cell properties [16, 17]. Consequently, the expression levels of Wnt/β-catenin target genes have been used as readout for the stemness of colon cancer cells [18]. Interestingly, silencing TFAM in DLD1 and HCT116 cells resulted in a decrease in Wnt target genes that are also known to be associated with colon cancer stem cells, including CD44, LGR5, TCF7, MYC, and CD133 (Fig. 3a, b). Moreover, we silenced TFAM in 293 T cells and detected similar reduction in Wnt target gene expression together with decreased mitochondrial gene expression and mtDNA copy number (Supplementary Fig. S3). Importantly, knockdown of TFAM significantly attenuated Wnt-simulated TOP-Flash reporter activity in 293 T cells (Supplementary Fig. S3), suggesting that mitochondrial respiration regulates the transcriptional activation of Wnt/β-catenin directly.
Fig. 3.

Inhibition of mitochondrial respiration decreases cancer stem cell function. a, b Inhibition of mitochondrial respiration reduced the expression of gene associated with colon cancer stem cells downstream of Wnt/β-catenin. The relative expression of CD44, LGR5, TCF7, MYC, and CD133 mRNA was determined by using qRT-PCR in control and TFAM knockdown DLD1 (a) and HCT116 (b) cells. Data represent the mean ± SD (n = 3, ¶p < 0.0001, §p < 0.001, *p < 0.01, and #p < 0.05). c Knockdown of TFAM inhibited the formation of tumor spheroids in suspension culture. HCT116 cells were seeded as single cell suspension in stem cell medium and the number of colonies formed was determined after 7 days. Data represent the mean ± SD (n = 4, #p < 0.05). d The expression of TFAM was silenced in tumor organoids derived from Apc/Kras mutant mice using two different lentiviral shRNAs. Single cell suspensions of control and TFAM knockdown cells were seeded in 3D Matrigel. Representative images of control and TFAM knockdown organoids are shown after 6 days in culture. Scale bar, 100 μm. e The number of organoids formed was quantified (total 2000 cells were seeded per group). Data represent the mean ± SD (n = 3, #p < 0.05). f The percentage of organoids with branching phenotype (as shown in d) was determined in control and TFAM knockdown organoids. Data represent the mean ± SD (n = 3, *p < 0.01 and #p < 0.05). g Control and TFAM knockdown organoids were subseeded and grown in 3D Matrigel for 3 days. The expression of TFAM as well as target genes downstream of Wnt/β-catenin (including Lgr5, Cd44, and Myc) or HIF1α (including Ldha and Slc2a1) was determined using qRT-PCR. Data represent the mean ± SD (n = 3, ¶p < 0.0001, *p < 0.01, and #p < 0.05)
Furthermore, we determined if the tumor initiation potential of colon cancer cells is regulated by mitochondrial respiration. Single cells of control and TFAM knockdown HCT116 cells were grown in the stem cell medium as suspension culture. We found that the numbers of tumor spheroids (colonies) formed were decreased in TFAM knockdown cells suggesting inhibition of colony formation ability (Fig. 3c). We have previously established a 3D tumor organoid model using tumor cells derived from Apc/Kras compound mutant mice [19]. The expression of TFAM was silenced using two different lentiviral shRNAs in the tumor organoids. Single cell suspensions of control and TFAM knockdown tumor cells were cultured in 3D Matrigel and the number of colonies formed was quantified (Fig. 3d). Consistently, the ability of TFAM knockdown cells to form tumor organoids in 3D was decreased (Fig. 3e). A large percentage of organoids derived from TFAM knockdown cells showed branching phenotype suggesting increased differentiation (Fig. 3d, f). Moreover, the expression of both Wnt and HIF1 target genes was significantly decreased in TFAM knockdown tumor organoids (Fig. 3g), suggesting decreased Wnt and HIF1 signaling in a physiologically relevant 3D system.
Mitochondrial deficiency induces metabolic reprograming in colon cancer cells
To further define the metabolic alterations induced by TFAM depletion, we profiled cellular metabolism using SIRM analysis. Control and TFAM knockdown HCT116 cells were cultured with uniformed-labeled (U-13C) glucose and the fate of labeled metabolites derived from 13C-glucose was traced using NMR and IC-MS analysis. The diagram shown in Fig. 4a illustrates a number of metabolites produced in the glycolysis and tricarboxylic acid (TCA) cycle that are detected in our analysis. We first measured the levels of 13C-glucose uptake and 13C-lactate production in cell media. Consistent with results shown in Fig. 3, knockdown of TFAM increased the utilization of 13C-glucose as well as the production of 13C-lactate and 13C-alanine, thus indicating increased glycolysis in cells with impaired mitochondrial respiration (Fig. 4b). Since mitochondrial OXPHOS is tightly coupled with the TCA cycle activity, we next measured the isotopologue patterns of metabolites that are derived from 13C-glucose via TCA cycle. Results from the fractional enrichment of 13C-isotopologues showed that the extent of 13C incorporation into the TCA cycle metabolites, including citrate, cis-aconitate, fumarate, malate, and aspartate, was largely decreased in TFAM knockdown cells (Fig. 4c, d), suggesting reduced utilization of glucose as a metabolic substrate through the TCA cycle in mitochondria. Note that the fractions of M + 2 isotopologues of citrate and cis-aconitate were initially increased in TFAM knockdown cells, whereas additional 13C-isotopologues were decreased (Fig. 4c), indicating that the incorporation of 13C-labeled carbons into TCA cycle metabolites gradually diminishes following each cycle. Nevertheless, the TFAM knockdown cells were able to derive a significant amount of TCA cycle metabolites from non-13C-labeled carbon sources as indicated by the enrichment of M + 0 fractions (Fig. 4c, d). Similarly, the fractional enrichment patterns of glutamate, α-KG and glutathione isotopologues showed that the M + 0 fractions were significantly increased whereas the 13C-isotopologues were decreased (Fig. 4e). These results suggest that the unlabeled fractions of glutamate and other TCA cycle metabolites are likely derived from glutamine via glutaminolysis in TFAM knockdown cells. This compensatory utilization of glutamine has also been reported in TFAM-deleted epidermal keratinocytes [20]. Collectively, mitochondrial deficiency alters the metabolic fate of glucose, in which it is mainly utilized within the glycolysis pathway without being fully metabolized via the TCA cycle in colon cancer cells.
Fig. 4.

Metabolic reprograming in TFAM knockdown colon cancer cells. Control and TFAM knockdown HCT116 cells were labeled with 13C6-glucose (Glc) for 24 h, and levels of metabolites in media and cells were analyzed. a Diagram showing the metabolic network connecting glycolysis with the TCA cycle. The metabolites shown were detected either by NMR in cell media or by IC-MS in cell extracts. b The levels of 13C-Glc, 13C-Lactate (Lac), and 13C-Alanine (Ala) were quantitatively determined by NMR analysis in media. Data represent the mean ± SD (*p < 0.01). c–e The fractional enrichments in the 13C isotopologues of metabolites derived from the TCA cycle, including citrate, cis-aconitate, fumarate, malate, aspartate, α-ketoglutarate (α-KG), glutamate and glutathione, as determined by IC-MS analysis. M + 0 (all carbons unlabeled e.g., 12C) to M + n isotopologues indicate the number of 13C atoms present in each particular metabolite. Data represent the mean ± SD (¶p < 0.0001, §p < 0.001, *p < 0.01, and #p < 0.05). All data shown were average measurements from three sets of independently labeled sh-control cells and two sets of sh-TFAM cells. A two-tailed unpaired t-test was used to determine the statistical differences
Mitochondrial respiration controls the activation of HIF1 and Wnt signaling
Given that the levels of TCA cycle metabolites are altered in TFAM knockdown cells, we investigated the functional effect of metabolic reprogramming induced by mitochondrial deficiency. α-KG is an important metabolite that serves as a cosubstrate for Fe(II)/α-KG-dependent dioxygenases [4]. We found in the SIRM analysis that the total cellular level of α-KG was significantly increased in TFAM knockdown cells (Fig. 5a), whereas the total levels of succinate and fumarate were not altered significantly (data not shown). The prolyl-hydroxylase domain (PHD) family of proteins are Fe(II)/α-KG-dependent HIF-hydroxylases that mediate the hydroxylation of proline residues to promote ubiquitination and degradation of HIF proteins in the presence of oxygen [21] (Fig. 5b). Since mitochondrial deficiency prevents HIF1α stabilization as shown in Fig. 2, we hypothesized that upregulation of α-KG as a result of metabolic reprogramming enhances PHD activity and subsequently HIF1α downregulation in TFAM knockdown cells. To test this, we silenced the expression of PHD2 in control and TFAM knockdown cells and determined if upregulation of HIF1α expression restores the activation of target genes downstream of HIF1 and Wnt/β-catenin. Indeed, while knockdown of TFAM reduced the expression of both Wnt (including LGR5, TCF7, and AXIN2) and HIF target genes (including LDHA and PDK1), PHD2 depletion largely rescued this defect (Fig. 5c, d). Similar results were obtained in control and TFAM knockdown cells treated with desferrioxamine (DFO), an iron chelator and inhibitor of PHD (Supplementary Fig. S4). To further confirm that the increased production of α-KG is responsible for suppressing HIF1α and Wnt/β-catenin activation, we treated HCT116 cells with octyl-α-KG, a cell-permeable precursor form of α-KG. The presence of α-KG reduced HIF1α protein expression and the rate of cell proliferation (Fig. 5e, f). In addition, the expression of both Wnt/β-catenin and HIF1α target genes was significantly decreased in α-KG treated cells (Fig. 5g). Together, our results identify α-KG as a signaling metabolite that regulates Wnt signaling through controlling HIF1α expression.
Fig. 5.

Inhibition of mitochondrial respiration suppresses Wnt/β-catenin signaling by enhancing α-KG-mediated degradation of HIF1α. a Total levels of α-KG were upregulated in TFAM knockdown HCT116 cells as measured by IC-MS. Data represent the mean ± SD (#p < 0.05). b Diagram showing that α-KG produced from the TCA cycle promotes PHD2-dependent degradation of HIF1α. c, d Stable control and TFAM knockdown DLD1 (c) and HCT116 (d) cells were infected with either control or sh-PHD2 lentivirus to generate TFAM and PHD2 double knockdown cells. The expression of target genes downstream of Wnt/β-catenin (including LGR5, TCF7, and AXIN2) or HIF1α (including LDHA and PDK1) was determined using qRT-PCR. Data represent the mean ± SD (n = 3, #p < 0.05). e HCT116 cells were treated with α-KG (1 mM) for 24 h and cell lysates were analyzed for the expression of HIF1α using Western blotting. f The presence of α-KG decreases cell proliferation. HCT116 cells were cultured in the presence or absence of α-KG for 24 h or 48 h and the number of cells was counted. Data represent the mean ± SD (n = 3, §p < 0.001 and *p < 0.01). g HCT116 cells were treated with α-KG for 48 h. The expression of Wnt/β-catenin (including LGR5, TCF7, and AXIN2) or HIF1α target genes (including LDHA and PDK1) was determined using qRT-PCR. Data represent the mean ± SD (¶p < 0.0001, §p < 0.001, and #p < 0.05)
Mitochondrial deficiency inhibits xenograft tumor growth and tumor initiation in vivo
We next determined the effect of TFAM downregulation on tumorigenesis in vivo. To this end, control and two different TFAM knockdown HCT116 cells were injected subcutaneously into NSG mice and the tumorigenesis process was monitored for 4 weeks. We found that silencing TFAM reduced the rate of tumor growth (Fig. 6a). Additionally, tumor sections stained with the Ki67 antibody showed that the percentage of Ki67-positive cells was decreased in TFAM knockdown tumors indicating decreased cell proliferation (Fig. 6b). Consistent with results obtained in vitro, the expression of genes associated with cancer stem cells, including CD44 and LGR5, was significantly decreased in TFAM knockdown tumors (Fig. 6c). In addition, the expression of MKI67 (Ki67) and LDHA mRNA was decreased confirming reduced proliferation and HIF1α activation (Fig. 6c). Furthermore, we performed tumor initiation experiments by injecting control and TFAM knockdown HCT116 cells at 100, 500, and 1000 cells/site into NSG mice. The number of tumors formed was determined after 3 months. Based on the extreme limiting dilution analysis (ELDA), the stem cell frequency was significantly decreased in TFAM knockdown cells (Fig. 6d), suggesting that mitochondrial deficiency inhibits tumor initiation. Collectively, our results demonstrate that mitochondrial respiration is required to sustain tumor growth and tumor initiation in vivo.
Fig. 6.

Knockdown of TFAM inhibits xenograft tumor growth and tumor initiation. a Control and TFAM knockdown HCT116 cells were injected subcutaneously into six NSG mice. The size of the tumors was measured every week starting at one week after injection. Data represent the mean ± SEM (n = 6, #p < 0.05 for both TFAM knockdown groups compared to sh-control group). b The percentage of Ki67-positive cells in tumors from the control and TFAM knockdown groups. Tumor sections from three mice of each group were stained and the numbers of Ki67-positive cells were quantified in eight randomly chosen areas in each tumor. Data represent the mean ± SD (n = 8, ¶p < 0.0001). c Downregulation of Wnt and HIF1α target genes in TFAM knockdown xenograft tumors. Tumor tissues from three mice of each group were analyzed for the expression of TFAM, CD44, LGR5, MKI67 (Ki67), and LDHA by using qRT-PCR. Data represent the mean ± SD (n = 3, ¶p < 0.0001, §p < 0.001, and *p < 0.01). d Tumor-initiation experiments were performed using control and TFAM knockdown HCT116 cells. Cells were mixed with Matrigel and subcutaneously inoculated into NSG mice at 100, 500, or 1,000 cells per site and total of eight injections were used for each cell line. The number of tumors formed was determined 3 months post inoculation. The stem cell frequency was calculated using extreme limiting dilution analysis (ELDA) (&p = 0.0002 as determined by Chi-square tests compared to sh-control)
Knockout of TFAM inhibits tumor initiation in Apc-mutant mice
Although knockdown of TFAM decreased tumor growth in vivo, the expression of TFAM was recovered to about 70% of the control level during the tumorigenesis process (Fig. 6c). This likely attenuates the effect of mitochondrial inhibition. To better assess the functional effect of TFAM-loss on colon cancer tumorigenesis in vivo, we crossed TFAMf/f mice to the Apcf/f/Vil-cre model to investigate the susceptibility of mitochondrial deficient mice to Apc-driven intestinal adenomas. Specifically, the following three cohorts of mice, including Apcf/+/Vil-Cre/TFAM+/+, Apcf/+/Vil-Cre/TFAMf/+, and Apcf/+/Vil-Cre/TFAMf/f (Apc/TFAM-WT, Apc/TFAM-Het, and Apc/TFAM-KO, respectively), were generated to monitor the tumor formation process. At 6 months of age, approximately 30–50 intestinal tumors were observed in Apc/TFAM-WT and Apc/TFAM-Het mice. However, the numbers of tumors were drastically decreased in Apc/TFAM-KO mice (Fig. 7a). In addition, IHC staining results showed that the rate of cell proliferation was reduced as indicated by decreased Ki67-positive cells in the few tumors found in Apc/TFAM-KO mice (Supplementary Fig. S5). Interestingly, IHC staining of tumor sections with the β-catenin antibody identified nuclear-localized β-catenin in numerous tumor cells in Apc/TFAM-WT mice; however, β-catenin was mainly membrane localized in Apc/TFAM-KO tumors (Fig. 7b). Furthermore, the expression of both HIF1 and Wnt/β-catenin target genes was significantly decreased in Apc/TFAM-KO tumors (Fig. 7c). To confirm that TFAM-loss reduces Wnt signaling, we deleted TFAM from TFAMf/f MEF cells using Cre-expressing adenovirus. The expression of mitochondrial-encoded genes and mtDNA copy number were largely decreased in TFAM knockout MEF cells. As a consequence, the expression of Wnt target genes was also downregulated (Supplementary Fig. S5). Taken together, our results show that genetic deletion of TFAM significantly inhibits the activation of Wnt signaling and tumorigenesis in Apc-driven mouse models of colon cancer.
Fig. 7.

Knockout of TFAM inhibits tumorigenesis in Apc-mutant mice. a The number of intestinal adenomas was counted in the following three cohorts of mice at 6 months: Apcf/+/TFAM+/+/Villin-Cre (Apc/TFAM-WT, n = 12), Apcf/+/TFAMf/+/Villin-Cre (Apc/TFAM-Het, n = 12), and Apcf/+/TFAMf/f/Villin-Cre (Apc/TFAM-KO, n = 11). Data represent the mean ± SD (¶p < 0.0001 compared to the Apc/TFAM-WT group). b TFAM-loss reduced nuclear localization β-catenin as detected by IHC staining using the anti-β-catenin antibody. The boxed regions in upper images were enlarged and shown at the bottom, and arrows indicate nuclear β-catenin staining. Scale bar, 50 μm. c Knockout of TFAM decreased the expression of genes associated with cancer stem cells. The relative expression of target genes downstream of Wnt/β-catenin (including Lgr5, Tcf7, and Axin2) or HIF1α (including LdhA and Pdk1) was determined using qRT-PCR in tumors derived from Apc/TFAM-WT and Apc/TFAM-KO mice. Tumor tissues from three mice per group were analyzed. Data represent the mean ± SD (¶p < 0.0001, §p < 0.001, and *p < 0.01 compared to Apc/TFAM-WT). d TCGA CRC RNA-seq dataset was used to analyze the expression of TFAM. The expression of TFAM in tumor vs. normal samples was compared using linear mixed models (p = 0.007). e The GSEA was performed using the TCGA CRC RNA-seq dataset to identify gene sets that have positive correlations with TFAM expression. Enrichment plots showed significant correlation of the TCA cycle and respiratory electron transport (NES = 1.84, FDR q-val = 0.003) and the signaling by Wnt (NES = 2.32, FDR q-val = 0.000) pathway with TFAM expression in CRC patients. f Diagram showing TFAM-mediated regulation of mitochondrial retrograde signaling in colon cancer cells. Our study here demonstrates that TFAM is required to maintain the mitochondrial integrity and TCA cycle activity. When the mitochondrial function is maintained, the production of TCA metabolite α-KG and α-KG-dependent PHD2 activity are kept at lower levels, which allows HIF1α stabilization under hypoxia and activation of Wnt/β-catenin-mediated cancer stem cell signaling
Moreover, we determined if TFAM gene expression is altered in colorectal cancer patients by analyzing the Cancer Genome Atlas (TCGA) RNA-seq dataset. Bioinformatics analysis of normal and tumor samples showed that the expression of TFAM gene is significantly increased in colorectal cancer (Fig. 7d). Additionally, the Gene Set Enrichment Analysis (GSEA) revealed that TFAM expression is positively associated with not only the TCA cycle and respiratory electron transport but also the Wnt signaling in colorectal cancer patients (Fig. 7e). These data support our findings that mitochondrial respiration is essential for enhancing signaling through the Wnt pathway. Collectively, our results support a model of mitochondrial retrograde signaling in which TFAM-dependent mitochondrial activity is required to maintain the activation of HIF1α and Wnt/β-cantenin signaling in colon cancer cells (Fig. 7f).
Discussion
Cancer cells are rapidly dividing cells that have increased demands for energy and macromolecules. To cope with these elevated requirements, cancer cells undergo major metabolic modifications [22, 23]. Here we identify mitochondrial metabolism as a key regulator of Wnt signaling and tumorigenesis in colon cancer. Silencing TFAM expression results in a decrease in mitochondrial respiration and an increase in glycolysis. Although this metabolic alteration allows the cells to fulfill their bioenergetic needs, Wnt signaling and cancer stem cell properties are significantly compromised. We uncover α-KG as a signaling metabolite to mediate the crosstalk between HIF1 and Wnt signaling. Our GSEA study identify additional gene sets related to cell cycle, DNA replication, RNA transcription, and other metabolic pathways that are positively associated with TFAM expression in colorectal cancer patients (Supplementary Fig. S6). Interestingly, the expression of TFAM is also negatively associated with gene sets related to immune response, receptor-mediated signal transduction, and cell-extracellular matrix interaction (Supplementary Fig. S6). Together, these data suggest that mitochondrial respiration plays an important role in regulating a number of cancer-related pathways.
Given the notion of tumor heterogeneity, it has been suggested that the metabolic programs employed by cancer stem cells are likely to be different from bulk tumor cells [24, 25]. However, recent studies comparing the metabolic differences between cancer stem cells and bulk tumor cells have yielded conflicting results depending on the tumor type and model system used [24]. In colon cancer, it has been shown that cancer stem cells isolated as CD133+/CD44+/LGR5+ cells from a small number of patients have increased mitochondrial activity compared to non-cancer stem cell population [26]. Our study here provides several lines of evidence supporting the role of mitochondrial respiration in promoting cancer stem cell properties: (i) TFAM-loss inhibits the expression of Wnt/β-catenin target genes; (ii) silencing TFAM decreases colony formation in vitro and tumor initiation in vivo; (iii) intestinal-specific deletion of TFAM inhibits Apc-driven tumorigenesis in mice; and (iv) the expression of TFAM gene is positively associated with Wnt signaling in human colorectal cancer patients. A recent study has shown that tamoxifen-induced TFAM-loss in TFAMf/f/Vil-Cre-ERT2 mice results in impaired intestinal stem cell renewal [27]. However, Apc/TFAM double KO mice in our study displayed normal cryp-villus structure in the non-tumor region of the intestine (data not shown). We cannot exclude the possibility that the WT escaper cells contribute to the maintenance of intestinal epithelium in our mouse model. Despite that escaper cells may attenuate the effect of TFAM KO, disruption of mitochondrial respiration largely prevents tumorigenesis of Apc-driven tumors in vivo.
Although a number of TCA cycle metabolites are altered in TFAM knockdown cells, results from SIRM analysis also reveal that the pentose phosphate pathway as well as purine and pyrimidine biosynthesis are not affected as the levels of phosphoribosyl pyrophosphate (PRPP), ATP and UMP remain unchanged (Supplementary Fig. S7). This finding suggests that the bioenergetic and biosynthetic function of mitochondria is compensated by glycolysis. Moreover, previous studies have shown that glutamine-dependent reductive carboxylation is upregulated in cells with impaired mitochondria [28]. To evaluate glutamine metabolism in TFAM knockdown cells, we performed 13C-glutamine tracing experiments. Consistent with results shown in Fig. 4, we found that the fraction of m + 5 glutamate was increased in TFAM knockdown cells supporting the notion that tumor cells with mitochondrial deficiency can produce glutamate via glutaminolysis (Supplementary Fig. S8a). However, we did not observe any increase in TCA metabolites that are produced by reductive carboxylation of glutamine. In fact, the fractions of citrate, fumarate, or malate isotopologues generated via reductive carboxylation were much smaller compared to those produced via oxidative metabolism (Supplementary Fig. S8b-d), thus suggesting that glutamine-dependent reductive carboxylation is not induced in TFAM knockdown cells. It is of particular interest for future studies to determine if the key enzymes needed to support the reductive metabolism of glutamine are insufficiently expressed in colon cancer cells used in our study.
It has been shown previously that heterozygous whole-body knockout of TFAM slightly increases the numbers of adenoma in ApcMin mice due to increased ROS [29]. However, we show here that heterozygous deletion of TFAM in mouse intestinal epithelial cells had no effect on Apc-driven tumorigenesis, suggesting that deletion of TFAM expression in other tissue types may contribute to the different phenotypes observed. Lack of mitochondria-generated ROS has been implicated as a mechanism for decreased HIF1α expression in a previous report [30]. We found that ROS levels were indeed reduced in TFAM knockdown colon cancer cells; however, inducing ROS production by treating cells with D-galactose and galactose oxidase was unable to rescue the expression of HIF or Wnt target genes (data not shown). Since cancer cells are known to produce elevated levels of ROS compared to normal cells [31, 32], it is possible that TFAM-loss-induced reduction in ROS is not sufficient to alter HIF1α expression. Furthermore, previous studies have suggested that TFAM may localize to the nucleus and regulate the transcription of nuclear-encoded genes [33, 34]. However, results from a recent genome-wide ChIP-seq analysis identify no evidence of TFAM binding to the nuclear genome, whereas extensive interaction between TFAM and the mitochondrial genome is detected [35]. Thus, it is unlikely that TFAM regulates HIF1 and Wnt target gene expression as a nuclear transcription factor. Instead, we identify α-KG as a key metabolite that connects TFAM-dependent regulation of mitochondrial metabolism to the HIF and Wnt pathway.
In summary, results from our study demonstrate the functional importance of mitochondrial metabolism in promoting tumorigenesis and Wnt signaling in colon cancer. By establishing a role of mitochondrial retrograde signaling in controlling cancer stem cell properties, our findings indicate that inhibition of mitochondrial respiration may provide an effective approach for targeting cancer stem cells.
Materials and methods
Cells and reagents
Human colon cancer cell lines DLD1 and HCT116 cells were cultured in DMEM and McCoy’s 5 A supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich) and 1% penicillin-streptomycin, respectively. These cells were purchased from ATCC and authenticated using short tandem repeat (STR) DNA profiling in March 2016 (Genetica). For culturing cells under hypoxic conditions, cells were placed in a sealed hypoxia incubator chamber (Stemcell Technologies) filled with 5% CO2, 1% O2, and 94% N2. Stable TFAM and PHD2 knockdown cells were generated using lentivirus-based RNAi [19, 36, 37]. The shRNA targeting sequences for human TFAM are as the following: 5’-GTAAGTTCTTACCTTCGATTT-3’ (a) and 5’-CGTGAGTATATTGATCCAGAA-3’ (b); for mouse TFAM: 5’-GCTGAGTGGAAAGCATACAAA-3’ (c) and 5’- AGGATTCGTTACGACAATGAA-3’ (d); and for PHD2: 5’-ACGCCACTGTAACGGGAAGCT-3’. The non-targeting control shRNA lentivirus plasmid (MISSION, SHC002), desferrioxamine (DFO), 2-Deoxy-D-glucose (2-DG), and octyl-α-KG were obtained from Sigma–Aldrich.
Mouse cohorts
TFAMf/f mice were previously described and maintained on a C57BL/6 background [10]. To produce animals used in this study, TFAMf/f mice were crossed with Apcf/f [38] and Villin-Cre mice to create intestinal epithelial cell-specific deletion of TFAM and Apc. TFAMf/f mice were generously provided by Dr. Navdeep Chandel and the other two mice strains were obtained from The Jackson Laboratory. The mice were then inter-crossed to produce three cohorts of animals including Apcf/+/TFAM+/+/Villin-Cre (Apc/TFAM-WT), Apcf/+/TFAMf/+/Villin-Cre (Apc/TFAM-Het), and Apcf/+/TFAMf/f/Villin-Cre (Apc/TFAM-KO). Both male and female mice are included in each group. Mice were sacrificed at the age of 6 months, and tumor burden was counted and intestine tissues were collected for histology and RNA analysis.
Quantitative RT-PCR
To measure relative gene expression by qRT-PCR, total RNA was isolated from human cancer cells, mouse tumor organoids, or mouse tumor tissues using the RNeasy Mini kit (Qiagen). Equal amounts of RNA were used as templates for the synthesis of cDNA using the High Capacity cDNA Reverse Transcription kit (Thermo). cDNA was then subjected to qPCR reactions using SYBR Green Master Mix (Thermo) and primers listed in Supplementary Table S1 using the StepOne RT-PCR system (Applied Biosystems). Additionally, to determine the expression of TFAM, SDHB, and PROM1 (CD133), gene-specific probes were purchased from Thermo and used in qPCR reactions. All values were normalized to the level of β-actin.
Seahorse extracellular flux analysis
The Seahorse XF96 Extracellular Flux Analyzer (Agilent) was used to measure the respiration activity of colon cancer cells as described previously [19, 39, 40]. Briefly, cells were seeded at the density of 3 × 104 cells per well in a XF96 plate approximately 16 h before the measurement. The glycolysis and mitochondrial stress tests were performed according to manufacturer’s protocol. The relative levels of glycolysis, glycolytic capacity, and glycolytic reserve were calculated based on ECAR data obtained in the glycolysis stress tests, whereas the relative levels of basal, maximal, ATP production-related respiration and reserved mitochondrial capacity were calculated based on OCR data obtained in the Mito stress tests.
In vitro colony formation assay
To determine the self-renewal capacity, 1000 single cell suspensions of HCT116 cells were seeded in non-adherent 24-well plates in StemPro hESC SFM medium supplied with 1% GlutaMAX, 2% StemPro hESC supplement, 1.8% BSA, 8 ng/mL FGF-basic, and 0.1 mM 2-mercaptoethanol (Thermo). After 6 days in culture, numbers of spheroids were counted under a light microscope.
Tumor organoids derived from Apcf/+/KrasLSL-G12D/Villin-Cre mice were generated and described previously [19]. To silence TFAM expression, tumor organoids were dissociated into small cell clusters using TrypLE (Thermo) and incubated with sh-TFAM lentivirus in suspension for 6 h in a 37 °C incubator. Cells were subsequently embedded in 33% Matrigel in 3D growth medium (Advanced DMEM/F12 supplemented with 1 × Glutamax, 1 × N-2, 1 × B-27, 1 mM N-Acetyl-L-cysteine, and 1% penicillin/streptomycin), and puromycin was added 2 days later to select for stable knockdown cells. For colony formation assays, tumor organoids were dissociated and single cell suspensions were seeded into 3D Matrigel. The number of tumor organoids formed after 6 days was counted and analyzed. For gene expression analysis, tumor organoids were cultured in 3D Matrigel for 3–4 days and collected for qRT-PCR.
NMR analysis
Stable control and TFAM knockdown HCT116 cells were cultured in glucose-free DMEM supplemented with 0.2% 13C6-glucose, 10% dialyzed FBS, and 1% penicillin-streptomycin for 24 h. At the end of incubation, media was removed for NMR analysis and cells were washed with cold PBS followed by extraction of polar metabolites for IC-MS analysis as described previously [41]. NMR spectra were obtained with a 3-mm inverse triple resonance HCN cold probe equipped Agilent DD2 14.1 T. The 1D proton PRESAT spectra were recorded at 288 K with an acquisition time of 2 s and a 4-s relaxation delay, during which the residual HOD resonance was irradiated. 1D 1H HSQC spectra were recorded with an acquisition time of 0.25 s and a 1.75-s relaxation delay with adiabatic decoupling. DSS-d6 at 25 nmol was used for internal chemical shift reference and absolute quantification. Raw data were processed using MNOVA (Mrestrelab Research) with 1 zerofilling for PRESAT data with apodization using an unshifted Gaussian 1-Hz line-broadening exponential. The HSQC data were processed with two zero fillings and apodization with an unshifted Gaussian and a 4-Hz line-broadening exponential. Baselines were corrected using a third order polynomial. Metabolites were assigned using an in-house database of standards [42]. Metabolites were quantified using the line fitting routines in MNOVA and normalized to the DSS resonance at 0 ppm. Isotopomers were quantified as absolute and fractional enrichments as previously described [43–45]. Assigned metabolites were normalized to mg protein determined by BCA analysis.
IC-MS analysis
Reconstituted polar fractions were analyzed on a Dionex ICS-5000 + ion chromatograph attached to an Orbitrap Fusion Tribrid mass spectrometer (Thermo). Operating resolution to acquire all 13C isotopologues was set to 500,000 (FWHM at m/z 200) on MS1 acquisition with a m/z range of 50–750 [41, 46]. The Orbitrap Fusion was tuned and calibrated according to the manufacturer’s default recommendations and a Dionex IonPac AS11-HC-4 μm RFIC&HPIC (2 × 200) mm column was used. Manual curation of peak area integration into Excel was performed with TraceFinder 3.3 (Thermo) software package. Peak areas were corrected for natural abundance distributions for each isotopologue and fractional enrichment of each metabolite per mg protein was calculated [47].
Xenograft tumorigenesis
All animal procedures were done using protocols approved by the University of Kentucky Animal Care and Use Committee. Six- to eight-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG, The Jackson Laboratory) mice were used. Both male and female mice of equal numbers were included in each group. For tumor initiation assay, 100, 500, or 1000 HCT116 cells were mixed with 5% Matrigel and injected subcutaneously. The number of tumors formed was determined 3 months post injection. For tumor growth assay, HCT116 cells were inoculated subcutaneously at 1 × 106 cells per injection. The tumor size was measured every week with a caliper, and the tumor volume was defined as (longest diameter) × (shortest diameter)2/2.
Statistical analysis
In experiments to assess the rate of cell proliferation, mRNA expression, glucose, lactate and ATP levels, and bioenergetic measurements were summarized using bar or line graphs and pairwise comparisons between different conditions were carried out using two-sample t-tests. A linear mixed model was employed to compare slope of xenograft tumor volume growth curves over time between groups. The relative mRNA expression results represent average of three separate qPCR experiments with four replicates for each gene in each experiment. For the Seahorse experiments, eight replicates were included for each cell line in each experiment and the experiments were repeated three times. Results shown in Figs. 2 and 3 were from one representative experiment. All other experiments were repeated three times and results shown represent the average of three experiments. The stem cell frequencies were calculated by extreme limiting dilution analysis (ELDA) [48] using analytic tools available at http://bioinf.wehi.edu.au/software/elda/.
The RNA-seq dataset from the TCGA colorectal cancer (CRC) study were used to determine the relative expression of gene in human CRC patients. The expression of TFAM in tumor vs. normal samples was compared using linear mixed models. All statistical analyses were performed using R (version 3.4.1). For the Gene Set Enrichment Analysis (GSEA), correlations between expressions of TFAM and other genes were quantified by Spearman’s correlation coefficient. The genes were then ordered from highest to lowest based on the correlation coefficient. This ranked list was inputted into the GSEA Desktop Application [49] to identify pathways that are associated with TFAM expression.
Supplementary information
Acknowledgements
This work was supported by R01CA133429 (TG), 1U24DK097215 (T.W.-M.F.) and 5R35CA197532 (NSC). The studies were conducted with support provided by the Redox Metabolism, Biospecimen Procurement and Translational Pathology, Flow Cytometry and Cell Sorting, and Biostatistics and Bioinformatics Shared Resource Facilities of the University of Kentucky Markey Cancer Center (P30CA177558) as well as the Center for Environmental and Systems Biochemistry (CESB) at the University of Kentucky.
Author contributions
Y-AW, TAB, and TG designed all aspects of the study; Y-AW, XX, TS, ATL, CW, HLW, and LT performed experiments and analysis; EB, TWMF, and NSC provided input into the project; Y-AW and TG wrote the manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by S. Kaufmann
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41418-018-0265-6) contains supplementary material, which is available to authorized users.
References
- 1.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014;12:34. doi: 10.1186/1741-7007-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13:878–90. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7:39–44. doi: 10.1016/j.mito.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 7.Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- 8.Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, et al. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet. 2004;13:935–44. doi: 10.1093/hmg/ddh109. [DOI] [PubMed] [Google Scholar]
- 9.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet. 1998;18:231–6. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 10.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–93. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26:1926–44. doi: 10.1101/gad.188292.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1alpha balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab. 2015;22:590–605. doi: 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 13.Vlashi E, Lagadec C, Vergnes L, Matsutani T, Masui K, Poulou M, et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc Natl Acad Sci USA. 2011;108:16062–7. doi: 10.1073/pnas.1106704108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–32. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–13. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 16.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–80. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 17.Vermeulen L, Morrissey E, van der Heijden M, Nicholson AM, Sottoriva A, Buczacki S, et al. Defining stem cell dynamics in models of intestinal tumor initiation. Science. 2013;342:995–8. doi: 10.1126/science.1243148. [DOI] [PubMed] [Google Scholar]
- 18.Zeuner A, Todaro M, Stassi G, De Maria R. Colorectal cancer stem cells: from the crypt to the clinic. Cell Stem Cell. 2014;15:692–705. doi: 10.1016/j.stem.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Wen YA, Xiong X, Harris JW, Zaytseva YY, Mitov MI, Napier DL, et al. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell death Dis. 2017;8:e2593. doi: 10.1038/cddis.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamanaka RB, Glasauer A, Hoover P, Yang S, Blatt H, Mullen AR, et al. Mitochondrial reactive oxygen species promote epidermal differentiation and hair follicle development. Sci Signal. 2013;6:ra8. doi: 10.1126/scisignal.2003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 22.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cairns RA, Harris I, McCracken S, Mak TW. Cancer cell metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:299–311. doi: 10.1101/sqb.2011.76.012856. [DOI] [PubMed] [Google Scholar]
- 24.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23:1124–34. doi: 10.1038/nm.4409. [DOI] [PubMed] [Google Scholar]
- 25.Mancini Rita, Noto Alessia, Pisanu Maria Elena, De Vitis Claudia, Maugeri-Saccà Marcello, Ciliberto Gennaro. Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene. 2018;37(18):2367–2378. doi: 10.1038/s41388-018-0141-3. [DOI] [PubMed] [Google Scholar]
- 26.Song IS, Jeong YJ, Jeong SH, Heo HJ, Kim HK, Bae KB, et al. FOXM1-induced PRX3 regulates stemness and survival of colon cancer cells via maintenance of mitochondrial function. Gastroenterology. 2015;149:1006–16 e9. doi: 10.1053/j.gastro.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 27.Srivillibhuthur M, Warder BN, Toke NH, Shah PP, Feng Q, Gao N, et al. TFAM is required for maturation of the fetal and adult intestinal epithelium. Dev Biol. 2018;439:92–101. doi: 10.1016/j.ydbio.2018.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woo DK, Green PD, Santos JH, D’Souza AD, Walther Z, Martin WD, et al. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/ + ) mice. Am J Pathol. 2012;180:24–31. doi: 10.1016/j.ajpath.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamanaka RB, Weinberg SE, Reczek CR, Chandel NS. The mitochondrial respiratory chain is required for organismal adaptation to hypoxia. Cell Rep. 2016;15:451–9. doi: 10.1016/j.celrep.2016.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–8. [PubMed] [Google Scholar]
- 32.Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell death Dis. 2016;7:e2253. doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe A, Arai M, Koitabashi N, Niwano K, Ohyama Y, Yamada Y, et al. Mitochondrial transcription factors TFAM and TFB2M regulate Serca2 gene transcription. Cardiovasc Res. 2011;90:57–67. doi: 10.1093/cvr/cvq374. [DOI] [PubMed] [Google Scholar]
- 34.Pastukh V, Shokolenko I, Wang B, Wilson G, Alexeyev M. Human mitochondrial transcription factor A possesses multiple subcellular targeting signals. FEBS J. 2007;274:6488–99. doi: 10.1111/j.1742-4658.2007.06167.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang YE, Marinov GK, Wold BJ, Chan DC. Genome-wide analysis reveals coating of the mitochondrial genome by TFAM. PLoS One. 2013;8:e74513. doi: 10.1371/journal.pone.0074513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Stevens PD, Liu J, Yang H, Wang W, Wang C, et al. PHLPP is a negative regulator of RAF1, which reduces colorectal cancer cell motility and prevents tumor progression in mice. Gastroenterology. 2014;146:1301–12. doi: 10.1053/j.gastro.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J, Weiss HL, Rychahou P, Jackson LN, Evers BM, Gao T. Loss of PHLPP expression in colon cancer: role in proliferation and tumorigenesis. Oncogene. 2009;28:994–1004. doi: 10.1038/onc.2008.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheung AF, Carter AM, Kostova KK, Woodruff JF, Crowley D, Bronson RT, et al. Complete deletion of Apc results in severe polyposis in mice. Oncogene. 2010;29:1857–64. doi: 10.1038/onc.2009.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wen YA, Xiong X, Zaytseva YY, Napier DL, Vallee E, Li AT, et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell death Dis. 2018;9:265. doi: 10.1038/s41419-018-0330-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiong X, Wen YA, Mitov MI, M CO, Miyamoto S, Gao T. PHLPP regulates hexokinase 2-dependent glucose metabolism in colon cancer cells. Cell Death Discov. 2017;3:16103. doi: 10.1038/cddiscovery.2016.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun RC, Fan TW, Deng P, Higashi RM, Lane AN, Le AT, et al. Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat Commun. 2017;8:1646. doi: 10.1038/s41467-017-01518-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan TW, Lane AN. Assignment strategies for NMR resonances in metabolomics research (eds. Lutz NW, Sweedler JV and Wevers RA). In: Methodologies for metabolomics: experimental strategies and techniques. New York: Cambridge University Press; 2013. p. 525–84.
- 43.Fan TW, Lane AN. Structure-based profiling of metabolites and isotopomers by NMR. Prog Nucl Magn Reson Spectrosc. 2008;52:69–117. doi: 10.1016/j.pnmrs.2007.03.002. [DOI] [Google Scholar]
- 44.Fan TW, Lane AN. NMR-based stable isotope resolved metabolomics in systems biochemistry. J Biomol NMR. 2011;49:267–80. doi: 10.1007/s10858-011-9484-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lane AN, Fan TW, Higashi RM. Isotopomer-based metabolomic analysis by NMR and mass spectrometry. Methods Cell Biol. 2008;84:541–88. doi: 10.1016/S0091-679X(07)84018-0. [DOI] [PubMed] [Google Scholar]
- 46.Fan TW, Warmoes MO, Sun Q, Song H, Turchan-Cholewo J, Martin JT, et al. Distinctly perturbed metabolic networks underlie differential tumor tissue damages induced by immune modulator beta-glucan in a two-case ex vivo non-small-cell lung cancer study. Cold Spring Harb Mol Case Stud. 2016;2:a000893. doi: 10.1101/mcs.a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moseley HN. Correcting for the effects of natural abundance in stable isotope resolved metabolomics experiments involving ultra-high resolution mass spectrometry. BMC Bioinformatics. 2010;11:139. doi: 10.1186/1471-2105-11-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 49.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.