

Abstract
Inherited genetic modifiers and pharmacologic agents that enhance fetal hemoglobin (HbF) expression reverse the clinical severity of sickle cell disease (SCD). Recent efforts to develop novel strategies of HbF induction include discovery of molecular targets that regulate γ-globin gene transcription and translation. The purpose of this study was to perform genome-wide microRNA (miRNA) analysis to identify genes associated with HbF expression in patients with SCD. We isolated RNA from purified reticulocytes for microarray-based miRNA expression profiling. Using samples from patients with contrasting HbF levels, we observed an eightfold upregulation of miR-144–3p (miR-144) and miR-144–5p in the low-HbF group compared with those with high HbF. Additional analysis by reverse transcription quantitative polymerase chain reaction confirmed individual miR-144 expression levels of subjects in the two groups. Subsequent functional studies in normal and sickle erythroid progenitors showed NRF2 gene silencing by miR-144 and concomitant repression of γ-globin transcription; by contrast, treatment with miR-144 antagomir reversed its silencing effects in a dose-dependent manner. Because NRF2 regulates reactive oxygen species levels, additional studies investigated mechanisms of HbF regulation using a hemin-induced oxidative stress model. Treatment of KU812 cells with hemin produced an increase in NRF2 expression and HbF induction that reversed with miR-144 pretreatment. Chromatin immunoprecipitation assay confirmed NRF2 binding to the γ-globin antioxidant response element, which was inhibited by miR-144 mimic treatment. The genome-wide miRNA microarray and primary erythroid progenitor data support a miR-144/NRF2-mediated mechanism of γ-globin gene regulation in SCD.
Sickle cell disease (SCD) is a genetic disorder caused by the βS-globin mutation leading to production of hemoglobin S, polymer formation under low oxygen conditions, and red blood cell sickling. The net outcome of this process is chronic hemolysis, oxidative stress, anemia, and vaso-occlusive episodes of pain and organ damage. The most effective treatment for SCD is fetal hemoglobin (HbF; α2γ2) induction, which inhibits sickle hemoglobin polymerization through the formation of hybrid molecules [1]. Hydroxyurea is the only Food and Drug Administration-approved drug that ameliorates the clinical symptoms of SCD through HbF induction and other beneficial properties such as increasing nitric oxide levels and anti-inflammatory effects [2,3]. Not all individuals respond to hydroxyurea therapy, so understanding the molecular mechanisms involved in γ-globin regulation to develop strategies for HbF induction is critical to the discovery of additional effective therapeutic options for SCD.
With completion of genome-wide association studies, single nucleotide polymorphisms (SNPs) associated with HbF levels in SCD and thalassemia patients [4–8] were discovered. Three genetic loci, including −158 Xmn1-HBG2, BCL11A at 2p15 and the HBS1L-MYB region, account for 30–50% of inherited variations in HbF levels in several populations [4–7]. The Xmn1-HBG2 locus contributes 13% of HbF variance in β-thalassemia populations, but this effect did not replicate in African American [7] or Tanzanian [8] people. The greatest effect on HbF expression is mediated by SNPs in the second intron of BCL11A leading to gene silencing [4,6,7,9]. Subsequent gene knockout confirmed a major repressor role of BCL11A in γ-globin gene silencing during hemoglobin switching [10] through KLF1 activation [11,12] and interaction with the corepressor SOX6 [13,14]. Furthermore, haploinsufficiency of KLF1 caused by SNPs in coding and noncoding DNA regions causes high HbF levels in humans [15]. Recent studies by Bauer et al. demonstrated that an erythroid-specific enhancer in the second intron of BCL11A [16] that regulates lineage-specific BCL11A activation, is an excellent target for the development of novel gene therapy for β-hemoglobinopathies.
The third loci affecting HbF expression is located in the HBS1L-MYB region 5′ of the repressor oncogene MYB [5]. Studies in primary erythroid cultures demonstrated binding of the transcription factors LDB1, Tal1, and KLF1 in the HBS1L-MYB region to control MYB expression [17]. Additional studies by Sankaran et al. demonstrated microRNA (miRNA) miR-15a and miR-16–1 enhance γ-globin expression through MYB silencing in a child with trisomy 13 [18].
Recent efforts have identified mechanisms of γ-globin gene expression that focus on posttranscriptional miRNA-mediated gene regulation. Azzouzi et al. demonstrated, in an miRNA screen of umbilical cord and peripheral blood reticulocytes, that miRNA-96 targets the open reading frame of the γ-globin mRNA molecule to silence γ-globin expression [19]. Additional work by Miller et al. [20] verified the ability of LIN28B to repress let-7 miRNA expression as a mechanism of HbF induction in tissue culture systems. Recently, we published data to support a role of miR34a in γ-globin activation [21] through STAT3 gene silencing. Our group previously demonstrated a negative role of STAT3 in γ-globin expression [22]. These studies expand the role of miRNA in γ-globin regulation, but additional targets remain to be discovered.
To this end, we performed genome-wide miRNA expression analysis using RNA isolated from the reticulocytes of individuals with SCD and contrasting high-and low-HbF levels. We observed significant differences in miR-144 between the two groups, along with other miRNA genes. Subsequent functional studies in normal and sickle erythroid progenitors confirmed the ability of miRNA-144 antagomir to mediate HbF induction while increasing NRF2 expression.
Methods
Subject recruitment and blood processing
After obtaining institutional review board approval and informed consent, blood samples were collected from patients with homozygous sickle cell anemia (HbSS) followed at Augusta University. None of the subjects received hydroxyurea therapy or transfusions before recruitment (Supplementary Table E1, online only, available at www.exphem.org). Medical record review was completed to obtain complete blood counts with differential, reticulocyte count, and HbF levels determined by high-performance liquid chromatography. Blood samples were processed by Ficoll-Histapaque separation of peripheral blood mononuclear cells (PBMCs) stored in dimethyl sulfoxide for primary erythroid cultures. From the same samples, red blood cells were processed on a MACS column with CD71+ MicroBeads (MACS, Miltenyi Biotec, Auburn, CA) to isolate reticulocytes for total RNA extraction using TRIzol (ThermoFisher).
Genome-wide miRNA microarray analysis
The quality of RNA was assessed using an Agilent 2100 Bioanalyzer followed by hybridization to the miRCURY LNA micro-RNA Array (Exiqon, Woburn, MA). Raw data were quantile normalized using a model-based correction algorithm (http://linus.nci.nih.gov/BRB-ArrayTools.html). miRNA gene expression profiling was conducted for SCD patients with HbF < 8.6% (low HbF) or HbF > 8.6% (high HbF) using principal component analysis (NIA Array Analysis Tool; https://lgsun.irp.nia.nih.gov/ANOVA/index.html). Microarray raw data were submitted to the Gene Expression Omnibus (GEO) under database accession number GGSE111356.
Reverse transcription - quantitative polymerase chain reaction (RT-qPCR) analysis
To quantify miR-144 levels, the miScript II RT and SYBRGreen PCR kit (Qiagen) were used and relative expression determined as described previously [21] to confirm miR-144 expression obtained by microarray for the 12 samples analyzed and in vitro functional studies. To quantify mRNA of γ-globin, β-globin, βS-globin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), we generated standard curves described previously [21]. Levels of NRF2, CD71, and CD235a were measured using the RT2-qPCR Primer Assay system (Qiagen, Valencia, CA) as described previously by our group [21]. All gene expression levels normalized to GAPDH mRNA.
Tissue culture
Human erythroid progenitors were generated from adult CD34+ stem cells (STEMCELL Technologies, Vancouver, BC) or sickle PBMCs in a two-phase culture system established in our laboratory [23,24]. During phase I, CD34+ stem cells were cultured in alpha minimum essential medium containing interleukin-3 (10 ng/mL), stem cell factor (10 ng/mL), and erythropoietin (2 IU/mL) to promote erythroid lineage commitment. On day 7, cells transitioned to phase II medium that was identical except that stem cell factor was removed. Erythroid progenitors were transfected on day 8 with human mature miR-144 or negative control mimics (Dharmacon, Lafayette, CO) by nucleofection using the Amaxa Human CD34+ Cell Nucleofector Kit. After 2 days, cells were harvested for flow cytometry, Western blot, and RT-qPCR analysis. In a second set of studies, we determined the effect of longer treatment using erythroid progenitors generated from normal CD34+ stem cells or sickle PBMCs treated on day 5 and then harvested on day 10 for gene expression and protein analysis.
KU812 cells maintained in Iscove’s modified Dulbecco’s medium supplemented with 10% fetal bovine serum were used for mechanistic studies. Cells were treated with 25–75 μmol/L hemin alone or pretreatment with 300 nmol/L miR-144 or negative control for 24 hours followed by 50 μmol/L hemin for 48 hours and then flow cytometry, Western blot, and RT-qPCR analysis completed.
Flow cytometry analysis
After the different treatments, cells were washed, fixed with 4% paraformaldehyde, and stained with fluorescein-isothiocyanate-conjugated anti-HbF (Thermo Fisher Scientific) or anti-CD235a and anti-CD71 antibodies (eBioscience, San Diego, CA); flow cytometry analysis was performed on an LSRII flow cytometer using gating parameters previously published by our group [23,24]. We routinely acquire 10,000 erythroid cells to quantify HbF positive cells (F-cells) shown in histograms.
To detect reactive oxygen species (ROS) levels, KU812 cells were incubated with 5 μmol/L dichlorodihydrofluorescein diacetate (DCF-DA) (Sigma-Aldrich) for 4 hours before harvest. The percentage of F-cells and DCF-positive cells were quantified using FACS Diva software.
Western blot analysis
Total protein was isolated and Western blot performed with 10–30 μg of protein [23,24] with HbF (sc-21756), HbS (sc-37–8 from Santa Cruz Biotechnology, Santa Cruz, CA), NRF2 (ab62352, Abcam), and tubulin (sc-53646; Santa Cruz) antibodies. The immunoblots were developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) and analyzed on a Fujifilm LAS-3000 gel imager (Stamford, CT) to acquire quantitative data.
Chromatin immunoprecipitation assay
Sickle erythroid progenitors were used for chromatin immunoprecipitation (ChIP) assay as described previously by our group [24]. Immunoprecipitations with anti-NRF2 and anti-TATA-binding protein (TBP) antibodies, along with an immunoglobulin G (IgG) control, were completed and chromatin isolated for qPCR analysis to quantify chromatin enrichment compared with input DNA.
Statistical analysis
The data are reported as the mean ± standard error of the mean of three to five replicates of independent experiments performed in triplicate. All data were analyzed by a two-tailed Student t test and p < 0.05 was considered statistically significant. Binary regression analysis determined the correlation between miR-144 levels obtained by microarray and RT-qPCR analysis.
Results
Individuals with SCD and contrasting HbF levels show differentially expressed miRNA genes
The role of miRNA genes in normal erythropoiesis [25,26] and globin expression has been demonstrated [19–21,27,28]. Therefore, the goal of the present study was to define novel miRNA genes differentially expressed in persons with SCD and contrasting high and low HbF levels. After obtaining informed consent, blood samples were collected from study subjects with confirmed HbSS genotype and clinical phenotype data (Supplementary Table E1, online only, available at www.exphem.org). We collected complete blood cell counts with differential, reticulocyte counts, and HbF levels (Table 1); HbF levels ranged from 0.1% to 30.6%. None of the other hematologic values was significantly different between the two groups except HbF, suggesting that the red blood cell turnover and hemolysis rates were similar between groups.
Table 1.
Summary of clinical phenotype data for sickle cell patients used in the miRNA analysisa
| High-HbF Group | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Subject # | Hgb (g/dl) | Hct (%) | Pits (× 103) |
WBC (× 103) |
Neutrophils ( × 103) |
Lymphocytes × ( 103) |
NRBC(%) | Reticulocyte Count (%) |
HbF (%) |
| 001 | 9.5 | 28.6 | 269 | 8.1 | 5.8 | 1.3 | 0 | 5.0 | 30.6 |
| 003 | 10.5 | 30.9 | 563 | 11.4 | 5.0 | 4.3 | 0 | 5.7 | 21.8 |
| 08A | 7.4 | 21.7 | 192 | 4.2 | 2.1 | 1.3 | 3 | 8.8 | 25.0 |
| 014 | 8.1 | 22.9 | 469 | 15.1 | 5.7 | 7.7 | 0 | 9.8 | 16.8 |
| 015 | 8.7 | 25.4 | 416 | 14.1 | 5.1 | 7.5 | 0 | 8.8 | 27.5 |
| 016 | 7.6 | 27.2 | 499 | 17.5 | 7.5 | 7.4 | 1 | 12.7 | 19.2 |
| Mean ± SEM 8.63 ± 0.49 Low-HbF Group |
26.11 ± 1.42 | 404.67 ± 60.17 | 11.7 ± 2.00 | 5.2 ± 0.72 | 4.917 ± 1.25 | 0.67 ± 0.49 | 8.467 ± 1.15 | 23.48 ± 2.12 | |
| 004 | 7.1 | 20.1 | 380 | 15.5 | 6.2 | 5.9 | 6 | 10.5 | 3.8 |
| 008 | 7.0 | 20.1 | 693 | 11.1 | 4.2 | 6.2 | 2 | 13.4 | 3.0 |
| 009 | 7.8 | 24.8 | 641 | 13.8 | 8.7 | 3.7 | 0 | 9.0 | 0.9 |
| 011 | 7.6 | 22.8 | 570 | 15.60 | 8.2 | 5.70 | 2 | 12.0 | 6.2 |
| 011A | 8.4 | 28.9 | 461 | 10.8 | 5.9 | 2.5 | 0 | 8.2 | 6.2 |
| 012A | 8.9 | 28.1 | 384 | 10.9 | 4.9 | 4.5 | 1 | 5.6 | 0.1 |
| Mean ± SEM 7.8 ± 1.30 | 24.13 ± 1.56 | 521 ± 54.35 | 12.95 ± 0.94 | 6.35 ± 0.73 | 4750 ± 0.59 | 1.83 ± 1.02 | 9.783 ± 1.42 | 3.37 ± 1.05 | |
| p valuesb | 0.1766 | 0.3698 | 0.1802 | 0.5943 | 0.2876 | 0.9067 | 0.1859 | 0.4353 | 0.0001* |
Shown are the values obtained for the individual complete blood counts and differential and reticulocyte counts for the 12 children and adults (011A and 012A) with HbSS included in the miRNA analysis. The mean ± standard error of the mean (SEM) is shown for each parameter. The Student t test was used to determine significant difference between the two study groups; p < 0.05 was considered statistically significant.
p values generated using the Student t test for data collected for each parameter for the two groups.Hgb=hemoglobin, Hct= hematocrit, Plts=platelet count, WBC=white blood cell count, NRBC=nucleated red blood cells
Twelve individuals were included in the miRNA microarray analysis (Figure 1A), including a high-HbF group (average HbF 23.48±2.12) and a low-HbF group (average HbF 3.37±1.02). Total RNA isolated from CD71+ reticulocytes was analyzed on the miRCURY LNA microRNA Array. After raw data normalization, principal component analysis identified 89 and 91 unique miRNA genes upregulated and downregulated, respectively, in the low-HbF group compared with the high-HbF group (Supplementary Table E2, online only, available at www.exphem.org). miR-144–3p (miR-144) and miR-144–5p expression was increased 7.96-fold (p = 0.0010) and 7.79-fold (p = 0.0037), respectively, in the low-HbF group compared with the high-HbF group. Other miRNA genes such as miR-96–5p and let-7b-5p implicated in globin gene regulation [19,20] showed enhanced expression in the low-HbF group, suggesting testable hypotheses that miRNA genes in this group might contribute to γ-globin transcription by silencing trans-activator DNA-binding proteins. Likewise, we identified the top miRNA genes downregulated in low-HbF group, such as miR-1, miR-5701, and miR-2116–3p (Supplementary Table E2, online only, available at www.exphem.org), that might silence repressors of γ-globin expression.
Figure 1.
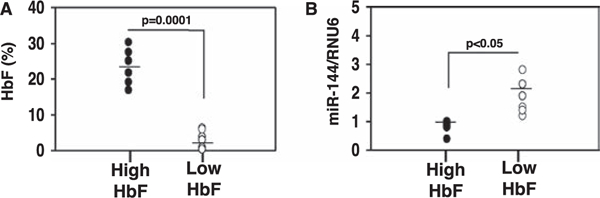
miR-144 was differentially express between individuals with SCD and low- or high-HbF phenotypes. Total RNA was isolated from the reticulocytes of 12 individuals with SCD and high HbF (n = 6) and low HbF (n = 6) for genome-wide miRNA gene expression profiling and RT-qPCR analysis (see Methods). (A) Mean HbF level for each study group is shown by the black horizontal lines. (B) Mature miR-144 expression levels were measured by RT-qPCR using RNA isolated from the 12 subjects and correlated with HbF levels after normalization by the internal control RNU6. Mean levels are shown (black line).
miR-144 levels observed by microarray analysis are confirmed by RT-qPCR
The oxidative stress conditions observed in sickle cell patients is associated with high miR-144 levels and severe anemia [29]; furthermore, NRF2 is a direct target of miR-144-mediated gene silencing. We and others [23,24,30] have demonstrated that NRF2 activates γ-globin gene transcription through binding the proximal promoter antioxidant response element (ARE), but data have not been published to demonstrate a connection between miR-144 and HbF expression. Therefore, using RNA isolated from reticulocytes, we quantified miR-144 levels by RT-qPCR to correlate with those obtained by microarray analysis (Table 2). Using regression statistical analysis, we observed an R2 value of 0.769, supporting a correlation between the miR-144 levels obtained by the two methods. Furthermore, similar to clinical HbF phenotypes, the miR-144 levels measured by RT-qPCR distributed between our high- and low-HbF groups (Figure 1B), supporting an association between miR-144 and HbF expression.
Table 2.
Summary of miR-144 levels for individual patients to test the correlation between miR-144 level in the microarray and RT-qPCR analyses
| Subject # | miR-144 Level Microarray |
miR-144/U6 Level qRT-PCR |
|---|---|---|
| 001 | 0.6081 | 0.8621 |
| 003 | 0.4275 | 0.403 |
| 08A | 0.0157 | 0.0826 |
| 014 | 0.0829 | 0.0362 |
| 015 | 0.4537 | 0.7195 |
| 016 | 0.1808 | 0.0882 |
| 004 | 1.3849 | 1.1565 |
| 008 | 0.8330 | 1.4985 |
| 009 | 0.4357 | 1.6691 |
| 011 | 0.5448 | 1.1845 |
| 011A | 1.618 | 1.5253 |
| 012A | 2.580 | 3.773 |
NRF2 gene silencing by miR-144 represses HbF expression in normal erythroid progenitors
To gain evidence of a mechanistic role of miR-144 in HbF regulation, we conducted studies in normal erythroid progenitors generated from CD34+ stem cells in our two-phase liquid culture system [23,24]. By day 7 in culture, erythroid progenitors showed an average 76.9% CD235a+ cells and 57.2% CD71+ cells. On day 8, progenitors were transfected with 200 nmol/L and 300 nmol/L miR-144 mimic or antagomir (anti-144) alone or combined treatment. We observed a 30–50% decrease in NRF2 mRNA levels after miR-144 overexpression (Figure 2A). Conversely, inhibition of endogenous miR-144 by antagomir alone produced a 1.4-fold increase in NRF2 gene expression (p < 0.001) above baseline (Figure 2A). For combination treatment, NRF2 silencing by miR-144 was inhibited by 300 nmol/L antagomir. To gain evidence for an association with globin gene transcription, the γ/γ+β mRNA ratio showed γ-globin silencing by miR-144 mimic (Figure 2B); moreover, antagomir produced a dose-dependent increase in γ-globin gene transcription by 1.3-fold and 2.0-fold, respectively. Analysis of the individual globin genes showed that miR-144 had no effects on β-globin gene expression (Supplementary Figure E1, online only, available at www.exphem.org). These data provide experimental evidence for γ-globin regulation in part by miR-144.
Figure 2.
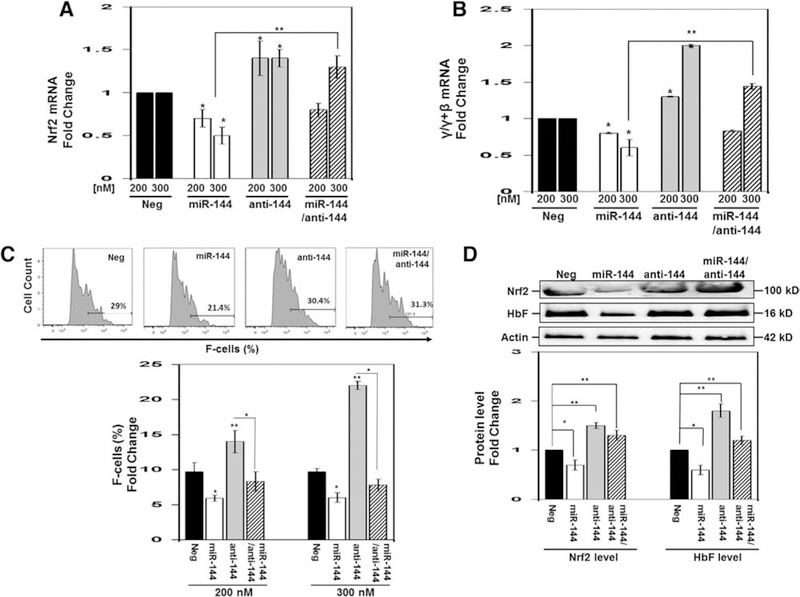
Overexpression of miR-144 repressed NRF2 and γ-globin gene expression in normal erythroid progenitors. Using CD34+ stem cells, adult erythroid progenitors were produced as described previously by our group [23,24] (see Methods for details). On day 8, we transfected cells with negative control, miR-144, and miR-144 antagomir (anit-144) mimics at 200 nmol/L and 300 nmol/L concentrations. In addition, combination treatments were conducted with 300 nmol/L each of miR-144 and antagomir. After 48 hours, cells were harvested for the different analyses. (A) RT-qPCR was performed to quantify NRF2 mRNA levels normalized to the internal control GAPDH. The negative mimic control (Neg) data were set to 1 to calculate the fold change in mRNA expression. Data are shown as the mean ± standard error of the mean with p < 0.05 considered statistically significant; *p < 0.05, **p < 0.01. (B) RT-qPCR was performed to quantify γ/γ+β-globin mRNA ratio calculated after the individual γ-globin and β-globin gene expression levels was normalized to the internal control GAPDH. See (A) for details of data analysis. (C) Representative histograms (top) of the flow cytometry analysis to determine the percentage of F-cells quantified in the graph using FACS Diva software. (D) Western blot analysis of NRF2 and HbF protein levels. Shown is a representative gel (top) where β-actin was the protein loading control. Densitometry analysis for the Western blot analysis was used to generate raw data shown in the graph for NRF2 and HbF.
Because miRNA molecules act primarily through post-transcriptional gene silencing, we conducted flow cytometry to determine the effects of miR-144 on HbF distribution by flow cytometry. Treatment with 200 nmol/L and 300 nmol/L miR-144 decreased F-cells from 9.7% to 6% (p < 0.05), respectively (Figure 2C). By contrast, the silencing effects of miR-144 was reversed by antagomir increasing F-cells a maximal 2.3-fold (22.3%) at the 300 nmol/L concentrations (p < 0.01). To quantify protein levels, we performed Western blot analysis in which treatment with miR-144 decreased NRF2 and HbF by 40% and 50%, respectively (Figure 2D). By contrast, antagomir reversed this effect increasing both NRF2 and HbF by 1.5-fold and 1.8-fold, respectively (p < 0.01). These data in erythroid progenitors support miR-144 as a repressor of HbF expression through NRF2 gene silencing.
Extended miR-144 antagomir treatment increases HbF expression in normal and sickle erythroid progenitors
To gain further evidence of the sustained effects of antagomir treatment on HbF induction under oxidative conditions, we conducted studies using normal and sickle erythroid progenitors generated from CD34+ stem cells and PBMCs treated for 5 days with 300 nmol/L miR-144 or antagomir (see Methods). To confirm overexpression, we achieved a twofold increase in miR-144 and 40% decrease in endogenous levels with antagomir treatment (Figure 3A). Similar to the 2 day treatment, antagomir produced a sustained increase in γ-globin expression of 2.3-fold and combination treatment reversed the 45% γ-globin gene silencing mediated by miR-144 mimic (Figure 3B). By contrast, changes in β-globin and α-globin expression were not significant. Interestingly, treatment with antagomir increase CD71 expression with no significant change in CD325a levels (Figure 3B). By flow cytometry analysis, miR-144 mimic and antagomir were shown to produce a 20% decrease and 2.2-fold (increase from 8% to 18% F-cells) increase in F-cells, respectively (Figure 3C). Western blot confirmed a decrease in NRF2 protein levels by miR-144 that was reversed by antagomir treatment (Figure 3D) along with HbF induction.
Figure 3.
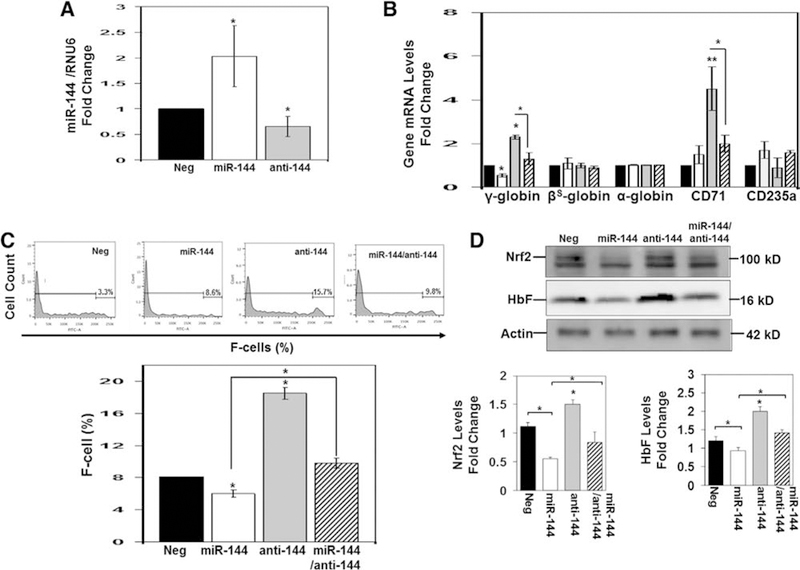
Extended treatment with miR-144 produces a sustained increase in HbF expression in normal erythroid progenitors. Using CD34+ stem cells, erythroid progenitors were produced, followed by treatment for 5 days with negative control (Neg), miR-144, antagomir (anit-144), or combination treatments at a 300 nmol/L concentration. (A) Mature miR-144 levels were measured by RT-qPCR and normalized to RNU6 (internal control). The Neg control data (black bar) were set to 1 to calculate the fold change in miR-144 mimic (white bar) or miR-144 antagomir (gray bar) expression. Data are shown as the mean ± standard error of the mean with p < 0.05 considered statistically significant. (B) RT-qPCR was used to quantify the genes shown after normalization to the internal control GAPDH. Shown is combination treatment for miR-144 and antagomir (striped bar). (C) Representative histograms (top) generated by flow cytometry analysis to determine the percentage of F-cells quantified using FACS Diva software to generate the graph. (D) Representative Western blot gel where β-actin was the protein loading control (top). Densitometry analysis was used to generate raw data shown in the graphs (bottom).
Similar studies were conducted with sickle erythroid progenitors. For untreated cells, we observed robust cell growth and viability, increased CD71 and CD235a expression over time, and a γ-globin to βS-globin switch around day 7 in culture (Supplementary Figure E2, online only, available at www.exphem.org). Similar to results obtained in normal erythroid progenitors, antagomir produced a sustained 2.0-fold increase in γ-globin expression without affecting βS-globin mRNA levels (Figure 4A). Western blot confirmed a decrease in NRF2 levels by miR-144 treatment that was reversed by antagomir with a significant 1.8-fold induction of HbF (Figure 4B). To further support HbF induction, F-cells levels increased 1.9-fold in sickle erythroid progenitors (Figure 4C).
Figure 4.
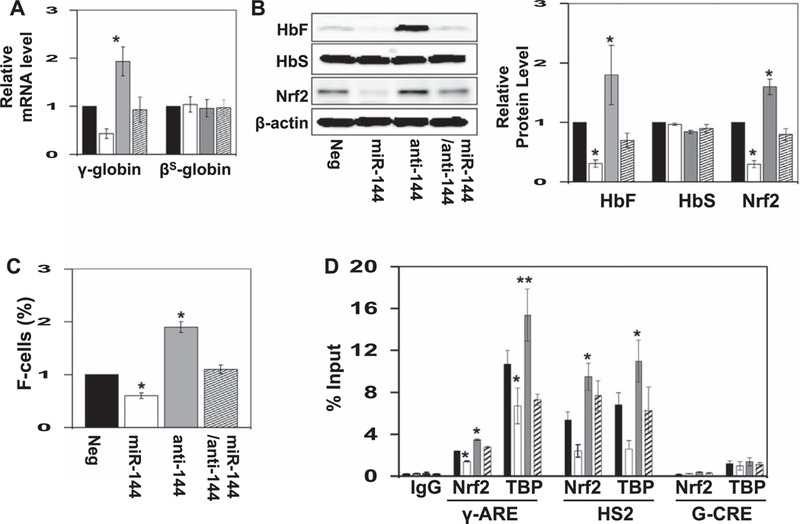
miR-144 treatment produces an increase in HbF expression in sickle erythroid progenitors. Using PBMCs, sickle erythroid progenitors were produced and treated with the various agents for 5 days (see Methods). (A) RT-qPCR was used to quantify the genes shown after normalization to the internal control GAPDH; data are shown as the mean ± standard error of the mean. (B) Western blot analysis of HbF, HbS, and NRF2 protein levels. Shown is a representative gel where β-actin was the protein loading control (left). Densitometry analysis for the western blot analysis was used to generate raw data shown in the graph. (C) F-cell levels quantified using FACS Diva software (see Methods). (D) Sickle erythroid progenitors were treated with the various reagents for 5 days and then ChIP assay was conducted with IgG, NRF2, and TBP antibody immunoprecipitation reactions. In the graph, data are shown for chromatin enrichment as the percentage of input DNA (% input), for negative control (black bar), miR-144 (white bar), miR-144 antagomir (gray bar), and combination treatment (striped bar). All data are shown as the mean ± standard error of the mean. Shown is the in vivo occupancy of the three proteins at the γ-globin ARE (γ-ARE), the locus control region hypersensitive site 2 (HS2), and negative control G-CRE region, not containing a known ARE-binding motif.
NRF2 binding in the γ-globin ARE is altered by miR-144 in sickle erythroid progenitors
To gain direct evidence of a molecular mechanism of γ-globin regulation by miR-144-mediated NRF2 silencing, we conducted DNA-binding studies to determine the effects on NRF2 binding to the γ-globin ARE [31,32]. In sickle erythroid progenitors, baseline NRF2 binding was inhibited 40% (p < 0.05) by miR-144, which was reversed by antagomir with 1.5-fold increased NRF2 binding in vivo (Figure 4D). We observed significant TBP binding in the proximal promoter region that increased 1.4-fold after antagomir treatment, supporting γ-globin transcription activation (Figure 4D). Likewise, NRF2 binding to the locus control region hypersensitive site 2 (HS2) was increased 1.8-fold (p < 0.01) by antagomir (Figure 4D) and miR-144 reduced occupancy by 60%. To confirm the specificity of in vivo NRF2 binding, negative control studies conducted in the Gγ-globin cyclic AMP response element (G-CRE). The level of NRF2 binding did not changed significantly in this region for any treatment condition (Figure 4D).
miR-144 represses NRF2 expression under oxidative conditions in KU812 cells
To gain additional evidence for a miR-144/NRF2-mediated mechanism of γ-globin regulation, we developed an oxidative stress model using KU812 cells treated with hemin. To measure ROS levels, DCF-DA staining and flow cytometry were conducted; the level of DCF-positive cells increased from 16% (untreated) to 50% (p < 0.01) at 75 μmol/L hemin (Figure 5A). Moreover, hemin produced a dose-dependent 4-fold (p < 0.01) increase in NRF2 and 2-fold (p < 0.05) increase in HbF levels (Figure 5B) and F-cells from 20% to 54% (p < 0.01) at the highest hemin concentration (Figure 5C). miR-144 targets the 3′-untranslated region of NRF2 to produce gene silencing [29]. Because miR-144 decreased NRF2 levels in sickle erythroid cells, we conducted studies to recapitulate the intracellular environment of sickle reticulocytes. KU812 cells were transfected with 300 nmol/L miR-144 or negative mimic control for 24 hours, followed by treatment with 50 μmol/L hemin for 48 hours. We confirmed a 12-fold increase in miR-144 in KU812 cells that was inhibited 25% by hemin (Figure 6A). In contrast to primary cells (Figure 2B), treatment with miR-144 alone did not alter γ-globin gene transcription significantly, but the ability of hemin to induce γ-globin gene transcription was inhibited 85% by miR-144 (Figure 6B). Under the same conditions, hemin produced a significant 4-fold increase in NRF2 and HbF (p < 0.05) expression (Figure 6C). Moreover, overexpression of miR-144 inhibited the ability of hemin to activate both targets by 50–80%.
Figure 5.
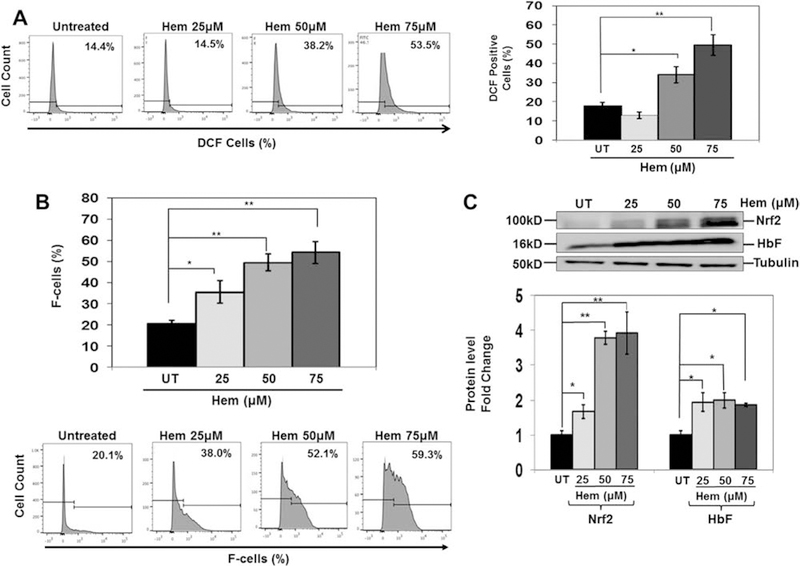
Oxidative stress conditions in KU812 cells alter NRF2 and HbF expression. KU812 cells treated with different hemin concentrations for 48 hours were harvested for analysis. (A) Shown are representative histograms of the percentage of DCF-positive cells determined by flow cytometry. Quantitative data were collected by FACS Diva software. (B) Representative histograms for F-cells analysis of the different treatment conditions (bottom). The quantitative data generated by FACS Diva were used to generate the graph (top). (C) Representative Western blots for NRF2 and HbF protein expression. Densitometry data were collected and used to generate the graph.
Figure 6.
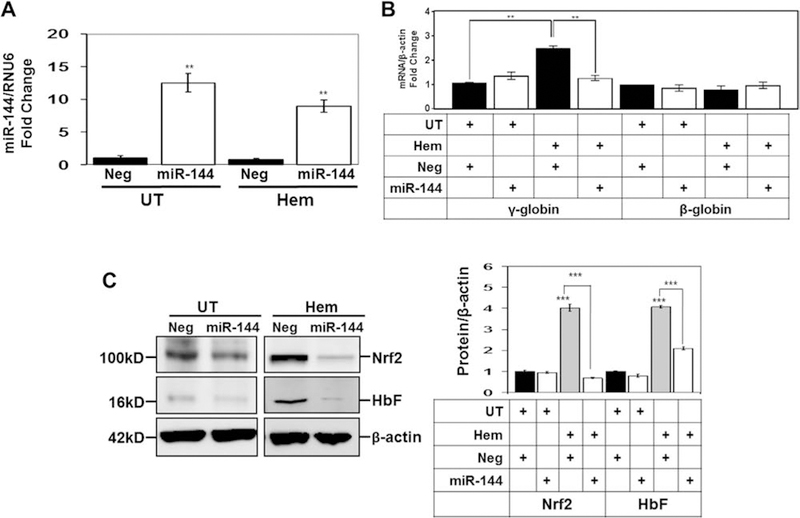
Pretreatment with miR-144 mimic blocks the ability of hemin to stimulate NRF2 and HbF expression. KU812 cells were transfected with 300 nmol/L miR-144 or Neg control mimic for 24 hours, followed by treatment with 50 μmol/L hemin for 48 hours, and then cells were harvested for analysis. (A) RT-qPCR to measured mature miR-144 expression normalized to RNU6 mRNA levels and the expression level for negative control was normalized to one. (B) RT-qPCR was used to quantify γ-globin and β-globin mRNA levels normalized to GAPDH. The table below the graph shows the different treatment conditions tested. UT=untreated; Hem=hemin; Neg=negative mimic control. (C) Western blot analysis of NRF2 and HbF expression was conducted and tubulin was used as the protein loading control. Shown are a representative gel (left) and the quantitative data generated by densitometry analysis (right). The table labels are the same as described in (B).
Discussion
Investigating molecular mechanisms involved in γ-globin gene regulation will facilitate the development of therapeutic agents to induce HbF for the treatment of β-hemoglobinopathies. To date, hydroxyurea is the most effective HbF inducer for treating SCD and β-thalassemia [35,36]. However, other agents such as decitabine [37], RN-1 [38], and bensarizide [39] that act by different molecular mechanisms are under investigation to expand the repertoire of drugs available to treat SCD. Our group demonstrated HbF induction in sickle erythroid progenitors by dimethyl fumarate through enhanced NRF2 expression [23,24,40].
Other promising molecules, including miRNA mimics and antagomirs that target human diseases such as cancer, diabetes, and polycythemia vera [33,34], are under development. Recent studies to expand approaches to inhibit hemoglobin switching have focused on posttranslational mechanisms mediated by miRNA molecules. Experimental data support a role of miR15a/16–1, miR-96, miR-34a, and let-7 in γ-globin regulation [18–21], although specific DNA mutations have not been demonstrated. Further evidence of the physiologic role of miRNA in HbF induction was illustrated by Walker et al., who showed that miR-151–3p and others correlated with the maximal tolerated dose of hydroxyurea in children with SCD [41]. The association of miR-486–3p and miR-210 with HbF expression in β-thalassemia patients [27,42] was verified; however, a direct causative relationship has not been established.
To discover additional targets involved in γ-globin regulation, we investigated variations of miRNA gene expression in reticulocytes isolated from individuals with SCD and contrasting low or high HbF levels. We observed higher miR-144 gene expression levels in the low-HbF group. Because we did not identify a predicted miR-144-binding site in the 3′-untranslated region of the γ-globin mRNA, our findings suggested that miR-144 might repress γ-globin indirectly by silencing a DNA trans-activator. Therefore, we conducted studies to test the hypothesis that repression of NRF2 by miR-144 is a mechanism of γ-globin silencing in SCD.
Interestingly, the top two upregulated miRNA genes, miR-144–5p and miR-144–3p, transcribe on the same hairpin loop DNA structure. We tested miR-144–3p (miR-144); however, involvement of miR-144–5p in γ-globin regulation was not excluded. We chose miR-144 because it correlates with severe anemia and reduced redox potential in adults with SCD [43] and is known to silence NRF2 gene expression through post-translational mechanisms [29].
Several miRNA genes [44–46] regulate erythropoiesis by inhibiting proliferation and differentiation [44,45] and by protecting erythroid cells from oxidative stress [46]. The bicistronic miR-144/miR-451 gene cluster transcribes as a primicroRNA (miR144/451) that is regulated by GATA-1 [47]. In fact, knockdown of miR-451 decreases erythroid differentiation in zebrafish and mice [47,48], but miR-144 knockdown had negligible effects on erythropoiesis [48].
The miRNA gene profiling conducted in sickle reticulocytes herein identified miR-144 as a highly differentially expressed gene in SCD patients with high and low HbF levels. Subsequent in vitro analysis in normal and sickle erythroid progenitors under oxidative stress support a miR-144/NRF2 model of γ-globin globin gene regulation (Figure 7). We observed NRF2 gene silencing by miR-144 overexpression and parallel repression of γ-globin gene transcription and HbF synthesis that reversed with anti-miR-144 antagomir treatment. Previous studies related miR-144 and NRF2 expression to oxidative stress, redox potential, and anemia in SCD [29,43]; however, a link between miR-144 and HbF expression has not been established. Although our studies do not exclude other mechanisms, antagomir treatment that directly block miR-144 in sickle erythroid progenitors supports a role of this pathway in γ-globin regulation.
Figure 7.
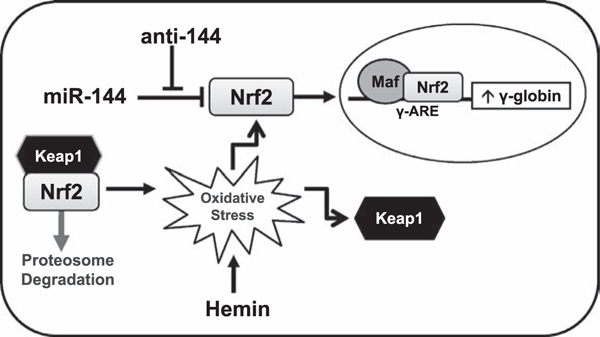
Model of miR-144/NRF2-mediated γ-globin gene silencing. Shown is a model of how miR-144 silences NRF2 gene expression and decreases binding in the γ-globin ARE, leading to gene repression and low HbF levels. Treatment with anti-144 (antagomir) reversed the negative effects of miR-144, enhancing NRF2 levels and translocation to the nucleus, where NRF2 binds the γ-globin ARE to stimulate transcription. Hemin treatment of KU812 cells created an oxidative stress model to recapitulate intracellular conditions in sickle erythrocytes and stimulate NRF2 expression. Recently, we demonstrated NRF2 interaction with its DNA-binding partners MAF to trans-activate γ-globin gene transcription [24].
Under low oxidative stress, NRF2 is sequestered in the cytoplasm by KEAP1 [31,49] and subjected to proteasome degradation. When oxidative stress conditions exist, KEAP1 is inactivated and NRF2 levels increase, prompting targeted activation of antioxidant proteins such as HMOX1 and NQO1, among others, to control cellular ROS levels. Because untreated KU812 cells have low ROS levels, we observed low NRF2 protein levels due to KEAP1 sequestration. However, after hemin treatment, ROS levels increased, which recapitulates chronic hemolysis and intracellular conditions of sickle erythrocytes that stimulated NRF2 expression and HbF synthesis, and pretreatment with miR-144 reversed these findings. Interestingly, hemin produced a dose-dependent increase in NRF2, whereas maximal HbF expression was observed at the lowest concentration, suggesting that NRF2 contributes in part to γ-globin gene activation. Furthermore, antagomir enhanced in vivo NRF2 and TBP occupancy in the γ-globin ARE and HS2 that was inhibited by miR-144. These findings support the ability of miR-144 to silence NRF2 expression and binding in the γ-globin ARE to regulate gene expression.
Conclusions
Oxidative stress and inflammation play a major role in the pathophysiology of SCD, so drugs that induce HbF and reduce oxidative stress have greater clinical efficacy [29,50,51,52]. There is mounting evidence that treatment with NRF2 activators in SCD mice reduces oxidative stress while inducing HbF [23,24,29,30,40]. Furthermore, we have demonstrated that NRF2 is required for HbF induction by dimethyl fumarate [23,40,53]. More recently, we verified the requirement for NRF2 in hemoglobin switching during embryonic mouse development using an SCD/NRF2 knockout mouse model [54]. These findings support the development of miRNA mimics or antagomirs as HbF inducers. Additional studies in the preclinical SCD mouse model are required to determine the efficacy of miR-144 antagomir as a therapeutic agent in vivo.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01 HL069234 to BSP and HL117684 to support training for XZ).
Footnotes
Conflict of interest disclosure
The authors declare no competing financial interests.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.10167j.exphem.2018.11.002.
References
- 1.Poillon WN, Kim BC, Rodgers GP, Noguchi CT, Schechter AN. Sparing effect of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S at physiologic ligand saturations. Proc Natl Acad Sci USA. 1993;90:5039–5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. [DOI] [PubMed] [Google Scholar]
- 3.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010;85:403–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39:1197–1199. [DOI] [PubMed] [Google Scholar]
- 5.Thein SL, Menzel S, Peng X, Best S, et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci U S A. 2007;104:11346–11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uda M, Galanello R, Sanna S, Lettre G, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA. 2008;105:1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bae HT, Baldwin CT, Sebastiani P, et al. Meta-analysis of 2040 sickle cell anemia patients: BCL11A and HBS1L-MYB are the major modifiers of HbF in African Americans. Blood. 2012; 120:1961–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mtatiro SN, Singh T, Rooks H, et al. Genome wide association study of fetal hemoglobin in sickle cell anemia in Tanzania. PLoS One. 2014;9:e111464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842. [DOI] [PubMed] [Google Scholar]
- 10.Xu J, Peng C, Sankaran VG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334:993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J, Bauer DE, Kerenyi MA, Vo TD, Hou S, Hsu YJ, Yao H, Trowbridge JJ, Mandel G, Orkin SH. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proc Natl Acad Sci USA. 2013;110:6518–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roosjen M, McColl B, Kao B, Gearing LJ, Blewitt ME, Vadolas J. Transcriptional regulators Myb and BCL11A interplay with DNA methyltransferase 1 in developmental silencing of embryonic and fetal β-like globin genes. FASEB J. 2014;28: 1610–1620. [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Sankaran VG, Ni M, et al. Transcriptional silencing of {gamma}-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010;24:783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou D, Liu K, Sun CW, Pawlik KM, Townes TM. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat Genet. 2010;42:742–744. [DOI] [PubMed] [Google Scholar]
- 15.Borg J, Papadopoulos P, Georgitsi M, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42:801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stadhouders R, Aktuna S, Thongjuea S, et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J Clin Invest. 2014;124:1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sankaran VG, Xu J, Byron R, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365: 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azzouzi I, Moest H, Winkler J, et al. MicroRNA-96 directly inhibits gamma-globin expression in human erythropoiesis. PLoS One. 2011. ;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee YT, de Vasconcellos JF, Yuan J, et al. LIN28B-mediated expression of fetal hemoglobin and production of fetal-like erythrocytes from adult human erythroblasts ex vivo. Blood. 2013;122:1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward CM, Li B, Pace BS. Original research: Stable expression of miR-34a mediates fetal hemoglobin induction in K562 cells. Exp Biol Med. 2016;241:719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foley HA, Ofori-Acquah SF, Yoshimura A, Critz S, Baliga BS, Pace BS. Stat3 beta inhibits gamma-globin gene expression in erythroid cells. J Biol Chem. 2002;277:16211–16219. [DOI] [PubMed] [Google Scholar]
- 23.Promsote W, Makala L, Li B, et al. Monomethylfumarate induces gamma-globin expression and fetal hemoglobin production in cultured human retinal pigment epithelial (RPE) and erythroid cells, and in intact retina. Invest Ophthalmol Vis Sci. 2014;55: 5382–5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu X, Li B, Pace BS. NRF2 mediates γ-globin gene regulation and fetal hemoglobin induction in human erythroid progenitors. Haematologica. 2017;102: e285–e288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim M, Tan YS, Cheng WC, Kingsbury TJ, Heimfeld S, Civin CI. MIR144 and MIR451 regulate human erythropoiesis via RAB14. Mol Med Rep. 2017;15:2495–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leecharoenkiat K, Tanaka Y, Harada Y, et al. Plasma micro-RNA-451 as a novel hemolytic marker for β0-thalassemia/HbE disease. Br J Haematol. 2015;168:583–597.25312678 [Google Scholar]
- 27.Lulli V, Romania P, Morsilli O, et al. MicroRNA-486–3p regulates γ-globin expression in human erythroid cells by directly modulating BCL11A. PLoS One. 2013;8:e60436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saki N, Abroun S, Soleimani M, et al. MicroRNA expression in β-thalassemia and sickle cell disease: A role in the induction of fetal hemoglobin. Cell J. 2016;17:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sangokoya C, Telen MJ, Chi JT. MicroRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood. 2010;116:4338–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macari ER, Lowrey CH. Induction of human fetal hemoglobin via the NRF2 antioxidant response signaling pathway. Blood. 2011;117:5987–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi A, Kang MI, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of NRF2. Mol Cell Biol. 2004; 24:7130–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itoh K, Chiba T, Takahashi S, et al. An NRF2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. [DOI] [PubMed] [Google Scholar]
- 33.Detassis S, Grasso M, Del Vescovo V, Denti MA. microRNAs make the call in cancer personalized medicine. Front Cell Dev Biol. 2017;5:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mascarenhas J, Roper N, Chaurasia P, Hoffman R. Epigenetic abnormalities in myeloproliferative neoplasms: a target for novel therapeutic strategies. Clin Epigenetics. 2011;2:197–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med.. 1995;332:1317–1322. [DOI] [PubMed] [Google Scholar]
- 36.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011;377: 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Molokie R, Lavelle D, Gowhari M, et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: A randomized phase 1 study. PLoS Med. 2017;14:e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui S, Lim KC, Shi L, et al. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice. Blood. 2015;126:386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boosalis MS, Sangerman JI, White GL, et al. Novel inducers of fetal globin identified through high throughput screening (HTS) are active in vivo in anemic baboons and transgenic mice. PLoS One. 2015;10:e0144660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishnamoorthy S, Pace B, Gupta D, et al. Dimethyl fumarate increases fetal hemoglobin, provides vascular protection and heme detoxification and corrects anemia in sickle cell disease. JCI Insight. 2017;2:96409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker AL, Steward S, Howard TA, et al. Epigenetic and molecular profiles of erythroid cells after hydroxyurea treatment in sickle cell anemia. Blood. 2011;118:5664–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lulli V, Romania P, Morsilli O, et al. MicroRNA-486–3p regulates γ-globin expression in human erythroid cells by directly modulating BCL11A. PLoS One. 2013;8:e60436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen SY, Wang Y, Telen MJ, Chi JT. The genomic analysis of erythrocyte microRNA expression in sickle cell diseases. PLoS One. 2008;3:e2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Listowski MA, Heger E, Boguslawska DM, et al. microRNAs: fine tuning of erythropoiesis. Cell Mol Biol Lett. 2013;18:34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Undi RB, Kandi R, Gutti RK. MicroRNAs as haematopoiesis regulators. Adv Hematol. 2013;2013:695754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Felli N, Pedini F, Romania P, et al. MicroRNA 223-dependent expression of LMO2 regulates normal erythropoiesis. Haematologica. 2009;94:479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dore LC, Amigo JD, Dos Santos CO, et al. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc Natl Acad Sci USA. 2008;105:3333–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rasmussen KD, Simmini S, Abreu-Goodger C, et al. The miR-144/451 locus is required for erythroid homeostasis. J Exp Med. 2010;207:1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by NRF2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Silva DG, Belini Junior E, de Almeida EA, Bonini-Domingos CR. Oxidative stress in sickle cell disease: an overview of erythrocyte redox metabolism and current antioxidant therapeutic strategies. Free Radic Biol Med. 2013;65:1101–1109. [DOI] [PubMed] [Google Scholar]
- 51.Kaddam L, Fadl-Elmula I, Eisawi OA, et al. Gum arabic as novel anti-oxidant agent in sickle cell anemia, phase II trial. BMC Hematol. 2017;17:4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Owusu-Ansah A, Choi SH, Petrosiute A, Letterio JJ, Huang AY. Triterpenoid inducers of NRF2 signaling as potential therapeutic agents in sickle cell disease: a review. Front Med. 2015;9:46–56. [DOI] [PubMed] [Google Scholar]
- 53.Belcher JD, Chen C, Nguyen J, et al. Control of oxidative stress and inflammation in sickle cell disease with the NRF2 activator dimethyl fumarate. Antioxid Redox Signal. 2017;26:748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu X, Xi C, Thomas B, Pace BS. Loss of NRF2 function exacerbates the pathophysiology of sickle cell disease in a transgenic mouse model. Blood. 131:558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.