

ABSTRACT
RhoGTPases are known regulators of intracellular actin dynamics that are important for maintaining endothelial barrier function. RhoA is most extensively studied as a key regulator of endothelial barrier function, however the function of the 2 highly homologous family-members (> 88%) RhoB and RhoC in endothelial barrier function is still poorly understood.
This study aimed to determine whether RhoA, RhoB and RhoC have overlapping or distinct roles in barrier function and permeability in resting and activated endothelium. By using primary endothelial cells in combination with siRNA transfection to establish individual, double or triple knockdown of the RhoA/B/C RhoGTPases, we found that RhoB, but not RhoA or RhoC, is in resting endothelium a negative regulator of permeability. Loss of RhoB accounted for an accumulation of VE-cadherin at cell-cell contacts. Thrombin-induced loss of endothelial integrity is mediated primarily by RhoA and RhoB. Combined loss of RhoA/B showed decreased phosphorylation of Myosin Light Chain and increased expression of VE-cadherin at cell-cell contacts after thrombin stimulation. RhoC contributes to the Rac1-dependent restoration of endothelial barrier function.
In summary, this study shows that these highly homologous RhoGTPases differentially control the dynamics of endothelial barrier function.
KEYWORDS: cytoskeleton, Endothelial barrier function, RhoA, RhoB, RhoC, RhoGTPases, VE-cadherin
Introduction
Endothelial cells (ECs) form the inner lining of the vasculature and act as a barrier between blood plasma and the underlying tissues. They play a critical role in many physiologic processes, such as the control of vasomotor tone, leukocyte transmigration, blood cell trafficking, thrombosis, permeability, angiogenesis and immunity.1 Consequently, endothelial dysfunction can lead to tissue damage and thus represents a hallmark of disease2,3 which needs to be carefully controlled. Evidence suggests that in ECs, proper regulation of intercellular junctions and the associated F-actin dynamics are among the most important factors controlling vascular homeostasis.4
Inter-endothelial cell-cell contacts comprise 2 types of junctional complexes, adherens junctions (AJs) and tight junctions (TJs). Belts of TJs are important for controlling the exchange of water, ions and small molecules over the endothelium and are most abundantly expressed in brain endothelium. In contrast, inter-endothelial sealing in other vascular and lymphatic endothelia are performed by AJs. AJs show increased dynamics, resulting in higher basal permeability as compared with TJs.2 AJs are mainly based on the homotypic adhesion molecule Vascular Endothelial cadherin (VE-cadherin), which clusters at endothelial cell-cell contacts in a calcium-dependent manner. The intracellular domain of VE-cadherin is linked to the F-actin cytoskeleton via adaptor protein such as α- and β-catenin.5 The actin cytoskeleton consists of F-actin filaments that are associated with myosin II and α-actinin, and are used to develop intracellular contractile forces. Differential signaling from VE-cadherin toward the actin cytoskeleton controls the opening and restoration of cell-cell contacts, making VE-cadherin one of the most essential adhesion molecules controlling endothelial integrity.6
Well-known regulators of VE-cadherin and the actin cytoskeleton are the Rho family of small guanine nucleotide triphosphatases (RhoGTPases). This family contains more than 20 members, of which Rac1, Cdc42 and RhoA are the most studied.7 Of these 3 members, Rac1 and Cdc42 promote endothelial barrier function, whereas RhoA induces contraction-driven endothelial hyperpermeability.8 There are 3 isoforms of RhoA, RhoB and RhoC, that share 88% amino acid identity and potentially all of them bind similar effector proteins such as Rho Kinase (ROCK).9 Previous research showed differential, critical roles for RhoA, RhoB and RhoC in cancer. RhoA stimulates cell cycle progression, migration and invasion, acting via the ROCK1 and ROCK2 effector proteins.10 In contrast, RhoB expression is reduced in various tumor cell types, suggesting RhoB acts as a tumor suppressor. RhoB promotes β1 integrin activation and in this way, increases the spreading and adhesion of tumor cells.11 RhoC interacts with the formin-like FMNL2 and FMNL3 effector proteins and induces lamellipodia extension which promotes cancer cell metastasis.12,13
RhoA is extensively studied as a key regulator of vascular leakage14 and leukocyte trans-endothelial migration.15 RhoA signals though ROCK kinases to induce F-actin stress fiber formation and acto-myosin-based contraction. This promotes a loss of VE-cadherin-mediated cell-cell contact which is a hallmark of vascular leakage.14,16 Several other studies addressed the role of RhoA in endothelial barrier function, showing that RhoA is activated upon stimulation with vaso-active agents such as thrombin or Vascular Endothelial Growth Factor (VEGF). RhoA activation leads to disrupted barriers due to the activation of ROCK, phosphorylation of Myosin Light Chain (MLC) and stress fiber formation.4,17,18 To date, there has been little published data on a specific role for RhoB or RhoC in endothelial barrier function and permeability. Recently, Reinhard et al. showed, by using FRET-based RhoGTPase bio-sensors, similar activation and localization profiles for RhoA and RhoC but spatiotemporally distinct activation of RhoB in endothelial cells.19 In addition, Marcos-Ramiro et al. showed that during inflammation, RhoB acts as a negative regulator of Rac1 membrane translocation, preventing Rac-1 mediated restoration of compromised endothelial integrity.20 Together, these studies indicate that RhoB plays a different role in endothelial cells in comparison to RhoA and RhoC.
This current study aimed to determine whether RhoA, RhoB and RhoC have overlapping or distinct roles in barrier function and permeability in resting and activated endothelium. Using primary Human Umbilical Vein Endothelial Cells (HUVECs), in combination with siRNA transfection to establish individual, double or triple knockdown of the RhoA/B/C RhoGTPases, we found that RhoB, but not RhoA or RhoC, is a negative regulator of permeability in resting endothelium. Thrombin-induced loss of endothelial integrity is mediated primarily by RhoA and RhoB, with RhoC contributing to the Rac1-dependent restoration of endothelial barrier function. Thus, these highly homologous GTPases differentially control the dynamics of endothelial barrier function.
Results
Loss of RhoB, but not RhoA or RhoC, improves basal endothelial barrier function
To study a potential differential role of RhoA, RhoB and RhoC in endothelial barrier regulation, we first investigated the effects of individual siRNA-mediated knockdowns of RhoA, RhoB and RhoC in HUVECs. The control cells in this study were transfected with non-targeting siRNA (siNT). Transfection with smart pool siRNAs resulted in the efficient deletion of RhoA, RhoB and RhoC at the protein level 72 hours after the start of transfection (77-98% knockdown) (Fig. 1A). Interestingly, knockdown of RhoB or of RhoC resulted in an almost 2-fold increased protein expression of RhoA and of RhoB, respectively, compared with the control cells. RhoC expression was less affected by the loss of either RhoA or RhoB (Fig. 1A). To further study the upregulation of RhoA and RhoB upon knockdown of RhoA, RhoB and RhoC, we blocked protein synthesis using cycloheximide. This treatment induced a time-dependent decrease in protein expression of RhoA and RhoB after 1 and 4 hours both in the control and siRNA transfected cells (Supplemental figure 1A and 1B). Expression of RhoC was again more stable and only slightly decreased in the control, RhoA and RhoB knockdown cells 4 hours after cycloheximide addition (Supplemental figure 1C). This data indicates that loss of the RhoA and RhoB GTPases leads to increased, compensatory expression of RhoB or RhoA GTPases, respectively, but that RhoC is not much affected.
Figure 1.
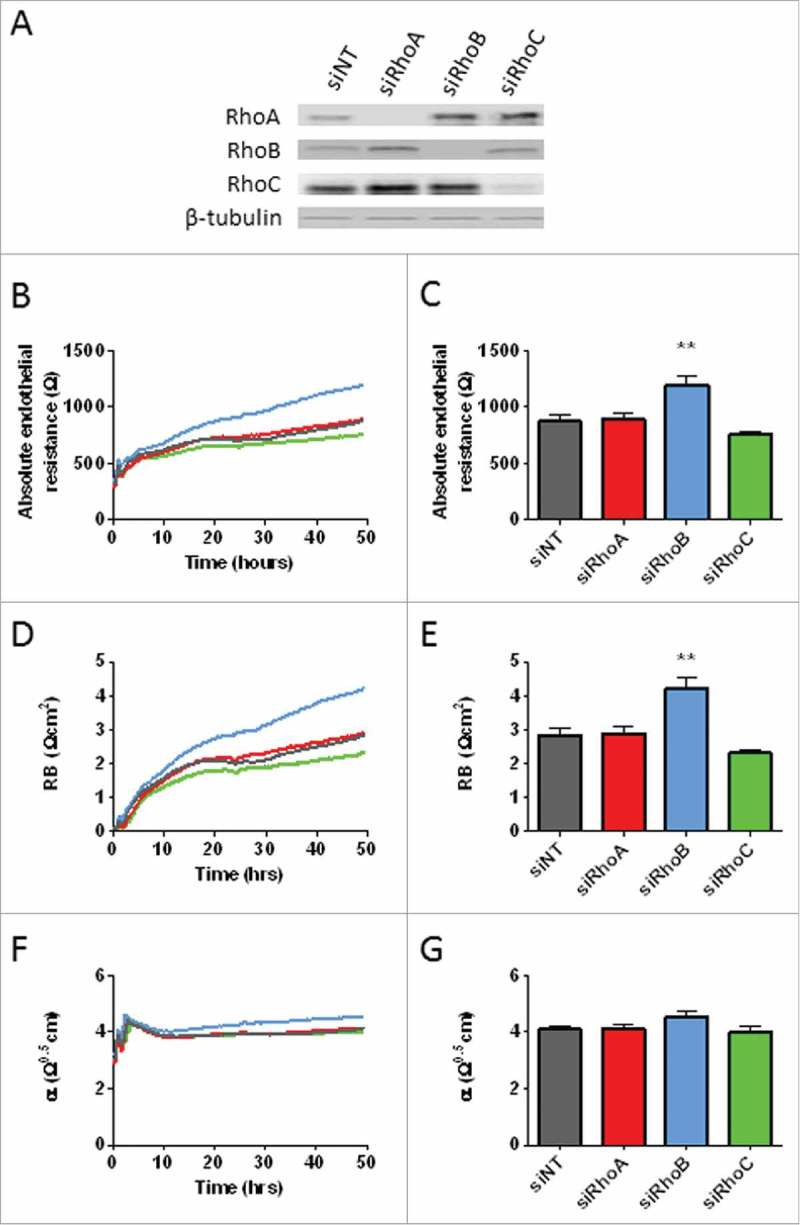
Effects of loss of RhoA, RhoB and RhoC on endothelial barrier function. (A) Western blot analysis of whole cell lysates collected from HUVECs 72 hours after transfection with siNT, siRhoA, siRhoB or siRhoC. Representative blots of 2 experiments are shown. Tubulin is included as loading control. (B) Effect of loss of RhoA, RhoB and RhoC on basal endothelial barrier function. (C) Basal endothelial barrier function at t = 72 hours. (D) Absolute endothelial resistance attributable to cell-cell adhesion (Rb) of RhoA, RhoB and RhoC knockdown cells. (E) Rb at t = 72 hours. (F) Absolute endothelial resistance attributable to cell-matrix adhesion (α) of RhoA, RhoB and RhoC knockdown cells. (E) α at t = 72 hours. All data represent average values (line graphs, representing barrier formation, Rb or α from medium change at 16 hours after transfection (t = 0) until the end of the experiment) or mean ± SEM (bar graphs) of N = 5 experiments performed in triplicates. **p < 0.01 Dunnett's post-hoc analysis of one-way ANOVA. siNT, non-targeting siRNA.
To evaluate the effects of loss of RhoGTPases on endothelial barrier regulation, we measured trans-endothelial electrical resistance in control versus RhoA, RhoB and RhoC knockdown HUVECs under basal conditions for 72 hours. Measurement of endothelial resistance showed that loss of RhoA did not change the endothelial barrier function compared with control cells, whereas RhoB knockdown significantly increased the endothelial resistance. Knockdown of RhoC tended to induce a slightly decreased endothelial barrier function, however this effect was not statistically significant (Fig. 1B and 1C). Resolving the endothelial resistance measurements into separate components reflecting cell-cell (Rb) and cell-matrix interaction (α),21 showed that knockdown of RhoA did not change cell-cell interactions. In contrast, RhoB knockdown significantly enhanced, whereas RhoC knockdown resulted in a small decrease in cell-cell interactions, 72 hours after transfection (Fig. 1D and 1E). None of these knockdowns significantly changed the cell-matrix interaction (Fig. 1F and 1G). These findings indicate that RhoA, RhoB and RhoC GTPases in part cross-regulate their expression levels in HUVECs and that of these 3, RhoB is the most dominant regulator of basal endothelial barrier function, through its control of cell-cell contacts.
RhoB is a negative regulator of basal endothelial integrity
To gain more information regarding functional interactions between RhoA, RhoB and RhoC in endothelial barrier function, we performed double and triple knockdowns of RhoA, RhoB and RhoC in HUVECs using siRNA smart pool combinations. Transfection with combinations of siRNAs resulted in a sufficient knockdown of RhoA, RhoB and RhoC, although the efficiency of transfection was slightly lower compared with single knockdown conditions (68-90% knockdown, Fig. 2A). Concomitant, knockdown of RhoA/C leads to an increased expression of RhoB (4-fold increase). This was also shown for RhoB/C knockdown which lead to a 4-fold increase in RhoA expression. Again, RhoC expression was not changed much after RhoA/B knockdown compared with control cells (Fig. 2A).
Figure 2.
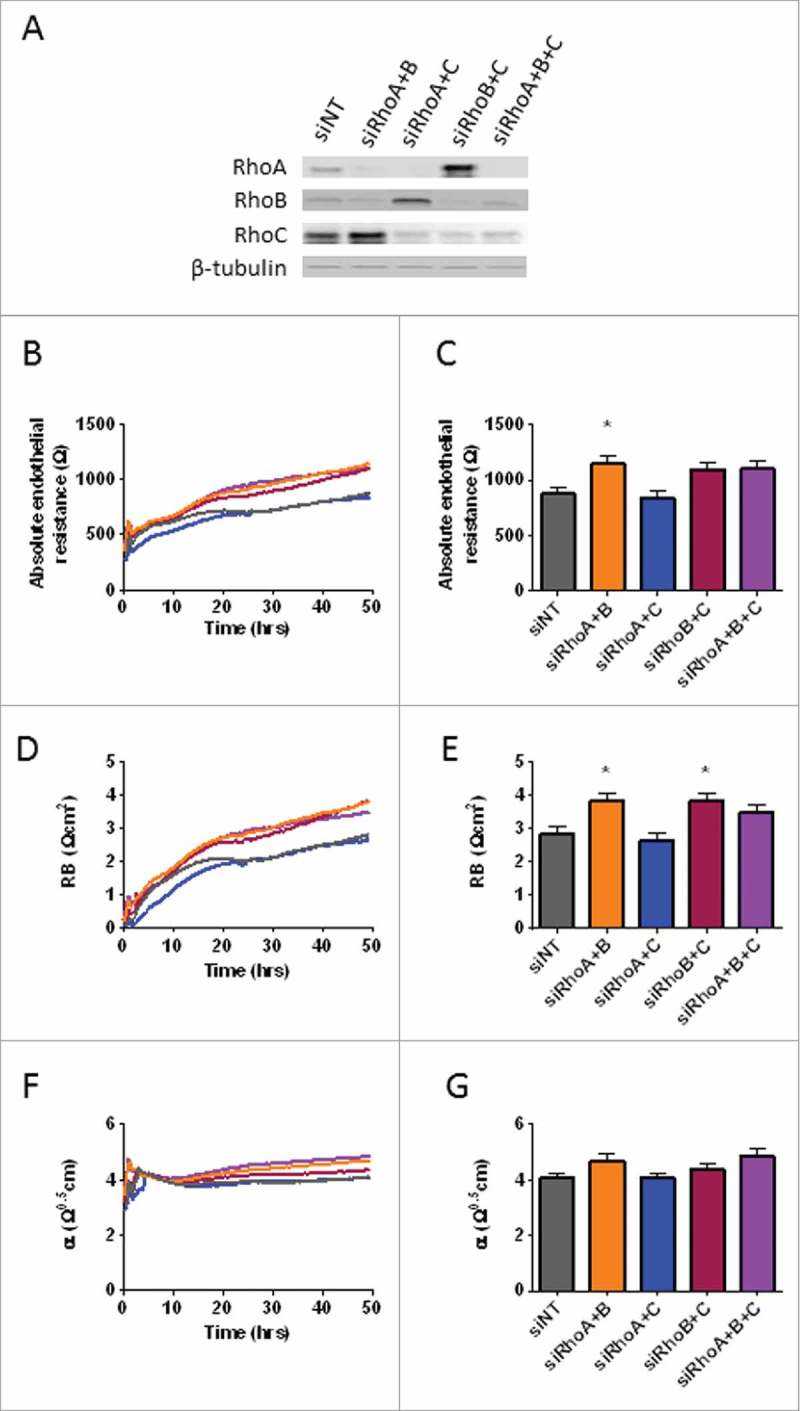
Effects of RhoA, RhoB and RhoC double and triple knockdowns on basal endothelial barrier function. (A) Western blot analysis of whole cell lysates collected from HUVECs 72 hours after transfection with siNT, siRhoA/B, siRhoA/C or siRhoB/C and siRhoA/B/C. Representative blots of 2 experiments are shown. Tubulin is included as loading control. (B) Effect of double and triple knockdown of RhoA, RhoB and RhoC on basal endothelial barrier function. (C) Basal endothelial barrier function at t = 72 hours. (D) Absolute endothelial resistance attributable to cell-cell adhesion (Rb) of RhoA, RhoB and RhoC double and triple knockdown cells. (E) RB at t = 72 hours. (F) Absolute endothelial resistance attributable to cell-matrix adhesion (α) of RhoA, RhoB and RhoC double and triple knockdown cells. (G) α at t = 72 hours. All data represent average values (line graphs, representing barrier formation, Rb or α from medium change at 16 hours after transfection (t = 0) until the end of the experiment) or mean ± SEM (bar graphs) of N = 5 experiments performed in triplicates. *p < 0.05 in Dunnett's post-hoc analysis of one-way ANOVA.
To study the effect of a double or triple knockdown of these GTPases on basal endothelial barrier regulation we measured electrical resistance of control vs. the double and triple knockdown of RhoA, RhoB and RhoC in HUVECs under basal conditions for 72 hours. Measurement of endothelial barrier function showed that knockdown of RhoA/B significantly increased basal barrier function, while knockdown of RhoA/C did not affect endothelial barrier function compared with the control cells. Combined knockdown of RhoB/C or triple knockdown showed again increased basal barrier function after 72 hours, although these values were not statistically significant (Fig. 2B and 2C). When comparing this data to the individual knockdowns of RhoA, RhoB and RhoC, we found that double knockdowns involving loss of RhoB showed increased basal barrier function similar to loss of only RhoB, implying that RhoA and RhoC may have a more limited contribution to the basal barrier function. Interestingly, combined knockdown of RhoB with either RhoA or RhoC or in combination (triple knockdown) did not show additive effects exceeding that of the RhoB knockdown (Supplemental figure 2A). When looking into separate components reflecting cell-cell (Rb) and cell-matrix (α) interaction, we found that cell-cell interaction was significantly increased for the knockdown of RhoA/B and RhoB/C. Knockdown of RhoA/C did not show any differences compared with the control cells, whereas the triple knockdown cells showed an increased cell-cell interaction in basal endothelial cells comparable to RhoA/B and RhoB/C knockdown albeit that this difference was not significant (Fig. 2D and 2E). Simultaneously, cell-matrix interaction was slightly increased in RhoA/B double knockdown and triple knockdown. However, also these effects were not statistically significant (Fig. 2F and 2G). Together, these findings indicate that among RhoA, RhoB and RhoC, RhoB is the main regulator of basal endothelial barrier function by negatively regulating cell-cell contacts.
RhoB knockdown increases VE-cadherin at cell-cell contacts
As ECIS measurements of transfected HUVEC's showed increased cell-cell interactions in the RhoB knockdown cells, we focused on the morphology of adherens junctions and the actin cytoskeleton in endothelial monolayers with a RhoA, RhoB or RhoC knockdown. Immunostaining of the adherens junction protein VE-cadherin in un-stimulated HUVEC's showed that all siRNA transfected monolayers form a barrier without intercellular gaps with a clear VE-cadherin staining at cell-cell contacts. When comparing the different knockdown conditions, we observed that RhoB and RhoC knockdown cells showed thicker VE-cadherin staining than control cells, whereas RhoA knockdown cells did not show differences. (Fig. 3A). We quantified these findings by measuring the Integrated Density of VE-cadherin staining per cell and measuring VE-cadherin-positive area per cell. Analysis of Integrated Density per cell showed that RhoB and RhoC knockdown cells have a significantly increased intensity of VE-cadherin staining while RhoA knockdown cells did not show a difference compared with the control cells (Fig. 3B). The area of VE-cadherin staining was significantly larger in RhoB knockdown cells while this area in RhoA and RhoC knockdown cells was not changed compared with the control cells (Fig. 3C). Western blot quantification of total VE-cadherin levels did not showed significant changes in total VE-cadherin levels for all conditions (Fig. 3E). Rhodamine-phalloidin staining for F-actin revealed that all knockdown conditions showed, cortical F-actin band and some cytoplasmic F-actin filaments similar to the control cells. Analysis of this staining showed that RhoB knockdown and RhoC knockdown cells have significant increased F-actin intensity compared with the control cells, while RhoA knockdown cells did not show any differences (Fig. 3D). Moreover, we did not detect any differences in cell size (Supplemental figure 3A). These findings indicate that loss of RhoB increased VE-cadherin at cell-cell junctions in endothelial cells, while the overall F-actin distribution was not changed.
Figure 3.
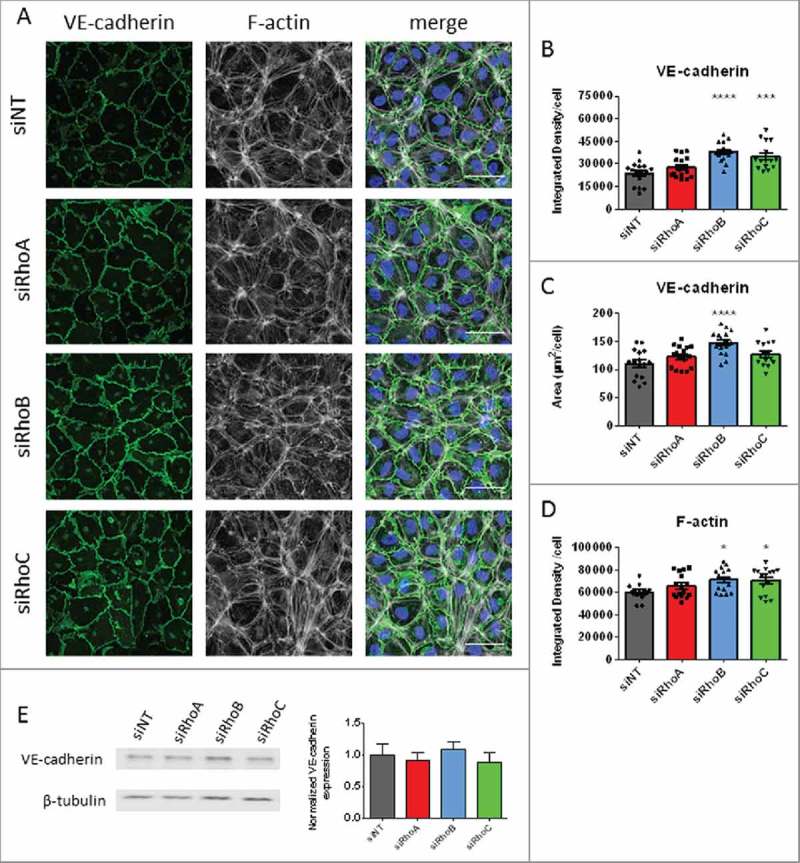
The effects of single knockdowns of RhoA RhoB and RhoC on basal endothelial morphology. (A) Immunofluorescent staining of VE-cadherin (green), F-actin (white) and nuclei (blue) in HUVECs for visualization of adherens junctions following loss of RhoA, RhoB and RhoC. Scale bars represent 50 μM. Representative pictures of 3-4 experiments. (B) VE-cadherin Integrated Density/cell; each point representing an individual measurement. (C) VE-cadherin area per cell. (D) F-actin Integrated Density per cell. (E) Western blot analysis for VE-cadherin of whole cell lysates collected from HUVECs 72 hours after transfection with siNT, siRhoA, siRhoB or siRhoC. Representative blots of 3 experiments are shown. Tubulin is included as loading control. Bar graphs represent mean ± SEM from 3 individual experiments all normalized to siNT. Data in panel B,C and D represent mean ± SEM (bar graphs) of 12–16 values from 4 independent experiments. *p < 0.05, ***p < 0.001, ****p < 0.0001 in Dunnett's post-hoc analysis of one-way ANOVA.
Double or triple knockdowns which include RhoB increased VE-cadherin intensity and area
ECIS measurements of double and triple knockdown combinations of RhoA, RhoB and RhoC showed either significantly increased, or a trend toward increased, cell-cell contact in resting endothelial cells. Immunostainings of the adherens junction protein VE-cadherin under these conditions showed that normal monolayers were formed with VE-cadherin clearly present at the cell-cell contacts (Fig. 4A). Comparable to RhoB single knockdown, the RhoA/B, RhoB/C and the triple knockdown situations showed more prominent VE-cadherin staining compared with control cells. Also, the honeycomb structure was more clear in these conditions. Images of the different conditions were quantified for Integrated Density per cell and VE-cadherin positive area per cell. Analysis of these data showed that all double and triple knockdown conditions showed an increased VE-cadherin intensity per cell. This significant difference was more pronounced when RhoB expression was reduced (Fig. 4B). The area per cell was significantly different in RhoA/B knockdown, RhoB/C knockdown and triple knockdown monolayers compared with the control cells. Here, double knockdown of RhoB/C showed the largest area of VE-cadherin per cell (Fig. 4C). Western blot quantification of total VE-cadherin levels showed a small increase of total VE-cadherin for RhoA/B and triple knockdown cells however this was nog significant (Fig. 4E). Rhodamine-phalloidin staining for F-actin revealed cortical actin rings in all knockdown conditions which were most pronounced in RhoA/C double knockdown cells. These RhoA/C double knockdown cells also showed more stress fiber formation compared with control cells, while stress fiber formation was almost completely lost in triple knockdown cells. RhoA/B and RhoB/C knockdown cells showed no large differences compared with control cells (Fig. 4A). Analysis of Integrated Density of F-actin per cell showed no significant differences between different conditions compared with the control cells, however there is a trend toward lower Integrated Density per cell for triple knockdown cells (Fig. 4D). It is important to note that all the knockdown combinations with RhoB showing increased VE-cadherin area per cell also showed a significantly decreased number of cells per image (Supplemental figure 3B), suggesting an increase in cell size. When comparing the data of single and double knockdowns, we found that lack of RhoB is the main factor driving increased Integrated Density of VE-cadherin per cell. Additional knockdown of RhoA and/or RhoC only slightly increased the integrated density per cell further (Supplemental figure 2B). However, additional knockdown of RhoA and or RhoC increased the cell size. Combined, this data indicates that RhoB is a central regulator of basal endothelial barrier function by modulating junctional distribution of VE-cadherin.
Figure 4.
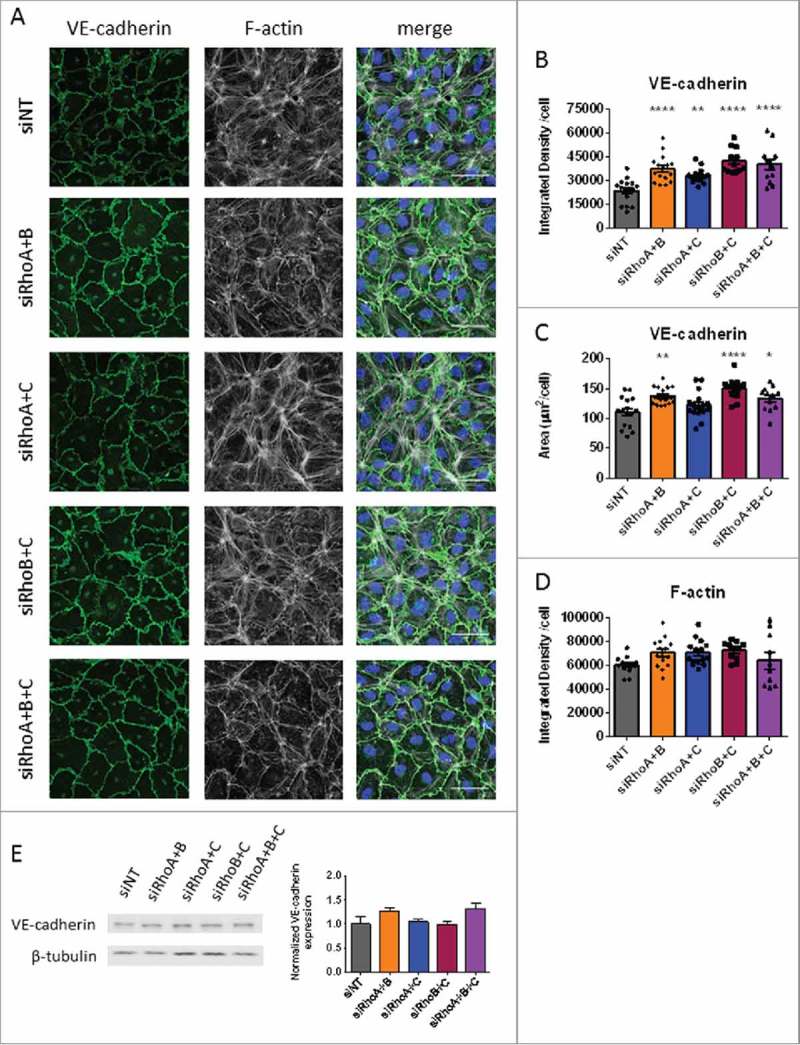
The effects of double and triple knockdowns on basal endothelial morphology. (A) Immunofluorescent staining of VE-cadherin (green), F-actin (white) and nuclei (blue) in HUVECs for visualization of adherens junctions presence after loss of (combinations of) RhoA, RhoB and RhoC. Scale bars represent 50 μM. Representative pictures of 3–4 experiments are shown. (B) VE-cadherin Integrated Density/cell, each point representing an individual measurement. (C) VE-cadherin area per cell. (D) F-actin Integrated Density per cell. (E) Western blot analysis for VE-cadherin of whole cell lysates collected from HUVECs 72 hours after transfection with siNT, siRhoA/B, siRhoA/C, siRhoB/C or siRhoA/B/C. Representative blots of 3 experiments are shown. Tubulin is included as loading control. Bar graph represent mean ± SEM from 3 individual experiments all normalized to siNT. Data in panel B,C and D represent mean ± SEM 12–16 values from 4 independent experiments (bar-graphs). *p < 0.05, **p < 0.01, ****p < 0.0001 in Dunnett's post-hoc analysis of one-way ANOVA.
Single knockdown of RhoA, RhoB or RhoC does not change thrombin induced hyper permeability
In the past, it was shown that RhoA is a key regulator of endothelial barrier disruption upon thrombin stimulation.22 We recently showed that besides RhoA, also RhoB and RhoC were similarly activated upon thrombin stimulation.19 Nevertheless, the specific roles of RhoB and RhoC in thrombin-induced hyper-permeability are still unknown.
Here, we studied the effect of thrombin stimulation in RhoA, RhoB and RhoC single knocked down HUVECs. Knockdown of RhoA or RhoB separately showed a slightly attenuated drop in resistance (i.e. loss of barrier function) compared with control cells, for RhoA knockdown this difference was significant. Knockdown of RhoC showed no differences in thrombin-induced reduction in resistance compared with control cells (Fig. 5A and 5B). The ability of the monolayer to recover from thrombin-induced loss in resistance also provides valuable information about the role of individual RhoGTPases in this response. We found that none of the single knockdown conditions showed a significantly different recovery after thrombin stimulation when compared with control cells (Fig. 5A and 5C). Pre-treatment with TNF-α for 7 hours lowered the basal barrier function and showed a differential % drop and % recovery after thrombin stimulation for all conditions (Supplemental figure 4A, 4C, 4E and 4G). To test the effect of thrombin on endothelial cells in a different way, we measured macromolecule passage over endothelial monolayers in a Transwell assay. Under basal conditions, we found, in agreement with the ECIS results on basal barrier function, that loss of RhoA did not change basal macromolecule permeability compared with control cells, where loss of RhoB reduced the macromolecule permeability and loss of RhoC induced a modest, but not significant increase in macromolecule permeability compared with the control (Fig. 5D).
Figure 5.
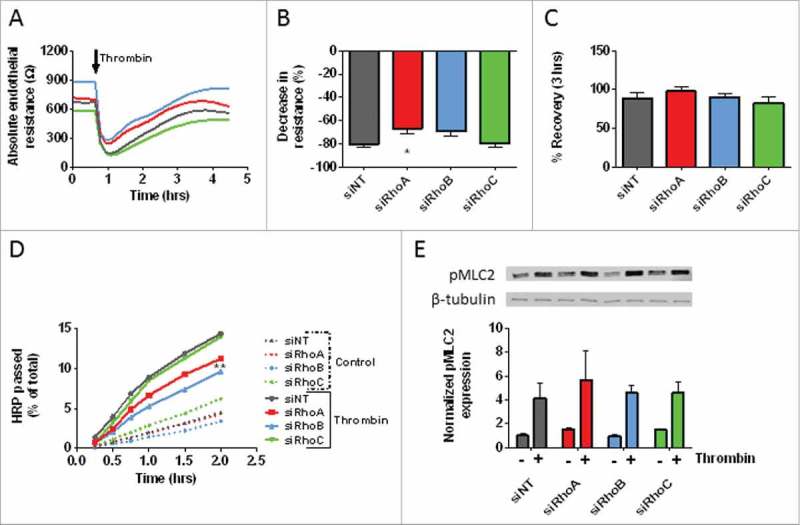
The effect of thrombin on HUVEC monolayers following siRNA-mediated loss of RhoA, RhoB and RhoC. (A) Thrombin-induced endothelial barrier disruption in cells transfected with RhoA, RhoB and RhoC siRNAs. Arrow indicates addition of Thrombin 1 U/mL. (B) The thrombin response (% decrease in resistance) in control vs. RhoA, RhoB and RhoC single knockdown HUVECs. Values represents the percentage drop at the lowest point of resistance following addition of thrombin. (C) The % recovery at 3 hours after thrombin relative to the start values in control vs. RhoA, RhoB and RhoC single knockdown HUVECs. (D) Time-dependent effects of RhoA, RhoB and RhoC knockdown on the passage of HRP across control and thrombin-stimulated HUVECs. Data represent average values of 3–5 experiments. (E) Western blot analysis of whole cell lysates showing the effect of thrombin stimulation in RhoA, RhoB and RhoC single knockdown HUVECs on the phosphorylation of MLC. Representative blots from 3 experiments are shown. Tubulin is included as loading control. Bar graph represents mean ± SEM from 3 individual experiments all normalized to siNT without thrombin. Panel A,B and C represent average values (line graph) or mean ± SEM (bar graph) of 5 experiments performed in duplicate or triplicate. *p < 0.05 in Dunnett's post-hoc analysis of one-way ANOVA. Panel D represent average values of 3–5 experiments performed in triplicates. **p < 0.01 in Dunnett's post-hoc analysis of repeated measures ANOVA.
After thrombin stimulation, we found that single knock-down of RhoA or RhoB reduced the thrombin-induced macromolecule permeability over the endothelial layer. Loss of RhoB caused significant lower thrombin-induced macromolecule permeability compared with control cells, whereas loss of RhoC did not affect permeability (Fig. 5D). A well-known downstream effector of RhoA is ROCK which, via phosphorylation of myosin-phosphatase, leads to increased phosphorylation of myosin light chain (pMLC) and eventually to stress fiber formation.22,23 For this study, we tested if loss of RhoA, RhoB or RhoC leads to different downstream signaling induced by thrombin. Single knockdown of RhoB did not change the phosphorylation of MLC under basal conditions, while the phosphorylation in RhoA and RhoC knockdown cells was slightly increased. The thrombin-induced increase in phosphorylation of MLC showed a slight increase for RhoA knockdown cells compared with control cells, although not significant. pMLC levels of RhoB and RhoC knockdown cells did not differ from control cells after thrombin stimulation (Fig. 5E). These findings indicate that single knockdowns of RhoA, RhoB or RhoC in endothelial monolayers were in-sufficient to attenuate the thrombin-induced hyper-permeability or phosphorylation of MLC.
Double knockdown of RhoA and RhoB protects against thrombin-induced loss of barrier function
Single knockdown of RhoA, RhoB or RhoC did not affect the response to thrombin significantly even though VE-cadherin intensity and area were increased in RhoB knockdown cells. Double and triple knockdown cells also showed differences in VE-cadherin intensity and area and we therefore tested the effect of thrombin induced permeability under these conditions as well.
Thrombin stimulation showed a significantly attenuated response in HUVEC monolayers in which RhoA/B or RhoA/B/C were downregulated, as compared with control cells. The decrease in percentage of thrombin-induced loss of resistance was reduced by 50% compared with control cells. Double knockdown of RhoA/C also showed a slightly attenuated response, however, this difference was not significant. Double knockdown of RhoB/C did not show a difference when compared with control cells, despite their higher starting values (Fig. 6A and 6B). When comparing the recovery after thrombin of double and triple knockdowns monolayers, we detected a slightly increased recovery for the RhoA/B and triple knockdown conditions, although this difference was not statistically significant. Combined loss of RhoA/C or RhoB/C did not show additional differences in recovery compared with control cells (Fig. 6A and 6C). When comparing this data to the single knockdown results, we found that double knockdowns which include a loss of RhoC did not have an additive effect to single knockdown of RhoA or RhoB. On the other hand, combined knockdown of RhoA/B and the triple knockdown condition resulted in an attenuated thrombin response (Supplemental figure 2C). Pre-treatment with TNF-α for 7 hours lowered the basal barrier function and resulted in a differential % drop and % recovery after thrombin stimulation for all conditions(Supplemental figure 4B, 4D, 4F and 4H).
Figure 6.
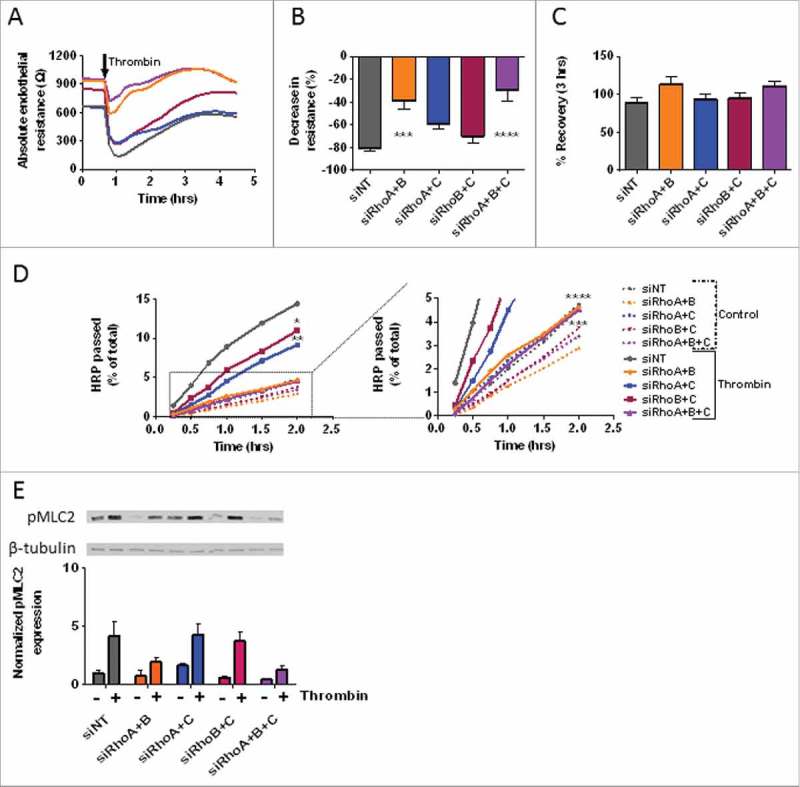
The effect of thrombin on monolayers following combined loss of RhoA, RhoB and RhoC. (A) Thrombin-induced endothelial barrier disruption in cells transfected with double and triple knockdowns of RhoA, RhoB and RhoC siRNAs. Arrow indicated addition of 1 U/mL thrombin. (B) The thrombin response (% decrease in resistance) in control vs. RhoA, RhoB and RhoC double and triple knockdown HUVECs. Values represent the % reduction in resistance at the lowest point after addition of thrombin. (C) The % recovery 3-hours after thrombin relative to the start values of control vs. RhoA, RhoB and RhoC double and triple knockdown HUVECs. (D) Time-dependent effects of RhoA, RhoB and RhoC double and triple knockdowns on the passage of HRP across control and thrombin-stimulated HUVECs. The graph on the right is a magnified portion of the left graph, as indicated by the dashed box and lines. (E) Western blot analysis of whole cell lysates showing the effect of thrombin stimulation in RhoA, RhoB and RhoC double and triple knockdown HUVECs on the phosphorylation of MLC. Representative blots from 3 experiments are shown. Tubulin is included as loading control. Bar graph represents mean ± SEM from 3 individual experiments all normalized to siNT without thrombin. Panel A, B, and C represent average values (line graph) or mean ± SEM (bar graph) of 5 experiments performed in duplicate or triplicate. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in Dunnett's post-hoc analysis of one-way ANOVA. Panel D represent average values of 3–5 experiments performed in triplicates. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in Dunnett's post-hoc analysis of repeated measures ANOVA.
By testing the effect of thrombin on macro-molecule permeability we found that all double knockdown and triple knockdown conditions have an attenuated response to thrombin stimulation, compared with control cells. Interestingly, knockdown of RhoA/B and the triple knockdown condition showed significantly lower macro-molecule leakage at all time points. The thrombin-induced permeability of these cells was even comparable to the basal leakage of control cells, indicating that RhoA and RhoB together fully account for the induction of thrombin-induced permeability, with no contribution of RhoC. Double knockdown of RhoA/C also showed protection after thrombin stimulation, however this was less significant. Loss of RhoB/C only showed a small significant protection compared with control cells. Basal leakage of RhoA/B, RhoB/C and triple knockdown monolayers was lower than control cells, however this difference was not significant (Fig. 6D). When comparing single knockdowns with double and triple knockdowns 2 hours after addition of thrombin we see that single knockdowns show only minor protection against thrombin-induced hyper-permeability. Additive knockdown of RhoC did not reduced the leakage significantly, however combined knockdown of RhoA/B and triple knockdowns showed a very protective phenotype (Supplemental figure 2D). To see if the expression of pMLC was altered, we performed western blot experiments on basal and thrombin-treated samples of cells from different conditions. We showed that loss of RhoA/B, RhoB/C and the triple knockdown conditions had lower basal pMLC levels compared with control cells, while RhoA/C knockdown cells showed an slight increase in basal pMLC level. When stimulated with thrombin, pMLC levels in RhoA/B and triple knockdown cells were only 50% of control levels, while RhoA/C and RhoB/C knockdown cells showed similar levels as control cells (Fig. 6E). These findings indicate that combined loss of RhoA/B protects against thrombin-induced permeability, which correlates with a markedly decreased phosphorylation of MLC.
Single knockdown of RhoA and RhoB cause decreased intercellular gap formation after thrombin stimulation
Basal endothelial barrier function was increased in RhoB knockdown cells due to increased basal VE-cadherin intensity and -area compared with control cells. Thrombin stimulation of cells in which RhoA or RhoB was reduced only showed minor protective effects compared with control cells. We therefore also used immunofluorescence for single knockdown cells after thrombin stimulation to document morphological changes under these conditions.
HUVECs with single knockdown of RhoA, RhoB or RhoC were stimulated with thrombin for 15 minutes and stained for VE-cadherin and F-actin. VE-cadherin staining showed that all knockdown conditions are affected by thrombin, with VE-cadherin showing a more activated and jagged distribution at adherens junctions. These structures were most clearly visible in control and RhoC knockdown cells and to a lesser extent in RhoA and RhoB single knockdown cells. The F-actin staining showed stress fiber formation in all conditions comparable to control cells (Fig. 7A). Quantitative analysis of images of VE-cadherin revealed that loss of RhoA, RhoB and RhoC induced an increase in VE-cadherin intensity after 15 minutes thrombin stimulation compared with control cells. Of these, the RhoB knockdown cells showed the biggest increase in intensity (Fig. 7B). These cells also showed an increased area of VE-cadherin compared with control cells, with RhoA and RhoC single knockdown cells showing no changes in the area occupied by VE-cadherin after thrombin stimulation (Fig. 7C). The intensity of F-actin staining was significantly increased for RhoB knockdown and RhoC knockdown cells, however the distribution of F-actin was not changed (Fig. 7D). Also, cells were all equal in size (Supplemental figure 3C).
Figure 7.
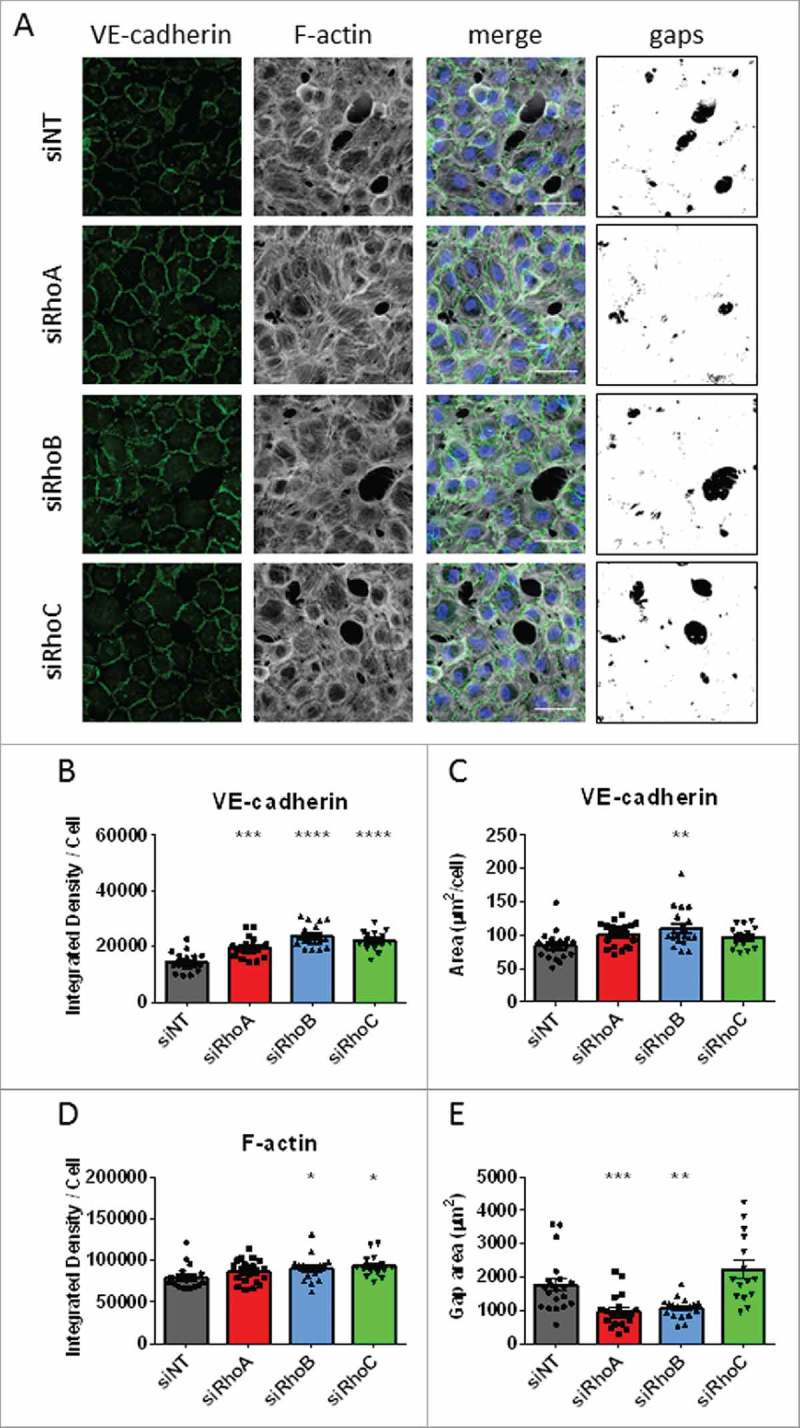
The effects of loss of RhoA RhoB and RhoC on endothelial morphology after thrombin stimulation. (A) Immunofluorescent staining of VE-cadherin (green), F-actin (white) and nuclei (blue) and overlay picture of gap size in HUVECs after loss of RhoA, RhoB and RhoC and stimulation with thrombin. Scale bars represent 50 μM. Representative pictures of 3–4 experiments are shown. (B) VE-cadherin Integrated Density/cell. (C) VE-cadherin area per cell. (D) F-actin Integrated Density per cell. (E) Total gap area after thrombin stimulation. All data represent mean ± SEM (bar graphs) of 15–20 values from 4 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 in Dunnett's post-hoc analysis of one-way ANOVA.
With the overlays of VE-cadherin, F-actin and pERK1/2 as a cytosolic marker, we visualized the intercellular gap size of the thrombin-treated monolayers. Measuring the gap size revealed that RhoA and RhoB knockdown cells show a significant decrease in intercellular gap area compared with control cells. The combined gap size of RhoC knockdown cells per field of view was slightly higher than control cells, however this was not significant (Fig. 7A and 7E). Analysis of the size of gaps showed that the main difference in total gap area of single knockdown cells were caused by the percentage of big gaps (Supplemental figure 5A). These findings indicate that upon stimulation with thrombin, the increased presence at junctions of VE-cadherin in RhoB knockdown cells remains detectable, which corresponds to the protection against intercellular gap formation. Although RhoA knockdown cells did not showed statistically increased accumulation of VE-cadherin at junctions it still protects against intercellular gap formation. Possibly by RhoB-mediated trafficking of Rac1 to the cell-cell contacts.
Double or triple knockdown conditions which include RhoA cause decreased intercellular gap formation after thrombin stimulation
Thrombin stimulation in ECIS and HRP experiments showed that double knockdown of RhoA/B and the triple knockdown were both protective for loss of barrier function. To gain more information about why this is the case, we imaged the morphological changes upon thrombin stimulation in double and triple knockdown cells. Immunofluorescent staining of VE-cadherin showed that stimulation with thrombin leads to a more activated phenotype. The cells showed a more jagged junctional distribution and the lining of the cells was sometimes discontinuous (Fig. 8A). Quantitative analysis of the intensity of VE-cadherin per cell showed that all double knockdown and the triple knockdown cells showed increased VE-cadherin intensity. This increase was most prominent in the triple knockdown condition (Fig. 8B). The area of junctional VE-cadherin was also significantly increased in all double knockdown and triple knockdown conditions; here the RhoA/B double and the triple knockdown cells showed the largest increase (Fig. 8C). F-actin morphology could also give an indication of cellular changes during thrombin stimulation. Phalloidin staining showed that the distribution of F-actin fibers is differentially organized in the different conditions. Zoomed images showed that control cells have a contractile ring and a diffuse F-actin staining in the cytosol, which was similar to the RhoB/C knockdown cells (Fig. 8A). Double and triple knockdowns which include RhoA (RhoA/B, RhoA/C, RhoA/B/C) showed more stress fiber like structures and less diffuse staining in the cytosol (Fig. 8A). Quantification of the F-actin intensity showed that RhoA/B and RhoA/C and the triple knockdowns have significantly increased F-actin per cell (Fig. 8D) which could be due to compensatory upregulation or incomplete downregulation of some of the GTPases (Fig. 2A). All double knockdown conditions also showed larger cells at 15 minutes after thrombin stimulation (Supplemental figure 3D).
Figure 8.
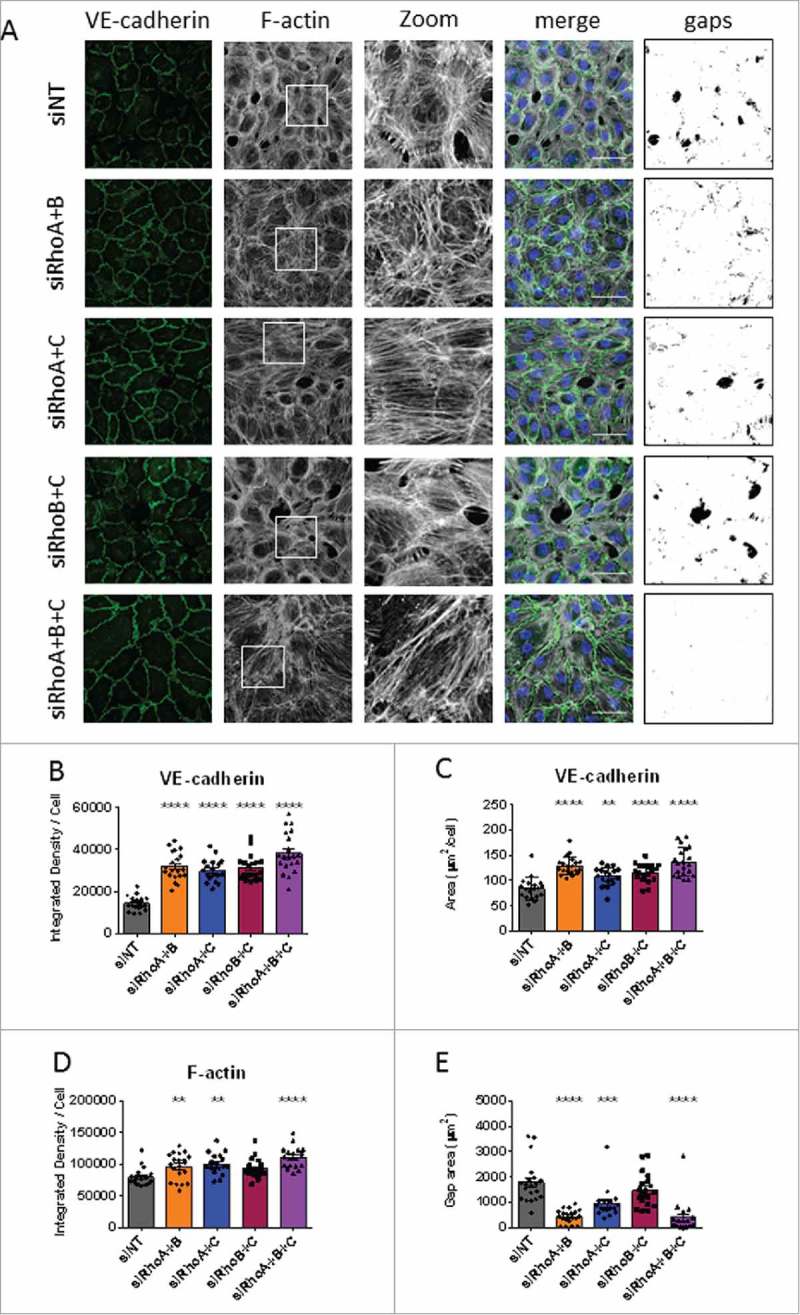
The effects of combined loss of RhoA, RhoB and RhoC on endothelial morphology after thrombin stimulation. (A) Immunofluorescent staining of VE-cadherin (green), F-actin (white) and nuclei (blue) and the corresponding image depicting gap size in HUVECs after siRNA-mediated loss of combinations of RhoA, RhoB and RhoC and stimulation with thrombin. Scale bars represent 50 μM. Representative pictures of 3–4 experiments are shown. (B) VE-cadherin Integrated Density/cell. (C) VE-cadherin area per cell. (D) F-actin Integrated Density per cell. E) Total gap area after thrombin stimulation. All data represent mean ± SEM (bar graphs) of 15–20 values from 4 independent experiments. **p < 0.01, ***p < 0.001, ****p < 0.0001 in Dunnett's post-hoc analysis of one-way ANOVA.
For double and triple knockdown conditions we also made overlays to measure intercellular gap size after thrombin stimulation. Analysis of gap size showed that RhoA/B and RhoA/C double knockdown and the triple knockdowns had significantly reduced gap areas compared with control cells. The largest decrease was found in RhoA/B double knockdown and the triple knockdown cells (Fig. 8A and 8E). Analysis of the distribution of gap area showed that in these 2 conditions the percentage of total area of larger gaps was significantly reduced while the percentage of total area of small gaps was significantly induced, which eventually results in smaller total gap area (Supplemental figure 5B). When comparing with the data of single knockdowns we found that double knockdowns had an additive effect on VE-cadherin intensity per cell, the triple knockdown even showed a larger effect (Supplemental figure 3E). With respect to gap area we show that loss of RhoA or RhoB already reduced gap formation. The combined loss of RhoA/B reduced gap formation even more, the loss of RhoA/C did not change the intercellular gap area and the loss of RhoB/C even made the gap area larger compared with RhoB knockdown cells (Supplemental figure 3F).
In summary, this data indicates that loss of RhoB accounted for increased VE-cadherin accumulation at cell-cell contacts after thrombin stimulation which partially leads to a protection against thrombin-induced permeability. However, loss of RhoA appeared as the common factor which mediates protection of intercellular gap formation.
Discussion
Here we show that RhoB, in addition to RhoA, is an important regulator of endothelial barrier function. Depletion of RhoB in resting, non-inflamed cells leads to increased VE-cadherin levels at cell-cell contacts which in turn promotes enhanced barrier function both in basal and thrombin-stimulated conditions. Also RhoB knockdown cells showed decreased phosphorylation of MLC, which correlates with decreased stress fiber formation. In combination with the loss of RhoA, the protective effect for thrombin-induced hyper-permeability is enlarged, while the combination with a loss of RhoC did not. This indicates that RhoB is an essential regulator of endothelial barrier function under basal conditions and that RhoA and RhoB are the main mediators of thrombin-induced hyper-permeability (Fig. 9).
Figure 9.
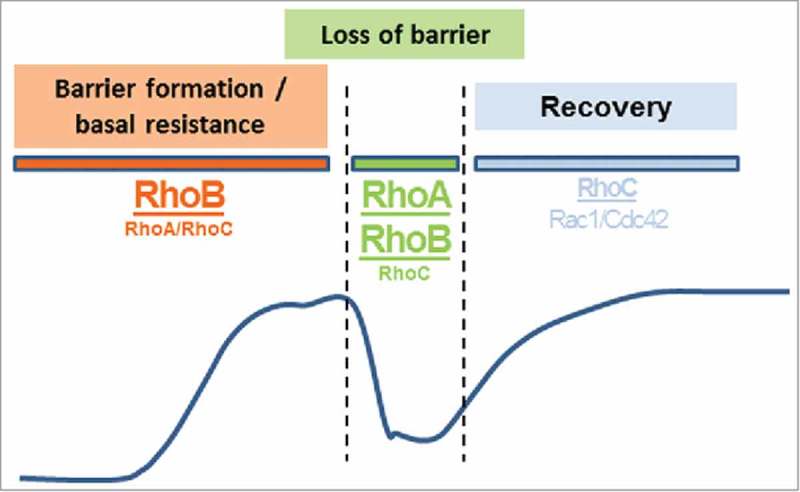
Proposed differential role of RhoA, RhoB and RhoC in endothelial barrier regulation. Under basal conditions, RhoB is a negative regulator of endothelial barrier maintenance, while upon stimulation with vasoactive agents, both RhoA and RhoB are important in mediating a transient loss of barrier function. For recovery of monolayer integrity after a barrier-disrupting stimulus, RhoC co-operates with Rac1 and Cdc42 to allow reformation of cell-cell contacts and barrier function. The line schematically depicts the corresponding ECIS-recorded changes in barrier function.
Our study is the first which reveals the importance of RhoB for regulation of barrier function in resting endothelium. Wojciak-Stothard et al.24 showed that under hypoxic conditions, the expression of RhoB was increased in pulmonary microvascular endothelial cells which was associated with decreased expression of VE-cadherin. Although our cells were not in a hypoxic condition, we showed a similar correlation with decreased RhoB expression and increased VE-cadherin expression. While Wojciak-Stothard et al. showed that upregulation of RhoB by hypoxia induces endothelial permeability and reduces VE-cadherin expression, we found exactly the opposite effect in resting, normoxic (i.e., 5% oxygen) HUVECs upon the downregulation of RhoB. In the same article, Wojciak-Stothard et al. found that knockdown of RhoA and RhoB individually or in combination, reduced pMLC activation in hypoxic conditions, and we found similar effects for thrombin stimulation in RhoA/B double knockdown cells.24 Importantly, our data show that loss of RhoB in HUVECs is protective for endothelial barrier function, indicating that in resting cells, RhoB signaling negatively affects endothelial integrity. This is surprising, given the fact that RhoA is upregulated consequent to the loss of RhoB. Yet, this increased RhoA cannot compensate for the loss of RhoB. In conditions where RhoB was downregulated, 4 findings suggest that Rac1/Cdc42-mediated stabilization of endothelial integrity becomes overt. This includes the improvement in barrier function, enhanced junctional VE-cadherin levels, an increase in cell area and decreased phosphorylation of MLC. Apparently, this Rac1/Cdc42 pathway is always on and is counteracted by RhoB but not by (elevated) RhoA in resting endothelium. It is unknown whether the different localization of RhoB activation (i.e., endosomal) vs. RhoA (plasma membrane) is relevant to this difference.19
Marcos-Ramiro et al. suggested that RhoC protects endothelial integrity in TNFα-treated cells, a conclusion based on thrombin-induced permeability, in the absence of significant effects on electrical barrier properties.20 In contrast, we found in resting cells little to no effects upon downregulation of RhoC, either as single knockdown or in combination with loss of RhoA. Most notably, we find that the relative contribution of the RhoGTPases to the control of barrier function is markedly different in thrombin treated vs. resting cells. In contrast to resting cells, where RhoA cannot compensate for RhoB, these GTPases are redundant in thrombin-induced permeability. This was already observed using single knockdowns, and this effect reached statistical significance upon combined loss of RhoA and RhoB. These data suggest that RhoA and RhoB serve different functions in resting cells while being redundant in the induction of thrombin-induced barrier loss.
Barrier recovery after thrombin treatment in TNFα-pretreated endothelium was recently claimed to be primarily dependent on RhoB, negatively controlling Rac1 recycling from endosomes to the cell border (Marcos-Ramiro et al.).20 A major difference with our study is that Marco-Ramiro et al. pre-treated the cells with TNF-α before analyzing the thrombin response. It was shown previously that TNF-α induces a marked upregulation of RhoB in endothelial cells.25 These authors showed that RhoB is essential for the inflammatory response of endothelial cells to TNFα.25 We performed a similar experiment with pre-treatment with TNF-α of our single, double and triple knockdown cells, and found an effect of TNF-α of 40–70% reduction in endothelial resistance. Adding thrombin showed similar results as found by Marcos-Ramiro et al. with increased recovery in the RhoB knockdown conditions (Supplemental figure 4). Thus, although the main messages of the study of Marcos-Ramiro et al. and ours are similar there are also important differences. Marcos-Ramiro et al. found, for example, that RhoB and RhoC single knockdown and all double and triple knockdown cells showed decreased TEER under basal conditions, while we found an increased barrier function for RhoB, RhoA/B, RhoB/C and triple knockdown cells. Also, the recovery after thrombin stimulation was different for these conditions when compared with TNF-α pre-treated HUVECs. Apart from possible differences in source and culture of HUVECs between our laboratories, a major difference appears to be the pre-treatment of TNF-α, which was used by Marcos-Ramiro, but not in our study.
Our results show evidence for only a limited role for RhoC in barrier recovery after thrombin stimulation. The observed differences were not significant but there appeared to be a consistent trend (e.g. slower recovery after thrombin stimulation, larger gaps in RhoC and RhoB/C knockdown cells). There are 2 mechanisms which might account for this effect. Firstly, Reinhard et al. showed that RhoC is co-localized with VE-cadherin at cell-cell contacts and that it disappears from cell-cell contacts upon stimulation with thrombin.19 The mechanism of this effect is unclear, but it could explain the reduced recovery in RhoC knockdown cells after thrombin stimulation. Secondly, knockdown of RhoB and RhoC together or RhoC alone leads to increased expression of respectively RhoA or RhoB. Overexpression of RhoA has been shown to induce an activated phenotype in endothelial cells. These cells show more F-actin stress fibers, an increased migration and also show an increase in cord-like structures in angiogenesis assays.26 Increased expression of RhoB was also shown to induce phosphorylation of MLC in endothelial cells.24 Although our RhoC and RhoB/C knockdown cells did not show clear activated phenotypes under basal conditions, loss of RhoC alone or RhoB and RhoC together induced the overexpression of RhoB and/or RhoA which could lead to increased stress fiber formation upon thrombin stimulation.
This study extends our knowledge of the individual, non-redundant roles of RhoA, RhoB and RhoC in endothelial barrier function. We showed that in resting endothelial cells, RhoB is most dominant in negatively regulating barrier function and that this could not be compensated by increased RhoA. Thrombin relies primarily on both RhoB and RhoA for induction of barrier loss while the recovery from such an insult requires RhoC to a limited extent, in addition to important roles for Rac1 and/or Cdc42 (Fig. 9). Additional work will be required to identify specific effector proteins that are activated downstream of each of these closely related GTPases. Since their effector domains are virtually identical27 the differential localization of the RhoGTPases and their activators19 as well as their different C-termini28 will likely be critical for differential functioning of RhoGTPases in endothelial cells.
Material & methods
Antibodies and siRNAs
The following antibodies were used: rabbit α(anti)RhoA (#2117), rabbit αRhoC (#3430), rabbit αβ-tubulin (#2128), rabbit αVE-cadherin XP (#2500), rabbit αphospho-Myosin Light Chain 2 (#3671), rabbit αp44/42 MAPK (ERK1/2) (#9102), mouse αphospho-p44/42 MAPK (ERK1/2) (#9106S) (Cell Signaling Technologies), rabbit αRhoB (#sc-180) (Santa Cruz Biotechnology) and αVE-cadherin (V1514) (Sigma).
For western blot, horseradish peroxidase-conjugated goat-anti-rabbit antibody (Dako) was used as a secondary antibody. For Immunofluorescence Alexa 488- secondary antibody (anti-rabbit) and Alexa 647 – secondary antibody (anti-mouse) (Both Invitrogen) were used.
The small interference RNAs (siRNAs) which were used are: ON-TARGET plus Non-targeting pool (siNT), ON-TARGET plus Human RHOA siRNA (siRhoA), ON-TARGET plus Human RHOB siRNA (siRhoB), ON-TARGET plus Human RHOC siRNA (siRhoC)(All Dharmacon/GE-healthcare).
Cell isolation and culture
The study was executed in accordance with the Declaration of Helsinki and was approved by the University Human Subjects Committee of the VU University Medical Center. For isolation of Human Umbilical Vein Endothelial Cells (HUVECs), umbilical cords from healthy donors were obtained from the Amstelland Ziekenhuis, Amstelveen. Cells were isolated from umbilical cord veins according to the protocol of Jaffe.29 Cells were cultured in 1% gelatin coated plates (Costar) with M199 medium containing 2 mMol/L L-glutamine, 100 U/mL penicillin, 100 mg/mL streptomycin (all Lonza) 10% heat-inactivated New Born Calf Serum from (Gibco), 10% heat-inactivated Human serum (Sanquin CLB), 5 U/mL heparin (Leo Pharmaceuticals products) and crude endothelial cell growth factor prepared from bovine brains. Cells were kept in incubators with 5% CO2 at 37 °C, with a medium change every other day. Confluent cells were washed with M199, trypsinized (0.05% trypsin, Gibco) and seeded in a 1:3 dilution. Passage 1 or 2 cells consisting of pools from 3 donors were used unless indicated otherwise, in all experiments.
Transfection with small interfering RNA
HUVECs were transfected with Dharmafect1, according to the manufacturer's protocol (Dharmacon/GE Healthcare). Cells were transfected with a final concentration of 25 nM siRNA and 25 nM Dharmafect1 in 10% NBCSi/M199 per condition. Transfection was done in 96 wells, 12 wells and 6 wells format on cells which were approximately 80% confluent. Transfection medium was replaced after 16 hours of transfection with regular culture medium to avoid toxicity. Transfection efficiency was evaluated by Western blot analysis of protein expression, and usually exceeded 85%.
Endothelial barrier function assays
Endothelial barrier function was measured with electrical cell-substrate impedance sensing (ECIS) and passage of Horse Radish Peroxidase (HRP). For ECIS measurements, cells were seeded 1:1 density on gelatin coated 96 wells ECIS plates containing gold intercalated electrodes (Applied Biophysics). 24 Hours after seeding, cells were transfected using Dharmafect 1 and siRNAs for 16 hours. 72 hours after transfection, cells were serum-starved with M199 supplemented with 1% Human Serum Albumin (HSA, Sanquin CLP) for 90 minutes. Subsequently, a thrombin mix was added to the wells with a final concentration of 1 U/mL (Sigma Aldrich). During the growth phase, resistance was measured at multiple frequencies to allow calculation for changes in cell-cell adhesion (Rb) and cell-matrix interaction (α).21 Rb is the resistivity of cell-cell contact to the current flow, and thereby an inverse measure of permeability. α is a measure for the impedance contribution arising from the cell-electrode junction. The assumption of the model is that cells on the electrode are circular and disk shaped, with an insulating membrane, hover over the electrode and are filled with electrolytes. Because the cell membrane is insulating, there are only 3 pathways for the current: a) between the ventral surface of the cells and electrode(α), b) between the cell-cell contacts (Rb), and c) through the cells (Impedance).30,31
For measurement of HRP passage, endothelial cells were transfected in a 12 wells plate 24 hours before passaging 2:1 to 1% gelatin-coated 0.33 cm2 polyester ThinCerts® cell culture inserts (Greiner Bio-one) with a pore-size of 3.0 µm. Approximately 72 hours after the start of transfection, cells were serum-starved with 1% HSA/M199 which was added to the filters for 60 minutes. Before stimulation, medium in the upper compartment was replaced with 1% HSA/M199 containing HRP 5 µg/mL (Sigma Aldrich) and 1 U/mL thrombin or a vehicle control. 1% HSA/M199 was added to the lower compartment. A sample was taken from the lower compartment at different time points. The HRP concentration was calculated by measuring absorption after adding TetraMethylBenzidine (Upstate/Millipore) and sulfuric acid.
Immunofluorescence imaging of cultured endothelial cells
Transfected cells were seeded on 2 cm2 glass coverslips, which were coated with 1% gelatin and crosslinked with 0.5% glutaraldehyde (Sigma Aldrich), approximately 24 hours after the start of transfection. Cells were grown for 48 hours with complete medium to reach the transfection time of 72 hours. After pre-incubating with 1% HSA/M199 for 1 hour, thrombin was added to the wells in a final concentration of 1 U/mL. After 15 minutes, cells were fixed with warm (37 °C) 4% paraformaldehyde (Sigma Aldrich) and put on ice for 15 minutes. The PFA was washed away with phosphate buffered saline (PBS). Subsequently, cells were permeabilized with 0.2% Triton X-100 in PBS (Sigma Aldrich) and stained with primary antibodies against VE-cadherin and pERK1/2 (in 0.1% HSA/PBS) overnight at 4°C. After washing 3 times, the cells were incubated with a FITC-labeled secondary antibody (anti-rabbit or anti-mouse 1:100 in 0.1% HSA/PBS) and acti stain phalloidin (direct staining, in 0.1% HSA/PBS (Tebu Bio)) at room temperature. After washing, the cells were incubated with DAPI (Thermo Fisher Scientific) at room temperature. Coverslips were mounted with Mowiol4-88/DABCO solution (Calbiochem, Sigma Aldrich). Confocal scanning laser microscopy was performed on a Leica TCS SP8 STED 3X (Leica Microsystems). A 63x oil objective with NA 1.4 was used to image the sample.
Images were analyzed and equally adjusted with ImageJ. To account for the variability of VE-cadherin and F-actin staining we made 4–5 images per knockdown condition using 4 independent HUVEC donor pools. We measured the Integrated Density per picture and corrected for the number of cells in the image. For analysis of VE-cadherin area, we measured the total area of VE-cadherin staining and divided this by the amount of cells on that same image. The gap area was measured by taking images with simultaneous detection of F-actin, VE-cadherin, Dapi and pERK1/2. All channels were equally adjusted and made binary. All parts which did not contain staining were considered as being a gap.
Protein analysis
For protein analysis, cells were seeded in 5 or 10 cm2 culture wells. After pre-incubation with 1% HSA/M199 for 1 hour, thrombin was added to a final concentration of 1 U/mL for 15 minutes. Subsequently, the cells were washed once with ice-cold PBS, and whole cell lysates were obtained by scraping the cells in the presence of 2x SDS sample buffer. Protein samples were loaded on 12.5% SDS-gels, electrophoresed and transferred to nitrocellulose membranes. Protein analysis was performed by incubation of the nitrocellulose membranes with antibodies for RhoA, RhoB, RhoC, pMLC, β-tubulin or ERK1/2. Bands were visualized with enhanced chemiluminescence (Amersham/GE-Healthcare) on a AI600 machine (Amersham/GE-Healthcare).
Statistical analysis
Data are presented as mean ± SEM, unless indicated otherwise. Statistical analysis was performed using a one way ANOVA with Dunnett's post-hoc test. Number of replicates (N) refers to the number of experiments performed with different endothelial cell donor pools, unless indicated otherwise. P-values <0.05 were considered statistically significant.
Supplementary Material
Abbreviations
- AJ
Adherens Junction
- EC
Endothelial cell
- HUVEC
Human Umbilical Vein Endothelial cell
- MLC
Myosin Light Chain
- RhoGTPases
Rho family of small guanine nucleotide triphosphatases
- ROCK
Rho Kinase
- siNT
Non-targeting siRNA
- TJ
Tight Junction
- VE-cadherin
Vascular Endothelial Cadherin
- VEGF
Vascular Endothelial Growth Factor
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank J. Kole and R. Musters (VU University Medical Center, The Netherlands) for their help with the microscope equipment.
Funding
This work was supported by the Rembrandt Institute for Cardio-vascular Research.
Author contributions
PLH, GPvNA and VWvH conceived the project and designed experiments. MCAP designed and performed experiments and analyzed data. JSMvB performed experiments. MCAP and PLH wrote the manuscript.
References
- [1].Aird WC. Endothelial cell heterogeneity. Cold Spring Harb Perspect Med 2012; 2(1):a006429; PMID:22315715; https://doi.org/ 10.1101/cshperspect.a006429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev 2006; 86(1):279–367; PMID:16371600; https://doi.org/ 10.1152/physrev.00012.2005 [DOI] [PubMed] [Google Scholar]
- [3].Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med 2010; 363(7):689–91; PMID:20818861; https://doi.org/ 10.1056/NEJMcibr1007320 [DOI] [PubMed] [Google Scholar]
- [4].Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res 2010; 87(2):243–53; PMID:20299335; https://doi.org/ 10.1093/cvr/cvq086 [DOI] [PubMed] [Google Scholar]
- [5].Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev Cell 2013; 26(5):441–54; PMID:24044891; https://doi.org/ 10.1016/j.devcel.2013.08.020 [DOI] [PubMed] [Google Scholar]
- [6].Abu Taha A, Schnittler HJ. Dynamics between actin and the VE-cadherin/catenin complex. Cell Adh Migr 2014; 8(2):125–35; PMID:24621569; https://doi.org/ 10.4161/cam.28243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hall A. Rho GTPases and the actin cytoskeleton. Science 1998; 279(5350):509–14; PMID:9438836; https://doi.org/ 10.1126/science.279.5350.509 [DOI] [PubMed] [Google Scholar]
- [8].Wojciak-Stothard B, Ridley AJ. Rho GTPases and the regulation of endothelial permeability. Vascul Pharmacol 2002; 39(4-5):187–99; PMID:12747959; https://doi.org/ 10.1016/S1537-1891(03)00008-9 [DOI] [PubMed] [Google Scholar]
- [9].Ridley AJ. RhoA, RhoB and RhoC have different roles in cancer cell migration. J Microsc 2013; 251(3):242–9; PMID:23488932; https://doi.org/ 10.1111/jmi.12025 [DOI] [PubMed] [Google Scholar]
- [10].Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett 2008; 582(14):2093–101; PMID:18460342; https://doi.org/ 10.1016/j.febslet.2008.04.039 [DOI] [PubMed] [Google Scholar]
- [11].Vega FM, Colomba A, Reymond N, Thomas M, Ridley AJ. RhoB regulates cell migration through altered focal adhesion dynamics. Open Biol 2012; 2(5):120076; PMID:22724071; https://doi.org/ 10.1098/rsob.120076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vega FM, Fruhwirth G, Ng T, Ridley AJ. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J Cell Biol 2011; 193(4):655–65; PMID:21576392; https://doi.org/ 10.1083/jcb.201011038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kitzing TM, Wang Y, Pertz O, Copeland JW, Grosse R. Formin-like 2 drives amoeboid invasive cell motility downstream of RhoC. Oncogene 2010; 29(16):2441–8; PMID:20101212; https://doi.org/ 10.1038/onc.2009.515 [DOI] [PubMed] [Google Scholar]
- [14].Szulcek R, Beckers CM, Hodzic J, de Wit J, Chen Z, Grob T, Musters RJ, Minshall RD, van Hinsbergh VW, van Nieuw Amerongen GP. Localized RhoA GTPase activity regulates dynamics of endothelial monolayer integrity. Cardiovasc Res 2013; 99(3):471–82; PMID:23536606; https://doi.org/ 10.1093/cvr/cvt075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Heemskerk N, Schimmel L, Oort C, van Rijssel J, Yin T, Ma B, van Unen J, Pitter B, Huveneers S, Goedhart J, et al.. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat Commun 2016; 7:10493; PMID:26814335; https://doi.org/ 10.1038/ncomms10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].van Nieuw Amerongen GP, Beckers CM, Achekar ID, Zeeman S, Musters RJ, van Hinsbergh VW. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler Thromb Vasc Biol 2007; 27(11):2332–9; PMID:17761936; https://doi.org/ 10.1161/ATVBAHA.107.152322 [DOI] [PubMed] [Google Scholar]
- [17].Carbajal JM, Schaeffer RC. RhoA inactivation enhances endothelial barrier function. Am J Physiol Gastrointest Liver Physiol 1999; 277(5):955–64; PMID:10564088 [DOI] [PubMed] [Google Scholar]
- [18].Mikelis CM, Simaan M, Ando K, Fukuhara S, Sakurai A, Amornphimoltham P, Masedunskas A, Weigert R, Chavakis T, Adams RH, et al.. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat Commun 2015; 6:6725; PMID:25857352; https://doi.org/ 10.1038/ncomms7725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Reinhard NR, van Helden SF, Anthony EC, Yin T, Wu YI, Goedhart J, Gadella TW, Hordijk PL. Spatiotemporal analysis of RhoA/B/C activation in primary human endothelial cells. Sci Rep 2016; 6:25502; PMID:27147504; https://doi.org/ 10.1038/srep25502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Marcos-Ramiro B, García-Weber D, Barroso S, Feito J, Ortega MC, Cernuda-Morollón E, Reglero-Real N, Fernández-Martín L, Durán MC, Alonso MA, et al.. RhoB controls endothelial barrier recovery by inhibiting Rac1 trafficking to the cell border. J Cell Biol 2016; 213(3):385–402; PMID:27138256; https://doi.org/ 10.1083/jcb.201504038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Moy AB, Winter M, Kamath A, Blackwell K, Reyes G, Giaever I, Keese C, Shasby DM. Histamine alters endothelial barrier function at cell-cell and cell-matrix sites. Am J Physiol Lung Cell Mol Physiol 2000; 278(5):L888–98; PMID:10781418 [DOI] [PubMed] [Google Scholar]
- [22].Van Nieuw Amerongen GP, Van Delft S, Vermeer MA, Collard JG, Van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability -role of rho kinase and protein tyrosine kinases. Circ Res 2000; 87:335–40; PMID:10948069; https://doi.org/ 10.1161/01.RES.87.4.335 [DOI] [PubMed] [Google Scholar]
- [23].Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al.. Regulation of myosin phophatase by rho and rho-associated kinase (Rho-Kinase). Science 1996; 273(5272):245–8; PMID:8662509; https://doi.org/ 10.1126/science.273.5272.245 [DOI] [PubMed] [Google Scholar]
- [24].Wojciak-Stothard B, Zhao L, Oliver E, Dubois O, Wu Y, Kardassis D, Vasilaki E, Huang M, Mitchell JA, Harrington LS, et al.. Role of RhoB in the regulation of pulmonary endothelial and smooth muscle cell responses to hypoxia. Circ Res 2012; 110(11):1423–34; PMID:22539766; https://doi.org/ 10.1161/CIRCRESAHA.112.264473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kroon J, Tol S, van Amstel S, Elias JA, Fernandez-Borja M. The small GTPase RhoB regulates TNFalpha signaling in endothelial cells. PLoS One 2013; 8(9):e75031; PMID:24086429; https://doi.org/ 10.1371/journal.pone.0075031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhao L, Xu G, Zhou J, Xing H, Wang S, Wu M, Lu YP, Ma D. The effect of RhoA on human umbilical vein endothelial cell migration and angiogenesis in vitro. Oncol Rep 2006; 15:1147–52; PMID:16596177 [PubMed] [Google Scholar]
- [27].Schaefer A, Reinhard NR, Hordijk PL. Toward understanding RhoGTPase specificity: Structure, function and local activation. Small GTPases 2014; 5(2):6; PMID:25483298; https://doi.org/ 10.4161/21541248.2014.968004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lam BD, Hordijk PL. The Rac1 hypervariable region in targeting and signaling: A tail of many stories. Small GTPases 2013; 4(2):78–89; PMID:23354415; https://doi.org/ 10.4161/sgtp.23310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. J Clin Invest 1973; 52(11):2745–56; PMID:4355998; https://doi.org/ 10.1172/JCI107470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Giaever I, Keese C. Micromotion of mammalian cells measured electrically. Proc Natl Acad Sci U S A 1991; 88:7896–900; PMID:1881923; https://doi.org/ 10.1073/pnas.88.17.7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lo C, Keese C, Giaever I. Cell-Substrate contact: Another factor may influence transepithelial electrical resistance of cell layers cultured on permeable Filters. Exp Cell Res 1999; 250:576–80; PMID:10413610; https://doi.org/ 10.1006/excr.1999.4538 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.