

ABSTRACT
We describe a bispecific dual-antagonist antibody against human B cell activating factor (BAFF) and interleukin 17A (IL-17). An anti-IL-17 single-chain variable fragment (scFv) derived from ixekizumab (Taltz®) was fused via a glycine-rich linker to anti-BAFF tabalumab. The IgG-scFv bound both BAFF and IL-17 simultaneously with identical stoichiometry as the parental mAbs. Stability studies of the initial IgG-scFv revealed chemical degradation and aggregation not observed in either parental antibody. The anti-IL-17 scFv showed a high melting temperature (Tm) by differential scanning calorimetry (73.1°C), but also concentration-dependent, initially reversible, protein self-association. To engineer scFv stability, three parallel approaches were taken: labile complementary-determining region (CDR) residues were replaced by stable, affinity-neutral amino acids, CDR charge distribution was balanced, and a H44-L100 interface disulfide bond was introduced. The Tm of the disulfide-stabilized scFv was largely unperturbed, yet it remained monodispersed at high protein concentration. Fluorescent dye binding titrations indicated reduced solvent exposure of hydrophobic residues and decreased proteolytic susceptibility was observed, both indicative of enhanced conformational stability. Superimposition of the H44-L100 scFv (PDB id: 6NOU) and ixekizumab antigen-binding fragment (PDB id: 6NOV) crystal structures revealed nearly identical orientation of the frameworks and CDR loops. The stabilized bispecific molecule LY3090106 (tibulizumab) potently antagonized both BAFF and IL-17 in cell-based and in vivo mouse models. In cynomolgus monkey, it suppressed B cell development and survival and remained functionally intact in circulation, with a prolonged half-life. In summary, we engineered a potent bispecific antibody targeting two key cytokines involved in human autoimmunity amenable to clinical development.
KEYWORDS: B cell activation factor, Interleukin-17, bispecific antibody, protein engineering, scFv
Introduction
Monoclonal antibodies (mAbs) inhibit their targets in a very selective way, thus reducing off-target side effect risk. However, the pathophysiology of autoimmune diseases is often multifactorial and thought to involve the complex interplay between environmental factors, T cells, B cells, dendritic cells, multiple cytokines, and varied genetics, leading to potential dysregulation of a variety of innate and adaptive immune responses. As such, simultaneous intervention on multiple fronts might be required to provide optimal therapeutic benefit. One approach to broaden the efficacy of new biologics is to engineer multiple binding specificities into a single molecule. The resulting blockade of distinct disease mediators and signaling cascades with a single drug can potentially lead to more profound disease suppression. Here, we describe the generation and preclinical characterization of a tetravalent bispecific antibody LY3090106 (tibulizumab) with potent inhibition of both B cell activating factor (BAFF) and interleukin-17A (IL-17).
LY3090106 is not an obligate bispecific, i.e., requiring dual target engagement to achieve biological activity, but rather a single agent combination of two independently acting bivalent antagonist moieties. In this respect, it can be thought of as a “four-headed-hammer” that can simultaneously knock out two distinct soluble ligands.1 Tabalumab and ixekizumab are formulated under strikingly discordant conditions.2,3 Incompatibility hinders co-formulation, potentially causing a suboptimal drug administration profile by requiring separate injections. A bispecific antibody approach mitigated these issues.
BAFF is involved in multiple aspects of immune system regulation, but is best known for its central role in B lymphocyte development and proliferation.4 Initially expressed as a membrane-bound form on various cell types, BAFF is subsequently cleaved, generating a soluble protein fragment.4 Dysregulated BAFF expression is thought to contribute to autoimmune diseases via effects on abnormal B-lymphocyte activation, proliferation, survival, and immunoglobulin secretion.5 Overexpression of BAFF in mouse models induces autoimmune disease mimicking rheumatoid arthritis (RA), systemic lupus erythematosus (SLE) and primary Sjögren’s syndrome (pSS).6 Likewise in humans, elevated serum BAFF levels are reported in SLE, RA, pSS, IgA nephropathy, and systemic sclerosis patient serum.7,8 BAFF therefore is a rational target for new therapies in B cell-driven autoimmune disorders, and BAFF-antagonizing mAbs have been tested in human clinical trials, including tabalumab9 and the US Food and Drug Administration (FDA)-approved belimumab.10,11
The IL-17 family of cytokines contains six structurally related isoforms: IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (IL-25), and IL-17F with distinct abilities to form homo- or heterodimers, leading to differences in specificity and signaling potencies.12,13 These cytokines are important for the control of infections, especially by extracellular fungi.14 Conversely, if unrestrained, they can contribute to the pathology of autoimmune and chronic inflammatory conditions. The prototypic member of this family is IL-17A, herein referred to as IL-17. T-helper cell 17 (Th17) are a subset of T cells, characterized by the production of the cytokine IL-17. In recent years, multiple studies have established the pathogenic involvement of Th17 cells and its cytokine IL-17 in autoimmune diseases, including psoriasis, psoriatic arthritis, RA, ankylosing spondylitis, and multiple sclerosis.15,16 Both IL-17 and its receptor have been targeted by an array of FDA-approved therapeutic mAbs, in several different autoimmune indications, including secukinumab,17 ixekizumab,18,19 and brodalumab.20,21
BAFF and IL-17 have been implicated singly in immunopathology in seemingly independent manners. However, BAFF affects both the adaptive and innate immune responses by activating B cells and promoting Th17 cell expansion. IL-17, in turn, is a central effector cytokine for BAFF-mediated pro-inflammatory effects. That simultaneous neutralization of BAFF and IL-17 might provide more than a simple “divide-and-conquer” therapeutic modality in autoimmune disease is evidenced by the cross-talk observed among these cytokines, and others, in multiple studies supporting a pathological axis involving multiple cell types.22–28
Myriad bispecific antibody formats have been described in recent years.29–31 In spite of the ingenious protein engineering solutions devised, inherent issues with chain pairing and assembly, expression and stability are still difficult challenges. Monovalent IgG-like bispecific formats are the most native structurally, but only possess a single binding site for each ligand. In the case of targeting soluble multimeric ligands, such as trimeric BAFF and dimeric IL-17, this can result in reduced binding avidity and conceivably require higher dose administration to match the potency of the mAb combination. This can pose a drug development challenge due to viscosity and formulation issues. Tetravalent bispecific antibody designs offer potential advantages in this regard. Herein we describe the systematic generation, characterization, and protein engineering of a novel tetravalent bispecific starting in a classical IgG – single-chain variable fragment (scFv) format.32,33 The IgG-scFv bispecific geometry can offer an advantage over other common tetravalent formats such as dual variable domain-IgG where the inner binding arm affinity is dependent on the variable region pairing, orientation, and linker length.34 The stabilized bispecific molecule LY3090106 displayed potent in vivo neutralization of both BAFF and IL-17 with IgG-like pharmacokinetic properties in non-human primates (NHP). Our results demonstrate solubility and stability issues can be mitigated by protein engineering, and that next-generation bispecific molecules may be developed with exemplary properties to enable clinical evaluation in autoimmune disorders.
Results
Design and characterization of tetravalent bispecific antibody
An IgG-scFv32 was engineered by fusion of an scFv derived from ixekizumab19 to the C-terminus of the HC of tabalumab.9 The scFv variable heavy region (VH) and variable kappa region (Vk) domains were connected and tethered to the mAb by glycine-rich flexible linkers. The bispecific molecule was expressed in human embryonic kidney 293 (HEK-293) cells and purified in two steps using standard Protein A-Sepharose and size exclusion chromatography (SEC) to remove aggregate (<3%). Once purified, it was characterized for ligand binding using surface plasmon resonance (SPR). Figure 1 shows that the bispecific simultaneously bound to both ligands regardless of the order of addition. The stoichiometries of BAFF trimer and IL-17 dimer binding to LY3090106 are 1.1 and 1.4, respectively. This implies that the BAFF trimer is bound predominantly in a bivalent mode, whereas IL-17 dimer binding involves a mixture of mono- and bivalent interactions. Identical stoichiometry was observed for each parental antibody.
Figure 1.
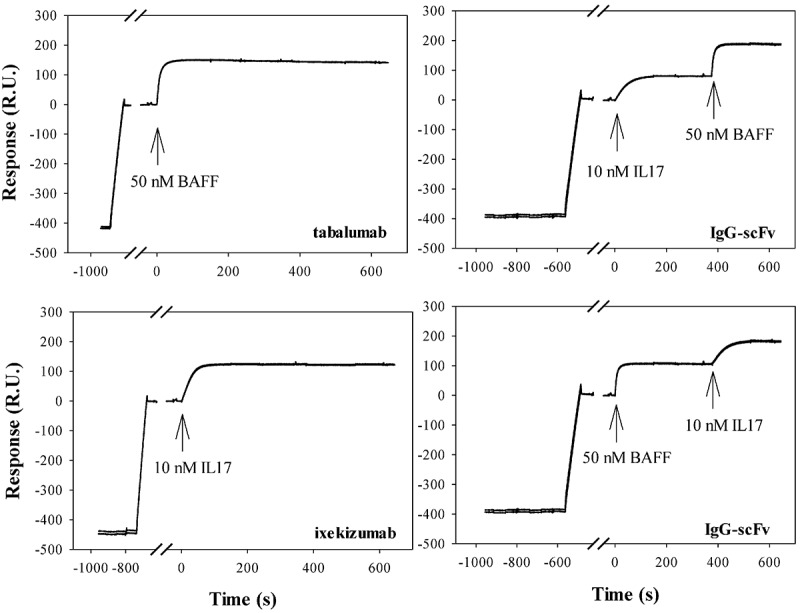
Biacore surface plasmon resonance demonstration of simultaneous target engagement by LY3090106. The IgG-scFv, or the parental antibodies, were captured by protein A immobilized onto a Biacore CM4 chip, followed by injections of either 10 nM IL-17, 50 nM BAFF, or both targets sequentially in either order. Binding stoichiometries were estimated based on the maximum signal changes from the binding of each target, relative to the signal response of the captured antibody (scaled by the relative molecular weights of antibody and target; the calculated stoichiometry values assume that IL-17 is predominantly dimeric, and that BAFF is predominantly trimeric).
Upon storage of the purified IgG-scFv, increased high molecular weight species (%HMW) were observed over time by analytical SEC (aSEC). The protein self-association and structural integrity were shown to be concentration, pH, and salt-dependent (data not shown). After limited concentration to 30 mg/mL in phosphate-buffered saline (PBS) formulation, the %HMW in the sample increased to greater than 15% following storage for only 2 days at 4°C. At the same concentration in 10 mM citrate pH 5 formulation, aggregation increased to over 35% and phase separation was visible. Moreover, accelerated stability studies conducted under stressed conditions (40°C, 1 month) showed chemical degradation including 4% clipping adjacent to the scFv tether, 13% oxidation of scFv heavy chain complementarity-determining region (CDR)-H2 Met53 and 12% deamidation of light chain CDR-L1 Asn30 (numbering according to Kabat).35 Neither of these modifications was observed for ixekizumab, suggesting a difference in the solvent exposure or local environment of these amino acids in the scFv context. Further development of the IgG-scFv molecule required significant protein engineering to mitigate these issues.
To fix the chemical instability the labile scFv amino acids were targeted for random mutagenesis and screening of phage-expressed antigen-binding fragments (Fabs) using a capture lift assay.36 Affinity neutral mutations identified in this manner were DNA-sequenced and unique clones expressed as periplasmic Fab in E.coli and analyzed by titration enzyme-linked immunosorbent assay (ELISA). Select mutations were examined by SPR, and preferred substitutions incorporated into the scFv context. In this way, chemically labile CDR-H2 Met53 and CDR-L1 Asn30 were replaced with Thr and Glu, respectively.
Electrostatic attractions between oppositely charged residues on the protein surface can contribute to protein self-association propensity.37–40 Calculation of the surface charge distribution potential for the parental scFv showed a distinct charge dipole created by an acidic VH patch and a basic Vk patch (Figure 2A). The Asn30Glu mutation described above removed a source of chemical instability but also introduced an acidic residue into the positively charged Vk surface region (Figure 2B). Additional mutagenesis was done to target the predominant negatively charged patch in VH. CDR-L1 contains an Asp at position 31. This amino acid was randomized by mutagenesis and variants screened to potentially identify charge and affinity neutral substitutions without success. A secondary approach was to replace the adjacent CDR residues with Arg or Lys. Ser28Lys showed slightly improved affinity by ELISA (data not shown) and, when combined with Asn30Glu, provided a more balanced surface electrostatic distribution profile (Figure 2C).
Figure 2.
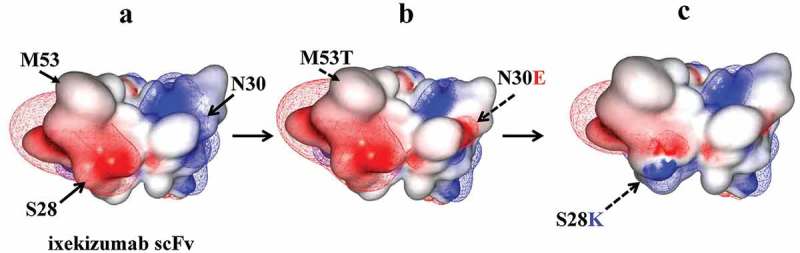
Electrostatic model showing the effect of scFv engineering to remove chemical liabilities and mitigate CDR surface charge imbalance. scFv models are shown with CDR residues facing outward. The Fv surface is mapped by the calculated electrostatic potential with isocontour value of ±1 kT/e using MOE. Positive and negative charged regions shown in blue and red, respectively.
The differential scanning calorimetry (DSC) profile of the purified scFv displayed a single thermal unfolding transition with a midpoint (Tm) of 73.1°C (Figure 3A). The relatively high Tm of the scFv implied that intrinsic thermal instability of either Fv was not the root cause of the observed protein aggregation. The concentration-dependent behavior of the purified scFv molecule was followed by aSEC. As shown in Figure 3B, the initial purified scFv at 0.55 mg/mL was largely monomeric in solution. After concentration to 5.2 mg/mL a higher order, presumably dimeric, species became apparent by aSEC. Further concentration of the scFv to 15 mg/mL revealed additional peaks, possibly reflecting dimeric, trimeric and tetrameric scFv oligomers. The reversibility of the scFv self-association was tested by dilution of the concentrated sample. As shown in Figure 3B, a 1:20 dilution resulted in nearly complete dissolution of the higher order species, indicative of largely reversible interactions under these conditions. Taken together, the high Tm and reversible self-association support a mechanism whereby transient “breathing” of the VH/VL interface can at high concentration lead to stochastic intermolecular interaction between neighboring molecules that have undergone a similar transition to an open scFv configuration. It should be noted that, while the scFv self-association was initially reversible, prolonged storage eventually led to irreversible aggregate formation (data not shown).
Figure 3.
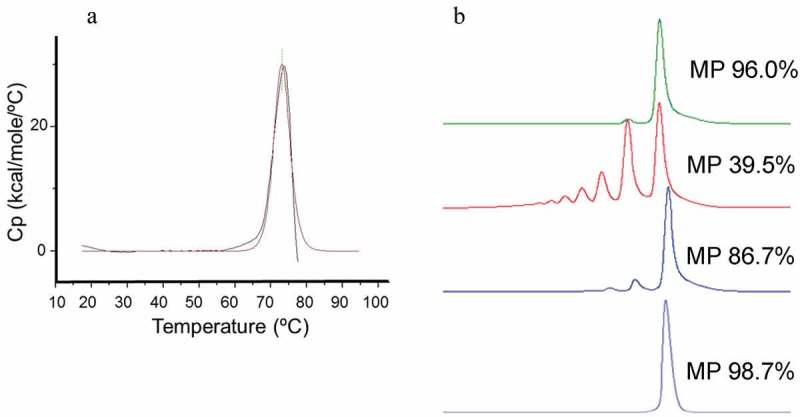
A. DSC analysis of anti-IL17 scFv without H44-L100 disulfide bond. A single thermal unfolding transition (Tm) of 73.1°C was determined. Figure 3B. aSEC HPLC analysis of anti-IL17 scFv concentration-dependent self-association. Main peak (MP) percentages were calculated for each concentration. 0.55 mg/mL (light blue), 5.2 mg/mL (dark blue), 15 mg/mL (red), 1:20 dilution of 15 mg/mL sample (0.75 mg/mL final; green).
Several engineering, or selection strategies, have been described to increase scFv interface stability.41–45 As originally described by Pastan and colleagues, one solution is the engineering of an interface disulfide bond.46–49 A H44-L100 disulfide bond was introduced, and the resulting scFv purified and characterized. Figure 4A shows the DSC profile of the disulfide-stabilized scFv. It displayed two thermal unfolding transitions with midpoints (Tm) of 75.3°C and 73.3°C, likely attributable to the unfolding of the individual VH and Vk domains, respectively. Thus, disulfide-stabilization had minimal effect upon the temperature-induced unfolding of the individual Fv domains. The concentration-dependent protein self-association of the H44-L100 scFv characterized by aSEC is shown in Figure 4B. In contrast to the parental molecule, the disulfide-stabilized scFv showed no detectable formation of higher order species and remained monomeric at concentrations up to 71 mg/mL. The DSC profile of the optimized IgG-scFv (Figure 4C) revealed a single thermal transition with a midpoint of 66.6°C similar to that of the tabalumab parent mAb (data not shown). Thus, we concluded that the optimized scFv in the bispecific context had a Tm of greater than or equal to this temperature.
Figure 4.
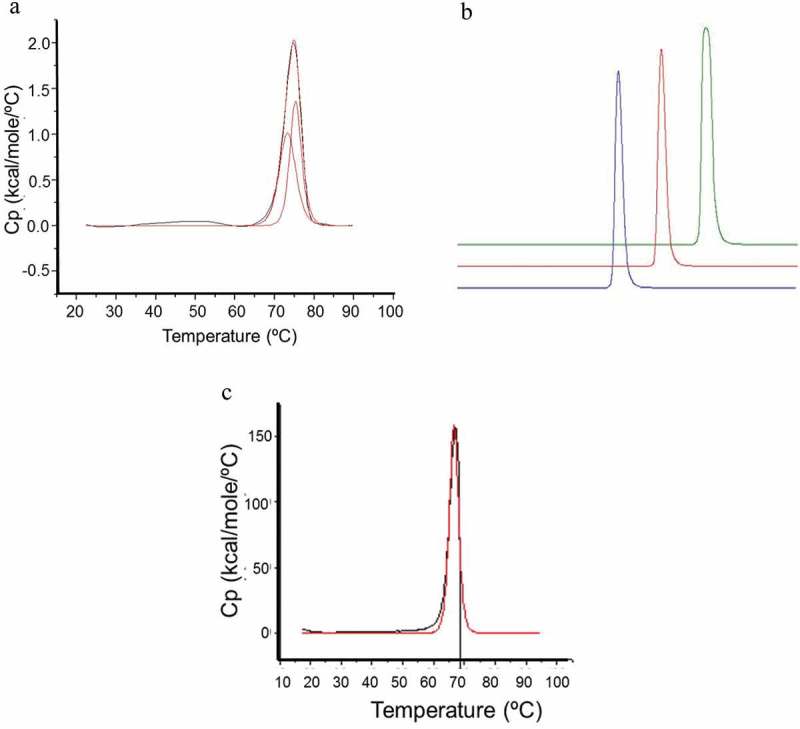
A) DSC analysis of H44-L100 anti-IL17 scFv. Two thermal unfolding transitions with midpoints (Tm) of 73.3°C and 75.3°C were determined. B) aSEC HPLC analysis of H44-L100 anti-IL17 scFv; 1 mg/mL (blue), 10 mg/mL (red), 71 mg/mL (green). C) DSC analysis of IgG-scFv. A single thermal unfolding with midpoint (Tm) of 66.6°C was determined.
Next, we compared the binding of 1-anilino-8-naphthalene sulfonate (ANS) to both parental and disulfide-stabilized scFv. ANS is thought to bind to hydrophobic (nonpolar) protein surfaces, primarily through its nonpolar anilinonaphthalene group, which is accompanied by an increase in fluorescence emission intensity and a blue shift of the peak maximum.50,51 ANS titrations were conducted to investigate whether the introduction of the H44-L100 disulfide bond could reduce the solvent exposure of the Fv hydrophobic interface residues. Figure 5 shows the fluorescence intensity data collected following titration of ANS from 0 to 300 µM to a protein solution containing 10 µM scFv in PBS buffer, pH 7.4. The parental scFv showed a striking increase in emission intensity following ANS binding. In contrast, the H44-L100 disulfide-stabilized scFv showed significantly less ANS fluorescence emission intensity, indicative of reduced exposure of hydrophobic residues. The H44-L100 scFv was also found to be resistant to pepsin-mediated hydrolysis, whilst peptide fragmentation was readily detected in the parental scFv sample (data not shown), presumably due to increased susceptibility of the molecule in its transient “open” state.
Figure 5.
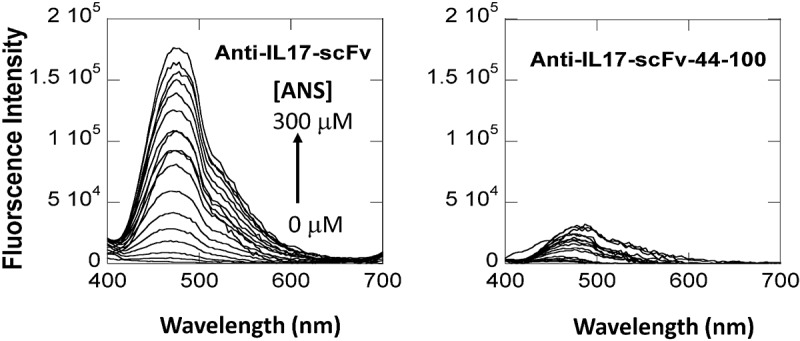
ANS binding to anti-IL-17 scFv (left panel) and H44-L100 anti-IL-17 scFv (right panel) as monitored by ANS fluorescence emission. Excitation wavelength at 360 nm. Emission scan from 400 to 700 nM. One nanometer excitation and emission slit widths.
The structure of the H44-L100 scFv (PDB id: 6NOU) was solved and aligned with the previously determined ixekizumab Fab structure (PDB id: 6NOV) (Figure 6).37 The scFv structure aligned well with the Fab structure, with an all-atom root-mean-squared deviation (rmsd) of 0.84 Ȧ (Pymol). The H44-L100 disulfide bond was efficiently formed without evidence of alternate conformations for either cysteine with both in preferred rotamers forming the disulfide. There was a small amount of negative density on the Cys44 sulfur that might indicate less than 100% disulfide formation. The most notable difference between the structures was observed immediately N-terminal to the H44-L100 disulfide in the VH domain (Figure 6, inset). HC Gln43, which was solvent exposed in the Fab structure, adopted an inward facing conformation and formed an H-bond with the backbone carbonyl of the LC Cys100. The CDRs of the two structures aligned with an all-atom rmsd of 0.52 Ȧ. The presence of the native VH-CH1 linker peptide appeared to push the AB loop of the VH further outward than observed in the scFv structure.
Figure 6.
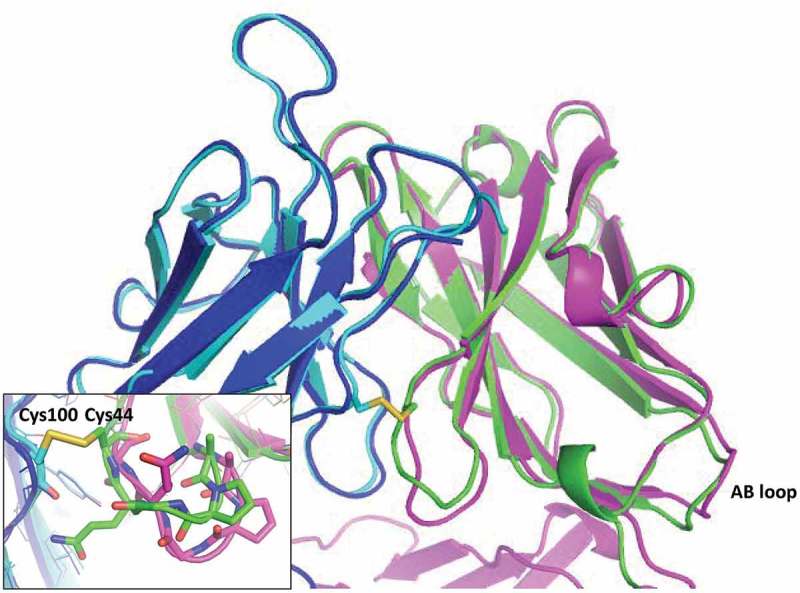
Structural comparison of ixekizumab Fab and H44-L100 anti-IL-17 scFv. Cartoon depiction of ixekizumab HC in magenta and LC in blue, scFv VH in green and Vk in cyan. The H44-L100 scFv disulfide is shown in sticks (inset). The AB loop of the VH domain of both molecules is indicated on the bottom right. The most significant structural rearrangement occurred immediately N-terminal to the VH Cys44 (inset).
Although the protein characterization was not done in the bispecific context, the combined data indicated that the introduced amino acid changes remedied several of the scFv-mediated stability issues associated with the original IgG-scFv molecule. The engineered scFv was introduced in the bispecific format by genetic fusion to the C-terminus of tabalumab HC, re-expressed and purified. The purified bispecific antibody was concentrated from ~1–2 mg/mL to 58 mg/mL and stored at 25°C. Samples analyzed over the course of four weeks by aSEC for %HMW growth indicated significantly increased stability relative to the original IgG-scFv (data not shown). Importantly, a full developability assessment determined that the engineered bispecific antibody LY3090106 had sufficient biophysical properties, including aggregation propensity and chemical stability, to support clinical development.
In vitro characterization of LY3090106
Antigen Binding: Binding affinity of the parent IgG-scFv, LY3090106, ixekizumab, and tabalumab to human IL-17 and BAFF ligands was determined by SPR. The results are summarized in Table 1. LY3090106 bound with relatively high affinity to both ligands under conditions of physiological pH and ionic strength, similar to each parental mAb measured in this study (Table 1) and consistent with values reported previously.9,19 LY3090106 bound to mouse BAFF with KD of 340 (±100) nM and did not bind to mouse IL-17. It bound to cynomolgus BAFF and cynomolgus IL-17 with KD of 26.8 (±0.9) and 19 (±11) pM, respectively.
Table 1.
In vitro binding affinity of LY3090106, the parent IgG-scFv, ixekizumab and tabalumab to BAFF and IL-17 determined using surface plasmon resonance. The KD values were calculated from the ratio of koff/kon for each individual measurement. The values reported for LY3090106 were obtained from averaging three independent measurements (n = 3) and error calculated using the standard deviations. The values reported for the other antibodies were from a single determination (n = 1).
| IL-17A | |||
| Antibody | kon (106 1/Ms) | koff (10–5 1/s) | KD (pM) |
| parent lgG-scFv | 1.94 | 2 | 10 |
| LY3090106 | 2.5 (±0.4) | 3.7 (±1.8) | 14 (±5) |
| ixekizumab | 6.7 | 1.2 | 2 |
| BAFF | |||
| Antibody | kon (106 1/Ms) | koff (10–5 1/s) | KD (pM) |
| parent lgG-scFv | 3.63 | 8.3 | 23 |
| LY3090106 | 2.3 (±0.9) | 12 (±0.5) | 60 (±50) |
| tabalumab | 3.26 | 8.1 | 25 |
IL-17 neutralization: Neutralization of IL-17 was tested using HT-29 cells, which produce CXCL1 upon stimulation with IL-17 (Figure 7A). LY3090106 antagonized the IL-17-induced secretion of CXCL1 in a concentration-dependent manner (closed circles), comparable to that observed with the positive control antibody (closed triangles). The average IC50 over three independent experiments for LY3090106 (2.00 ± 0.21 nM) was similar to what was observed with ixekizumab (1.86 ± 0.22 nM; mean ± SEM). Moreover, the antagonist activity was not influenced by the presence of a saturating concentration of BAFF (Figure 7A, open symbols). Thus, LY3090106 effectively antagonized IL-17 and was unobstructed by the presence of saturating pre-bound BAFF.
Figure 7.
In vitro neutralization of IL-17 and BAFF. A) LY3090106 (circles) dose-dependently inhibited IL-17-induced CXCL1 production by HT-29 cells in the absence (closed symbols) or presence (open symbols) of BAFF. Positive control (triangles) and negative isotype control (squares) is shown. B) LY3090106 (circles) dose-dependently inhibited proliferation of T1165 cells induced by BAFF in the absence (closed symbols) or presence (open symbols) of IL-17. Positive control (triangles) and negative isotype control (squares) is shown. All results are mean ± SEM.
BAFF neutralization: Neutralization of BAFF was tested using T1165 cells, which proliferate in response to BAFF (Figure 7B). LY3090106 blocked BAFF-induced proliferation in a concentration-dependent manner (closed circles), comparable to that observed with the positive control antibody tabalumab (closed triangles), whereas the negative control antibody (closed squares) did not inhibit proliferation. The average IC50 over two independent experiments for LY3090106 (0.064 ± 0.021 nM) was similar to that observed with tabalumab (0.071 ± 0.002 nM; mean ± SEM). Moreover, the antagonist activity was not influenced by the presence of a saturating amount of pre-bound IL-17 (Figure 7B, open symbols). Thus, LY3090106 simultaneously bound and blocked both BAFF and IL-17.
In vivo target engagement in mice
IL-17 neutralization: Mice were injected subcutaneously (SC) with indicated antibodies two days prior to administration of IL-17. Injection of human IL-17 in mice resulted in an increase in serum levels of CXCL1 (Figure 8A, PBS versus IL-17), which was significantly reduced (P < .01, ANOVA) in the presence of LY3090106 relative to the isotype control antibody. The level of reduction in CXCL1 with LY3090106 was comparable to that observed with the positive control anti-IL-17 antagonist antibody ixekizumab. Thus, LY3090106 effectively antagonized the biological effects induced by human IL-17 in the mouse.
Figure 8.
In vivo neutralization of IL-17 and BAFF. A) LY3090106 inhibited IL-17-induced CXCL1 production in mice. Each circle represents an individual animal; horizontal line represents the group mean. Positive control and LY3090106 have significantly lower CXCL1 levels compared to negative isotype control. B) LY3090106 significantly reduced the number of splenic B cells in huBAFF-transgenic mice (compared to negative control antibody) similar to the positive control. Each circle represents an individual animal; horizontal line represents the group mean.
BAFF neutralization: LY3090106 binds mouse BAFF with significantly reduced affinity compared to human BAFF, complicating its use in preclinical disease models. It has been established by previous research in our laboratory that mice carrying a transgene encoding soluble human BAFF have an abnormally high number of splenic B lymphocytes that can be reduced to non-BAFF-transgenic mice levels by tabalumab.9 A single injection of LY3090106 in BAFF-transgenic mice resulted in a significant reduction in the number of B cells compared to the negative control group, comparable to that observed with the positive control tabalumab. (Figure 8B) Thus, LY3090106 effectively neutralized the biological effects induced by human BAFF in the mouse.
In vivo PK/PD in cynomolgus monkey
Having demonstrated that LY3090106 inhibited its human ligands in vivo, we evaluated the structural integrity and the functional efficacy in NHP. Serum pharmacokinetic (PK) profiles of LY3090106 were characterized following a single intravenous (IV) administration of 0.3, 1, 5 or 20 mg/kg and single SC administration of 5 mg/kg. The PK based on the total IgG ELISA (Figure 9) was non-linear over the dose range of 0.3 to 20 mg/kg, with serum concentrations declining in a concentration-dependent fashion. Consistent with nonlinear kinetics, mean AUC0-∞ increased supra-proportionally with the IV dose from 0.3 to 20 mg/kg, mean clearance (CL) decreased approximately twofold and mean elimination half-life increased from approximately 4 to 10 days, which is likely due to target-mediated elimination of LY3090106. A similar result was seen for tabalumab in humans that we speculate resulted from binding to cell membrane‐associated BAFF.52 SC bioavailability was estimated to be 94% by noncompartmental analysis with maximal concentrations observed at 4 days post dose. To investigate whether LY3090106 remained intact as a bispecific molecule in circulation, two additional antigen-capture assays were performed. The immunoreactivity profiles over time of these assays were largely superimposable with the total IgG ELISA (Figure 9). This demonstrated that LY3090106 remained functionally intact in vivo and possessed a half-life in the range of a standard mAb without obvious indication of anti-drug antibody formation.
Figure 9.
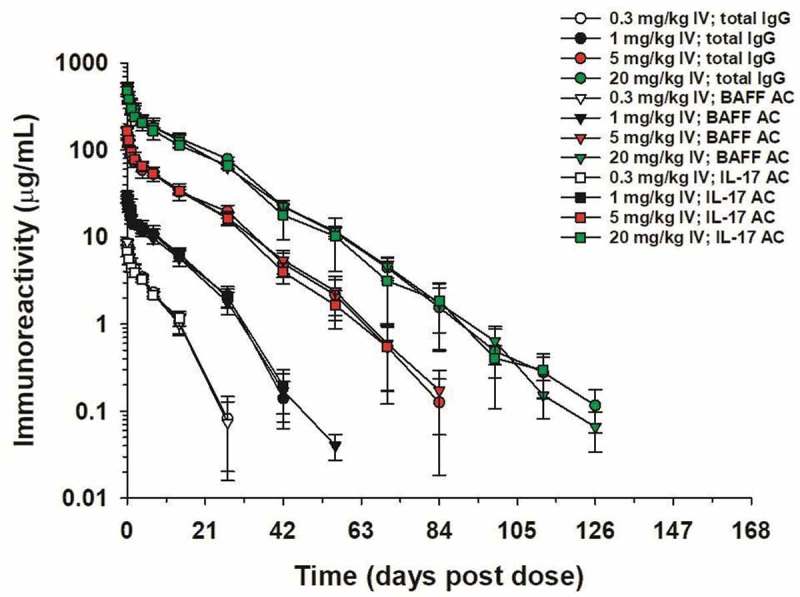
Serum concentrations of LY3090106 in male cynomolgus monkeys following a single IV bolus administration of 0.3, 1, 5 or 20 mg/kg as measured by ELISA. The total IgG, BAFF antigen capture, and IL-17 antigen capture curves are nearly superimposable indicating LY3090106 remained functionally intact in vivo. Data are the mean ± standard deviation (n = 3–4/group).
To determine the ability of LY3090106 to bind IL-17 and BAFF in vivo, total levels of these ligands (i.e., bound plus free) were measured using a drug-tolerant target engagement assay. When bound to LY3090106, the levels of both IL-17 and BAFF increased as the bound ligands assumed the half-life of the bispecific antibody. As shown in Figure 10, the total concentration of BAFF and IL-17 increased following a single IV dose of LY3090106 and remained elevated for the duration of the experiment (28 days). This tracked with the PK profile of LY3090106 (Figure 9) and demonstrated that LY3090106 bound both IL-17 and BAFF in NHP.
Figure 10.
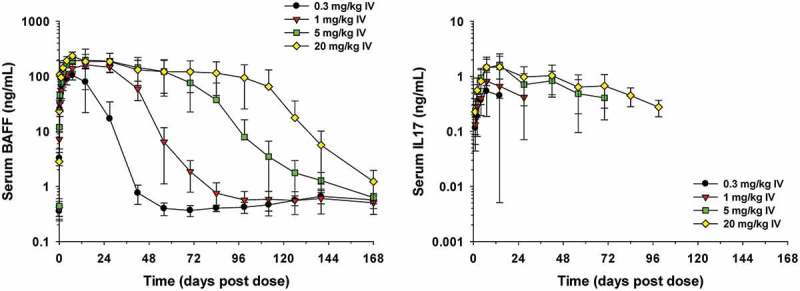
Serum concentrations of total BAFF or total IL-17 in male cynomolgus monkeys following a single IV bolus administration of 0.3, 1, 5 or 20 mg/kg of LY3090106. Data are the mean ± standard deviation (n = 3–4/group).
There is no known pharmacodynamic (PD) marker for the neutralization of IL-17 in healthy NHP. However, neutralization of BAFF is known to reduce the number of circulating B cells in both cynomolgus monkeys and humans. Therefore, the relative change in the number of B cells was investigated as a PD marker for LY3090106. We observed a dose-dependent decrease in the number of B cells in the circulation following a single injection with LY3090106 (Figure 11). The B cell decrease was fairly rapid and prolonged, with a maximal inhibition of ~50–60% observed after about 2 months in the 5 mg/kg IV dose group. This level of long-term B cell reduction is consistent to what was observed following administration of tabalumab at a similar dose.9 No significant decrease in the B cell numbers was observed in the 0.3 mg/kg group, whereas a partial decrease and complete recovery to baseline were observed in the 1 mg/kg group. These data confirmed the expected biological effect associated with the neutralization of BAFF and demonstrated the ability of LY3090106 to effectively antagonize BAFF in NHP.
Figure 11.
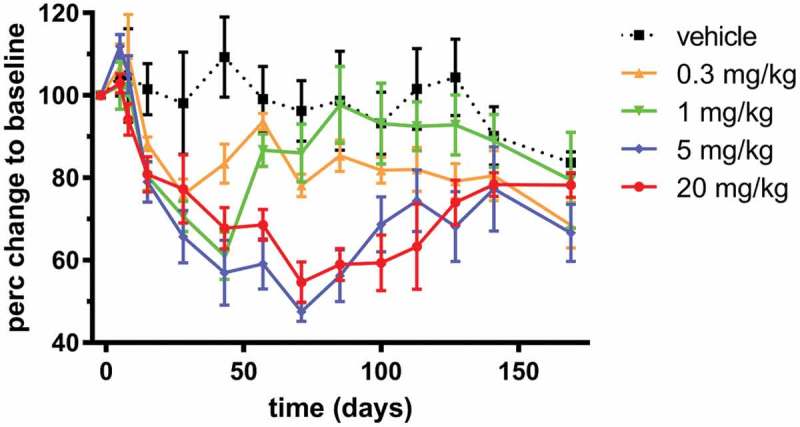
Analysis of changes in B cell numbers in cynomolgus monkeys. Changes in CD20 positive, CD3 negative B lymphocytes over time following a single injection with LY3090106 as indicated. Changes in B cell numbers at each time point were expressed relative to each animals’ baseline value (the average of day −6 and −1 pre-dose) and mean ± SEM are shown.
Discussion
Ixekizumab (Taltz®) is a humanized high-affinity IL-17 antagonist mAb with fully human germline frameworks IGHV1-69 and IGKV2D-29.19 Systematic biophysical characterization of a panel of isolated human Fv domains by means of GdnHCl-induced unfolding showed that representative VH1/Vk2 families possess relatively high stability (behind only VH3/Vk3) and yield of soluble monomer, without requiring refolding following periplasmic expression in E. coli.53 Consistent with these studies, the ixekizumab-derived scFv showed high thermal stability with a single-concerted melting transition observed at 73.1°C (Figure 3A). Despite its apparent conformational stability, the scFv displayed a strong propensity for protein self-association mediated by concentration-dependent non-covalent interactions.
In general, the biophysical properties of antibody fragments such as scFv are hard to predict and can vary widely. Problems with expression, solubility, and stability represent major challenges that can preclude their development as therapeutics. A major cause of scFv instability is attributed to the VH/VL interface. It can be speculated that there is a lack of evolutionary pressure for high-affinity VH/VL interaction due to the additional binding energy normally provided by the constant domain associations in the mAb context.54 Moreover, the plasticity of a generic interface is a critical feature to enable the pairing of different Fv framework and CDR combinations during the generation of a diverse immunoglobulin repertoire, suggesting a constraining attribute of antibody evolution.55 Studies of VH/VL packing geometry have shown residues in both the frameworks and the CDRs, especially located near the ends of CDR-H3 and CDR-L3, contribute inter-domain VH/VL contacts.56,57 Analysis of 200 Fab structures showed on average that approximately 1570 (± 160) Ȧ2 of solvent-accessible surface is buried upon the formation of the VH/VL interface, of which 70% is non-polar.54 Even with a footprint of this size, the dissociation constant between the VH and VL domains is weak, measured in the 10−8 M to 10−5 M range,57,58 and as such is often not of sufficient magnitude to maintain stable inter-domain contacts even when these are held proximal by a peptide linker.
In the case of the IL-17 scFv, interface dynamics were observed indirectly by two methods: comparison of the concentration-dependence of the SEC profiles and ANS dye binding titrations for molecules with and without a H44-L100 disulfide bond. The concentration-dependence of the size exclusion profile for the parental scFv (Figure 3B) indicated that the monomer peak was reduced by ~50% at a concentration of ~13.8 mg/mL. In contrast, the H44-L100 scFv remained monomeric up to 71 mg/mL (Figure 4B), implying that the disulfide bond was efficiently formed and functionally prevented scFv oligomer formation. Covalent linkage of the variable domains also resulted in significantly reduced ANS fluorescence emission intensity relative to the parental scFv (Figure 5). ANS is a convenient probe of protein structure because it is essentially non-fluorescent in water, but highly fluorescent when dissolved in non-polar solvent or bound to hydrophobic patches in proteins with partially folded or molten globule states.59 The differential fluorescence profiles are consistent with more efficient burial of the highly non-polar interface surfaces in the H44-L100 scFv. However, some ANS binding was still observed for the stabilized scFv. We cannot unequivocally state whether this was due to less than quantitative disulfide formation or interaction of ANS with regions outside of the VH/Vk interface. The consequences of the surface charge-balancing mutations on the scFv solution behavior in the absence of the disulfide were not studied using either methodology. However, when analogous mutations were incorporated into a closely related mAb, reductions in viscosity and liquid-liquid phase separation were observed.37 In short, as evidenced herein and elsewhere,46–49,60 the disulfide was critical to reduce “interface breathing dynamics”, thereby preventing scFv oligomerization, whereas remodeling the dominant surface charge patches discouraged CDR-mediated self-interaction at high protein concentration. The relatively high Tm of the parental scFv obviated the need for thermal stability engineering. Interestingly, Bhatta et al. recently showed Fab-dsFv/dsscFv with lower Fv Tms generally displayed higher monomer levels.61 Thus, the high Tm of our parental IL-17 scFv might have contributed to its inherent oligomerization propensity.
Comparison of the crystal structure of the ixekizumab Fab37 and H44-L100 scFv (Figure 6) showed the CDR loops to be nearly superimposable (0.52Ȧ rmsd). This was unremarkable given the similar affinity for IL-17 between the two molecules. Calculation of the surface area of the VH/Vk interface showed that approximately 1779 Ȧ2 of solvent-accessible surface was buried, on the high end of the normal range for measured Fabs.50 The structural interface was further examined for overall chemical incompatibility or obvious steric clashes. However, without the structure of the unpaired domains, it is difficult to discern the energetic penalties incurred during the resolution of subtle repulsive forces during interface formation. Regardless, single CDR amino acids can have a dramatic effect on scFv stability. For example, Tu et al. identified a single CDR-L3 Ser89Ala mutation that was sufficient to improve thermal stability and reduce aggregation by the elimination of a polar side chain in a hydrophobic environment and removal of an unsatisfied hydrogen bond donor/acceptor.62 Similarly, ixekizumab scFv has an analogous-buried serine at CDR-L3 position 98. However, it satisfied its hydrogen bonding capability through a 2.9Ȧ H-bond to a buried water molecule interacting with Thr105 of the VH. The presence of interface water molecules reduces specific contacts between side chains, increasing non-specific characteristics. Examination of the remainder of the interface failed to reveal any obvious cause for instability. The hydroxyl of heavy chain Tyr99 is buried but interacts with the same water molecule. We did not determine its contribution to stability via mutagenesis, as Tyr99 is important for high-affinity antigen binding and was not amenable to change during affinity maturation. Thus, there exists a trade-off between high-affinity antigen interaction and interface stability that is magnified in the scFv context.
The stabilized IgG-scFv (LY3090106) was extensively characterized in functional assays for ligand binding and target neutralization. Not unpredictably given the molecular geometry of the IgG C-terminal scFv fusion, it simultaneously bound both ligands and antagonized in vitro BAFF-induced B cell proliferation and IL-17-induced chemokine secretion. In vivo PD models in either wild type or human BAFF transgenic mice indicated LY3090106 possessed parental-like IL-17 and BAFF ligand neutralization activities. In cynomolgus monkey, it remained functionally intact in circulation and possessed a half-life consistent with a standard mAb without indication of anti-drug antibody response. Moreover, LY3090106 bound cynomolgus BAFF and IL-17 indicated by increased in vivo levels of both measured using a drug-tolerant target engagement assay. Finally, LY3090106 elicited a dose-dependent decrease in B cell number, indicative of cynomolgus BAFF neutralization in vivo. Together, these results demonstrated the engineering of a highly potent and stable bispecific molecule.
In summary, LY3090106 has suitable drug-like biophysical properties, and amenability for large-scale manufacturing without need for further stability engineering. It was tested in a human clinical trial entitled “A Multiple Ascending Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of LY3090106 in Subjects with Sjögren’s Syndrome” (ClinicalTrials.gov Identifier: NCT02614716). Optimistically, future clinical trials in autoimmune diseases will demonstrate a profound benefit of simultaneous BAFF and IL-17 inhibition.
Materials and methods
Engineering of bispecific IgG-scfv
Anti-IL-17 was chosen as the scFv component of the IgG-scFv because the previously measured Fab Tm of tabalumab (64–65°C) is on the low end of the typical range for IgGs.63 Anti-IL-17 VH and Vk chain genes were amplified by PCR from a plasmid encoding ixekizumab. V-region specific 5ʹ-biotinylated forward primers and 5ʹ-phosphorylated reverse primers were designed with added DNA sequence complementary to a parental scFv template. Single-stranded nucleic acids were isolated by negative selection using streptavidin magnetic beads, annealed to an uracil-containing scFv single-stranded phage template followed by second strand synthesis and electroporation into E.coli XLO. The scFv was assembled in the VH-Vk domain order connecting the variable domains by a 20 amino acid glycine-serine rich linker of the following sequence: GGGGSGGGGSGGGGSGGGGS. To create the IgG-scFv construct, the IL-17 scFv was fused to the anti-BAFF IgG4 (S228P)64 heavy chain C-terminus by overlapping PCR, restriction digestion of the PCR products, and three-fragment ligation into mammalian expression vector using a 15 amino acid tether of the following sequence: GGGGSGGGGTGGGGS. Additional linker sequences predicted to form a secondary structural element (e.g., QPDGGSGGSGGGSGGSKADYEKHKV) were tested, but showed increased susceptibility to proteolysis during purification (data not shown) and abandoned. Separate plasmids encoding the anti-BAFF HC-IL-17 scFv fusion and parental anti-BAFF LC were used for transient transfection of HEK-293 cells.
Kunkel mutagenesis65 was used to target CDR amino acids that exhibited chemical instability (oxidation and deamidation) in the anti-IL-17 scFv context. Oligonucleotide was synthesized to specifically deplete the labile Met and Asn codons, mixed and used to generate randomization libraries that were screened for IL-17A affinity using a capture lift assay and biotinylated antigen.36,66 Positive phage plaques identified by this approach were picked, sequenced and unique clones expressed as periplasmic Fab. Titration ELISA using IL-17A coated plates was used to measure the relative binding affinities of the Fab variants and absolute affinity confirmed by SPR. The affinity neutral CDR-H2 Met53Thr and CDR-L1 Asn30Glu were subsequently introduced in the IgG-scFv context using quick-change mutagenesis (Agilent cat#200524).
Expression and purification
Bispecific antibodies were expressed in mammalian cell culture (HEK-293 or Chinese hamster ovary cells). Bispecific antibody-containing cell culture supernatants were harvested and clarified media was purified in two steps by affinity chromatography using Protein A-Sepharose™ (GE Healthcare) and Superdex 200 SEC.
Anti-IL-17 scFv molecules were expressed in HEK-293E cells. After the media were concentrated and filtered, scFv was captured by CaptoMMC column and further purified by HIC Phenyl Sepharose HP column, followed by Superdex 75 SEC in PBS buffer. The protein concentration of purified protein samples was determined by measuring the optical density (OD) at 280 nm. Purity and molecular weight of bispecific antibodies were analyzed by SDS-PAGE in the presence and absence of a reducing agent (5 mM 1,4-dithiotreitol (DTT)).
Stability of purified scfv and bispecific antibody
The purified scFv sample was concentrated with Amicon Ultra centrifugal filters (Ultracel-10K). After concentration, the samples were analyzed by aSEC using a TSKgel G3000SWxL column (Tosoh Bioscience). The post-concentration recovery and percentage of the monomeric main peak were calculated after samples were concentrated to 15 mg/mL for anti-IL-17 scFv and 71 mg/mL for anti-IL-17 H44-L100. After concentration, the recovery of anti-IL-17 scFv was 38%, and recovery of H44-L100 was 95%. Recovery % is defined as total protein amount (concentration x volume) in solution before/after concentration. The stability of scFv with or without a H44-L100 disulfide bond was also analyzed as a function of storage time at 4°C.
The aggregation of IgG-scFv samples after concentration or storage was analyzed by aSEC using an Agilent 1100 system and TSKgel G3000SWxL column. PBS containing 0.35 M NaCl, pH 7.4 was used as the mobile phase running at 0.5 mL/min for 35 min. A volume of l µL of the concentrated antibody was injected into the column, and the detection measured at 280 nm. Chromatograms were analyzed using ChemStation and % HMW calculated from the ratio of area under the curve (AUC) of the peaks eluted before the monomer peak to total AUC. Samples were analyzed for %HMW at initial concentration and different time points of incubation post concentration.
Electrospray mass spectrometry
The intact mass of the purified BAFF/IL-17 IgG-scFv was measured with and without partial reduction using DTT. Intact mass analysis was completed on an Agilent 6210 electrospray using an ethylene-bridged hybrid C4 column (Waters) and 0.2% formic acid, 0.05% trifluoroacetic acid (TFA) in water/0.2% formic acid, 0.04% TFA in acetonitrile gradient for HPLC elution into the mass spectrometer. The mass spectrometry (MS) scan was from 500 m/z to 4000 m/z at a rate of 1 scan/s. External calibration was used for mass calibration, and Mass Hunter Max Entropy was used for mass deconvolution. The error is expressed as percent (%) difference between the theoretical and measured masses.
Enzymatic digest analysis
Fifty micrograms of each sample were dried under vacuum (not to dryness), resuspended in 8 M guanidine and allowed to denature for 30 min at 37°C. Freshly prepared 0.1 M DTT was added to each sample to a final concentration of 10 mM, followed by incubation at 37°C for 30 min. The reduction was quenched by addition of freshly prepared iodoacetamide to a final concentration of 15 mM and incubated at room temperature for 30 min in the dark. Prior to digestion, samples were desalted and buffer exchanged into digestion buffer (25 mM Tris, 20 mM CaCl2, pH 7.5) using Zeba micro desalting spin columns prepared according to manufacturer’s procedure. Trypsin was added to a final protein concentration ratio of 1:20 (w: w), and digested for 4 h at 37°C. The digest was quenched by the addition of 1 µl of formic acid and analyzed with an Agilent 6530 ESI-Q-TOF run in positive ion mode with an MS scan from 350 to 1850m/z and an MS/MS scan from 50 to 2200m/z. The reversed phase separation was on an Agilent 1200 HPLC using a Zorbax 1.8 µm C18 (2.1 x 50 mM) column with a flow rate of 0.25 ml/min and gradient elution. Mobile phase consisted of 0.2% formic acid in water and acetonitrile. Mass Hunter and BioConfirm software was used to align the tryptic peptides to the protein sequence and to check for potential chemical modifications. Post-translational modification quantitation was by extracted ion chromatogram. The tryptic digest and subsequent LC/MS analysis showed two hotspots of chemical degradation following incubation of the purified IgG-scFv under stressed conditions at 40°C for one month in PBS. Oxidation (13%) of HC CDR-H2 Met53 and deamidation (12%) of LC CDR-L1 Asn30 was observed (data not shown). Neither of these modifications was seen for the parental molecule.
Binding affinity to IL-17 and BAFF
Binding affinity and stoichiometry of the IgG-scFv to human IL-17 and human BAFF were determined using SPR on a Biacore 2000 instrument primed with HBS-EP+ (GE Healthcare, 10 mM Hepes pH 7.4 + 150 mM NaCl + 3 mM EDTA + 0.05% surfactant P20) running buffer and analysis temperature set at 25°C. Except as noted, all reagents and materials were from BIAcore AB (Uppsala, Sweden). A CM5 chip containing immobilized protein A (generated using standard NHS-EDC amine coupling) on all four flow cells (Fc) was employed as capture methodology. The affinity of Fc for protein A is of similar magnitude (pM range) as LY3090106 for its ligands. Therefore, the apparent SPR off-rates of the IgG-scFv-antigen complexes should be considered greater than or equal to the actual values. Samples were prepared at a concentration of 10 µg/mL by dilution into running buffer. Human IL-17 or human BAFF were sourced from Eli Lilly and Company reference standards and prepared at final concentrations of 20.0, 10.0, 5.0, 2.5, 1.25 and 0 (blank) nM by dilution into running buffer. Each analysis cycle consisted of (1) capturing antibody samples on separate flow cells (Fc2, Fc3, and Fc4), (2) injection of 250 µL (300-sec) of human IL-17 or human BAFF over all Fc at 50 µL/min, (3) return to buffer flow for 20 min to monitor dissociation phase, (4) regeneration of chip surfaces with a 5 µL (30-sec) injection of glycine, pH 1.5, (5) equilibration of chip surfaces with a 10 µL (60-sec) injection of HBS-EP+. Data were processed using standard double referencing and fit to a 1:1 binding model using Biacore 2000 Evaluation software version 4.1 to determine the association rate (kon, M−1 s−1 units), dissociation rate (koff, s−1 units), and Rmax (RU units). The equilibrium dissociation constant (KD) was calculated from the relationship KD = koff/kon.
Simultaneous binding of IL-17 and BAFF
A BIAcore 2000 instrument was used to determine whether human IL-17 and human BAFF can bind to the IgG-scFv simultaneously. All measurements were performed at 25°C. HBS-EP+ buffer (150 mM sodium chloride, 3 mM EDTA, 0.05% (w/v) surfactant P-20, and 10 mM HEPES, pH 7.4) was used as the running buffer and sample buffer. Protein A was immobilized on flow cells 1 and 2 of a CM4 sensor chip using an amine coupling kit. The bispecific antibody was first captured on flow cell 2, followed by injection of human IL-17 at 20 nM for 5 min to saturate IL-17 binding site. After binding of IL-17, human BAFF at 50 nM was injected for 5 min and additional binding signal was observed. Chip surface was then regenerated using 10 mM glycine pH 1.5. The same process was repeated except with a reverse order of human IL-17 and human BAFF addition. The stoichiometry was calculated to determine the level of saturation of human IL-17 or human BAFF binding to the IgG-scFv.
Differential scanning calorimetry
To compare the conformational stability of anti-IL-17 scFv, anti-IL-17 H44-L100 scFv, and LY3090106 thermal stability (Tm) of samples were measured using a TA Instruments NanoDSC (TA Instruments, New Castle, DE) equipped with an autosampler. Samples diluted to 0.8 mg/ml in PBS were heated from 20°C to 110°C at a rate of 1°C/min under 45 psi of pressure. Sample scans were buffer blank-subtracted, converted to molar heat capacity, and fit to a two-state-scaled model to obtain Tm.
Crystallization and structure determination
Anti-IL-17 scFv H44-L100 concentrated to 10.4 mg/mL was set up in vapor diffusion sitting drops at a ratio of 1:1 with a well solution of 2.8 M sodium acetate, pH 7.0. Crystals were transferred to a cryo-protectant consisting of well solution supplemented with 25% glycerol, prior to freezing in liquid nitrogen. X-ray diffraction data were collected at the Advanced Light Source (Lawrence Berkeley National Laboratory, Berkeley, CA), beamline 8.3.1 at 100°K using 1.115879 Angstrom X-rays. Data were integrated and reduced using MOSFLM and the CCP4 suite of programs. Initial phases were generated using Phaser (CCP4).67,68 The crystal structure was modeled using COOT, further refined using BUSTER and validated using MolProbity.
Surface properties of ixekizumab and its mutants
The crystal structure of the Fv domain of ixekizumab was used for structural modeling of its surface properties, including hydrophobicity and charge distribution. Homology models of mutants were generated in the Molecular Operating Environment (MOE) (Chemical Computing Group, Montreal, Canada), based on the crystal structure and subject to the same analysis. Solvent accessible surface area (SASA) and surface exposure (%) for each residue were calculated based on the static model structure using the protein properties analysis module (MOE). Spatial aggregation propensity (SAP) was computed according to the method in the BIOVIA Discovery Studio 4.0 software (San Diego, CA). Environment setting of Amber10: EHT force field was applied before electrostatic potential computing by BIOVIA Discovery Studio 4.0 software.
ANS binding to anti-IL-17 scFv with or without H44-L100 disulfide bond
ANS titrations were conducted to investigate whether disulfide bond stabilization reduced the exposure of the variable heavy and light chain hydrophobic interface residues. Anti-IL-17 scFv with or without an H44-L100 disulfide bond was diluted to 0.28 mg/mL in PBS buffer at pH 7.4. ANS was prepared as 1 mM stock solution in PBS buffer. The fluorescence emission spectrum was collected at 25°C from 400 nm to 700 nm, with a step size of 3 nm following excitation at 360 nm. Fluorescence measurements were made using an ISS PC1 fluorometer (Champaign, IL) equipped with a xenon lamp. The emission spectrum was recorded as a function of increasing concentration of ANS from 2 μM to 300 μM. Sequential ANS additions were performed followed by a brief mixing step, incubation, and collection of the emission spectra at approximately 2 min. intervals. A similar protocol was followed for the parent and H44-L100 scFv. The background of the buffer was subtracted from the sample spectrum at each corresponding concentration of ANS.
Pepsin-mediated proteolysis
The susceptibility to proteases such as pepsin can be used to assess the relative conformational stability of proteins in solution. Samples of the parental and H44-L100 scFv molecules were prepared and incubated with 50 µL of pepsin beads on ice for 1, 5 and 10 min followed by LC-MS analysis to detect peptide fragments generated following protease treatment. Under these conditions, the H44-L100 scFv was resistant to pepsin-mediated hydrolysis.
Functional in vitro assays
BAFF: The murine plasmacytoma cell line T1165.17 naturally expresses the receptor for BAFF and is dependent on BAFF for survival and growth. The ability of LY3090106 to inhibit BAFF (150 pM) dependent proliferation was measured across a wide concentration range (4.1 pM to 1 nM). The assay was performed as described.9 To determine the effect of IL-17 co-engagement, the assay was repeated in the presence of 15.6 nM of IL-17. Anti-BAFF or human IgG4 isotype were used as positive and negative controls, respectively. Data are expressed as % inhibition, using wells without BAFF as the minimum and wells with 150 pM BAFF as the maximum responses. The concentration where 50% of the BAFF-induced response is inhibited (IC50) by either bispecific antibody or the positive control was calculated using a 4-parameter sigmoidal fit of the data (SigmaPlot).
IL-17: The human colorectal adenocarcinoma epithelial cell line HT-29 naturally expresses the IL-17 receptor and produces CXCL1 upon stimulation with IL-17. The ability of anti-IL-17 or LY3090106 to inhibit IL-17 (3.75 nM)-induced CXCL1 production was measured across a wide concentration range (2.64 pM to 41.2 nM) after 48-h incubation using a commercial ELISA (R&D Systems). To determine whether BAFF would alter the inhibition, the assay was repeated in the presence of 1.25 nM of BAFF. Anti-IL-17 or human IgG4 isotype were used as positive and negative controls, respectively. Data are expressed as % of maximum amount of CXCLl produced (with IL-17 alone being 100%). The concentration where 50% of the IL-17-induced response is inhibited (IC50) by either bispecific antibody or the positive control was calculated using a 4-parameter sigmoidal fit of the data (GraphPad Prism).
Functional in vivo assays
BAFF: Mice transgenic for human BAFF were generated as described.9 Human BAFF transgenic mice have elevated numbers of B cells compared to wild type mice. Mice (n = 5 per group) were injected IP with either a single dose of LY3090106 (660 µg/mouse) or a positive (tabalumab, 500 µg/mouse) or negative (huIgG4 isotype control, 500 µg/mouse) control antibody. Eight days later, serum and spleens were collected. A single cell suspension of spleen cells was prepared, and the total number of leukocytes was determined after lysis of the red blood cells. The relative percentage of B cells was determined by flow cytometry using the cell surface marker B220 and used to calculate the total number of B cells per spleen.
IL-17: Female C57Bl6 mice (n = 8 per group) were injected SC with either LY3090106 (66 µg/mouse) or a positive (ixekizumab, 50 µg/mouse) or negative (huIgG4 isotype control, 50 µg/mouse) control antibody. Two days later, mice received a single intraperitoneal injection of human IL-17 (3 µg/mouse), and 2 h later serum was collected and the concentration of CXCL1 was determined by a commercial ELISA (R&D Systems, Cat# DY453), following the manufacturer’s instructions.
In vivo exposure: To quantitate exposure, antibody levels in serum were analyzed by a human IgG-specific ELISA. Briefly, microtiter plates were coated with an antibody capturing human Fc (Jackson ImmunoResearch, 109–005-098) and incubated overnight at 4°C. Plates were washed, blocked with casein, and 100 µL of serum (serial dilutions) was added. Plates were incubated for 2 h at room temperature (RT), washed, and a horseradish peroxidase-labeled detection antibody (anti-human IgG, Jackson ImmunoResearch, 109–035-098) was added. Plates were incubated for 1 h at RT, washed and developed using tetramethylbenzidine substrate and read using a microtiter plate reader. A standard curve of purified antibody was used to calculate the plasma concentration.
Cynomolgus monkey PK/PD
To determine the PK/PD profiles of LY3091016, male cynomolgus monkeys were injected IV once with different doses (0.3, 1, 5, or 20 mg/kg) of LY3090106 (n = 4/group) or vehicle (PBS; n = 3). One group (n = 2) received a single 5 mg/kg subcutaneous dose to evaluate bioavailability. PK samples were collected pre-dose, and 0.25, 8, and 24 h post-dose and on study days 3, 5, 8, 15, 28, 43, 57, 71, 85, 100, 113, 127, 141, and 169. Blood was allowed to clot, and serum was collected after centrifugation and frozen until time of analysis. Serum samples were analyzed in three separate assays: total IgG and BAFF or IL-17 antigen capture assays. Briefly, each well of a microtiter 96-well plate was coated with goat anti-human kappa antibody (Southern Biotech, Birmingham, AL, 2060–01), BAFF antigen or IL-17 antigen (Eli Lilly and Company reference standards). Standards and controls were prepared in 100% cynomolgus serum using LY3090106. Standards, controls, and samples were diluted with a minimum required dilution (MRD) of 1:100 in 100% mouse serum and were added to the coated ELISA plates after blocking and washing. After standards, controls and samples were incubated on the plates, the plates were washed and LY3090106 was detected with mouse anti-human IgG Fc-horseradish peroxidase antibody (Southern Biotech, Birmingham, AL, 9040–05) using TMB Microwell Peroxidase Substrate System (KPL, Gaithersburg, MD) for a colorimetric response. Plates were read at 450 nm with a reference of 630 nm. Immunoreactivity was determined from known amounts of LY3090106 using a 5-parameter algorithm (StatLia, version 3.2; Brendan Technologies, Carlsbad, CA) for all three assays. The standard curve ranges for the total IgG, BAFF and IL-17 antigen capture ELISAs were 10–2500 ng/mL, 15–700 ng/mL and 75–2500 ng/mL, respectively. The upper limits of quantitation were defined as 2000 ng/mL for total IgG and IL-17 capture and 625 ng/mL for BAFF. The lower limits of quantitation were defined as 25 ng/mL for total IgG and BAFF capture, and 100 ng/mL for IL-17.
Serum concentration-time data following IV administration were described using a model-independent method according to the statistical moment theory using WinNonLin® Enterprise version 5.3 software package (Pharsight, A Certara™ Company, St. Louis, MO). The parameters calculated included the maximum serum concentration (Cmax), area under the curve (AUC0-∞), clearance (CL) and elimination half-life (T1/2).
Serum samples were also analyzed to determine total serum BAFF and IL-17 levels utilizing a modified sandwich electrochemiluminescence immunoassay format. In the assays, streptavidin pre-coated microplates were blocked with 1% bovine serum albumin. Following incubation, plates were washed and coated with either biotin-labeled anti-human BAFF or biotin-labeled anti-human IL-17 antibody (eBioscience, San Diego, CA; biotinylated in house) for the BAFF or IL-17 target assay, respectively. Standards were prepared in dilution buffer containing excess LY3090106 using either BAFF or IL-17. Controls and sample dilutions were made up in 100% cynomolgus serum and diluted 1:4 in dilution buffer. Standards, controls, and samples were added to the coated plates after washing. After incubation with standards, controls, and samples containing the analyte, plates were washed and bound BAFF or IL-17 antigen was detected using a ruthenium-labeled anti-human IgG4 antibody (Southern Biotech, Birmingham, Al, 9200–01; ruthenium-labeled in house). The antigen levels were determined from standard curves prepared from known amounts of BAFF or IL-17 antigen using a 5-parameter algorithm (StatLia, version 3.2; Brendan Technologies, Carlsbad, CA). The standard curve range was 5.08–300,000 pg/mL for BAFF and 13.7–30,000 pg/mL for IL-17. Values were reported through the entire curve range for both the BAFF and IL-17 target assays (1:4 MRD).
PD samples were collected pre-dose (days −6 and −1) and on study days 5, 8, 15, 28, 43, 57, 71, 85, 100, 113, 127, 141, and 169. Samples were processed for immunophenotyping by flow cytometry and a separate tube was used for routine hematology. Immunophenotyping results were enumerated as absolute number of B cell (CD20 positive, CD3 negative; cells/µL) and change relative to baseline was calculated for each animal, where baseline was the average of the day −6 and −1 values.
Abbreviations
- ANS
1-Anilino-8-naphthalene sulfonate
- aSEC
analytical size exclusion chromatography
- AUC
area under the concentration curve
- BAFF
B cell activating factor
- CDR
complementarity-determining region
- CL
clearance rate
- DSC
differential scanning calorimetry
- ELISA
enzyme-linked immunosorbent assay
- Fab
antigen-binding fragment
- HC
heavy chain
- HEK-293
human embryonic kidney cells
- Interleukin-17A
IL-17
- LC
light chain
- IV
intravenous
- LY2127399
tabalumab, anti-BAFF IgG4
- LY2439821
ixekizumab, Taltz®, anti-IL-17 IgG4
- LY3090106
tibulizumab, stabilized IgG-scFv bispecific
- LC-MS
liquid chromatography–mass spectrometry
- NHP
non-human primate
- PBS
phosphate-buffered saline
- PD
pharmacodynamic
- PK
pharmacokinetic
- pSS
primary Sjögren’s syndrome
- RA
rheumatoid arthritis
- RMSD
root mean squared deviation
- RT
room temperature
- SC
subcutaneous
- scFv
single-chain variable fragment
- SLE
systemic lupus erythematosus
- SPR
surface plasmon resonance
- T-helper cell 17
Th17
- VH
variable heavy region
- Vk
variable kappa region
- %HMW
percentage of high molecular weight species
Disclosure of interest
During the execution of this work, all authors were employees and stockholders of Eli Lilly and Company. No other conflicts of interest are disclosed.
References
- 1.Mullard A. Bispecific antibody pipeline moves beyond oncology. Nat Rev Drug Discov. 2017;16:666–68.doi: 10.1038/nrd.2017.187. [DOI] [PubMed] [Google Scholar]
- 2.Breslin WJ, Hilbish KG, Martin JA, Halstead CA, Edwards TL. Developmental toxicity and fertility assessment in rabbits with tabalumab: a human igg4 monoclonal antibody. Birth Defects Res B Dev Reprod Toxicol. 2015;104:117–28.doi: 10.1002/bdrb.21147. [DOI] [PubMed] [Google Scholar]
- 3.Taltz prescribing information. 2016. https://pi.lilly.com/us/taltz-uspi.pdf
- 4.Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, Ambrose C, Lawton P, Bixler S, Acha-Orbea H, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189:1747–56.doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosello S, Pers JO, Rochas C, Devauchelle V, De Santis M, Daridon C, Saraux A, Ferraccioli GF, Youinou P. BAFF and rheumatic autoimmune disorders: implications for disease management and therapy. Int J Immunopathol Pharmacol. 2007;20:1–8.doi: 10.1177/039463200702000101. [DOI] [PubMed] [Google Scholar]
- 6.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, Browning JL. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–710.doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsushita T, Hasegawa M, Yanaba K, Kodera M, Takehara K, Sato S. Elevated serum BAFF levels in patients with systemic sclerosis: enhanced BAFF signaling in systemic sclerosis B lymphocytes. Arthritis Rheum. 2006;54:192–201.doi: 10.1002/art.21526. [DOI] [PubMed] [Google Scholar]
- 8.Seyler TM, Park YW, Takemura S, Bram RJ, Kurtin PJ, Goronzy JJ, Weyand CM. BLyS and APRIL in rheumatoid arthritis. J Clin Invest. 2005;115:3083–92.doi: 10.1172/JCI25265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manetta J, Bina H, Ryan P, Fox N, Witcher DR, Kikly K. Generation and characterization of tabalumab, a human monoclonal antibody that neutralizes both soluble and membrane-bound B-cell activating factor. J Inflamm Res. 2014;7:121–31.doi: 10.2147/JIR.S67751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, León MG, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–31.doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- 11.Stohl W. Therapeutic targeting of the BAFF/APRIL axis in systemic lupus erythematosus. Expert Opin Ther Targets. 2014;18:473–89.doi: 10.1517/14728222.2014.888415. [DOI] [PubMed] [Google Scholar]
- 12.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–67.doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hot A, Zrioual S, Toh ML, Lenief V, Miossec P. IL-17A- versus IL-17F-induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in rheumatoid synoviocytes. Ann Rheum Dis. 2011;70:341–48.doi: 10.1136/ard.2010.132233. [DOI] [PubMed] [Google Scholar]
- 14.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–89.doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 15.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–98.doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 16.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600.doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanford M, McKeage K. Secukinumab: first global approval. Drugs. 2015;75:329–38.doi: 10.1007/s40265-015-0359-0. [DOI] [PubMed] [Google Scholar]
- 18.Markham A. Ixekizumab: first Global Approval. Drugs. 2016;76:901–05.doi: 10.1007/s40265-016-0579-y. [DOI] [PubMed] [Google Scholar]
- 19.Liu L, Lu J, Allan BW, Tang Y, Tetreault J, Chow CK, Barmettler B, Nelson J, Bina H, Huang L, et al. Generation and characterization of ixekizumab, a humanized monoclonal antibody that neutralizes interleukin-17A. J Inflamm Res. 2016;9:39–50.doi: 10.2147/JIR.S100940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jinna S, Strober B. Anti-interleukin-17 treatment of psoriasis. J Dermatolog Treat. 2016;27:311–15.doi: 10.3109/09546634.2015.1115816. [DOI] [PubMed] [Google Scholar]
- 21.Greig SL. Brodalumab: first Global Approval. Drugs. 2016;76:1403–12.doi: 10.1007/s40265-016-0634-8. [DOI] [PubMed] [Google Scholar]
- 22.Lai Kwan Lam Q, Ko King Hung O, Bj Z, Lu L. Local BAFF gene silencing suppresses Th17-cell generation and ameliorates autoimmune arthritis. Proc Natl Acad Sci U S A. 2008;105:14993–98.doi: 10.1073/pnas.0806044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francois A, Gombault A, Villeret B, Alsaleh G, Fanny M, Gasse P, Adam SM, Crestani B, Sibilia J, Schneider P, et al. B cell activating factor is central to bleomycin- and IL-17-mediated experimental pulmonary fibrosis. J Autoimmun. 2015;56:1–11.doi: 10.1016/j.jaut.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Elicabe RJ, Silva JE, Dave MN, Lacoste MG, Tamashiro H, Blas R, Munarriz A, Rabinovich GA, Di Genaro MS. Association between IL-17 and IgA in the joints of patients with inflammatory arthropathies. BMC Immunol. 2017;18:8.doi: 10.1186/s12865-017-0189-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elela MA, Gawdat HI, Hegazy RA, Fawzy MM, Abdel Hay RM, Saadi D, Shaker O. B cell activating factor and T-helper 17 cells: possible synergistic culprits in the pathogenesis of Alopecia Areata. Arch Dermatol Res. 2016;308:115–21.doi: 10.1007/s00403-016-1617-z. [DOI] [PubMed] [Google Scholar]
- 26.Munari F, Fassan M, Capitani N, Codolo G, Vila-Caballer M, Pizzi M, Rugge M, Della Bella C, Troilo A, D’Elios S, et al. Cytokine BAFF released by Helicobacter pylori-infected macrophages triggers the Th17 response in human chronic gastritis. J Immunol. 2014;193:5584–94.doi: 10.4049/jimmunol.1302865. [DOI] [PubMed] [Google Scholar]
- 27.Lopez P, Rodriguez-Carrio J, Caminal-Montero L, Mozo L, Suarez A. A pathogenic IFNalpha, BLyS and IL-17 axis in Systemic Lupus Erythematosus patients. Sci Rep. 2016;6:20651.doi: 10.1038/srep20651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambataro D, Sambataro G, Dal Bosco Y, Polosa R. Present and future of biologic drugs in primary Sjogren’s syndrome. Expert Opin Biol Ther. 2017;17:63–75.doi: 10.1080/14712598.2017.1235698. [DOI] [PubMed] [Google Scholar]
- 29.Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9:182–212.doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67:95–106.doi: 10.1016/j.molimm.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 31.Weidle UH, Tiefenthaler G, Weiss EH, Georges G, Brinkmann U. The intriguing options of multispecific antibody formats for treatment of cancer. Cancer Genomics Proteomics. 2013;10:1–18. [PubMed] [Google Scholar]
- 32.Coloma MJ, Morrison SL. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol. 1997;15:159–63.doi: 10.1038/nbt0297-159. [DOI] [PubMed] [Google Scholar]
- 33.Orcutt KD, Ackerman ME, Cieslewicz M, Quiroz E, Slusarczyk AL, Frangioni JV, Wittrup KD. A modular IgG-scFv bispecific antibody topology. Protein Eng Des Sel. 2010;23:221–28.doi: 10.1093/protein/gzp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Digiammarino EL, Harlan JE, Walter KA, Ladror US, Edalji RP, Hutchins CW, et al Ligand association rates to the inner-variable-domain of a dual-variable-domain immunoglobulin are significantly impacted by linker design. MAbs. 2011;3:487–94.doi: 10.4161/mabs.3.5.16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kabat EA, W T T, Perry H, Gottesman K, Foeller C. Sequences of proteins of immunological interest. 5th ed Bethesda (MD): U.S. Dept. of Health and Human Services, Public Health Service, National Institutes of Health; 1991. [Google Scholar]
- 36.Watkins JD, Beuerlein G, Wu H, McFadden PR, Pancook JD, Huse WD. Discovery of human antibodies to cell surface antigens by capture lift screening of phage-expressed antibody libraries. Anal Biochem. 1998;256:169–77.doi: 10.1006/abio.1997.2523. [DOI] [PubMed] [Google Scholar]
- 37.Chow CK, Allan BW, Chai Q, Atwell S, Lu J. Therapeutic antibody engineering to improve viscosity and phase separation guided by crystal structure. Mol Pharm. 2016;13:915–23.doi: 10.1021/acs.molpharmaceut.5b00817. [DOI] [PubMed] [Google Scholar]
- 38.Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, Balthazor B, Conley TR, Sek D, Allen MJ. Rational design of viscosity reducing mutants of a monoclonal antibody: hydrophobic versus electrostatic inter-molecular interactions. MAbs. 2015;7:212–30.doi: 10.4161/19420862.2014.985504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Austerberry JI, Dajani R, Panova S, Roberts D, Golovanov AP, Pluen A, van der Walle CF, Uddin S, Warwicker J, Derrick JP, et al. The effect of charge mutations on the stability and aggregation of a human single chain Fv fragment. Eur J Pharm Biopharm. 2017;115:18–30.doi: 10.1016/j.ejpb.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 40.Du Q, Damschroder M, Pabst TM, Hunter AK, Wang WK, Luo H. Process optimization and protein engineering mitigated manufacturing challenges of a monoclonal antibody with liquid-liquid phase separation issue by disrupting inter-molecule electrostatic interactions. MAbs. 2019;11:789–802.doi: 10.1080/19420862.2019.1599634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Groves MA, Amanuel L, Campbell JI, Rees DG, Sridharan S, Finch DK, Lowe DC, Vaughan TJ. Antibody VH and VL recombination using phage and ribosome display technologies reveals distinct structural routes to affinity improvements with VH-VL interface residues providing important structural diversity. MAbs. 2014;6:236–45.doi: 10.4161/mabs.27261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Igawa T, Tsunoda H, Kikuchi Y, Yoshida M, Tanaka M, Koga A, Sekimori Y, Orita T, Aso Y, Hattori K, et al. VH/VL interface engineering to promote selective expression and inhibit conformational isomerization of thrombopoietin receptor agonist single-chain diabody. Protein Eng Des Sel. 2010;23:667–77.doi: 10.1093/protein/gzq034. [DOI] [PubMed] [Google Scholar]
- 43.Masuda K, Sakamoto K, Kojima M, Aburatani T, Ueda T, Ueda H. The role of interface framework residues in determining antibody V(H)/V(L) interaction strength and antigen-binding affinity. Febs J. 2006;273:2184–94.doi: 10.1111/j.1742-4658.2006.05232.x. [DOI] [PubMed] [Google Scholar]
- 44.Worn A, Pluckthun A. Different equilibrium stability behavior of ScFv fragments: identification, classification, and improvement by protein engineering. Biochemistry. 1999;38:8739–50.doi: 10.1021/bi9902079. [DOI] [PubMed] [Google Scholar]
- 45.Nieba L, Honegger A, Krebber C, Pluckthun A. Disrupting the hydrophobic patches at the antibody variable/constant domain interface: improved in vivo folding and physical characterization of an engineered scFv fragment. Protein Eng. 1997;10:435–44. [DOI] [PubMed] [Google Scholar]
- 46.Reiter Y, Brinkmann U, Kreitman RJ, Jung SH, Lee B, Pastan I. Stabilization of the Fv fragments in recombinant immunotoxins by disulfide bonds engineered into conserved framework regions. Biochemistry. 1994;33:5451–59. [DOI] [PubMed] [Google Scholar]
- 47.Reiter Y, Brinkmann U, Lee B, Pastan I. Engineering antibody Fv fragments for cancer detection and therapy: disulfide-stabilized Fv fragments. Nat Biotechnol. 1996;14:1239–45.doi: 10.1038/nbt1096-1239. [DOI] [PubMed] [Google Scholar]
- 48.Weatherill EE, Cain KL, Heywood SP, Compson JE, Heads JT, Adams R, Humphreys DP. Towards a universal disulphide stabilised single chain Fv format: importance of interchain disulphide bond location and vL-vH orientation. Protein Eng Des Sel. 2012;25:321–29.doi: 10.1093/protein/gzs021. [DOI] [PubMed] [Google Scholar]
- 49.Brinkmann U, Reiter Y, Jung SH, Lee B, Pastan I. A recombinant immunotoxin containing a disulfide-stabilized Fv fragment. Proc Natl Acad Sci U S A. 1993;90:7538–42.doi: 10.1073/pnas.90.16.7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Royer CA. Fluorescence spectroscopy. Methods Mol Biol. 1995;40:65–89.doi: 10.1385/0-89603-301-5:65. [DOI] [PubMed] [Google Scholar]
- 51.Hawe A, Sutter M, Jiskoot W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm Res. 2008;25:1487–99.doi: 10.1007/s11095-007-9516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Witcher J, Fleischmann R, Chindalore VL, Hansen RJ, Hu L, Radtke D, Voelker J, Gomez E, McColm J. Pharmacokinetics and safety of single doses of tabalumab in subjects with rheumatoid arthritis or systemic lupus erythematosus. Br J Clin Pharmacol. 2016;81:908–17.doi: 10.1111/bcp.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ewert S, Huber T, Honegger A, Pluckthun A. Biophysical properties of human antibody variable domains. J Mol Biol. 2003;325:531–53. [DOI] [PubMed] [Google Scholar]
- 54.Rothlisberger D, Honegger A, Pluckthun A. Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J Mol Biol. 2005;347:773–89.doi: 10.1016/j.jmb.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 55.Wang N, Smith WF, Miller BR, Aivazian D, Lugovskoy AA, Reff ME, Glaser SM, Croner LJ, Demarest SJ. Conserved amino acid networks involved in antibody variable domain interactions. Proteins. 2009;76:99–114.doi: 10.1002/prot.22319. [DOI] [PubMed] [Google Scholar]
- 56.Bujotzek A, Dunbar J, Lipsmeier F, Schafer W, Antes I, Deane CM, Georges G. Prediction of VH-VL domain orientation for antibody variable domain modeling. Proteins. 2015;83:681–95.doi: 10.1002/prot.24756. [DOI] [PubMed] [Google Scholar]
- 57.Herold EM, John C, Weber B, Kremser S, Eras J, Berner C, Deubler S, Zacharias M, Buchner J. Determinants of the assembly and function of antibody variable domains. Sci Rep. 2017;7:12276.doi: 10.1038/s41598-017-12519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pluckthun A. Mono- and bivalent antibody fragments produced in Escherichia coli: engineering, folding and antigen binding. Immunol Rev. 1992;130:151–88. [DOI] [PubMed] [Google Scholar]
- 59.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, Gilmanshin RI. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–28.doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 60.Reiter Y, Brinkmann U, Webber KO, Jung SH, Lee B, Pastan I. Engineering interchain disulfide bonds into conserved framework regions of Fv fragments: improved biochemical characteristics of recombinant immunotoxins containing disulfide-stabilized Fv. Protein Eng. 1994;7:697–704. [DOI] [PubMed] [Google Scholar]
- 61.Humphreys PBaDP. Relative contribution of framework and cdr regions in antibody variable domains to multimerisation of Fv- and scFv-containingbispecific antibodies. Antibodies. 2018;7:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tu C, Terraube V, Tam AS, Stochaj W, Fennell BJ, Lin L, Stahl M, LaVallie ER, Somers W, Finlay WJJ, et al. A Combination of Structural and Empirical Analyses Delineates the Key Contacts Mediating Stability and Affinity Increases in an Optimized Biotherapeutic Single-chain Fv (scFv). J Biol Chem. 2016;291:1267–76.doi: 10.1074/jbc.M115.688010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun. 2007;355:751–57.doi: 10.1016/j.bbrc.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 64.Labrijn AF, Buijsse AO, van Den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, Killestein J, Polman CH, Aalberse RC, Schuurman J, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. 2009;27:767–71.doi: 10.1038/nbt.1553. [DOI] [PubMed] [Google Scholar]
- 65.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–92.doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huse WD, Sastry L, Iverson SA, Kang AS, Alting-Mees M, Burton DR, Benkovic SJ, Lerner RA. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science. 1989;246:1275–81. [DOI] [PubMed] [Google Scholar]
- 67.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74.doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42.doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Taltz prescribing information. 2016. https://pi.lilly.com/us/taltz-uspi.pdf