

ABSTRACT
Inflammatory bowel disease, encompassing both ulcerative colitis and Crohn’s disease, is characterized by chronic, relapsing-remitting gastrointestinal inflammation of unknown etiology. SHIP deficient mice develop fully penetrant, spontaneous ileitis at 6 weeks of age, and thus offer a tractable model of Crohn’s disease-like inflammation. Since disruptions to the microbiome are implicated in the pathogenesis of Crohn’s disease, we conducted a 16S rRNA gene survey of the ileum, cecum, colon, and stool contents of SHIP+/+ and SHIP−/− mice. We predicted that diversity and compositional changes would occur after, and possibly prior to, the onset of overt disease. No differences were found in alpha diversity, but significant changes in beta diversity and specific commensal populations were observed in the ileal compartment of SHIP deficient mice after the onset of overt disease. Specifically, reductions in the Bacteroidales taxa, Muribaculum intestinale, and an expansion in Lactobacillus were most notable. In contrast, expansions to bacterial taxa previously associated with inflammation, including Bacteroides, Parabacteroides, and Prevotella were observed in the ilea of SHIP deficient mice prior to the onset of overt disease. Finally, antibiotic treatment reduced the development of intestinal inflammation in SHIP−/− mice. Thus, our findings indicate that SHIP is involved in maintaining ileal microbial homeostasis. These results have broader implications for humans, since reduced SHIP protein levels have been reported in people with Crohn’s disease.
KEYWORDS: SHIP, intestinal inflammation, ileitis, Crohn’s disease, microbiome
Introduction
Inflammatory bowel disease, consisting primarily of ulcerative colitis and Crohn’s disease, is characterized by chronic, relapsing and remitting, or progressive gastrointestinal inflammation of unknown etiology. Current thinking is that complex genetic and environmental factors converge to cause a dysregulated host immune response to commensal microbes.1 The intestinal microbiome (i.e. the microbes, their genomes, and the microenvironment with which they interact) has been extensively implicated in the pathogenesis of IBD. Compared to the microbiome in a healthy intestine, people with IBD often display dysbiosis characterized by reduced bacterial diversity, enriched Enterobacteriaceae, and depleted members of Bacteroidetes and Firmicutes.2
Germ-free rodent models of intestinal inflammation have shown that bacteria are necessary for the development of spontaneous inflammation.3,4 Additionally, specific taxa have been shown to promote intestinal inflammation in genetically susceptible mice.5 Other taxa have been shown to protect from intestinal inflammation through the production of anti-inflammatory metabolites.6,7 Until recently, there has been a paucity of evidence to establish the temporality of microbiome changes in the context of intestinal inflammation. Glymenaki and colleagues reported that microbiome structure and function alterations can occur prior to the onset of colitis in genetically susceptible mice.8,9
The SH2-domain containing inositol 5´-phosphatase (SHIP) is a hematopoietic-specific lipid phosphatase involved in regulating immune activation [Reviewed in ref. 10]. In mice, SHIP deficiency causes spontaneous inflammation and fibrosis limited to the distal ileum, reminiscent of ileal Crohn’s disease in humans.11 This mouse model is particularly relevant to humans because SHIP protein levels and activity have been found to be reduced in people with Crohn’s disease.12
Herein, we hypothesized that SHIP deficiency leads to changes in the intestinal microbiome. We also hypothesized that changes would precede overt ileitis because dysbiosis caused by SHIP deficiency could initiate or exacerbate inflammation. Indeed, antibiotic treatment reduced the development of ileal inflammation in SHIP deficient mice. Our 16S rRNA gene profiling results suggest that SHIP-deficiency causes subtle changes specifically to the ileal microbiome, which are amplified after the onset of overt inflammation.
Results
SHIP deficiency results in spontaneous ileitis
Prior to investigating potential perturbations to the intestinal microbiome in SHIP−/− mice, we wanted to demonstrate that these mice develop spontaneous ileitis. SHIP± mice were bred to generate SHIP+/+ and SHIP−/− littermates for experiments. SHIP−/− mice developed gross inflammation in the distal ileum beginning at six weeks of age, which affected all SHIP−/− mice by eight weeks of age, but not co-housed SHIP+/+ mice (Figure 1(a)). Histopathological examination of 8-week-old mice revealed elevated lamina propria immune cell infiltration, villi and goblet cell hyperplasia, and muscle thickening in inflamed portions of the SHIP−/− distal ileum (Figure 1(b,c)). Histological damage scoring, which has been used to assess active inflammation, chronic inflammation, and disruption in villous architecture in SAMP1/Yit mice13 demonstrated significantly elevated scores for all of these parameters in the 8-week-old SHIP−/− mice compared to their SHIP+/+ counterparts (Figure 1(d)). SHIP−/− mice also had significantly higher MPO activity in ileal tissue homogenates compared to SHIP+/+ mice (Figure 1(e)). We also report significantly increased weight (in g) per length (in cm) in the ilea of SHIP−/− mice compared to SHIP+/+ mice, which may result from increased weight caused by oedema as well as decreased length caused by muscle contraction in the inflamed, 8-week-old SHIP−/− mice (Figure 1(f)). Finally, we asked if SHIP deficiency affects intestinal transit time by measuring the passage time of an orogastrically-gavaged bolus of red carmine dye and observed no significant difference (p > 0.05) between genotypes at 8 weeks of age (Figure 1(g)).
Figure 1.
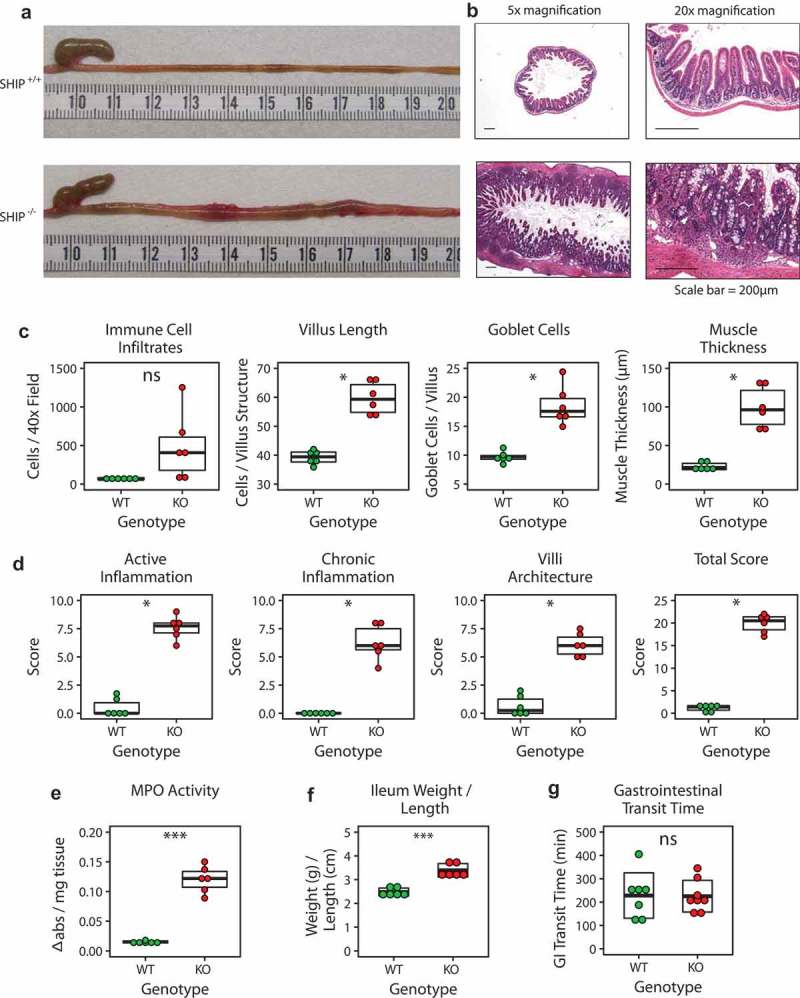
SHIP deficiency causes spontaneous ileal inflammation in mice but gastrointestinal transit time is unaffected. (a) Gross and (b) histological appearance of 8-week-old SHIP+/+ and SHIP−/− ilea. (c) Immune cell numbers, villus length, goblet cell numbers, and muscle thickness in 8-week-old SHIP+/+ and SHIP−/− ilea. N = 6 mice per genotype; ns = not significantly different, *p < 0.006 comparing SHIP+/+ to SHIP−/− genotypes using an unpaired Student’s t-test and Welch’s t-test with Bonferroni correction for multiple comparisons. (d) Histological scoring for inflammation including active inflammation, chronic inflammation, villi architecture, and total score in 8-week-old SHIP+/+ and SHIP−/− mice ilea. N = 6 mice per genotype; *p < 0.00625 comparing SHIP+/+ to SHIP−/− genotypes using an unpaired Student’s t-test, Wilcoxon test, and Welch’s t-test with Bonferroni correction for multiple comparisons. (e) MPO activity in full-thickness ileal tissue homogenates from SHIP+/+ and SHIP−/− mice. N = 6 mice per genotype; ***p < 0.001 comparing SHIP+/+ to SHIP−/− genotypes using an unpaired Student’s t-test and Welch’s t-test with Bonferroni correction for multiple comparisons. (f) Weight (in g) per length (in cm) of the inflamed ileum or corresponding location in SHIP−/− and SHIP+/+ mice, respectively. N = 6 mice per genotype; ***p < 0.001 comparing SHIP+/+ to SHIP−/− genotypes using a Welch’s t-test. (g) Gastrointestinal transit time in 8-week-old SHIP+/+ and SHIP−/− mice (p > 0.05). N = 7–8 mice per genotype; ns = not significantly different comparing SHIP+/+ to SHIP−/− genotypes using an unpaired Student’s t-test.
Ship-associated inflammation is not caused by overt bacterial overgrowth
To determine if active disease is caused by a bacterial overgrowth, we quantified total bacteria in the ileum, cecum, colon, and stool from 8-week-old SHIP+/+ and SHIP−/− mice. We estimated bacterial load by using qPCR to enumerate total 16S rRNA gene copies followed by gene copy number correction at the phylum level. We found no difference (p > 0.00625) in the estimated number of bacteria at the site of inflammation in the ileum, nor in the other intestinal compartments (Figure 2).
Figure 2.
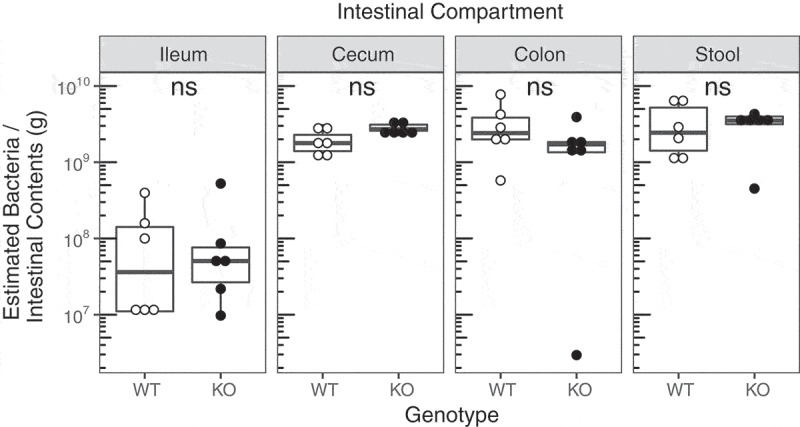
SHIP deficiency in mice does not change estimated intestinal bacterial loads. Bacterial 16S gene number adjusted for gene copy number in the ileum, cecum, colon, and stool contents of 8-week-old SHIP+/+ and SHIP−/− mice. N = 6 mice per genotype and intestinal compartment; ns = not significantly different comparing SHIP+/+ and SHIP−/− genotypes using an unpaired Student’s t-test and Wilcoxon test with Bonferroni correction for multiple comparisons.
16S rRNA gene metadata
We sequenced the V4-V5 region of the 16S rRNA gene of bacterial DNA extracted from intestinal contents to investigate potential alterations to the microbial composition of 4- and 8-week-old SHIP+/+ and SHIP−/− mice. A total of 96 samples were acquired from 24 mice (6 × 8-week-old SHIP+/+, 6 × 8-week-old SHIP−/−, 6 × 4-week-old SHIP+/+, and 6 × 4-week-old SHIP−/− with ileum, cecum, colon, and stool samples taken from each mouse). Next generation sequencing followed by sequence processing with the Divisive Amplicon Denoising Algorithm 2 (DADA2) pipeline (v1.6.0) yielded a mean of 23,223.96 (SD 8139.91) high-quality reads per sample (Fig. S1A), which was comparable to the output from the QIIME (v1.8.0) pipeline (Fig. S1B). To normalize our data for diversity computations, we subsampled reads to 5500 sequences per sample resulting in the loss of one poor quality sample. To guide our sampling depth, we plotted a rarefaction curve depicting a plateau in the number of observed OTUs as sampling depth increases to 5000 sequences per sample (Fig. S1C). 3490 unique sequence variants (herein referred to as operational taxonomic units; OTUs) were identified and assigned taxonomy with DADA2 and the Ribosomal Database Project (RDP) bacterial classifier (v11.5).
SHIP deficiency did not affect α diversity, but changed β diversity in the ileum
First, we evaluated the bacterial α diversity (i.e. richness and evenness) of 8-week-old SHIP+/+ and SHIP−/− mouse ileal contents. We used QIIME to measure the number of observed OTUs, Simpson’s index, and Faith’s phylogenetic diversity. With each of these measurements, we found no differences (p > 0.004) between 8-week-old SHIP+/+ and SHIP−/− mice ileal diversity (Figure 3(a)) or that in other intestinal compartment samples (Table S1). Though means of number of OTUs and phylogenetic diversity were higher in 8-week-old SHIP−/− ileal samples compared to SHIP+/+, no significant differences were observed. To identify changes in the bacterial β diversity (i.e. population structural changes), we used the phyloseq R package (v1.21.3) to conduct principal coordinates analyses of three distinctly informative metrics. No structural differences (p > 0.004) were seen in either unweighted or weighted UniFrac distances (Figure 3(b)). However, Bray-Curtis dissimilarity between 8-week-old genotypes reached statistical significance (p = 0.002) with Adonis testing and Bonferroni correction (Figure 3(b)). Differences were confined to the ileum (Table S2).
Figure 3.
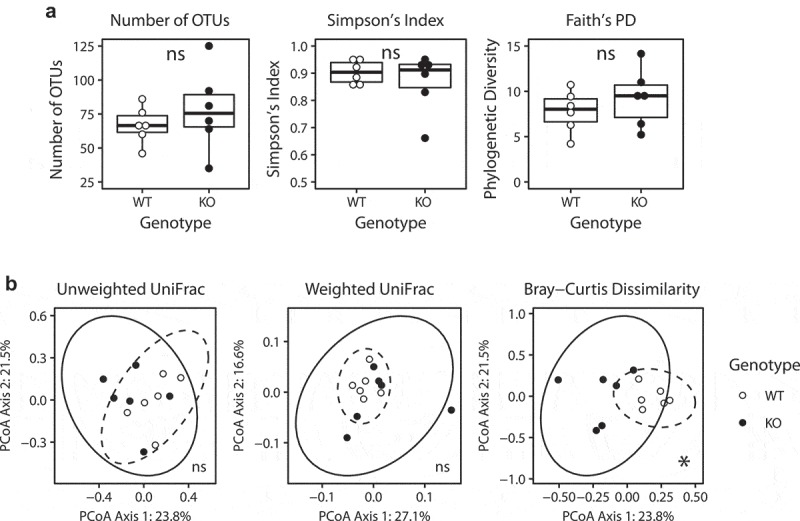
SHIP deficiency in mice does not affect α diversity, but changes β diversity in the intestinal microbiota. (a) Measures of α diversity in SHIP+/+ and SHIP−/− mice ileal contents. ns = not significantly different using an unpaired Student’s t-test, Welch’s t-test, and Wilcoxon test with Bonferroni correction for multiple comparisons. (b) Principal coordinates analysis plots of β diversity in SHIP+/+ and SHIP−/− mice ileal contents including unweighted UniFrac distances, (ns = not significantly different using Adonis with Bonferroni correction), weighted UniFrac distances (ns = not significantly different using Adonis with Bonferroni correction), and Bray-Curtis dissimilarity (*p < 0.004 using Adonis, with Bonferroni correction for multiple comparisons). Ellipses indicate t-distribution 95% confidence intervals in (b). N = 6 mice per genotype.
SHIP deficiency alters specific intestinal microbial populations
Since we detected changes to the bacterial community structure in 8-week-old SHIP−/− mice, we next asked if specific bacteria or bacterial communities were driving these changes. At the phylum level, we observed fluctuations in the relative abundance of bacteria between 8-week-old SHIP+/+ and SHIP−/− mouse ileal contents. While SHIP−/− mice had reduced bacteria in the Bacteroidetes phylum and a compensatory increase in Proteobacteria (Figure 4(a)), these changes were superficial and driven by a single sample (Fig. S2). To identify whether changes were driven by specific taxa at lower phylogenetic levels, we used two biomarker discovery tools. First, after obtaining a classification accuracy of 92%, the random forests R program (v4.6–12) identified the top ten sequences contributing to this accuracy (Figure 4(b)). Next, linear discriminant analysis (LDA) effect size (LEfSe) revealed seven discriminating sequences with an LDA score > 2. These coincided with seven of the ten taxa identified with random forests (Figure 4(c)). Of these ten sequences identified, eight belonged to the order Bacteroidales, while one was classified as Lactobacillus, and another was classified as Desulfovibrio (Table 1). Abundances in individual mice are shown in Fig. S3.
Figure 4.
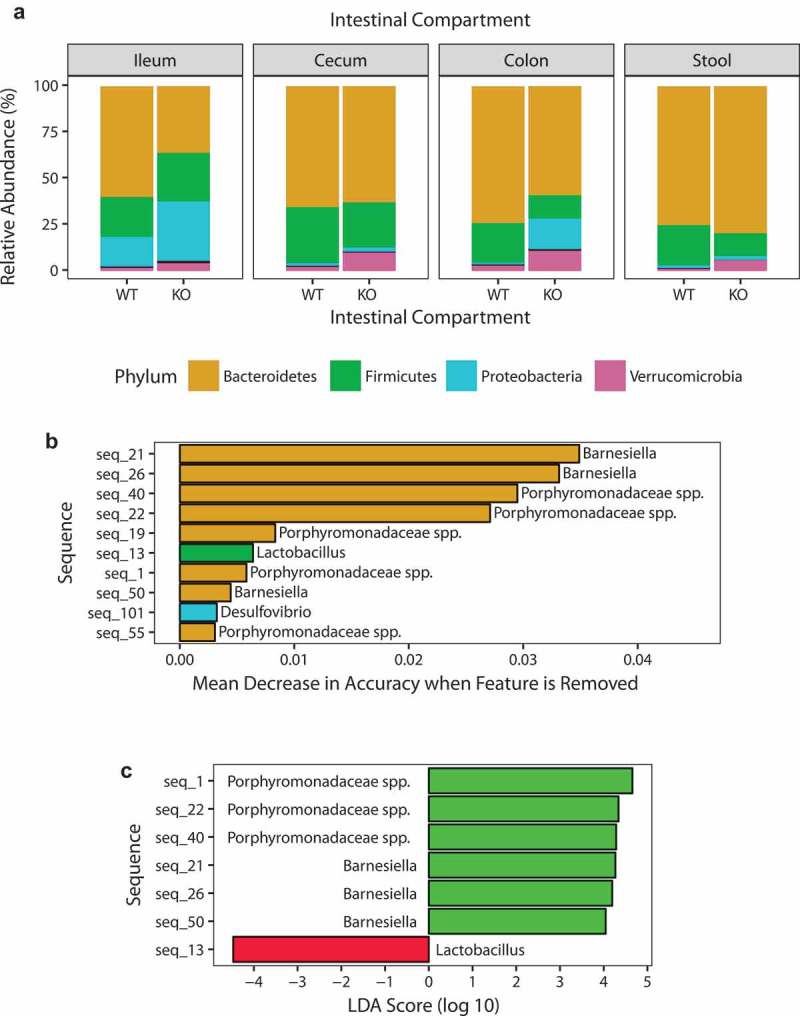
SHIP deficiency in mice causes changes to specific taxa in the ileal microbiota. (a) Phylum-level relative abundances of taxa in SHIP+/+ and SHIP−/− mice ileum, cecum, colon, and stool contents. N = 5–6 mice per genotype and intestinal compartment. (b) Identification of discriminating taxa using random forests. (c) Identification of discriminating taxa using linear discriminant analysis effect size. Green bars indicate taxa more associated with SHIP+/+ mice; Red bars indicate taxa more associated with SHIP−/− mice. N = 6 mice per genotype in (b) and (c).
Table 1.
Discriminating taxa identified in the 8-week-old SHIP+/+ and SHIP−/− mouse ilea. Top ten discriminating taxa in 8-week-old SHIP+/+ and SHIP−/− mice ileal contents. Taxa are listed in descending order according to their LDA effect size. Presented are their randomForest (RF) weights and relative abundance in %. Grey bars indicate taxa, which are lower in relative abundance in SHIP−/− mice compared to SHIP+/+ mice. N = 6 mice per genotype.
| RF |
LDA |
Abundance (%) |
||||||
|---|---|---|---|---|---|---|---|---|
| Seq_ID | Phylum | Order | Family | Genus | Weight | Effect Size | WT | KO |
| Seq_1 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | 0.00582 | 4.6565 | 10.4970 | 2.4182 | |
| Seq_13 | Firmicutes | Lactobacillales | Lactobacillaceae | Lactobacillus | 0.0064 | −4.4730 | 2.2576 | 7.2576 |
| Seq_22 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | 0.02711 | 4.3391 | 2.7515 | 0.3758 | |
| Seq_40 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | 0.02949 | 4.2862 | 1.9061 | 0.2121 | |
| Seq_21 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | Barnesiella | 0.03489 | 4.2686 | 3.3848 | 0.6394 |
| Seq_26 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | Barnesiella | 0.03314 | 4.1963 | 2.8455 | 0.4818 |
| Seq_50 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | Barnesiella | 0.00445 | 4.0462 | 1.0727 | 0.1424 |
| Seq_19 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | 0.00834 | NA | 1.4152 | 0.6848 | |
| Seq_101 | Proteobacteria | Desulfovibrionales | Desulfovibrionaceae | Desulfovibrio | 0.00324 | NA | 0.0091 | 0.2273 |
| Seq_55 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | 0.00307 | NA | 0.4424 | 0.2152 | |
Together, these results converge to suggest that the most profound changes to the SHIP−/− ileal microbiota occur to relatively abundant taxa belonging to the Bacteroidales family Porphyromonadaceae. By cross-referencing the identified taxa sequences with NCBI BLAST, we identified that the unclassified taxa belonging to the family Porphyromonadaceae shared up to 100% sequence identity in the V4-V5 region of the 16S rRNA gene with the recently classified mouse gut commensal, Muribaculum intestinale (Table 2).
Table 2.
NCBI BLAST identification of species in the 8-week-old SHIP+/+ and SHIP−/− mouse ilea. Top ten discriminating taxa in 8-week-old SHIP+/+ and SHIP−/− mice ileal contents assigned taxonomy at the species and strain level. Presented are the sequence identities corresponding to what percentage of the sequenced 16S V4-V5 region matches taxa in the NCBI database. Grey bars indicate taxa, which are lower in relative abundance in SHIP−/− mice compared to SHIP+/+ mice. N = 6 mice per genotype.
| Seq_ID | Ident. | Species | Strain |
|---|---|---|---|
| Seq_13 | 100% | Lactobacillus hominis | |
| Seq_22 | 100% | Muribaculum intestinale | YL27 |
| Seq_101 | 99% | Desulfovibrio fairfieldensis | CCUG 45958 |
| Seq_40 | 99% | Muribaculum intestinale | YL27 |
| Seq_19 | 95% | Muribaculum intestinale | YL27 |
| Seq_21 | 95% | Muribaculum intestinale | YL27 |
| Seq_26 | 95% | Muribaculum intestinale | YL27 |
| Seq_55 | 92% | Muribaculum intestinale | YL27 |
| Seq_1 | 90% | Muribaculum intestinale | YL27 |
| Seq_50 | 90% | Muribaculum intestinale | YL27 |
SHIP deficiency does not affect short chain fatty acid production
Since certain microbial metabolites are associated with intestinal inflammation, we investigated if the ileal microbiome of SHIP−/− mice undergoes functional changes. After performing gas chromatography on ileal contents, we did not detect differences (p > 0.00833) in the concentrations of any of the short chain fatty acids (SCFAs) measured (Figure 5).
Figure 5.
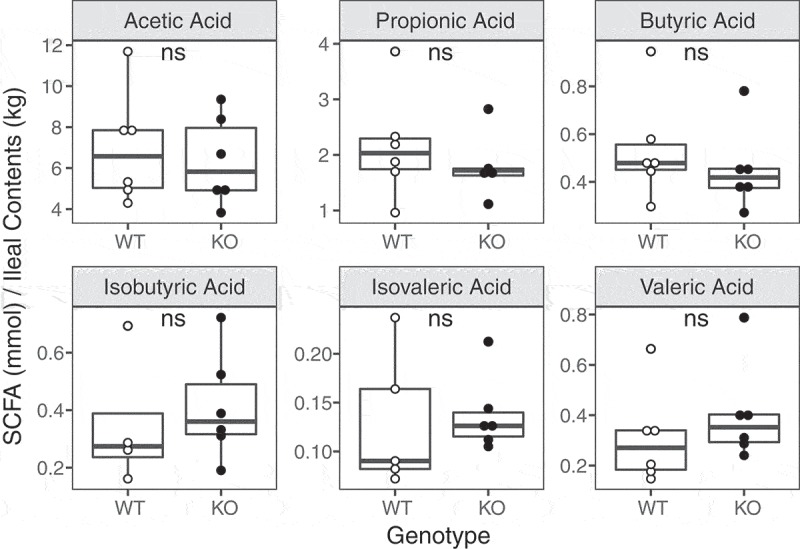
SHIP deficiency in mice does not cause changes to ileal short chain fatty acid concentrations. (a) Short chain fatty acid concentrations in the ileal contents of 8-week-old SHIP+/+ and SHIP−/− mice. N = 4–6 mice per genotype; ns = not significantly different using an unpaired Student’s t-test and Wilcoxon test with Bonferroni correction for multiple comparisons.
Subtle changes occur to bacteroidales bacteria prior to overt inflammation in SHIP−/− mice
Since significant changes were detectable in the 8-week-old SHIP−/− mice microbiota (after the onset of inflammation), we next asked if changes occurred prior to overt inflammation. 4-week-old SHIP−/− mice did not have significantly increased histological parameters that are present in the 8-week-old SHIP−/− mice including immune cell infiltration, villus length, goblet cell numbers, or muscle thickening (Figure 6(a)). Similarly, histopathology scoring revealed no evidence of acute or chronic inflammation or disruption in villous architecture in 4-week-old SHIP−/− mice compared to their wild type littermates (Figure 6(b)). To rule out sub-clinical inflammation as a contributor to any microbiome changes in 4-week-old SHIP−/− mice, we also measured MPO activity and IL-1β concentrations in 4-week-old SHIP−/− mice as these are sensitive measures of the inflammation that characterizes the 8-week-old SHIP−/− mice (Figure 1)12. MPO activity was not elevated in 4-week-old SHIP−/− mice relative to their wild type littermates (Figure 6(c)). Similarly, IL-1β concentrations were not elevated in the ileal of 4-week-old SHIP−/− mice compared to their wild type littermates (Figure 6(d)). Together, this suggests that the ileal inflammation that we have described in 8-week-old SHIP−/− mice is not present in mice at 4 weeks of age. As in 8-week-old mice, there were also no significant differences in estimated ileal bacterial load between 4-week-old SHIP+/+ and SHIP−/− mice (Figure 6(e), Table S3). We applied our 16S rRNA gene analysis to SHIP+/+ and SHIP−/− mouse ileal contents from 4-week-old mice, prior to the onset of inflammation, and found no significant differences (p > 0.004) in α or β diversity (Figure 6(f,g), Table S4 and S5).
Figure 6.
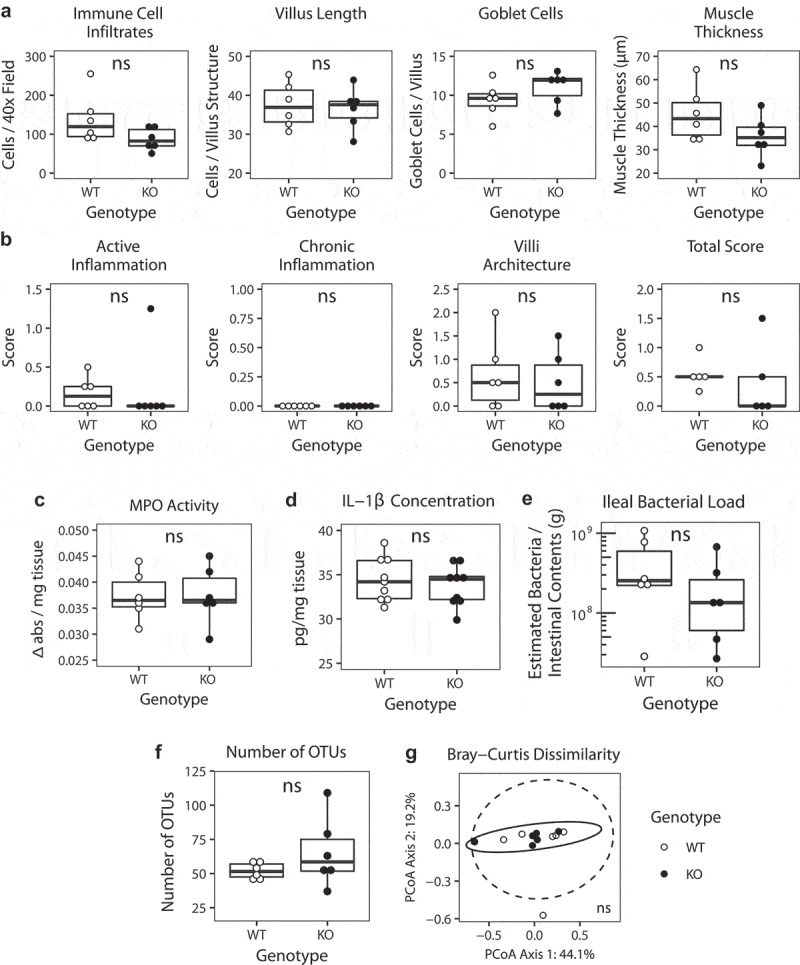
In 4-week-old mice, SHIP deficiency does not cause overt inflammation or changes to estimated ileal bacterial load, or α or β diversity.(a) Immune cell numbers, villus length, goblet cell numbers, and muscle thickness in 4-week-old SHIP+/+ and SHIP−/− ilea. N = 6 mice per genotype; ns = not significantly different using an unpaired Student’s t-test with Bonferroni correction for multiple comparisons. (b) Histological scoring for inflammation including active inflammation, chronic inflammation, villi architecture, and total score in 4-week-old SHIP+/+ and SHIP−/− mice ilea. N = 6 mice per genotype; ns = not significantly different comparing SHIP+/+ to SHIP−/− genotypes using an unpaired Student’s t-test and Wilcoxon test with Bonferroni correction for multiple comparisons. (c) MPO activity in full-thickness ileal tissue homogenates from 4-week-old SHIP+/+ and SHIP−/− mice. N = 6 mice per genotype; ns = not significantly different comparing SHIP+/+ to SHIP−/− genotypes using an unpaired Student’s t-test. (d) IL-1β concentrations in full-thickness ileal tissue homogenates from 4-week-old SHIP+/+ and SHIP−/− mice. N = 9 mice per genotype; ns = not significantly different comparing SHIP+/+ to SHIP−/− genotypes using an unpaired Student’s t-test. (e) Estimated ileal bacterial load in 4-week-old SHIP+/+ and SHIP−/− mice. N = 6 mice per genotype; ns = not significantly different using an unpaired Student’s t-test and Wilcoxon test with Bonferroni correction for multiple comparisons. (f) Number of observed OTUs in the ileal contents of 4-week-old SHIP+/+ and SHIP−/− mice. N = 6 mice per genotype; ns = not significantly different using an unpaired Student’s t-test and Welch’s test with Bonferroni correction for multiple comparisons. (g) Principal coordinates analysis plot of Bray-Curtis dissimilarity of taxa of 4-week-old SHIP+/+ and SHIP−/− ileal contents. N = 6 mice per genotype; ns = not significantly different using an Adonis test with Bonferroni correction for multiple comparisons. Ellipses indicate t-distribution 95% confidence intervals in (g).
However, in 4-week-old SHIP−/− mice, subtle changes (eight sequences) were detectable with random forests (Figure 7(a)) and LEfSe with an LDA score > 2 (Figure 7(b) and Table 3). While an unclassified Porphyromonadaceae taxon sharing 89% sequence identity in the 16S rRNA gene V4-V5 region with Muribaculum intestinale was reduced (LDA score = 3.698) in the ilea of 4-week-old SHIP−/− mice, seven sequences were increased including taxa identified as members of the Parabacteroides, Bacteroides, and Prevotella genera (Table 4). Abundances in individual mice are shown in Fig. S4. Taken together, these results indicate that subtle changes occur to the relative abundance of specific bacterial taxa prior to overt ileitis, but not the overall structure of the ileal microbiome.
Figure 7.
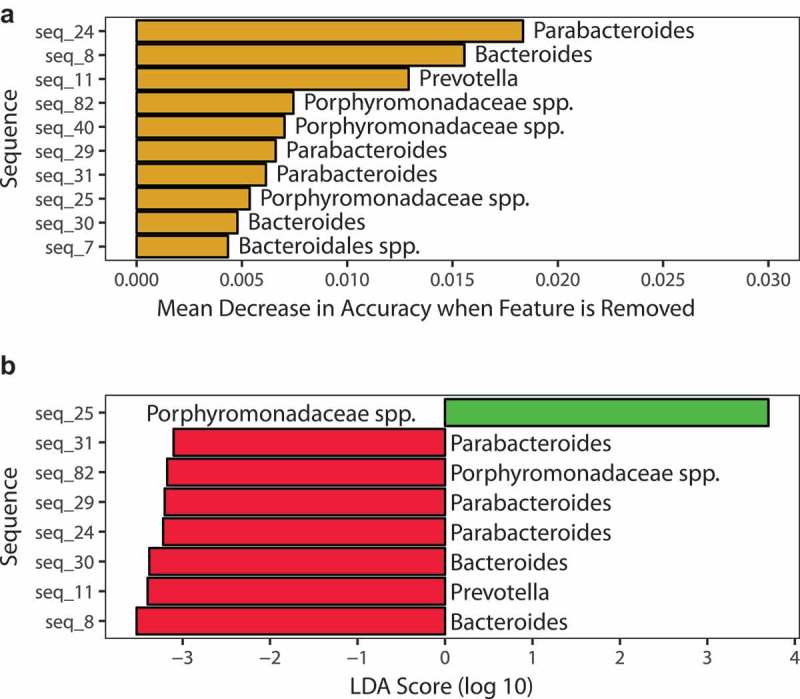
SHIP deficiency in mice causes subtle changes to specific taxa and KEGG orthologs in the ileal microbiome prior to the onset of overt inflammation. (a) Identification of discriminating taxa using random forests. (b) Identification of discriminating taxa using linear discriminant analysis effect size. Green bars indicate taxa more associated with SHIP+/+ mice; Red bars indicate taxa more associated with SHIP−/− mice. N = 6 mice per genotype in each analysis.
Table 3.
Discriminating taxa identified in the 4-week-old SHIP+/+ and SHIP−/− mouse ilea. Top ten discriminating taxa in 4-week-old SHIP+/+ and SHIP−/− mice ileal contents. Taxa are listed in descending order according to their LDA effect size. Presented are their randomForest weights and relative abundance (%) in SHIP+/+ and SHIP−/− mice. Grey bars indicate taxa, which are lower in relative abundance in SHIP−/− mice compared to SHIP+/+ mice. N = 6 mice per genotype.
| RF |
LDA |
Abundance (%) |
||||||
|---|---|---|---|---|---|---|---|---|
| Seq_ID | Phylum | Order | Family | Genus | Weight | Effect Size | WT | KO |
| Seq_25 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | 0.005369463 | 3.6980 | 1.8121 | 0.8303 | |
| Seq_8 | Bacteroidetes | Bacteroidales | Bacteroidaceae | Bacteroides | 0.015566777 | −3.5270 | 0.0061 | 0.6030 |
| Seq_11 | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | 0.012918708 | −3.4000 | 0.0061 | 0.4515 |
| Seq_30 | Bacteroidetes | Bacteroidales | Bacteroidaceae | Bacteroides | 0.004791188 | −3.3812 | 0.0030 | 0.4667 |
| Seq_24 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | Parabacteroides | 0.018348165 | −3.2232 | 0.0000 | 0.1758 |
| Seq_29 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | Parabacteroides | 0.00660434 | −3.2054 | 0.0091 | 0.1636 |
| Seq_82 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | 0.007449255 | −3.1769 | 0.0121 | 0.1455 | |
| Seq_31 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | Parabacteroides | 0.006141053 | −3.1030 | 0.0212 | 0.1515 |
| Seq_40 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | 0.007024298 | NA | 0.5939 | 0.4030 | |
| Seq_7 | Bacteroidetes | Bacteroidales | 0.004341233 | NA | 0.0121 | 0.1182 | ||
Table 4.
NCBI BLAST identification of discriminating taxa in the 4-week-old SHIP+/+ and SHIP−/− mouse ilea. Top ten discriminating taxa of 4-week-old SHIP+/+ and SHIP−/− mice ileal contents assigned taxonomy at the species and strain level. Presented are the sequence identities corresponding to what percentage of the sequenced 16S V4-V5 region matches taxa in the database. Grey bars indicate taxa, which are lower in relative abundance in SHIP−/− mice compared to SHIP+/+ mice.
| Seq_ID | Ident. | Species | Strain |
|---|---|---|---|
| Seq_30 | 100% | Bacteroides ovatus | ATC 8483 |
| Seq_24 | 99% | Parabacteroides distasonis | ATCC 8503 |
| Seq_40 | 99% | Muribaculum intestinale | YL27 |
| Seq_29 | 99% | Parabacteroides distasonis | ATCC 8503 |
| Seq_31 | 99% | Parabacteroides distasonis | ATCC 8503 |
| Seq_8 | 97% | Bacteroides uniformis | ATCC 8492 |
| Seq_11 | 90% | Prevotella copri | DSM 18205 |
| Seq_25 | 89% | Muribaculum intestinale | YL27 |
| Seq_82 | 85% | Muribaculum intestinale | YL27 |
| Seq_7 | 85% | Solitalea canadensis | DSM 3403 |
Antibiotic treatment reduces intestinal inflammation in SHIP−/− mice
Finally, given changes in the ileal microbiota in 8-week-old SHIP−/− mice in 4-week-old SHIP−/− mice, we next asked whether antibiotic treatment could ameliorate ileal inflammation in SHIP−/− mice. SHIP+/+ and SHIP−/− mice were given ciprofloxacin and metronidazole in their drinking water from 4 to 8 weeks of age and pathology was assessed in mice after treatment (at 8 weeks of age). SHIP+/+ mice were not significantly affected by antibiotic treatment (Figure 8). Antibiotic treatment reduced gross pathology in SHIP−/− mice (Figure 8(a)). Antibiotic treatment also improved intestinal architecture evident in H&E stained tissue sections (Figure 8(b)). Specifically, lamina propria immune cell infiltration, villi and goblet cell hyperplasia, and muscle thickening were reduced in ileal cross-sections of antibiotic-treated SHIP−/− mice compared to sham-treated littermates (Figure 8(c)). Total histological damage scores, which include measures of active inflammation, chronic inflammation, and disruption of villus architecture, were also reduced in antibiotic treated SHIP−/− mice compared to sham-treated littermates (Figure 8(d)). These results suggest that the microbiome contributes to spontaneous Crohn’s-disease like ileal inflammation in SHIP−/− mice.
Figure 8.
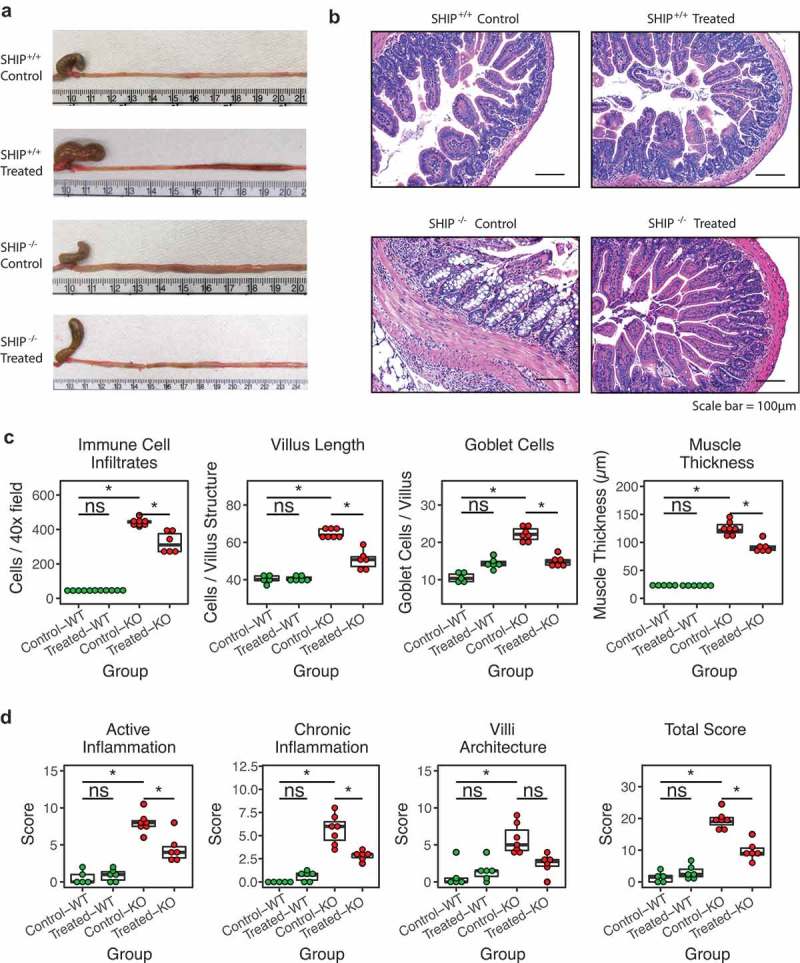
Antibiotic treatment ameliorates intestinal inflammation in SHIP deficient mice. (a) Gross and (b) histological appearance (H&E stains) of 8-week-old SHIP+/+ and SHIP−/− ilea from mice that are untreated (control) or treated with antibiotics (ABx) from 4 to 8 weeks of age. (c) Immune cell numbers, villus length, goblet cell numbers, and muscle thickness in 8-week-old SHIP+/+ and SHIP−/− ilea from mice that were untreated or treated with antibiotics from 4 to 8 weeks of age. N = 6 mice per genotype; ns = not significantly different, *p < 0.0021 comparing SHIP+/+ to SHIP−/− genotypes and comparing antibiotic treated to sham treated mice for each genotype. Comparisons were made using an ANOVA with Tukey’s HSD and Bonferroni correction for multiple comparisons. (d) Histological scoring for inflammation including active inflammation, chronic inflammation, villi architecture, and total score in 8-week-old SHIP+/+ and SHIP−/− mice ilea from mice that were untreated or treated with antibiotics from 4 to 8 weeks of age. N = 6 mice per genotype; ns = not significantly different, *p < 0.0021 comparing SHIP+/+ to SHIP−/− genotypes and comparing antibiotic treated to sham treated mice for each genotype. Comparisons were made using an ANOVA and Tukey’s HSD and Bonferroni correction for multiple comparisons.
Discussion
Here, we demonstrated that spontaneous ileitis in SHIP-deficient mice is dependent, at least in part, on the microbiome as it is ameliorated in SHIP−/− mice by treatment with antibiotics. Ileitis is associated with pronounced alterations to the ileal bacteria taxonomic composition. Reductions to members of the Bacteroidales order occur following overt inflammation, whereas specific genera are enriched prior to disease onset, including Bacteroides, Parabacteroides, and Prevotella.
In addition to the SHIP-deficient mouse, two other tractable mouse models of spontaneous ileitis have been previously reported. SAMP1/YitFc mice develop spontaneous ileitis, ileal fibrosis, skin lesions, cecum inflammation, and perianal disease.13
The involvement of susceptibility loci indicates a polygenic, idiopathic etiology. Such a complex model offers value for testing the efficacy of novel treatments, but is challenging to interrogate the pathogenic roles of specific genes. TNFΔARE mice also exhibit spontaneous TNF-driven transmural inflammation of the distal ileum, though without fibrosis.14
However, since the incidence of primary nonresponse to anti-TNFα therapy is 13–33% in clinical practice,15 it becomes important to interrogate different facets of the complex IBD immunogenetic landscape. Knockout of the SHIP gene (Inpp5d) yields a tractable model of spontaneous, fully penetrant ileal inflammation and fibrosis. To our knowledge, we are the first to report whether bacterial compositional shifts occur in different intestinal compartments prior to and after the onset of spontaneous ileitis in mice. Herein, we also predict and characterize potentially pathophysiological features of the SHIP-deficient mouse.
Increases and decreases in gastrointestinal transit time have been associated with Crohn’s disease16 and dysbiosis,17 respectively. Specifically, reductions in small bowel motility have been found in Crohn’s disease patients; increased motility has been associated with microbial dysbiosis, suggesting that a faster transit time selects for fast growing bacterial species.17 However, mice receiving a high fat diet display intestinal dysbiosis that leads to increased colonic transit time/decreased motility.18 When considering the impact of SHIP deficiency, it is also important to note that SHIP may act through the endocannabinoid system to control intestinal muscle tone. Cannabinoid receptor type 1 (CB1R) inhibits gastrointestinal motility in humans and mice,19 and CB1R is activated more strongly by anandamide (AEA) than phosphoanandamide (pAEA) in the mouse brain.20 Since SHIP is needed to dephosphorylate pAEA to AEA,21 we would predict that SHIP deficient mice would exhibit reduced gastrointestinal transit time. However, our observations revealed no net change, which could be explained by the opposing effects of SHIP’s role in the endocannabinoid system versus inflammation-associated dysbiosis.
A higher bacterial load has been observed in colon biopsies from people with active Crohn’s disease and ulcerative colitis,22 and a similar effect has been seen in rodents. Carboxymethylcellulose-treated IL-10-deficient mice and TNBS- and DSS- treated rats develop small and large bowel bacterial overgrowth and inflammation.23,24 In contrast, no change is seen in the 16S rRNA gene copy count of the colonic mucus compartment of mdr1a-deficient mice, which develop spontaneous colitis.8 As in mdr1a deficient mice, SHIP deficient mice do not display bacterial overgrowth. Observed differences in estimated bacterial load and its contributions to disease development may reflect differences in pathogenic mechanisms of disease and need to be determined experimentally in different mouse models. Given that there were no differences in estimated bacterial load or transit time (exposure to bacterial constituents) in SHIP deficient mice, we next examined the composition of intestinal flora in SHIP+/+ and SHIP−/− mice.
α diversity is an ecological description of an individual community, defined by richness and evenness. In human IBD patient microbiotas, α diversity is typically reduced.25–27 In contrast, SHIP deficient mice do not have reduced α diversity. Our results are consistent with the unaffected microbial diversity seen in mdr1a-deficient8 and TNFΔARE 28 mice. Like people with IBD, SAMP1/YitFc exhibit higher Shannon diversity in their fecal metagenome, although this may not be reflective of diversity in their ileal compartment.29 Since reductions in α diversity are more pronounced in human IBD patients than mouse models, our findings support the idea that confounding factors like diet30 or antibiotic use,31 perhaps combined with inflammatory disease, contribute to, or drive, the reduction in diversity in humans. This is also consistent with the observation that people with ulcerative colitis display reduced bacterial diversity in their ilea, suggesting that non-inflammatory processes contribute to changes to bacterial diversity.32
In ecology, β diversity is used to compare overall community compositions, and can be used to characterize dysbiosis. Community shifts can be measured with or without accounting for the abundance or phylogenetic relatedness of taxa. Bray-Curtis dissimilarity in the SHIP+/+ and SHIP−/− mice is statistically significantly different, which indicates that differences observed in community structure are driven by fluctuations in highly abundant taxa rather than by the types of taxa present, since UniFrac distances were not different. Since mice from this study were cohoused and supplied the same diet, we would not expect to see profound differences in the types of taxa present. Importantly, we found that disruptions to β diversity were confined to the ileum, highlighting how analysis of stool samples may not be sensitive to disease-associated dysbiosis. In practice, a recent report achieved a classification accuracy of 79% in discriminating healthy from active Crohn’s disease stool samples, but this included samples from which disease was localized to the colon.33 To our knowledge, no study has assessed the performance of a disease-state classifier using stool samples from only patients with ileal Crohn’s disease.
SHIP deficient mice have reduced relative representation of taxa from the Porphyromonadaceae family.34 This is consistent with reduced Porphyromonadaceae, including Barnesiella and Odoribacter genera, that have been observed in people with IBD.32,35 In contrast, enrichment in the Bacteroidales order, which includes the Porphyromonadaceae family, were recently reported in pooled fecal samples from SAMP1/YitFc mouse, which also develop ileitis.29 The closely related Muribaculum genus is also enriched in mice with T cell-induced colitis, making the role of this bacterial group in murine models complicated.36 Upon deeper inspection, it is clear that unclassified members of the families S24-7 and Porphyromonadaceae have been reported to be misidentified in the literature and ought to belong to the family Muribaculaceae, which currently consists of one characterized species, Muribaculum intestinale.37 Importantly, taxonomic classification of our data with two different 16S databases, RDP and Greengenes, resulted in the highest abundant Bacteroidales bacterial taxa being classified as Porphyromonadaceae and S24-7, respectively, of which many of the discriminating taxa we identified share between 90 to 100% sequence identity in the 16S rRNA gene V4-V5 region with M. intestinale. In addition to the apparent heterogeneity within this clade, this highlights the potential misidentification in other studies. Extending our results to studies where S24-7 taxa were prominent revealed reductions in T-bet−/− Rag2−/− mice,38 TNFΔARE mice,39 DSS-treated mice, and Salmonella-infected mice.40 In the latter study, NCBI BLAST revealed S24-7 sequences have highest identity with M. intestinale. The discordant identification of prominent taxa belonging to the Bacteroidales order underscores the need to harmonize taxonomic identification methods before comparisons across studies can be made. Nevertheless, M. intestinale is generally associated with a healthy phenotype and so we predict it to exhibit commensal behaviour.
Lactobacillus hominis, which is increased in 8-week-old SHIP-deficient mice ilea, has not yet been reported, although other members of the Lactobacillus genus have exhibited disparate roles in intestinal inflammation in rodents. For instance, L. acidophilus41 and L. plantarum42 improve intestinal inflammation in DSS-treated mice, while L. fermentum potently induces TLR signaling and IL-1β production in germ-free rat colons,43 and higher levels of Lactobacillus have been associated with Salmonella-induced inflammation in mice.40 Hence, Lactobacillus exemplifies the functional variability seen within species of the same genus.
We also identified Desulfovibrio fairfieldensis as increased in the overt disease state in SHIP−/− mice. Desulfovibrio is a genus of sulfate-reducing bacteria (SRB) and has been associated with CD in humans.44,45 Mechanistically, it is posited that generation of sulphide from sulfate is toxic to colonocytes.46 However, despite research on the presence and role of SRB in the ileum having been warranted,35,47-51 ###it has not been reported prior to this study.
Our finding of subtle ileal dysbiosis preceding the onset of overt inflammation is consistent with observations made in the mdr1a-deficient mouse.8 Specific expansions of Bacteroides, Parabacteroides, and Prevotella, seen in the SHIP deficient mouse ileum prior to disease onset, have also been previously associated with intestinal inflammation. Bacteroides ovatus is associated with an increased serum antibody response in people with IBD, although its role in disease pathogenesis remains unclear.52 Parabacteroides taxa are enriched in Crohn’s disease patients,53 and associated with non-inflamed tissue,54 which may be captured in the SHIP−/− mouse model of ileal inflammation because inflammation is discontinuous. Opposing roles of this taxa have been found in mice as well, where P. distasonis is colitis-predisposing in Pglyrp-deficient mice, but P. distasonis cell lysates attenuate DSS-induced colitis.55 Prevotella copri, which is increased in SHIP-deficient mice prior to disease onset, is enriched in healthy IBD-patient relatives and posited to predispose genetically susceptible individuals to disease.56 In mice, P. copri increases sensitivity to DSS-induced colitis.57-61 ###Taken together, these data indicate potentially inflammatory and/or disease-predisposing roles for specific Bacteroidales taxa.
Finally, we report that antibiotic treatment reduced gross and histopathological measures of inflammation in SHIP−/− mice. Together, our data suggest that compositional changes that occur to the ileal microbiome of SHIP-deficient mice before the onset of overt inflammation contribute to and exacerbate inflammation. Members of the Bacteroidales order were associated with disease, whereas Muribaculum species were depleted in inflamed conditions, and Bacteroides, Parabacteroides, and Prevotella were enriched preceding overt inflammation. We also found that dysbiosis was confined to the site of disease, underscoring how stool samples may not accurately predict compositional shifts in intestinal compartments proximal to the colon. An emerging overarching theme supported by our and others’ data is that bacterial taxa of the same genera and species exhibit disparate roles in intestinal inflammation, which may be explained by strain-level functional differences, host-specific interactions, and host microenvironment conditions.
Taken together, our findings suggest that SHIP is involved in ileal microbial homeostasis, and that SHIP-associated inflammation amplifies microbial compositional shifts. We established that inflammation is not associated with gastrointestinal transit time or bacterial overgrowth, but insight into the mechanism by which SHIP alters microbial populations is worth exploring. Further study is also needed to elucidate whether specific bacteria are protective or pathogenic in the SHIP deficient mouse and Crohn’s disease patients.
Materials and methods
Mice
All animal procedures were performed in accordance with ethical guidelines set forth by the Canadian Council on Animal Care and were approved by the University of British Columbia Animal Care Committee (protocol numbers A17-0071, A17-0061, and A17-0277). Mice heterozygous for SHIP expression (Inpp5d±) on a mixed C57BL/6 x 129Sv background (F2 generation) were bred to generate SHIP+/+ and SHIP−/− progeny. Mice were weaned at 3 weeks of age. Same sex SHIP+/+ and SHIP−/− littermates were co-housed upon weaning; if SHIP+/+ and SHIP−/− littermates were opposite sex, mice were singly housed until used for experiments at 4 or 8 weeks of age. Groups of mice compared in experiments contained balanced numbers of males and females in each group. Mice were bred in-house at the BC Children’s Hospital research institute Animal Care Facility and maintained in specific-pathogen free conditions. 12 hour light cycles (600h on/1800h off) were maintained throughout the study. Mice were provided autoclaved drinking water, irradiated Teklad Global 18% Protein Rodent Diet pellets (Envigo, 2918), and Performance bedding and enrichment (BioFresh).
Intestinal transit time
8-week-old SHIP+/+ and SHIP−/− mice were orogastrically gavaged with a semiliquid 0.5% methylcellulose (Sigma-Aldrich, M0512) solution containing 6% w/v carmine red (Sigma-Aldrich, C1022) bolus, as previously described62. Gavage was performed with a straight 22 gauge needle with a 1.25 mm diameter ball. Administered fluid volumes of 0.1 mL/kg body weight were determined for each mouse. Mice were isolated from each other with access to food and water in separate cages and observed every 5 minutes upon gavage for up to 6 hours beginning at 1800h. Fecal pellets were obtained and smeared on white paper to test for the presence of red dye. Intestinal transit time was calculated as the time difference between oral gavage and detection of red dye in fecal pellets.
Myeloperoxidase (MPO) activity
MPO activity was measured in full-thickness ileal tissue homogenates from mice using the Myeloperoxidase Colorimetric Activity Assay Kit, according to the manufacturer’s instructions (Sigma-Aldrich, MAK068-1KT).
Il-1β ELISA
IL-1β was measured in full-thickness ileal tissue homogenates from 4-week-old SHIP+/+ and SHIP−/− mice by ELISA, according to the manufacturer’s instructions (BD Biosciences, 557953).
Quantitative PCR (qPCR)
Purified DNA obtained from intestinal contents was used for qPCR to quantitate 16S rRNA genes, as adapted from the protocol designed by the Surette laboratory. Briefly, an Escherichia coli DNA (Affymetrix/USB, J14380) standard was created using serial dilutions from 180 ng to 0.001 ng. Universal 16S rRNA gene primers (926F forward 5´- AAACTCAAAKGAATTGACGG – 3´, 1062R reverse 5´ – CTCACRRCACGAGCTGAC – 3´) were used to quantitate the 16S gene copy count of each sample using a CFX96 Real-Time System and C1000 Thermocycler (BioRad, Hercules, CA). ΔCt values were obtained for all samples in triplicate. 16S gene copy counts were normalized to each sample’s average 16S gene copy count to infer total bacteria quantities. To identify average 16S gene copy counts per sample, a sample-specific modifier was calculated by weighting the average 16S gene copy count/cell of each phyla contained in the sample according to the formula: where R is the normalization value, A is the normalized phylum abundance, C is the phylum-level 16S gene copy count and p is the number of phyla. Phylum-level 16S gene copy counts were obtained from the ribosomal RNA operon copy number database.63 Sample 16S gene copy count values ranged from 2.91 to 6.11 (mean = 3.9).
Histopathology
1 cm segments of mouse ilea, ceca, and colons were excised and fixed in 10% formalin at 4°C for 24 hours and transferred to 70% ethanol until paraffin processing for histological analysis. 5 µm thick sections of each organ were stained with hematoxylin and eosin (H&E). Slides were visualized and photographed with an Axioscope 7 microscope fixed with an AxioCamMR3 Digital Camera (Zeiss, Oberkochen, Germany). Photographs were scored for histological features described by two individuals blinded to experimental condition. Measurements included immune cell infiltrates per 40x magnified field, goblet cell, and total cell count per villus structure, and cross-sectional muscle thickness. Mean values were taken from 6 fields per section from 6 sections for each mouse. Histological damage was also scored in tissue sections by two individuals blinded to experimental condition. Active inflammation, chronic inflammation, and villus architecture were scored independently and added together to generate a total score for histological damage, as previously described.13
Microbial DNA extraction
Immediately prior to euthanasia, mice were isolated and fecal pellets were placed in sterile, pre-weighed tubes on ice. Upon euthanasia, mouse ileal, cecal, and colonic contents were extracted and stored in sterile, pre-weighed tubes. Samples were frozen and stored at −80°C until DNA extraction. DNA was isolated with the PowerSoil DNA isolation kit (MO BIO, 12888), according to the manufacturer’s protocol. Briefly, samples were homogenized with high velocity bead beating, cells were lysed by mechanical and proprietary chemical methods, and bacterial genomic DNA was captured on a silica membrane in a spin column. DNA was washed and eluted from the membrane and stored at −80°C until quantification. DNA was quantified with the NanoDrop 1000 spectrophotometer (ThermoFisher, Waltham, MA) and samples showed acceptable DNA yield (>1ng/µL) and purity (A260/280 ratio ~ 1.8).
16S gene rrna library preparation and sequencing
Isolated intestinal content DNA amplicon fragments were PCR-amplified using the high-fidelity Phusion polymerase (ThermoFisher, M0530S) and fusion primers including the adaptors, barcodes, and 16S V4-V5 region (F: 5´-GTGYCAGCMGCCGCGGTAA-3´; R: 5´-AAACTYAAAKRAATTGRCGG-3´). PCR products were verified with a high-throughput 96-well E-gel (Invitrogen, G8700801). PCR reactions were pooled, cleaned, and normalized using a high-throughput SequalPrep 96-well plate kit (ThermoFisher, A1051001). Samples were multiplexed and run on the Illumina MiSeq (Victoria, BC) platform using 300 + 300 bp paired-end V3 chemistry to generate FASTQ files. This work was performed by the Integrated Microbiome Resource (Dalhousie University, Halifax, NS).
FASTQ processing
FASTQ files were processed using the Microbiome Helper v2.1.0 package wrapping the DADA2 v1.4 pipeline.64,65 Sequences were pre-processed to discard primer sequences and inspected for quality scores. Low quality sequences were trimmed to a maximum length of 270 bases for forward reads and 180 bases for reverse reads and limited to a maximum of 3 and 7 expected errors in forward and reverse reads, respectively. Reads were truncated at the first instance of a quality score of less than or equal to 2. The DADA2 algorithm was applied to infer error models from reads and identify sequence variants with a random seed set to 4124. Paired-end reads were stitched together before chimera checking and taxonomic assignment with reference to Ribosomal Database Project taxonomic training data set 16 (release 11.5). A biom file was generated for bioinformatic analysis with QIIME (v1.8.0)66 and its dependencies, and the phyloseq R package (v1.21.3).67 Rarefaction plots revealed that diversity plateaued at approximately 5500 reads per sample, so sub-sampling was performed to this depth for diversity, classification, and differential abundance analyses.
Statistical analysis
Shapiro-Wilks and Levene’s tests were used to assess the normality of distributions and homogeneity of variances, and Student’s t-tests, Welch’s t-tests, Wilcoxon tests were employed accordingly with R (v3.4.3). ANOVA with Tukey’s Honest Significant Difference post-hoc test were used where applicable with R. α diversity data (Number of Observable OTUs, Simpon’s Index (1-Dominance), and Faith’s Phylogenetic Distance) were obtained from the alpha.diverstiy.py script in QIIME. β diversity metrics including Bray-Curtis dissimilarity, and unweighted and weighted UniFrac distance68 principal coordinates analysis plots were assessed using phyloseq. The vegan R package (v.2.4–6)69 function Betadisper was used to assess homogeneity of variance of distance matrices, which were then analyzed for statistical significance using the adonis function with 999 permutations. A Bonferroni correction for family-wise multiple testing was used.
Since sequenced data typically deviate from a Gaussian distribution, non-parametric analysis is often used for differential abundance testing of taxa. However, compositional data exists in a simplex, rather than Euclidian space, and is not suitable for testing without transformation.70 Log-transformations permit testing without violating test assumptions, but cannot handle zero counts, which are abundant in sequenced data.71 Pseudocounts, which are small place holder values (typically < 1) solve the problem of inflated zeroes, but can affect data clustering if chosen incorrectly.72 Biomarker discovery tools, like random forests and LEfSe, use non-traditional statistical approaches to circumvent this chain of problems and identify discriminating taxa or features for practical applications. Random forests create a predictive model that determines the importance of taxa or other features based on how much they contribute to the model’s classification accuracy. This method has been previously used to classify disease state and identify discriminating taxa in a mouse exercise model,73 human multiple sclerosis patients,74 and human IBD patients.33,75–77 Further, LEfSe uses traditional testing only to filter out taxa or features that are unlikely to be important for classification. Subsequently, linear discriminant analysis employs dimensionality reduction and maximization of differences between groups to identify discriminating features based on their effect size.
The randomForest R package (v4.6–14)78 was used to classify disease state. Briefly, rare features defined as present in less than 20% of samples were removed. Input tables were formatted for classification of state and a random seed was set at 151. Model fit was assessed for significance with a permutation test with 1000 permutations. Accuracy estimation was assessed with leave-one-out cross-validation using the Classification And Regression Training (caret) R package (v6.0–79).79 The top ten discriminating features driving model performance were identified and plotted. Linear Discriminant Analysis Effect Size (LEfSe)80,81 ###was employed to corroborate random forests findings. Briefly, a Kruskal-Wallis test identified significantly differentially abundant features between groups, where if the null hypothesis was rejected (p < 0.05) features underwent linear discriminant analysis to predict their effect size. LDA scores above 2 were considered as important in discriminating genotypes unless otherwise noted. All graphs were plotted with ggplot2 (v2.2.1).
SCFA analysis
Ileal contents were extracted from SHIP+/+ and SHIP −/− mice and stored at −80°C until SCFA quantitation. Samples were used for gas chromatography with the TRACE 1310 Gas Chromatograph coupled to a Flame Ionization Detector (ThermoFisher, Waltham, MA), as previously described.82 This work was performed by Microbiome Insights (Vancouver, BC).
FASTQ accession number
These sequence data have been submitted to the EMBL-EBI European Nucleotide Archive database, accession number ena-STUDY-BCCHR-25–04-2018–23:01:38:230–20.
Antibiotic treatment
Four-week-old SHIP+/+ and SHIP−/− mice were provided ad libitum access to autoclaved drinking water containing 0.66 mg/mL ciprofloxacin (Sigma-Aldrich, 17850), 2.5 mg/mL metronidazole (Sigma-Aldrich, M3761), and 20 mg/mL Ice Blue KOOL-AID (Kraft Foods), as previously described.83 Control mice (no antibiotics) were provided with sterile drinking water containing 20 mg/mL Ice Blue KOOL-AID only. Drinking water was replaced and cages were changed every 2–3 days under sterile conditions. After 4 weeks of treatment, mice were euthanized and ilea were excised for gross and histopathological analyses.
Funding Statement
This work was supported by the Canadian Institutes of Health Research [MOP-133607].
Authorship
PAD performed experiments, data analyses, and manuscript preparation. SCM maintained the mouse colony. CT, JPS, and SCM assisted with experiments and performed data acquisition including blinded histopathological scoring. LMS generated hypothesis and contributed to experimental design and manuscript preparation.
Acknowledgments
LMS is the recipient of a biomedical scholar award from the Michael Smith Foundation for Health Research and an investigatorship from the BC Children’s Hospital research institute. This work was supported by an operating grant awarded to LMS from the Canadian Institutes of Health Research (CIHR; MOP-133607).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary Material
Supplemental data for this article can be accessed on publisher’s website.
References
- 1.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134(2):577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 2.Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun. 2017;8(1):1784. doi: 10.1038/s41467-017-01973-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernández-Sueiro JL, Balish E, Hammer RE. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. 1994;180(6):2359–2364. doi: 10.1084/jem.180.6.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL, Dunne WM, Allen PM, Stappenbeck TS. Commensal Bacteroides Species Induce Colitis in Host-Genotype-Specific Fashion in a Mouse Model of Inflammatory Bowel Disease. Cell Host Microbe. 2011;9(5):390–403. doi: 10.1016/j.chom.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L, Wilson JE, Koenigsknecht MJ, Chou W-C, Montgomery SA, Truax AD, Brickey WJ, Packey CD, Maharshak N, Matsushima GK, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol. 2017;18(5):541–551. doi: 10.1038/ni.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geirnaert A, Calatayud M, Grootaert C, Laukens D, Devriese S, Smagghe G, De Vos M, Boon N, Van de Wiele T. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci Rep. 2017;7. doi: 10.1038/s41598-017-11734-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glymenaki M, Singh G, Brass A, Warhurst G, McBain AJ, Else KJ, Cruickshank SM. Compositional changes in the Gut Mucus microbiota precede the onset of colitis-induced inflammation. Inflamm Bowel Dis. 2017;23(6):912–922. doi: 10.1097/MIB.0000000000001118. [DOI] [PubMed] [Google Scholar]
- 9.Glymenaki M, Barnes A, Hagan SO, Warhurst G, McBain AJ, Wilson ID, Kell DB, Else KJ, Cruickshank SM. Stability in metabolic phenotypes and inferred metagenome profiles before the onset of colitis-induced inflammation. Sci Rep. 2017;7(1):8836. doi: 10.1038/s41598-017-08732-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobranowski P, Sly LM. SHIP negatively regulates type II immune responses in mast cells and macrophages. J Leukoc Biol. 2018. doi: 10.1002/JLB.3MIR0817-340R. [DOI] [PubMed] [Google Scholar]
- 11.McLarren KW, Cole AE, Weisser SB, Voglmaier NS, Conlin VS, Jacobson K, Popescu O, Boucher J-L, Sly LM. SHIP-deficient mice develop spontaneous intestinal inflammation and arginase-dependent fibrosis. Am J Pathol. 2011;179(1):180–188. doi: 10.1016/j.ajpath.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ngoh EN, Weisser SB, Lo Y, Kozicky LK, Jen R, Brugger HK, Menzies SC, McLarren KW, Nackiewicz D, van Rooijen N, et al. Activity of SHIP, which prevents expression of interleukin 1β, is reduced in patients with crohn’s disease. Gastroenterology. 2016;150(2):465–476. doi: 10.1053/j.gastro.2015.09.049. [DOI] [PubMed] [Google Scholar]
- 13.Burns RC, Rivera-Nieves J, Moskaluk CA, Matsumoto S, Cominelli F, Ley K. Antibody blockade of ICAM-1 and VCAM-1 ameliorates inflammation in the SAMP-1/Yit adoptive transfer model of Crohn’s disease in mice. Gastroenterology. 2001;121(6):1428–1436. doi: 10.1053/gast.2001.29568. [DOI] [PubMed] [Google Scholar]
- 14.Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, Chieppa M, Arseneau KO, Ley K, Cominelli F. The SAMP1/YitFc mouse strain: a spontaneous model of Crohn’s disease-like ileitis. Inflamm Bowel Dis. 2011;17(12):2566–2584. doi: 10.1002/ibd.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding NS, Hart A, Cruz PD. Systematic review: predicting and optimising response to anti-TNF therapy in Crohn’s disease – algorithm for practical management. Aliment Pharmacol Ther. 2016;43(1):30–51. doi: 10.1111/apt.13445. [DOI] [PubMed] [Google Scholar]
- 16.Bickelhaupt S, Pazahr S, Chuck N, Blume I, Froehlich JM, Cattin R, Raible S, Bouquet H, Bill U, Rogler G, et al. Crohn’s disease: small bowel motility impairment correlates with inflammatory-related markers C-reactive protein and calprotectin. Neurogastroenterol Motili. 2013;25(6):467–e363. doi: 10.1111/nmo.12088. [DOI] [PubMed] [Google Scholar]
- 17.Vandeputte D, Falony G, Vieira-Silva S, Tito RY, Joossens M, Raes J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut. 2016;65(1):57–62. doi: 10.1136/gutjnl-2015-309618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anitha M, Reichardt F, Tabatabavakili S, Nezami BG, Chassaing B, Mwangi S, Vijay-Kumar M, Gewirtz A, Srinivasan S. Intestinal dysbiosis contributes to the delayed gastrointestinal transit in high-fat diet fed mice. Cell Mol Gastroenterol Hepatol. 2016;2(3):328–339. doi: 10.1016/j.jcmgh.2015.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esfandyari T, Camilleri M, Ferber I, Burton D, Baxter K, Zinsmeister AR. Effect of a cannabinoid agonist on gastrointestinal transit and postprandial satiation in healthy human subjects: a randomized, placebo-controlled study. Neurogastroenterol Motili. 2006;18(9):831–838. doi: 10.1111/j.1365-2982.2006.00834.x. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, Chan AC, Zhou Z, Huang BX, Kim H-Y, et al. A biosynthetic pathway for anandamide. Proc Natl Acad Sci. 2006;103(36):13345–13350. doi: 10.1073/pnas.0601832103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, Wang L, Harvey-White J, Huang BX, Kim H-Y, Luquet S, Palmiter RD, Krystal G, Rai R, Mahadevan A, et al. Multiple Pathways Involved in the Biosynthesis of Anandamide. Neuropharmacology. 2008;54(1):1–7. doi: 10.1016/j.neuropharm.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vrakas S, Mountzouris KC, Michalopoulos G, Karamanolis G, Papatheodoridis G, Tzathas C, Gazouli M. Intestinal bacteria composition and translocation of bacteria in inflammatory bowel disease. PLoS One. 2017;12(1):e0170034. doi: 10.1371/journal.pone.0170034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swidsinski A, Ung V, Sydora BC, Loening-Baucke V, Doerffel Y, Verstraelen H, Fedorak RN. Bacterial overgrowth and inflammation of small intestine after carboxymethylcellulose ingestion in genetically susceptible mice. Inflamm Bowel Dis. 2009;15(3):359–364. doi: 10.1002/ibd.20763. [DOI] [PubMed] [Google Scholar]
- 24.Hernández GA, Appleyard CB. Bacterial load in animal models of acute and chronic ‘reactivated’ colitis. Digestion. 2003;67(3):161–169. doi: 10.1159/000071296. [DOI] [PubMed] [Google Scholar]
- 25.Quince C, Ijaz UZ, Loman N, Eren AM, Saulnier D, Russell J, Haig SJ, Calus ST, Quick J, Barclay A, et al. Extensive modulation of the fecal metagenome in children with crohn’s disease during exclusive enteral nutrition. Am J Gastroenterol. 2015;110(12):1718–1729. doi: 10.1038/ajg.2015.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walters WA, Xu Z, Knight R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 2014;588(22):4223–4233. doi: 10.1016/j.febslet.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagao-Kitamoto H, Shreiner AB, Gillilland MG, Kitamoto S, Ishii C, Hirayama A, Kuffa P, El-Zaatari M, Grasberger H, Seekatz AM, et al. Functional characterization of inflammatory bowel disease–associated gut dysbiosis in gnotobiotic mice. Cell Mol Gastroenterol Hepatol. 2016;2(4):468–481. doi: 10.1016/j.jcmgh.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaubeck M, Clavel T, Calasan J, Lagkouvardos I, Haange SB, Jehmlich N, Basic M, Dupont A, Hornef M, von Bergen M, et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut. 2016;65(2):225–237. doi: 10.1136/gutjnl-2015-309333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodriguez-Palacios A, Harding A, Menghini P, Himmelman C, Retuerto M, Nickerson KP, Lam M, Croniger CM, McLean MH, Durum SK, et al. The artificial sweetener splenda promotes gut proteobacteria, dysbiosis, and myeloperoxidase reactivity in crohn’s disease–like ileitis. Inflamm Bowel Dis. 2018;24(5):1005–1020. doi: 10.1093/ibd/izy060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mosca A, Leclerc M, Hugot JP. Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem? Front Microbiol. 2016;7. doi: 10.3389/fmicb.2016.00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langdon A, Crook N, Dantas G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016;8. doi: 10.1186/s13073-016-0294-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alipour M, Zaidi D, Valcheva R, Jovel J, Martínez I, Sergi C, Walter J, Mason AL, Wong GK-S, Dieleman LA, et al. Mucosal barrier depletion and loss of bacterial diversity are primary abnormalities in paediatric ulcerative colitis. J Crohn’s Colitis. 2016;10(4):462–471. doi: 10.1093/ecco-jcc/jjv223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tedjo DI, Smolinska A, Savelkoul PH, Masclee AA, van Schooten FJ, Pierik MJ, Penders J, Jonkers DMAE. The fecal microbiota as a biomarker for disease activity in Crohn’s disease. Sci Rep. 2016;6:35216. doi: 10.1038/srep35216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Wu Y, Wang J, Wu G, Long W, Xue Z, Wang L, Zhang X, Pang X, Zhao Y, et al. Accelerated dysbiosis of gut microbiota during aggravation of DSS-induced colitis by a butyrate-producing bacterium. Sci Rep. 2016;6:27572. doi: 10.1038/srep27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reinoso Webb C, Den Bakker H, Koboziev I, Jones-Hall Y, Rao Kottapalli K, Ostanin D, Furr KL, Mu Q, Luo XM, Grisham MB. Differential susceptibility to T cell-induced colitis in mice: role of the intestinal microbiota. Inflamm Bowel Dis. 2018;24(2):361–379. doi: 10.1093/ibd/izx014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garzetti D, Brugiroux S, Bunk B, Pukall R, McCoy KD, Macpherson AJ, Stecher B. High-Quality Whole-Genome Sequences of the Oligo-Mouse-Microbiota Bacterial Community. Genome Announc. 2017;5(42). doi: 10.1128/genomeA.00758-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rooks MG, Veiga P, Wardwell-Scott LH, Tickle T, Segata N, Michaud M, Gallini CA, Beal C, van Hylckama-Vlieg JE, Ballal SA, et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J. 2014;8(7):1403–1417. doi: 10.1038/ismej.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roulis M, Bongers G, Armaka M, Salviano T, He Z, Singh A, Seidler U, Becker C, Demengeot J, Furtado GC, et al. Host and microbiota interactions are critical for development of murine Crohn’s-like ileitis. Mucosal Immunol. 2016;9(3):787–797. doi: 10.1038/mi.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borton MA, Sabag-Daigle A, Wu J, Solden LM, O’Banion BS, Daly RA, Wolfe RA, Gonzalez JF, Wysocki VH, Ahmer BMM, et al. Chemical and pathogen-induced inflammation disrupt the murine intestinal microbiome. Microbiome. 2017;5. doi: 10.1186/s40168-017-0264-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park J-S, Choi JW, Jhun J, Kwon JY, Lee B-I, Yang CW, Park S-H, Cho M-L. Lactobacillus acidophilus improves intestinal inflammation in an acute colitis mouse model by regulation of Th17 and treg cell balance and fibrosis development. J Med Food. 2018;21(3):215–224. doi: 10.1089/jmf.2017.3990. [DOI] [PubMed] [Google Scholar]
- 42.Yokota Y, Shikano A, Kuda T, Takei M, Takahashi H, Kimura B. Lactobacillus plantarum AN1 cells increase caecal L. reuteri in an ICR mouse model of dextran sodium sulphate-induced inflammatory bowel disease. Int Immunopharmacol. 2018;56:119–127. doi: 10.1016/j.intimp.2018.01.020. [DOI] [PubMed] [Google Scholar]
- 43.Anderson RC, Ulluwishewa D, Young W, Ryan LJ, Henderson G, Meijerink M, Maier E, Wells JM, Roy NC. Human oral isolate Lactobacillus fermentum AGR1487 induces a pro-inflammatory response in germ-free rat colons. Sci Rep. 2016;6:20318. doi: 10.1038/srep20318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loubinoux J, Bronowicki J-P, Pereira IAC, Mougenel J-L, Faou LEA. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol Ecol. 2002;40(2):107–112. doi: 10.1111/j.1574-6941.2002.tb00942.x. [DOI] [PubMed] [Google Scholar]
- 45.Fite A. Identification and quantitation of mucosal and faecal desulfovibrios using real time polymerase chain reaction. Gut. 2004;53(4):523–529. doi: 10.1136/gut.2003.031245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roediger WEW, Duncan A, Kapaniris O, Millard S. Reducing sulfur compounds of the colon impair colonocyte nutrition: implications for ulcerative colitis. Gastroenterology. 1993;104(3):802–809. doi: 10.1016/0016-5085(93)91016-B. [DOI] [PubMed] [Google Scholar]
- 47.Haller D. Author’s response. Gut. 2012;61(2):324. doi: 10.1136/gutjnl-2011-300227.21450695 [DOI] [Google Scholar]
- 48.Sharpton T, Lyalina S, Luong J, Pham J, Deal EM, Armour C, Gaulke C, Sanjabi S, Pollard KS. Development of inflammatory bowel disease is linked to a longitudinal restructuring of the Gut Metagenome in mice. mSystems. 2017;2(5):e00036–17. doi: 10.1128/mSystems.00036-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwab C, Berry D, Rauch I, Rennisch I, Ramesmayer J, Hainzl E, Heider S, Decker T, Kenner L, Müller M, et al. Longitudinal study of murine microbiota activity and interactions with the host during acute inflammation and recovery. ISME J. 2014;8(5):1101–1114. doi: 10.1038/ismej.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palmela C, Chevarin C, Xu Z, Torres J, Sevrin G, Hirten R, Barnich N, Ng SC, Colombel J-F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut. 2018;67(3):574–587. doi: 10.1136/gutjnl-2017-314903. [DOI] [PubMed] [Google Scholar]
- 51.Tchaptchet S, Fan T-J, Goeser L, Schoenborn A, Gulati AS, Sartor RB, Hansen JJ. Inflammation-induced acid tolerance genes gadAB in luminal commensal escherichia coli attenuate experimental colitis. Infect Immun. 2013;81(10):3662–3671. doi: 10.1128/IAI.00355-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saitoh S, Noda S, Aiba Y, Takagi A, Sakamoto M, Benno Y, Koga Y. Bacteroides ovatus as the predominant commensal intestinal microbe causing a systemic antibody response in inflammatory bowel disease. Clin Diagn Lab Immunol. 2002;9(1):54–59. doi: 10.1128/CDLI.9.1.54-59.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imhann F, Vich Vila A, Bonder MJ, Fu J, Gevers D, Visschedijk MC, Spekhorst LM, Alberts R, Franke L, van Dullemen HM, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2018;67(1):108–119. doi: 10.1136/gutjnl-2016-312135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zitomersky NL, Atkinson BJ, Franklin SW, Mitchell PD, Snapper SB, Comstock LE, Bousvaros A. Characterization of adherent bacteroidales from intestinal biopsies of children and young adults with inflammatory bowel disease. PLoS One. 2013;8(6):e63686. doi: 10.1371/journal.pone.0063686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kverka M, Zakostelska Z, Klimesova K, Sokol D, Hudcovic T, Hrncir T, Rossmann P, Mrazek J, Kopecny J, Verdu EF, et al. Oral administration of parabacteroides distasonis antigens attenuates experimental murine colitis through modulation of immunity and microbiota composition. Clin Exp Immunol. 2011;163(2):250–259. doi: 10.1111/j.1365-2249.2010.04286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jacobs JP, Goudarzi M, Singh N, Tong M, McHardy IH, Ruegger P, Asadourian M, Moon B-H, Ayson A, Borneman J, et al. A disease-associated microbial and metabolomics state in relatives of pediatric inflammatory bowel disease patients. Cell Mol Gastroenterol Hepatol. 2016;2(6):750–766. doi: 10.1016/j.jcmgh.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife. 2013;2. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Young NM, Foote SJ, Wakarchuk WW. Review of phosphocholine substituents on bacterial pathogen glycans: synthesis, structures and interactions with host proteins. Mol Immunol. 2013;56(4):563–573. doi: 10.1016/j.molimm.2013.05.237. [DOI] [PubMed] [Google Scholar]
- 59.Moustafa A, Li W, Anderson EL, Wong EHM, Dulai PS, Sandborn WJ, Biggs W, Yooseph S, Jones MB, Venter JC, et al. Genetic risk, dysbiosis, and treatment stratification using host genome and gut microbiome in inflammatory bowel disease. Clin Transl Gastroenterol. 2018;9(1):e132. doi: 10.1038/ctg.2017.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, Laughlin RC, Gomez G, Wu J, Lawhon SD, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science (New York, N.Y.). 2013;339(6120):708–711. doi: 10.1126/science.1232467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schumann S, Alpert C, Engst W, Loh G, Blaut M. Dextran sodium sulfate-induced inflammation alters the expression of proteins by intestinal escherichia coli strains in a gnotobiotic mouse model. Appl Environ Microbiol. 2012;78(5):1513–1522. doi: 10.1128/AEM.07340-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dey N, Wagner VE, Blanton LV, Cheng J, Fontana L, Haque R, Ahmed T, Gordon JI. Regulators of gut motility revealed by a gnotobiotic model of diet-microbiome interactions related to travel. Cell. 2015;163(1):95–107. doi: 10.1016/j.cell.2015.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stoddard SF, Smith BJ, Hein R, Roller BRK, Schmidt TM. rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015;43(D1):D593–D598. doi: 10.1093/nar/gku1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Comeau AM, Douglas GM, Langille MGI. Microbiome helper: a custom and streamlined workflow for microbiome research. mSystems. 2017;2(1):e00127–16. doi: 10.1128/mSystems.00127-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14(6):927–930. doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- 70.Xia Y, Sun J. Hypothesis testing and statistical analysis of microbiome. Genes Dis. 2017;4(3):138–148. doi: 10.1016/j.gendis.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Hara RB, Kotze DJ. Do not log-transform count data. Methods in Ecol Evol. 2010;1(2):118–122. doi: 10.1111/j.2041-210X.2010.00021.x. [DOI] [Google Scholar]
- 72.Costea PI, Zeller G, Sunagawa S, Bork P. A fair comparison. Nat Methods. 2014;11(4):359. doi: 10.1038/nmeth.2897. [DOI] [PubMed] [Google Scholar]
- 73.Lamoureux EV, Grandy SA, Langille MGI. Moderate Exercise Has Limited but Distinguishable Effects on the Mouse Microbiome. mSystems. 2017;2(4). doi: 10.1128/mSystems.00006-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen J, Chia N, Kalari KR, Yao JZ, Novotna M, Soldan MMP, Luckey DH, Marietta EV, Jeraldo PR, Chen X, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep. 2016;6:28484. doi: 10.1038/srep28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shaw KA, Bertha M, Hofmekler T, Chopra P, Vatanen T, Srivatsa A, Prince J, Kumar A, Sauer C, Zwick ME, et al. Dysbiosis, inflammation, and response to treatment: a longitudinal study of pediatric subjects with newly diagnosed inflammatory bowel disease. Genome Med. 2016;8. doi: 10.1186/s13073-016-0331-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Papa E, Docktor M, Smillie C, Weber S, Preheim SP, Gevers D, Giannoukos G, Ciulla D, Tabbaa D, Ingram J, et al. Non-invasive mapping of the gastrointestinal microbiota identifies children with inflammatory bowel disease. PLoS One. 2012;7(6):e39242. doi: 10.1371/journal.pone.0039242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, et al. Inflammation, antibiotics, and diet as environmental stressors of the Gut microbiome in pediatric crohn’s disease. Cell Host Microbe. 2015;18(4):489–500. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 79.Kuhn M. Building predictive models in R using the caret package. J Stat Softw. 2008;28(5):1–26. doi: 10.18637/jss.v028.i05.27774042 [DOI] [Google Scholar]
- 80.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao G, Nyman M, Jönsson JÅ. Rapid determination of short-chain fatty acids in colonic contents and faeces of humans and rats by acidified water-extraction and direct-injection gas chromatography. Biomed Chromatogr. 2006;20(8):674–682. doi: 10.1002/bmc.580. [DOI] [PubMed] [Google Scholar]
- 83.Kang SS, Bloom SM, Norian LA, Geske MJ, Flavell RA, Stappenbeck TS, Allen PM. An antibiotic-responsive mouse model of fulminant ulcerative colitis. PLoS Med. 2008;5(3):e41. doi: 10.1371/journal.pmed.0050041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.