

ABSTRACT
IgA antibodies have broad potential as a novel therapeutic platform based on their superior receptor-mediated cytotoxic activity, potent neutralization of pathogens, and ability to transcytose across mucosal barriers via polymeric immunoglobulin receptor (pIgR)-mediated transport, compared to traditional IgG-based drugs. However, the transition of IgA into clinical development has been challenged by complex expression and characterization, as well as rapid serum clearance that is thought to be mediated by glycan receptor scavenging of recombinantly produced IgA monomer bearing incompletely sialylated N-linked glycans. Here, we present a comprehensive biochemical, biophysical, and structural characterization of recombinantly produced monomeric, dimeric and polymeric human IgA. We further explore two strategies to overcome the rapid serum clearance of polymeric IgA: removal of all N-linked glycosylation sites creating an aglycosylated polymeric IgA and engineering in FcRn binding with the generation of a polymeric IgG-IgA Fc fusion. While previous reports and the results presented in this study indicate that glycan-mediated clearance plays a major role for monomeric IgA, systemic clearance of polymeric IgA in mice is predominantly controlled by mechanisms other than glycan receptor clearance, such as pIgR-mediated transcytosis. The developed IgA platform now provides the potential to specifically target pIgR expressing tissues, while maintaining low systemic exposure.
KEYWORDS: Asialoglycoprotein receptor (ASGPR), N-linked glycan, polymeric IgG receptor (pIgR), serum half-life, transcytosis
Introduction
A recent renaissance in immunoglobulin A (IgA) research has highlighted multiple potential therapeutic applications and unique mechanisms of action for both monomeric and polymeric immunoglobulin A (IgA) antibodies compared to the traditional IgG-based therapeutics.1–3 In oncology, monomeric and polymeric anti-epidermal growth factor receptor (EGFR) and anti-CD20 IgAs have demonstrated superior tumor cell killing compared to IgG, driven by FcαRI-mediated cytotoxicity or more effective receptor binding and downmodulation.4–6 The cytotoxic activity of IgA could be further increased via dual engagement of both FcγR and FcαRI by IgG/A fusion or hybrid molecules.7,8 For infectious disease, IgA multivalent target engagement enabled superior antigen binding and neutralization in influenza infection models.9 Additionally, human IgA dimer (dIgA) could be effectively delivered to the kidney lumen in a polycystic kidney disease mouse model via binding to the polymeric immunoglobulin receptor (pIgR), whereas IgG molecules could not.10 Harnessing the specific transcytosis activity of IgA could potentially allow access to therapeutic targets within the luminal side of mucosal tissues that are inefficiently targeted by current IgG therapeutics.2,10,11
Contrary to other human immunoglobulin classes, IgA has the unique ability to naturally exist as both monomeric and polymeric soluble species, whereas only polymeric IgA can bind to pIgR for subsequent transcytosis.1 Oligomerization of IgA is facilitated by an 18-residue C-terminal extension of the heavy chain (HC) called the tailpiece and the 137-amino acid joining chain (JC). The penultimate residue of the IgA tailpiece, Cys471, of the first IgA monomer mediates disulfide bond formation with Cys15 of the JC, while Cys471 of the second IgA monomer mediates disulfide bond formation with Cys69 of the JC (Figure 1A,B) to form a covalent IgA dimer that is held together by a single JC.14,15 As each IgA monomer is composed of two HCs, each with a tailpiece, the IgA dimer has two unpaired Cys471 residues through which additional IgA monomers could be linked. Indeed, higher order IgA oligomers such as trimers, tetramers, and pentamers have been reported.9 Whereas serum IgA is predominantly monomeric, polymeric IgAs are produced by plasma cells in the lamina propria.1 The presence of the JC in polymeric IgA is required for binding pIgR on the basolateral side of the epithelium and for active transport to the apical side of mucosal tissues.16 Upon transcytosis, the extracellular domain of pIgR is proteolytically cleaved, creating what is known as the secretory component (SC), which remains covalently attached to the polymeric IgA heavy chain through a disulfide bond between Cys467 in pIgR and Cys311 in one HC.17,18 This complex is deemed secretory IgA (sIgA), the main determinant of mucosal immunity.19,20
Figure 1.
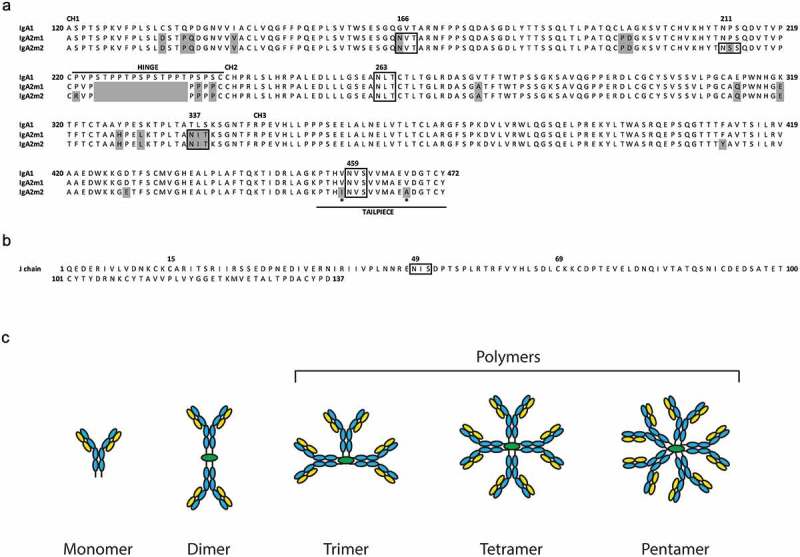
Protein sequences of human IgA heavy chain constant domains and J chain.
(A) Alignment of protein sequences for the human heavy chain constant domains CH1, CH2, CH3, hinge,12 and tailpiece of IgA1, IgA2m1 and IgA2m2.13 Mismatches relative to the IgA1 sequence are highlighted gray, N-linked glycosylation motifs are boxed, and asterisks indicate amino acid differences in IgA2m2 from IgA1 and IgA2m1 in the tailpiece. (B) Protein sequence of the human J chain with the N-linked glycosylation motif boxed. (C) Schematic of IgA oligomeric states with light chain (LC, yellow), heavy chain (HC, blue), and joining chain (JC, green). IgA polymers represent trimer, tetramer, and pentamer species.
In humans, there are two IgA isotypes, IgA1 and IgA2, distinguished by a 13-residue extension in the hinge region of the IgA1 HC that is absent in IgA2 molecules.3 Both isotypes are abundant in all organs and tissues, except in the intestines where IgA2 is predominant and in the serum where IgA1 monomer is found almost exclusively.21 There are three allotypes of IgA2: m1,22 m213 and mn.23 The m2 and mn allotypes form canonical light chain (LC)-HC disulfides, whereas the presence of a proline at position 221 of the HC in IgA2m1 results in LC-LC disulfide bond formation instead.24 Mutation of proline 221 in the IgA2m1 allotype to arginine (P221R), which is found in the m2 and mn allotypes, restores the canonical LC-HC linkage.6,24 Sequence identity between the IgA1 and IgA2 isotypes is quite high (~90%) and even higher amongst the IgA2 allotypes, with only six residue differences between m1 and m2 and two residue differences between either m1 or m2 with mn.23
Production of recombinant monomeric IgA is more challenging than that of the well-established IgG molecule, as IgA typically suffers from poor expression and heterogeneous glycosylation. For example, transient expression levels for monomeric IgA have been reported for human IgA1 or IgA2m1 isotypes at 30–70 µg/L.25 To overcome these low production yields, stable cell lines have increased expression levels of monomeric IgA1, IgA2m1 and IgA2m2 to 110–190 mg/L.26 Whereas human IgG1 typically has only two N-linked glycosylation sites, one in each CH2 domain, human IgA contains multiple glycosylation sites that can be susceptible to glycan heterogeneity.3 IgA1 has multiple O-linked glycosylation sites in the hinge region and also two N-linked glycosylation sites in the HC constant domain. While IgA2 molecules are not modified by O-linked glycans, they do contain either four (IgA2m1) or five (IgA2m2 and IgA2mn) N-linked glycosylation sites (Figure 1A, boxed)1,2, and the JC also contains one N-linked glycosylation site (Figure 1B, boxed). Assembly of the three polypeptide chains (LC, HC, and JC) leads to multiple oligomeric states and further contributes to the overall complexity of recombinant polymeric IgA (Figure 1C).26,27 With the increasing size of an IgA oligomer comes not only an increased number of glycosylation sites but also the potential for more glycan heterogeneity. To date, most studies have focused on recombinant monomeric IgA, likely due to the added complexities of polymeric IgA production.
IgA has previously been shown to have a short circulating half-life (<1 day to ~4 days) in multiple species.3,28 This is not surprising given that, unlike IgG, IgA does not bind the neonatal receptor, FcRn, and therefore, cannot undergo endosomal recycling and escape from lysosomal degradation.29 In addition to the lack of FcRn binding, immature N-linked glycans can also contribute to shorter serum half-lives of recombinant IgA by making them susceptible targets of carbohydrate-specific, endocytic receptors such as the asialoglycoprotein receptor (ASGPR)5,30 and mannose receptor.31,32 These scavenging receptors, which are highly concentrated in the liver, recognize glycoproteins bearing incompletely sialylated N-linked glycans and remove them from circulation.33,34
To investigate the therapeutic potential of recombinant IgA oligomers, we expressed, purified, and characterized both dimeric and tetrameric IgA molecules. We confirmed their functional properties by measuring binding to antigen, FcαRI and pIgR, as well as by demonstrating their transcytosis capability in vitro. However, in vivo we observed that our polymeric IgA antibodies demonstrated fast serum clearance in mice. Other studies have reported rapid clearance of recombinant monomeric IgA molecules, and successful approaches to overcome rapid catabolism include removal of N-linked glycosylation sites through mutagenesis,6 increasing sialylation of the N-linked glycans by strain engineering,26 and FcRn-mediated half-life extension strategies.7,25,35 As previous studies have mainly focused on monomeric IgA, we applied two different approaches to overcome rapid serum clearance of polymeric IgA in mice. We engineered an aglycosylated polymeric IgA2m2, as well as a dimeric IgG1-IgA2m1 Fc fusion. Both formats bound antigen and pIgR, were able to transcytose in vitro and successfully demonstrated increased serum exposure in mice, with the IgG1-IgA2m1 Fc fusion showing the most improved exposure. To our knowledge, this is the first report of an IgA oligomer that has been engineered to exhibit improved pharmacokinetic (PK) properties in mice.
Results
Analysis of factors affecting IgA oligomer formation
Recombinant production of monomeric IgA is well understood and can be achieved by coexpression of LC and HC, similar to the production of IgG. In contrast, the assembly of polymeric IgA requires coexpression of LC, HC, and JC, and the resulting IgA oligomeric states are less well characterized. To gain a better understanding of the assembly process of IgA oligomers, we characterized the expression of various human IgA isotypes and allotypes, including IgA1, IgA2m1, IgA2m1.P221R (disulfide stabilized LC-HC pairing)36 and IgA2m2 (Figure 1A). The murine variable domains of an anti-mouse interleukin-13 (mIL-13) antibody were cloned as chimeras with the human kappa LC and IgA HC constant domains. The chimeric LCs and HCs were then coexpressed in the presence and absence of the human JC (Figure 1B). After affinity purification with Protein L, IgA produced in the absence of cotransfected JC yielded relatively pure monomer from 30 mL Expi293T transient expressions (data not shown). In these experiments, cells were transfected with equal mass quantities of LC and HC DNA. In contrast, transfection of equal mass quantities of LC, HC and JC DNA produced a variety of oligomeric species, corresponding to IgA monomer, dimer, and polymer that contains three to five IgA monomers (Figures 1C and Figure 2A–C). IgA1, IgA2m1, and IgA2m1.P221R were found to produce predominantly dimeric IgA (Figure 2A,B and data not shown, respectively), while IgA2m2 produced roughly equal amounts of dimer and polymer (Figure 2C). A similar distribution of oligomers was observed in Chinese hamster ovary (CHO) transient expressions upon scale up to the liter scale (data not shown). Separating IgA dimer from polymer by secondary purification proved challenging at the larger scale. In an attempt to bias assembly towards dimer formation, we increased the amount of JC DNA relative to both LC and HC DNA amounts to promote increased JC expression levels. This resulted in a decrease in the relative percentage of polymer species (Figure 2A-C). Conversely, decreasing the amount of JC DNA relative to both LC and HC DNA amounts resulted in an increased percentage of higher order polymer (trimer/tetramer/pentamer). The ability to influence oligomeric species based upon the JC DNA amount was most pronounced for the IgA2m2 species.
Figure 2.
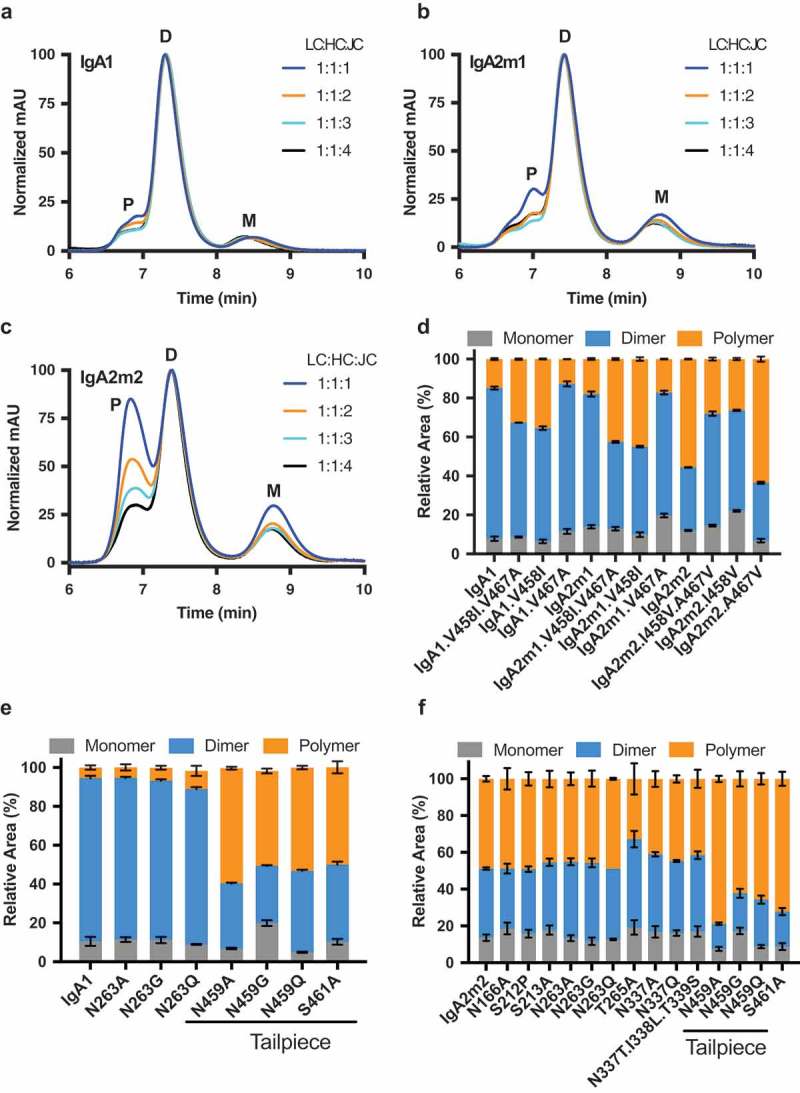
The oligomeric state of recombinantly produced IgA is affected by the amount of J chain DNA used in transfection and the heavy chain tailpiece sequence.
(A-C) Overlay of normalized analytical size-exclusion chromatograms of affinity-purified IgA from small-scale transient transfections performed with varying ratios of light chain (LC), heavy chain (HC), and joining chain (JC) DNA for the following isotypes/allotypes: (A) IgA1 (B) IgA2m1 or (C) IgA2m2. Monomer (M), dimer (D), and polymer (P) peaks are indicated. Values were normalized based on the highest signal of each chromatogram. (D-F) Relative amounts of monomer, dimer, and polymer (trimer/tetramer/pentamer) species produced for IgA variants, quantified by analytical SEC. (D) The effect of mutations in the IgA tailpiece of IgA1, IgA2m1, and IgA2m2 at positions 458 and 467 on polymer formation. The effect of mutations which remove N-linked glycosylation sites in (E) IgA1 or (F) IgA2m2 on polymer formation.
To understand why IgA2m2 has a higher propensity to form larger oligomers than IgA1 or IgA2m1, we compared the amino acid sequences of the HC tailpieces for the different isotypes/allotypes. While the sequences of the IgA1 and IgA2m1 tailpieces are identical, IgA2m2 differs by two residues. Residues 458 and 467 are both valines in IgA1 and IgA2m1, whereas IgA2m2 has an isoleucine and alanine at these positions, respectively (Figure 1A, asterisks). Therefore, we investigated whether these two amino acid differences could explain the unique predisposition of recombinant IgA2m2 to form larger oligomers. Indeed, when isoleucine was substituted for valine at position 458 in IgA1 or IgA2m1, more polymer was produced, and this was independent of alanine or valine at residue 467 (Figure 2D). Conversely, when position 458 in IgA2m2 was changed from isoleucine to valine, the content of polymeric species was reduced in favor of increased dimer content. Glycosylation is also known to play a role in oligomerization,37 so we made mutations to remove each N-linked glycosylation site in IgA1 and IgA2m2. Four separate mutations (N459A/G/Q or S461A) that removed the N-linked glycosylation site in the tailpiece of IgA1 or IgA2m2 also increased the amount of polymer produced, while mutations to remove glycosylation sites outside the tailpiece did not alter oligomer formation (Figure 2E and F, respectively). Therefore, in addition to modulating the DNA ratios in transfection, IgA polymer formation can be increased by having isoleucine at tailpiece residue 458 or preventing N-linked glycosylation of the IgA tailpiece.
Large-scale purification and biophysical characterization of IgA monomers and oligomers
Using insights into IgA oligomer formation gained through small-scale expression, we scaled up monomeric, dimeric and tetrameric IgA using CHO transient expression. We isolated the monomeric and dimeric species of IgA1, IgA2m1, IgA2m1.P221R, and IgA2m2, as well as the tetrameric species of IgA2m2 (Figure 3A). Non-reduced sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of these samples shows predominant bands of molecular weights consistent with the expected masses of ~150 kDa, ~310 kDa, and ~610 kDa for an IgA monomer, dimer, and tetramer, respectively (Figure 3B). These expected masses are based on the amino acid sequence without glycosylation and assume incorporation of one JC per oligomer. Molar masses of the purified oligomeric species were also measured by size-exclusion chromatography-multi-angle light scattering (SEC-MALS) and found to be consistent with the expected masses of dimeric and tetrameric IgA (Table S1). The reduced SDS-PAGE analysis of the purified IgA samples confirmed the presence of LC and HC bands for monomers at ~25 kDa and ~55 kDa, respectively, whereas in the oligomeric samples a band for JC just below 25 kDa was also detected (Figure 3B). The identity of the LC, HC, and JC were additionally confirmed by mass spectrometry after reduction and enzymatic deglycosylation (data not shown). Negative stain electron microscopy (EM) was also used to further validate the oligomeric state of the isolated species. Negative stain images of the IgA2m2 dimer (Figure 3C) and tetramer (Figure 3D) confirm the presence of two or predominantly four IgA molecules, respectively. In the dimer, two IgA molecules are linked tail-to-tail by their Fc domains into an elongated particle, whereas in the tetramer, interactions between four Fc domains give rise to a compact complex of four IgA molecules. Raw images of both samples showed the presence of well-behaved, monodisperse particles (Figure S1).
Figure 3.
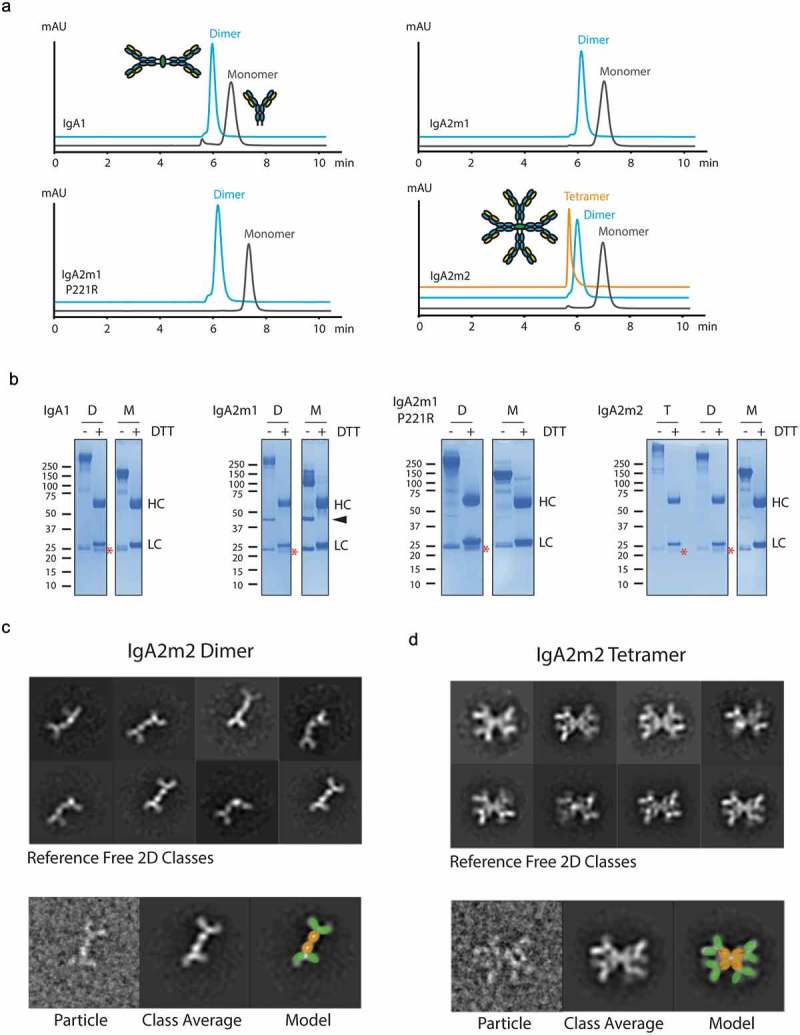
Biophysical and structural characterization of recombinant IgA oligomers.
(A) Overlay of analytical size-exclusion chromatograms of purified IgA1, IgA2m1, IgA2m1 P221R, and IgA2m2 monomers, dimers, and tetramer. (B) SDS-PAGE analysis of non-reduced (-DTT) and reduced (+DTT) IgA1, IgA2m1, IgA2m1 P221R, and IgA2m2 monomers (M), dimers (D) and tetramer (T). Heavy chain (HC), light chain (LC), and joining chain (*) are indicated in reduced samples, and the LC-LC dimer of IgA2m1 is indicated with an arrowhead. (C-D, upper panels) Reference-free 2D classes from negative stain electron microscopy for (C) IgA2m2 dimer or (D) IgA2m2 tetramer. (C-D, lower panels) A raw image particle compared to its assigned 2D class is presented next to a model of IgA superimposed on the 2D class with the Fc domains (orange) and Fab fragments (green) highlighted.
The anti-mIL-13 IgA monomers, dimers, and tetramer bound murine IL-13 with similar affinity as the anti-mIL-13 IgG1 (Table 1), indicating that regions of the antigen-binding fragment (Fab) are properly folded and functional in the recombinant IgAs. As expected, mouse and human pIgR binding was only seen for the IgA oligomers, while both monomeric and oligomeric anti-mIL-13 IgA bound with similar affinity to human FcαRI (Table 1). Due to pIgR binding capabilities, all IgA oligomers, but not monomers, were capable of transcytosis in vitro using a Madin-Darby canine kidney (MDCK) cell line ectopically expressing human pIgR (Figures 4A and 7E). Additionally, the IgA monomers and oligomers all showed increased stability compared to the anti-mIL-13 human IgG1 and similar or increased stability compared to the IgG1 Fab, as measured by differential scanning fluorimetry (DSF) (Figure 4B).
Table 1.
Binding affinity of anti-mouse IL-13 IgA and IgG molecules to antigen and receptors.
| KD (nM) |
||||
|---|---|---|---|---|
| Mouse IL-13 | Mouse pIgR | Human pIgR | Human FcαRI | |
| IgA1 Monomer | 0.78 ± 0.05 | NB | NB | 425 ± 7 |
| IgA2m1 Monomer | 1.09 ± 0.03 | NB | NB | 429 ± 6 |
| IgA2m1 P221R Monomer | 1.16 ± 0.01 | NB | NB | 443 ± 8 |
| IgA2m2 Monomer | 0.70 ± 0.01 | NB | NB | 455 ± 5 |
| IgA1 Dimer | 0.34 ± 0.01 | 2.66 ± 1.45 | 8.80 ± 0.55 | 369 ± 3 |
| IgA2m1 Dimer | 0.18 ± 0.01 | 2.54 ± 0.15 | 5.05 ± 0.88 | 499 ± 7 |
| IgA2m1 P221R Dimer | 0.84 ± 0.01 | 5.59 ± 0.03 | 15.5 ± 0.10 | 462 ± 4 |
| IgA2m2 Dimer | 0.81 ± 0.01 | 2.45 ± 0.98 | 13.9 ± 0.10 | 597 ± 5 |
| IgA2m2 Tetramer | 0.97 ± 0.02 | 0.69 ± 0.05 | 1.93 ± 0.02 | 533 ± 5 |
| IgG1 | 0.88 ± 0.05 | NB | NB | NB |
NB: No Binding. All experiments were performed at least n = 3.
Figure 4.
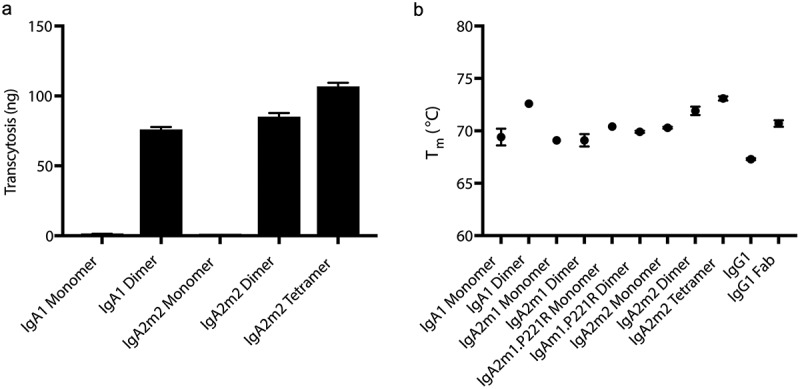
Recombinantly produced IgA oligomers are stable and functional in vitro.
(A) In vitro transcytosis of anti-mIL-13 hIgA monomers, dimers and tetramer in MDCK cells transfected with human pIgR. IgA oligomers transcytose, while monomers do not. (B) Thermostability of anti-mIL-13 IgAs, IgG1, and IgG1 Fab are measured by differential scanning fluorimetry (DSF). Only one melting transition was observed for all samples.
Figure 7.
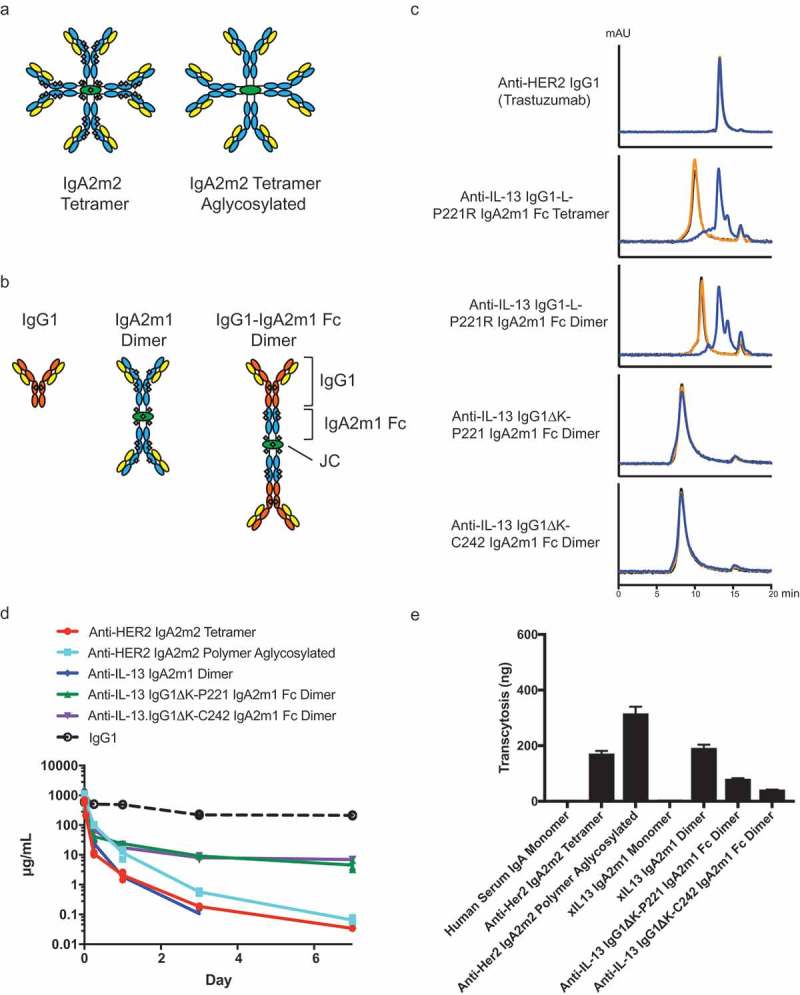
IgG1-IgA2m1 Fc fusions and aglycosylated IgA2m2 show improved serum exposures compared to wild-type IgA in vivo and demonstrate the ability to transcytose in vitro.
(A) Schematic of IgA2m2 tetramer with light chain (LC, yellow), heavy chain (HC, blue) and joining chain (JC, green) with 41 N-linked glycosylation sites (gray) (left) or aglycosylated (right). (B) Schematic of IgG1, IgA2m1 dimer, or IgG1-IgA2m1 Fc dimer formats with LC (yellow), IgG1 HC (orange), IgA2m1 HC (blue), JC (green), and N-linked glycosylation (gray). (C) Analytical SEC of iodinated IgG1-IgA2m1 Fc dimers or tetramer after 0 h (black), 24 h (orange) or 96 h (blue) incubation in mouse plasma. The initial IgG1-L-P221R IgA2m1 Fc tetramer and dimer show degradation similar to the peak of anti-HER2 IgG1 (Trastuzumab) control, whereas the reengineered IgG1K-P221 IgA2m1 Fc or IgG1K-C242 IgA2m1 Fc dimers are stable. (D) Serum-time concentration profiles of IgA or IgG in mice. The overall serum exposures of Balb/c mice administered with a single 30 mg/kg IV dose of IgA molecules. The in-house concentration data of a typical human IgG1 (anti-gD) previously dosed as a single intravenous (IV) injection at 30 mg/kg are shown as a dashed line. All mice were bled retro-orbitally or via cardiac puncture under isoflurane to evaluate serum concentration profile. (E) In vitro transcytosis of hIgA in MDCK cells transfected with human pIgR.
Pharmacokinetic profiles and biodistribution of recombinant IgA
We then studied the serum concentration-time profiles of our recombinant IgA oligomers, and determined those to be comparable to previously reported data using recombinant monomers (Figure 5A and Table 2).5,26,38 Very rapid serum clearance (>200 mL/day/kg) was observed after a single administration of recombinant IgA oligomers. Serum purified human IgA monomer exhibited slower overall clearance, and a serum PK profile generally in line with that previously reported for a highly sialylated IgA monomer.26
Figure 5.
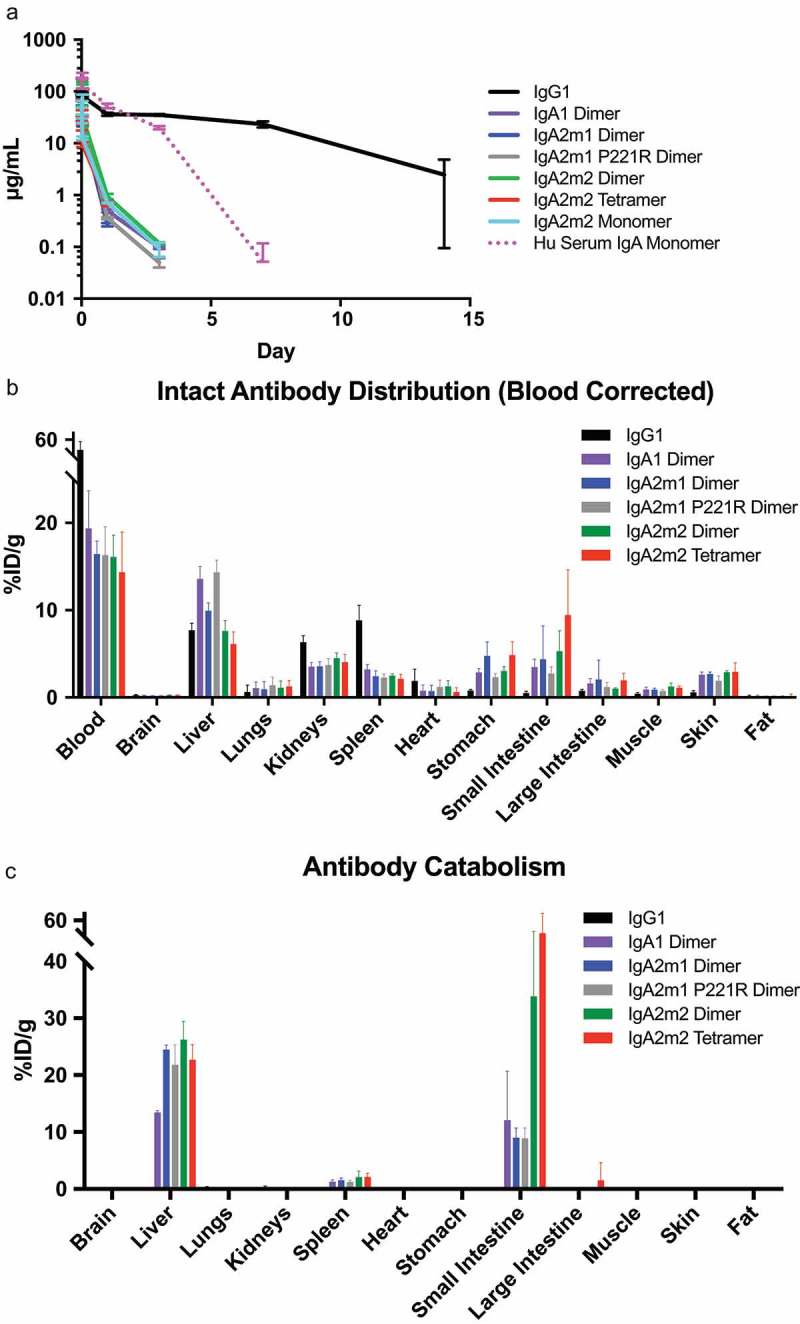
Recombinant IgA oligomers demonstrate rapid serum clearance in vivo.
(A) Serum-time concentration profiles of IgA or IgG in mice. The overall serum exposures of Balb/c mice administered with a single 5 mg/kg intravenous (IV) dose of IgA or IgG molecules at 5 min, 15 min, 30 min, 1 h, 1 day, 3 day, 7, and 14 days post dose. All mice were bled retro-orbitally under isoflurane to evaluate serum concentration profile. Human serum IgA monomer was administered at 10 mg/kg and is shown as a dashed line. (B-C) Tissue distribution of IgA or IgG in mice at 1 h post injection. All graphs are means ± SEM for each group with n = 4. %ID/g: Injected Dose/gram. (B) Concentrations of intact antibodies were subtractive blood normalized per tissue, except blood, as125I (%ID/g tissue). (C) Concentrations of catabolized antibody values were determined by subtracting the125I (%ID/g tissue) from the1111In (%ID/g tissue).
Table 2.
Pharmacokinetic parameter estimates (Mean) after a 5 mg/kg IV bolus of IgA monomers/oligomers to Balb/C mice.
| Cmax (µg/mL) | AUClast (day*µg/mL) | CL (mL/day/kg) | Half Life (Days) | |
|---|---|---|---|---|
| IgA2m2 Monomer | 76.4 | 8.70 | 573 | 0.36 |
| IgA1 Dimer | 85.2 | 14.6 | 341 | 0.31 |
| IgA2m1 Dimer | 92.5 | 13.4 | 373 | 0.26 |
| IgA2m1 P221R Dimer | 108 | 17.6 | 285 | 0.27 |
| IgA2m2 Dimer | 145 | 20.9 | 239 | 0.31 |
| IgA2m2 Tetramer | 60.5 | 6.10 | 788 | 0.22 |
| Human Serum IgA Monomer# | 203 | 203 | 45.7 | 0.97 |
| IgG1 | 100 | 339 | 14.3 | 2.89 |
AUClast = area under the concentration–time curve, last measurable concentration; CL = clearance; Cmax = maximum concentration observed; IV = intravenous; #Dosed 10 mg/kg IV bolus to Balb/c mice. Note: As sparse PK analysis was performed for all mouse PK data, data from individual mice per group (n = 3) was pooled and SD was not reported.
In addition to characterizing the serum concentration-time profiles, we performed a radiolabeled biodistribution study in mice with dual I-125- and In-111-labeled antibodies. The dual tracer approach allowed us to distinguish between intact antibody prior to lysosomal degradation (I-125) and internalized/catabolized antibody (In-111 minus I-125) as previously described (Figure 5B,C).39–41 Briefly, iodine rapidly diffuses out of cells and is cleared after iodinated antibodies undergo lysosomal degradation, while In-111-labeled antibodies show intercellular accumulation of In-111 adducts following lysosomal degradation. Since the IgA antibodies cleared so rapidly compared to the IgG1, direct comparisons of tissue distribution data were difficult since raw tissue values represented both interstitial and vascular concentrations. Therefore, intact antibody distribution data were blood corrected as previously described to represent only interstitial concentrations in tissues.42 We observed slight enrichment of intact IgA oligomers compared to IgG1 after 1 h in the liver, stomach, small intestine, large intestine, and skin (all pIgR-expressing tissues43,44), albeit at low levels (Figure 5B). We also saw high levels of IgA degradation in the liver and small intestine, across the formats studied after 1 h of dosing (Figure 5C).
To account for the difference in total blood concentrations observed between the formats, the ratio of individual tissue to plasma concentrations was also described (Figure S2). After 1 day, we saw almost no intact IgA antibody left in the tissues (Figure S3) and detected the greatest catabolism in the liver (Figure S4), although catabolism in the small intestine may not have been detected as In-111 does not accumulate very well at later time points in intestinal cells.45 We hypothesized that reducing degradation and eventual clearance mechanisms of IgA could further improve uptake of IgA molecules into mucosal tissue compared with IgG.
Sialylation content on the N-linked glycans of monomeric IgA molecules has been reported to negatively correlate with antibody clearance via specific glycan receptors.26 Thus, we analyzed our IgA molecules to determine their overall sialylation content. We classified our glycans into categories based on the level of processing, with complex and sialylated being the most desired for our IgA molecules (Figure 6A). Our recombinantly produced dimers of IgA1, IgA2m1, IgA2m1.P221R, IgA2m2 as well as IgA2m2 monomer and tetramer were about 20–50% sialylated (Figure 6B and Table S2). This indicates that the IgA molecules contain incompletely processed glycans that could be recognized by glycan receptors. Additionally, we examined the sialylation content at each site on the IgA2m1 dimer and found that all sites, including the site on the JC, contained incompletely processed glycans, suggesting the incomplete glycan processing does not occur at only one specific site (Figure 6C and Table S3). In stark contrast to our recombinant IgA molecules, IgA purified from human serum has a sialylation content of 95% (Figure 6B and Table S2), and was monomeric as determined by SEC-MALS (data not shown). As serum IgA is known to be predominantly monomeric,21 it may be enriched for highly sialylated molecules since sialylation content positively correlates with the systemic exposure of antibodies. We hypothesized that this increased sialylation level of the human purified IgA monomer would correlate with decreased serum clearance of the molecule in mice relative to our recombinant IgA monomer. Indeed, we demonstrated this to be true, suggesting that binding to specific glycan receptors in the liver may be an important clearance mechanism for IgA monomer (Figure 5A).
Figure 6.
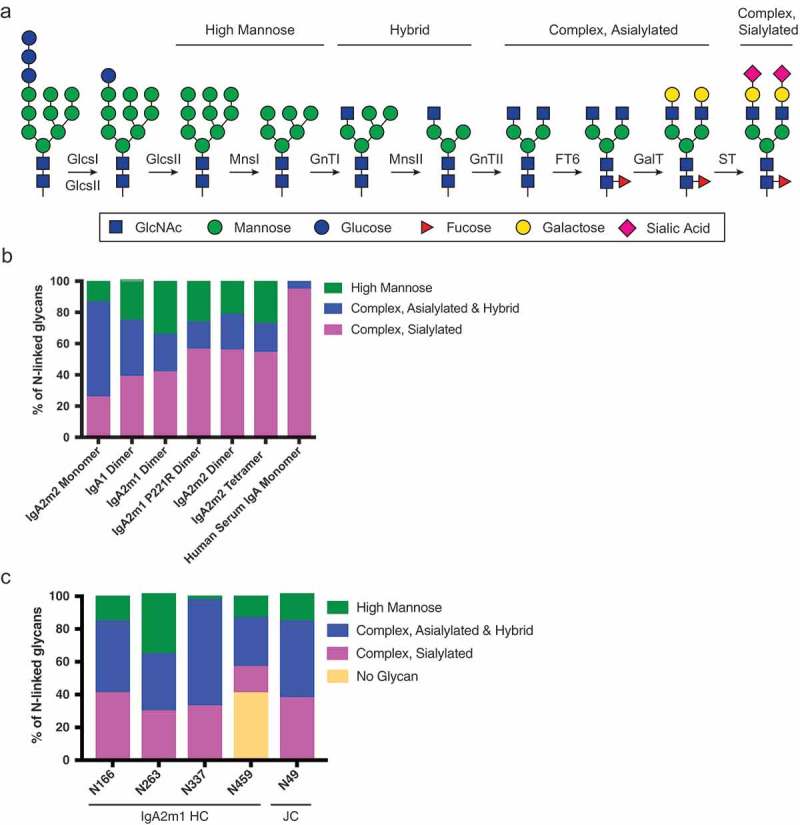
Incomplete glycosylation of recombinant IgA molecules.
(A) Schematic of N-linked glycan processing. (B) Global N-linked glycan analysis of recombinant IgA and IgA purified from human serum. Glycan analysis was done by mass spectrometric analysis after antibody deglycosylation and subsequent glycan enrichment. While human serum IgA shows greater than 90% sialylation, all recombinantly expressed IgA molecules have less than 60% sialylation. (C) Site-specific N-linked glycan analysis of the IgA2m1 dimer reveals heterogenous glycan composition between the different N-linked glycosylation sites on the IgA2m1 heavy chain (HC) and joining chain (JC).
Improving the pharmacokinetic profile of recombinant IgA through glycosylation site and FcRn engineering
We took two parallel approaches to reduce the clearance of recombinant polymeric IgA in mice. First, we removed by mutagenesis all five N-linked glycosylation motifs in IgA2m2 and the single site in the JC to produce a molecule without glycans (aglycosylated) (Figure 7A). An aglycosylated IgA polymer will not be recognized by glycan receptors, which allowed us to study PK independent of glycan receptor-mediated clearance mechanisms. For removal of the N-linked glycosylation motifs N-X-S/T in IgA2m2, we mutated the N to A/G/Q or the S/T to A or reverted this to the non-glycosylated IgA1 sequence in the three instances this occurs (Figure 1A). We also mutated the JC residue N49 to A/G/Q or S51 to A (data not shown). We found the individual IgA2m2 mutations N166A, S212P, N263Q, N337T.I338L.T339S, and N459Q, and the N49Q JC mutation to give the highest levels of transient expression (data not shown), but the combination of mutations to remove all five glycosylation sites in IgA2m2 resulted in poor expression and the further addition of the J chain N49Q mutation completely abolished it (data not shown). In our experience, chimeric immunoglobulins often show lower expression levels in mammalian cells relative to those from a single species. We, therefore, switched the murine anti-mIL13 variable domains to humanized anti-human epidermal growth factor receptor 2 (HER2) variable domains to generate humanized IgAs, but this did not improve transient expression levels (data not shown). Therefore, we produced a CHO targeted integration (TI) stable cell line to increase the expression level of the aglycosylated human anti-HER2 IgA2m2 polymer, which was purified as a mixture of oligomeric species.
Second, we engineered two IgG1-IgA Fc fusions in order to exploit FcRn binding as a way to reduce lysosomal degradation (Figure 7B). Previous studies suggested that this approach rescued IgA monomer serum clearance to levels comparable to IgG1.7,35 Initially, we made dimeric and tetrameric versions of the previously reported IgG1-IgA2m1 P221R Fc fusion,35 but observed that these displayed instability in mouse plasma at 4 days (Figure 7C). The primary truncation product eluted in analytical SEC at a similar time to the full-length anti-HER2 IgG1 (trastuzumab), suggesting that this instability was caused by endoproteases cleaving at the IgG1-IgA2m1 P221R Fc junction. We inspected the amino acid sequence of the junction and identified a stretch of positively charged residues that resembled a furin-like cleavage site (Figure S5). To mitigate proteolytic cleavage, we eliminated these positively charged residues at the junction by removal of the C-terminal K447 of the IgG1 heavy chain and started the IgA2m1 Fc with either P221, the native IgA2m1 residue (instead of the P221R mutation), or C242, which deletes the IgA2m1 hinge (Figure S5). The C242 Fc start was also included because it was the first residue of the IgA1 Fc crystal structure construct, so was presumed to be a stable truncated protein.46 When the reengineered IgG1K-P221 IgA2m1 Fc and IgG1K-C242 IgA2m1 Fc fusions were produced as dimers, both were found to be stable in mouse plasma for up to 4 days (Figure 7C).
The engineered IgA molecules were further characterized by global glycan content, antigen binding, and receptor binding. The aglycosylated anti-HER2 IgA2m2 polymer indeed had no glycosylation, while the anti-mIL-13 IgG1K-P221 IgA2m1 Fc and IgG1K-C242 IgA2m1 Fc fusions contained only ~20% complex, sialylated glycans (Figure S6 and Table S4). The aglycosylated anti-HER2 IgA2m2 polymer was found to have similar binding affinity to human (h)HER2, murine (m)pIgR, and hpIgR as the glycosylated IgA2m2 tetramer, while it did not bind the IgA-specific hFc receptor, hFcαRI (Table 3). Interestingly, an IgA2m2 tetramer lacking glycosylation on the IgA2m2 HC, but retaining glycosylation on the J-chain was also unable to bind hFcαRI (data not shown), suggesting that glycosylation of the IgA HC is required for receptor binding. Both the anti-mIL-13 IgG1K-P221 IgA2m1 Fc and IgG1K-C242 IgA2m1 Fc dimers had similar binding affinities to mIL-13, mFcRn, and hFcRn as the anti-mIL-13 IgG1 (Table 3), as well as similar binding affinities to mpIgR, hpIgR, and hFcαRI as an IgA2m1 dimer (Tables 1 and 3). Thus, the IgG1-IgA2m1 Fc fusions retain the desired attributes of both IgG and polymeric IgA.
Table 3.
Binding affinity of IgG1-IgA2m1 Fc fusion dimers and aglycosylated IgA2m2 polymer to antigens and receptors.
| KD (nM) |
|||||||
|---|---|---|---|---|---|---|---|
| Mouse IL-13 | Human HER2 | Mouse pIgR | Human pIgR | Mouse FcRn* pH 6.0 | Human FcRn* pH 6.0 | Human FcαRI | |
| Anti-HER2 IgA2m2 Tetramer | NB | 0.21 ± 0.08 | 0.35 ± 0.01 | 0.50 ± 0.06 | NB | NB | 1,590 ± 200 |
| Anti-HER2 IgA2m2 Polymer Aglycosylated | NB | 0.27 ± 0.07 | 0.55 ± 0.10 | 0.72 ± 0.06 | NB | NB | NB |
| Anti-IL-13 IgG1K- P221 IgA2m1 Fc Dimer | 1.46 ± 0.33 | NB | 3.32 ± 1.07 | 7.67 ± 0.42 | 6,800 ± 556 | 8,400 ± 294 | 1,070 ± 69.8 |
| Anti-IL-13 IgG1K- C242 IgA2m1 Fc Dimer | 1.38 ± 0.15 | NB | 3.32 ± 1.62 | 4.41 ± 1.72 | 7,400 ± 830 | 9,900 ± 838 | 938 ± 93.9 |
| Anti-IL-13 IgG1 | 1.15 ± 0.17 | NB | NB | NB | 7,800 ± 499 | 9,800 ± 1, 081 | NB |
| Human Serum IgA Monomer | NB | NB | NB | NB | NB | NB | 1,750 ± 92.9 |
NB: No Binding. *KD was calculated using steady-state kinetics. All experiments were performed at least n = 3.
We then measured the in vitro pIgR-mediated transcytosis and serum concentration-time profiles in mice of both of these newly generated formats. The IgG1-IgA2m1 Fc fusions showed the most marked improvements in the overall IgA serum-exposures in mice (Figure 7D and Table 4), yet the lowest levels of in vitro transcytosis compared to the aglycosylated IgA2m2 polymer, which showed the highest level of in vitro transcytosis (Figure 7E).
Table 4.
Pharmacokinetic parameter estimates (Mean) after a 30 mg/kg IV bolus of IgA oligomers to Balb/C mice.
| Cmax (µg/mL) | AUClast (day*µg/mL) | CL (mL/day/kg) | Half Life (Days) | |
|---|---|---|---|---|
| Anti-HER2 IgA2m2 Tetramer | 618 | 48.9 | 613 | 0.87 |
| Anti-HER2 IgA2m2 Polymer Aglycosylated | 1064 | 159 | 321 | 0.68 |
| Anti-IL-13 IgG1∆K-P221 IgA2m1 Fc Dimer | 963 | 176 | 237 | 3.42 |
| Anti-IL-13 IgG1∆K-C242 IgA2m1 Fc Dimer | 740 | 177 | 192 | 1.94 |
| Anti-IL-13 IgA2m1 Dimer | 1372 | 115 | 431 | 0.41 |
AUClast = area under the concentration–time curve, last measurable concentration; CL = clearance; Cmax = maximum concentration observed; IV = intravenous. Note: As sparse PK analysis was performed for all mouse PK data, data from individual mice per group were pooled and SD was not reported.
Discussion
IgA has the potential to extend the therapeutic reach of monoclonal antibodies beyond the current functionalities provided by IgG. In part, this is enabled by the versatility of IgA to form both monomeric and oligomeric species. Substantial progress has recently been made on the recombinant production of monomeric IgA,3,47–49 providing a robust path to isolate well-characterized material with increased sialylation content of the N-linked glycans that has resulted in improved serum clearance.26 While the strong cytotoxic properties of monomeric IgA are an attractive feature for oncology indications, polymeric IgA is required to reach targets beyond epithelial barriers via pIgR-mediated transcytosis. Prior to our work, only mixtures of recombinantly made monomer and oligomers were used for in vivo experiments studying transcytosis.10,30 We established a robust expression and purification route allowing the enrichment of dimeric and tetrameric IgA. In particular, modulating the amount of JC DNA used in the transfection or the glycosylation state of the IgA tail region was able to influence the distribution of oligomeric species. Interestingly, the N-linked glycosylation site on the IgA tail is the only site that is extremely conserved among species (Figure S7), suggesting this may be a way to control higher order IgA oligomer formation in vivo. For purification of recombinant IgA, we used Protein L affinity chromatography followed by HPLC-SEC and were readily able to separate dimer from tetramer.
It has been previously demonstrated that increasing the level of sialylation on recombinantly expressed IgA monomer reduces serum clearance by overcoming glycan receptor-mediated catabolism.26 As an alternative strategy, we eliminated the contribution of glycan to the serum clearance of oligomeric IgA and produced fully aglycosylated IgA2m2 polymer. The lack of N-linked glycans did not affect binding to pIgR, as assessed by surface plasmon resonance. Surprisingly, in our in vitro MDCK transcytosis assay, we observed significantly improved transcytosis compared to glycosylated tetramer or dimer. This improvement was not observed in a previous study with human IgA1 dimer that had N-linked glycans removed from the antibody Fc region, but still had the carbohydrate present on the murine J-chain.37 One explanation might be that the lack of glycan completely eliminates the binding to glycan receptors, thus providing unperturbed and more efficient transcytosis via pIgR binding. Interestingly, binding of tetrameric IgA2m2 to FcαRI was dependent on glycosylation, which is in sharp contrast to the lack of effect on binding observed for monomeric IgA2m1 that was engineered to contain a reduced number of glycosylation sites.6 Although we did not evaluate the cytotoxic effects of polymeric IgA in this study, an IgA therapeutic that can transcytose without activating FcαRI is very desirable for inflammatory diseases, as this would prevent pro-inflammatory responses from neutrophil migration.50
Glycosylated IgA oligomers and monomers produced recombinantly in our study cleared rapidly from serum similar to what was previously observed.26,37 Therefore, to better understand the contribution of having glycan, we generated an IgA polymer without glycans (aglycosylated) and directly compared its in vivo PK profile to the glycosylated version. The aglycosylated IgA2m2 polymer showed no appreciable difference in overall mouse serum exposure compared to the glycosylated polymer (<twofold). This suggests that having N-linked glycan plays a minimal role in contributing to the clearance of IgA polymers in mice. While further studies are necessary to understand why IgA oligomer without glycans does not improve serum clearance, we speculate that pIgR-mediated transcytosis or clearance may play a significant role in determining the overall serum concentrations and the fate of polymeric IgA in mice. Indeed, the equilibrium binding affinity of tetrameric IgA to pIgR is at least in the picomolar range allowing for efficient binding to the abundant pIgR receptor, followed by transcytosis. However, a detailed biodistribution study looking at the tissue distribution profile will be needed to better interpret the serum concentration-time profiles of the molecules and the disposition of aglycosylated polymeric IgA. One important caveat to studying PK properties and biodistribution of a polymeric IgA molecule in rodents is that expression patterns of pIgR differ between rodents and humans, potentially confounding eventual clinical translation. In particular, high expression of pIgR in the hepatocytes of rodents and rabbits have been associated with biliary clearance mechanisms of polymeric IgA,34 not thought to occur in humans, where pIgR expression is found instead in cells of the bile duct.33 Therefore, the exact role that pIgR plays in the biodistribution and clearance of a polymeric IgA molecule in mice needs to be separately evaluated.
A separate strategy that we took to avoid accelerated serum clearance and improve exposure was via engineering IgG1-IgA2m1 Fc fusions. The ability to bind FcRn may enable recycling in the endosome, avoiding sorting for lysosomal degradation. Previous reports with monomeric IgG-IgA Fc fusions reported serum clearance comparable to IgG.7,35 In addition, an improvement in PK properties was observed for monomeric IgA that was fused to albumin binding peptides.25 Since we did not observe a large contribution by removing glycans from the IgA polymers, we produced glycosylated fusion proteins. While our dimeric IgG1-IgA2 Fc fusions showed improved overall serum exposures, it was not comparable to IgG as observed for the monomeric IgG-IgA Fc fusions.7,35 We demonstrated by surface plasmon resonance that the dimeric IgG1-IgA2m1 Fc fusions can bind FcRn and pIgR, albeit having reduced in vitro transcytosis in our MDCK model. While the binding to FcRn extended the terminal half-life (Table 4), IgG1-IgA2m1 Fc dimer interaction with pIgR may have provided a clearance mechanism, particularly in the early phase, which resulted in pIgR-mediated transcytosis or clearance, contributing to the reduced serum concentrations compared to IgG. More follow up in vivo studies are needed to understand whether the in vitro assays we developed to measure pIgR-mediated transcytosis can be used as an accurate predictor of tissue partitioning in vivo and how changes in serum exposures observed with the novel IgA formats correlate with tissue distribution and partitioning. Of note, healthy mice, rhesus macaques, and humans all have a high pIgR expression in the intestine, so it will be interesting to see if this correlates to the best tissue delivery.43,44,51
IgA in serum is constituted predominantly of IgA1 monomer secreted from bone marrow cells, while polymeric IgA2 is secreted from plasma cells in the lamina propria at the location of transcytosis.1 The high affinity between polymeric IgA and pIgR may naturally lead to fast scavenging of polymeric IgA from circulation, providing effective clearance of harmful antigens from the circulation as IgA-antigen complexes.52 In a therapeutic setting, this may be exploited to restrict drug activity to a defined tissue, something that may be of particular benefit when agonizing cytokine receptors. The fast clearance of polymeric IgA via pIgR from serum in mice is further supported by the approximately 20-fold increased IgA concentrations in the serum of pIgR-deficient non-obese diabetic (NOD) mice.53 As only polymeric IgA, but not IgA monomer, binds pIgR for transcytosis, this increased concentration of IgA in serum must be reflective of increased polymeric IgA concentrations. Future biodistribution studies will show if removing clearance through glycan receptors contributes to more selective binding to pIgR. Regardless, characterization of an aglycosylated IgA therapeutic molecule is easier compared to a highly glycosylated counterpart. In addition, finding a balance between serum exposure and transcytosis is essential to be able to efficiently deliver a therapeutic molecule to the mucosa. The combination of an aglycosylated IgA molecule that binds FcRn may provide a promising path forward for an IgA therapeutic.
Materials and methods
Plasmid cloning and sequence alignments
Antibody variable domain sequences used include a humanized anti-human HER2 antibody54 and a murine anti-murine IL-13 antibody (Genentech). Protein sequences of human IgA constant heavy chains IgA1, IgA2m1, and IgA2m2, other IgA species, and human J chain were obtained from UniProt (www.uniprot.org) or NCBI (www.ncbi.nlm.nih.gov/protein). P221R is a mutation in IgA2m1 that stabilizes the light chain-heavy chain disulfide as previously reported.24 Genes encoding a fusion of the antibody variable domains to the human light chain and human IgA1, IgA2m1, and IgA2m2 heavy chain constant domains were synthesized and cloned into the mammalian pRK vector.55 Site-directed mutagenesis was used to introduce point mutations. All plasmids were sequence-verified. Sequence alignments were done using GSeqWeb (Genentech) and Excel (Microsoft).
Small-scale antibody expression and purification
Expi293T™ cells were transiently transfected at the 30 mL scale with 15 µg of DNA of both LC and HC for IgA monomers or a total of 30 µg of DNA of varying ratios of LC, HC, and JC for IgA oligomers.56,57 IgAs were affinity purified in batch with Protein L (GE Healthcare) as all antibodies contained kappa light chains. Proteins were washed with phosphate-buffered saline (PBS), eluted with 50 mM phosphoric acid pH 3.0, and neutralized with 20x PBS pH 11. Protein L eluate was characterized by analytical SEC-HPLC (Tosoh Bioscience LLC TSKgel SuperSW3000 column, Thermo Scientific Dionex UltiMate 3000 HPLC). A constant volume was loaded on the column, and the area under each curve was quantitated using Chromeleon Chromatography Data System software (Thermo Scientific).
Large-scale antibody expression and purification
IgA, IgG, and IgG-IgA Fc fusions were transiently expressed in CHO DP12 cells as previously described.58 For low expressing clones, TI stable cell lines were generated. IgG and IgG-IgA Fc fusions were affinity-purified using MabSelect Sure (GE Healthcare), followed by SEC with a HiLoad Superdex 200 pg column (GE Healthcare). IgAs were affinity-purified using Capto L (GE Healthcare) followed by SEC. For IgA samples where DNA ratios successfully biased expression to mainly one oligomeric state, a HiLoad Superdex 200 pg column (GE Healthcare) was used for SEC. For IgA samples containing complex mixtures of oligomers, a 3.5 μm, 7.8 mm x 300 mm Xbridge Protein BEH 450 Å SEC column (Waters) was used for better separation of dimer and tetramer peaks.
SEC-MALS
Polymeric IgAs were run over a 3.5 μm, 7.8 mm x 300 mm Xbridge Protein BEH 200 Å SEC column (Waters) and directly injected onto a DAWN HELEOS/Optilab T-rEX II (Wyatt) MALS detector for molar mass determination and polydispersity measurement.
Differential scanning fluorimetry
DSF was performed as described previously.59
In vitro transcytosis assay
MDCKII cells (European Collection of Authenticated Cell Cultures, Salisbury, U.K.) cells were transduced with retrovirus containing cDNA coding for the human pIgR gene (Retro-X, Takara Bio; OriGene, Rockville, MD). Expression of the pIgR gene was confirmed by qRT-PCR and Western Blotting. MDCKII cells expressing pIgR were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin (Thermo Fisher, Carlsbad, CA), and 2 µg/ml Puromycin (Takara Bio, Mountain View, CA). For the transcytosis assay, cells were seeded on 0.4 µm Millicell 24-well cell culture insert (Millipore, Burlington, MA) and cultured for 4 days. On the day of the experiment, the cells were washed twice with FluoroBrite DMEM (Thermo Fisher), and 6 µg of IgA molecules was added to the basolateral compartments. After 24 h of incubation, media from both apical and basolateral compartments were collected for analysis by ELISA.
Electron microscopy
Purified anti-mIL-13 IgA2m2 dimer and tetramer samples were first crosslinked by incubating in 0.015% glutaraldehyde (Polysciences, Inc.) for 10 min at room temperature. Once fixed, the samples were diluted using Tris-buffered saline to achieve a concentration of 10 ng/µL. Then, 4 µl of each sample was incubated for 40 s on freshly glow discharged 400 mesh copper grids covered with a thin layer of continuous carbon before being treated with 2% (w/v) uranyl acetate negative stain (Electron Microscopy Sciences). IgA dimers and tetramers were then imaged using a Tecnai Spirit T12 (Thermo Fisher) operating at 120 keV, at a magnification of 25,000x (2.2 Å/pixel). Images were recorded using a Gatan 4096 × 4096 pixel CCD camera under low dose conditions. About 5000 particles for both IgA dimer and tetramers were then selected and extracted using the e2boxer.py software within the EMAN2 package60 using a 128-pixel particle box size. Reference-free 2D classification, within the RELION image software package61 was used to generate averaged images of both samples.
Antibody kinetics
A 96 × 96 array-based Surface Plasmon Resonance (SPR) imaging system (Carterra USA) was used to analyze the kinetics of purified IgA, IgG-IgA Fc fusions, or IgG. Antibodies were diluted at 10 μg/ml in 10 mM sodium acetate buffer pH 4.5 and using amine coupling, were directly immobilized onto a SPR sensorprism CMD 200M chip (XanTec Bioanalytics, Germany) using a Continuous Flow Microspotter. Antigens diluted in running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 0.05% Tween 20, 1 mM EDTA) were injected at various concentrations for 3 minutes and allowed to dissociate for 10 minutes, with regeneration between cycles using 10 mM glycine pH 2.5. Antigens were from R&D Systems (mIL-13, 413-ML-025/CF; mpIgR, 2800-PG-050; hpIgR, 2717-PG-050; hFcaRI, 3939-FA-050), Sino Biologicals (hHER2, 10004-H08H), or Genentech, co-expressed with species specific beta-2 microglobulin (m/hFcRn). The data was processed with the Wasatch kinetic software tool.
Global N-linked glycan composition analysis (LC-MS analysis)
Ten μg of each IgA sample were denatured with 8 M guanidine HCl at 1:1 volume ratio and reduced with 100 mM dithiothreitol (DTT) for 10 min at 95°C. Samples were diluted with 100 mM Tris HCl, pH 7.5, to a final concentration of 2 M guanidine HCl, followed by overnight N-linked deglycosylation at 37°C with 2 μl of P0705S PNGase F (New England BioLabs). After deglycosylation, 150 ng of each sample were injected onto an Agilent 1260 Infinity LC system and eluted by an isocratic gradient of 2% to 32% solvent B (solvent A: 99.88% water containing 0.1% formic acid and 0.02% trifluoroacetic acid (TFA); solvent B: 90% acetonitrile containing 9.88% water plus 0.1% formic acid and 0.02% TFA). The HPLC system was coupled via an Agilent G4240A Chip Cube MS system to a G6520B Q-TOF mass spectrometer. The samples were glycan enriched and separated using porous-graphitized carbon columns built within a G4240-64025 mAb-Glyco chip in the Chip Cube MS system. Data acquisition: 1.9 kV spray voltage; 325°C gas temperature; 5 l/min drying gas flow; 160 V fragmentor voltage; 65 V skimmer voltage; 750 V oct 1 RF Vpp voltage; 400 to 3000 m/z scan range; positive polarity; MS1 centroid data acquisition using extended dynamic range (2 GHz) instrument mode; 333.3 ms/spectrum and 3243 transients/spectrum. Acquired mass spectral data were searched against a glycan library in the Agilent MassHunter Qualitative Analysis software utilizing a combination of accurate mass with a mass tolerance of 10 ppm and expected retention time for glycan identification. N-linked glycans were label-free quantified relative to all identified N-linked glycans within each sample based on the AUC in the extracted compound chromatogram of each glycan.
N-linked glycopeptide site mapping analysis (LC-MS/MS analysis)
Ten μg of IgA was reduced with 10 mM DTT at 37°C for 1 h, alkylated with 10 mM iodoacetamide at room temperature for 20 min, digested with 0.2 μg of trypsin (Promega) and 0.2 μg of chymotrypsin (Thermo Fisher Scientific) separately at 37°C overnight, quenched with 0.1% TFA and subjected clean up with C18 (3M Empore C18 extraction disks) stage-tip (50% acetonitrile, 49.9% water, 0.1% TFA). A quantity of 200 fmol of the sample was injected onto a Waters NanoAcquity UPLC system via an autosampler and separated at 45°C on a Waters Acquity M-Class BEH C18 column (0.1 mm × 100 mm, 1.7 μm resin). A gradient of 2% to 40% solvent B was used for elution (solvent A: 99.9% water, 0.1% formic acid; solvent B 99.9% acetonitrile, 0.1% formic acid).
Separated peptides were analyzed on-line via nanospray ionization into an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) using the following parameters for data acquisition: 60000 resolution; 375–1600 m/z scan range; positive polarity; centroid mode; 1 m/z isolation width with 0.25 activation Q and 10 ms activation time and a collisional voltage of 35. Data were collected in data-dependent mode in which precursor ions being analyzed in the Orbitrap and the top 15 most abundant ions were sent to the ion trap for fragmentation. Acquired mass spectral data were searched against the candidate protein sequence using Protein Metrics Byonic software and analyzed in Protein Metrics Byologic software. Peptide identification for each glycosylation site was manually validated based on a combination of MS2 fragmentation spectra, extracted ion chromatograms (XIC), and retention time. N-linked glycopeptides were label-free quantified relative to the unmodified peptide by the integration of peak areas in the XICs.
Mouse studies
Female Balb/C mice (6–8 weeks old) were obtained by Charles River laboratories. Upon arrival, all mice were maintained in a pathogen-free animal facility under a standard 12 h light/12 h dark cycle at 21°C room temperature with access to food and water ad libitum. All mice received a single intravenous (IV) injection of either IgG or IgA antibody. Blood samples (150–200 µL) were collected via either via retro-orbital sinus or cardiac puncture under isoflurane anesthesia at various times post injection. Samples were collected into serum separator tubes. The blood was allowed to clot at ambient temperature for at least 20 min. Clotted samples were maintained at room temperature until centrifuged, commencing within 1 h of the collection time. Each sample was centrifuged at a relative centrifugal force of 1500–2000 x g for 5 min at 2–8°C. The serum was separated from the blood sample within 20 min after centrifugation and transferred into labeled 2.0-mL polypropylene, conical-bottom microcentrifuge tubes.
Only animals that appeared to be healthy and that were free of obvious abnormalities were used for the study. All animal work performed was reviewed and approved by Genentech’s Institutional Animal Care and Use Committee.
IgA ELISA for transcytosis and pharmacokinetic studies
IgA antibody levels were measured by sandwich ELISA. Wells of 384-microtiter plates were coated overnight at 4°C with 2 μg/ml of goat anti-human Kappa antibody (SouthernBiotech, Cat# 2060–01) in 25 μl of coating buffer (0.05 M sodium carbonate, pH 9.6), followed by blocking with 50 μl of 0.5% bovine serum albumin (BSA) in PBS for 2 h at 37°C. Samples (25 μl) diluted in sample buffer (1x PBS, pH 7.4, 0.5% BSA, 0.35 M NaCl, 0.05% Tween20, 0.25% CHAPS, 5 mM EDTA) were then added to the blocked plates and incubated for 2 h at room temperature. After incubation, 25 μl of horseradish peroxidase-conjugated goat anti-human IgA (SouthernBiotech, Cat# 2053–05) were added and incubated for 1 h at room temperature. The plates were then incubated with 25 μl of 3,3ʹ,5,5ʹ-tetramethylbenzidine (Moss, Cat# TMBE-1000) for 15 min, and the reaction was stopped with 25 μl 1M H3PO4. Absorbance was measured at 450 nm with a reduction at 630 nm using a plate reader. Between steps, plates were washed six times with 200 μl of washing buffer (0.05% Tween-20 in PBS). As a reference for quantification, a standard curve was established using serially diluted stock material (20 ng/ml-0.15 ng/ml) for each IgA molecule. The IgA ELISA tolerates biological matrices up to 10% mouse serum and 10% tissue lysates.
Radiochemistry
Iodine-125 [125I]was obtained as sodium iodide in 0.1 N sodium hydroxide from Perkin Elmer (Boston, MA). 1 mCi of 125I (~3 µL) was used to label randomly through tyrosine residues at a specific activity of ~10 μCi/μg with 125I, using the indirect Iodogen method (Pierce Chemical Co., Rockford, IL). Radiosynthesis of 111In labeled antibodies (~8 μCi/μg) was achieved through incubation of 111In and 1,4,7,10-tetraazacyclododecane−1,4,7,10-tetraacetic acid (DOTA)-conjugated (randomly through lysines) antibody in 0.3 M ammonium acetate pH 7 at 37°C for 1 h. Purification of all radioimmunoconjugates was achieved using NAP5 columns equilibrated in PBS and confirmed by SEC.
Antibodies were radioiodinated using an indirect iodogen addition method.62 The radiolabeled proteins were purified using NAP5™ columns (GE Healthcare Life Sciences, cat. 17–0853-01) pre-equilibrated in PBS. Following radioiodination, the labeled antibodies were characterized by SEC-HPLC to compare to the unlabeled antibodies. Samples were injected onto an Agilent 1100 series HPLC (Agilent Technology, Santa Clara, CA) and a Yarra SEC-3000, 3 µM 300 mm x 7.8 mm (Phenomenex, Torrance, CA, cat. 00H-4513-K0) size exclusion columns connected in series and eluted with PBS pH 7.0 at a flow rate of 0.8 mL/min for 20 min. Elution was monitored by absorption at 280 nm and by measuring the radioactivity of the eluted fractions in an in-line Gabi gamma counter (Elysia-Raytest, Germany).
Tissue distribution study design and analysis
The protocol, housing, and anesthesia were approved by the Institutional Animal Care and Use Committees of Genentech Laboratory Animal Resources, in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care regulations.
Female BALB-c mice in a 20–30 g body weight range and 6–7 weeks age range were obtained from Jackson/West (CA). Six groups of 12 mice each were used for this study. To prevent thyroid sequestration of 125I 100 µL of 30 mg/mL of sodium iodide was intraperitoneally administered 1 and 24 h prior to dosing. All mice received a single IV injection consisting of a mixture of 125I- and 111In-labeled antibodies (5 μCi of each) plus the respective unmodified antibodies for a total dose of 5 mg/kg. Cohorts of four mice were bled retro-orbitally under Isoflurane (inhalation to effect) at 5 min, 15 min, 30 min, 1 h, 4 h, 12 h, 1 day, 2 days, and 3 days after injection. At 1 h, 1 day, and 3 days; four animals were euthanized under anesthesia of ketamine (75−8 mg/kg)/xylene (7.5−15 mg/kg) by thoracotomy. The following tissues collected, rinsed in cold PBS, blotted dry, weighed and frozen: Brain, liver, lung, kidney, spleen, heart, stomach, small intestine, muscle, skin, fat, large intestine. Sample radioactivity was counted for radioactivity using a 1480 WIZARD™ Gamma Counter in the energy windows for11,n (245 keV; decay t1/2 = 2.8 days) and1I (35 keV; decay t1/2 = 59.4 days) with automatic background and decay correction. Data were analyzed and graphed using GraphPad Prism (version 7.00 for Windows, GraphPad Software, San Diego California USA, www.graphpad.com).
Mouse plasma stability
Mouse plasma (with anti-coagulant Lithium Heparin) was obtained from BioIVT (Westbury, NY), and a buffer control was made by mixing BSA (Sigma-Aldrich; St. Louis, MO, cat. A2058) with PBS (PBS + 0.5% BSA). Radiolabeled antibodies were mixed into mouse plasma or buffer control at 5 µCi of radiolabeled tracer, and then incubated in an incubator set at 37°C with 5% CO2. At the set time point of 0, 24, and 96 h of incubation, the samples were removed from the incubator and stored at −80°C freezer until analysis.
The samples were analyzed by SEC-HPLC method described above with a 1:1 sample dilution in PBS. The result chromatograms were compared between the time points to monitor the changes from the parent peak at time zero.
Abbreviations
- ASGPR
asialoglycoprotein receptor
- CHO
Chinese hamster ovary
- dIgA
dimeric IgA
- FcαRI
Fc alpha receptor 1
- FcγR
Fc gamma receptor
- FcRn
neonatal Fc receptor
- LC
light chain
- HC
heavy chain
- JC
joining chain
- sIgA
secretory IgA
- PK
pharmacokinetic
- pIgR
polymeric immunoglobulin receptor
Acknowledgments
We thank the Research Materials Group for mammalian cell expression, especially George Dutina for making the stable CHO cell lines. We thank the Antibody Production Group for purification and characterization of IgA antibodies, including Yi Xia who cloned the stable cell constructs. We appreciate Yoshu Joy Xie for assistance with molecular biology and Kam Hon Hoi for his help with the sequence alignment figure.
Author Contributions
TNL, SR, JAZ, MLM, and CS devised the conceptual framework of the study. TNL performed in vitro antibody characterization. FF, SV, SL, and AG carried out small-scale protein purification and analysis. JAZ and MLM performed large-scale protein purification and analysis. AE and CC performed EM experiments. EC developed and performed in vitro transcytosis experiments. DBP performed and analyzed ELISA data from in vitro transcytosis assays and non-radioactive PK experiments. SR coordinated and analyzed all non-radioactive PK studies. DM and AB performed and analyzed radioactive PK studies. WP and WS analyzed glycan occupancy by mass spectrometry. VY conducted the mouse plasma stability experiments. TNL, MLM, and CC made the figures. TNL, SR, MLM, and CS wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest
All authors are current or former employees of Genentech, Inc., a member of the Roche Group, and may hold stock and options. This work was funded and conducted by Genentech, Inc.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Yoo EM, Morrison SL.. IgA: an immune glycoprotein. Clin Immunol. 2005;116:3–10. doi: 10.1016/j.clim.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 2.Bakema JE, van Egmond M.. Immunoglobulin A: A next generation of therapeutic antibodies? MAbs. 2011;3:352–61. doi: 10.4161/mabs.3.4.16092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leusen JHW. IgA as therapeutic antibody. Mol Immunol. 2015;68:35–39. doi: 10.1016/j.molimm.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Pascal V, Laffleur B, Debin A, Cuvillier A, van Egmond M, Drocourt D, Imbertie L, Pangault C, Tarte K, Tiraby G, et al. Anti-CD20 IgA can protect mice against lymphoma development: evaluation of the direct impact of IgA and cytotoxic effector recruitment on CD20 target cells. Haematologica. 2012;97:1686–94. doi: 10.3324/haematol.2011.061408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boross P, Lohse S, Nederend M, Jansen JHM, van Tetering G, Dechant M, Peipp M, Royle L, Liew LP, Boon L, et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med. 2013;5:1213–26. doi: 10.1002/emmm.201201876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohse S, Meyer S, Meulenbroek LAPM, Jansen JHM, Nederend M, Kretschmer A, Klausz K, Möginger U, Derer S, Rösner T, et al. An anti-EGFR IgA that displays improved pharmacokinetics and myeloid effector cell engagement in vivo. Cancer Res. 2016;76:403–17. doi: 10.1158/0008-5472.CAN-16-0584. [DOI] [PubMed] [Google Scholar]
- 7.Li B, Xu L, Tao F, Xie K, Wu Z, Li Y, Li J, Chen K, Pi C, Mendelsohn A, et al. Simultaneous exposure to FcγR and FcαR on monocytes and macrophages enhances antitumor activity in vivo. Oncotarget. 20178:39356–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelton W, Mehta N, Charab W, Lee J, Lee C-H, Kojima T, Kang TH, Georgiou G. IgGA: a “cross-isotype” engineered human Fc antibody domain that displays both IgG-like and IgA-like effector functions. Chem Biol. 2015;21:1603–09. doi: 10.1016/j.chembiol.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki T, Kawaguchi A, Ainai A, Tamura S-I, Ito R, Multihartina P, Setiawaty V, Pangesti KNA, Odagiri T, Tashiro M, et al. Relationship of the quaternary structure of human secretory IgA to neutralization of influenza virus. Proc Natl Acad Sci USA. 2015;112:7809–14. doi: 10.1073/pnas.1503885112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsan EE, Matsushita T, Rezaei M, Weimbs T. Exploitation of the polymeric immunoglobulin receptor for antibody targeting to renal cyst lumens in polycystic kidney disease. J Biol Chem. 2015;290:15679–86. doi: 10.1074/jbc.M114.607929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borrok MJ, DiGiandomenico A, Beyaz N, Marchetti GM, Barnes AS, Lekstrom KJ, Phipps SS, McCarthy MP, Wu H, Dall’acqua WF, et al. Enhancing IgG distribution to lung mucosal tissue improves protective effect of anti–pseudomonas aeruginosa antibodies. JCI Insight. 2018;3. doi: 10.1172/jci.insight.97941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brezski RJ, Georgiou G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol. 2016;40:62–69. doi: 10.1016/j.coi.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Toraño A, Putnam FW. Complete amino acid sequence of the alpha 2 heavy chain of a human IgA2 immunoglobulin of the A2m (2) allotype. Proc Natl Acad Sci USA. 1978;75:966–69. doi: 10.1073/pnas.75.2.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zikan J, Mestecky J, Kulhavy R, Bennett JC. The stoichiometry of J chain in human secretory dimeric IgA. Mol Immunol. 1986;23:541–44. doi: 10.1016/0161-5890(86)90117-3. [DOI] [PubMed] [Google Scholar]
- 15.Halpern MS, Koshland ME. The stoichiometry of J chain in human secretory IgA. J Immunol. 1973;111:1653–60. [PubMed] [Google Scholar]
- 16.Wu C-T, Davis PA, Luketic VA, Gershwin ME. A review of the physiological and immunological functions of biliary epithelial cells: targets for primary biliary cirrhosis, primary sclerosing cholangitis and drug-induced ductopenias. Clin Dev Immunol. 2004;11:205–13. doi: 10.1080/17402520400004177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fallgreen-Gebauer E, Gebauer W, Bastian A, Kratzin HD, Eiffert H, Zimmermann B, Karas M, Hilschmann N. The covalent linkage of secretory component to IgA. Structure of sIgA. Biol Chem Hoppe-Seyler. 1993;374:1023–28. doi: 10.1515/bchm3.1993.374.7-12.1023. [DOI] [PubMed] [Google Scholar]
- 18.Bastian A, Kratzin H, Fallgren-Gebauer E, Eckart K, Hilschmann N. Intra- and inter-chain disulfide bridges of J chain in human S-IgA. Adv Exp Med Biol. 1995;371A:581–83. [DOI] [PubMed] [Google Scholar]
- 19.Mantis NJ, Rol N, Corthésy B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011;4:603–11. doi: 10.1038/mi.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansen F-E, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor and IgA transport: new advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011;4:598–602. doi: 10.1038/mi.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerr MA. The structure and function of human IgA. Biochem J. 1990;271:285–96. doi: 10.1042/bj2710285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuzukida Y, Wang CC, Putnam FW. Structure of the A2m(1) allotype of human IgA–a recombinant molecule. Proc Natl Acad Sci USA. 1979;76:1104–08. doi: 10.1073/pnas.76.3.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chintalacharuvu KR, Raines M, Morrison SL. Divergence of human alpha-chain constant region gene sequences. A novel recombinant alpha 2 gene. J Immunol. 1994;152:5299–304. [PubMed] [Google Scholar]
- 24.Chintalacharuvu KR, Morrison SL. Residues critical for H-L disulfide bond formation in human IgA1 and IgA2. J Immunol. 1996;157:3443–49. [PubMed] [Google Scholar]
- 25.Meyer S, Nederend M, Jansen JHM, Reiding KR, Jacobino SR, Meeldijk J, Bovenschen N, Wuhrer M, Valerius T, Ubink R, et al. Improved in vivo anti-tumor effects of IgA-Her2 antibodies through half-life extension and serum exposure enhancement by FcRn targeting. MAbs. 2016;8:87–98. doi: 10.1080/19420862.2016.1196521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rouwendal GJ, van der Lee MM, Meyer S, Reiding KR, Schouten J, de Roo G, Egging DF, Leusen JH, Boross P, Wuhrer M, et al. A comparison of anti-HER2 IgA and IgG1 in vivo efficacy is facilitated by high N-glycan sialylation of the IgA. MAbs. 2016;8:74–86. doi: 10.1080/19420862.2016.1196521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brunke C, Lohse S, Derer S, Peipp M, Boross P, Kellner C, Beyer T, Dechant M, Royle L, Liew LP, et al. Effect of a tail piece cysteine deletion on biochemical and functional properties of an epidermal growth factor receptor-directed IgA2m(1) antibody. MAbs. 2013;5:936–45. doi: 10.4161/mabs.22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Challacombe SJ, Russell MW. Estimation of the intravascular half-lives of normal rhesus monkey IgG, IgA and IgM. Immunology. 1979;36:331–38. [PMC free article] [PubMed] [Google Scholar]
- 29.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 30.Rifai A, Fadden K, Morrison SL, Chintalacharuvu KR. The N-glycans determine the differential blood clearance and hepatic uptake of human immunoglobulin (Ig)A1 and IgA2 isotypes. J Exp Med. 2000;191:2171–82. doi: 10.1084/jem.191.12.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SJ, Evers S, Roeder D, Parlow AF, Risteli J, Risteli L, Lee YC, Feizi T, Langen H, Nussenzweig MC. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science. 2002;295:1898–901. doi: 10.1126/science.1069540. [DOI] [PubMed] [Google Scholar]
- 32.Heystek HC, Moulon C, Woltman AM, Garonne P, van Kooten C. Human immature dendritic cells efficiently bind and take up secretory IgA without the induction of maturation. J Immunol. 2002;168:102–07. doi: 10.4049/jimmunol.168.1.102. [DOI] [PubMed] [Google Scholar]
- 33.Tomana M, Kulhavy R, Mestecky J. Receptor-mediated binding and uptake of immunoglobulin A by human liver. Gastroenterology. 1988;94:762–70. doi: 10.1016/0016-5085(88)90252-1. [DOI] [PubMed] [Google Scholar]
- 34.Daniels CK, Schmucker DL, Jones AL. Hepatic asialoglycoprotein receptor-mediated binding of human polymeric immunoglobulin A. Hepatology. 1989;9:229–34. doi: 10.1002/(ISSN)1527-3350. [DOI] [PubMed] [Google Scholar]
- 35.Borrok MJ, Luheshi NM, Beyaz N, Davies GC, Legg JW, Wu H, Dall’acqua WF, Tsui P. Enhancement of antibody-dependent cell-mediated cytotoxicity by endowing IgG with FcαRI (CD89) binding. MAbs. 2015;7:743–51. doi: 10.1080/19420862.2015.1047570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chintalacharuvu KR, Morrison SL. Production and characterization of recombinant IgA. Immunotechnology. 1999;4:165–74. doi: 10.1016/S1380-2933(98)00012-8. [DOI] [PubMed] [Google Scholar]
- 37.Chuang PD, Morrison SL. Elimination of N-linked glycosylation sites from the human IgA1 constant region: effects on structure and function. J Immunol. 1997;158:724–32. [PubMed] [Google Scholar]
- 38.Lohse S, Loew S, Kretschmer A, Jansen JHM, Meyer S, Broeke Ten T, Rösner T, Dechant M, Derer S, Klausz K, et al. Effector mechanisms of IgA antibodies against CD20 include recruitment of myeloid cells for antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. Br J Haematol. 2017;112:4170. [DOI] [PubMed] [Google Scholar]
- 39.Boswell CA, Tesar DB, Mukhyala K, Theil F-P, Fielder PJ, Khawli LA. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjugate Chem. 2010;21:2153–63. doi: 10.1021/bc100261d. [DOI] [PubMed] [Google Scholar]
- 40.Mandikian D, Takahashi N, Lo AA, Li J, Eastham-Anderson J, Slaga D, Ho J, Hristopoulos M, Clark R, Totpal K, et al. Relative target affinities of T-cell-dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol Cancer Ther. 2018;17:776–85. doi: 10.1158/1535-7163.MCT-17-0657. [DOI] [PubMed] [Google Scholar]
- 41.Rajan S, Mandikian D, Baruch A, Gelzleichter TR, Stevens D, Sonoda J, Cowan K, Boswell CA, Stefanich E. Preclinical pharmacokinetic characterization of an adipose tissue-targeting monoclonal antibody in obese and non-obese animals. MAbs. 2017;9:1379–88. doi: 10.1080/19420862.2017.1373923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boswell CA, Mundo EE, Ulufatu S, Bumbaca D, Cahaya HS, Majidy N, Van Hoy M, Schweiger MG, Fielder PJ, Prabhu S, et al. Comparative physiology of mice and rats: radiometric measurement of vascular parameters in rodent tissues. Mol Pharmaceutics. 2014;11:1591–98. doi: 10.1021/mp400748t. [DOI] [PubMed] [Google Scholar]
- 43.Asano M, Saito M, Suguro H, Nomura H, Inage T, Moro I. Active synthesis of mouse polymeric immunoglobulin receptor in the epithelial cells of the distal urinary tubule in kidney. Scand J Immunol. 2004;60:267–72. doi: 10.1111/sji.2004.60.issue-3. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Yang GB. Alteration of polymeric immunoglobulin receptor and neonatal Fc receptor expression in the gut mucosa of immunodeficiency virus-infected rhesus macaques. Scand J Immunol. 2016;83:235–43. doi: 10.1111/sji.2016.83.issue-4. [DOI] [PubMed] [Google Scholar]
- 45.Boswell CA, Mundo EE, Firestein R, Zhang C, Mao W, Gill H, Young C, Ljumanovic N, Stainton S, Ulufatu S, et al. An integrated approach to identify normal tissue expression of targets for antibody-drug conjugates: case study of TENB2. Br J Pharmacol. 2013;168:445–57. doi: 10.1111/j.1476-5381.2012.02138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herr AB, Ballister ER, Bjorkman PJ. Insights into IgA-mediated immune responses from the crystal structures of human FcalphaRI and its complex with IgA1-Fc. Nature. 2003;423:614–20. doi: 10.1038/nature01685. [DOI] [PubMed] [Google Scholar]
- 47.Dicker M, Maresch D, Strasser R. Glyco-engineering for the production of recombinant IgA1 with distinct mucin-type O-glycans in plants. - PubMed - NCBI. Bioengineered. 2016;7:484–89. doi: 10.1080/21655979.2016.1201251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vasilev N, Mark Smales C, Schillberg S, Fischer R, Schiermeyer A. Developments in the production of mucosal antibodies in plants. Biotechnol Adv. 2016;34(2):77–87. [DOI] [PubMed] [Google Scholar]
- 49.Virdi V, Juarez P, Boudolf V, Depicker A. Recombinant IgA production for mucosal passive immunization, advancing beyond the hurdles. Cell Mol Life Sci. 2016;73(3):535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aleyd E, Heineke MH, van Egmond M. The era of the immunoglobulin A Fc receptor FcαRI; its function and potential as target in disease. Immunol Rev. 2015;268:123–38. doi: 10.1111/imr.12337. [DOI] [PubMed] [Google Scholar]
- 51.Krajci P, Solberg R, Sandberg M, Øyen O, Jahnsen T, Brandtzaeg P. Molecular cloning of the human transmembrane secretory component (poly-Ig receptor) and its mRNA expression in human tissues. Biochem Biophys Res Commun. 1989;158:783–89. [DOI] [PubMed] [Google Scholar]
- 52.Shroff KE, Meslin K, Cebra JJ. Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect Immun. 1995;63:3904–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simpfendorfer KR, Strugnell RA, Brodnicki TC, Wijburg OLC. Increased autoimmune diabetes in pIgR-deficient NOD mice is due to a “Hitchhiking” interval that refines the genetic effect of Idd5.4. PLoS One. 2015;10:e0121979. doi: 10.1371/journal.pone.0121979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA. 1992;89:4285–89. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eaton DL, Wood WI, Eaton D, Hass PE, Hollingshead P, Wion K, Mather J, Lawn RM, Vehar GA, Gorman C. Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochemistry. 1986;25:8343–47. doi: 10.1021/bi00374a001. [DOI] [PubMed] [Google Scholar]
- 56.Bos AB, Duque JN, Bhakta S, Farahi F, Chirdon LA, Junutula JR, Harms PD, Wong AW. Development of a semi-automated high throughput transient transfection system. J Biotechnol. 2014;180:10–16. doi: 10.1016/j.jbiotec.2014.03.027. [DOI] [PubMed] [Google Scholar]
- 57.Bos AB, Luan P, Duque JN, Reilly D, Harms PD, Wong AW. Optimization and automation of an end-to-end high throughput microscale transient protein production process. Biotechnol Bioeng. 2015;112:1832–42. doi: 10.1002/bit.25601. [DOI] [PubMed] [Google Scholar]
- 58.Wong AW, Baginski TK, Reilly DE. Enhancement of DNA uptake in FUT8-deleted CHO cells for transient production of afucosylated antibodies. Biotechnol Bioeng. 2010;106:751–63. doi: 10.1002/bit.22659. [DOI] [PubMed] [Google Scholar]
- 59.Lombana TN, Dillon M, J III B, Spiess C. Optimizing antibody expression by using the naturally occurring framework diversity in a live bacterial antibody display system. Sci Rep. 2015;5:17488. doi: 10.1038/srep17488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 61.Scheres SHW. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–30. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chizzonite R, Truitt T, Podlaski FJ, Wolitzky AG, Quinn PM, Nunes P, Stern AS, Gately MK. IL-12: monoclonal antibodies specific for the 40-kDa subunit block receptor binding and biologic activity on activated human lymphoblasts. J Immunol. 1991;147:1548–56. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.