

ABSTRACT
Although peroxide and leachable metal-induced chemical modifications are among the most important quality attributes in bioprocess development, there is no mainstream characterization method covering all common modifications theoretically possible on therapeutic proteins that also gives consistent results quickly. Here, we describe a method for rapid and consistent global characterization of leachable metals- or peroxide-stressed immunoglobulin (Ig) G1 monoclonal antibodies (mAbs). Using two independent protease digestions, data-independent acquisition and data-dependent acquisition liquid chromatography high-resolution mass spectrometry, we monitored 55 potential chemical modifications on trastuzumab, a humanized IgG1 mAb. Processing templates including all observed peptides were developed on Skyline to consistently monitor all modifications throughout the stress conditions for both enzymatic digestions. The Global Characterization Data Processing Site, a universal automated data processing application, was created to batch process data, plot modification trends for peptides, generate sortable and downloadable modification tables, and produce Jmol code for three-dimensional structural models of the analyzed protein. In total, 53 sites on the mAb were found to be modified. Oxidation rates generally increased with the peroxide concentration, while leachable metals alone resulted in lower rates of modifications but more oxidative degradants. Multiple chemical modifications were found on IgG1 surfaces known to interact with FcɣRIII, complement protein C1q, and FcRn, potentially affecting activity. The combination of Skyline templates and the Global Characterization Data Processing Site results in a universally applicable assay allowing users to batch process numerous modifications. Applying this new method to stability studies will promote a broader and deeper understanding of stress modifications on therapeutic proteins.
KEYWORDS: Global characterization, leachable metals, peroxides, oxidative stress, post-translational modifications (PTM), monoclonal antibody (mAb), immunoglobulin G1 (IgG1), quantitative proteomics, high-resolution mass spectrometry (HRMS), data-independent acquisition (DIA), data-dependent acquisition (DDA), processing automation, proteomics
Introduction
Monoclonal antibodies (mAbs) are a major class of biological pharmaceuticals widely prescribed for various kinds of cancer or immune-mediated diseases.1-3 Understanding how post-translational modifications (PTMs) occur and affect mAb activity is essential to the biotherapeutics industry. Oxidizer- or leachable metal-induced chemical modifications are among the most common changes that can occur to a protein during process development.4 These modifications may affect yield,5 overall drug purity, aggregate percentage,6-8 bioavailability, and efficacy.9,10 As the field of protein bottom-up characterization has matured, modifications caused by reactive oxygen species (ROS) and metals have been increasingly defined. Previous studies using a therapeutic IgG1 have shown that peroxide (H2O2) stress could modify up to 9% of the entire protein sequence and generate cascades of degradants.11 Metals are known to primarily catalyze degradation through Fenton or Fenton-like pathways on Cys, His, Tyr, Arg, Lys, Thr, and Pro.4,12,13 The reactive sulfhydryls and thioether of Cys and Met are capable of di- or tri-oxidation.14,15 Aromatic amino acids Tyr, Phe, and especially Trp alone have over nine commonly reported oxidative degradants (Table 1), formed through quinone- or kynurenine-derivative related pathways.14,16,17 The oxidation of His generates various open-ring imidazole degradants. Other positively charged amino acids, such as Lys and Arg, may be converted to ɣ- glutamic-semialdehyde or 2-amino-adipic semialdehyde. The polar or acidic side chains of Thr, Asn, Gln, Asp, and Glu could undergo deamidation, pyroglutamate formation, or decarboxylation.12,14,15,18 Moreover, stress conditions can cause carbonylation or mono-oxidation on the aliphatic groups of almost all amino acids.12,19
Table 1.
Variable modificaitons monitored in the global characterization assay.
| Cysteine 12,13 | Tryptophan 12-15 | Methionine 12,13 | Histidine 12,13, 16-18 | Tyrosine 17-19 | Phenylalanine 17, 18 | Leucine/Isoleucine 17, 18 | Valine 17, 18 | Serine 17, 18 |
|---|---|---|---|---|---|---|---|---|
| carbamidomethylation (+57.0516)a | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) |
| oxidation (+15.9949) | dioxidation (+31.9898) | dioxidation (+31.9898) | dioxidation (+31.9898) | dioxidation (+31.9898) | dioxidation (+31.9898) | carbonylation (+13.9793) | carbonylation (+13.9793) | carbonylation (+13.9793) |
| dioxidation (+31.9898) | trioxidation (+47.9847) | trioxidation (+47.9847) | dopa-derrived quinone (+13.9793) | |||||
| trioxidation (+47.9847) | kynurenin (+3.9949) | ring opening 1 (-23.0160) | quinone (+29.9742) | |||||
| hydroxykynurenin (+19.9898) | ring opening 2 (-22.0320) | |||||||
| trptoline (+12.0000) | ring opening 3 (+4.9790) | |||||||
| quinone (+29.9742) | ring opening 4 (-10.0320) | |||||||
| unsaturated tryptophandione (+27.9585) | hydroxy-dioxidiation (+34.0055) | |||||||
| hydroxytryptophandione (+43.9534) |
| Threonine 12, 13, 17, 18 | Asparagine 17, 18 | Aspartic acid 17, 18 | Glutamine 17, 18 | Glutamic Acid 12, 13, 17, 18 | Arginine 17, 18 | Lysine 17, 18 | Proline 17, 18 |
|---|---|---|---|---|---|---|---|
| oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) | oxidation (+15.9949) |
| carbonylation (+13.9793) | demaidation (+0.9849) | decarboxylation (-30.0105) | demaidation (+0.9849) | carbonylation (+13.9793) | carbonylation (+13.9793) | carbonylation (+13.9793) | |
| pyro-glutamate (-18.0105) | decarboxylation (-30.0105) | GluSA (-43.0534) | |||||
| pyro-glutamate (-18.0105) |
The following Skyline filters were used: precursor ions, 5 ppm mass error, 0.95 isotope dot product; product ions, 10 ppm mass errors. Up to 3 modifications per peptide and a maximum of 4 miscleavages were monitored. Very few tryptic peptides above 1 miscleavages or chymotryptic peptides above 2 miscleavages were observed.
aCysteine carbamidomethylation was selected as variable modification during peptide search on Skyline, so that other cysteine modifications could be searched at equal weight. But it is selected as constant modification during data analysis on the Global Characterization Data Processing Site, so that it is not calculated as a stressed modification.
Despite the wealth of individual footprinting research focusing on individual classes of modifications or some combination thereof,11,20-22 stress studies in the pharmaceutical industry have often centered around Met oxidations or Asn deamidations.23,24 This excludes the numerous potential modification sites that may affect various important physiological functions of implicated mAbs. For example, the interaction site between IgG1 and complement C1q does not contain a single Met or Asn.25,26 In addition, multiple reports suggest that oxidations on Met H256 and Met H432 reduce the binding affinity between IgG1 and the neonatal Fc receptor (FcRn),9,27,28 while crystal structures and site-directed mutagenesis studies indicate that these two Met residues do not directly contact with FcRn or change binding affinity significantly when mutated to Ala.29,30 This suggests that modifications or degradation of other amino acids may also be responsible for that loss of binding affinity, but are currently invisible to basic bottom-up characterization techniques that search for only a few modifications on a limited number of residues. To address these discrepancies and eventually predict binding behavior changes of IgG1 with various receptors, a much deeper and wider view of chemical modifications on mAbs is needed.
In this study, fully formulated trastuzumab, a humanized IgG1 mAb, were stressed with H2O2 or leachable metals and digested by trypsin or chymotrypsin (Figure 1). Iron (Fe), Nickel (Ni), and Chromium (Cr) were used because they are the most common leachable metals from stainless-steel equipment used in various upstream and downstream processes.31 Peroxides are commonly introduced into biopharmaceutics from raw materials or excipients.4 Oxidizer (H2O2) and the leachable metals listed above were used in high concentrations to induce the maximum combination of modifications and stress-test our method (Table 1), and the data processing templates were built in Skyline (http://Skyline.ms/).32 The resulting combination of modifications were prohibitive for manual processing. The Global Characterization Data Processing Site was coded in Python with the capabilities of aligning data from multiple digestions, creating interactive modification time plots, generating sortable and downloadable modification tables, and producing Jmol code for visualizing the percentage of modifications on a three-dimensional (3-D) molecular model (Figure 1). The utility of the site is independent of the number or type of digestive enzymes, protein subunits, chemical modifications of interest, and the numbers of sample replicates. Thus, the site was designed to be commonly applicable for modification data processing of any protein or protein complex. We used trastuzumab as the study material to demonstrate the capabilities of the Global Characterization Data Processing Site because it is a well-known and well-characterized mAb that is globally marketed. Defining and refining this global characterization assay has obvious benefits for biopharmaceutical processing, as it could improve quality controls, enhance visibility in lot-to-lot comparison studies, and assess the potential impact of chemical stresses on physiological functions of protein drugs.
Figure 1.
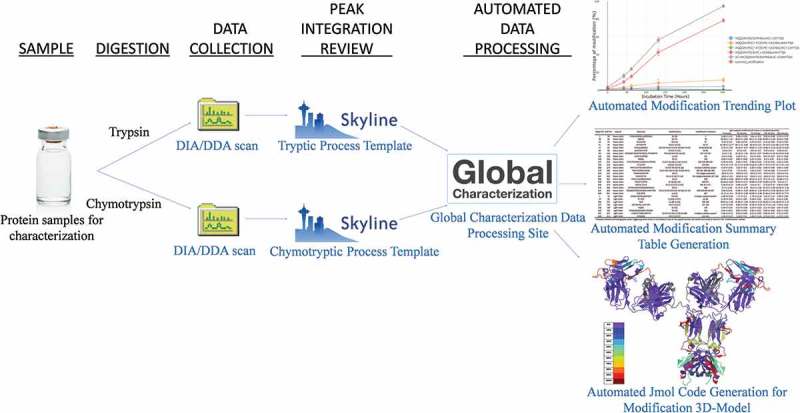
Workflow for global characterization of chemical modifications on therapeutic proteins.
Results
Data-independent acquisition vs. data-dependent acquisition
Data-independent acquisition (DIA) and data-dependent acquisition (DDA) were performed and compared across the tryptic and chymotryptic digestions of stressed trastuzumab to maximize confidence in identifying modifications. Despite slight differences in ion collection window settings (DIA mass spectrometry MS/MS collection window was 25 m/z vs 1 m/z for DDA), DIA and DDA resulted in almost identical peptide fragment profiles and peak area comparisons (two representative peptides are shown in Figure 3, panels a-l). Setting mass error filters for precursor and product ions at 5 and 10 ppm increased the identification accuracy for both acquisition methods. The noticeable difference between DIA and DDA scans was the number of data points across the precursor and product ion peaks: DDA collects more precursor MS scans at the expense of product MS/MS scans. For example, the tryptic peptide DTLM[+16]ISR scanned by DDA had 25 MS spectra on the precursor liquid chromatography (LC) peak (Figure 3(d)) and 3 MS/MS spectra, which poorly defined the shape of the product ion peak (Figure 3(e)). In contrast, the same peptide monitored by DIA had 13 MS data points on the precursor peak (Figure 3(a)) and 8 MS/MS scans (Figure 3(b)). The DIA monitoring offered enough MS/MS scans that its product peak shape and LC elution time can closely align with its corresponding precursor peak (Figure 3(a,b)). Despite this, the product ion spectra profiles generated by DIA (Figure 3(c)) and DDA (Figure 3(f)) were quite similar.
Figure 3.
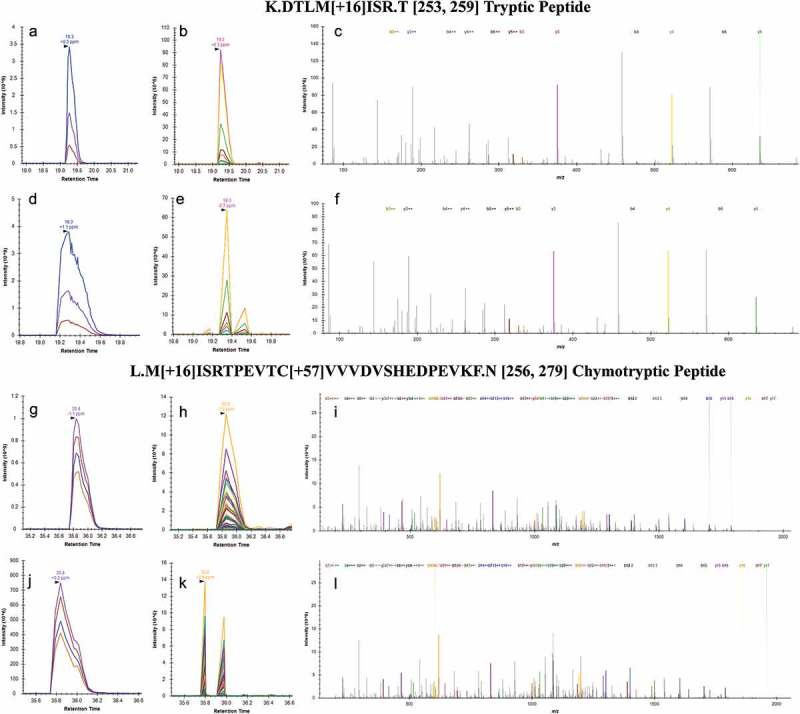
DIA versus DDA scanning for peptide identification. The elution of tryptic peptide DTLM[+16]ISR, was monitored under LC-DIA-MS precursor (a) and LC-DIA-MS/MS product (b) chromatograms. The same peptide was also observed in precursor (d) and product (e) chromatograms under DDA mode. The product ion spectra generated by DIA (c) and DDA (f) were very comparable to each other. A chymotryptic peptide (M[+16]ISRTPEVTC[+57]VVVDVSHEDPEVKF) on the overlapping protein sequence also revealed the same methionine oxidation. Its DIA precursor (g) and product (h) chromatograms were also compared to the precursor (j) and product (k) chromatograms from the DDA scan, with similar product ion spectra (I from DIA and L from DDA). Generations of modification combinations on in silico digested peptides and peptide searching from raw MS files were conducted with Skyline.
Similar trends were observed with the chymotryptic peptide on the overlapping protein sequence (M[+16]ISRTPEVTC[+57]VVVDVSHEDPEVKF): the DIA had 14 MS scans on the precursor peak compared to 16 points from the DDA scan (Figure 3(g,j)); and 8 MS/MS data points from the DIA compared to 3 from the DDA monitoring (Figure 3(h,k)). Regardless, the integrated LC-MS peak areas generated by both methods are comparable as reflected by the tight error bars across various peptides shown in Figure 4. Consistent observations are also reflected in chymotryptic digestions as well, which are illustrated in Figure 3(g–l).
Figure 4.
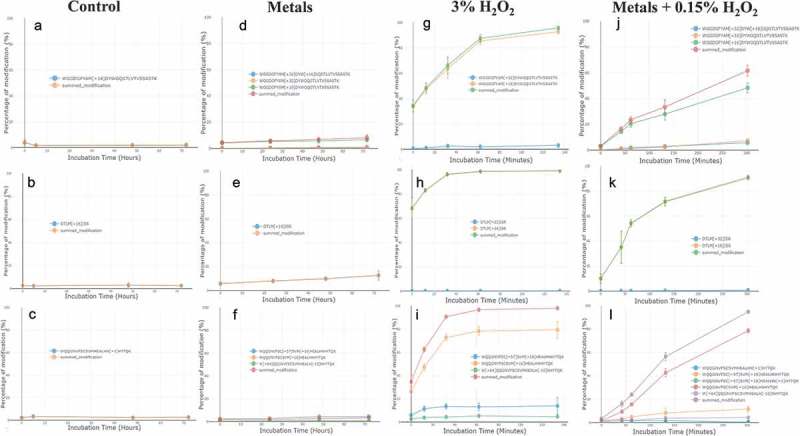
Examples of modification differences between four tested conditions. Trastuzumab was incubated with either no stressor as a negative control (a, b, and c), leachable metals (d, e, and f), H2O2 (g, h, and i), and combination of metals and H2O2 (j, k, and l). Dots and error bars represent the means and standard deviations of four LC-MS scans. Figures were generated by the Global Characterization Data Processing Site.
Manual vs. automated data processing
Probing more modifications generates tremendous amounts of data. To speed up data analysis, ensure reproducibility, and eliminate analysis error, we designed the Global Characterization Data Processing Site. To our knowledge, this is the first program that processes up to two independent digestion data files of any chemical or enzymatic digestions, allows for any number of subunits in a protein/protein complex, allows any number or any kind of PTM, and uses data from any number of replicates.
Major functions of the Global Characterization Data Processing Site were validated by comparing the output with manual processing (see Supplemental Material). Manual calculation and figure-formatting took approximately 20 min per plot, whereas calculations and figure/table/3-D molecular model generation each took less than a minute on the site. The Site provides flexibilities in changing plot scales between linear and logarithmic, zooming in and out, and has a single click to download function so that figures may be easily exported.
The Global Characterization Data Processing Site also generates a sortable summary table (Table 2) for all peptides in both digestion files according to the peptide selection rules (Materials and Methods) defined after the files have been loaded. The table includes peptide starting and ending positions, all modifications and their positions on the peptide, and modification means and standard deviations at each sampling time point. The table is sortable by amino acid, allowing the user a clear idea of the frequency of each modification. Automated data processing using the Global Characterization Data Processing Site yields the same results as manual data analysis, and ensures data processing quality and reproducibility while dramatically reducing the data analysis burden. A video tutorial and demonstration of the Global Characterization Data Processing is available online (https://www.youtube.com/watch?v=atwfpfsiB5w&feature=youtu.be and https://github.com/YaoChen1/Global-Characterization-Data-Processing-Site).
Table 2.
Modification summary tableb of metals + H2O2 co-stressed trastuzumab.
| Begin Pos | End Pos | Subunit | Sequence | Modifications | Modification Positions | Trastuzumab peptide modification % mean ± standard deviationc |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2 min | 42 min | 62 min | 132 min | 302 min | ||||||
| 1 | 19 | Heavy chain | EVQLVESGGGLVQPGGSLR | E[−18] | 1 | 3 ± 1 | 3 ± 0.6 | 3 ± 0.4 | 4 ± 0.6 | 4 ± 0.7 |
| 39 | 43 | Heavy chain | QAPGK | Q[−17] | 39 | 18 ± 1 | 20 ± 3 | 21 ± 5 | 24 ± 5 | 21 ± 3 |
| 44 | 50 | Heavy chain | GLEWVAR | W[+16] | 47 | 0.0 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.2 ± 0.0 |
| 51 | 59 | Heavy chain | IYPTNGYTR | N[+1] Y[+16] | 55 57 | 62 ± 13 | 69 ± 4 | 82 ± 8 | 79 ± 3 | 68 ± 4 |
| 77 | 87 | Heavy chain | NTAYLQMNSLR | M[+16] N[+1] Y[+14] M[+32] Y[+16] | 83 84 80 83 80 | 9 ± 2 | 17 ± 3 | 18 ± 2 | 19 ± 2 | 14 ± 3 |
| 88 | 98 | Heavy chain | AEDTAVYYCSR | C[+57] Y[+16] | 96 94 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.0 |
| 99 | 124 | Heavy chain | WGGDGFYAMDYWGQGTLVTVSSASTK | M[+32] W[+16] M[+16] | 107 110 107 | 3 ± 1 | 16 ± 1 | 24 ± 3 | 34 ± 6 | 62 ± 4 |
| 125 | 136 | Heavy chain | GPSVFPLAPSSK | L[+16] | 131 | 0.2 ± 0.1 | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.4 ± 0.1 | 0.6 ± 0.1 |
| 162 | 166 | Heavy chain | NSGAL | N[+1] | 162 | 4 ± 0.1 | 5 ± 0.1 | 5 ± 0.4 | 6 ± 0.3 | 4 ± 0.1 |
| 227 | 252 | Heavy chain | THTCPPCPAPELLGGPSVFLFPPKPK | C[+57] H[+34] L[+14] C[+57] | 230 228 238 233 | 0.1 ± 0.0 | 0.2 ± 0.0 | 0.4 ± 0.0 | 1 ± 0.5 | 3 ± 0.5 |
| 253 | 259 | Heavy chain | DTLMISR | M[+32] M[+16] | 256 256 | 10 ± 4 | 35 ± 13 | 54 ± 3 | 72 ± 3 | 91 ± 2 |
| 260 | 278 | Heavy chain | TPEVTCVVVDVSHEDPEVK | C[+57] H[+16] H[−22] | multiple oxidative peaksa | 0.1 ± 0.0 | 0.3 ± 0.0 | 0.3 ± 0.0 | 0.5 ± 0.1 | 0.7 ± 0.0 |
| 279 | 292 | Heavy chain | FNWYVDGVEVHNAK | N[+1] W[+16] H[−23] Y[+32] H[+5] | 290 281 289 282 289 | 5 ± 0.7 | 8 ± 0.5 | 8 ± 0.5 | 8 ± 0.8 | 7 ± 0.8 |
| 293 | 305 | Heavy chain | TKPREEQYNSTYR | N[+1] | 301 (deglycosylation) | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 |
| 297 | 305 | Heavy chain | EEQYNSTYR | N[+1] E[−18] Y[+16] | 301 (deglycosylation) 297 304 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 | 100 ± 0.0 |
| 306 | 321 | Heavy chain | VVSVLTVLHQDWLNGK | N[+1] | 319 | 50 ± 9 | 59 ± 1.0 | 60 ± 0.8 | 68 ± 1 | 56 ± 1 |
| 349 | 359 | Heavy chain | EPQVYTLPPSR | E[−30] Q[+14] | 349 351 | 42 ± 5 | 21 ± 4 | 21 ± 1.0 | 14 ± 5 | 12 ± 2 |
| 365 | 374 | Heavy chain | NQVSLTCLVK | N[+1] C[+57] C[+32] | 365 371 371 | 0.4 ± 0.1 | 0.5 ± 0.1 | 0.5 ± 0.1 | 0.8 ± 0.1 | 0.5 ± 0.1 |
| 375 | 396 | Heavy chain | GFYPSDIAVEWESNGQPENNYK | N[+1] | 388 | 9 ± 2 | 12 ± 3 | 13 ± 2 | 15 ± 4 | 10 ± 2 |
| 421 | 443 | Heavy chain | WQQGNVFSCSVMHEALHNHYTQK | N[+1] C[+57] M[+16] W[+64] H[−10] | 438 429 432 42 437 | 3 ± 0.4 | 16 ± 2 | 24 ± 1 | 56 ± 3 | 95 ± 1 |
| 444 | 451 | Heavy chain | SLSLSPGK | L[+14] | 447 | 41 ± 3 | 45 ± 6 | 42 ± 4 | 34 ± 5 | 28 ± 8 |
| 1 | 18 | Light chain | DIQMTQSPSSLSASVGDR | M[+16] | multiple oxidative peaksa | 0.7 ± 0.1 | 2 ± 0.2 | 2 ± 0.2 | 3 ± 1 | 4 ± 1.0 |
| 19 | 24 | Light chain | VTITCR | C[+32] C[+57] | 23 23 | 0.1 ± 0.0 | 0.2 ± 0.1 | 0.2 ± 0.0 | 0.2 ± 0.0 | 0.1 ± 0.0 |
| 104 | 107 | Light chain | VEIK | E[−30] I[+14] | 105 106 | 38 ± 1 | 32 ± 2 | 32 ± 3 | 29 ± 1 | 34 ± 1 |
| 127 | 142 | Light chain | SGTASVVCLLNNFYPR | N[+1] C[+57] | 137 134 | 7 ± 1.0 | 9 ± 0.4 | 10 ± 0.5 | 11 ± 1 | 8 ± 0.2 |
| 146 | 149 | Light chain | VQWK | W[+16] | 148 | 0.0 ± 0.0 | 0.2 ± 0.0 | 0.2 ± 0.0 | 0.3 ± 0.0 | 0.4 ± 0.0 |
| 150 | 169 | Light chain | VDNALQSGNSQESVTEQDSK | V[+16] | 163 | 0.0 ± 0.0 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.1 ± 0.0 | 0.1 ± 0.0 |
| 191 | 207 | Light chain | VYACEVTHQGLSSPVTK | C[+57] H[+16] C[+16] C[+48] | multiple oxidative peaksa | 0.3 ± 0.2 | 0.2 ± 0.0 | 0.2 ± 0.0 | 0.5 ± 0.3 | 0.7 ± 0.1 |
| 208 | 214 | Light chain | SFNRGEC | N[+1] C[+57] | 210 214 | 1 ± 0.4 | 2 ± 0.6 | 2 ± 0.4 | 3 ± 0.5 | 2 ± 0.4 |
aMultiple oxidized chromatographic peaks were present suggesting more mono- or di-oxidation sites on the peptide other than the amino acid residues shown in the “Modifications” column
bThe table was automatically generated and downloaded from the Global Characterization Data Processing Site and formatted in Excel
cAll percentage of modifications and standard deviations were rounded to one decimal place when the number is less or equal to 1% or zero decimal when above 1%
Comparing different stress conditions
Figure 4 shows examples of modification trends (automatically generated by the Global Characterization Data Processing Site) for three representative peptides, demonstrating differences among the tested conditions. The no stress control (Figure 4(a–c)) showed no changes across the 72-h incubation time. Although these experiments were conducted in the presence of peroxide scavengers (20 mM histidine, 0.02% PS80), oxidative species were still capable of modifying the protein over time. Trastuzumab incubated with metals (Figure 4(d–f)) demonstrated linear, slow, and steady increases in modification. While 3% of hydrogen peroxide rapidly oxidized many sites, reaching close to 100% summed modifications on the three illustrated peptides (Figure 4(g–i)).
Multiple reports have suggested that metals promote oxidation of proteins through catalyzing oxygen free-radical generation from peroxides.4,6,14,33 Unexpectedly, the combined condition of metals and H2O2 did not lead to a faster or higher overall level of oxidation (Figure 4(j-l)). The oxidation rate appears primarily determined by the concentration of H2O2, and is minimally impacted by the presence of metals in the tested time span.
Although the overall rate of oxidation did not increase in the combined condition, it was noted that the tri-oxidized peptide H99-H124 was formed only under both stresses with metals and was not present with 3% H2O2 incubation.
Figure 5 and Table 2, both generated automatically by the Global Characterization Data Processing Site, give a global view of modifications at different time points. The rainbow gradient in Figure 5 shows modification quantity at the beginning (control) and end points of each stress condition. The 3-D molecular model of other stress time points is available in the Supplemental Materials. In Figure 5, multiple peptides (the deglycosylation site Asn H301was considered 100% modified) show some level of deamidation and pyroglutamate formation at the start of the control condition, providing a baseline of modification likely due to the higher temperature incubation during protein denaturation and deglycosylation steps.
Figure 5.
IgG1 structure with mapped modification levels at initial and ending time points of three stress conditions. Color code represents percentage means calculated from quadruplicated scans (3 DIA + 1 DDA) of triplicated tryptic and chymotryptic digestions. IgG1 was mapped to construct of 1HZH by the Global Characterization Data Processing Site.
Among all 52 modifications detected across the various conditions studied (Table 2), a higher percentage of modified peptides was observed in the complementarity-determining region (CDR) loops, and the Fc portion (Figure 5). The modified regions correlate well with the water molecule distributed area in the 1HZH X-ray crystal structure (Figure 6), suggesting that water accessible sites are generally more prone to modification under the tested oxidative stresses.
Figure 6.
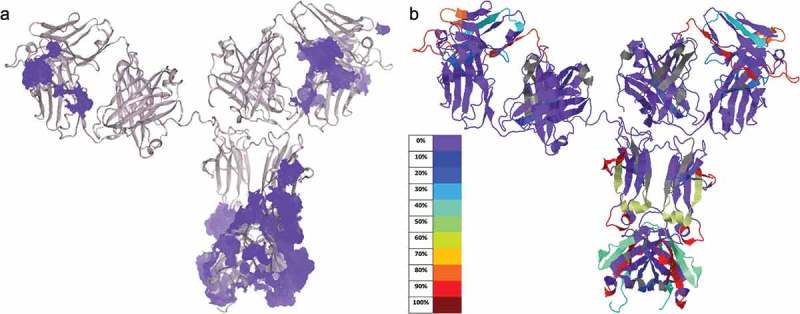
Comparing modifications from oxidative stress to water accessible areas on IgG1. (a) Model of IgG1 structure 1HZH showing water molecules (light purple) recorded in the PDB file. (b) Modifications of mAb from 3% H2O2 treatment at 132 min were mapped onto the 1HZH structure by the Global Characterization Data Processing Site.
Discussion
Antibodies accomplish their physiological functions through specific binding to targets on the CDR, and through their association, via the CH2 or CH3 domains, with complement proteins, FcRn, or Fc receptors (FcɣR). The interaction of an antibody with complement proteins initiates attack on pathogenic cell membranes and enhances inflammation. Its union with FcɣR activates various cell-mediated immunity. Additionally, attachment to FcRn recycles antibodies in the bloodstream and is responsible for the half-life of antibody pharmaceuticals.34 Beyond in vivo functionality, production and purification of antibodies relies on their strong affinities to bacterial proteins A and G. As a result, chemical modifications may change antibody binding sites, affecting potency, quality, or yield.4
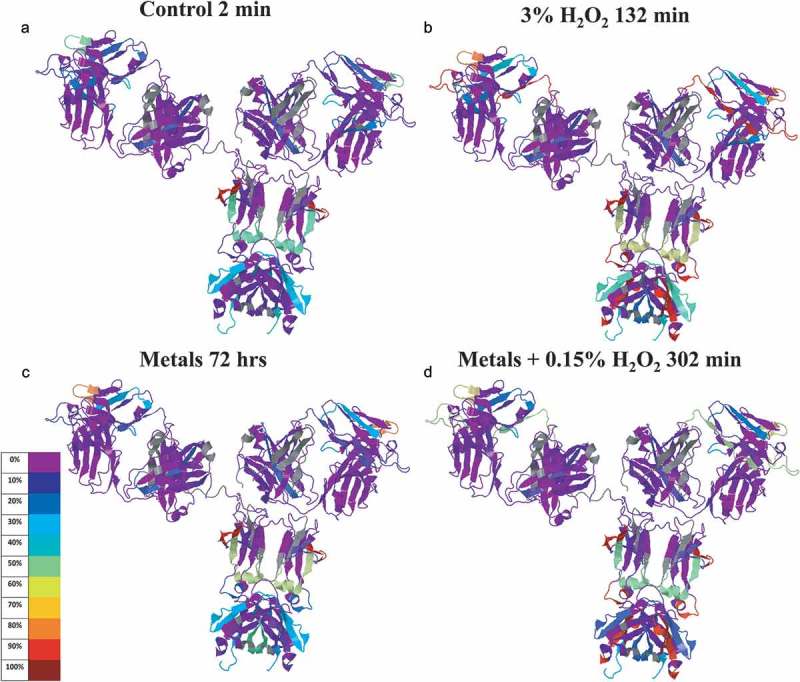
The IgG Fc fragment − FcɣRIII complex crystal structure reported by Sondermann et. al. describes the interface between these two molecules in detail.35 The most important binding-determining region on the Fc was the area from LeuH238 to SerH243, which provides six hydrogen bonds that link to FcɣRIII. Other studies also show that mutagenesis on Leu H238 (to Ala) reduced IgG binding affinity to all FcɣR.29 In our study, Leu H238 carbonylation was observed in both H2O2 and H2O2 plus metal incubations (Figure 2). Although Leu H238 carbonylation rarely has been monitored in the past, our data combined with historic studies indicate that it should be included in biotherapeutic stability studies because it may affect the binding of all FcɣR.
Figure 2.
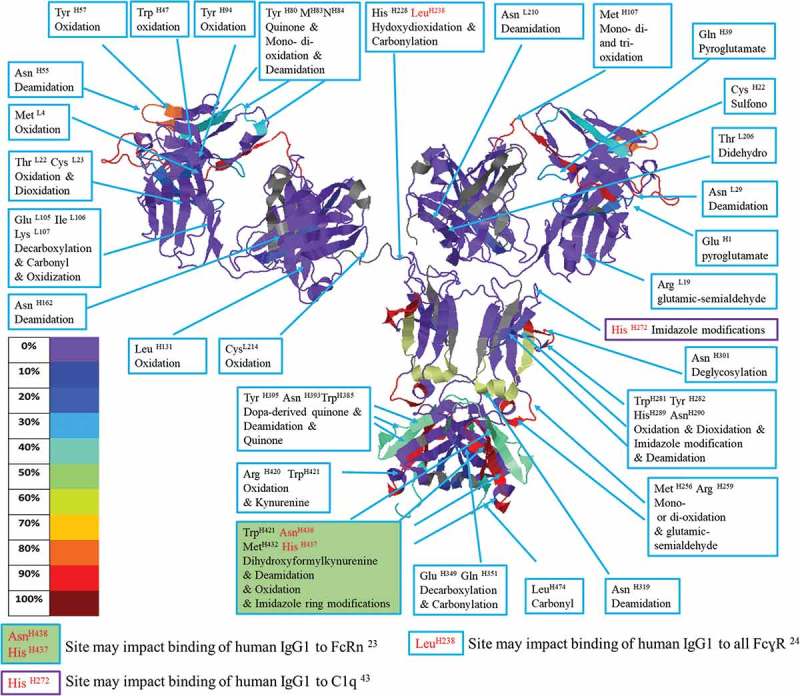
IgG1 structure with mapped modification levels and annotated degraded residues. A model of IgG1 stressed under 3% H2O2 for 132 min constructed using the structure of 1HZH by Jmol and the Global Characterization Data Processing Site.
In a comprehensive study, Shields et. al. individually mutated every solvent-exposed residue in the IgG1 CH2 and CH3 domains and tested the mutant binding affinities to FcɣRI, FcɣRII, FcɣRIII, and FcRn. None of the Met site-directed mutations caused significant changes of IgG1 affinity to any tested receptors. Specifically, Met H256 and Met 358 mutations (Met 358 is not in trastuzumab) had no effect on binding to FcRn or any FcɣR. Additionally, the crystal structure of an FcRn/heterodimeric Fc complex showed that the pH-sensitive binding between Fc and FcRn was predominately controlled by His 433 and His 435 (equivalent His H437 H439 in this study) and did not note the importance of any Met residues to binding.29,30 Nevertheless, multiple subsequent studies have claimed that oxidations on Met H256, Met 358, or Met H432 are responsible for reduced affinity to FcRn or FcɣRs.9,27,28 Although Met oxidation is common under oxidative stresses, studies that do not monitor additional modifications on residues known to be part of binding interfaces may miss the actual PTM that causes altered binding affinity.12,19 Both the Shields et. al. mutagenesis study and the Martin et. al. crystal structure indicated that His 433, Asn 434, and His 435 (equivalent to His H437 H439 and Asn H438 in this study) are among the key binding sites to FcRn. In our experiment, two elevated imidazole modifications were observed on His H437 when trastuzumab was incubated with metals + H2O2 (4.8%) or only with metals (4.2%), and Asn H438 was found to be deamidated consistently in metals + H2O2 stressed conditions (Figure 4). Although we cannot conclude that these specific modifications on His H437 and Asn H438 are the common cause of reduced binding to FcRn under oxidative stress, we do prescribe monitoring these modifications in future studies.
The mutation of His H272 to Phe has been shown to enhance complement-dependent cytotoxicity through its interaction with complement protein C1q.36 In our study, we observed this residue to be modified to 2-oxo-imidazole in both H2O2 and metals + H2O2 conditions, which could potentially modulate antibody-C1q binding. Met H256 and Met H432, which impact protein A and G binding affinity,5,37 were found to be up to 90% oxidized (Figures 2, 4, 5 and Table 2).
The modifications discussed here may be responsible for the changes in binding affinities observed in previous oxidative stress studies that only searched for more common, but perhaps less physiologically relevant oxidations. We suggest that this global characterization method reveals modifications that have long been hiding in plain sight, unobserved by current standard techniques.
In summary, our global characterization assay uses conventional ultra-high-performance liquid chromatography high-resolution mass spectrometry (UHPLC HR-MS) to monitor 55 degradants on 18 amino acids of trypsin and chymotrypsin digested trastuzumab, stressed by H2O2 or leachable metals. Dual digestions enhanced the sequence coverage and modification visibility. Peptides were selected allowing up to 3 modifications per peptide, a maximum of 4 miscleavages, 5 ppm precursor and 10 ppm product ion mass filters, three rounds of false positive eliminations, and cross-validation of peptides from two digests. Fifty-two modification sites were found on 14 amino acids. Skyline data processing templates were created for fast and consistent modification searches. The Global Characterization Data Processing Site was programmed for fast, accurate, and automatic plotting of modification trends, generation of modification summary tables, and production of the 3-D molecular models.
Several modifications were observed in the known binding sites of IgG1 Fc to FcɣRs, complement protein C1q, and FcRn (Figure 2) and suggest future experiments should target the potential connection between these degradations and interactions with corresponding binding proteins.
Overall, this fast, consistent, and accurate assay provides unprecedented visibility into chemical modifications of fully formulated trastuzumab, and furnishes the foundations for a new standard in stability and stress studies of biotherapeutics. We hope that by using this assay, new observations may be used to better predict binding behavior between IgG1 and its many targets and receptors.
Materials and methods
Chemicals and reagents
All reagents were of analytical grade or purer. For the convenience of dilutions, pure water density was approximated as 1 kg/L for all weight/volume (w/v) or weight/weight (w/w) concentrations. Trastuzumab (humanized IgG1DB00072) was produced and formulated in 20 mM histidine 0.02% (w/v) PS80 pH 6.0 on site at Catalent Biologics (Bloomington, IN). Ammonium bicarbonate for LC-MS, CrCl2, FeCl3, NiCl2, and peroxide (H2O2) 30% (w/w) in water were acquired from Sigma-Aldrich (St. Louis, MO). LC-MS grade acetonitrile, formic acid, and water with 0.1% formic acid were purchased from Fisher Chemical (Fair Lawn, NJ). Single use iodoacetamide (IAA) and dithiothreitol (DTT) was purchased from Thermo Scientific (Rockford, IL). Sequencing-grade modified trypsin and sequencing grade chymotrypsin was purchased from Promega Corporation (Madison, WI). Rapid™ PNGase F was purchased from New England Biolab Inc (Ipswich, MA).
Protein stress conditions
Formulated trastuzumab was stressed under the following conditions: a. incubation with 3% (w/v) H2O2 for 2, 12, 32, 63, 132 min; b. 1 ppm Fe, 100 ppb Ni, 100 ppb Cr mixture for 0, 24, 48, and 72 h; c. 1 ppm FeCl3, 100 ppb NiCl2, 100 ppb CrCl2 mix, 0.15% (w/v) H2O2 for 2, 42, 62, 132, and 302 min, and d, no stressor for control for 0, 5, 48, and 72 h. The sampling time points above were titrated during scouting/method-establishing experiments (same stress conditions, longer stress time points, single digestion for each time point, see Supplemental Materials). All conditions were incubated at 25°C (±1°C). Each sample was buffer exchanged three times into formulation buffer (400 µL, 20 mM Histidine, 0.02% (w/v) PS80, pH 6.0) with 10 kDa Millipore Amicon molecular weight cutoff filter with centrifugation (14,000 × g, 10 min each cycle). Before the first buffer exchange, every sample was centrifuged at 14,000 × g for 2 min and spiked with 3x the volume of formulation buffer. Thus, the first time point for all conditions was considered 2 min. All samples were titrated back to approximately 1 mg/mL with formulation buffer and stored at −80°C before digestion.
Sample reduction, deglycosylation, and digestion
The use of multiple digestion enzymes to enhance sequence visibility and coverage has been reported.38-41 Here, we digest each stressed sample separately in triplicate with either trypsin or chymotrypsin. 200 µg of protein was added to 100 µL of 20 mM DTT, 100 mM NH4HCO3 pH 8.0, and incubated at 56°C for 45 min. After brief cooling, reduced proteins were mixed with 25 mM IAA in 100 µL of 100 mM NH4HCO3 pH 8.0, and incubated at room temperature in the dark for 30 min. The alkylation reaction was quenched as the samples were buffer exchanged into 100 mM NH4HCO3 pH 8.0 by centrifuging for 10 min at 14,000 × g in a 10 kDa Amicon filter. After the centrifugation, the remaining 40 µL of the sample above the filter was spiked with 10 µL Rapid PNGase F buffer and 2 µL Rapid PNGase F, and then incubated at 50°C for 15 min. The deglycosylated proteins were buffer exchanged again into 100 mM NH4HCO3 pH 8.0 by centrifuging for 15 min at 14,000 × g in a 10 kDa Amicon filter. The retained sample was recovered by brief centrifugation (4000 × g for 3 min) into protein LoBind® vials, where they were spiked with either 10 µg of trypsin or chymotrypsin and reconstituted to approximately 200 µL with 100 mM NH4HCO3 pH 8.0. The digestion lasted 4 h at 37°C with 600 RPM orbital shaking. Digested samples were kept in a 4°C autosampler and analyzed within 48 h.
LC-MS analysis
Chymotrypsin cleaves at more sites than trypsin, and thus generally creates shorter peptides that elute earlier than tryptic peptides in reverse phase chromatography (see scouting experiments in Supplemental Materials). To ensure coverage of all peptides, two chromatographic elution gradients and two DIA methods were tailored for both tryptic and chymotryptic digests. Either an 80- or 70-min gradient on Waters BEH C18, 2.1 × 150 mm, 1.7 µm column was used for the digests of trypsin or chymotrypsin, respectively. Detailed LC methodology is available in the Supplemental Materials.
Each digestion was run on a Thermo Scientific Q-Exactive Plus (Extended Mass Resolution) mass spectrometer. The mass spectrometer was calibrated and tuned every 72 h. Every digest was monitored under DIA mode, and one of each triplicate digestion set was scanned under DDA mode. For trypsin digests: MS scan collects precursor ions from 300 to 2000 atomic mass unit (amu) at a resolution of 70,000; MS/MS scans 350–1650 m/z on 54 isolation windows, 25 m/z span per isolation window, 0.4 m/z overlap per isolation, 27 loop counts per MS scan, a resolution of 17,500, and HCD collision energy of 28. For chymotryptic peptide monitoring: precursor ion scan was the same as above; MS/MS collected product ions from 225 to 1210 m/z on 40 isolation windows, 25 m/z span per window, 0.4 m/z overlap, 20 loop counts, at the resolution of 17,500, and HCD collision energy of 28. Detailed instrument settings are in the Supplemental Materials.
One of each triplicate digest set was monitored using a standard DDA method. MS scan collects precursor ions from 200 to 2000 amu at resolution of 70,000; AGC target of 1e6, maximum injection time at 100 ms; MS/MS scan collects product ions at a resolution of 35,000; AGC target of 5e5, maximum injection time of 500 ms, 20 loop counts, 1 m/z isolation window, 28 NCE collision energy, 2e3 minimum AGC target, 4e4 intensity threshold, and a 20 s dynamic exclusion.
Experimental design
Scouting experiments were conducted to optimize LC-MS conditions, choose stress time courses, and generate Skyline templates for peptide identification. The scouting experiments are described in the Supplemental Materials. Briefly, fully formulated trastuzumab was stressed by H2O2, leachable metals, a combination of both, or incubated as a negative control with no stressors. Aliquots of samples were taken at least four time points and digested independently by trypsin or chymotrypsin. Data from scouting experiments (supplemental materials) are results of duplicate LC-MS runs of a single digestion.
The final global characterization experiments included a non-stressed control condition, additional intermediate time points, and triplicate digestions with four (3 DIA + 1 DDA) LC-MS analyses per condition.
Data analysis
Amino acids were counted from the N- to C-terminus of the trastuzumab sequence (DB00072) (EU numbering was not used in this report). To be consistent, referenced works that used EU numbering were all converted. Skyline32 -Daily (64-bit) 4.2 was used for peptide-spectrum matching. For transition settings, precursor ions with up to 3 charges were searched with a 5 ppm mass error tolerance, and b and y product ions with up to 2 charges were searched with a 10 ppm window. Light chain and heavy chain were searched separately to reduce the computational burden. All combinations of modifications were generated using Skyline or Skyline-Daily following importing of FASTA sequences (https://www.drugbank.ca/drugs/DB00072).
To accommodate the large data volume, both chains (light or heavy) of each digestion went through four levels of searches. All modifications searched were variable (Table 1). Cysteine carbamidomethylation was selected as a variable modification during peptide search on Skyline, so that other cysteine modifications could be searched with equal weight in Skyline. Cysteine carbamidomethylation was then selected as a constant modification during data analysis on the Global Characterization Data Processing Site, so that it is not calculated as a stressed modification (Table 1). Variable modifications and the number of miscleavages settings vary in each round of search and are detailed in Supplemental Material.
Peptides of both chains that were positively identified after all four rounds of searches were combined (by pooling identified peptide transitions from different Skyline search files and deleting the redundant identifications) into one Skyline data processing template. At least two experienced MS specialists then cross reviewed surviving peptides according to the false positive prevention criteria detailed in Supplemental Materials. The processing templates generated were used for rapid and consistent global characterization in the experiments described in this study. Following LC-MS peak integration adjustments and cross-review, csv or excel files exported from the Skyline templates were imported to Global Characterization Data Processing Site V0.1 (beta) for automatic PTM trend plotting, sortable PTM summary table generation, and 3-D structural model generation on Jmol (Figure 1). Both the Skyline templates and the source code of the Global Characterization Data Processing Site were uploaded to https://github.com/YaoChen1/Global-Characterization-Data-Processing-Site.
Global characterization data processing site
Manually processing all the PTM data, plotting peptide modifications, and typing scripts in Jmol for molecular modification visualization would take approximately two months’ time for each stress condition. Thus, the Global Characterization Data Processing Site was built to automate all the work above, and generate a sortable summary table, with completion of all tasks within 4 min or less.
The Global Characterization Data Processing Site was coded on Python 3.6. Multiple packages were used and are detailed in https://github.com/YaoChen1/Global-Characterization-Data-Processing-Site. Users can import any type of enzymatic or chemical digestion data. Either one or two independent digestions of the same protein can be imported. After data import, users are then prompted to choose subunits of the target protein for data processing. In the case of mAbs, the user could choose either or both light and heavy chains for processing; if an 80S ribosome complex were to be analyzed, the user could choose any number of the 82 ribosomal subunits. The Site is designed to process modification data for any protein or protein complex with any numbers of subunits.
The percentage of each modified peptide is calculated as: peak area of modified peptide divided by the sum of peak areas of all forms of this peptide, where peak areas are the summed peak areas from all detected charge states of MS1 precursor ions. The Global Characterization Data Processing Site can process any number of experimental replicates. The modification plots and error bars are means and standard deviations of replicates at each sampling time point. In our experiments, modification mean and standard deviations at each time point come from a total of four MS runs on triplicate digestions, including one DIA MS analysis for each digestion plus one DDA from one of the triplicate digestion sets.
The Global Characterization Data Processing Site can also handle any number of miscleavages. If multiple overlapping non-miscleaved/miscleaved peptides that cover the same area on the protein sequence are found, the site will follow the preferred peptide selection rule, which maximizes total coverage and gives preference to less miscleaved peptides. The percentage of modifications of the preferred peptides can be visualized in trending plots, summarized into a sortable and downloadable table, and condensed into a Jmol code that encodes the 3-D structural model of the analyzed protein (Figures 2, 5, and 6(b)). The redundant peptides, deselected by the peptide selection rule, are also plotted, but their %PTMs are not considered in the table or the 3-D structural model. If data files from two independent digestions are uploaded, peptides from both files are sorted by the peptide selection rules. The site maps out the protein sequence that is not covered (gap sequence) from peptides in the primary digestion file and finds gap-filling peptides in the preferred peptides of the secondary digestion file to increase the global characterization map coverage. The gap-filling peptides are also plotted, and are included in the summary table as well as in the Jmol code for 3-D structural model.
Users are also able to interact with the PTM trending plots: change the axis scales between linear or logarithmic, zoom in or out at any axial number, or download the plot as high-resolution images.
Each function of the site was validated by comparison with the manually processed results calculated using Microsoft Excel 2013 and GraphPad Prism 7.03 during scouting experiments.
For more detailed tutorial on the Global Characterization Data Processing Site, visit https://www.youtube.com/watch?v=atwfpfsiB5w&feature=youtu.be
Data representation and figure generation
Automated data calculation and kinetics plots were created on the Global Characterization Data Processing Site V0.1 (beta). Global modifications were mapped onto the crystal structure of IgG1 (PDB number: 1HZH) by Jmol 14.27.2, with the Jmol code automatically generated by Global Characterization Data Processing Site V0.1 (beta). The amino acid sequence of trastuzumab was aligned with the sequences of 1HZH using Global Characterization Data Processing Site V0.1 (beta) to ensure the positional correctness of modifications onto the crystal structures shown in Figures 1, 2, 5, and 6. The water molecule rendered 1HZH structure was generated using The Protein Imager online molecular viewer (https://3dproteinimaging.com).
Acknowledgments
The authors greatly appreciate the cell culture and purification teams who supplied the trastuzumab for analytical studies at Catalent Biologics, Bloomington. We appreciate Bettina Kehoe, who graciously proofread, and grammar edited this manuscript. We are also grateful that Brendan MacLean and Nicolas Shulman of the University of Washington upgraded the Skyline maximum number of FASTA imported peptides to 200,000 and the maximum number of transitions to 5,000,000, facilitating data processing.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Harris M. Monoclonal antibodies as therapeutic agents for cancer. Lancet Oncol. 2004;5:292–302. doi: 10.1016/S1470-2045(04)01467-6. [DOI] [PubMed] [Google Scholar]
- 2.Reichert JM. Monoclonal antibodies as innovative therapeutics. Curr Pharm Biotechnol. 2008;9:423–30. doi: 10.2174/138920108786786358. [DOI] [PubMed] [Google Scholar]
- 3.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. 2010;10:301–16. doi: 10.1038/nri2761. [DOI] [PubMed] [Google Scholar]
- 4.Wang W, Ignatius AA, Thakkar SV. Impact of residual impurities and contaminants on protein stability. J Pharm Sci. 2014;103:1315–30. doi: 10.1002/jps.23931. [DOI] [PubMed] [Google Scholar]
- 5.Gaza-Bulseco G, Faldu S, Hurkmans K, Chumsae C, Liu H. Effect of methionine oxidation of a recombinant monoclonal antibody on the binding affinity to protein A and protein G. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;870:55–62. doi: 10.1016/j.jchromb.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Nguyen TH, Schöneich C, Borchardt RT. Aggregation and precipitation of human relaxin induced by metal-catalyzed oxidation. Biochemistry. 1995;34:5762–72. doi: 10.1021/bi00017a008. [DOI] [PubMed] [Google Scholar]
- 7.Khossravi M, Shire SJ, Borchardt RT. Evidence for the involvement of histidine A(12) in the aggregation and precipitation of human relaxin induced by metal-catalyzed oxidation. Biochemistry. 2000;39:5876–85. doi: 10.1021/bi9924720. [DOI] [PubMed] [Google Scholar]
- 8.Luo Q, Joubert MK, Stevenson R, Ketchem RR, Narhi LO, Wypych J. Chemical modifications in therapeutic protein aggregates generated under different stress conditions. J Biol Chem. 2011;286:25134–44. doi: 10.1074/jbc.M110.160440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R, et al. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol. 2011;48:860–66. doi: 10.1016/j.molimm.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 10.Wei Z, Feng J, Lin HY, Mullapudi S, Bishop E, Tous GI, Casas-Finet J, Hakki F, Strouse R, Schenerman MA, et al. Identification of a single tryptophan residue as critical for binding activity in a humanized monoclonal antibody against respiratory syncytial virus. Anal Chem. 2007;79:2797–805. doi: 10.1021/ac062311j. [DOI] [PubMed] [Google Scholar]
- 11.Deperalta G, Alvarez M, Bechtel C, Dong K, McDonald R, Ling V. Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. MAbs. 2013;5:86–101. doi: 10.4161/mabs.22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu G, Chance MR. Radiolytic modification of sulfur-containing amino acid residues in model peptides: fundamental studies for protein footprinting. Anal Chem. 2005;77:2437–49. doi: 10.1021/ac0484629. [DOI] [PubMed] [Google Scholar]
- 13.Kumar S, Zhou S, Singh SK. Metal ion leachates and the physico-chemical stability of biotherapeutic drug products. Curr Pharm Des. 2014;20:1173–81. doi: 10.2174/13816128113199990063. [DOI] [PubMed] [Google Scholar]
- 14.Stadtman ER. Oxidation of free amino acids and amino acid residues in proteins by radiolysis and by metal-catalyzed reactions. Annu Rev Biochem. 1993;62:797–821. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 15.Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–18. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- 16.Plowman JE, Deb-Choudhury S, Grosvenor AJ, Dyer JM. Protein oxidation: identification and utilisation of molecular markers to differentiate singlet oxygen and hydroxyl radical-mediated oxidative pathways. Photochem Photobiol Sci. 2013;12:1960–67. doi: 10.1039/c3pp50182e. [DOI] [PubMed] [Google Scholar]
- 17.Perdivara I, Deterding LJ, Przybylski M, Tomer KB. Mass spectrometric identification of oxidative modifications of tryptophan residues in proteins: chemical artifact or post-translational modification? J Am Soc Mass Spectrom. 2010;21:1114–17. doi: 10.1016/j.jasms.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jefferis R. Posttranslational modifications and the immunogenicity of biotherapeutics. J Immunol Res. 2016;2016:5358272. doi: 10.1155/2016/5358272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu G, Chance MR. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal Chem. 2005;77:4549–55. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Shen W, Rempel D, Monsey J, Vidavsky I, Gross ML, Bose R., et al. Carboxyl-group footprinting maps the dimerization interface and phosphorylation-induced conformational changes of a membrane-associated tyrosine kinase. Mol Cell Proteomics. 2011;10:M110.005678. doi: 10.1074/mcp.M110.005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Charvátová O, Foley BL, Bern MW, Sharp JS, Orlando R, Woods RJ. Quantifying protein interface footprinting by hydroxyl radical oxidation and molecular dynamics simulation: application to galectin-1. J Am Soc Mass Spectrom. 2008;19:1692–705. doi: 10.1016/j.jasms.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collier TS, Diraviyam K, Monsey J, Shen W, Sept D, Bose R. Carboxyl group footprinting mass spectrometry and molecular dynamics identify key interactions in the HER2-HER3 receptor tyrosine kinase interface. J Biol Chem. 2013;288:25254–64. doi: 10.1074/jbc.M113.474882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowak CK, Cheung JM, Dellatore S, Katiyar A, Bhat R, Sun J, Ponniah G, Neill A, Mason B, Beck A, et al. Forced degradation of recombinant monoclonal antibodies: A practical guide. MAbs. 2017;9:1217–30. doi: 10.1080/19420862.2017.1368602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Li X, Liu YH, Richardson D, Li H, Shameem M, et al. Simultaneous monitoring of oxidation, deamidation, isomerization, and glycosylation of monoclonal antibodies by liquid chromatography-mass spectrometry method with ultrafast tryptic digestion. MAbs. 2016;8:1477–86. doi: 10.1080/19420862.2016.1196521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, Meng Y G., Mulkerrin M G., et al. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol. 2000;164:4178–84. doi: 10.4049/jimmunol.164.8.4178. [DOI] [PubMed] [Google Scholar]
- 26.Thommesen JE, Michaelsen TE, Løset G, Sandlie I, Brekke OH. Lysine 322 in the human IgG3 C(H)2 domain is crucial for antibody dependent complement activation. Mol Immunol. 2000;37:995–1004. doi: 10.1016/S0161-5890(01)00010-4. [DOI] [PubMed] [Google Scholar]
- 27.Bertolotti-Ciarlet A, Wang W, Lownes R, Pristatsky P, Fang Y, McKelvey T, et al. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc gamma receptors. Mol Immunol. 2009;46:1878–82. doi: 10.1016/j.molimm.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 28.Pan H, Chen K, Chu L, Kinderman F, Apostol I, Huang G. Methionine oxidation in human IgG2 Fc decreases binding affinities to protein A and FcRn. Protein Sci. 2009;18:424–33. doi: 10.1002/pro.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 30.Martin WL, West AP, Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 2001;7:867–77. [DOI] [PubMed] [Google Scholar]
- 31.Zhou S, Evans B, Schöneich C, Singh SK. Biotherapeutic formulation factors affecting metal leachables from stainless steel studied by design of experiments. AAPS PharmSciTech. 2012;13:284–94. doi: 10.1208/s12249-011-9747-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–68. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stadtman ER, Oliver CN. Metal-catalyzed oxidation of proteins. Physiological consequences. J Biol Chem. 1991;266:2005–08. [PubMed] [Google Scholar]
- 34.Schroeder HW, Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. 2010;125:S41–52. doi: 10.1016/j.jaci.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406:267–73. [DOI] [PubMed] [Google Scholar]
- 36.Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs. 2010;2:181–89. doi: 10.4161/mabs.2.5.13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sauer-Eriksson AE, Kleywegt GJ, Uhlén M, Jones TA. Crystal structure of the C2 fragment of streptococcal protein G in complex with the Fc domain of human IgG. Structure. 1995;3:265–78. doi: 10.1016/S0969-2126(01)00157-5. [DOI] [PubMed] [Google Scholar]
- 38.Tsiatsiani L, Heck AJ. Proteomics beyond trypsin. Febs J. 2015;282:2612–26. doi: 10.1111/febs.13287. [DOI] [PubMed] [Google Scholar]
- 39.van der Post S, Thomsson KA, Hansson GC. Multiple enzyme approach for the characterization of glycan modifications on the C-terminus of the intestinal MUC2mucin. J Proteome Res. 2014;13:6013–23. doi: 10.1021/pr500874f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chiva C, Ortega M, Sabidó E. Influence of the digestion technique, protease, and missed cleavage peptides in protein quantitation. J Proteome Res. 2014;13:3979–86. doi: 10.1021/pr500294d. [DOI] [PubMed] [Google Scholar]
- 41.Biringer RG, Amato H, Harrington MG, Fonteh AN, Riggins JN, Hühmer AF. Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap LC-MS/MS. Brief Funct Genomic Proteomic. 2006;5:144–53. doi: 10.1093/bfgp/ell026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.