

ABSTRACT
Biotherapeutics may contain a multitude of different post-translational modifications (PTMs) that need to be assessed and possibly monitored and controlled to ensure reproducible product quality. During early development of biotherapeutics, unexpected PTMs might be prevented by in silico identification and characterization together with further molecular engineering. Mass determinations of a human IgG1 (mAb1) and a bispecific IgG-ligand fusion protein (BsAbA) demonstrated the presence of unusual PTMs resulting in major +80 Da, and +16/+32 Da chain variants, respectively. For mAb1, analytical cation exchange chromatography demonstrated the presence of an acidic peak accounting for 20%. A + 79.957 Da modification was localized within the light chain complementarity-determining region-2 and identified as a sulfation based on accurate mass, isotopic distribution, and a complete neutral loss reaction upon collision-induced dissociation. Top-down ultrahigh resolution MALDI-ISD FT-ICR MS of modified and unmodified Fabs allowed the allocation of the sulfation to a specific Tyr residue. An aspartate in amino-terminal position-3 relative to the affected Tyr was found to play a key role in determining the sulfation. For BsAbA, a + 15.995 Da modification was observed and localized to three specific Pro residues explaining the +16 Da chain A, and +16 Da and +32 Da chain B variants. The BsAbA modifications were verified as 4-hydroxyproline and not 3-hydroxyproline in a tryptic peptide map via co-chromatography with synthetic peptides containing the two isomeric forms. Finally, our approach for an alert system based on in-house in silico predictors is presented. This system is designed to prevent these PTMs by molecular design and engineering during early biotherapeutic development.
KEYWORDS: Biotherapeutics, fab purification, MALDI-ISD FT-ICR, hydroxyproline, mass spectrometry, PTM, post-translational modification, sulfotyrosine
Introduction
The development of biotherapeutics requires extensive characterization of the molecular purity, homogeneity, integrity, assessment of potential critical quality attributes (pCQAs), including the analysis of heterogeneities from post-translational modifications (PTMs). PTMs occur at distinct amino acid side chains or peptide linkages, and well-known chemical antibody modifications include deamidation, isomerization, oxidation, reduction, glycation, cysteinylation, and trisulfides. Common heterogeneities due to enzyme-catalyzed PTMs of therapeutic antibodies include Fc domain N-glycosylation, heavy chain C-terminal lysine cleavage and proline amidation, as well as incomplete removal of signal peptides.1 Some modifications are added shortly after translation is completed, others occur after folding during passage through the Golgi apparatus, upon bioprocessing, storage, stress (e.g., elevated temperatures, oxidation, light) or after administration.2 Enzyme-catalyzed PTMs account for a substantial number of different modifications, and involve enzymes like kinases, phosphatases, hydroxylases, and transferases, which add or remove functional groups, proteins, peptides, lipids, or oligosaccharides to amino acid side chains. Occasionally, unexpected enzyme-catalyzed PTMs, such as hydroxylysine, glutamine-linked glycosylation, and non-consensus Asn-glycosylation, have been reported in constant domains or framework regions of IgGs.3–5 Also, complementarity-determining regions (CDRs), which have inherent variability and solvent exposure, have been identified with unusual enzyme-catalyzed PTMs like O-fucosylation and sulfotyrosine.6,7 Further uncommon enzyme-catalyzed PTMs (based on the experience and knowledge from IgGs and monoclonal antibodies (mAbs)) have been reported in non-IgG domains and linkers of antibody-fusion proteins.8–12
In therapeutic proteins, PTMs are especially critical if they negatively influence drug potency or drug safety. Even if PTMs just increase the product heterogeneity of the desired molecules, when identified they need to be diminished or removed by manufacturing process optimizations or further protein engineering, or they at least have to be monitored and controlled to demonstrate batch consistency or comparability of manufactured clinical material. While N-glycosylation sites are usually predictable due to the consensus sequence NXS/T, N-glycosylation of the antigen-binding fragment (Fab) is present in the CDRs of some therapeutic antibodies and increase the product heterogeneity further.13 The same is true for cysteinylations in CDRs.14 Although PTMs observed in natural IgGs might pose a lower safety risk for mAbs, failure to detect low-level PTMs of <1% abundance may represent a safety risk due to potential direct or indirect effects on immunogenicity.2,15 In these cases, PTMs are considered relevant pCQAs. Another aspect is the risk of not fulfilling predefined release criteria due to medium-level PTMs of >1% abundance influencing e.g., charge heterogeneity, which is widely analyzed during batch release and/or for the direct comparison of product batches to ensure drug quality and bio-process consistency.
Technically, the main challenges to the study of PTMs are the development of specific detection, characterization and purification methods. These technical obstacles are being overcome with a variety of reliable characterization methods. For example, suitable and non-inducing tryptic digestion methods for analytical detection and characterization have been reported for the assessment of deamidations and succinimide formation,16,17 trisulfide quantitation,18 disulfide bond analysis,19 or the availability of electron-transfer/higher-energy collision dissociation (EThcD) to give more complete fragmentation and more reliable phosphorylation site localization compared to electron transfer dissociation (ETD) or higher-energy collision dissociation (HCD) alone.20 Recently, matrix-assisted laser desorption ionization (MALDI) was combined with in-source decay (ISD) fragmentation and ultrahigh resolution Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometry (MS) for fast characterization of mAbs providing complementary sequence information compared to other MS-based techniques.21 Other approaches have focused on the development of structure-based prediction tools for the identification of, for example, deamidation, isomerization, and oxidation hot spots in mAbs22–24 or other in silico PTM prediction tools.25–27
Herein, we describe the integration of innovative PTM analysis and in silico identification tool for the detection and verification of 4-hydroxyproline (4Hyp) and sulfotyrosine (sTyr) in antibody-based therapeutics. These two PTMs are technically challenging to verify, and have the potential to be present at substantial levels in biotherapeutics produced in Chinese hamster ovary (CHO) cells. 4Hyp is difficult to distinguish from its isomeric form 3-hydroxyproline (3Hyp), and sTyr is challenging to localize, as the modified amino acid is very labile using common MS/MS technologies. Here, we demonstrate the identification of 4Hyp based on chromatographic separation of peptides containing the hydroxyproline isomers, and the use of top-down ultrahigh resolution MALDI-ISD FT-ICR MS to allocate the sulfation. We present the strategy we recommend to identify and monitor these uncommon PTMs during early development of biotherapeutics.
Results
mAb1 and BsAbA contain major mass variants
The molecular masses of mAb1 (a monoclonal human IgG1) and BsAbA (a bispecific IgG-fusion protein) expressed in CHO cells were analyzed by electrospray ionization quadrupole time-of-flight mass spectrometry (ESI-QTOF-MS). In addition to the expected mass of mAb1 (142735 Da), an unknown +80 Da variant (142815 Da) was present at 21% in the deconvoluted mass spectrum of the N-deglycosylated antibody (Figure 1a). Upon further reduction, the +80 Da modification could be allocated to the light chain (Lc) at 10% (Figure 1b). Assuming a binominal distribution, 10% modified Lc at the reduced level would result in 18% of mAb1 having one +80 Da modification and 1% with both Lcs carrying the +80 Da modification at the intact antibody level (see supplementary Figure S1). The 21% +80 Da variant for the intact N-deglycosylated antibody (Figure 1a) indicates that the modifications on the two Lcs are independent.
Figure 1.
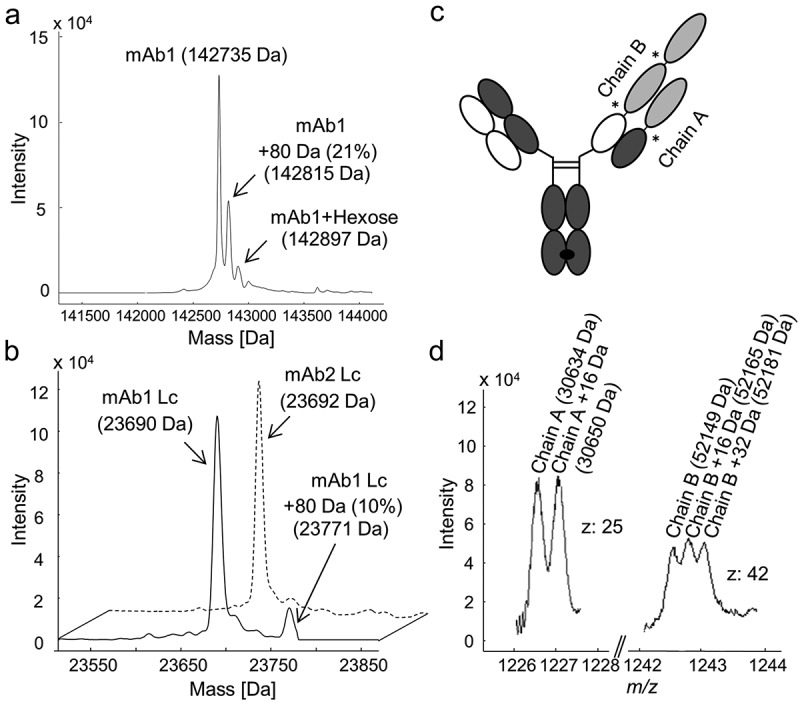
Intact and reduced mass analysis of human IgG1s mAb1 and mAb2, and bispecific IgG-fusion protein BsAbA expressed in Chinese hamster ovary cells. Deconvoluted mass spectra of (a) the N-deglycosylated mAb1, and (b) the reduced light chain (Lc) of mAb1 (solid line) and mAb1-related antibody mAb2 (dotted line). (c) Schematic illustration of BsAbA consisting of four different chains and based on the CrossMabCH1-CL format including three 4–1BB ligand domains (light grey) allocated to chain A and B. * denotes glycine-serine linkers. (d) Mass spectrum of the N-deglycosylated and reduced BsAbA (chain A, charge state z: 25; chain B, charge state z: 42).
BsAbA consists of four different chains and is based on the CrossMabCH1-CL format28,29 including three 4-1BB ligand domains allocated to two molecular chains A and B (Figure 1c) comparable to the molecules reported by Claus et al.30 In the mass spectrum of the N-deglycosylated and reduced BsAbA, two chains with the expected mass of 30634 Da (chain A) and 52149 Da (chain B) were present, as well as an unknown +16 Da variant of chain A (30650 Da), and +16 Da (52165 Da) and +32 Da (52181 Da) variants of chain B (Figure 1d). The +16 Da variant of chain A was quantified to ~50% relative abundance, and the +16 Da and +32 Da variants of chain B were quantified to ~33% each.
The +80 Da modification makes mAb1 acidic
We wondered whether the +80 Da mAb1 variant could be explained by a phosphorylation or a sulfation (both +80 Da), an amino acid sequence variant, or a more complex combination of various events. Analyzed by cation exchange chromatography (CIEX), mAb1 demonstrated the presence of a significant acidic variant peak that was quantified to 20% by peak integration (Figure 2a), matching the 21% +80 Da variant of the intact mAb1 detected by ESI-MS (Figure 1a). To determine whether the +80 Da modification of the mAb1 Lc was in fact causing the presence of the acidic peak, the antibody was cleaved into Fabs by a limited endoprotease Lys-C digestion and purified by a combination of MabSelect SuRe affinity chromatography to remove the Fc domain and CH1 affinity chromatography. A main Fab peak and a second more acidic Fab peak were separated and isolated by CIEX (Figure 2b). Subsequently, the isolated fragments were re-analyzed by CIEX (Figure 2c) and ESI-MS (Figure 2d). The mass analysis of the isolated Fabs confirmed that only the acidic eluting Fab contained the +80 Da modification. The Fabs were also isolated to obtain close to 100% non-sulfated and sulfated portions to determine if there was any functional effect of the modification and to allow a differential analysis by MS. A surface plasmon resonance-based target binding assay did not demonstrate any functional impact of the modification (data not shown).
Figure 2.
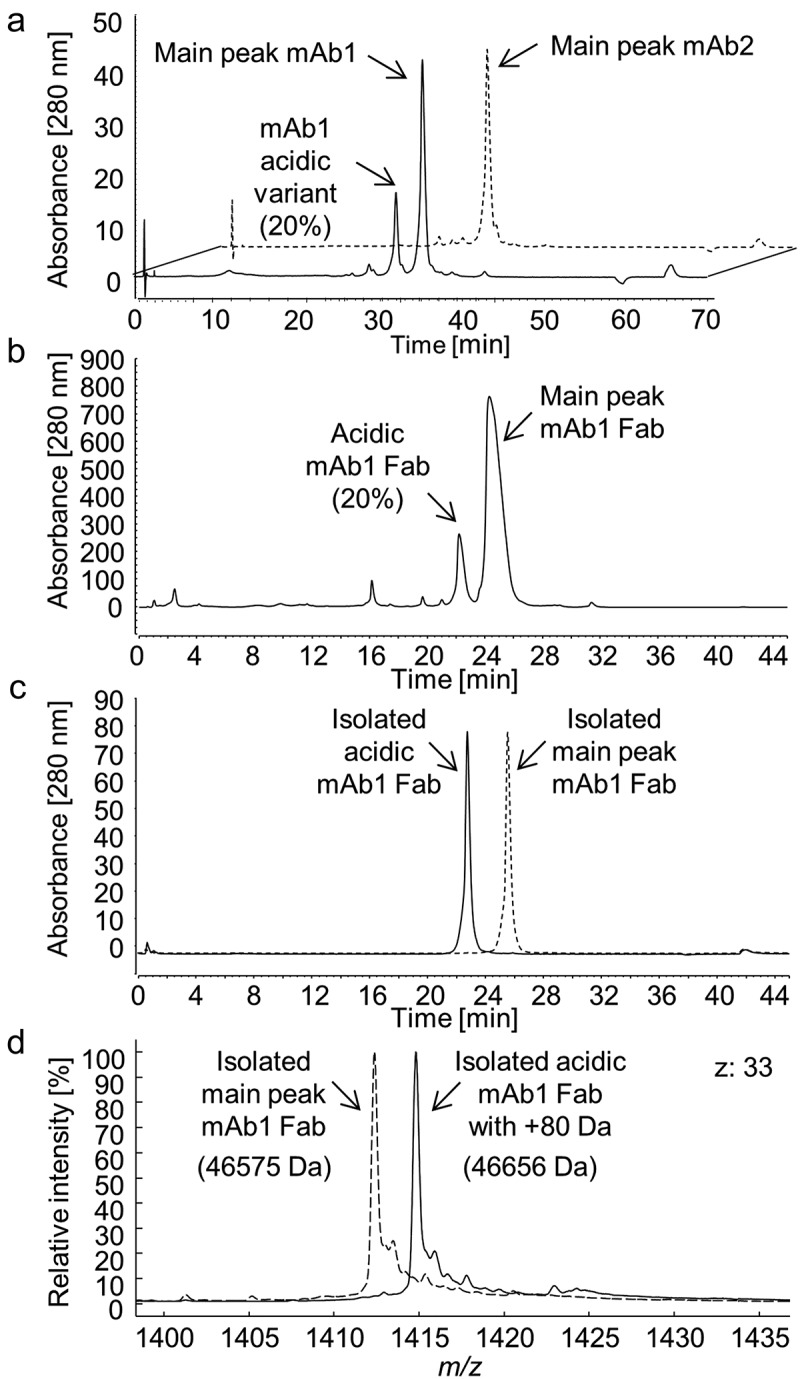
Cation exchange chromatography of (a) the intact mAb1 (solid line) and mAb2 (dotted line), (b) CH1 affinity purified antigen-binding (Fab) domains succeeding a limited endoproteinase Lys-C digest of mAb1 and removal of fragments with an Fc domain by MabSelect SuRe affinity chromatography, and (c) isolated acidic and main peak mAb1 Fabs. (d) Mass spectra (charge state z: 33) of the isolated Fabs with (acidic Fab, solid lines; 46656 Da) and without (main peak Fab, dotted lines; 46575 Da) the +80 Da modification.
LC-MS/MS peptide mapping of mAb1
In addition, mAb1 was analyzed by LC-MS/MS peptide mapping to assess and interpret the +80 Da variant. Evaluation of the LC-MS data from a chromatographic separation of a tryptic digest on a reverse-phase C18 column proved the presence of a mAb1 peptide (LIYSASDLDYGVPSR) with a mass corresponding to a + 79.957 Da modification. On a C18 column, the modified peptide co-eluted with the unmodified peptide (data not shown), and a relative quantification by integration of the EIC chromatograms indicated only 4.6% modified peptide, which did not match the 10% relative abundance determined by ESI-QTOF-MS for the N-deglycosylated/reduced mAb1 (Figure 1b). We speculated whether co-elution on the reverse-phase C18 column caused reduced ionization efficiency (ion suppression) of the modified peptide. Notably, when analyzed using a mixed-mode hybrid reverse-phase C18 column carrying a low-level surface charge, the modified and unmodified tryptic peptides were chromatographically separated, with the modified peptide (41.3 min versus 38.3 min) being more retained on the charged surface hybrid column (Figure 3a). A relative quantitative evaluation (charge states z: 2 and 3) of the unmodified and the modified peptide determined the modification to be present at 9.4% relative abundance, which is in accordance with the 10% modified Lc as determined by ESI-MS (Figure 1b).
Figure 3.
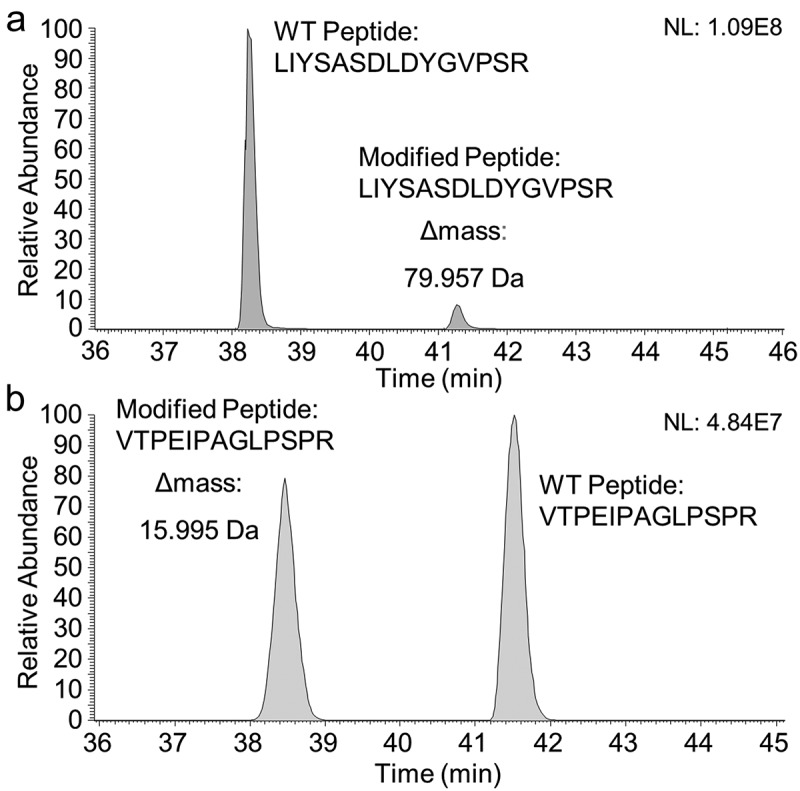
(a) Extracted ion current (EIC) chromatogram of the unmodified (elution time 38.3 min, z: 2 and 3 and m/z window 559.6096–552.6184 + 828.4097–828.4229; MA: 1121769959 (90.6%)) and the +79.957 Da modified (elution time 41.3 min, z: 2 and 3 and m/z window 579.2621–579.2713 + 868.3879–868.4017; MA: 116324914 (9.4%)) tryptic peptides (LIYSASDLDYGVPSR) of mAb1 separated on a mixed-mode hybrid CSH130 C18 column. (b) EIC chromatogram of the +15.995 Da modified (elution time 38.4 min, charge states z: 2 and 3 and m/z = 450.5772–450.5932 + 675.3647–675.3807; manually integrated peak area (MA): 728231181 (46%)) and the unmodified (elution time 41.5 min, z: 2 and 3 and m/z = 445.2459–445.2619 + 667.3666–677.3826; MA: 869604154 (54%)) tryptic peptides (VTPEIPAGLPSPR) of BsAbA separated on a BEH300 C18 reverse-phase column. A relative comparison of the integrated EIC chromatograms quantified the +79.957 Da and +15.995 Da modifications to 9.4% and 46%, respectively. NL, normalized intensity level.
Detailed evaluation of the CID-MS/MS data of the identified modified and unmodified mAb1 tryptic peptides demonstrated close to identical fragment ion profiles for both peptides featuring complete neutral loss of the +79.957 Da modification (data not shown). However, based on the accurate mass, the +79.957 Da modification suggested the presence of a sulfation (SO3, monoisotopic mass: +79.9568 Da) rather than a phosphorylation (HPO3, monoisotopic mass: +79.9663 Da). Also, the isotopic distribution with relative abundance of the second, third, and fourth isotopic peak of 91.4%, 50.0%, and 20.4% peak area relative to the monoisotopic peak was found to be in agreement with sulfation (theoretical distribution: second, 91.197%; third, 51.295%; and fourth isotopes, 21.463%) rather than phosphorylation (theoretical distribution: second, 90.422%; third, 46.164%; and fourth isotopes, 17.100%). Treatment with alkaline phosphatase as described in Tyshchuk et al.11 did not remove the +79.957 Da modification from the tryptic peptide (data not shown). In addition, it is known that the sulfoester bond is labile when fragmented by CID causing a complete neutral loss of 80 Da from the precursor,31,32 which is in line with the above-mentioned experimental results. Without having the position identified, all these observations together with the acidic elution of the modified Fab (Figure 2) pointed towards sulfation. Sulfation adds an acidic group to either a tyrosine, a serine or a threonine residue, with sTyr being most commonly observed.33–35 Of note, the modified tryptic peptide contained two tyrosine and three serine residues (LIYSASDLDYGVPSR) as potential sulfation sites, and further experiments were performed to identify the modified residue, including several alternative digests. However, no single or combined set of digests allowed us to unambiguously determine the site of the mAb1 sulfation.
Various fragmentation techniques, including ETD, HCD, EThcD in positive ion mode and CID and HCD in negative ion mode, were tested but did not provide diagnostic-sulfated fragment ions that would allow identification of the sulfation site (data not shown). Also, spiking experiments with synthetic peptides containing sTyr (sY) in positions 3 (LIsYSASDLDYGVPSR) and 10 (LIYSASDLDsYGVPSR) and LC-MS using the charged surface hybrid column did not exclude any of the tyrosine residues as the site of sulfation. Both synthetic peptides co-eluted with the modified tryptic peptide (data not shown).
Identification of the sulfation site of mAb1 by MALDI-ISD FT-ICR mass spectrometry
To unambiguously determine the site of the mAb1 sulfation, several fragmentation techniques were tested, including MALDI-PSD-TOF MS in negative ion mode with different matrices, as well as derivatization of the carboxyl-groups close to the sulfation by dimethylamidation or ethyl esterification (hypothesis: protons provided by carboxyl groups close to sulfotyrosine might enhance the sulfate loss), and ESI-FT-ICR-ETD MS/MS in positive ion mode of the synthetic peptides. However, all methods suffered from complete neutral loss of the SO3 unit and/or low signal intensity. The site of sulfation was finally identified by characterization of the non-sulfated and sulfated intact Fab fragments of mAb1 (Figure 2(c,d)) by ultrahigh resolution MALDI-ISD coupled to FT-ICR MS. The interpretation of MALDI-ISD FT-ICR MS spectra was based on the assignment of c-type and z + 1-type fragment ions, solely. These fragment types were the most abundant ions in the spectra, and, since they are generated via a radical-mediated mechanism, as for ETD, were considered the most valuable ions for the characterization of a labile PTM such as sulfation. The results from this analysis are summarized in Figure 4. While the non-sulfated c50 fragment ion was detected in the spectra of both non-sulfated and sulfated Fab at m/z 5544.907, the sulfated c50 fragment ion at m/z 5624.864, was not detected (Figure 4(a–c)). Similarly, sulfation was also not detected on Ser51 and Ser53 (LIYSASDLDYGVPSR) (Figure S2). The non-sulfated c57 fragment ion was detected in the spectrum of the non-sulfated Fab at m/z 6296.210 (Figure 4d). However, the sulfated c57 fragment ion of m/z 6376.167 was clearly verified in the ISD spectrum of the sulfated Fab (Figure 4e and Figure S3). In combination, these data allowed the localization of sulfation to Tyr57 (LIYSASDLDYGVPSR). In addition, the intense c-type fragments c65*-c67* (covering the framework region C-terminal to the CDR2, asterisks denoting sulfated fragment ions) of the sulfated Fab light chain were in line with sulfation at Tyr57 (Figure S4).
Figure 4.
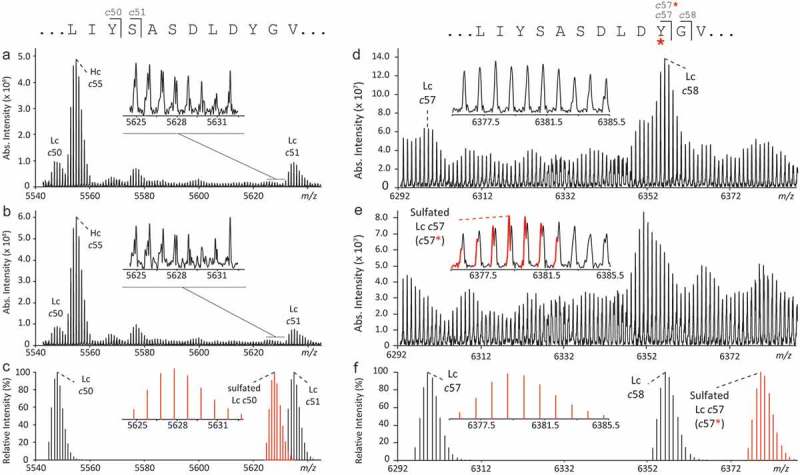
Identification of the sulfation site by top-down MALDI-ISD-FT-ICR MS of antigen-binding (Fab) domains. A comparison was made between the fragmentation spectra generated from the mAb1 Fab without (a and d) and with (b and e) sulfation. In the m/z-range 5540–5640, detected fragment ions (a and b) matched the theoretical isotopic distributions of c-type fragment ions (c) of the light chain (Lc) without sulfation at Tyr50. In the m/z-range 6292–6390, clear differences were observed between the ISD spectra generated from Fab portion (d) without and (e) with sulfation. The presence of the sulfation on Tyr57 of the Lc was confirmed by the detection of the fragment c57 with sulfation (i.e., c57*) at m/z 6376.14 (inset in e) which was in good agreement with the calculated theoretical isotopic distribution (f). Increased confidence in the identification of sulfated c57 fragment was obtained from absorption mode visualization (Figure S3). Larger sulfated c-type fragments were detected at higher m/z (Figure S4).
Figure 5.
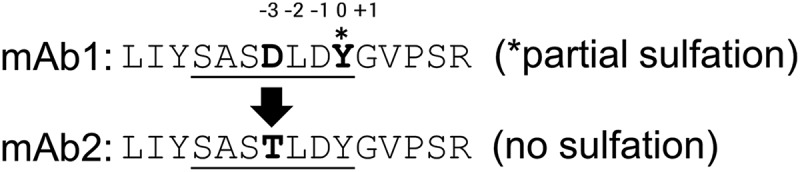
The light chain complementarity-determining region-2 (underlined, according to Kabat et al.37) of mAb1 and mAb2 differ in position −3 relative to the sulfated tyrosine (sY) in mAb1 by an aspartate (mAb1) and a threonine residue (mAb2).
Aspartate in amino-terminal position-3 is involved in determining the sulfation of mAb1
In searching for structural motifs that would instruct Tyr57 sulfation, we analyzed a second antibody, mAb2, which shows high sequence homology to mAb1. Notably, MS analysis after reduction did not indicate any +80 Da variant of the light chain (Figure 1b), and when analyzed by CIEX, mAb2 did not contain a distinct acidic variant as compared to mAb1 (Figure 2a). The amino acid sequences of the light and heavy chains of mAb1 and mAb2 are 98.6% and 99.8% identical, respectively, with the following few amino acid differences between the Lcs: Asn31 to Thr31, Lys33 to Arg33 (both CDR-1), and Asp54 to Thr54 (CDR-2) (mAb1 to mAb2, respectively). The modified mAb1 tryptic peptide thereby contains an aspartate (Asp54) in position 7 (LIYSASDLDYGVPSR), whereas the corresponding mAb2 tryptic peptide contains a threonine in the same position (LIYSASTLDYGVPSR).
As expected from intact ESI-MS and CIEX, when analyzed by LC-MS/MS peptide mapping, the tryptic peptide (LIYSASTLDYGVPSR) of mAb2 was not present as a + 79.957 Da variant or with any other modifications (data not shown). Data generated by Lin et al.36 support that acidic residues near the tyrosines, especially on the amino-terminal side promote sulfation individually and contribute quantitatively and independently to the affinity between the modification site and tyrosyl sulfotransferase. This indicates that the aspartate residue in the amino-terminal position-3 relative to the affected tyrosine residue may be critical for the susceptibility of mAb1 to sulfation (Figure 5). The additional amino acid differences between the Lcs of mAb1 and mAb2 are spatially not as close to the sulfated Tyr57 as Asp54 and likely have no impact on the mAb1 sulfation.
LC-MS/MS peptide mapping of BsAbA identifies a pro residue as modification site
For the BsAbA variants of +16 Da and +32 Da observed by reduced total mass analysis we speculated whether these could be attributed to mono- and di-oxidations or hydroxylations. To further characterize the +16 and +16/+32 Da variants of the BsAbA chains A and B, respectively, the bispecific IgG-fusion protein was digested by trypsin and analyzed by LC-MS/MS peptide mapping. Evaluation of the MS data located a + 15.995 Da modification on a peptide with the sequence VTPEIPAGLPSPR. Relative quantification by extracted ion current (EIC) chromatograms (charge states z: 2 and 3) of the modified and more hydrophilic peptide (38.4 min) and the unmodified peptide (41.5 min) demonstrated the BsAbA peptide to be modified to 46% (Figure 3b). No peptide bearing a + 32 Da modification could be identified by analyzing the tryptic BsAbA peptides. Neither did an additional digestion with thermolysin followed by LC-MS/MS analysis indicate any +32 Da modified peptides.
Using the PEAKS studio software, tandem mass spectrometry by collision-induced dissociation (CID) on the modified and double-charged BsAbA peptide demonstrated the following fragments ions to contain the +15.995 Da modification: b10+ (m/z 991.5564) to b12+ (m/z 1175.6311), and y4+ (m/z 472.2514) to y12+ (m/z 1250.5738) (Figure 6). In addition, identical y2+ (modified peptide: m/z 272.1717) and y3+ (modified peptide: m/z 359.2037), and b2+ (modified peptide: m/z 201.1239) to b9+ (modified peptide: m/z 878.4987) fragment ions were detected for both peptides, locating the +15.995 Da modification of the BsAbA tryptic peptide to the proline residue in position 10 (VTPEIPAGLPSPR). The observed mass difference between the y4+ and y3+ ions of 113.0477 Da was found to be in perfect agreement with the presence of a hydroxyproline (theoretical mass increment of 113.0477 Da) in position 10, and virtually ruled out alternative explanations such as proline to leucine and/or isoleucine (113.0841 Da) sequence variants. Also, identical misincorporations in both chain A and B of BsAbA were unlikely unless proline limitation during fermentation was playing a role. Of note, the +15.995 Da modified peptide appeared to be less hydrophobic than the unmodified tryptic peptide as evidenced by less retention in RP-LC (Figure 3b). In contrast, both leucine and isoleucine misincorporations would be expected to increase the hydrophobicity of the modified peptide.
Figure 6.
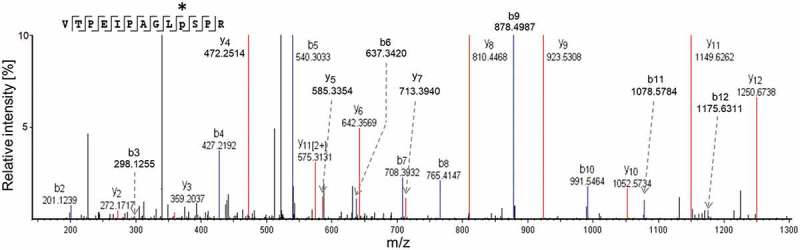
Orbitrap MS/MS data obtained by collision-induced dissociation of the doubly protonated +15.995 Da modified tryptic peptide (VTPEIPAGLPSPR) of BsAbA. Mass differences between the b10+ (m/z 991.5464) to b12+ (m/z 1175.6311), and y4+(m/z 472.2514) to y12+(m/z 1250.6738) fragments of the modified peptide and the corresponding theoretical ions of the unmodified peptide were identified. In addition, no modified b2+ (m/z 201.1239) to b9+ (m/z 878.4987), y2+(m/z 272.1717), and y3+(m/z 359.2037) fragment ions for the modified peptide were detected, implying the +15.995 Da modification to be localized to the proline residue in position 10 (*).
The modified peptide is part of the C-terminus of the three ligand domains of BsAbA (Figure 1c). The mass variants observed by intact ESI-QTOF-MS agree with the fact that the identified peptide is present once in chain A containing one of the three identical ligand domains and in duplicate in chain B containing two of the ligand domains of BsAbA. The +32 Da variant of chain B (Figure 1d) thereby corresponds to both peptides/proline residues being modified. Also, the 46% relative quantification at peptide level (Figure 3b) correlates with the signal intensities of the +16 and +32 Da variants of the reduced and deglycosylated BsAbA (Figure 1d).
Spiking of synthetic peptides proves the presence of 4Hyp in BsAbA and excludes 3Hyp, Leu, and Ile
Hydroxyproline is known to exist in two isomeric forms, trans-3- (3Hyp) and trans-4-hydroxyproline (4Hyp)38,39 with the same nominal mass of 113 Da as leucine and isoleucine. Leucine and isoleucine are successfully distinguished in MS3 experiments (e.g., ETD-HCD) by secondary fragmentation of z-ions to generate Cß–Cγ bond cleavages and to produce w-ions for the specific side chains.40,41 A similar approach has been applied to differentiate 3Hyp and 4Hyp using high energy CID mass spectrometry.38 However, when performing different MS3 fragmentation experiments using an Orbitrap Fusion Lumos, we were not able to generate the w-ions for the specific Hyp side chains (data not shown). We therefore tested whether we could determine the identity of the +15.995 Da modification by spiking of synthetic peptides containing 4Hyp (VTPEIPAGL4HypSPR), 3Hyp, (VTPEIPAGL3HypSPR), leucine (VTPEIPAGLLSPR), and isoleucine (VTPEIPAGLISPR) to the BsAbA tryptic digest with subsequent analysis by LC-MS. EICs of the unmodified and modified tryptic peptides with and without spiking of the synthetic peptides are shown in Figure 7 (charge states z: 2 and 3). In a spiking experiment with the synthetic 4Hyp-containing peptide, the area of the EIC peak eluting at 38.3–38.5 min and representing the modified peptide increased eightfold (Figure 7e), demonstrating the co-elution of the +15.995 Da modified tryptic peptide of BsAbA and the synthetic 4Hyp peptide. With elution times of 38.3–38.5 min (Figure 7(a–e)) and 39.8 min (Figure 7d) of the 4Hyp and 3Hyp-containing peptides, respectively, the spiking experiment demonstrates that both synthetic hydroxyproline-containing peptides are less hydrophobic on the reverse-phase column relative to the unmodified peptide (41.3–41.5 min) as expected (Figure 7). Both the synthetic isoleucine (46.5 min) and leucine (48.3 min) containing peptides eluted later than the unmodified peptide (Figure 7(b,c)). In conclusion, the spiking experiments demonstrated a chromatographic separation of the two peptides containing the isomeric hydroxyprolines and clearly allowed identification of the BsAbA modification as 4Hyp.
Figure 7.
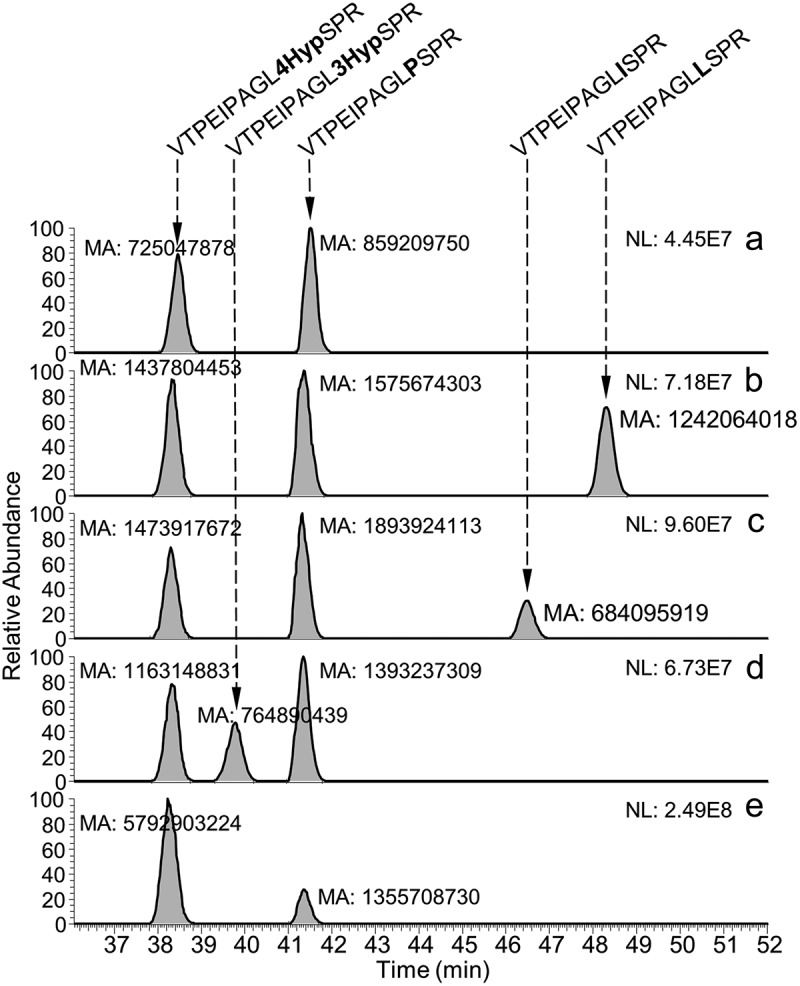
Identification of the modified proline residue as 4-hydroxyproline by spiking of synthetic peptides to the tryptic digest of BsAbA. Extracted ion current chromatograms (charge states z: 2 and 3) of (a) the +15.995 Da modified (38.4 min) and unmodified (41.5 min) tryptic peptides (VTPEIPAGLPSPR) without spiking, and spiked with synthetic peptides containing (b) leucine (VTPEIPAGLLSPR, 48.3 min), (c) isoleucine (VTPEIPAGLISPR, 46.5 min), (d) 3-hydroxyproline (VTPEIPAGL3HypSPR, 39.8 min), and (e) 4-hydroxyproline (VTPEIPAGL4HypSPR, 38.3 min). The peak area of the +15.995 Da modified tryptic peptide increased when the tryptic digest was spiked with the synthetic peptide containing 4-hydroxyproline. NL, normalized intensity level; MA, manually integrated peak area.
In silico identification to hint at the potential presence of sTyr and 4Hyp at early development stage
Early in silico identification of the 4Hyp and sTyr formations could have prevented the heterogeneities of BsAbA and mAb1. On this basis, we developed in-house in silico models based on peptide sequences extracted from the Swiss-Prot database and, in the case of sTyr, also the physicochemical and biochemical properties of various selected amino acids (see Material and Methods). As the focus of our research is the development of therapeutic proteins for human applications, all non-mammalian sequences were excluded in this development, especially as they don’t show the typical mammalian 42pattern around PTM prone positions. For both sTyr and 4Hyp, we obtained very good model qualities (Supplementary Table SI) with respect to all considered performance measures. In particular, we outperformed the sTyr in silico identification performance reported by Yang43 in terms of sensitivity, while keeping specificity at highest level, especially with undersampling.
First, we applied our random forest (RF) and k-nearest neighbor (kNN) models to the sequence RLIYSASDLDYGVPSRFSGSG comprising the mAb1 CDR-2, and all other Tyr residues found in the protein sequence of mAb1. In total, 18 Tyr residues were present. None of the Tyr residues were predicted as modified with a probability of ≥50%, but the position RLIYSASDLDYGVPSRFSGSG had the highest probability for modification among all 18 sites tested, both for RF (pPTM = 0.18) and kNN (pPTM = 0.33), while the similar position RLIYSASTLDYGVPSRFSGSG of mAb2 ranked in the middle for RF (pPTM = 0.11), and lowest for kNN (pPTM = 0). Thus, given our prior suspicion regarding the presence of a sulfation, the algorithms gave the correct information about the location of the PTM, despite the very difficult setting (modified and unmodified sequence differ only at one position). In comparison, the computational tyrosine sulfation site predictor SulfoSite44 predicts the modified tyrosine residue mAb1 with a probability of 95% and the identical but unmodified position in mAb2 with a probability of 75%. In the case of the 4Hyp modification in BsAbA, the kNN had a 50% probability for the affected residue, which was the highest probability observable for all sites and methods in the chain A sequence. Thus, the in-silico tool again identified the correct location. Sequence logos illustrating the most conserved amino acids around sTyr and 4-Hyp residues based on the mammalian peptide sequences we extracted from the Swiss-Prot database are shown in Figure 8.
Figure 8.
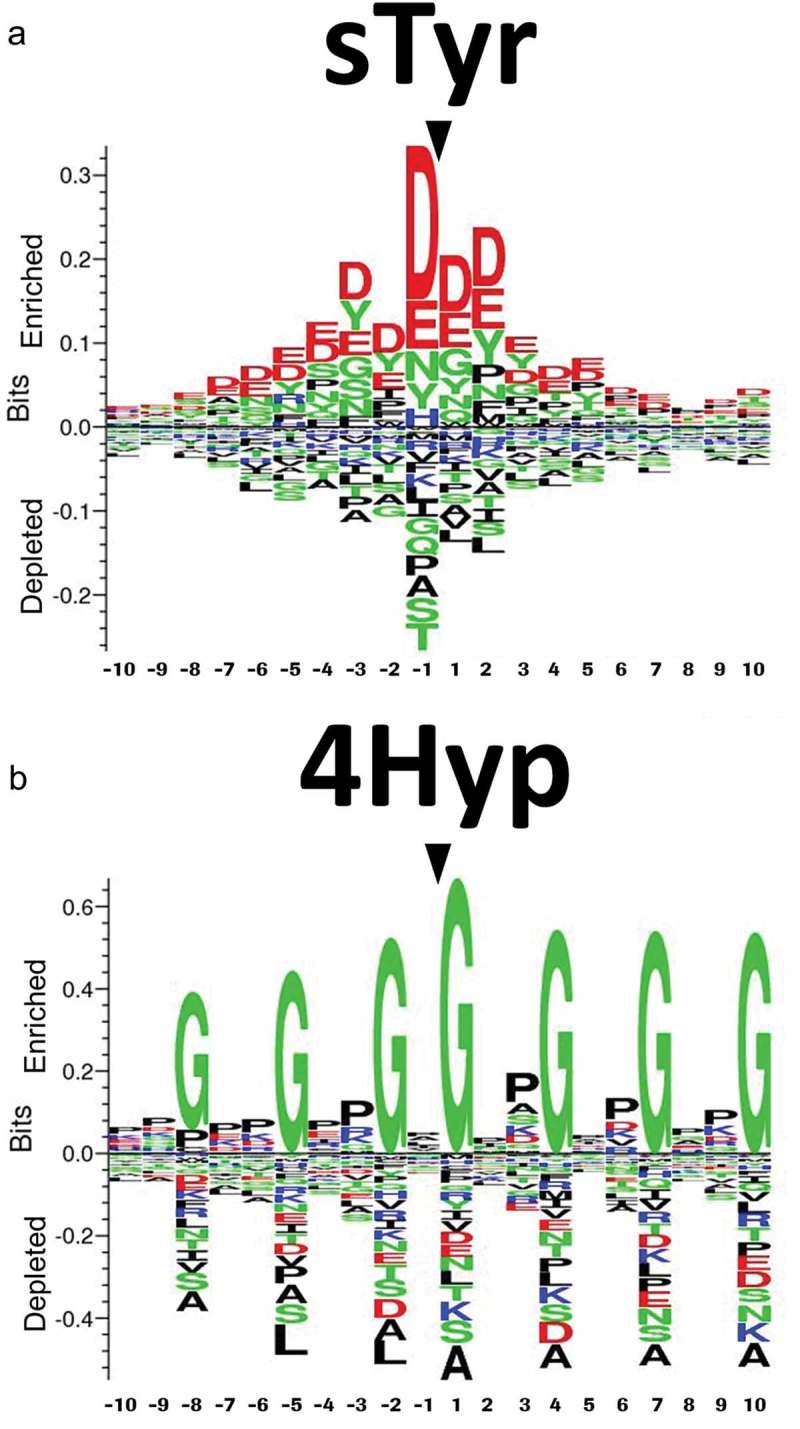
Sequence logos showing the most conserved amino acids around (a) sulfotyrosine (sTyr) and (b) 4-hydroxyproline (4Hyp) residues based on mammalian peptide sequences extracted from the Swiss-Prot database. For scaling reasons, the positions of the central sTyr and 4Hyp residues were marked after creating the logos using Seq2Logo.
Discussion
Several enzyme-catalyzed PTMs to antibodies and other biotherapeutics are well known, while others are more unexpected. Some enzyme-catalyzed PTMs have been reported at significant levels in therapeutic proteins transiently expressed in human embryonic kidney (HEK) cells during early development. However, once the proteins are stably expressed in CHO cells, the very same PTMs have been found to be present at low or very low levels. For example, the O-xylosylations of glycine-serine linkers have been reported to be 10-fold higher in fusion proteins expressed in HEK compared to CHO cells.10 This observation matches our experience with several different fusion proteins in development. We also reported 11% phosphoserine in a glycine-serine linker of a fusion protein transiently expressed in HEK cells compared to <1% when stably expressed in CHO cells.11 Evaluation of further batches of the fusion protein from CHO cells has demonstrated levels <0.1% (unpublished data). Other than cultivation and feeding conditions, the root cause for these quantitative differences between the two expression systems may include differences in drug productivity, and in the expression level, activity, or localization of enzymes involved, and the availability of important co-factors.
However, unexpected PTMs are not always at low levels in CHO-produced proteins. Here, we report the detection of substantial levels of positional 4Hyp and sTyr in two proteins expressed in CHO cells resulting in significant mass and for sTyr charge variant heterogeneities. If a charge variant is influenced by critical process parameters and not kept at a certain level, it might cause out-of-specification events revealing lack of process control. The accumulation of PTMs for bi- or multivalent molecules is non-linear, as pictured in the supplementary Figure S1. Consequently, the expected influence on an ion exchange chromatography profile is also non-linear.
Both 4Hyp and sTyr were technically challenging to characterize and verify. The identification of the 4Hyp was finally accomplished by chromatographic separation of peptides containing the two isomeric forms of hydroxyproline. To our knowledge, the differentiation of the two isomeric forms of hydroxyproline by spiking of synthetic peptides has not previously been reported. In the case of the sTyr, all fragmentation methods tested, including ETD, EThcD, and negative ion mode CID, did not allow determination of the position of the sulfation of mAb1. Finally, a novel top-down MALDI-ISD FT-ICR MS method21 recently developed for the characterization of the amino acid sequence of mAbs was successfully applied to intact Fab fragments, proving the applicability of this powerful ultrahigh resolution method for the analysis of sulfated proteins and therapeutic proteins in general. To our knowledge, this is the first application of MALDI-ISD FT-ICR MS for the characterization of sulfated mAbs. Antibody CDR sulfation was first reported by Zhao et al.7 using positive ion mode ETD. Often, however, sulfations have been reported to be undetectable using conventional positive ion mode fragmentation techniques due to poor ionization efficiency and sulfate loss preceding peptide backbone fragmentation.35,45
Recently, hydroxyproline was reported at a similar high level (40%) in a partially rigid G4P linker as part of an Fc-growth factor fusion protein stably expressed in CHO cells.9 Also, the level of sTyr recently reported in the Lc CDR-1 of a CHO-derived antibody was determined to a relatively high (~20% at the peptide level, corresponding to ~40% at intact level).7 Together, these observations and the data presented here demonstrate that CHO cells are generally capable of introducing high levels of hydroxyproline and sulfotyrosine in therapeutic proteins, which thereby significantly increase the heterogeneity of the molecules. In a very recent study by Hou et al.,12 the impact of specific nutrients on phosphoserine (~20%) and hydroxylysine (~25%) of a CHO platform, fed batch-produced Fc-fusion protein was reported. Although the exact influence of the nutrients is unknown, increased vitamin C, ferric citrate, and niacinamide feeding rates and a decreased cysteine feeding rate reduced the phosphorylation level to ~3%. An increase in the niacinamide and cysteine feeding rates reduced the hydroxylation level to ~10%. Whether similar reductions are possible with 4Hyp and sTyr is unknown. However, as the biopharmaceutical industry often uses standardized fed-batch cultivation and feeding strategies, molecule-specific major adaptations are mostly undesired.
In recent years, several companies have reported the development of structure-based tools for the prediction of chemical PTMs like deamidation, isomerization, and oxidation hot spots in therapeutic antibodies.22–24 Also, several open-source web-based predictors have been developed to suggest positions of enzyme-catalyzed PTMs based on consensus sequences/logos (e.g., www.expasy.org/proteomics, www.cbs.dtu.dk/databases/PTMpredictions, www.modpred.org/). Some of these predictors are available as downloads, but often they are not optimal for therapeutic proteins because they are not exclusively based on human or mammalian protein sequences. Nevertheless, in the biopharmaceutical industry, it is not customary to upload amino acid sequence information of early development biotherapeutics to non-regulated external websites. With the detection and verification of substantial levels of the enzyme-catalyzed PTMs in mAb1 and BsAbA, we therefore decided to pursue a prevention strategy by developing in silico tools for sTyr and 4Hyp and several other enzyme-catalyzed PTMs found in mammalian proteins. The objective was to obtain an early warning as to where enzyme-catalyzed PTMs may occur in drug candidates, thereby avoiding the possibility that PTMs at low relative abundance may stay undiscovered or are uncovered in later phases of drug development, when it may be too late to alter the molecule. Furthermore, suggested positions for PTMs will help guide a positional analytical approach and minimize time-consuming analysis, which may delay drug development.
As with several other PTMs, the activity of prolyl hydroxylases and tyrosyl sulfotransferases is governed by the adjacent amino acids relative to the affected residues. Examination of modified tyrosines has revealed that sulfotyrosines are characterized by acidic amino acids in the immediate environment, especially on the amino-terminal side of the affected tyrosine, with the majority possessing a negative residue in the −1 position as the most important single determinant.36,46,47 Also, the sequence logo we generated for sTyr in mammalian proteins illustrates that acidic amino acids surround the sTyr (Figure 8a). Bundgaard et al.47 suggested that acidic residues in the positions −1 and possibly −3 enhance sulfation, although they are not required to obtain a partial sulfation. When comparing the position of the sTyr in mAb1 with the sequence logo, the aspartates in the positions −1 and −3 relative to the modified Tyr (RLIYSASDLDYGVPSRFSGSG) are likely both involved in determining the sTyr in mAb1. As the Asp in position −1 alone did not cause tyrosine sulfation in mAb2, the Asp in position −3 in mAb1 is likely essential for the affinity of tyrosyl sulfotransferase for this PTM to occur in mAb1. Moreover, the Gly residue in position +1 of mAb1 is enriched in the sTyr sequence logo.
The sequence logo we generated for 4Hyp in mammalian proteins implies key roles for Gly residues in the positions −8, −5, −2, +1, +4, +7, and +10 relative to the 4Hyp, and several enriched positions for Pro (Figure 8b). Similar observations have been reported for predictors based not exclusively on mammalian proteins.48,49 When comparing the position of the 4Hyp in the 4-BB ligand domains of BsAbA with the sequence logo we generated for 4Hyp in mammalian proteins (Figure 8b), the Gly in position −2 relative to the modified proline (VTPEIPAGL4HypSPR) is likely critical for the proline hydroxylation of BsAbA. Also, the Pro residues in −7, −4 and +2 are enriched in the 4Hyp sequence logo. To our knowledge, 4Hyp in human 4-1BBL has not been reported. As BsAbA involves 4-1BBL domains with carboxy-terminal glycine-serine linkers (VTPEIPAGL4HypSPRSEGGGGSGGGGS; see also Figure 1c), the linker Gly residue in position +7 might be involved in determining the 4Hyp of BsAbA. This could indicate that the specific sequence of BsAbA determines the presence of the 4Hyp.
Materials and methods
Enzymes, peptides, and proteins
Endoproteases and alkaline phosphatase were purchased from Promega. Synthetic peptides (95–98% HPLC-purity) were synthesized by Biosyntan GmbH. PNGase F was obtained from Custom Biotech, Roche Diagnostics GmbH. BsAbA was transiently expressed in CHO cells. mAb1 was stably expressed in CHO cells and purified from 250 L platform fed-batch fermentations.
Intact and reduced mass analysis by ESI-QTOF-MS
One hundred micrograms BsAbA or mAb1 were Fc-deglycosylated by adding 45 U PNGase F, and 100 mM sodium phosphate buffer to a final volume of 230 μL, followed by incubation at 37°C for 16 h. Reduction was done by adding 115 μL 100 mM tris(2-carboxyethyl)phosphine in 4 M guanidine hydrochloride to 115 μL (50 μg) Fc-deglycosylated protein followed by incubation at 37°C for 30 min. The samples were desalted by HPLC on a self-packed Sephadex G25 (Amersham Biosciences) 5 × 250 mm column at room temperature using an 8 min isocratic gradient with 40% acetonitrile with 2% formic acid (v/v) at 1 mL/min. By monitoring the UV absorption at 280 nm, the protein peaks were collected using a fraction collector. Approx. 50 μg protein was injected. Total masses were determined by ESI-QTOF-MS on a maXis 4G QTOF mass spectrometer (Bruker Daltonik) equipped with a TriVersa NanoMate source (Advion). Calibration was performed with sodium iodide (Tof G2-Sample Kit 2; Waters). For the deglycosylated and deglycosylated/reduced proteins, data acquisition was performed at m/z 1000–4000 (ISCID: 130.0 eV), and m/z 600–2000 (ISCID: 0.0 eV), respectively. Data acquisition with Fabs was done at m/z 900–3000 (ISCID: 0.0 eV). The raw mass spectra were evaluated and transformed into individual relative molar masses using an in-house developed software tool. The quantitative evaluation of the mass spectra was performed by summing up contributions of m/z ion intensities of all charge states forming the dominant part (larger than 20%) of the charge state envelope as observed for the most abundant individual product mass. Then, all peak contributions (fitted as Gaussians) of all signals in these charge states were used to calculate the relative contents of the individual species.
Analytical cation exchange chromatography of mAb1
Cation exchange chromatography was performed on a 4 × 250 mm ProPac WCX-10 column (Thermo Fisher Scientific) using 20 mM MES, pH 6.0 (eluent A) and 20 mM MES, 500 mM NaCl, pH 6.0 (eluent B), 1 mL/min flow rate, 25°C column temperature, and the following gradient: 75% eluent A [0–5 min], 25% to 56% eluent B [5–57 min], 56% to 100% eluent B [57–58 min], 100% eluent B [58–63 min], 0% to 75% eluent A [63–64 min], 75% eluent A [64–70 min]. Fifty micrograms of mAb1 was injected and detected by 280 nm absorbance.
Isolation of modified and unmodified mAb1 Fabs
One hundred and sixty milligrams of mAb1 were diluted to 1 mg/mL in 0.2 x phosphate-buffered saline, 150 mM NH4Ac, pH 7.0 and incubated 10 min at 37°C. 3.3 mg Lys-C was added and the digest incubated at 37°C for 1.5 h. Following, the cleaved Fabs were isolated by a combination of two connected affinity chromatography columns under standard conditions. The mixture of Lys-C cleaved antibody, Fc-domain, and the Fabs was passed through both chromatography columns where the remaining non-cleaved antibody plus the cleaved Fc-domain were retained on MabSelect™ SuRe™ (GE Healthcare), whereas all cleaved Fabs were captured with CaptureSelect™ IgG-CH1 (Thermo Fisher Scientific). After acidic elution of the Fabs with 150 mM acetic acid, the pH value was adjusted with 1 M Tris to pH 5.5 and a size exclusion polishing step with Superdex™ 200 (GE Healthcare) in 20 mM His/HisHCl, 140 mM NaCl at pH 6.0 applied.
To separate the Fab mixture containing acidic and main peak Fabs, the sample was concentrated to >40 mg/mL with centrifugal spin columns and buffer exchanged to 10 mM sodium phosphate, pH 7.0. Multiple runs using a CIEX ProPac™ WCX-10 analytical column (Thermo Fisher Scientific) with 10 mM sodium phosphate, pH 7.0 (eluent A) and 10 mM sodium phosphate, 500 mM NaCl, pH 7.0 (eluent B) and a gradient of 0–38% eluent B within 9.5 column volumes at a flow rate of 1 mL/min led to a baseline separation of both Fab species. With this method, 50 µg purified Lys-C cleaved Fabs were injected multiple times and detected by 280 nm absorbance. Separated peaks were pooled, concentrated, aliquoted and stored at −80°C. The concentrations of the Fabs were determined by UV280 nm and using the molar extinction coefficients calculated on the basis of the amino acid sequences. The purified Fabs were reanalyzed by analytic CIEX and ESI-QTOF-MS.
LC-MS/MS peptide mapping
The antibodies were denatured and reduced in 0.3 M Tris-HCl pH 8, 6 M guanidine-HCl and 20 mM dithiothreitol (DTT) at 37°C for 1 h, and alkylated by adding 40 mM iodoacetic acid (C13: 99%) (Sigma-Aldrich) at room temperature in the dark for 15 min. Excess iodoacetic acid was inactivated by adding DTT to 40 mM. The alkylated fusion protein was buffer exchanged using NAP5 gel filtration columns, and a proteolytic digestion with trypsin was performed in 50 mM Tris-HCl, pH 7.5 at 37°C for 16 h. The reaction was stopped by adding formic acid to 0.4% (v/v). Thermolysin digests were performed as described by Tyshchuk et al.11 Digested samples were stored at −80°C and analyzed by UPLC-MS/MS using a nanoAcquity UPLC (Waters) and an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). About 2.5 µg digested fusion protein was injected in 5 µL. Chromatographic separation was performed by reversed-phase on a BEH300 C18 column (1 × 150 mm, 1.7 µm) or a CSH130 C18 column, 1 × 150 mm, 1.7 µm (Waters) using mobile phase A and B containing 0.1% (v/v) formic acid in UPLC grade water and acetonitrile, respectively, 60 µL/min flow rate, 50°C column temperature, and the following gradient: 1% mobile phase B [0–3 min], 1% to 40% mobile phase B [3–93 min], 40% to 99% mobile phase B [93–94 min], 99% mobile phase B [94–96 min], 99% to 1% mobile phase B [96–97 min], and 1% mobile phase B [97–105 min]. Two injections of mobile phase A were performed between sample injections using a similar 50 min gradient up to 99% mobile phase B to prevent carry-over between samples. Synthetic peptides were spiked into to digests at different levels.
High-resolution MS spectra were acquired with the Orbitrap mass analyzer, and detection of CID, HCD, or ETD MS/MS fragment ion spectra in the ion trap with dynamic exclusion enabled (repeat count of 1, exclusion duration of 15 s (±10 ppm)). The Orbitrap Fusion Lumos was used in the data-dependent mode. CID essential MS settings were: full MS (AGC: 2 × 105, resolution: 1.2 × 105, m/z range: 300–2000, maximum injection time: 100 ms), MS/MS (AGC: 5.0 × 103, maximum injection time: 100 ms, isolation window (m/z): 2). HCD essential MS settings were: full MS (AGC: 4 × 105, resolution: 1.2 × 105, m/z range: 300–2000, maximum injection time: 50 ms), MS/MS OT (AGC: 5.0 × 105, maximum injection time: 500 ms, isolation window: 2), Orbitrap resolution was 15 × 103, MS/MS IT (AGC: 1.0 × 104, maximum injection time: 100 ms, isolation window: 2). ETD essential MS settings were: full MS (AGC: 2.0 × 105, resolution: 6.0 × 105, m/z range: 300–2000, maximum injection time: 100 ms). MS/MS IT (reaction time: 60–80 ms, reagent target: 1.0 × 106, maximum reagent injection time: 200 ms, AGC: 1.0 × 105, maximum injection time: 150 ms, isolation window: 2, supplemental activation was used without and with 10–35% collision energy). Orbitrap resolution was 15 × 103 or 30 × 103. MS/MS IT (AGC: 5.0 × 104, maximum injection time: 100–150 ms, isolation window: 2). Normalized collision energy was set to CID: 35% (activation q: 0.25); HCD: 28%; ETD: 15–35%.
A complementary EThcD method based on HCD and ETD as data-dependent fragmentation techniques involved full scan MS acquired with the Orbitrap mass analyzer, and parallel detection of ETD and HCD fragment ion spectra in the ion trap and Orbitrap mass analyzer, respectively. A fixed cycle time was set for the full scan with as many as possible data-dependent MS/MS scans. Full MS: same setting as for HCD. For HCD, the MS/MS was detected in the ion trap and the settings were as follows: MS/MS (AGC: 1.0 × 104, maximum injection time: 100 ms, isolation window: 2, charge state 1–3). Normalized collision energy: 28%. ETD: reaction time was set to 60 ms, 1 × 105 reagent target, reagent injection time: 200 ms, charge state 3–8. Supplemental activation collision energy was set to 30%. The AGC target was set to 5.0 × 104, Orbitrap resolution was 1.5 × 104, the precursor isolation window was 2 and the maximum injection time was set to 500 ms.
High-resolution MS spectra were acquired with the Orbitrap mass analyzer in negative mode, and parallel detection of CID or HCD MS/MS fragment ion spectra in the ion trap and Orbitrap with dynamic exclusion enabled as targeted mass experiment. For negative ion mode analysis mobile phases containing 5–10 mM ammonium acetate, 0.1% (v/v) triethylamine, or 5 mM ammonium formate in UPLC grade water and acetonitrile were used. CID essential MS settings were: full MS (AGC: 2 × 105, resolution was used 6.0 × 105, 1.2 × 105, 2.4 × 105, m/z range: 300–2000, maximum injection time: 100 ms), MS/MS IT (AGC: 1.35 × 105, maximum injection time: 100–200 ms, isolation window: 2). Normalized collision energy was set to 5–45%, activation q: 0.25, isolation window: 2. HCD essential MS settings were: full MS (AGC: 4 × 105, resolution: 1.2 × 105, m/z range: 300–2000, maximum injection time: 50 ms), MS/MS OT (AGC: 1.0 × 105, maximum injection time: 500 ms, isolation window: 2). Normalized collision energy was set to 10–35% additional with stepped collision energy. Orbitrap resolution was 15 × 103. MS/MS IT (AGC: 1.0 × 104, maximum injection time: 100 ms, isolation window: 2). Normalized collision energy was set to 10–35%.
MS/MS data evaluation
Analysis of the Orbitrap MS/MS data and the PTM identification was performed using the PEAKS studio 8.0, 8.5 and X software (Bioinformatics Solutions Inc.). Manual data interpretation and quantification were performed using the Xcalibur Qual Browser v.4.0 (Thermo Fisher Scientific). The Protein Calculator (Thermo Fisher Scientific) was used to calculate theoretical masses, and the XICs were generated with the most intense isotope mass using a mass tolerance of 5 ppm.
Top-down MALDI-ISD FT-ICR mass spectrometry of isolated mAb1 Fabs
MALDI-ISD FT-ICR MS experiments were performed on a 15 T solariX XR system equipped with a CombiSource and a ParaCell (Bruker Daltonics). MS measurements were performed as previously reported with some modification.21 Briefly, 1 μL of each sample was spotted onto a ground steel MALDI target plate together with 1 μL of 1,5-diaminonaphthalene (saturated in 50% ACN, 49.9% H2O, 0.1% formic acid). For each spot, an average spectrum was obtained from the acquisition of 200 spectra in the m/z-range 2023–30,000 with 512 k data points. Absorption mode spectra were obtained using AutoVectis software suite (Spectroswiss) and evaluated using mMass data miner.50
Development and testing of sTyr and 4Hyp prediction tools
In silico prediction was attempted with two machine learning approaches, the kNN and the RF algorithms. For that purpose, public data from Swiss-Prot were used as training reference with non-mammalian and completely redundant sequences removed. Residues for which PTMs were annotated were treated as PTM-positive, residues in the same records with no annotation or explicit exclusion of PTM (rarely reported) were treated as PTM-negative. The dataset used was trimmed to 21 amino acid long peptides, with the target amino acid (P or Y, respectively) located at position 11 with the adjacent N- and C-terminal 10 amino acid positions. The dataset for sTyr contained 6304 peptides (out of which 588 contained sTyr at position 11), the dataset for 4Hyp contained 4055 peptides (with 299 4Hyp examples). Features used for modeling included the peptide sequences and in the case of sTyr also physicochemical and biochemical properties of various selected amino acids as provided at ftp://ftp.genome.jp/pub/db/community/aaindex/aaindex1.51 In addition, we created for both PTM-types an undersampled dataset with an equal modified to unmodified sequence ratio. For training, 75% of peptides were used (with the ratio of modified to unmodified sequences left constant), and the remaining 25% were used for testing. After assessment of model quality as given in Supplementary Table SI, the models were used for predicting the 4Hyp and sTyr in BsAbA and mAb1, respectively, which were previously unseen by the prediction tools.
Acknowledgments
We thank Eva Greiter and Björn Mautz for experimental support, the Roche Penzberg management for continuous support, and Yury O. Tsybin (Spectroswiss, Lausanne, Switzerland) and David P. A. Kilgour (Nottingham Trent University, Nottingham, U.K.) for providing AutoVectis software and training support.
Declaration of interest statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Beck A, Liu H.. Macro- and micro-heterogeneity of natural and recombinant IgG antibodies. Antibodies. 2019;8(1):18. doi: 10.3390/antib8010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. 2016:1298473. doi: 10.1155/2016/1298473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie Q, Moore B, Beardsley RL. Discovery and characterization of hydroxylysine in recombinant monoclonal antibodies. MAbs. 2016;8(2):371–78. doi: 10.1080/19420862.2015.1122148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valliere-Douglass JF, Kodama P, Mujacic M, Brady LJ, Wang W, Wallace A, Yan B, Reddy P, Treuheit MJ, Balland A. Asparagine-linked oligosaccharides present on a non-consensus amino acid sequence in the CH1 domain of human antibodies. J Biol Chem. 2009;284(47):32493–506. doi: 10.1074/jbc.M109.014803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valliere-Douglass JF, Eakin CM, Wallace A, Ketchem RR, Wang W, Treuheit MJ, Balland A. Glutamine-linked and non-consensus asparagine-linked oligosaccharides present in human recombinant antibodies define novel protein glycosylation motifs. J Biol Chem. 2010;285(21):16012–22. doi: 10.1074/jbc.M109.096412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valliere-Douglass JF, Brady LJ, Farnsworth C, Pace D, Balland A, Wallace A, Wang W, Treuheit MJ, Yan B. O-fucosylation of an antibody light chain: characterization of a modification occurring on an IgG1 molecule. Glycobiology. 2009;19(2):144–52. doi: 10.1093/glycob/cwn116. [DOI] [PubMed] [Google Scholar]
- 7.Zhao J, Saunders J, Schussler SD, Rios S, Insaidoo FK, Fridman AL, Li H, Liu YH. Characterization of a novel modification of a CHO-produced mAb: evidence for the presence of tyrosine sulfation. MAbs. 2017;9(6):985–95. doi: 10.1080/19420862.2017.1332552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spahr C, Kim JJ, Deng S, Kodama P, Xia Z, Tang J, Zhang R, Siu S, Nuanmanee N, Estes B, et al. Recombinant human lecithin-cholesterol acyltransferase Fc fusion: analysis of N- and O-linked glycans and identification and elimination of a xylose-based O-linked tetrasaccharide core in the linker region. Protein Sci. 2013;22(12):1739–53. doi: 10.1002/pro.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Spahr C, Gunasekaran K, Walker KW, Shi SD. High-resolution mass spectrometry confirms the presence of a hydroxyproline (Hyp) post-translational modification in the GGGGP linker of an Fc-fusion protein. Mabs. 2017;9(5):812–19. doi: 10.1080/19420862.2017.1325556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spencer D, Novarra S, Zhu L, Mugabe S, Thisted T, Baca M, Depaz R, Barton C. O-xylosylation in a recombinant protein is directed at a common motif on glycine-serine linkers. J Pharm Sci. 2013;102(11):3920–24. doi: 10.1002/jps.23733. [DOI] [PubMed] [Google Scholar]
- 11.Tyshchuk O, Völger HR, Ferrara C, Bulau P, Koll H, Mølhøj M. Detection of a phosphorylated glycine-serine linker in an IgG-based fusion protein. MAbs. 2017;9(1):94–103. doi: 10.1080/19420862.2016.1236165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou Y, Su H, Luo Z, Li M, Ma X, Ma N. Nutrient optimization reduces phosphorylation and hydroxylation level on an Fc-fusion protein in a CHO fed-batch process. Biotechnol J. 2018;3:e1700706. doi: 10.1002/biot.201700706. [DOI] [PubMed] [Google Scholar]
- 13.Huang L, Biolsi S, Bales KR, Kuchibhotla U. Impact of variable domain glycosylation on antibody clearance: an LC/MS characterization. Anal Biochem. 2006;349(2):197–207. doi: 10.1016/j.ab.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 14.McSherry T, McSherry J, Ozaeta P, Longenecker K, Ramsay C, Fishpaugh J, Allen S. Cysteinylation of a monoclonal antibody leads to its inactivation. MAbs. 2016;8(4):718–25. doi: 10.1080/19420862.2016.1160179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jefferis R. Posttranslational modifications and the immunogenicity of biotherapeutics. J Immunol Res. 2016;(2016:5358272. doi: 10.1155/2016/5358272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang HZ, Nichols A, Liu D. Direct identification and quantification of aspartyl succinimide in an IgG2 mAb by RapiGest assisted digestion. Anal Chem. 2009;81(4):1686–92. doi: 10.1021/ac802708s. [DOI] [PubMed] [Google Scholar]
- 17.Diepold K, Bomans K, Wiedmann M, Zimmermann B, Petzold A, Schlothauer T, Mueller R, Moritz B, Stracke JO, Mølhøj M, et al. Simultaneous assessment of Asp isomerization and Asn deamidation in recombinant antibodies by LC-MS following incubation at elevated temperatures. PLoS One. 2012;7(1):e30295. doi: 10.1371/journal.pone.0030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornell C, Karanjit A, Chen Y, Jacobson F. A high-throughput hydrophilic interaction liquid chromatography coupled with a charged aerosol detector method to assess trisulfides in IgG1 monoclonal antibodies using tris(2-carboxyethyl)phosphine reaction products: tris(2-carboxyethyl)phosphine-oxide and tris(2-carboxyethyl)phosphine-sulfide. J Chromatogr A. 2016;1457:107–15. doi: 10.1016/j.chroma.2016.06.037. [DOI] [PubMed] [Google Scholar]
- 19.Zhang W, Marzilli LA, Rouse JC, Czupryn MJ. Complete disulfide bond assignment of a recombinant immunoglobulin G4 monoclonal antibody. Anal Biochem. 2002;311(1):1–9. doi: 10.1016/S0003-2697(02)00394-9. [DOI] [PubMed] [Google Scholar]
- 20.Frese CK, Zhou H, Taus T, Altelaar AF, Mechtler K, Heck AJ, Mohammed S. Unambiguous phosphosite localization using electrontransfer/higher-energy collision dissociation (EThcD). J Proteome Res. 2013;12:1520–25. doi: 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.YEM VDB, Kilgour DPA, Tsybin YO, Srzentić K, Fornelli L, Beck A, Wuhrer M, Nicolardi S. Structural analysis of monoclonal antibodies by ultra-high resolution MALDI in-source decay FT-ICR mass spectrometry. Anal Chem. 2018. doi: 10.1021/acs.analchem.8b04515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Mølhøj M, Kuentzer J, Klostermann S, Schoch J, Voelger HR, et al. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS One. 2014;9(6):e100736. doi: 10.1371/journal.pone.0100736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan Q, Huang M, Lewis MJ, Hu P. Structure based prediction of asparagine deamidation propensity in monoclonal antibodies. MAbs. 2018;10(6):901–12. doi: 10.1080/19420862.2018.1478646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sankar K, Hoi KH, Yin Y, Ramachandran P, Andersen N, Hilderbrand A, McDonald P, Spiess C, Zhang Q. Prediction of methionine oxidation risk in monoclonal antibodies using a machine learning method. MAbs. 2018;10(8):1281–90. doi: 10.1080/19420862.2018.1518887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenhaber B, Eisenhaber F. Prediction of posttranslational modification of proteins from their amino acid sequence. Methods Mol Biol. 2010;609:365–84. doi: 10.1007/978-1-60327-241-4_21. [DOI] [PubMed] [Google Scholar]
- 26.Gianazza E, Parravicini C, Primi R, Miller I, Eberini I. In silico prediction and characterization of protein post-translational modifications. J Proteomics. 2016;134:65–75. doi: 10.1016/j.jprot.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 27.Mohabatkar H, Rabiei P, Alamdaran M. New achievements in bioinformatics prediction of post translational modification of proteins. Curr Top Med Chem. 2017;17(21):2381–92. doi: 10.2174/1568026617666170328100908. [DOI] [PubMed] [Google Scholar]
- 28.Klein C, Schaefer W, Regula JT. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. MAbs. 2016;8(6):1010–20. doi: 10.1080/19420862.2016.1197457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Regula JT, Imhof-Jung S, Mølhøj M, Benz J, Ehler A, Bujotzek A, Schaefer W, Klein C. Variable heavy-variable light domain and Fab-arm CrossMabs with charged residue exchanges to enforce correct light chain assembly. Protein Eng Des Sel. 2018;31(7–8):289–99. doi: 10.1093/protein/gzy021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, Albrecht R, Herter S, Schlenker R, Hüsser T, et al. Tumor-targeted 4-1BB agonists for combination with T-cell bispecific antibodies as off-the-shelf therapy. Sci Transl Med. 2019;11(496):pii: eaav5989. doi: 10.1126/scitranslmed.aav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi X, Huang Y, Mao Y, Naimy H, Zaia J. Tandem mass spectrometry of heparan sulfate negative ions: sulfate loss patterns and chemical modification methods for improvement of product ion profiles. J Am Soc Mass Spectrom. 2012;23(9):1498–511. doi: 10.1007/s13361-012-0429-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen G, Zhang Y, Trinidad JC, Dann C 3rd. Distinguishing sulfotyrosine containing peptides from their phosphotyrosine counterparts using mass spectrometry. J Am Soc Mass Spectrom. 2018;29(3):455–62. doi: 10.1007/s13361-017-1854-1. [DOI] [PubMed] [Google Scholar]
- 33.Niehrs C, Beisswanger R, Huttner WB. Protein tyrosine sulfation, 1993–an update. Chem Biol Interact. 1994;92(1–3):257–71. doi: 10.1016/0009-2797(94)90068-X. [DOI] [PubMed] [Google Scholar]
- 34.Medzihradszky KF, Darula Z, Perlson E, Fainzilber M, Chalkley RJ, Ball H, Greenbaum D, Bogyo M, Tyson DR, Bradshaw RA, et al. O-sulfonation of serine and threonine: mass spectrometric detection and characterization of a new posttranslational modification in diverse proteins throughout the eukaryotes. Mol Cell Proteomics. 2004;3(5):429–40. doi: 10.1074/mcp.M300140-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Stone MJ, Chuang S, Hou X, Shoham M, Zhu JZ. Tyrosine sulfation: an increasingly recognised post-translational modification of secreted proteins. N Biotechnol. 2009;25:299–317. [DOI] [PubMed] [Google Scholar]
- 36.Lin WH, Larsen K, Hortin GL, Roth JA. Recognition of substrates by tyrosylprotein sulfotransferase. Determination of affinity by acidic amino acids near the target sites. J Biol Chem. 1992;267:2876–79. [PubMed] [Google Scholar]
- 37.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest. Bethesda (MD): U.S. department of health and human service; 1991. [Google Scholar]
- 38.Kassel DB, Biemann K. Differentiation of hydroxyproline isomers and isobars in peptides by tandem mass spectrometry. Anal Chem. 1990;62(15):1691–95. doi: 10.1021/ac00214a032. [DOI] [PubMed] [Google Scholar]
- 39.Srivastava AK, Khare P, Nagar HK, Raghuwanshi N, Srivastava R. Hydroxyproline: A potential biochemical marker and its role in the pathogenesis of different diseases. Curr Protein Pept Sci. 2016;17(6):596–602. doi: 10.2174/1389203717666151201192247. [DOI] [PubMed] [Google Scholar]
- 40.Johnson RS, Martin SA, Biemann K, Stults JT, Watson JT. Novel fragmentation process of peptides by collision-induced decomposition in a tandem mass spectrometer: differentiation of leucine and isoleucine. Anal Chem. 1987;59(21):2621–25. doi: 10.1021/ac00148a019. [DOI] [PubMed] [Google Scholar]
- 41.Lebedev AT, Damoc E, Makarov AA, Samgina TY. Discrimination of leucine and isoleucine in peptides sequencing with Orbitrap fusion mass spectrometer. Anal Chem. 2014;86(14):7017–22. doi: 10.1021/ac501200h. [DOI] [PubMed] [Google Scholar]
- 42.Smeeton NC. Early history of the kappa statistic. Biometrics. 1985;41:795. [Google Scholar]
- 43.Yang ZR. Predicting sulfotyrosine sites using the random forest algorithm with significantly improved prediction accuracy. BMC Bioinformatics. 2009;10:361. doi: 10.1186/1471-2105-10-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang WC, Lee TY, Shien DM, Hsu JB, Horng JT, Hsu PC, Wang TY, Huang HD, Pan RL. Incorporating support vector machine for identifying protein tyrosine sulfation sites. J Comput Chem. 2009;30(15):2526–37. doi: 10.1002/jcc.21258. [DOI] [PubMed] [Google Scholar]
- 45.Monigatti F, Hekking B, Steen H. Protein sulfation analysis – a primer. Biochim Biophys Acta. 2006;1764(12):1904–13. doi: 10.1016/j.bbapap.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 46.Hortin G, Folz R, Gordon JI, Strauss AW. Characterization of sites of tyrosine sulfation in proteins and criteria for predicting their occurrence. Biochem Biophys Res Commun. 1986;141(1):326–33. doi: 10.1016/S0006-291X(86)80372-2. [DOI] [PubMed] [Google Scholar]
- 47.Bundgaard JR, Vuust J, Rehfeld JF. New consensus features for tyrosine O-sulfation determined by mutational analysis. J Biol Chem. 1997;272(35):21700–05. doi: 10.1074/jbc.272.35.21700. [DOI] [PubMed] [Google Scholar]
- 48.Shi SP, Chen X, Xu HD, Qiu JD. PredHydroxy: computational prediction of protein hydroxylation site locations based on the primary structure. Mol Biosyst. 2015;11(3):819–25. doi: 10.1039/c4mb00646a. [DOI] [PubMed] [Google Scholar]
- 49.Li S, Lu J, Li J, Chen X, Yao X, Xi L. HydPred: a novel method for the identification of protein hydroxylation sites that reveals new insights into human inherited disease. Mol Biosyst. 2016;12(2):490–98. doi: 10.1039/c5mb00681c. [DOI] [PubMed] [Google Scholar]
- 50.Strohalm M, Hassman M, Kosata B, Kodicek M. mMass data miner: an open source alternative for mass spectrometric data analysis. Rapid Commun Mass Spectrom. 2008;22(6):905–08. doi: 10.1002/rcm.3444. [DOI] [PubMed] [Google Scholar]
- 51.Kawashima S, Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res. 2000;28(1):374. doi: 10.1093/nar/28.1.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.