

ABSTRACT
Biotherapeutic proteins are commonly dosed at high concentrations into the blood, which is an inherently complex, crowded solution with substantial protein content. The effects of macromolecular crowding may lead to an appreciable level of non-specific hetero-association in this physiological environment. Therefore, developing a method to characterize the diverse consequences of non-specific interactions between proteins under such non-ideal, crowded conditions, which deviate substantially from those commonly employed for in vitro characterization, is vital to achieving a more complete picture of antibody function in a biological context. In this study, we investigated non-specific interactions between human serum albumin (HSA) and two monoclonal antibodies (mAbs) by static light scattering and determined these interactions are both ionic strength-dependent and mAb-dependent. Using biolayer interferometry (BLI), we assessed the effect of HSA on antigen binding by mAbs, demonstrating that these non-specific interactions have a functional impact on mAb:antigen interactions, particularly at low ionic strength. While this effect is mitigated at physiological ionic strength, our in vitro data support the notion that HSA in the blood may lead to non-specific interactions with mAbs in vivo, with a potential impact on their interactions with antigen. Furthermore, the BLI method offers a high-throughput advantage compared to orthogonal techniques such as analytical ultracentrifugation and is amenable to a greater variety of solution conditions compared to nuclear magnetic resonance spectroscopy. Our study demonstrates that BLI is a viable technology for examining the impact of non-specific interactions on specific biologically relevant interactions, providing a direct method to assess binding events in crowded conditions.
KEYWORDS: Protein non-ideality, non-specific interactions, light scattering, biolayer interferometry, monoclonal antibody, human serum albumin
Introduction
Proper analysis of in vitro binding equilibria is necessarily constrained to concentrations near the dissociation constant, typically on the order of micromolar and below. Under these conditions, proteins behave essentially as ideal molecules and any non-specific interactions can easily be ignored. Increasing the protein concentration to arbitrarily high values leads to macromolecular crowding, where complex formation is no longer linear with respect to protein concentration and the binding equations derived from the law of mass action are better expressed in terms of thermodynamic activity rather than concentration.1 The magnitude of the activity coefficient depends on the composition of the whole solution; non-ideality arises not only from elevated levels of the protein itself but also from the presence of non-interacting macromolecules and co-solutes. With regard to a biological environment, the blood is a complex, crowded solution composed of hundreds of different molecules. An understanding of the non-specific interactions between proteins under such non-ideal conditions is vital to achieving a more complete picture of protein function in a biological context. Measurement of protein activity in a crowded environment is therefore of utmost importance, and methods such as analytical ultracentrifugation (AUC) and nuclear magnetic resonance (NMR) have been utilized previously to examine molecular behavior in crowded solutions.2–7 However, these methods present disadvantages: AUC is a time-consuming method with experiments that can take days, and NMR poses limitations with regard to ionic strength of samples. We present here an alternative approach, using biolayer interferometry (BLI), which provides a higher throughput approach than either AUC or NMR, while offering a greater degree of flexibility in measuring the impact of solution crowding on protein activity. This convenient approach is more amendable to screening larger numbers of test articles in a shorter period of time.
The effects of macromolecular crowding on the thermodynamic and kinetic properties of proteins are remarkably complex and difficult to predict.8–11 A principal and unavoidable consequence is steric exclusion, also referred to as the excluded volume effect, which generally leads to a greater potential for macromolecular association in order to increase the volume available for all molecules. The various physicochemical attributes of proteins, including size, shape, surface and inherent charge properties, and solvation state, contribute to the net non-specific interactions. Furthermore, electrostatic interactions, van der Waals forces, charge anisotropy (local dipole moments), and hydrophobic interactions modulate the overall effect, possibly in opposing manners. Lastly, non-specific interactions depend greatly on solution conditions (e.g., pH and ionic strength; inert co-solutes) and in certain cases may change from net repulsive to attractive interactions.12–14 Thus, in a solution containing otherwise non-interacting proteins, the non-ideality that stems from high protein concentration may lead to an appreciable level of hetero-association or may maintain the solutes in a more disperse distribution.
The consequences of thermodynamic non-ideality are manifold. From the perspective of antibody manufacturing and formulation, in which the final presentation of the molecule frequently exceeds 100 g/L, non-ideality has been shown to alter a variety of protein solution phenomena, including viscosity, solubility, phase separation, and self-association.15–18 With respect to specific environments of biological systems, including the intracellular milieu, the extracellular matrix, and circulating blood, macromolecular crowding not only affects binding equilibria, but also reaction rates, protein folding and isomerization, protein–protein interactions, and overall cellular homeostasis.19–21 For example, theoretical modeling of cellular osmotic equilibrium, which affects osmotic transport into and out of the cell, requires consideration of non-ideal intracellular thermodynamics due to the crowded cellular environment.22 Additionally, macromolecular crowding can influence cellular pathology; accelerated amyloid formation has been demonstrated to occur in crowded environments.23–25 The composition of physiological environments often interferes with the analytical methods typically used to characterize virial coefficients. Thus, the phenomenon of thermodynamic non-ideality and the consequences that follow are of both practical and biological significance.
Although highly complex at the molecular level, the deviation from ideality that is observed from moderately high levels of protein concentration (on the order of 10 g/L) may be conveniently expressed with the second osmotic virial coefficient.1 Self (B22) and cross (B23) virial coefficients characterize weak, non-specific protein–protein interactions in solutions containing single and multiple protein species, respectively. Multi-angle static light scattering (MALS) is a first principles analytical method that allows determination of molar mass for a variety of macromolecules, including proteins, in the ideal limit. Static light scattering methods are also commonly used to determine the second virial coefficient, which reflects net interactions (protein-protein and protein-solute) and excluded volume effects for all species in solution, from the concentration dependence of molar mass.26 In composition-gradient multi-angle light scattering (CG-MALS), the light scattering detector is placed downstream of an automated syringe pump system capable of simultaneously injecting up to three different solutions, each containing different molecules, as necessary.27 In this batch mode, the weight-average molar mass of all solutes in solution is determined and can provide a quantitative analysis of binding interactions with limited prior knowledge. Several implementations of CG-MALS have been developed to characterize specific and non-specific interactions between proteins and other macromolecules. For non-specific protein–protein interactions, the CG-MALS system has the advantage of extracting both self-virial coefficients as well as the cross-virial term from a single experiment. The robustness of the technique, in addition to the well-established analysis algorithm used, enables efficient and relatively straightforward characterization of interactions in protein solutions over a range of concentrations.28
The CG-MALS method is highly convenient for determining the degree and nature of non-specific interactions between two species; however, the data analysis becomes more cumbersome and less precise for such systems as the concentrations exceed 10 g/L. This necessitated the pursuit of an alternate method that could extend the concentration range into physiologically relevant concentrations, as well as expand our studies to include the impact of non-specific interactions on specific, functional binding events. BLI is a label-free optical technique for measurement of specific macromolecular interactions, including determination of kinetics and binding affinity.29–31 BLI analyzes the interference pattern of white light reflected from an internal reference layer as well as a layer of immobilized protein on a biosensor tip (i.e., the biolayer). Binding events increase the number of molecules on the biolayer, producing a shift in the interference pattern that can be monitored in real-time. This method has been used to assess protein–protein interactions,32 protein–ligand interactions,33 protein-nucleic acid interactions,34,35 and small-molecule and peptide screening,36 among others.
In this study, we aimed to develop a novel method to measure the impact of non-specific interactions on mAb:antigen interactions in crowded solutions, using human serum albumin (HSA) to demonstrate these principles in a simplified system. Albumin constitutes a majority of the volume fraction in serum, at a physiological concentration range of 35–50 g/L, and is negatively charged at physiological pH, which may lead to electrostatic association (or repulsion) with biotherapeutics bearing a net positive (or negative) charge or solvent-exposed surface. To this end, we investigated non-specific interactions between HSA and two recombinant fully human IgG4 monoclonal antibodies (mAb1 and mAb2) that bind the same antigen, first in a binary (HSA and mAb) system using CG-MALS, then in a ternary interaction (HSA, mAb, antigen) system with BLI. These mAbs are highly similar in sequence apart from the complementarity-determining region (CDR), which target different epitopes on the antigen. The binary system enabled us to demonstrate with well-established light scattering methodologies that non-specific interactions between HSA and mAbs at sub-physiological protein concentrations are both ionic strength-dependent as well as mAb-specific. To further elucidate the effects of these interactions on the functional properties of the mAbs, we used BLI in a non-standard manner to assess antigen binding by mAbs from low to physiological HSA concentrations. The BLI results correlated with the CG-MALS data, supporting that this novel use of the BLI in the presence of high HSA concentrations can directly assess the impact of non-specific interactions due to crowding on a highly specific, functional interaction such as antibody:antigen binding. The results obtained also support the hypothesis that high concentrations of HSA in the blood serum leads to non-specific interactions with mAbs, with a potential impact on antibody function. While the effect is particularly apparent at low ionic strength, it is mitigated at physiological ionic strength for this particular set of mAbs; however, this trend does not necessarily extend to all other mAb:antigen systems. By utilizing this approach at an early stage of development of a biotherapeutic, the effects of non-specific interactions can be easily detected; conversely, this type of investigation can also alleviate concern for unanticipated consequences in vivo. Using the BLI platform with an adapted analysis in a simple, controlled system, we demonstrate that the functional impact of non-specific interactions can be determined, setting the stage for exploring the breadth of consequences macromolecular crowding and protein non-ideality may exhibit in more complex solutions.
Results
Ionic strength dependence of mAb1/HSA and mAb2/HSA non-specific interactions
Non-specific interactions between HSA and each mAb at different ionic strengths were examined using CG-MALS, which is a well-established approach to determine the cross-virial coefficient (CVC). This approach was applied to determine both the degree and nature of non-specific interactions between HSA and the mAbs prior to analysis by BLI in order to best interpret the data. Several investigators have pointed out, based on rigorous thermodynamic principles, that the virial coefficient determined from static light scattering is not a pure self, or cross, interaction parameter, but rather it is convolved with protein:co-solute (i.e., buffer or electrolyte) interactions.26,37,38 Therefore, the preferred convention is to refer to the virial coefficient from light scattering analysis as A2 in order to distinguish it from the molal condition (B22). Provided the proteins are not highly charged and the co-solutes are simple buffers and electrolytes, numerical differences between A2 (used here) and B22 are minimal. Similarly, the CVC from light scattering measurements, or A23, is an indicator of the nature and degree of non-specific interactions between two species, and was measured for mAb1/HSA and mAb2/HSA interactions in buffered solutions containing 10–750 mM NaCl (Figure 1). A negative value for A23 indicates attractive forces between the two species, while a positive value indicates repulsive forces. At a concentration of 10 mM NaCl, both mAb1/HSA and mAb2/HSA exhibited attractive forces, with stronger forces observed between mAb1/HSA compared to mAb2/HSA. This phenomenon was mitigated with increasing ionic strength. At physiological ionic strength (~137 mM NaCl), the non-specific interactions between mAb1 and HSA were slightly attractive while those between mAb2 and HSA were slightly repulsive. The results show both the ionic-strength dependence and mAb-dependence of non-specific interactions with HSA. To determine the role of electrostatics in these interactions, we assessed the molecules by ion exchange chromatography.
Figure 1.
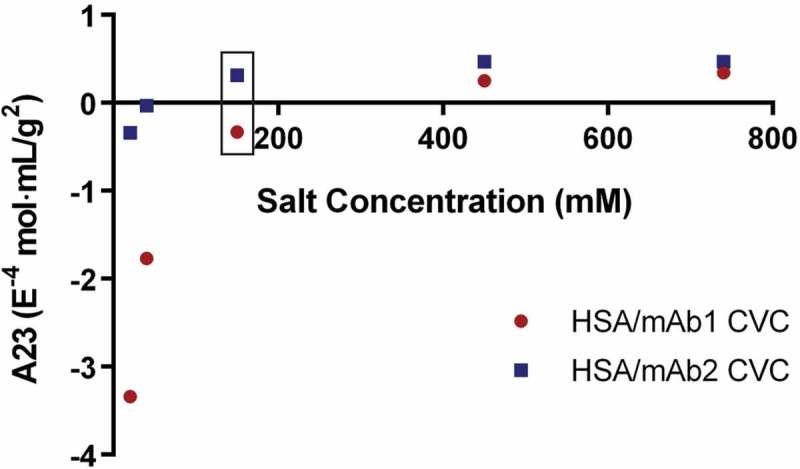
Ionic strength dependence of mAb1/HSA and mAb2/HSA cross-interactions measured by CG-MALS. Cross-virial coefficients (A23) were determined by CG-MALS for interactions between 10 g/L HSA and 10 g/L mAb1 (red) or mAb2 (blue) at increasing concentrations of NaCl in phosphate buffer. Negative values for CVC indicate attractive forces between the molecules, while positive CVC values indicate repulsive forces between the molecules. The box indicates physiological ionic strength where the CVC values for mAb1 and mAb2 are negative and positive, respectively.
Chromatographic methods show that mAb1 and mAb2 have different surface properties
The surface properties of mAb1 and mAb2 were examined to determine whether the differing degree of non-specific interaction with HSA could be driven by long-range charge–charge interactions between the molecules. We subsequently performed weak cation exchange chromatography to more specifically assess the surface properties of the two mAbs. These experiments showed that the retention time of mAb1 (~13 min.) was much longer than for mAb2 (~6 min.), indicating stronger interactions with the charged column resin (Figure 2). Similarly, we assessed the chromatographic profile for HSA, which eluted at a very low retention time (~2 min.) compared to the two mAbs, indicating a more acidic surface charge. Assessment of the mAbs with hydrophobic interaction chromatography showed minimal differences in the elution volume (data not shown), indicating that the differences in non-specific interaction between the mAbs and HSA is likely electrostatic in nature rather than due to a hydrophobic interaction. Together, these data support the hypothesis that surface properties of each mAb could play a significant role in the degree and nature of the non-specific interactions with HSA. To further assess these interactions, as well as their impact on functional properties of the mAbs, we used BLI to examine binding affinity of the mAbs to antigen in the absence and presence of HSA.
Figure 2.
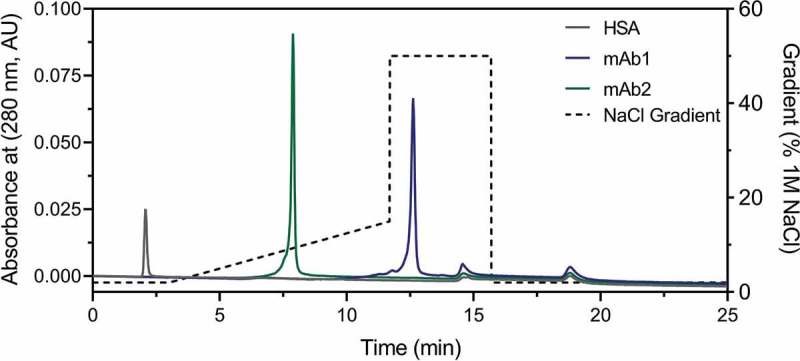
Weak cation exchange chromatography elution profiles for mAbs and HSA. Each mAb and HSA were assessed by analytical chromatography on a weak cation exchange column equilibrated with 200 mM MES, 20 mM NaCl, pH 6.5. A gradient was applied from 20 to 500 mM NaCl and is represented by a dashed line as a percentage of a 1 M NaCl solution. Representative elution profiles for HSA (grey), mAb1 (blue), and mAb2 (green) are shown.
The two mAbs bind to biotinylated antigen with a similar binding affinity using biolayer interferometry
In order to assess the effect of physiologically relevant levels of HSA on the binding properties of the two mAbs, we used BLI to monitor association of the mAbs to their common antigen. To test our system and reagents, we performed standard affinity measurement experiments using anti-human IgG Fc capture (AHC) biosensor tips, loading mAb1 or mAb2 onto the tip, and measured antigen binding (Figure S2) with either unmodified antigen or biotinylated antigen in low and physiological salt conditions. The data from these experiments are summarized in Table S1, and show strong similarity to previously generated Biacore surface plasmon resonance (SPR) data (data not shown) with regard to kon, koff, and KD.
We also performed standard avidity measurement experiments with antigen-loaded biosensor tips to detect antibody binding. To do this, we site-specifically biotinylated the antigen and loaded it onto the streptavidin-coated biosensor tip. By immobilizing the smaller antigen rather than the mAb, the change in response (in nm) resulting from binding of antibody gave a more pronounced signal, and thus improved signal to noise. Figure 3 shows the results from the standard BLI experiment performed to determine binding affinity for each mAb under ideal solution conditions. The slow dissociation kinetics do not enable an accurate estimate of apparent KD, but the tight binding indicates sub-nanomolar functional binding avidity, which is consistent with Biacore SPR data (not shown).
Figure 3.
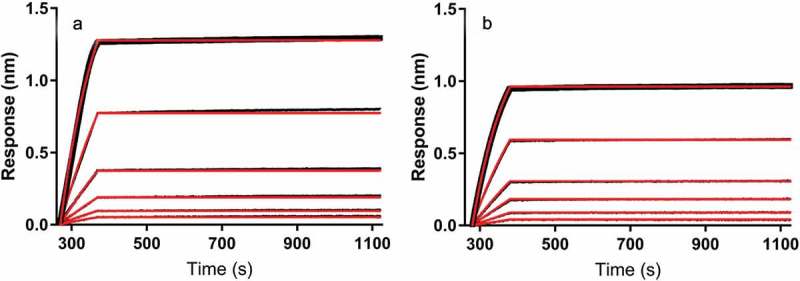
Binding of mAb to biotinylated antigen at 137 mM NaCl in absence of HSA measured by BLI. Binding of mAb1 (a) and mAb2 (b) to biotinylated antigen was observed using biolayer interferometry at physiological salt concentration in phosphate buffer. Change in wavelength in nanometers (response, nm) is plotted as a function of time to indicate changes in thickness of the biolayer due to binding events. Association and dissociation steps are shown for 1.25 nM, 2.5 nM, 5 nM, 10 nM, 20 nM, and 40 nM mAb. Black traces indicate raw data and red traces indicate fitted curves. Data traces are aligned at the mAb association step, and reference data was subtracted from all sample traces.
Having established that the kinetic binding experiments performed under ideal solution and experimental conditions produced consistent results to previously performed Biacore SPR studies (data not shown) and that the prepared reagents were fully active, we then examined mAb binding to biotinylated antigen in the presence of high, physiologically relevant concentrations of HSA. While these experiments would ideally be performed closer to the approximate theoretical physiological dosing concentration of ~550 nM, the upper limit for equilibrium binding experiments with BLI requires using a mAb concentration that is closer to 10-fold above the KD. Due to tight affinity of these mAbs and the response level of binding to antigen, which allows for monitoring of decreases and increases in signal, we performed these experiments at a mAb concentration of 40 nM.
The overall effect of HSA on mAb binding to antigen is ionic-strength dependent and mAb specific
To investigate the effect of HSA on mAb-antigen binding, we employed a BLI experiment under non-ideal solution conditions and analyzed steady state, end-point data. By monitoring the steady-state response (nm) level after a lengthy (20 min) association step with each mAb, we determined the level of binding to antigen achieved in the presence of HSA at or near equilibrium. Kinetic (on and off-rate) analysis was not performed because of the avidity format and the added complexity imparted by increasing concentrations of HSA. A control was also performed with only HSA in the absence of antibody (data not shown) to demonstrate that HSA does not interact with the antigen. Figure 4 shows sensorgrams for mAb1 and mAb2 in the presence and absence of 10 g/L HSA at either 10 mM or 137 mM NaCl. These data qualitatively show the difference in the effect of HSA has at the two ionic strength conditions for the two mAbs, demonstrating both the ionic-strength dependence and differences in mAb interactions with HSA. At low salt, the effect of HSA is greater on mAb1 than on mAb2, reflected in the difference in response upon addition of HSA. At physiological salt, the impact of 10 g/L HSA is minimal on either mAb interaction with antigen. These results correlate with the CG-MALS data described above, and reveal a potential effect on functional properties of mAbs.
Figure 4.
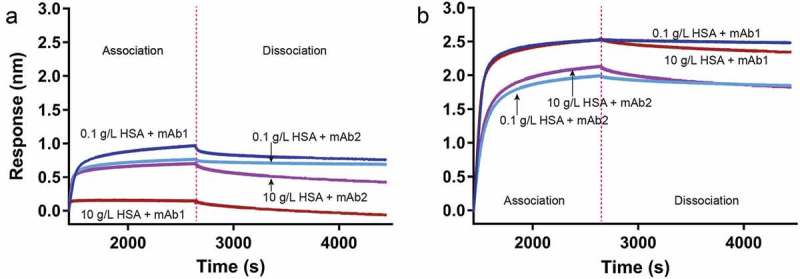
Overall effect of HSA on mAb binding to antigen measured by BLI is ionic-strength dependent. Binding of 40 nM mAb1 and mAb2 to biotinylated antigen in the absence and presence of HSA was observed by biolayer interferometry at 10 (panel a) and 137 mM NaCl (panel b) in phosphate buffer. Change in wavelength (response, nm) as a function of time indicates binding events, and only the mAb association and dissociation steps are shown. Biotinylated antigen was loaded onto SA tips as described above. Binding of mAb1 was assessed in the absence (dark blue) and presence (brown) of 10 g/L HSA, and mAb2 binding was assessed in the absence (light blue) and presence (purple) of 10 g/L HSA. A minimum of 0.1 g/L HSA is required to prevent non-specific binding to the biosensor tip. Data traces are aligned at the mAb association step following a baseline measurement in equivalent concentrations of HSA. Traces for samples containing 10 g/L HSA were corrected for a change in signal upon transitioning from association to dissociation due to the change in refractive index of the solution.
The effect of increasing HSA concentrations is further illustrated in the low and physiological salt conditions (Figure 5), where the response level at equilibrium of various HSA concentrations was normalized to the 0.1 g/L HSA level. At 10 mM NaCl, mAb2 had a modest decrease in response (i.e., decrease in antigen binding) with increasing HSA concentrations (~20%), while mAb1 exhibited a more dramatic decrease (~40%; Figure 5(a), Table S2). At 137 mM NaCl, both mAbs showed a modest increase in antigen binding at HSA concentrations under 20 g/L; at higher HSA concentrations, the effect of HSA on antigen binding is greater for mAb1 compared to mAb2 (Figure 5(b), Table S3). In comparison to the response of mAb binding in the absence of HSA (denoted by the dotted line in Figure 5(b)), the signal for mAb1 was ~10% reduced while the signal for mAb2 was ~6% enhanced in the physiological HSA range (35–50 g/L, Table S4). The observed binding events were specific to antigen binding; a control mAb (mAb3) that does not bind this antigen showed no increase in signal, or binding, in the presence of 0.1–50 g/L HSA (Figure S3).
Figure 5.
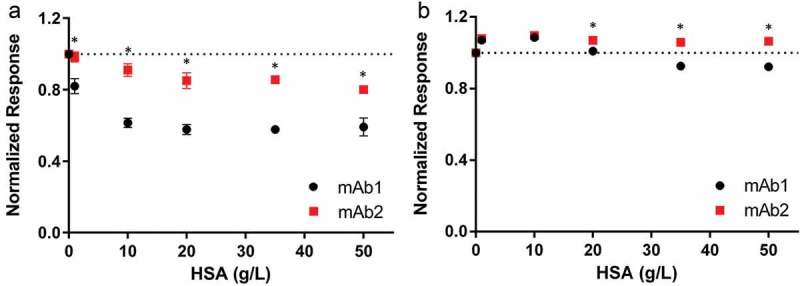
Effect of HSA on mAb binding to antigen measured by BLI is ionic-strength dependent and mAb-specific. Binding of mAb1 (black) and mAb2 (red) to biotinylated antigen in the presence of increasing HSA concentrations was observed by biolayer interferometry at 10 (panel a) and 137 (panel b) mM NaCl. The normalized response (to binding at 0.1 g/L HSA) is shown as a function of HSA concentration. The dotted line indicates the normal at 1.0, in order to illustrate the relationship of the data points to this line. A minimum of 0.1 g/L HSA is required to prevent non-specific binding to the biosensor tip. Experiments were performed in triplicate, and the mean value and standard deviation are shown. One-way ANOVA was performed at each HSA concentration and p-values <0.05 are indicated with an asterisk. All data are summarized in Tables S2–4.
The crowding agent ficoll 70 does not produce the same effect on mAb binding to antigen
To determine whether the observed effect of HSA on mAb/antigen binding could be attributed to non-specific interactions between mAb and HSA or to a more general macromolecular crowding effect, we assessed mAb binding to antigen in the presence of equivalent concentrations of Ficoll 70, a highly soluble polysaccharide frequently used as a crowding agent.21 Ficoll 70 is a colorless ~70 kDa polymer that does not interact specifically with proteins. We observed little to no change in the normalized response of mAb1 and mAb2 binding to antigen at 10 and 137 mM NaCl at concentrations of Ficoll 70 equivalent to those used in HSA experiments (0–50 g/L, Figure 6, Tables S5–7). Binding was also assessed at concentrations of Ficoll 70 more representative of those used in crowding studies (100–300 g/L, Figure 6). As expected, the slow kinetics of mAb binding to antigen in these concentrations of Ficoll 70, particularly for mAb1, required an extension of the association phase to achieve near-equilibrium response levels (overall time was limited to ~3 h to prevent any evaporation of well solutions; Figure 6, panels C and D). For both low and physiological salt concentrations, a minimal and highly similar effect on antigen binding was observed for all Ficoll concentrations. These data suggest that the effect on antigen binding observed with HSA is likely due to electrostatic interactions between HSA and the mAb, rather than the more general phenomenon of excluded volume effects.
Figure 6.
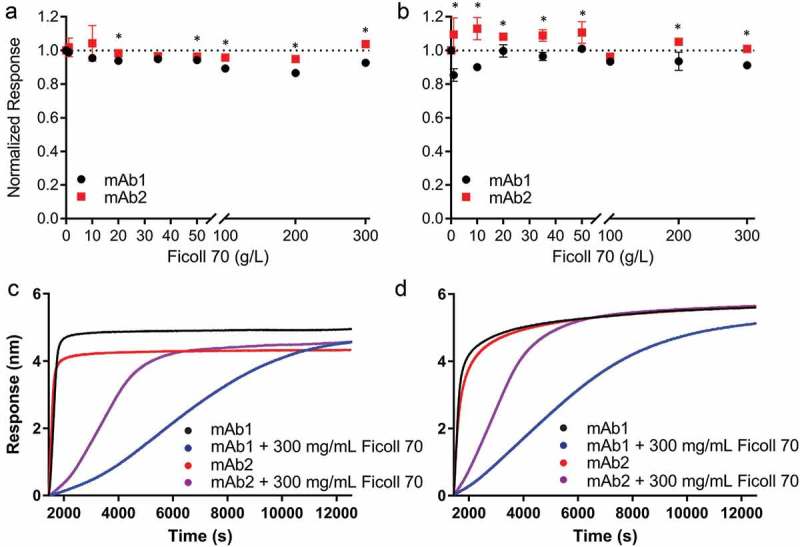
Effect of Ficoll 70 on mAb binding to antigen measured by BLI. Binding of mAb1 (black circles) and mAb2 (red squares) to biotinylated antigen in the presence of increasing Ficoll 70 concentrations was observed by biolayer interferometry at 10 mM (panel a) and 137 mM (panel b) NaCl. The normalized response (to binding at 0.1 g/L Ficoll 70) is plotted as a function of Ficoll 70 concentration. The dotted line indicates the normal at 1.0, in order to illustrate the relationship of the data points to this line. Experiments were performed in triplicate, and the mean value and standard deviation are shown. One-way ANOVA was performed at each HSA concentration and p-values <0.05 are indicated with an asterisk. All data are summarized in Tables S5–7. Slow binding kinetics required an extension of the time for mAb association to antigen at very high Ficoll 70 concentrations (200 g/L and above, panels c and d). Binding of mAbs to biotinylated antigen in the presence of 300 g/L Ficoll was observed by biolayer interferometry at 10 mM (panel c) and 137 mM (panel d) NaCl. The aligned responses in nm for mAb1 (red) and mAb2 (black) in the absence of Ficoll 70 are shown as a function of time for an extended association time (~3 h). Addition of 300 g/L Ficoll 70 to mAb1 (blue) and mAb2 (purple) show comparatively slowed binding kinetics.
Discussion
Macromolecular crowding is ubiquitous in biology. The resulting non-ideal interactions between proteins in crowded solutions are predicted to profoundly affect protein behavior and function.10,39,40 The specific nature of these highly non-linear effects is often difficult to predict, as evidenced by divergent conclusions in several reports.41,42 A limited number of studies using different macromolecular crowding agents have shown considerable consequences for equilibrium constants and reaction rates, often on the order of several logs.10,41,43–45 Together, this highlights the need for techniques capable of readily providing information on the effect of non-ideality in conditions closely replicating physiological environments. Here, we examined how physiological concentrations of albumin-affected mAb function with two complimentary techniques, CG-MALS and BLI. CG-MALS, a powerful and well-established tool that enables measurement of the CVC between two species, was used to obtain an initial understanding of non-specific interactions in the systems. We then utilized a BLI method, with steady-state analysis adapted for non-ideal solution conditions, to first replicate our CG-MALS results, and then extend these observations by performing equilibrium measurements of antigen binding under physiological concentrations of HSA. While orthogonal methods such as AUC with fluorescence detection can measure specific interactions in non-ideal conditions,7,46 BLI is advantageous as a convenient and high-throughput method to assess binding interactions with the inherent flexibility to test many different conditions at high concentrations of crowding agents, and can therefore provide information about binding in various environments in a small set of experiments. This approach is an easy and efficient way to eliminate mAbs or other molecules from consideration during the screening process, early in discovery research.
The physicochemical complexity of the solvent-accessible surface areas presented by different proteins plays a fundamental role in the diversity of non-specific macromolecular interactions. At moderate protein concentrations (≤10 g/L), where the excluded volume effect is less prominent, electrostatics are likely the dominant intermolecular force.13 Consistent with this notion, both mAb1 and mAb2, with different experimentally observed basic isoelectric points (differing by ~0.65 pH units), were shown to interact with HSA, which has an acidic isoelectric point, using CG-MALS. These are not specific interactions, but instead non-specific interactions between HSA and each antibody. For both antibodies, the magnitude of interactions with HSA were shown to be mitigated upon increasing ionic strength, further suggesting the primary force between the molecules is electrostatic, as electrostatic interactions can effectively be screened with increasing ionic strength.47,48 Interestingly, near physiological ionic strength, mAb1 continued to exhibit attractive interactions with HSA, but mAb2 exhibited slightly repulsive interactions with HSA. While the two-component system used in CG-MALS does not fully reflect the complexity of physiological conditions and uses concentrations below physiological for technical reasons, the analysis suggests that the degree and nature of non-specific interactions between proteins may affect biological function. As the antibodies differ only in the CDR, it is possible that the difference in the weak interactions with HSA occur at this region. Furthermore, these non-specific interactions are protein-dependent, indicating the potential for a vast spectrum of functional and structural behavior in a physiological environment, and possibly explain occasional differences observed between in vitro results and pharmacokinetic and clinical results.
Molecular dynamics simulations of synthetic and protein crowders have shown that the effect of crowding on the structure, dynamics, and interactions of proteins within a biological network may facilitate transient interactions that can affect functionality.9 Indeed, it has been hypothesized that evolutionary pressure minimizes non-specific protein–protein interactions to reduce complexity and the potential for protein promiscuity.49,50 In addition, several unrelated studies suggest electrostatics are the primary driver of non-specific interactions.8,12,51–53 The CG-MALS and chromatography data presented here further expand on these studies and highlight the importance of understanding surface charge properties of proteins and the potential effects of electrostatic interactions arising from those charges, as we demonstrate that non-specific interactions can affect functional interactions such as antibody:antigen binding events.
Using a dip and read design rather than microfluidics, BLI is more conducive to studying the effects of non-specific interactions induced by high solute concentrations on highly specific functional interactions, such as antibody:antigen binding. By immobilizing the antigen and using mAb solutions that contained increasing concentrations of HSA, we were able to extend the conditions used for CG-MALS to examine both increased HSA concentration and use the method to establish a ternary interaction system, albeit in an avidity-based format. Importantly, the impact of physiological HSA concentrations on antigen binding was shown to be greater for mAb1 than for mAb2 at low ionic strength, whereas at physiological salt levels, the effect of HSA was considerably diminished. Although the magnitude of the difference between the mAbs at physiological ionic strength is considerably less than observed in 10 mM salt, it is statistically meaningful at 20 g/L and above given the precision within the replicates. While current technical issues with solution evaporation necessitate BLI testing at ambient temperature rather than at physiological temperature (instrument now in development), our results clearly suggest the BLI approach described here is capable of replicating and expanding on the data from light-scattering methods, which were also performed at ambient temperature due to technical issues. Moreover, the non-specific interactions observed with CG-MALS at low ionic strength indeed have a functional impact on antibody:antigen interactions, and this effect appears to plateau at moderate HSA concentration; for example, 50 g/L HSA does not have an appreciably larger impact on binding than 35 g/L. It remains unclear if this is a general trend for most therapeutic mAbs or other biotherapeutics, and continued investigation is needed.
A study performed using the biosensor platform KinExA that tested mAbs associating with their native unpurified antigens in serum54 demonstrated that some mAbs show the same apparent affinity in buffer or serum, while others show differences in apparent affinity. These results further our understanding of macromolecular crowding mediated by protein co-solutes. Several proteins including lysozyme, RNase A, albumin, and reconstituted E. coli cytosol have been used as macromolecular crowding agents, often with contrasting results. For example, the self-association of apo-myoglobin was found to be enhanced in crowded RNase A solutions, but not in crowded HSA solutions.55 Conversely, dimerization of the A34F mutant of GB1 was enhanced with 100 g/L bovine serum albumin (BSA) and diminished in 50 g/L lysozyme.56 The authors point to the differences in charge state, relative to that of A34F, as the principal driver of the observed differences in dissociation constants. Furthermore, weak hetero-interactions in concentrated BSA/SH3 domain solutions slowed the translational diffusion of both proteins well beyond that expected for the solution viscosity.57 This result likely stems from transient binding events on a timescale comparable or faster than translational diffusion. Taken together with the results presented here, it is clear that transient interactions can have an effect on high affinity (nM-pM) interactions, such as antibody:antigen binding events, as well.
Synthetic polymers such as PEG, dextran, or Ficoll, are frequently used as crowding agents; however, the aim of the investigation is typically protein folding or stability.9,58–63 Relatively few studies have been published on the effects polymers have on heterogeneous protein–protein interactions.9,45,64,65 Here again, there is no clear consensus regarding the true effects of synthetic polymers, and it appears the net outcome is specific to the system of interest. Schreiber and colleagues showed minimal effects of PEG and dextran on interactions between barnase and barstar or between β-lactamase with its protein inhibitor, while Liang and co-workers showed considerable effects of polymer crowding on catalase-superoxide dismutase association.45,66 Although the impact of high HSA concentration on antibody:antigen binding could simply be due to excluded volume effects, equivalent experiments performed in the presence of the polysaccharide Ficoll 70 rather than HSA yielded different results, showing little to no effect on binding even at high concentrations. The difference is particularly apparent at low ionic strength, which showed a significant mAb-specific decrease in binding activity in the presence of HSA. In Ficoll crowded solutions, the effect is minimal and similar for the two mAbs. This suggests that the effect of HSA cannot be explained purely by effects on excluded volume; the complexity of biological systems (i.e., the surface properties of proteins) plays a significant role in biological processes. Investigations with additional systems of interest are likely to help refine the model of protein macromolecular crowding.
Not surprisingly, high Ficoll concentrations (100 g/L and above) slowed the apparent binding for both antibodies, as assessed by the time required to achieve a steady state condition; however, the effect was more pronounced on mAb1 than mAb2. This suggests an additional protein-specific effect on the binding properties of the system at Ficoll concentrations similar to those used for typical crowding studies in addition to the high solution viscosity. The specific reason for the different effects of high Ficoll concentration on the two mAbs is unclear but could be attributed to differences in preferential interactions, either binding or exclusion.67,68 Notably, the effects of Ficoll on each mAb were fairly consistent between low and high ionic strength. Previous studies suggest that Ficoll can variably affect the thermal stability and conformational dynamics of molecules.69–72 and differences in the CDRs between the two mAbs could contribute to varied Ficoll-induced effects on structure or dynamics, with a concomitant effect on binding. An investigation using maltose-binding protein showed that Ficoll can bind to the protein and compete with binding of the natural ligand, maltose.73 This demonstrates a direct effect of crowding agents on protein–ligand interactions and the potential consequences of competition from other macromolecules. However, the effects observed with Ficoll were based on the time required to reach equilibrium, rather than on the equilibrium response itself, which demonstrates that polymer (Ficoll) and protein (HSA) crowding agents do not necessarily generate the same result. These potential differences are likely to be dependent on the inherent physicochemical properties associated with the molecules being examined. An additional investigation is necessary to develop a better understanding of this complex phenomenon; furthermore, protein hydration or preferential exclusion/interaction studies may clarify the mechanism behind this discrepancy. Polymer crowders do not consistently produce an effect on ligand binding. Substitution of a protein crowder for the polymers in this system might have a profoundly different effect on the thermodynamics, as this would introduce additional complexity with electrostatic interactions and other non-ideal behavior.
The complexity and volume occupancy of biological solutions impose substantial deviations from ideal behavior on constituent molecules. An understanding of the non-ideal behavior and non-specific interactions of proteins under such conditions is vital to achieving a more complete and accurate picture of protein function. Here, we demonstrated a high-throughput approach to characterize the functional impact of non-specific protein–protein interactions using BLI, which allows screening of a large number of test articles in a relatively short time using minimal material. Specifically, we investigated the impact that physiological concentrations of albumin have on antibody-antigen binding. Two different antibodies that bind the same antigen were affected differently by the presence of albumin, suggesting that biotherapeutics may exhibit a range of non-specific interactions in defined systems with albumin. Although the effect was highly mitigated at physiological ionic strength, we cannot conclude that this is a universal feature of mAbs or biotherapeutics, in general. Given the potential consequences of non-specific interactions on mAb binding to antigen or other molecules, we are currently investigating additional antibody-antigen systems, as well as solution conditions more similar to serum, to further explore this phenomenon. Assessment of mAb binding to other biologically relevant molecules, such as neonatal Fc receptor (FcRn), in the presence of HSA, is paramount. Recycling of both IgG and HSA to the bloodstream from acidic endosomes is facilitated by FcRn in a pH-dependent manner,74 and while this process is well-understood, the potential interplay between IgG and HSA that we observed suggests a more complex process.75 Lastly, as a largely unexplored area of biotherapeutic development, characterizing non-specific interactions relevant to the indication and route of administration could serve as an important discriminator among a pool of lead candidate molecules. The application of BLI technology to a variety of biological and drug discovery problems is expanding.76–78 As an early discovery research screening tool, BLI can be used to more quickly eliminate candidates from the pipeline and can be beneficial in diversifying the types of assays used in discovery research. The approach described here is an important tool that can be used in conjunction with other biophysical methods, such as NMR and AUC, to better investigate crowded solution phenomena.
Materials and methods
Materials and reagent preparation
All mAbs used in this study were research grade and produced at Regeneron Pharmaceuticals, Inc. in the PreClinical Manufacturing and Process Development Department (Tarrytown, NY). All antibodies are fully human IgG4 molecules and contain the mutation S108P in the hinge region in order to recreate the IgG1 hinge sequence to stabilize IgG4 dimer formation, and were produced in Regeneron’s proprietary cell line cloned from Chinese hamster ovary cells. Lyophilized HSA, Ficoll 70, and solution components were purchased from Sigma-Aldrich (St. Louis, MO) or VWR (Radnor, PA) and were the highest grade available.
Monomeric HSA was prepared by dissolving lyophilized HSA in phosphate buffer (1.8 mM KH2PO4, 10 mM Na2HPO4, 2.7 mM KCl, pH 7.4) supplemented with 10 mM NaCl, and purified with a HiLoad 26/100 Superdex 200 size-exclusion column (GE Healthcare, Little Chalfont, UK) equilibrated in the same buffer. Following purification, HSA was concentrated to ~100–130 g/L using a centrifugal filter with a 10 kDa cutoff (Amicon, Billerica, MA). The antibodies were prepared in a similar manner for the CG-MALS experiment. HSA concentration was determined with a SoloVPE spectrophotometer at UVλ=280 nm using an extinction coefficient of 35,700 M−1cm−1. For BLI measurements, the stock solution (1 g/L) of mAb was prepared by diluting a high concentration mAb formulation (>50 g/L) into phosphate buffer supplemented with 10 mM NaCl, and used at a final concentration of 40 nM in equilibrium experiments. Protein concentrations were determined at UVλ=280 nm using an extinction coefficient of 103,555 M−1cm−1 for mAb1 and 100,700 M−1cm−1 for mAb2. The stock solution (600 mM) of Ficoll 70 was prepared by dissolving lyophilized Ficoll 70 into phosphate buffer supplemented with 10 mM NaCl and gently rotated overnight to facilitate solubilization. The antigen was biotinylated with biotin-hydrazide (Thermo Fisher, Waltham, MA) following the manufacturer’s labeling protocol.
The glycoprotein antigen was biotinylated on the single glycan with biotin-hydrazide (Thermo Fisher, Waltham, MA) following the manufacturer’s labeling protocol. Briefly, a solution of 8 g/L sodium meta-periodate (Sigma-Aldrich) was made using 0.1 M sodium acetate pH 4.7 and mixed with antigen, rotating in foil at room temperature for 15 min, followed by quenching with 1% (v/v) glycerol. Oxidized antigen was eluted through a Superdex 75 Increase 10/300 column (GE Healthcare, Little Chalfont, UK) in 0.1 M sodium phosphate pH 6.0. Fractions containing antigen were pooled and concentrated, then incubated with a 10-fold molar excess of biotin-hydrazide for 2 h at room temperature. Labeled antigen was eluted through the same column equilibrated with phosphate buffer at pH 7.4 supplemented with 10 mM NaCl.
Weak cation exchange chromatography
Weak cation exchange chromatography was performed on a ProPac WCX-10 (4 mm × 250 mm) liquid chromatography column (Thermo Fisher) equilibrated with 200 mM MES, 20 mM NaCl, pH 6.5. Proteins were injected neat and 10 μg of each sample was applied to the column on an ACQUITY UPLC system (Waters, Milford, MA) at a flow rate of 0.5 mL/min. A gradient ranging from 20 to 500 mM NaCl was used for protein elution.
Composition gradient multi-angle light scattering
All proteins were dialyzed overnight against the appropriate buffer; all buffers were passed through a 0.02 μm filter and all protein samples were passed through 0.1 μm Anotop 25 Plus syringe filters (Whatman, Maidstone, UK) and vacuum degassed at ~25 Torr for 10 min prior to use. Initial protein stock solutions were manually diluted to approximately 10 g/L prior to filtration. A Calypso composition gradient system in conjunction with a miniDAWN TREOS MALS photometer and an Optilab T-rEX in-line differential refractometer (Calypso system and both detectors from Wyatt Technology, Santa Barbara, CA) was employed to collect static light scattering measurements of HSA, mAb, and mixtures thereof using a cross-over gradient scheme. Briefly, the Calypso pump system was programmed to automatically dilute and inject HSA from 1 to 10 g/L concentrations in 1 g/L increments (10 injections, or steps). Upon injecting undiluted (10 g/L) HSA, the concentration of HSA was reduced by 10% as the concentration of mAb was increased 10% (the cross-over period) in a series of 10 injections. After injecting undiluted mAb (10 g/L), its concentration was reduced in 1 g/L increments through a series of nine injections. While this concentration does not reflect physiological conditions, it is the maximum optimal concentration recommended for determination of the CVC. At each step, a 2 mL bolus of appropriately diluted/mixed sample was injected to fully saturate the detector flow cells; data were acquired for 90 s under quiescent conditions before creating and injecting a subsequent concentration/mixture step. Baseline measurements were obtained immediately before and after the gradient program. After confirming a lack of significant angular-dependent light scattering, only data from the 90° light scattering detector was used in the analysis. Instrument control, data acquisition, and data analysis27,79 were all performed with Calypso software (Wyatt Technology).
Biolayer interferometry
BLI experiments were performed using an Octet Red96 with Streptavidin (SA, cat. number 18–5019) or anti-human IgG Fc capture (AHC, cat. number 18–5064)-coated biosensor tips (ForteBio, Menlo Park, CA). The 96-well plates were filled with 200 μL of solution (buffer, antigen, HSA, or mAb) and agitated at 1000 rpm, and all experiments were temperature controlled at 25°C. Higher temperatures were avoided due to evaporation of solutions. For all experiments, SA or anti-human Fc tips were hydrated in phosphate buffer, pH 7.4, supplemented with low (10 mM NaCl) or physiological (137 mM NaCl) salt concentrations for 20 min at room temperature. All buffers and sample solutions described contained 0.1 g/L HSA unless otherwise noted. Baseline subtraction was performed with tips dipped into buffer in the absence of analyte.
Standard experiments measuring antigen binding to antibody-loaded tips were performed using AHC tips. Following a baseline measurement of AHC tips in phosphate buffer containing low or physiological salt concentrations supplemented with 0.1 g/L HSA for 2 min, the tips were incubated in 2.5 μg/mL antibody to achieve ~0.6 nm response. Antibody-loaded tips were then dipped into buffer to remove excess mAb for 2 min, followed by a 100–300 s association step with various concentrations of unlabeled antigen, typically 2.5–50 nM. The tips were dipped into buffer for 750 s for the dissociation step. The same procedure was followed for biotinylated antigen, at 10 mM and 137 mM NaCl, for both mAbs.
Standard binding experiments measuring antibody binding to antigen-loaded tips were performed using SA tips. Following a baseline measurement of SA tips in the low or physiological salt phosphate buffer solution containing 0.1 g/L HSA for 2 min, the tips were incubated in 5 μg/mL biotinylated antigen to achieve ~0.6 nm response. Antigen-loaded tips were then dipped into buffer to remove excess antigen for 2 min, followed by a 900 s association step with various concentrations of unlabeled antigen, typically 2.5–50 nM. The tips were dipped into buffer for 1800–3600 s for the dissociation step. The same procedure was followed for 10and 137 mM NaCl, for both mAbs.
For steady state analysis of mAb binding to antigen in the presence of up 50 g/L HSA, an additional incubation step in HSA was required. Following antigen loading, sensors were dipped into wells containing 0.1–50 g/L HSA for equilibration for ~15 min, followed by a 2-min incubation in fresh solution with the same composition to establish a new baseline due to a slight increase in signal upon HSA incubation (see Figure S1, step B). Sensors were then dipped into wells containing 40 nM mAb plus 0.1–50 g/L HSA for 20 min as a second association step. For all equilibrium experiments, signal response (nm) at the completion of the mAb binding to antigen association step was used as the metric. No kinetic analysis was performed for any experiments measuring mAb:antigen binding in the presence of HSA >0.1 g/L. The data were normalized to 1.0 by dividing the raw response (in nm) obtained for each HSA concentration by the raw response of mAb:antigen binding in 0.1 g/L HSA. All experiments were performed in triplicate. One-way analysis of variance (ANOVA) was performed using JMP software (SAS Institute, Cary, NC) at each condition to assess whether differences between mAb1 and mAb2 were statistically significant through determination of p-values.
Experiments containing Ficoll 70 were performed in a similar manner, substituting HSA with Ficoll 70. For experiments containing 200 g/L Ficoll 70 or above, a longer association time for mAb binding to antigen was required due to increased viscosity (~1.7–3 h) in order to achieve equilibrium.
Abbreviations
- AUC
Analytical ultracentrifugation
- BLI
Biolayer interferometry
- CDR
Complementarity-determining region
- CG-MALS
Composition gradient multi-angle light scattering
- CVC
Cross virial coefficient
- FcRn
neonatal Fc receptor
- HAS
human serum albumin
- IgG
Immunoglobulin G
- mAb
monoclonal antibody
- NMR
nuclear magnetic resonance
- SPR
Surface plasmon resonance
Acknowledgments
The authors would like to thank Thomas Daly and Erica Pyles for their helpful discussions, critical review of the manuscript, and valuable input. The authors would also like to acknowledge the Pre-Clinical Manufacturing and Process Development Department at Regeneron for generation of materials used in this study.
Disclosure summary
All authors are employees and shareholders of Regeneron Pharmaceuticals with the exception of Michael Marlow, who is an employee of Boehringer Ingelheim. The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Neal BL, Asthagiri D, Lenhoff AM.. Molecular origins of osmotic second virial coefficients of proteins. Biophys J. 1998;75:2469–77. doi: 10.1016/S0006-3495(98)77691-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heddi B, Phan AT.. Structure of human telomeric DNA in crowded solution. J Am Chem Soc. 2011;133:9824–33. doi: 10.1021/ja200786q. [DOI] [PubMed] [Google Scholar]
- 3.Martorell G, Adrover M, Kelly G, Temussi PA, Pastore A. A natural and readily available crowding agent: NMR studies of proteins in hen egg white. Proteins. 2011;79:1408–15. doi: 10.1002/prot.22967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rivas G, Fernandez JA, Minton AP. Direct observation of the self-association of dilute proteins in the presence of inert macromolecules at high concentration via tracer sedimentation equilibrium: theory, experiment, and biological significance. Biochemistry. 1999;38:9379–88. doi: 10.1021/bi990355z. [DOI] [PubMed] [Google Scholar]
- 5.Rivas G, Minton AP. Non-ideal tracer sedimentation equilibrium: a powerful tool for the characterization of macromolecular interactions in crowded solutions. J Mol Recognit. 2004;17:362–67. doi: 10.1002/jmr.708. [DOI] [PubMed] [Google Scholar]
- 6.Pielak GJ, Li C, Miklos AC, Schlesinger AP, Slade KM, Wang G-F, Zigoneanu IG. Protein nuclear magnetic resonance under physiological conditions. Biochemistry. 2009;48:226–34. doi: 10.1021/bi8018948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright RT, Hayes DB, Stafford WF, Sherwood PJ, Correia JJ. Characterization of therapeutic antibodies in the presence of human serum proteins by AU-FDS analytical ultracentrifugation. Anal Biochem. 2018;550:72–83. doi: 10.1016/j.ab.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Elcock AH. Prediction of functionally important residues based solely on the computed energetics of protein structure. J Mol Biol. 2001;312:885–96. doi: 10.1006/jmbi.2001.5009. [DOI] [PubMed] [Google Scholar]
- 9.Candotti M, Orozco M. The differential response of proteins to macromolecular crowding. PLoS Comput Biol. 2016;12:e1005040. doi: 10.1371/journal.pcbi.1005154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minton AP. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J Biol Chem. 2001;276:10577–80. doi: 10.1074/jbc.R100005200. [DOI] [PubMed] [Google Scholar]
- 11.Zimmerman SB, Minton AP. Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu Rev Biophys Biomol Struct. 1993;22:27–65. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Witham S, Alexov E. On the role of electrostatics in protein-protein interactions. Phys Biol. 2011;8:035001. doi: 10.1088/1478-3975/8/3/035001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elcock AH, McCammon JA. Calculation of weak protein-protein interactions: the pH dependence of the second virial coefficient. Biophys J. 2001;80:613–25. doi: 10.1016/S0006-3495(01)76042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanco MA, Perevozchikova T, Martorana V, Manno M, Roberts CJ. Protein-protein interactions in dilute to concentrated solutions: alpha-chymotrypsinogen in acidic conditions. J Phys Chem B. 2014;118:5817–31. doi: 10.1021/jp412301h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salinas BA, Sathish HA, Bishop SM, Harn N, Carpenter JF, Randolph TW. Understanding and modulating opalescence and viscosity in a monoclonal antibody formulation. J Pharm Sci. 2010;99:82–93. doi: 10.1002/jps.21797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Connolly BD, Petry C, Yadav S, Demeule B, Ciaccio N, Moore JM, Shire SJ, Gokarn YR. Weak interactions govern the viscosity of concentrated antibody solutions: high-throughput analysis using the diffusion interaction parameter. Biophys J. 2012;103:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J, Nguyen MD, Andya JD, Shire SJ. Reversible self-association increases the viscosity of a concentrated monoclonal antibody in aqueous solution. J Pharm Sci. 2005;94:1928–40. doi: 10.1002/jps.20347. [DOI] [PubMed] [Google Scholar]
- 18.Raut AS, Kalonia DS. Pharmaceutical perspective on opalescence and liquid-liquid phase separation in protein solutions. Mol Pharm. 2016;13:1431–44. doi: 10.1021/acs.molpharmaceut.5b00937. [DOI] [PubMed] [Google Scholar]
- 19.Spitzer J. From water and ions to crowded biomacromolecules: in vivo structuring of a prokaryotic cell. Microbiol Mol Biol Rev. 2011;75:491–506. second page of table of contents. doi: 10.1128/MMBR.00010-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Den Berg J, Boersma AJ, Poolman B. Microorganisms maintain crowding homeostasis. Nat Rev Microbiol. 2017;15:309–18. doi: 10.1038/nrmicro.2017.17. [DOI] [PubMed] [Google Scholar]
- 21.Zhou HX, Rivas G, Minton AP. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–97. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ross-Rodriguez LU, Elliott JA, McGann LE. Non-ideal solution thermodynamics of cytoplasm. Biopreserv Biobank. 2012;10:462–71. doi: 10.1089/bio.2012.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatters DM, Minton AP, Howlett GJ. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J Biol Chem. 2002;277:7824–30. doi: 10.1074/jbc.M110429200. [DOI] [PubMed] [Google Scholar]
- 24.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT Jr.. Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 25.Munishkina LA, Cooper EM, Uversky VN, Fink AL. The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J Mol Recognit. 2004;17:456–64. doi: 10.1002/jmr.699. [DOI] [PubMed] [Google Scholar]
- 26.Alford JR, Kendrick BS, Carpenter JF, Randolph TW. Measurement of the second osmotic virial coefficient for protein solutions exhibiting monomer-dimer equilibrium. Anal Biochem. 2008;377:128–33. doi: 10.1016/j.ab.2008.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Some D, Kenrick S. Characterization of protein-protein interactions via static and dynamic light scattering In: Cai J, editor. Protein Interactions. London (UK): InTech Open; 2012. p. 401–426. [Google Scholar]
- 28.Some D, Pollastrini J, Cao S. Characterizing reversible protein association at moderately high concentration via composition-gradient static light scattering. J Pharm Sci. 2016;105:2310–18. doi: 10.1016/j.xphs.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Abdiche Y, Malashock D, Pinkerton A, Pons J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the octet. Anal Biochem. 2008;377:209–17. doi: 10.1016/j.ab.2008.03.035. [DOI] [PubMed] [Google Scholar]
- 30.Fang Y, Li G, Ferrie AM. Non-invasive optical biosensor for assaying endogenous G protein-coupled receptors in adherent cells. J Pharmacol Toxicol Methods. 2007;55:314–22. doi: 10.1016/j.vascn.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Rich RL, Myszka DG. Survey of the year 2006 commercial optical biosensor literature. J Mol Recognit. 2007;20:300–66. doi: 10.1002/jmr.862. [DOI] [PubMed] [Google Scholar]
- 32.Shah NB, Duncan TM. Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects. J Visualized Exp. 2014 Feb;18(84):e51383. doi: 10.3791/51383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frenzel D, Willbold D, Kourentzi K. Kinetic titration series with biolayer interferometry. PLoS One. 2014;9:e106882. doi: 10.1371/journal.pone.0106882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park S, Phukan PD, Zeeb M, Martinez-Yamout MA, Dyson HJ, Wright PE. Structural basis for interaction of the tandem zinc finger domains of human muscleblind with cognate RNA from human cardiac troponin T. Biochemistry. 2017;56:4154–68. doi: 10.1021/acs.biochem.7b00484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sultana A, Lee JE. Measuring protein-protein and protein-nucleic acid interactions by biolayer interferometry. Curr Protoc Protein Sci. 2015;79:19–25. [DOI] [PubMed] [Google Scholar]
- 36.Wartchow CA, Podlaski F, Li S, Rowan K, Zhang X, Mark D, Huang KS. Biosensor-based small molecule fragment screening with biolayer interferometry. J Comput Aided Mol Des. 2011;25:669–76. doi: 10.1007/s10822-011-9439-8. [DOI] [PubMed] [Google Scholar]
- 37.Deszczynski M, Harding SE, Winzor DJ. Negative second virial coefficients as predictors of protein crystal growth: evidence from sedimentation equilibrium studies that refutes the designation of those light scattering parameters as osmotic virial coefficients. Biophys Chem. 2006;120:106–13. doi: 10.1016/j.bpc.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 38.Winzor DJ, Deszczynski M, Harding SE, Wills PR. Nonequivalence of second virial coefficients from sedimentation equilibrium and static light scattering studies of protein solutions. Biophys Chem. 2007;128:46–55. doi: 10.1016/j.bpc.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Hu Z, Jiang J, Rajagopalan R. Effects of macromolecular crowding on biochemical reaction equilibria: a molecular thermodynamic perspective. Biophys J. 2007;93:1464–73. doi: 10.1529/biophysj.107.104646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei J, Dobnikar J, Curk T, Song F, Yan Y-B. The effect of attractive interactions and macromolecular crowding on crystallins association. PLoS One. 2016;11:e0151159. doi: 10.1371/journal.pone.0151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuznetsova IM, Turoverov KK, Uversky VN. What macromolecular crowding can do to a protein. Int J Mol Sci. 2014;15:23090–140. doi: 10.3390/ijms151223090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minton AP. Implications of macromolecular crowding for protein assembly. Curr Opin Struct Biol. 2000;10:34–39. [DOI] [PubMed] [Google Scholar]
- 43.Minton AP. Molecular crowding: analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzymol. 1998;295:127–49. doi: 10.1016/s0076-6879(98)95038-8. [DOI] [PubMed] [Google Scholar]
- 44.Kim JS, Yethiraj A. Effect of macromolecular crowding on reaction rates: a computational and theoretical study. Biophys J. 2009;96:1333–40. doi: 10.1016/j.bpj.2008.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiao M, Li HT, Chen J, Minton AP, Liang Y. Attractive protein-polymer interactions markedly alter the effect of macromolecular crowding on protein association equilibria. Biophys J. 2010;99:914–23. doi: 10.1016/j.bpj.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wright RT, Hayes D, Sherwood PJ, Stafford WF, Correia JJ. AUC measurements of diffusion coefficients of monoclonal antibodies in the presence of human serum proteins. Eur Biophys J. 2018;47:709–22. doi: 10.1007/s00249-018-1319-x. [DOI] [PubMed] [Google Scholar]
- 47.Roberts D, Keeling R, Tracka M, van der Walle CF, Uddin S, Warwicker J, Curtis R. Specific ion and buffer effects on protein-protein interactions of a monoclonal antibody. Mol Pharm. 2015;12:179–93. doi: 10.1021/mp500533c. [DOI] [PubMed] [Google Scholar]
- 48.Roberts D, Keeling R, Tracka M, van der Walle CF, Uddin S, Warwicker J, Curtis R. The role of electrostatics in protein-protein interactions of a monoclonal antibody. Mol Pharm. 2014;11:2475–89. doi: 10.1021/mp5002334. [DOI] [PubMed] [Google Scholar]
- 49.Johnson ME, Hummer G. Nonspecific binding limits the number of proteins in a cell and shapes their interaction networks. Proc Natl Acad Sci U S A. 2011;108:603–08. doi: 10.1073/pnas.1010954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deeds EJ, Ashenberg O, Gerardin J, Shakhnovich EI. Robust protein protein interactions in crowded cellular environments. Proc Natl Acad Sci U S A. 2007;104:14952–57. doi: 10.1073/pnas.0702766104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gunasekaran K, Pentony M, Shen M, Garrett L, Forte C, Woodward A, Ng SB, Born T, Retter M, Manchulenko K, et al. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285:19637–46. doi: 10.1074/jbc.M110.117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Persson BA, Jonsson B, Lund M. Enhanced protein steering: cooperative electrostatic and van der Waals forces in antigen-antibody complexes. J Phys Chem B. 2009;113:10459–64. doi: 10.1021/jp904541g. [DOI] [PubMed] [Google Scholar]
- 53.Wlodek ST, Shen T, McCammon JA. Electrostatic steering of substrate to acetylcholinesterase: analysis of field fluctuations. Biopolymers. 2000;53:265–71. doi:. [DOI] [PubMed] [Google Scholar]
- 54.Bee C, Abdiche YN, Pons J, Rajpal A, Kaveri S. Determining the binding affinity of therapeutic monoclonal antibodies towards their native unpurified antigens in human serum. PLoS One. 2013;8:e80501. doi: 10.1371/journal.pone.0080501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zorrilla S, Rivas G, Acuna AU, Lillo MP. Protein self-association in crowded protein solutions: a time-resolved fluorescence polarization study. Protein Sci. 2004;13:2960–69. doi: 10.1110/ps.04809404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kyne C, Crowley PB. Short arginine motifs drive protein stickiness in the escherichia coli cytoplasm. Biochemistry. 2017;56:5026–32. doi: 10.1021/acs.biochem.7b00731. [DOI] [PubMed] [Google Scholar]
- 57.Rothe M, Gruber T, Groger S, Balbach J, Saalwachter K, Roos M. Transient binding accounts for apparent violation of the generalized Stokes-Einstein relation in crowded protein solutions. Phys Chem Chem Phys. 2016;18:18006–14. doi: 10.1039/c6cp01056c. [DOI] [PubMed] [Google Scholar]
- 58.McGuffee SR, Elcock AH, Briggs JM. Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput Biol. 2010;6:e1000694. doi: 10.1371/journal.pcbi.1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mittal S, Singh LR, Subramanyam R. Denatured state structural property determines protein stabilization by macromolecular crowding: a thermodynamic and structural approach. PLoS One. 2013;8:e78936. doi: 10.1371/journal.pone.0078936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou HX. Polymer crowders and protein crowders act similarly on protein folding stability. FEBS Lett. 2013;587:394–97. doi: 10.1016/j.febslet.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Batra J, Xu K, Qin S, Zhou HX. Effect of macromolecular crowding on protein binding stability: modest stabilization and significant biological consequences. Biophys J. 2009;97:906–11. doi: 10.1016/j.bpj.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Senske M, Tork L, Born B, Havenith M, Herrmann C, Ebbinghaus S. Protein stabilization by macromolecular crowding through enthalpy rather than entropy. J Am Chem Soc. 2014;136:9036–41. doi: 10.1021/ja503205y. [DOI] [PubMed] [Google Scholar]
- 63.Hong J, Gierasch LM. Macromolecular crowding remodels the energy landscape of a protein by favoring a more compact unfolded state. J Am Chem Soc. 2010;132:10445–52. doi: 10.1021/ja103166y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phillip Y, Schreiber G. Formation of protein complexes in crowded environments–from in vitro to in vivo. FEBS Lett. 2013;587:1046–52. doi: 10.1016/j.febslet.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kozer N, Kuttner YY, Haran G, Schreiber G. Protein-protein association in polymer solutions: from dilute to semidilute to concentrated. Biophys J. 2007;92:2139–49. doi: 10.1529/biophysj.106.097717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Phillip Y, Sherman E, Haran G, Schreiber G. Common crowding agents have only a small effect on protein-protein interactions. Biophys J. 2009;97:875–85. doi: 10.1016/j.bpj.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arakawa T, Timasheff SN. Preferential interactions of proteins with solvent components in aqueous amino acid solutions. Arch Biochem Biophys. 1983;224:169–77. doi: 10.1016/0003-9861(83)90201-1. [DOI] [PubMed] [Google Scholar]
- 68.Timasheff SN. Protein-solvent preferential interactions, protein hydration, and the modulation of biochemical reactions by solvent components. Proc Natl Acad Sci U S A. 2002;99:9721–26. doi: 10.1073/pnas.122225399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qu Y, Bolen DW. Efficacy of macromolecular crowding in forcing proteins to fold. Biophys Chem. 2002;101–102:155–65. [DOI] [PubMed] [Google Scholar]
- 70.Sasahara K, McPhie P, Minton AP. Effect of dextran on protein stability and conformation attributed to macromolecular crowding. J Mol Biol. 2003;326:1227–37. doi: 10.1016/s0022-2836(02)01443-2. [DOI] [PubMed] [Google Scholar]
- 71.Stagg L, Zhang SQ, Cheung MS, Wittung-Stafshede P. Molecular crowding enhances native structure and stability of alpha/beta protein flavodoxin. Proc Natl Acad Sci U S A. 2007;104:18976–81. doi: 10.1073/pnas.0705127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tokuriki N, Kinjo M, Negi S, Hoshino M, Goto Y, Urabe I, Yomo T. Protein folding by the effects of macromolecular crowding. Protein Sci. 2004;13:125–33. doi: 10.1110/ps.03288104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miklos AC, Sumpter M, Zhou H-X, Riggs PD. Competitive interactions of ligands and macromolecular crowders with maltose binding protein. PLoS One. 2013;8:e74969. doi: 10.1371/journal.pone.0074969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vaughn DE, Bjorkman PJ. Structural basis of pH-dependent antibody binding by the neonatal Fc receptor. Structure. 1998;6:63–73. [DOI] [PubMed] [Google Scholar]
- 75.Wang W, Lu P, Fang Y, Hamuro L, Pittman T, Carr B, Hochman J, Prueksaritanont T. Monoclonal antibodies with identical Fc sequences can bind to FcRn differentially with pharmacokinetic consequences. Drug Metab Dispos. 2011;39:1469–77. doi: 10.1124/dmd.111.039453. [DOI] [PubMed] [Google Scholar]
- 76.Verzijl D, Riedl T, Parren P, Gerritsen AF. A novel label-free cell-based assay technology using biolayer interferometry. Biosens Bioelectron. 2017;87:388–95. doi: 10.1016/j.bios.2016.08.095. [DOI] [PubMed] [Google Scholar]
- 77.Kaminski T, Gunnarsson A, Geschwindner S. Harnessing the versatility of optical biosensors for target-based small-molecule drug discovery. ACS Sens. 2017;2:10–15. doi: 10.1021/acssensors.6b00735. [DOI] [PubMed] [Google Scholar]
- 78.Yang D, Singh A, Wu H, Kroe-Barrett R. Determination of high-affinity antibody-antigen binding kinetics using four biosensor platforms. J Visualized Exp. 2017. doi: 10.3791/55659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Some D. Light-scattering-based analysis of biomolecular interactions. Biophys Rev. 2013;5:147–58. doi: 10.1007/s12551-013-0107-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.