

ABSTRACT
Forced degradation experiments of monoclonal antibodies (mAbs) aid in the identification of critical quality attributes (CQAs) by studying the impact of post-translational modifications (PTMs), such as oxidation, deamidation, glycation, and isomerization, on biological functions. Structure-function characterization of mAbs can be used to identify the PTM CQAs and develop appropriate analytical and process controls. However, the interpretation of forced degradation results can be complicated because samples may contain mixtures of asymmetrically and symmetrically modified mAbs with one or two modified chains. We present a process to selectively create symmetrically and asymmetrically modified antibodies for structure-function characterization using the bispecific DuoBody® platform. Parental molecules mAb1 and mAb2 were first stressed with peracetic acid to induce methionine oxidation. Bispecific antibodies were then prepared from a mixture of oxidized or unoxidized parental mAbs by a controlled Fab-arm exchange process. This process was used to systematically prepare four bispecific mAb products: symmetrically unoxidized, symmetrically oxidized, and both combinations of asymmetrically oxidized bispecific mAbs. Results of this study demonstrated chain-independent, 1:2 stoichiometric binding of the mAb Fc region to both FcRn receptor and to Protein A. The approach was also applied to create asymmetrically deamidated mAbs at the asparagine 330 residue. Results of this study support the proposed 1:1 stoichiometric binding relationship between the FcγRIIIa receptor and the mAb Fc. This approach should be generally applicable to study the potential impact of any modification on biological function.
KEYWORDS: Monoclonal antibody, structure function characterization, methionine oxidation, duobody®, asymmetric antibodies, mass spectrometry, single chain modification, Fc, protein A separation
Introduction
Monoclonal antibodies (mAbs) are among the fastest growing class of therapeutic molecules used to treat human diseases such as cancer, neurodegeneration, and inflammatory disease.1−4 Typical therapeutic mAbs are from the immunoglobulin G (IgG) class, which consists of two heavy chain (HC) sequences and two light chain sequences connected by disulfide bonds.5 Antigen binding is generally governed by three complementary-determining regions located on the variable region of the heavy and light chains. The Fc region is responsible for effector functions, such as antibody-dependent cell-mediated cytotoxicity (ADCC)6,7 and extended serum half-life.8 Cell culture bioreactor conditions, downstream purification steps, and formulation conditions can generate post-translational modifications (PTMs), which results in a complex mixture of product variants2,9 potentially affecting biological function.4 PTMs that affect antigen binding10 and Fc effector functions11 are considered critical quality attributes (CQAs) that must be controlled for product release.12,13
6Forced degradation studies, such as light, heat,14,15 or chemical stress16,17 are used to aid in CQA assessment.18 Modifications to a mAb through forced degradation studies can occur asymmetrically on one chain or symmetrically on both chains, which may have different biological impacts. For example, the neonatal Fc receptor binds both HCs of a mAb in a 2:1 stoichiometric relationship,19 which implies that IgG mAbs retain FcRn binding even if one chain is functionally inactive. However, FcγRIIIa binds to both chains simultaneously, and binding could be significantly impacted by modification of one chain.20-22 It is typically not possible to selectively prepare chain-specific modifications in forced degradation studies because most modifications are randomly added to either chain. Although separation techniques can be employed to isolate asymmetrically modified molecules from the mixture, the resolution is usually poor making the isolation of enriched fractions difficult.23 The selective preparation of symmetrically and asymmetrically modified antibodies would allow the experimental evaluation of predicted structural models and provide additional understanding of structure–function relationships.
Bispecific antibodies (BsAb)24,25 provide the opportunity to study the structure–function relationship of asymmetrically modified mAbs.26 The “knobs-into-holes” bispecific technique was used to prepare glycosylated heterodimeric antibodies to characterize afucosylation asymmetry in cell-based bioassays.22 Similar strategies can be used to expand the study of symmetrically and asymmetrically modified mAbs to include oxidation, deamidation, and other PTMs. The DuoBody® technology generates bispecific molecules through a controlled Fab-arm exchange (FAE) redox reaction of two parental homodimeric antibodies.27 The parental mAbs are expressed separately and pooled prior to selective reduction of the hinge region disulfide bonds. Chains are recombined to a bispecific molecule, which is facilitated by the amino acid substitutions in the CH3 domain to drive preferential chain association of a heterodimer.28,29 The DuoBody® retains the structural integrity of homodimeric mAbs, and Fc effector functions are preserved.
In this study, symmetrically and asymmetrically oxidized BsAb were prepared by FAE using the DuoBody® platform30 by combining oxidized parental mAb1 with non-oxidized parental mAb2.31 The oxidized and non-oxidized parental mAbs were pooled to produce four individual combinations of symmetrically or asymmetrically modified BsAb (AA, AB, BA, and BB). This process establishes a platform to create symmetrically and asymmetrically modified mAbs for more selective studies that involve other mAb modification, such as deamidation, glycation, or glycosylation.
Results
Preparation and purity of oxidized BsAbs
Symmetrically and asymmetrically oxidized BsAbs were generated using the DuoBody® FAE process28,30 (Figure 1). The mAb1 and mAb2 parental molecules were chemically oxidized using peracetic acid to induce complete methionine oxidation of the most sensitive sites on all chains (Figure 1(a)). As shown in Figure 1(b), four BsAb products were created by FAE using non-oxidized (A) or oxidized (B) mAb1 and mAb2: a symmetrically non-oxidized control BsAb1 (AA), asymmetrically oxidized BsAb2 (AB) and BsAb3 (BA), and symmetrically oxidized BsAb4 (BB). Each BsAb product was purified using two chromatography steps and exchanged into formulation buffer for characterization.
Figure 1.
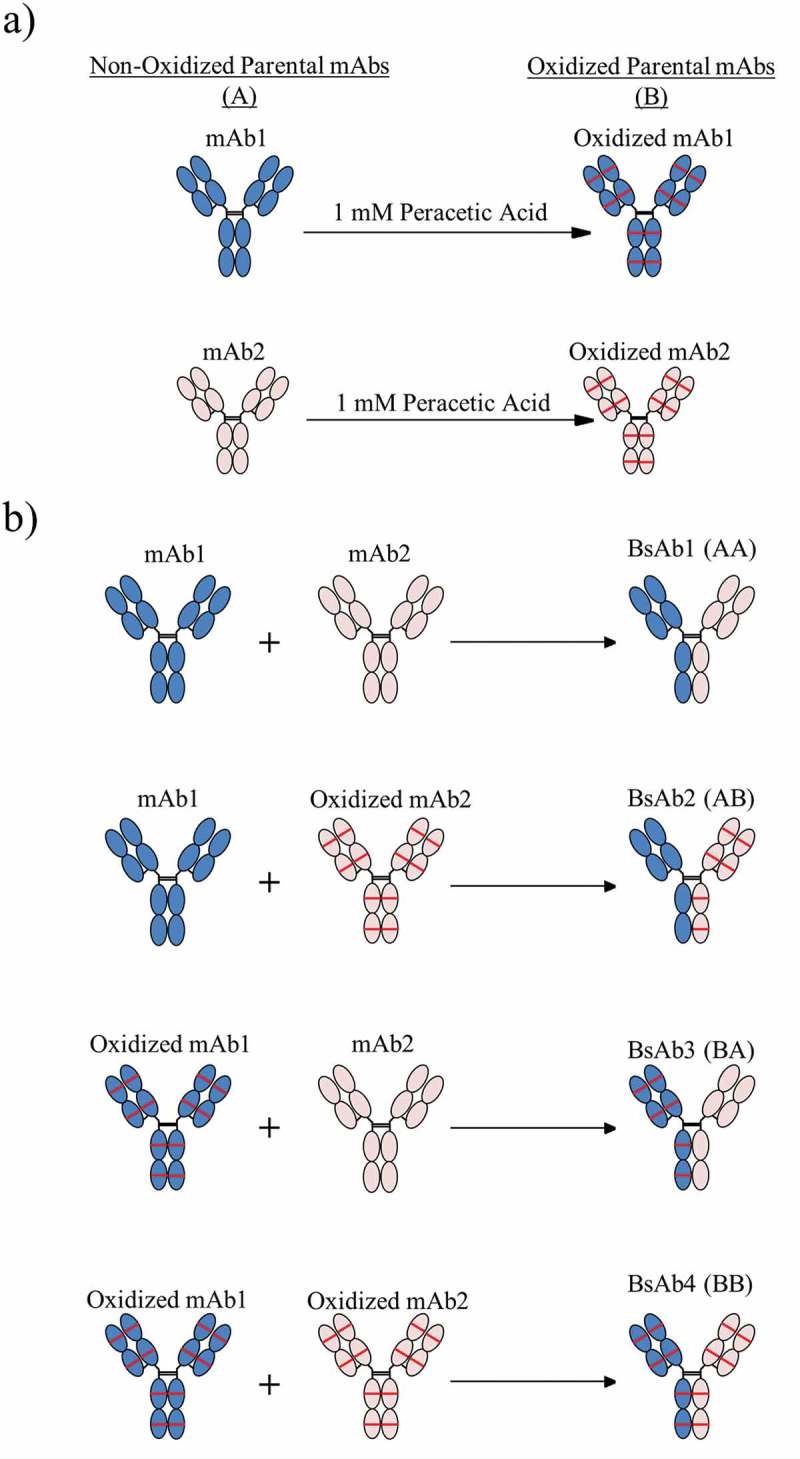
The process for creating asymmetrically oxidized bispecific molecules (BsAb) is presented. (a) Peracetic acid treatment of mAb1 (blue) or mAb2 (peach) resulted in methionine oxidation (illustrated as red lines) throughout the molecule. (b) Combinations of non-oxidized (A) and/or oxidized (B) mAb1 and mAb2 were pooled prior to Fab arm exchange to generate control BsAb1 (AA), asymmetrically oxidized BsAb2 (AB) and BsAb3 (BA), and symmetrically oxidized BsAbs (BB). All BsAbs were purified to the final product prior to analytical characterization.
The purity of all four BsAb products was assessed by hydrophobic interaction chromatography (HIC), size-exclusion chromatography (SEC), and non-reduced capillary SDS electrophoresis (cSDS). FAE efficiency was assessed by HIC, which separated residual homodimeric parental mAb1 and mAb2 from the heterodimeric BsAb products. All BsAb products eluted between 7.7 and 8.0 minutes, while excess parental mAb1 and mAb2 eluted as a pre-peak and post-peak, respectively (Figure 2(a)). The formation of the correct BsAb was confirmed by HIC retention time and intact molecular weight analysis, which showed the formation of intact heterodimers with no light chain swapping (data not shown). The purities of the BsAb1, BsAb2, BsAb3, and BsAb4 products by HIC were 88.3%, 93.1%, 90.5%, and 93.1%, respectively (Table 1), which indicated that extensive oxidation did not affect the FAE process. The residual, parental mAbs were removed from the BsAb samples by downstream processing as shown in Figure 2(b). The purities of the final BsAb1, BsAb2, BsAb3, and BsAb4 products were >98% by HIC analysis (Table 1).
Figure 2.
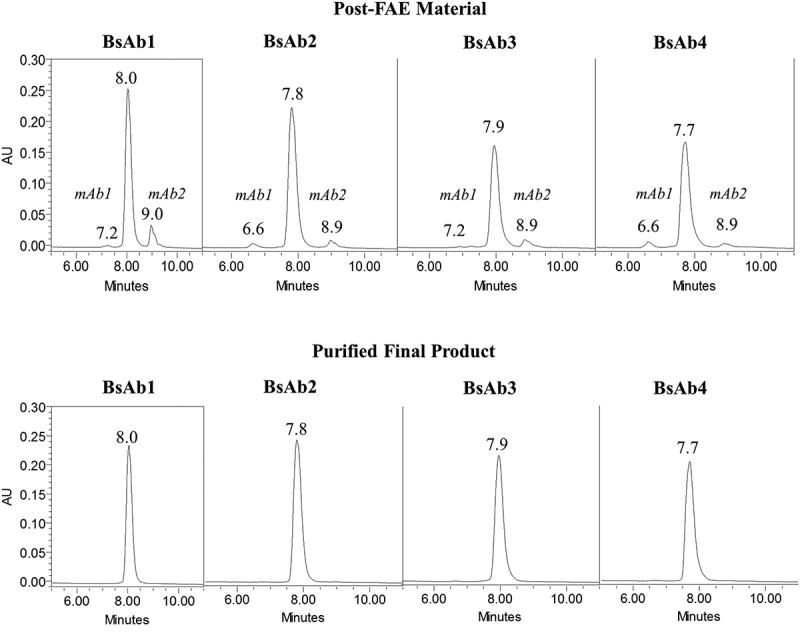
HIC chromatograms of BsAb products. Chromatograms for post-FAE reaction intermediates and final-purified products are shown in the top and bottom panels, respectively.
Table 1.
Purity and free thiol levels for the BsAb intermediates and purified final products.
| Bispecific producta |
BsAb1 |
BsAb2 |
BsAb3 |
BsAb4 |
||||
|---|---|---|---|---|---|---|---|---|
| mAb 1 |
Oxidation (-) |
Oxidation (+) |
Oxidation (-) |
Oxidation (+) |
||||
| mAb 2 |
Oxidation (-) |
Oxidation (-) |
Oxidation (+) |
Oxidation (+) |
||||
| Assay | Post-FAEb | Final formulationc | Post-FAEb | Final formulationc | Post-FAEb | Final formulationc | Post-FAEb | Final formulationc |
| HIC | ||||||||
| BsAb | 88.3% | 99.7% | 93.1% | 99.2% | 90.5% | 99.0% | 93.1% | 98.7% |
| Parental mAb | 11.7% | 0.3% | 6.9% | 0.7% | 9.5% | 1.0% | 6.9% | 1.3% |
| NR-cSDS | ||||||||
| Purity | 95.9% | 96.8% | 90.5% | 92.4% | 87.6% | 90.1% | 85.1% | 88.3% |
| LMWS | 3.9% | 3.2% | 9.3% | 7.6% | 12.0% | 9.9% | 14.5% | 11.7% |
| SEC | ||||||||
| Monomer | n/a | 99.7% | n/a | 99.5% | n/a | 99.1% | n/a | 99.1% |
| LMWS | n/a | 0.0% | n/a | 0.1% | n/a | 0.3% | n/a | 0.3% |
| HMWS | n/a | 0.3% | n/a | 0.4% | n/a | 0.6% | n/a | 0.6% |
| Free Thiold | n/a | 0.7% | n/a | 1.1% | n/a | 1.0% | n/a | 1.0% |
aBispecific Product is synthesized from respective oxidized (+) or unoxidized (-) mAb1 and mAb 2
bMaterial after Fab-arm exchange reaction prior to downstream purification steps
cFinal product after purification
dCalculated as % free thiol/cysteine residue.
All values are expressed as a relative abundance
HMWS = high molecular weight species
LMWS = low molecular weight species
The levels of aggregates and fragments in the BsAb preparations were assessed by SEC and cSDS. The purity of all four BsAbs was >99% by SEC analysis (Table 1). The purity of the control BsAb1 was 96.8% by non-reduced cSDS analysis with 3.2% low molecular weight (LMW) impurities. The purities of asymmetrically oxidized BsAb2 and BsAb3, and BsAb4 by non-reduced cSDS were lower than BsAb1 (92.4%, 90.1%, and 88.3% IgG, respectively) with higher levels of LMW species. The primary impurities were free heavy and light chains based on cSDS migration times and intact molecular weight analysis, which suggested incomplete disulfide bond formation during the FAE process step. These results were consistent with a previous report showing high levels of methionine oxidation can affect the disulfide bond structure of human IgG.32
To further assess the disulfide structure, the BsAb final products were evaluated by the Ellman’s assay and non-reduced peptide mapping. Non-reduced peptide map data showed the expected disulfide linkages were present in all BsAb products (data not shown). The levels of free thiols were 0.7%, 1.1%, 1.0%, and 1.0% in BsAb1, BsAb2, BsAb3, and BsAb4, respectively (Table 1). Overall, these results demonstrated that the structural integrity and purity of the BsAb batches were sufficient for characterization.
Structural characterization of oxidized BsAbs
The methionine oxidation levels of the parental mAbs and BsAb products were characterized by peptide mapping and subunit mass spectrometry (MS) analysis. Peptide mapping analysis of the peracetic acid-treated parental mAb1 and mAb2 confirmed the primary sites of oxidation to be HC Met254, Met360, and Met430 in the Fc region (data not shown). The level of oxidation at all three sites was ≥99.9%. For subunit MS analysis, samples were digested with EndoS and IdeS33 followed by reduction with dithiothreitol (DTT) to generate free light chain, Fd’, and single-chain Fc subunits (scFc) and analyzed by high-performance liquid chromatography (HPLC)-MS.34 MS analysis was limited to the scFc subunits since this region contained the most sensitive methionine sites. As shown in Figure 3(a), a single-deconvoluted mass peak was detected at 23,957 Da by subunit analysis of mAb1, which corresponded to the non-oxidized scFc subunit. Subunit analysis of oxidized mAb1 showed a single-deconvoluted mass peak detected at 24,006 Da, which corresponded to the scFc subunit with three oxidized methionine residues. Similarly, oxidized mAb2 showed a single-deconvoluted mass peak at 24067 Da by subunit analysis, which corresponds to the mass of an scFc subunit with three oxidized methionine residues. These results indicated that the oxidized parental mAbs were fully oxidized at HC residues Met254, Met360, and Met430 in the Fc region.
Figure 3.
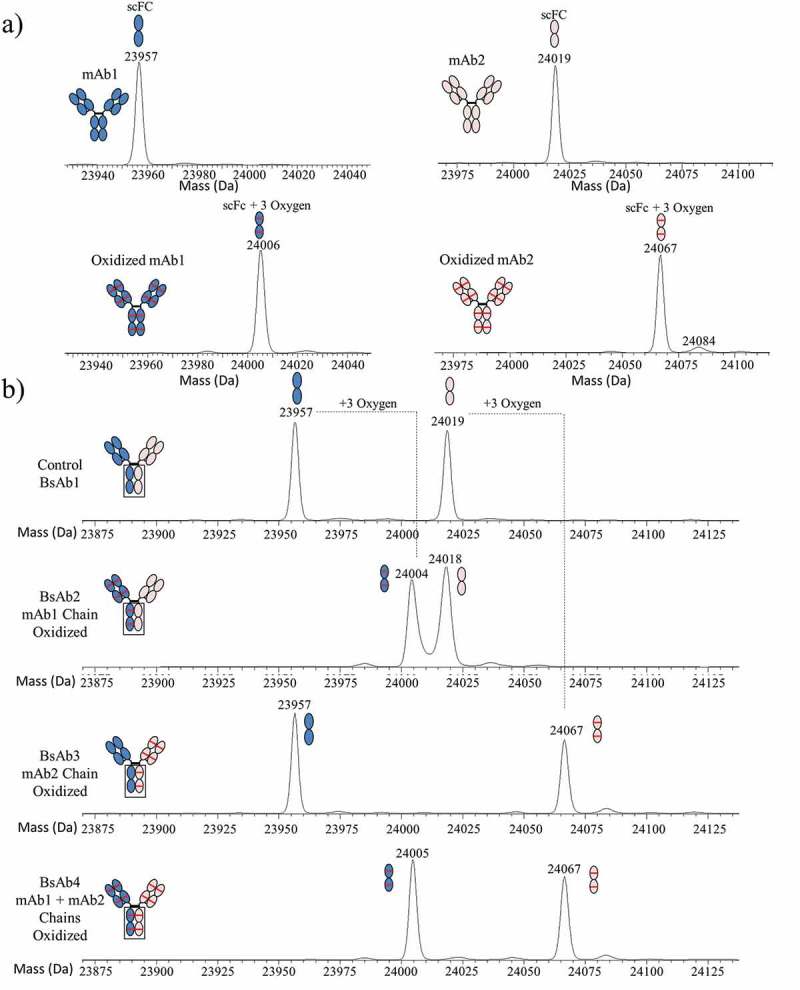
Subunit MS analysis of mAb and BsAb. (a) The deconvoluted spectra for control and oxidized mAb1 and mAb2 are shown in the left and right panels, respectively. (b) The deconvoluted spectra for BsAb1, BsAb2, BsAb3, and BsAb4 are shown in order from top to bottom. mAb1, mAb2, and methionine oxidation are shown with blue fill, peach fill, and red lines, respectively.
The BsAb products were also assessed by peptide mapping and subunit analysis to characterize methionine oxidation in the Fc region. The levels of HC Met254 oxidation measured by peptide mapping were 5.7%, 56.4%, 57.6%, and 99.4% for BsAb1, BsAb2, BsAb3, and BsAb4, respectively (Table 2). These results suggested that BsAb2 and BsAb3 were asymmetrically oxidized, but chain-specific oxidation of mAb1 HC and mAb2 HC in each BsAb material was not retained by peptide map. However, subunit MS analysis34 could be used to verify the formation of asymmetrically oxidized BsAb2 and BsAb3 at residues HC Met254, Met360, and Met430 because the integrity of the mAb1 and mAb2 HCs were preserved, and the amino acid point mutations in the Fc region produced two scFc subunits with unique mass values that could be distinguished by subunit MS. As shown in Figure 3(b), deconvoluted mass peaks of 23,957 Da and 24,019 Da were detected for the BsAb1 control, which corresponded to the expected scFc subunit masses of non-oxidized mAb1 and mAb2. Subunit MS analysis of BsAb2 showed deconvoluted masses of 24,004 Da and 24,018 Da, which corresponded to the expected scFc subunit masses of oxidized mAb1 with three oxidized residues and the non-oxidized scFc subunit from mAb2, respectively. These results confirmed the successful synthesis of an asymmetrically oxidized molecule because all oxidation sites were confirmed on the mAb1 chain. Similarly, the deconvoluted masses of BsAb3 were determined to be 23,957 Da and 24,067 Da by subunit MS analysis, which corresponded to scFc subunit masses expected for the non-oxidized scFc subunit from mAb1 and the oxidized scFc subunit from mAb2 with three oxidized residues, respectively. Subunit MS analysis of BsAb4 showed deconvoluted masses of 24,005 Da and 24,067 Da, which corresponded to the expected scFc subunit masses of mAb1 and mAb2 with three oxidized residues each, respectively.
Table 2.
Methionine 254 oxidation levels and Fc binding results for BsAb final products.
| Duobody® compositiona |
Met 254 oxidation (peptide map)b |
Fc binding assayc |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Material | mAb1 | mAb2 | mAb1 | mAb2 | Final BsAb product | FcRn | FcγRI | FcγRIIa | FcγRIIIa |
| BsAb1 | Native | Native | 3.8% | 3.0% | 5.7% | 100.0% | 100.0% | 100.0% | 100.0% |
| BsAb2 | Native | Oxidized | 3.8% | 99.9% | 56.4% | 44.4% | 97.6% | 94.3% | 102.1% |
| BsAb3 | Oxidized | Native | 99.9% | 3.0% | 57.6% | 54.3% | 120.2% | 89.7% | 106.4% |
| BsAb4 | Oxidized | Oxidized | 99.9% | 99.9% | 99.4% | 8.6% | 128.6% | 112.6% | 125.5% |
aBsAb molecule prepared from native or oxidized parental mAb1 or mAb2.
bValue expressed as a relative abundance of oxidized Met 254.
cBinding results are normalized relative to the respective control BsAb1 results.
FcRn binding results for the BsAbs
As shown in Table 2, HC Met254 oxidation levels on the mAb1 and mAb2 HC measured by peptide mapping were 3.8% and 3.0% for BsAb1, 3.8% and 99.9% for BsAb2, 99.9% and 3.0% for BsAb3 and 99.9% and 99.4% for BsAb4. Oxidation of the Met254 residue in the Fc of IgG disrupts antibody binding to FcRn.16,35 The effects of symmetrically and asymmetrically oxidized HC Met254 on FcRn binding were evaluated using a time-resolved fluorescence resonance energy transfer (TR-FRET) competitive binding assay.36 The FcRn binding for BsAb2 was 44.4% of the FcRn binding for control BsAb1, suggesting that the non-oxidized mAb1 chain of BsAb2 is still capable of independently binding to the FcRn (Table 2). Similarly, the FcRn binding of asymmetrically oxidized BsAb3 was 54.3% of the FcRn binding for control BsAb1. Symmetrically oxidized BsAb4 had minimal FcRn binding (<10%) due to the near 100% oxidation of Met254 on both HCs. The nearly 50% FcRn binding of asymmetrically oxidized BsAb2 and BsAb3 provides direct evidence for a 2:1 binding stoichiometry between the FcRn receptor and the Fc region of an antibody, which is consistent with previously presented results using point mutations to block FcRn binding.19
Protein Abinding results for the BsAbs
The impact of asymmetrically oxidized Met254 on Protein A binding was analyzed using Protein A affinity chromatography (PAAC).33 PAAC analysis of intact BsAb1 showed a main peak with a retention time of 35.5 min corresponding to native BsAb1 (Figure 4). Asymmetric oxidation of HC Met254 on a single HC (BsAb2 and BsAb3) produced a shift in retention time to ~29 min, while oxidation of HC Met254 on both chains (BsAb4) produced a further shift in retention time to 22.8 min. Together, these results indicated that Fc oxidation reduced the binding affinity to Protein A.
Figure 4.
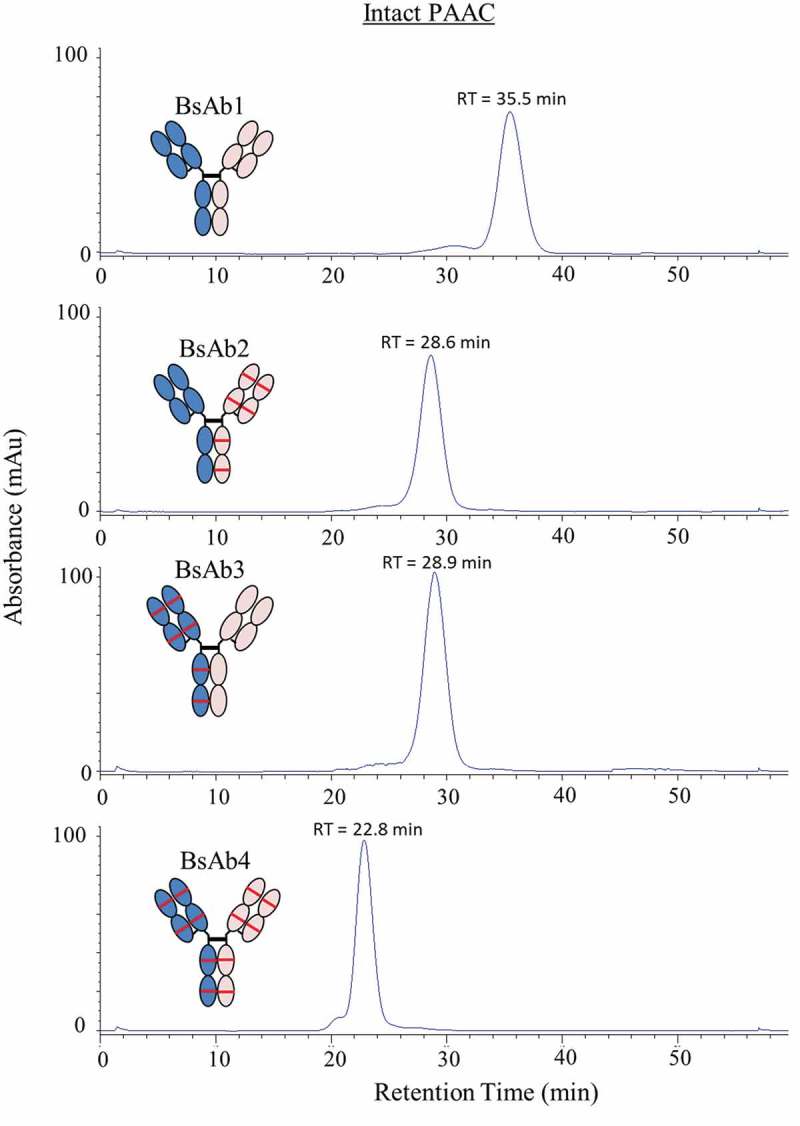
Protein A affinity chromatography chromatograms for BsAb1, BsAb2, BsAb3, and BsAb4 intact IgG. Protein absorbance was monitored at 280 nm.
Preparation of asymmetric heat-stressed BsAbto evaluate the impact of deamidation on FcγRIIIa binding
To demonstrate the suitability of this approach to study additional PTMs, new sets of BsAb products were prepared from unstressed and heat stressed parental mAb1 and mAb2 at pH 5.5, which causes deamidation at HC Asn330 in the Fc region.15 Control BsAb5 with little deamidation (<2%), asymmetrically deamidated BsAb6 and BsAb7, and symmetrically deamidated BsAb8 were prepared from heat-stressed mAb1 and mAb2, which were incubated for 4 months at 40°C (Figure 5(a)). As shown in Figure 5(b), HC Asn330 deamidation levels on the mAb1 and mAb2 HC measured by peptide mapping were 0.7% and 0.8% for BsAb5, 0.7% and 40.2% for BsAb6, and 39.6% and 0.8% for BsAb7, respectively. FcγRIIIa binding for the asymmetrically deamidated BsAb6 and BsAb7 were 64.4% and 66.3% of the FcγRIIIa binding observed in control BsAb5, respectively. Deamidation levels for the asymmetrically modified BsAb6 and BsAb7 correlated very well with the FcγRIIIa binding results. That is, deamidation levels of approximately 40% on one HC translated to approximately a 40% decrease in FcγRIIIa binding, which demonstrated a 1:1 stoichiometry of binding (IgG to FcγRIIIa). The results showed that Asn330 deamidation on a single HC was sufficient to affect mAb binding to FcγRIIIa. The levels of FcγRIIIa binding would likely have been lower if the levels of deamidation levels on each HC were higher, but HC Asn330 deamidation occurs very slowly even at 40°C.
Figure 5.
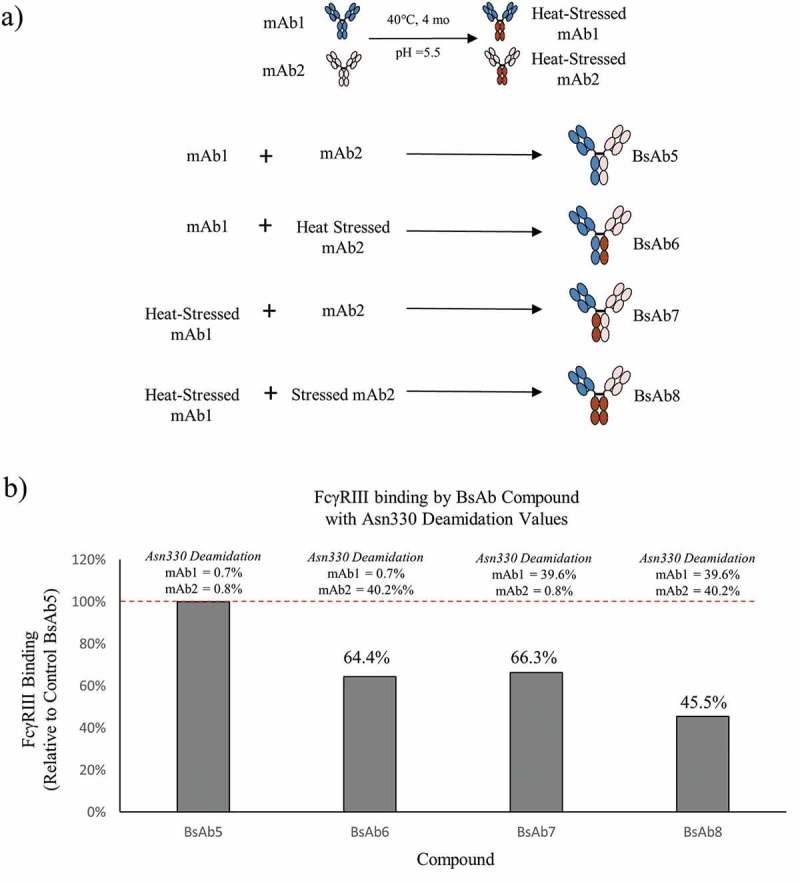
The process for creating bispecific antibodies with asymmetric deamidation at Asn330 is presented. (a) mAb1 (blue) and mAb2 (peach) were stressed at 40°C at pH 5.5 to induce selective deamidation at the Asn330 residue in the Fc Region (brown with red lines). Combinations of native or stressed mAb1 and mAb2 were pooled prior to Fab-arm exchange to generate control BsAb5, asymmetrically deamidated BsAb6 and BsAb7, and symmetrically deamidated BsAb8. (b) The bar plot illustrates the levels of FcγRIIIa binding of BsAb6, BsAb7, and BsAb8 relative to the control BsAb5 sample. The HC Asn330 deamidation values for the mAb1 and mAb2 arms of each BsAb are shown above the corresponding bar.
Deamidation levels of parental mAbs did not yield 100% deamidation at Asn330. Therefore, BsAb8 was composed of a mixed population of both asymmetric and symmetrically deamidated molecules, which is expected to show decreased FcγRIIIa binding relative to BsAb6 and BsAb7. The Asn330 deamidation levels of mAb1 and mAb2 were 39.6% and 40.2%, respectively. We expect approximately 64% of the BsAb8 population (16% containing symmetrically deamidated chains and 48% containing asymmetric deamidation) to be deamidated at Asn330 on one or two HCs based on the probabilities of a stochastic process and the known chain association combinations resulting from the FAE.30 This calculated value agrees with the observed FcγRIIIa binding result of 45.5% and explains the decreased binding observed in BsAb8 compared to FcγRIIIa results for asymmetrically deamidated mAbs, BsAb6, and BsAb7.
Discussion
Structure-function characterization of mAbs is required to identify CQAs that may affect antigen binding,37 Fc effector function,38,39 pharmacokinetics,40,41 immunogenicity,42 and stability43 so that the corresponding degradation pathways can be effectively controlled during production and storage of drug products.35,44,45 Forced degradation experiments are typically used to identify CQAs but can yield mixtures of asymmetrically and symmetrically modified mAbs, which are difficult to isolate and purify.23,46 Thus, it is not generally possible to experimentally evaluate the impact of chain-specific PTMs on biological function.
Controlling chain symmetry of PTMs would also not be possible using in vivo co-expression strategies where bispecific molecules are constructed within the cells.47 We demonstrated the ability to create, isolate, and characterize preparations of symmetrically and asymmetrically modified bispecific DuoBody® molecules.28,48 The complementary mutations of K409R and F405L are designed to destabilize the CH3 interface and favor heterodimerization.30 The Duobody® bispecific platform offers the advantage to control in vitro assembly of asymmetrically modified molecules using a controlled FAE of stressed or unstressed parental mAb molecules. We evaluated the impact of symmetric and asymmetric oxidation and deamidation on Fc binding to demonstrate the utility of this approach. The results of structure–function studies with these symmetrically and asymmetrically modified antibodies were used to validate structural modeling analysis and provide a clearer interpretation of forced degradation results. In addition, symmetrically and asymmetrically modified antibodies can be used to evaluate the impact on analytical methods such as Protein A binding.
It is well known that oxidation of HC Met254 directly affects the binding of human IgG1 to the FcRn receptor.16,36 Analysis from hydrogen-deuterium exchange shows that methionine oxidation disrupts the interface of the CH2 and CH3 domains, inducing a conformational change that impacts binding to FcRn.35,49 Additionally, a 2:1 binding stoichiometry has been proposed based on crystallography analysis.50-52 The FcRn binding results for asymmetrically oxidized BsAb2 and BsAb3 presented in this report were approximately 50% of the FcRn binding results for the control BsAb1, which provides experimental support for a 2:1 FcRn:IgG binding ratio.19 Additionally, the near 50% decrease in FcRn binding occurred regardless of which chain was oxidized, suggesting that FcRn can bind independently to either chain. The impact of chain-specific HC Met254 oxidation on Protein A binding was also assessed using Protein A affinity chromatography (PAAC). PAAC is an affinity-based chromatography method that was used to evaluate the binding between the CH2–CH3 interface of the Fc region and Protein A.53 HC Met254 oxidation reduces the binding between Protein A and the Fc, which resulted in earlier elution by PAAC.54 Results of this study indicated that symmetrically oxidized mAbs eluted earlier than asymmetrically oxidized mAbs, and Protein A can bind independently to either HC. This provided experimental support for a 2:1 Protein A:IgG binding ratio.
BsAb products were also created using heat stress to demonstrate additional applications of asymmetrically modified mAbs. Deamidation induced by thermal stress is a common degradation pathway that can affect the bioactivity of a mAb.55 Site-specific deamidation of HC Asn330 (VSNK motif) can be induced by heat stress under mildly acidic pH conditions while having minimal impact to the other Asn residues in the Fc region.15 Crystal structure analysis of a complex formed between soluble FcγRIIIa and human IgG1 Fc shows a 1:1 stoichiometric binding ratio.56 Our experimental data provides evidence that Asn330 deamidation on a single HC directly affects FcγRIIIa binding and deamidation levels for the asymmetrically modified mAbs measured by peptide map correlate very well with FcγRIIIa binding results. These results suggest that Asn330 deamidation on one HC is sufficient to inhibit FcγRIIIa binding to an IgG, which is consistent with a 1:1 stoichiometric ratio from crystal structure analysis.
Although we performed these studies using symmetrically and asymmetrically oxidized and deamidated BsAb using the DuoBody® process, we believe this technique provides a platform to study any modification type and the impact of chain symmetry on the biological function provided the primary sequence incorporates mutations in the CH3 domain to drive selective re-association during FAE. This technique can provide insight into unknown mechanisms of actions involving more complex modalities. Kinetic studies demonstrate dissociation of the parental mAbs containing the single point mutation in the CH3 domain controls the rate of the controlled FAE27 during the formation of a BsAb. HC association may be controlled using two homodimeric mAbs through single point mutations to create a hydrophobic binding pocket between the CH3-CH3 domains of mAb1 and mAb2 chains.57,58 The conversion of a homodimeric mAb to a heterodimeric bispecific molecule allows a clear assessment of individual chains using analytical methods. In addition to forced degradation, glycoengineering could also be employed to generate asymmetrically glycosylated IgG molecules to study the impact on effector function mechanisms.7,59,60 This allows the study of asymmetry in carbohydrate structure to investigate the effects of afucosylation on ADCC and complement-dependent cytotoxicity cell-based bioassays for oncology-related effector function competent mAbs.22,61-63 Characterization of asymmetrically modified molecules can provide new insight and understanding to the complex structure–function relationship between PTMs and biological activity.
Materials and methods
IdeS (FabRICATOR®) was purchased from Genovis. Trypsin was purchased from Promega. All other reagents and material were purchased from Thermo Scientific or Sigma-Aldrich unless otherwise stated. Reagents were analytical reagent grade or mass spectrometry grade and were used without further purification or manipulation. In house, MilliQ water was used to prepare aqueous solutions. IgG mAb1 and mAb2 and corresponding BsAb products were manufactured by Janssen Research and Development, LLC.
Preparation of asymmetrically modified bispecific molecules
The parental IgG1 antibodies, mAb1 and mAb2, were expressed in Chinese hamster ovary cells produced in individual fed-batch bioreactor systems. Antibodies were captured using Protein A affinity chromatography and adjusted to pH 6.5. Aliquots of mAb1 and mAb2 were incubated with 1 mM peracetic acid for 2 h at 30°C to selectively induce methionine oxidation or adjusted to pH 5.5 and incubated at 40°C for 4 months to selectively induce HC Asn330 deamidation. Reaction mixtures were buffer exchanged into a tris-neutralized sodium acetate buffer (pH 6.5) to remove excess reagent. BsAbs were generated through controlled FAE.30 Briefly, mAb1 and mAb1 were combined, treated with 2-mercaptoethylamine hydrochloride, and incubated overnight at 26°C. Reducing agent was removed via buffer exchange, and BsAb products were purified using two polishing chromatography steps.50,51 BsAb products were loaded onto a Capto Adhere MMAEX chromatography column (GE Healthcare) followed by Capto Phenyl ImpRes (GE Healthcare) HIC.64 BsAb products were buffer exchanged to formulation buffer and stored frozen at −70°C until further analyses.
Size-exclusion chromatography
BsAb products were injected onto a Tosoh TSKgel BioAssist G3SWXL, PEEK HPLC column (7.8 x 300 mm, 5 µm, 25°C column temperature). Isocratic separation was performed using an Agilent 1200 series HPLC system with sodium phosphate buffer (pH 6.8) at a flow rate of 0.7 mL/min. Eluted protein was detected using an online ultraviolet (UV) detector at 280 nm.
Hydrophobic interaction chromatography
BsAb samples were diluted to a protein concentration of 2 mg/mL and evaluated using an Agilent 1200 series HPLC with a Tosoh TSKgel Butyl-NPR column (4.6 x 350 mm, 2.5 µm, 25°C column temperature). Protein was eluted using a 12-min gradient from 25 mM sodium phosphate, 1.8 M ammonium sulfate, pH 7.0 (mobile phase A) to 25 mM sodium phosphate, pH 7.0 (mobile phase B). Eluted protein was detected by UV absorbance at 280 nm.
Capillary sodium dodecyl sulfate electrophoresis
BsAb material was treated with N-ethylmaleimide in bis-tris/citrate buffer (25 mM, pH 7.0) containing 1% sodium dodecyl sulfate and incubated for 5 min at 75°C. Denatured samples were cooled and injected electrokinetically (5 kV). Protein separation was performed using a Beckman Coulter PA800 system with a capillary and a photodiode array detector at 220 nm.
Peptide mapping
BsAb and mAb parental compounds were denatured in 6 M guanidine, 50 mM tris, 5 mM ethylenediaminetetraacetic acid, pH 8.0 at room temperature. Proteins were reduced with 1 M DTT for 1 h at 37°C followed by alkylation with 1 M sodium iodoacetate for 1 h at 25°C. Samples were exchanged into digestion buffer (50 mM Tris HCl, pH 7.0) using a NAP5TM column (GE Healthcare). The protein eluate was digested with trypsin for 2 h at 37°C. and the resulting peptides were analyzed by HPLC-MS/MS using an Agilent 1200 or Agilent 1260 HPLC system coupled an online mass spectrometer. The HPLC separation was performed using a gradient of 0.1% trifluoroacetic acid (TFA) in water to 0.1% TFA in acetonitrile on a BioSuite C18 column (250x2.1 mm, 40°C). Eluted peptides were subject to online electrospray ionization and detected using a Thermo Fisher Orbitrap XL mass spectrometer or a Q-Exactive mass spectrometer. The levels of modified amino acids were quantitated by peak area integration of the extracted ion chromatograms, and the results were expressed as a percentage of the total peptide peak area (modified + unmodified).
Subunit mass spectrometry
BsAb products were diluted to 1 mg/mL in 100 mM tris, pH 7.5 and incubated with EndoS (100 units/μL) and IdeS (1 unit/μg) for 30 min at 37°C. Samples were reduced with 1 M DTT for 15 min at 37°C and quenched with 10% (v/v) 1 M HCl. IdeS digested material was analyzed using a Waters Acquity I-Class HPLC system and separated using Waters BEH300 C4 column (2.1x50 mm, 60°C) using a gradient of 0.1% formic acid in acetonitrile (200 μL/min). Eluted material was subject to electrospray ionization and detected using a Xevo G2-XS mass spectrometer in positive ion mode for an m/z range of 800–3000. Data files were acquired using MassLynx software and deconvoluted using the MaxEnt1 algorithm. Oxidation was quantitated using the peak height intensities of the deconvoluted and centered spectra.
Fc receptor binding assays
Characterization of the binding of the Fc region of BsAb variants to Fc receptors FcγRI, FcγRIIa, FcγRIIIa, and FcRn was assessed by using a competitive TR-FRET assay. In these methods, binding was assessed by the ability of unlabeled BsAb to compete with a fixed concentration of europium-labeled antibody for binding to the relevant Cy5-labeled Fc receptor. Briefly, FcγRII, FcγRIIIa or FcRn receptor was added to varying concentrations of unlabeled BsAb sample and incubated at room temperature for 90 min in the dark. Following incubation, TR-FRET was measured using a spectrophotometer plate reader (Perkin Elmer, Victor3) with an excitation/emission wavelength of 615 nm and 665 nm. The relative binding activity was then calculated from the mean EC50 value of replicate dose-response curves generated with BsAb and expressed as a percentage of the mean EC50 value of dose-response curves generated with reference material. All TR-FRET binding methods have been validated in accordance with the International Council on Harmonization guidelines.
Protein Aaffinity chromatography
High-performance Protein A chromatography was performed on intact BsAb products diluted to 5 mg/mL with Dulbecco’s phosphate-buffered saline (DPBS) and loaded onto a Protein A column (POROS A/G, 4.6 × 50 mm). Separation was performed using an Agilent 1200 series HPLC system with a flow rate of 1mL/min. Bound IgG was eluted from the Protein A column using a 40-min gradient of 100 mM acetic acid in 100 mM NaCl, pH 2.8. Protein eluate was detected by UV absorption at 280 nm.
Abbreviations
- ADCC
antibody-dependent cell-mediated cytotoxicity
- BsAb
bispecific antibody
- CQA
critical quality attribute
- cSDS
capillary SDS electrophoresis
- DPBS
Dulbecco’s phosphate-buffered saline
- DTT
dithiothreitol
- FAE
Fab-arm exchange
- HC
heavy chain
- HIC
hydrophobic interaction chromatography
- HMWS
high molecular weight species
- HPLC
high performance liquid chromatography
- IgG
immunoglobulin G
- LMW(S)
low molecular weight (species)
- mAbs
monoclonal antibody
- MS
mass spectrometry
- PAAC
Protein A affinity chromatography
- PTM
post-translational modification
- scFc
single-chain Fc
- SDS
sodium dodecyl sulfate
- SEC
size-exclusion chromatography
- TFA
trifluoroacetic acid
- TR-FRET
time-resolved fluorescence resonance energy transfer
- UV
ultraviolet
Acknowledgments
The authors acknowledge Dr. Carmelata Chitikila for reviewing bioassay data and Dr. Qingrong Yan for providing structural modeling review. The authors would like to thank Dr. Mehul Patel for his continuing support of exploring novel technologies for biopharmaceutical development.
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Ecker DM, Jones SD, Levine HL.. The therapeutic monoclonal antibody market. mAbs. 2015;7(1):9–14. doi: 10.4161/19420862.2015.989042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianferani S.. Characterization of therapeutic antibodies and related products. Anal Chem. 2013;85(2):715–36. doi: 10.1021/ac3032355. [DOI] [PubMed] [Google Scholar]
- 3.Buss NA, Henderson SJ, McFarlane M, Shenton JM, de Haan L.. Monoclonal antibody therapeutics: history and future. Curr Opin Pharmacol. 2012;12(5):615–22. doi: 10.1016/j.coph.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Beck A, Sanglier-Cianferani S, Van Dorsselaer A. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal Chem. 2012;84(11):4637–46. doi: 10.1021/ac3002885. [DOI] [PubMed] [Google Scholar]
- 5.Thompson NJ, Rosati S, Heck AJ. Performing native mass spectrometry analysis on therapeutic antibodies. Methods. 2014;65(1):11–17. doi: 10.1016/j.ymeth.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–46. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 7.Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr Opin Immunol. 2008;20(4):471–78. doi: 10.1016/j.coi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 8.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol. 2006;6(5):343–57. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Wang G, Song T, Lebrilla CB, Heck AJR. Resolving the micro-heterogeneity and structural integrity of monoclonal antibodies by hybrid mass spectrometric approaches. mAbs. 2017;9(4):638–645. doi: 10.1080/19420862.2017.1290033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qi P, Volkin DB, Zhao H, Nedved ML, Hughes R, Bass R, Yi SC, Panek ME, Wang D, Dalmonte P, et al. Characterization of the photodegradation of a human IgG1 monoclonal antibody formulated as a high-concentration liquid dosage form. J Pharm Sci. 2009;98(9):3117–30. doi: 10.1002/jps.21617. [DOI] [PubMed] [Google Scholar]
- 11.Hmiel LK, Brorson KA, Boyne MT. 2nd, Post-translational structural modifications of immunoglobulin G and their effect on biological activity. Anal Bioanal Chem. 2015;407(1):79–94. doi: 10.1007/s00216-014-8108-x. [DOI] [PubMed] [Google Scholar]
- 12.Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci. 2008;97(7):2426–47. doi: 10.1002/jps.21180. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Pan H, Chen X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev. 2009;28(1):147–76. doi: 10.1002/mas.20190. [DOI] [PubMed] [Google Scholar]
- 14.Pace AL, Wong RL, Zhang YT, Kao YH, Wang YJ. Asparagine deamidation dependence on buffer type, pH, and temperature. J Pharm Sci. 2013;102(6):1712–23. doi: 10.1002/jps.23529. [DOI] [PubMed] [Google Scholar]
- 15.Zhang YT, Hu J, Pace AL, Wong R, Wang YJ, Kao YH. Characterization of asparagine 330 deamidation in an Fc-fragment of IgG1 using cation exchange chromatography and peptide mapping. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;965:65–71. doi: 10.1016/j.jchromb.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 16.Bertolotti-Ciarlet A, Wang W, Lownes R, Pristatsky P, Fang Y, McKelvey T, Li Y, Li Y, Drummond J, Prueksaritanont T, et al. Impact of methionine oxidation on the binding of human IgG1 to Fc Rn and Fc gamma receptors. Mol Immunol. 2009;46(8–9):1878–82. doi: 10.1016/j.molimm.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Viski K, Gengeliczki Z, Lenkey K, Baranyane Ganzler K. Parallel development of chromatographic and mass-spectrometric methods for quantitative analysis of glycation on an IgG1 monoclonal antibody. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1032:198–204. doi: 10.1016/j.jchromb.2016.04.043. [DOI] [PubMed] [Google Scholar]
- 18.Hawe A, Wiggenhorn M, van de Weert M, Garbe JH, Mahler HC, Jiskoot W. Forced degradation of therapeutic proteins. J Pharm Sci. 2012;101(3):895–913. doi: 10.1002/jps.22812. [DOI] [PubMed] [Google Scholar]
- 19.Abdiche YN, Yeung YA, Chaparro-Riggers J, Barman I, Strop P, Chin SM, Pham A, Bolton G, McDonough D, Lindquist K, et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. mAbs. 2015;7(2):331–43. doi: 10.1080/19420862.2015.1008353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okazaki A, Shoji-Hosaka E, Nakamura K, Wakitani M, Uchida K, Kakita S, Tsumoto K, Kumagai I, Shitara K. Fucose depletion from human IgG1 oligosaccharide enhances binding enthalpy and association rate between IgG1 and FcgammaRIIIa. J Mol Biol. 2004;336(5):1239–49. doi: 10.1016/j.jmb.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SH, Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277(30):26733–40. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 22.Shatz W, Chung S, Li B, Marshall B, Tejada M, Phung W, Sandoval W, Kelley RF, Scheer JM. Knobs-into-holes antibody production in mammalian cell lines reveals that asymmetric afucosylation is sufficient for full antibody-dependent cellular cytotoxicity. mAbs. 2013;5(6):872–81. doi: 10.4161/mabs.26307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ha S, Ou Y, Vlasak J, Li Y, Wang S, Vo K, Du Y, Mach A, Fang Y, Zhang N. Isolation and characterization of IgG1 with asymmetrical Fc glycosylation. Glycobiology. 2011;21(8):1087–96. doi: 10.1093/glycob/cwr047. [DOI] [PubMed] [Google Scholar]
- 24.Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;20(7):838–47. doi: 10.1016/j.drudis.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Sedykh SE, Prinz VV, Buneva VN, Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Des Devel Ther. 2018;12:195–208. doi: 10.2147/DDDT.S151282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, Gunasekaran K, Wang W, Razinkov V, Sekirov L, Leng E, Sweet H, Foltz I, Howard M, Rousseau AM, et al. Asymmetrical Fc engineering greatly enhances antibody-dependent cellular cytotoxicity (ADCC) effector function and stability of the modified antibodies. J Biol Chem. 2014;289(6):3571–90. doi: 10.1074/jbc.M113.513366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goulet DR, Orcutt SJ, Zwolak A, Rispens T, Labrijn AF, de Jong RN, Atkins WM, Chiu ML. Kinetic mechanism of controlled Fab-arm exchange for the formation of bispecific immunoglobulin G1 antibodies. J Biol Chem. 2018;293(2):651–61. doi: 10.1074/jbc.RA117.000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gramer MJ, van Den Bremer ET, van Kampen MD, Kundu A, Kopfmann P, Etter E, Stinehelfer D, Long J, Lannom T, Noordergraaf EH, et al. Production of stable bispecific IgG1 by controlled Fab-arm exchange: scalability from bench to large-scale manufacturing by application of standard approaches. mAbs. 2013;5(6):962–73. doi: 10.4161/mabs.26233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labrijn AF, Meesters JI, Priem P, de Jong RN, van Den Bremer ET, van Kampen MD, Gerritsen AF, Schuurman J, Parren PW. Controlled Fab-arm exchange for the generation of stable bispecific IgG1. Nat Protoc. 2014;9(10):2450–63. doi: 10.1038/nprot.2014.169. [DOI] [PubMed] [Google Scholar]
- 30.Labrijn AF, Meesters JI, de Goeij BE, van Den Bremer ET, Neijssen J, van Kampen MD, Strumane K, Verploegen S, Kundu A, Gramer MJ, et al. Efficient generation of stable bispecific IgG1 by controlled Fab-arm exchange. Proc Natl Acad Sci U S A. 2013;110(13):5145–50. doi: 10.1073/pnas.1220145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Protection, M. D. o. E. Hydrogen peroxide, peracetic acid, and sodium percarbonate. Boston (MA): Massachusetts Department of Agricultural Resources; 2010, 1–15.
- 32.Liu D, Ren D, Huang H, Dankberg J, Rosenfeld R, Cocco MJ, Li L, Brems DN, Remmele RL Jr.. Structure and stability changes of human IgG1 Fc as a consequence of methionine oxidation. Biochemistry. 2008;47(18):5088–100. doi: 10.1021/bi702238b. [DOI] [PubMed] [Google Scholar]
- 33.An Y, Zhang Y, Mueller HM, Shameem M, Chen X. A new tool for monoclonal antibody analysis: application of IdeS proteolysis in IgG domain-specific characterization. mAbs. 2014;6(4):879–93. doi: 10.4161/mabs.28762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sokolowska I, Mo J, Dong J, Lewis MJ, Hu P. Subunit mass analysis for monitoring antibody oxidation. mAbs. 2017;9(3):498–505. doi: 10.1080/19420862.2017.1279773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mo J, Yan Q, So CK, Soden T, Lewis MJ, Hu P. Understanding the impact of methionine oxidation on the biological functions of IgG1 antibodies using hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2016;88(19):9495–502. doi: 10.1021/acs.analchem.6b01958. [DOI] [PubMed] [Google Scholar]
- 36.Gao X, Ji JA, Veeravalli K, Wang YJ, Zhang T, McGreevy W, Zheng K, Kelley RF, Laird MW, Liu J, et al. Effect of individual Fc methionine oxidation on FcRn binding: met252 oxidation impairs FcRn binding more profoundly than Met428 oxidation. J Pharm Sci. 2015;104(2):368–77. doi: 10.1002/jps.24136. [DOI] [PubMed] [Google Scholar]
- 37.Haberger M, Bomans K, Diepold K, Hook M, Gassner J, Schlothauer T, Zwick A, Spick C, Kepert JF, Hienz B, et al. Assessment of chemical modifications of sites in the CDRs of recombinant antibodies: susceptibility vs. functionality of critical quality attributes. mAbs. 2014;6(2):327–39. doi: 10.4161/mabs.27876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5:520. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Mol Immunol. 2011;48(6–7):860–66. doi: 10.1016/j.molimm.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Putnam WS, Prabhu S, Zheng Y, Subramanyam M, Wang YM. Pharmacokinetic, pharmacodynamic and immunogenicity comparability assessment strategies for monoclonal antibodies. Trends Biotechnol. 2010;28(10):509–16. doi: 10.1016/j.tibtech.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Goetze AM, Schenauer MR, Flynn GC. Assessing monoclonal antibody product quality attribute criticality through clinical studies. mAbs. 2010;2(5):500–07. doi: 10.4161/mabs.2.5.12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Beers MM, Bardor M. Minimizing immunogenicity of biopharmaceuticals by controlling critical quality attributes of proteins. Biotechnol J. 2012;7(12):1473–84. doi: 10.1002/biot.201200065. [DOI] [PubMed] [Google Scholar]
- 43.Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Stability of protein pharmaceuticals: an update. Pharm Res. 2010;27(4):544–75. doi: 10.1007/s11095-009-0045-6. [DOI] [PubMed] [Google Scholar]
- 44.Alt N, Zhang TY, Motchnik P, Taticek R, Quarmby V, Schlothauer T, Beck H, Emrich T, Harris RJ. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biol J Int Assoc Biol Standardization. 2016;44(5):291–305. doi: 10.1016/j.biologicals.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 45.Wang W, Singh S, Zeng DL, King K, Nema S. Antibody structure, instability, and formulation. J Pharm Sci. 2007;96(1):1–26. doi: 10.1002/jps.20727. [DOI] [PubMed] [Google Scholar]
- 46.Jefferis R. Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev Drug Discov. 2009;8(3):226–34. doi: 10.1038/nrd2804. [DOI] [PubMed] [Google Scholar]
- 47.Krah S, Sellmann C, Rhiel L, Schroter C, Dickgiesser S, Beck J, Zielonka S, Toleikis L, Hock B, Kolmar H, et al. Engineering bispecific antibodies with defined chain pairing. N Biotechnol. 2017;39(Pt B):167–73. doi: 10.1016/j.nbt.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 48.Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9(2):182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang A, Hu P, MacGregor P, Xue Y, Fan H, Suchecki P, Olszewski L, Liu A. Understanding the conformational impact of chemical modifications on monoclonal antibodies with diverse sequence variation using hydrogen/deuterium exchange mass spectrometry and structural modeling. Anal Chem. 2014;86(7):3468–75. doi: 10.1021/ac404130a. [DOI] [PubMed] [Google Scholar]
- 50.Popov S, Hubbard JG, Kim J, Ober B, Ghetie V, Ward ES. The stoichiometry and affinity of the interaction of murine Fc fragments with the MHC class I-related receptor, FcRn. Mol Immunol. 1996;33:521–30. [DOI] [PubMed] [Google Scholar]
- 51.West AP Jr., Bjorkman PJ. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor(,). Biochemistry. 2000;39:9698–708. [DOI] [PubMed] [Google Scholar]
- 52.Raghavan M, Wang Y, Bjorkman PJ. Effects of receptor dimerization on the interaction between the class I major histocompatibility complex-related Fc receptor and IgG. Proc Natl Acad Sci U S A. 1995;92(24):11200–04. doi: 10.1073/pnas.92.24.11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sjodahl J. Structural studies on the four repetitive Fc-binding regions in protein A from Staphylococcus aureus. Eur J Biochem. 1977;78:471–90. [DOI] [PubMed] [Google Scholar]
- 54.Gaza-Bulseco G, Faldu S, Hurkmans K, Chumsae C, Liu H. Effect of methionine oxidation of a recombinant monoclonal antibody on the binding affinity to protein A and protein G. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;870(1):55–62. doi: 10.1016/j.jchromb.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 55.Phillips JJ, Buchanan A, Andrews J, Chodorge M, Sridharan S, Mitchell L, Burmeister N, Kippen AD, Vaughan TJ, Higazi DR, et al. Rate of asparagine deamidation in a monoclonal antibody correlating with hydrogen exchange rate at adjacent downstream residues. Anal Chem. 2017;89(4):2361–68. doi: 10.1021/acs.analchem.6b04158. [DOI] [PubMed] [Google Scholar]
- 56.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature. 2000;406(6793):267–73. doi: 10.1038/35018508. [DOI] [PubMed] [Google Scholar]
- 57.Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J, Brinkmann U, Kettenberger H, Regula JT, Schaefer W. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs. 2012;4(6):653–63. doi: 10.4161/mabs.21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Labrijn AF, Rispens T, Meesters J, Rose RJ, Den Bleker TH, Loverix S, van Den Bremer ET, Neijssen J, Vink T, Lasters I, et al. Species-specific determinants in the IgG CH3 domain enable Fab-arm exchange by affecting the noncovalent CH3-CH3 interaction strength. J Immunol. 2011;187(6):3238–46. doi: 10.4049/jimmunol.1003336. [DOI] [PubMed] [Google Scholar]
- 59.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103(11):4005–10. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX. Modulating IgG effector function by Fc glycan engineering. Proc Natl Acad Sci U S A. 2017;114(13):3485–90. doi: 10.1073/pnas.1702173114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pace D, Lewis N, Wu T, Gillespie R, Leiske D, Velayudhan J, Rohrbach A, Connell-Crowley L. Characterizing the effect of multiple Fc glycan attributes on the effector functions and FcgammaRIIIa receptor binding activity of an IgG1 antibody. Biotechnol Prog. 2016;32(5):1181–92. doi: 10.1002/btpr.2300. [DOI] [PubMed] [Google Scholar]
- 62.Lanier LL, Kipps TJ, Phillips JH. Functional properties of a unique subset of cytotoxic CD3+ T lymphocytes that express Fc receptors for IgG (CD16/Leu-11 antigen). J Exp Med. 1985;162(6):2089–106. doi: 10.1084/jem.162.6.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kellner C, Derer S, Valerius T, Peipp M. Boosting ADCC and CDC activity by Fc engineering and evaluation of antibody effector functions. Methods. 2014;65(1):105–13. doi: 10.1016/j.ymeth.2013.06.036. [DOI] [PubMed] [Google Scholar]
- 64.Manzke O, Tesch H, Diehl V, Bohlen H. Single-step purification of bispecific monoclonal antibodies for immunotherapeutic use by hydrophobic interaction chromatography. J Immunol Methods. 1997;208:65–73. [DOI] [PubMed] [Google Scholar]