

Abstract
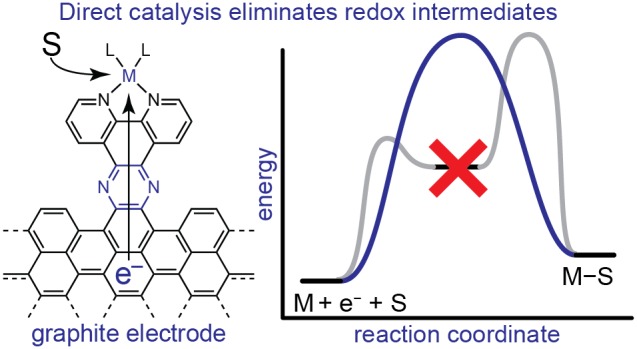
The efficient interconversion of electrical and chemical energy requires the intimate coupling of electrons and small-molecule substrates at catalyst active sites. In molecular electrocatalysis, the molecule acts as a redox mediator which typically undergoes oxidation or reduction in a separate step from substrate activation. These mediated pathways introduce a high-energy intermediate, cap the driving force for substrate activation at the reduction potential of the molecule, and impede access to high rates at low overpotentials. Here we show that electronically coupling a molecular hydrogen evolution catalyst to a graphitic electrode eliminates stepwise pathways and forces concerted electron transfer and proton binding. Electrochemical and X-ray absorption spectroscopy data establish that hydrogen evolution catalysis at the graphite-conjugated Rh molecule proceeds without first reducing the metal center. These results have broad implications for the molecular-level design of energy conversion catalysts.
Introduction
The efficient interconversion of electrical and chemical energy requires molecular-level control over critical redox transformations involving small-molecule substrates such as O2, H2O, and H2 at or near electrode surfaces.1−33 Discrete molecular catalysts typically proceed through mediated pathways in which the catalyst is oxidized or reduced prior to substrate activation. As an example, a molecular catalyst for the hydrogen evolution reaction (HER), M, could proceed through a pathway in which M is reduced by two e– to generate a M2– species that then binds a proton to make a M–H intermediate. In this simplified mechanism, subsequent proton transfer (PT) would generate H2 and regenerate the catalyst (Figure 1a). If proton transfer to the M2– species is rate-limiting, this mechanism would give rise to the free energy diagram depicted in Figure 1b. For this mechanism, varying the applied potential (Eapplied) modulates the driving force for reducing M to M2–, but does not affect the driving force for protonating M2– to generate M–H (ΔGM–H). Thus, for this sequence, the maximum driving force for substrate conversion is pinned by E°(M0/2–) irrespective of the potential applied to the electrode, Eapplied. While there are many pathways by which a hydrogen evolution catalyst may operate,34,35 to the best of our knowledge, all reported molecular HER catalysts proceed through stepwise redox mediation pathways in which one or more electron transfer (ET) steps precede a chemical rate-determining step for binding a proton.4,5,14,28,29,31,33,34,36−40 Due to the ubiquity of these stepwise mediated pathways, molecular electrocatalyst design has focused on optimizing the reduction potential of the molecule and correlating this value to the overall reaction rate.1,2
Figure 1.
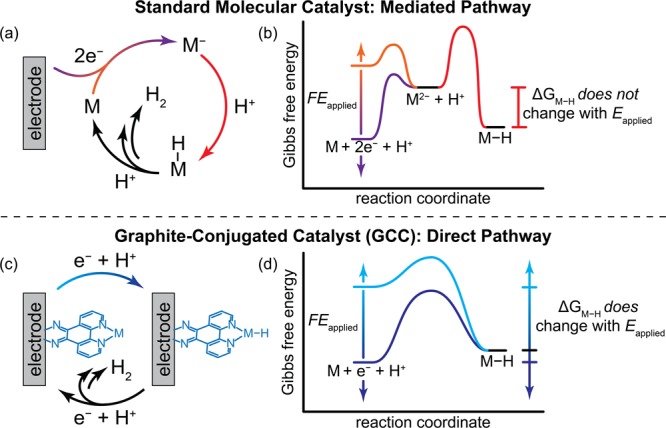
Mechanistic pathways and corresponding free energy diagrams for hydrogen evolution catalysis at a molecule vs graphite-conjugated catalyst. (a) Molecular catalyst (M) mediates the conversion of 2 H+ and 2 e– to H2 through a stepwise pathway involving two-electron transfer followed by substrate activation. (b) Free energy diagram for the catalytic cycle in part a. (c) Graphite-conjugated M catalyzes the conversion of 2 H+ and 2 e– to H2 directly through an M–H intermediate on the surface. (d) Free energy diagram for the catalytic cycle in part c. F is Faraday’s constant, Eapplied is the applied potential, and ΔGs is the driving force for forming the M–H intermediate.
These stepwise mediated pathways impose several inherent drawbacks. Taking the pathway in Figure 1a as an example, we note the following: (1) The energy of the reduced catalyst is necessarily higher than that of the M–H intermediate; (2) the maximum rate of catalysis is pinned because changes in Eapplied beyond E°(M0/2–) do not further increase the rate of H+ activation; (3) the minimum necessary value of E°(M0/2–) is dependent on the pKa of the proton donor, so changing the electrolyte environment can require a redesign of the catalyst to augment E°(M0/2–); and (4) the rate of substrate activation typically scales with the effective overpotential dictated by the redox couple, impeding simultaneous access to low overpotentials and high rates.43−45 While we are using the HER pathway in Figure 1a as an example, we stress that similar constraints exist for all mediated pathways, i.e., pathways in which outer-sphere ET from the electrode and substrate activation occur in separate elementary steps. Thus, these constraints exist, to varying degrees, in all molecular electrocatalysis.
In principle, these challenges can be overcome by designing catalysts that proceed exclusively through pathways involving concerted ET and substrate activation. However, even in elementary steps, concerted pathways are rarely observed due to the slow nuclear motion of the substrate relative to electron transfer and the prohibitively high activation barrier associated with a ternary transition state.24 Despite extensive studies of concerted proton- and halide-coupled electron transfer elementary steps,46−49 to the best of our knowledge, rate-determining steps in which ET and substrate activation are concerted have not been observed in molecular electrocatalysis. Even molecular catalysts that preorganize PT via moieties in the secondary coordination sphere proceed through stepwise pathways.6,8,14,33
We have previously shown that conjugating a molecule to a graphitic carbon electrode through an aromatic pyrazine linkage engenders strong electronic coupling between the electrode and the appended molecular unit.50−200 This observation raises the tantalizing possibility that these graphite-conjugated catalysts (GCCs) may carry out substrate activation without being constrained by the redox intermediates ubiquitous in molecular electrocatalysis. Here, we demonstrate that electronically coupling a molecular hydrogen evolution catalyst to an electrode forces a direct pathway in which ET and PT are concerted by eliminating access to stepwise redox mediation pathways (Figure 1c and d). Specifically, we conjugate a known Rh-based molecular HER catalyst to glassy carbon electrodes and compare the electrokinetic profile to that of the molecular analogue. We selected the HER as a rigorous test reaction because direct, unmediated hydrogen evolution pathways must proceed via rate-limiting concerted proton–electron transfer (CPET) steps, which are readily identifiable experimentally. Additionally, we directly probe the valence state of the Rh-based GCC (GCC-Rh) during catalysis using in situ X-ray absorption spectroscopy. We show that hydrogen evolution catalysis at the GCC proceeds exclusively via CPET without first reducing the metal center (Figure 1c). Consequently, the driving force for H+ activation is no longer pinned by the reduction potential of the molecular analogue, but is instead directly modulated by the applied potential (Figure 1d).
Results and Discussion
Synthesis of Molecular Rh Catalyst and GCC-Rh
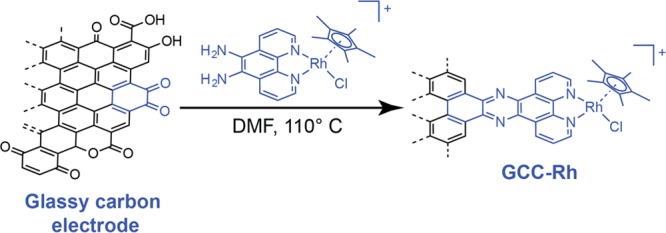
Water-soluble [RhCp*(bpds)Cl]− (Cp* = pentamethylcyclopentadienyl, bpds = bathophenanthrolinedisulfonate) was synthesized by modification of a literature procedure,53 and GCC-Rh electrodes were prepared via treatment of carbon surfaces with [RhCp*(phenda)Cl]+ (phenda = 5,6-diamino-1,10-phenanthroline) following procedures described previously (Scheme 1).52 (Full synthetic details are provided in the Supporting Information.)
Scheme 1. Synthesis of GCC-Rh.
RhCp*(bpds)OH2 is only active at low pH
The {RhCp*(bpy)} fragment is a known catalyst for HER in low-pH aqueous electrolytes54 and nonaqueous electrolytes with strong acids.40 For RhCp*(bpds)OH2, formed by aquation of [RhCp*(bpds)Cl]−,55 we also observe HER catalysis at pH 1 (0.1 M HClO4) (Figure 2a), with an onset of 0.05 mA cm–2 at −0.45 V vs the normal hydrogen electrode (NHE). At pH 4 (0.1 M sodium formate), catalytic onset shifts by −0.2 V, and an irreversible RhIII/I prefeature is observed at −0.45 V vs NHE (Figure S1). We attribute the prefeature to the formation of a Rh hydride, Rh–H, as has been invoked previously.40 Under these conditions, HER catalysis is mediated by subsequent reduction of the Rh–H species. By pH 7, we no longer observe catalysis out to −0.9 V vs NHE. Based on the onset potentials observed in these cyclic voltammograms (CVs), we have qualitatively denoted the region of HER catalysis for RhCp*(bpds)OH2 in orange in Figure 3.
Figure 2.

pH-dependence of hydrogen evolution catalysis at RhCp*(bpds)OH2 vs GCC-Rh. Cyclic voltammograms (CVs) (10 mV s–1) of 0.3 mM RhCp*(bpds)OH2 in (a) 0.1 M perchloric acid (pH 1), (b) 0.1 M sodium phosphate (pH 7), and (c) 0.1 M sodium hydroxide (pH 13). CVs (10 mV s–1) of GCC-Rh (blue) and GCC-phenazine (black) in (d) 0.1 M perchloric acid (pH 1), (e) 0.1 M sodium phosphate (pH 7), and (f) 0.1 M sodium hydroxide (pH 13).
Figure 3.
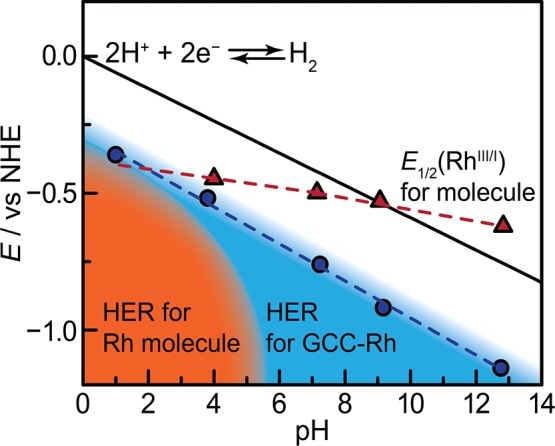
Potential vs pH (Pourbaix) diagram. The black line denotes the thermodynamic potential of hydrogen evolution. The red triangles mark E1/2(RhIII/I). The dotted red line is a guide to the eye and shows extrapolated values for E1/2(RhIII/I) in the pH range in which the hydrogen evolution reaction (HER) occurs. The orange region denotes the potential-pH region in which RhCp*(bpds)OH2 catalyzes HER. The blue region, which includes the orange region, denotes the potential-pH region in which GCC-Rh catalyzes HER, and the blue circles mark a per-site turnover frequency of 1 s–1. The dotted blue line is a best fit line with a slope of −68 mV/pH unit.
At more alkaline pH values, RhCp*(bpds)OH2 is not an HER catalyst. Instead, CVs recorded at pH 7 (0.1 M sodium phosphate), pH 9 (0.1 M sodium borate), and pH 13 (0.1 M sodium hydroxide) only display quasi-reversible RhIII/I waves at −0.50 V, –0.53 V, and −0.62 V vs NHE, respectively (Figure 2b and 2c, Figure 3 (red triangles), and Figures S2–S4). We attribute the slight decrease in the RhIII/I reduction potential, E1/2(RhIII/I), with increasing pH to a two-electron reduction coupled to loss of OH– in more basic conditions. Together the data show that HER catalysis at low pH is replaced by catalytically inert RhIII/I redox processes in alkaline media.
The fact that HER from RhCp*(bpds)OH2 is only observed in low-pH electrolytes is a consequence of the dramatic change in the strength of the proton donor in solution vs the weak pH-dependence of the reduction potentials of the molecule. We note that in our experiments, the proton donor is likely the buffer, and the pKa of the buffer closely matches the pH at which each experiment was conducted. Thus, the black line in Figure 3 is a good approximation of the thermodynamic potential for HER in each electrolyte.56,57 As the pH increases, the acidity of the donor decreases, but the basicity of the RhI intermediate and the subsequently formed Rh–H species remain the same. This mismatch leads to a progressive decrease in driving force for proton transfer to the catalyst, and eventually catalysis shuts off.
GCC-Rh Is Active Across the Entire pH Range
In contrast to the molecule, GCC-Rh catalyzes HER across all pH conditions. CVs of GCC-Rh in 0.1 M HClO4 display a redox wave at −0.03 V vs NHE. This wave shifts −59 mV/pH unit (Figure S5) and is attributed to the two-proton, two-electron reduction of the pyrazine linkage to dihydropyrazine, in line with previous studies.50,52 Irrespective of pH, the pyrazine wave is followed by a catalytic wave for HER that onsets well before the background of a phenazine-modified electrode (Figure 2d–2f and Figure S6). Notably, we do not observe redox features that can be attributed to the RhIII/I couple at any pH. At pH 1, 4, 7, 9, and 13, HER occurs with a turnover frequency (TOF) of 1 s–1 at overpotentials of 0.30, 0.29, 0.33, 0.38, and 0.38 V, respectively (Figures 3, blue dots, and Figures S7–S11; see Supporting Information for details of TOF calculations). The catalytic onset shifts by −68 mV/pH unit, which is similar to the shift of the overall thermodynamic potential of HER. Remarkably, the catalytic activity of GCC-Rh does not correlate to E1/2(RhIII/I) for RhCp*(bpds)OH2 at all; we observe catalysis even under conditions in which E1/2(RhIII/I) for the molecule lies at a 0.13 V underpotential to HER (pH 13 on Figure 3). This unprecedented behavior demonstrates that GCCs circumvent the known scaling relationship between E1/2 and the rate of a molecular electrocatalyst.43−45
Rh Sites Are the Active Sites in GCC-Rh
A series of studies establishes that the molecular Rh center is the active site for HER at GCC-Rh across the pH range. X-ray photoelectron spectroscopy (XPS) data establish the fidelity of the Rh sites after HER catalysis (Figures S12–S18). High-resolution XPS data collected after 10 min of catalysis in each electrolyte show N/Rh ratios at or near the expected value of 4:1 (Table S1) and Rh 3d5/2 peaks centered around 309.3 eV, consistent with RhIII (Table S2).58,59 This Rh binding energy is 2.3 eV positive of the expected 3d5/2 peak for Rh0,59,60 suggesting that there is no formation of Rh nanoparticles during electrolysis. Together, these data demonstrate that the Rh sites maintain their molecular fidelity even under electrocatalytic conditions. Furthermore, the catalytic activity of GCC-Rh in 0.1 M HClO4 was immediately suppressed upon introduction of CO to the electrolyte (Figure S19), whereas CO had a negligible effect on the background activity of the electrode (Figure S20). These observations indicate that CO selectively and irreversibly poisons the Rh sites and that these are the sites of catalysis.
Conjugation to Graphite Is Essential for the Unique Activity of GCC-Rh
GCC-Rh contains a phenazine linkage that is redox active unto itself. To probe the role of the phenazine unit in the catalytic activity of the molecule, we also examined the redox behavior of a water-soluble Rh molecule containing a phenazine moiety fused to the phenanthroline ligand, [RhCp*(dppz-SO3)OH2]+ (dppz-SO3 = dipyrido[3,2-a:2′,3′-c]phenazine-11-sulfonate) across the pH range (Figures S21–S23). Phenazine redox waves are observed in addition to the RhIII/I wave, but the overlap of the two features prevents unambiguous assignment at all pH values. Nonetheless, the presence of the phenazine moiety does not dramatically alter the catalytic behavior of the molecule. Like those of RhCp*(bpds)OH2, CVs of [RhCp*(dppz-SO3)OH2]+ display catalytic current at pH 1 and pH 3, but display no catalytic activity at pH 7 or pH 13 (Figures S21–S23). These observations indicate that the mere presence of a phenazine moiety in the ligand backbone is insufficient to lead to HER catalysis across the pH range and that the graphite itself plays the critical role in defining the divergent reactivity of GCC-Rh.
GCC-Rh Proceeds through Rate-Limiting Concerted Proton–Electron Transfer
Mechanistic investigations provide a basis for understanding why GCC-Rh can catalyze HER over the entire pH range, while the molecule cannot. To probe the mechanism of HER catalysis at GCC-Rh, we collected steady-state current density vs potential (Tafel) data at pH 1 (0.1 M HClO4, Figure 4a) and pH 13 (0.1 M NaOH, Figure S24). Control experiments establish that these data are not subject to transport limitations or significant deactivation over the course of data collection (Figures S25–S28) and therefore reflect the intrinsic activation-controlled kinetics of HER. At pH 1 and pH 13, we observe Tafel slopes of 110 mV dec–1 and 200 mV dec–1, respectively. These slopes correspond to transfer coefficients, α, of 0.5 and 0.3 and are consistent with rate-limiting first-electron transfer from the catalyst resting state.18,61−64 Importantly, the Tafel data exclude a mechanism in which the Rh center is reduced by one or two electrons prior to rate-limiting activation of H+, as those pathways would give rise to α values of exactly 1 or 2, respectively.65 (See Supporting Information for derivations of α values corresponding to different mechanistic pathways.) Furthermore, CV data collected in 1 M perchloric acid establish an H/D kinetic isotope effect (KIE) of 2.0 ± 0.1 (Figures 4b and S29–S30; see Supporting Information for details on computing the H/D KIE), indicating that proton transfer is also involved in the rate-limiting step.66,67 Together, the kinetic data are consistent with rate-limiting CPET for HER catalysis at GCC-Rh (Scheme 2). In Scheme 2, we invoke the formation of a R−H bond, but we acknowledge that PT could occur to the Cp* ring as well.40,201 Irrespective of the site of PT, the data indicate that changes in the applied potential directly impact the free energy of H+ activation (Figure 1d).
Figure 4.
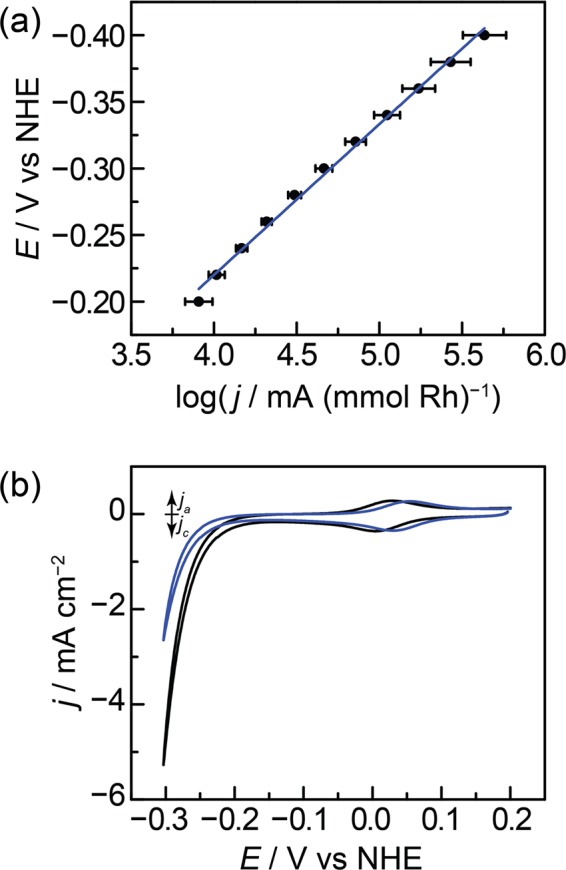
(a) Potential vs activation-controlled current density for HER at GCC-Rh in 0.1 M HClO4. The Tafel slope is 110 mV dec–1. (b) Cyclic voltammograms (5 mV s–1) of GCC-Rh in 1 M HClO4 in H2O (black) and D2O (blue).
Scheme 2. Proposed Mechanism for HER at GCC-Rh.
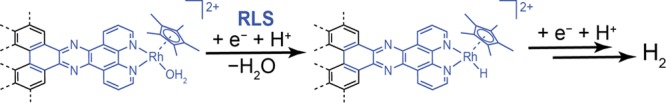
“RLS” denotes the rate-limiting step.
Rh valency in GCC-Rh Remains Constant during Catalysis
To directly probe the valency of the GCC-Rh sites during HER, we collected in situ X-ray absorption near edge structure (XANES) spectra. The XANES edge is sensitive to the oxidation state and coordination environment of the element being probed; indeed, there is a significant difference in both the position and shape of the Rh K-edge in the model complexes [RhIIICp*(phen)Cl]+ (phen = phenanthroline) and RhICp*(phen) (Figure S31).52 In situ XANES data collected on GCC-Rh samples at the open circuit potential (∼0.65 V vs NHE in 0.1 M HClO4 and ∼0.15 V vs NHE in 0.1 M NaOH) and during HER catalysis in 0.1 M HClO4 (−0.55 V vs NHE) and 0.1 M NaOH (−1.16 V vs NHE) are all identical (Rh K-edge of 23,229.0 eV) (Figures 5 and S32). These potentials are >0.1 V and 0.54 V more negative than E1/2(RhIII/I) for the molecule at pH 1 and pH 13, respectively. Remarkably, even over this vast potential range, we do not observe a RhI species during catalysis.
Figure 5.
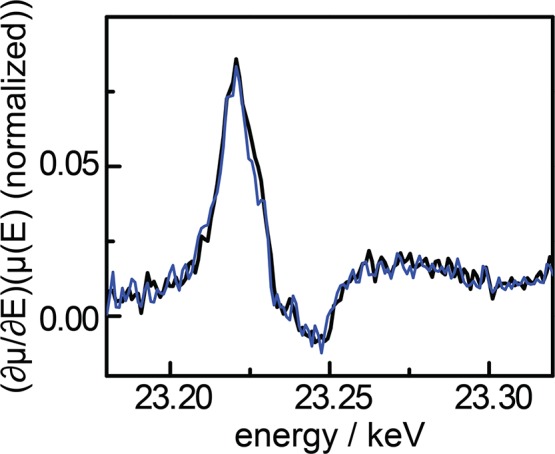
First derivative plot of in situ Rh K-edge X-ray absorption near edge structure spectra of GCC-Rh in 0.1 M HClO4 at the open circuit potential (∼0.65 V vs NHE) (black) and −0.55 V vs NHE (blue).
Mechanistic Model
The unique catalytic behavior of GCC-Rh can be fully accounted for by the strong electronic coupling between the Rh center and the graphite electrode. A cartoon model comparing ET at RhCp*(bpds)OH2 with ET at GCC-Rh is shown in Figure 6. In each panel, the electronic structure of the glassy carbon electrode consists of filled states (gray) and unfilled states (beige). The transition between the filled and unfilled states is the Fermi level of the electrode, EF, which is the potential directly measured or applied by a potentiostat. EF has both “chemical” contributions that depend on the composition of the electrode material and can be approximated by the work function and also has contributions from the electrostatic potential difference between the electrode and the solution.61 In this paper, we refer to the “electrostatic potential” explicitly, and all other uses of the word “potential” refer to electrochemical potential. The dotted red lines denote the electrostatic potential drop between EF and the solution. The potential of zero free charge, EPZFC, is the electrochemical potential at which there is no electrostatic potential drop at the interface and is related to the work function of the electrode.61 The top two panels in Figure 6 show outer-sphere reduction of RhCp*(bpds)OH2, in which the Rh molecule lies outside of the electrical double layer (EDL). For a glassy carbon electrode, EPZFC is positive of the reduction potential of the molecule (E1/2(RhIII/I)), and therefore, when EF is equal to EPZFC, there is insufficient driving force for ET (top left). Applying a more negative potential to the electrode raises EF and generates an interfacial electric field that shifts the energies of the electronic states of the electrode relative to the species in solution (top right).61 When the applied potential approaches E1/2(RhIII/I)), electrons cross the double layer to reduce the Rh molecule, and electrons from the external circuit fill the resulting holes in the graphite electrode, giving rise to the observed current. If the electrolyte contains a strong enough proton donor (i.e., in low-pH electrolytes), PT occurs to the RhI species to form a Rh–H intermediate, which can then go on to mediate HER catalysis.
Figure 6.
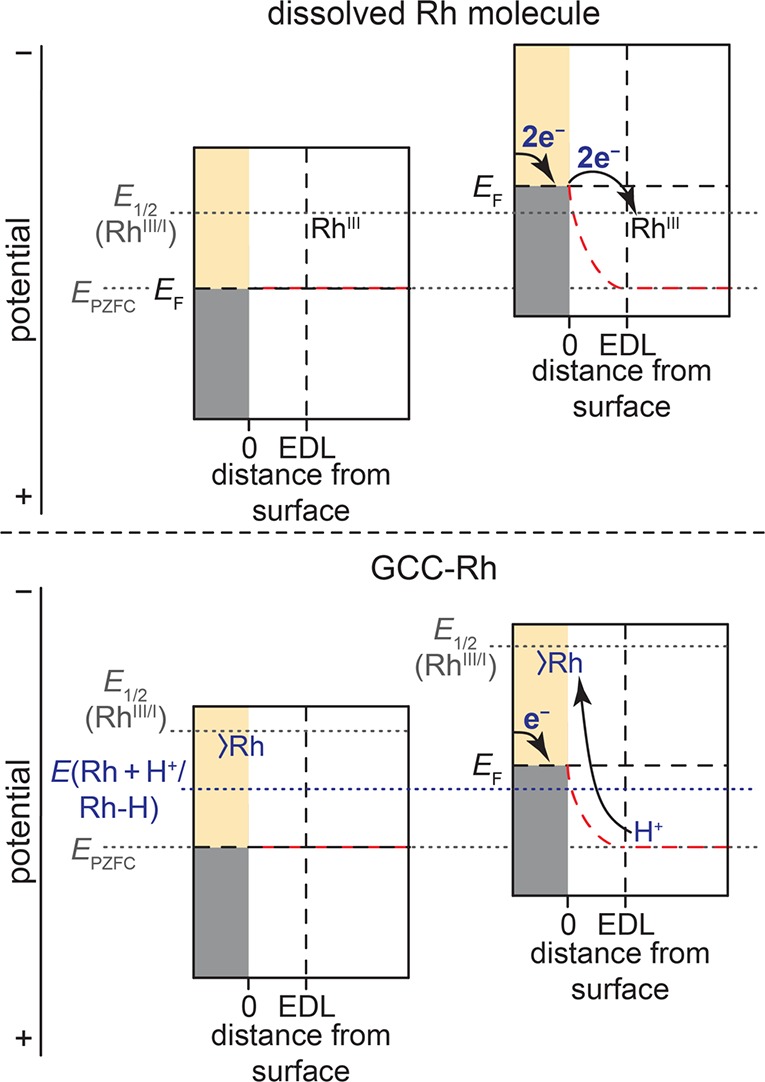
Proposed interfacial free energy diagrams for unmodified electrodes with dissolved RhCp*(bpds)OH2 molecules (top) and GCC-Rh electrodes (bottom). In each panel, the gray denotes the filled band states of the electrode, the beige denotes the unfilled band states, and the dotted horizontal black line between the filled and unfilled states denotes the Fermi level of the electrode, EF. The approximate edge of the electrical double layer, EDL, is denoted by a vertical dotted black line. The reduction potential of the molecule (E1/2(RhIII/I)) and the potential for formation of a Rh–H species at the GCC site (E(Rh + H+/Rh–H)) are depicted with dotted gray and blue lines, respectively. The electrostatic potential across the EDL is indicated by the dotted red line, and the potential of zero free charge (EPZFC), at which the electrostatic potential drop vanishes, is also indicated with a dotted gray line. For RhCp*(bpds)OH2, varying EF drives outer-sphere two-electron transfer to the Rh. For GCC-Rh (bottom), varying EF shifts the electrostatic potential of the surface, simultaneously shifting the energy levels of the Rh acceptor states, and preventing Rh-centered reduction. The electrostatic potential of the surface drives proton transfer to Rh with internal electron rearrangement to make the Rh–H bond.
In GCC-Rh, rather than residing outside of the EDL, the Rh site is strongly electronically coupled to the electrode and resides inside the EDL.52 Consequently, as EF is varied from EPZFC (Figure 6, bottom left), the orbitals of the conjugated Rh center shift along with the electronic states of the solid (Figure 6, bottom right). Thus, changes in the applied potential at the electrode do not alter the driving force for ET between the graphite electrode and the Rh sites, and outer-sphere reduction of the Rh species never occurs.52 This phenomenon explains why we do not observe RhIII/I redox features in CVs of GCC-Rh. It also explains why we observe overlapping XANES spectra across the entire 1.8 V range examined, and why there is no change in the XANES edge even at potentials 0.54 V more negative than E1/2(RhIII/I). The fact that outer-sphere ET cannot occur between the Rh sites and the electrode in GCC-Rh explicitly excludes all reaction pathways that proceed through reduction of the conjugated Rh center prior to Rh–H bond formation.
This phenomenon does not exclude catalysis at GCC sites that proceeds through inner-sphere ET pathways. Although varying the potential does not alter the driving force for ET between the conjugated site and the electrode, it does alter the driving force for ion transfer between the GCC site and the solution.52 In the context of hydrogen evolution catalysis, this means that when the potential is sufficiently negative (Figure 6, bottom), the interfacial electric field drives protons across the double layer to the conjugated Rh site. When the proton binds to the Rh site, the strong electronic coupling provided by the conjugated linkage ensures that electron flow is concerted with H+ transfer (Figure 1c). Importantly, in this model, binding one positively charged proton per Rh site leads to obligatory compensatory one-electron transfer from the external circuit in order to hold the potential of the electrode constant. This net proton–electron stoichiometry for forming Rh–H bonds is consistent with the rate-determining one-proton, one-electron CPET step indicated by the Tafel data (Figure 4a). The proton–electron stoichiometry in the elementary CPET steps is a reflection of the electrosorption valence of the proton, which is the net charge passed from the external circuit per adsorption event.61 Following one-electron, one-proton interfacial CPET, we invoke the formation of a Rh–H intermediate that we still view as a metal hydride. The two electrons localized in the Rh–H bond can come from rapid internal rearrangement of electrons from the band states of the solid68 even though this net process only involves transfer of one electron from the external circuit to balance the adsorption of one proton. We also note that the subsequent protonation of the Rh–H to form H2 involves transfer of a second proton across the double layer and, correspondingly, will lead to a second electron transfer from the external circuit, thereby completing the overall two-electron stoichiometry of HER. We note that, in this sense, the mechanism for HER at GCC-Rh is identical to the mechanism for HER at bulk metal electrodes such as Ag and Au, which also proceed through unmediated one-proton, one-electron CPET steps.18,35,63,64,69,70
The fact that outer-sphere ET cannot occur between the Rh sites and the electrode in GCC-Rh renders the outer-sphere reduction potentials of the molecular fragment irrelevant in determining the driving force for forming the GCC-Rh–H intermediate. We have previously demonstrated that PCET reactions occur at GCC sites irrespective of the redox properties of the molecular analogue.71 In particular, we have shown that, for GCCs, the driving force for PCET is given by the intrinsic affinity of the conjugated site for binding H+and the magnitude of the interfacial electric field that attracts H+ to the GCC site.71 In the context of GCC-Rh, negative polarization of the electrode increases the interfacial field strength and consequently increases the driving force for H+ binding to the GCC-Rh site (Figure 6 bottom right). Thus, unlike for the Rh molecule, the driving force for PCET to GCC-Rh is directly dependent on Eapplied. Increasing the overpotential by applying a more negative potential raises the Fermi level of the electrode, directly increasing the driving force for Rh–H bond formation (Figure 6 and Figure 1d) and the rate of catalysis. Similarly, changing the pKa of the proton donor directly changes the driving force for forming the Rh–H bond. For GCC-Rh, this change in proton transfer driving force can be fully compensated for by the increased interfacial electric field strength experienced by the Rh center at more negative applied potentials. This critical distinction is what makes GCC-Rh active for HER catalysis across the entire pH range. This phenomenon cannot occur in a typical redox mediation pathway because, as highlighted in Figure 1a, the basicity of the operative reduced Rh molecule is intrinsic to its coordination environment and redox potential, but is independent of Eapplied.
Importantly, this direct, unmediated electrocatalytic mechanism is a result of the strongly coupled aromatic linkage rather than the primary coordination environment or metal ion present in the active site, and therefore any conjugated molecular catalyst is expected to proceed through pathways in which substrate activation is concerted with electron flow. Indeed, this mechanistic model explains the previously reported promotion of CO2 reduction catalysis at conjugated Re(phen)(CO)3Cl (GCC-Re) relative to the molecular analogue.51 The molecular Re catalyst is believed to proceed exclusively through pathways involving stepwise ET and substrate bond rearrangement.72−74 These pathways lead to a 60 mV dec–1 scaling in overpotential and a reaction rate that reaches a maximum beyond the reduction potential of the molecule. In stark contrast, GCC-Re displays a 150 mV dec–1 scaling in overpotential and reaction rate, which continues to increase >0.5 V past the plateau current for the molecular catalyst.51 These mechanistic features for CO2 reduction at GCC-Re resemble those observed for HER at GCC-Rh, indicating that increasing the electrode potential directly increases the driving force for substrate bond activation across diverse reactions and metal centers. In this respect, GCC active sites behave like metallic solids, but with an unprecedented degree of molecular control.
Conclusion
The unique combination of metallic electronic structure and molecular-level control over the active site in GCCs has broad implications for catalyst design. The metallic electronic structure of the solid eliminates the outer-sphere reduction potential of the molecular fragment as a design consideration. The ability to tune the local environment6,8,14,33 of the active site at the molecular level enables synthetic optimization of the metal-substrate bond strength and kinetics of substrate activation. Together, these factors enable the rational synthesis of molecularly precise energy conversion catalysts that maximize reaction rate and energetic efficiency.
Acknowledgments
We gratefully acknowledge Joshua Wright for assistance with the XAS experiments. We acknowledge Cyrille Costentin, Jeffrey Warren, Jay Winkler, and Michael L. Pegis for fruitful discussions. This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Science Program, under Award Number DE-SC0014176. S.O. and C.J.K. were supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1122374. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. MRCAT operations are supported by the Department of Energy and the MRCAT member institutions. XPS investigations made use of Shared Experimental Facilities supported in part by the MRSEC Program of the National Science Foundation under Award No. DMR-1419807. Y.S. acknowledges the Sloan Foundation and the Research Corporation for Science Advancement (Cottrell Award).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b04981.
Full experimental details, turnover frequency calculations, XPS data and fitting, derivations of transfer coefficients, H/D kinetic isotope calculation, XAS data, and additional electrochemical data (PDF)
Author Present Address
† Department of Chemistry, University of California, Berkeley, California 94720, United States
The authors declare the following competing financial interest(s): M.N.J., C.J.K., S.O., and Y.S. are inventors on patent application 15/236,963, submitted by the Massachusetts Institute of Technology, that covers the graphite-conjugated materials described in this work.
Supplementary Material
References
- Reece S. Y.; Nocera D. G. Proton-Coupled Electron Transfer in Biology: Results from Synergistic Studies in Natural and Model Systems. Annu. Rev. Biochem. 2009, 78 (1), 673–699. 10.1146/annurev.biochem.78.080207.092132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S.; Iordanova N. Theoretical Studies of Proton-Coupled Electron Transfer Reactions. Biochim. Biophys. Acta, Bioenerg. 2004, 1655 (1–3), 29–36. 10.1016/j.bbabio.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Roubelakis M. M.; Bediako D. K.; Dogutan D. K.; Nocera D. G. Proton-Coupled Electron Transfer Kinetics for the Hydrogen Evolution Reaction of Hangman Porphyrins. Energy Environ. Sci. 2012, 5 (7), 7737. 10.1039/c2ee21123h. [DOI] [Google Scholar]
- Rountree E. S.; Dempsey J. L. Reactivity of Proton Sources with a Nickel Hydride Complex in Acetonitrile: Implications for the Study of Fuel-Forming Catalysts. Inorg. Chem. 2016, 55 (10), 5079–5087. 10.1021/acs.inorgchem.6b00885. [DOI] [PubMed] [Google Scholar]
- Rountree E. S.; Martin D. J.; McCarthy B. D.; Dempsey J. L. Linear Free Energy Relationships in the Hydrogen Evolution Reaction: Kinetic Analysis of a Cobaloxime Catalyst. ACS Catal. 2016, 6 (5), 3326–3335. 10.1021/acscatal.6b00667. [DOI] [Google Scholar]
- Carver C. T.; Matson B. D.; Mayer J. M. Electrocatalytic Oxygen Reduction by Iron Tetra-Arylporphyrins Bearing Pendant Proton Relays. J. Am. Chem. Soc. 2012, 134 (12), 5444–5447. 10.1021/ja211987f. [DOI] [PubMed] [Google Scholar]
- Rosenthal J.; Nocera D. G. Role of Proton-Coupled Electron Transfer in O-O Bond Activation. Acc. Chem. Res. 2007, 40 (7), 543–553. 10.1021/ar7000638. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Drouet S.; Robert M.; Savéant J. M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338 (6103), 90–94. 10.1126/science.1224581. [DOI] [PubMed] [Google Scholar]
- Wilson A. D.; Fraze K.; Twamley B.; Miller S. M.; DuBois D. L.; DuBois M. R. The Role of the Second Coordination Sphere of [Ni(PCy2NBz2)2](BF4)2 in Reversible Carbon Monoxide Binding. J. Am. Chem. Soc. 2008, 130 (3), 1061–1068. 10.1021/ja077328d. [DOI] [PubMed] [Google Scholar]
- DuBois M. R.; DuBois D. L. Development of Molecular Electrocatalysts for CO2 Reduction and H2 Production/Oxidation. Acc. Chem. Res. 2009, 42 (12), 1974–1982. 10.1021/ar900110c. [DOI] [PubMed] [Google Scholar]
- Mora S. J.; Odella E.; Moore G. F.; Gust D.; Moore T. A.; Moore A. L. Proton-Coupled Electron Transfer in Artificial Photosynthetic Systems. Acc. Chem. Res. 2018, 51 (2), 445–453. 10.1021/acs.accounts.7b00491. [DOI] [PubMed] [Google Scholar]
- Wuttig A.; Yaguchi M.; Motobayashi K.; Osawa M.; Surendranath Y. Inhibited Proton Transfer Enhances Au-Catalyzed CO2-to-Fuels Selectivity. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (32), E4585–E4593. 10.1073/pnas.1602984113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S.; Soudackov A. V. Proton-Coupled Electron Transfer in Solution, Proteins, and Electrochemistry. J. Phys. Chem. B 2008, 112 (45), 14108–14123. 10.1021/jp805876e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M. L.; Stewart M. P.; Bullock R. M.; DuBois M. R.; DuBois D. L. A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s–1 for H2 Production. Science 2011, 333 (6044), 863–866. 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- Chang C. J.; Loh Z.; Shi C.; Anson F. C.; Nocera D. G. Targeted Proton Delivery in the Catalyzed Reduction of Oxygen to Water by Bimetallic Pacman Porphyrins. J. Am. Chem. Soc. 2004, 126 (32), 10013–10020. 10.1021/ja049115j. [DOI] [PubMed] [Google Scholar]
- Chang C. J.; Chng L. L.; Nocera D. G. Proton-Coupled O-O Activation on a Redox Platform Bearing a Hydrogen-Bonding Scaffold. J. Am. Chem. Soc. 2003, 125 (7), 1866–1876. 10.1021/ja028548o. [DOI] [PubMed] [Google Scholar]
- Nichols E. M.; Derrick J. S.; Nistanaki S. K.; Smith P. T.; Chang C. J. Positional Effects of Second-Sphere Amide Pendants on Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins. Chem. Sci. 2018, 9 (11), 2952–2960. 10.1039/C7SC04682K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M. N.; Surendranath Y. Donor-Dependent Kinetics of Interfacial Proton-Coupled Electron Transfer. J. Am. Chem. Soc. 2016, 138 (9), 3228–3234. 10.1021/jacs.6b00167. [DOI] [PubMed] [Google Scholar]
- Wuttig A.; Liu C.; Peng Q.; Yaguchi M.; Hendon C. H.; Motobayashi K.; Ye S.; Osawa M.; Surendranath Y. Tracking a Common Surface-Bound Intermediate during CO2-to-Fuels Catalysis. ACS Cent. Sci. 2016, 2 (8), 522–528. 10.1021/acscentsci.6b00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y.; Hall A. S.; Surendranath Y. Tuning of Silver Catalyst Mesostructure Promotes Selective Carbon Dioxide Conversion into Fuels. Angew. Chem. 2016, 128 (49), 15508–15512. 10.1002/ange.201607942. [DOI] [PubMed] [Google Scholar]
- Hall A. S.; Yoon Y.; Wuttig A.; Surendranath Y. Mesostructure-Induced Selectivity in CO2 Reduction Catalysis. J. Am. Chem. Soc. 2015, 137 (47), 14834–14837. 10.1021/jacs.5b08259. [DOI] [PubMed] [Google Scholar]
- Schreier M.; Yoon Y.; Jackson M. N.; Surendranath Y. Competition between H and CO for Active Sites Governs Copper-Mediated Electrosynthesis of Hydrocarbon Fuels. Angew. Chem., Int. Ed. 2018, 57 (32), 10221–10225. 10.1002/anie.201806051. [DOI] [PubMed] [Google Scholar]
- Li C. W.; Ciston J.; Kanan M. W. Electroreduction of Carbon Monoxide to Liquid Fuel on Oxide-Derived Nanocrystalline Copper. Nature 2014, 508 (7497), 504–507. 10.1038/nature13249. [DOI] [PubMed] [Google Scholar]
- Mayer J. M. Proton-Coupled Electron Transfer: A Reaction Chemist’s View. Annu. Rev. Phys. Chem. 2004, 55 (1), 363–390. 10.1146/annurev.physchem.55.091602.094446. [DOI] [PubMed] [Google Scholar]
- Ledezma-Yanez I.; Wallace W. D. Z.; Sebastián-Pascual P.; Climent V.; Feliu J. M.; Koper M. T. M. Interfacial Water Reorganization as a pH-Dependent Descriptor of the Hydrogen Evolution Rate on Platinum Electrodes. Nat. Energy 2017, 2 (4), 17031. 10.1038/nenergy.2017.31. [DOI] [Google Scholar]
- Kas R.; Kortlever R.; Milbrat A.; Koper M. T. M.; Mul G.; Baltrusaitis J. Electrochemical CO2 Reduction on Cu2 O-Derived Copper Nanoparticles: Controlling the Catalytic Selectivity of Hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16 (24), 12194–12201. 10.1039/C4CP01520G. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Li C. W.; Kanan M. W. Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au Nanoparticles. J. Am. Chem. Soc. 2012, 134 (49), 19969–19972. 10.1021/ja309317u. [DOI] [PubMed] [Google Scholar]
- Weinberg D. R.; Gagliardi C. J.; Hull J. F.; Murphy C. F.; Kent C.; Westlake B. C.; Paul A.; Ess D. H.; McCafferty D. G.; Meyer T. J. Proton-Coupled Electron Transfer. Chem. Rev. 2012, 112 (7), 4016–4093. 10.1021/cr200177j. [DOI] [PubMed] [Google Scholar]
- Huynh M. H. V; Meyer T. J. Proton-Coupled Electron Transfer. Chem. Rev. 2007, 107 (11), 5004–5064. 10.1021/cr0500030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nocera D. G. Chemistry of Personalized Solar Energy. Inorg. Chem. 2009, 48 (21), 10001–10017. 10.1021/ic901328v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Proton-Coupled Electron Transfer: Moving Together and Charging Forward. J. Am. Chem. Soc. 2015, 137 (28), 8860–8871. 10.1021/jacs.5b04087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveant J. M.Elements of Molecular and Biomolecular Electro-Chemistry: An Electrochemical Approach to Electron Transfer Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, 2006. [Google Scholar]
- DuBois M. R.; DuBois D. L. The Roles of the First and Second Coordination Spheres in the Design of Molecular Catalysts for H2 Production and Oxidation. Chem. Soc. Rev. 2009, 38 (1), 62–72. 10.1039/B801197B. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Dridi H.; Savéant J. M. Molecular Catalysis of H2 Evolution: Diagnosing Heterolytic versus Homolytic Pathways. J. Am. Chem. Soc. 2014, 136 (39), 13727–13734. 10.1021/ja505845t. [DOI] [PubMed] [Google Scholar]
- Koper M. T. M. Activity Volcanoes for the Electrocatalysis of Homolytic and Heterolytic Hydrogen Evolution. J. Solid State Electrochem. 2016, 20 (4), 895–899. 10.1007/s10008-015-3036-z. [DOI] [Google Scholar]
- Dempsey J. L.; Brunschwig B. S.; Winkler J. R.; Gray H. B. Hydrogen Evolution Catalyzed by Cobaloximes. Acc. Chem. Res. 2009, 42 (12), 1995–2004. 10.1021/ar900253e. [DOI] [PubMed] [Google Scholar]
- Tsay C.; Yang J. Y. Electrocatalytic Hydrogen Evolution under Acidic Aqueous Conditions and Mechanistic Studies of a Highly Stable Molecular Catalyst. J. Am. Chem. Soc. 2016, 138 (43), 14174–14177. 10.1021/jacs.6b05851. [DOI] [PubMed] [Google Scholar]
- Tsay C.; Ceballos B. M.; Yang J. Y. pH-Dependent Reactivity of a Water-Soluble Nickel Complex: Hydrogen Evolution vs Selective Electrochemical Hydride Generation. Organometallics 2019, 38 (6), 1286–1291. 10.1021/acs.organomet.8b00558. [DOI] [Google Scholar]
- Elgrishi N.; McCarthy B. D.; Rountree E. S.; Dempsey J. L. Reaction Pathways of Hydrogen-Evolving Electrocatalysts: Electrochemical and Spectroscopic Studies of Proton-Coupled Electron Transfer Processes. ACS Catal. 2016, 6 (6), 3644–3659. 10.1021/acscatal.6b00778. [DOI] [Google Scholar]
- Quintana L. M. A.; Johnson S. I.; Corona S. L.; Villatoro W.; Goddard W. A.; Takase M. K.; VanderVelde D. G.; Winkler J. R.; Gray H. B.; Blakemore J. D. Proton−Hydride Tautomerism in Hydrogen Evolution Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (23), 6409–6414. 10.1073/pnas.1606018113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costentin C.; Savéant J. M. Towards an Intelligent Design of Molecular Electrocatalysts. Nat. Rev. Chem. 2017, 1 (11), 0087. 10.1038/s41570-017-0087. [DOI] [Google Scholar]
- Costentin C.; Drouet S.; Robert M.; Savéant J. M. Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis. J. Am. Chem. Soc. 2012, 134 (27), 11235–11242. 10.1021/ja303560c. [DOI] [PubMed] [Google Scholar]
- Hu X.; Brunschwig B. S.; Peters J. C. Electrocatalytic Hydrogen Evolution at Low Overpotentials by Cobalt Macrocyclic Glyoxime and Tetraimine Complexes. J. Am. Chem. Soc. 2007, 129 (29), 8988–8998. 10.1021/ja067876b. [DOI] [PubMed] [Google Scholar]
- Wakerley D. W.; Reisner E. Development and Understanding of Cobaloxime Activity through Electrochemical Molecular Catalyst Screening. Phys. Chem. Chem. Phys. 2014, 16 (12), 5739–5746. 10.1039/C4CP00453A. [DOI] [PubMed] [Google Scholar]
- Pegis M. L.; McKeown B. A.; Kumar N.; Lang K.; Wasylenko D. J.; Zhang X. P.; Raugei S.; Mayer J. M. Homogenous Electrocatalytic Oxygen Reduction Rates Correlate with Reaction Overpotential in Acidic Organic Solutions. ACS Cent. Sci. 2016, 2 (11), 850–856. 10.1021/acscentsci.6b00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savéant J. M. Concerted Proton-Electron Transfers: Fundamentals and Recent Developments. Annu. Rev. Anal. Chem. 2014, 7 (1), 537–560. 10.1146/annurev-anchem-071213-020315. [DOI] [PubMed] [Google Scholar]
- Savéant J. M. Evidence for Concerted Pathways in Ion-Pairing Coupled Electron Transfers. J. Am. Chem. Soc. 2008, 130 (14), 4732–4741. 10.1021/ja077480f. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Tronic T. a.; Mayer J. M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev. 2010, 110 (12), 6961–7001. 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima T.; Drisdell W.; Yano J.; Surendranath Y. Graphite-Conjugated Pyrazines as Molecularly Tunable Heterogeneous Electrocatalysts. J. Am. Chem. Soc. 2015, 137 (34), 10926–10929. 10.1021/jacs.5b06737. [DOI] [PubMed] [Google Scholar]
- Oh S.; Gallagher J. R.; Miller J. T.; Surendranath Y. Graphite-Conjugated Rhenium Catalysts for Carbon Dioxide Reduction. J. Am. Chem. Soc. 2016, 138 (6), 1820–1823. 10.1021/jacs.5b13080. [DOI] [PubMed] [Google Scholar]
- Jackson M. N.; Oh S.; Kaminsky C. J.; Chu S. B.; Zhang G.; Miller J. T.; Surendranath Y. Strong Electronic Coupling of Molecular Sites to Graphitic Electrodes via Pyrazine Conjugation. J. Am. Chem. Soc. 2018, 140 (3), 1004–1010. 10.1021/jacs.7b10723. [DOI] [PubMed] [Google Scholar]
- Kaminsky C. J.; Wright J.; Surendranath Y. Graphite-conjugation enhances porphyrin electrocatalysis. ACS Catal. 2019, 9 (4), 3667–3671. [Google Scholar]
- Kolle U.; Grutzel M. Organometallic Rhodium(III) Complexes as Catalysts for the Photoreducton of Protons to Hydrogen on Colloidal TiO2. Angew. Chem., Int. Ed. Engl. 1987, 26 (6), 567–570. 10.1002/anie.198705671. [DOI] [Google Scholar]
- Caix C.; Chardon-Noblat S.; Deronzier A.; Ziessel R. Electrochemical Generation of a Metal-Hydride Complex [(C5Me5)Ir(L)H]+ (L = 2,2’-Bipyridine). The Electrochemical Behaviour. J. Electroanal. Chem. 1993, 362 (1–2), 301–304. 10.1016/0022-0728(93)80035-G. [DOI] [Google Scholar]
- Huang J.; Antonietti M.; Liu J. Bio-Inspired Carbon Nitride Mesoporous Spheres for Artificial Photosynthesis: Photocatalytic Cofactor Regeneration for Sustainable Enzymatic Synthesis. J. Mater. Chem. A 2014, 2 (21), 7686–7693. 10.1039/C4TA00793J. [DOI] [Google Scholar]
- Bonin J.; Costentin C.; Robert M.; Routier M.; Savéant J. M. Proton-Coupled Electron Transfers: pH-Dependent Driving Forces? Fundamentals and Artifacts. J. Am. Chem. Soc. 2013, 135 (38), 14359–14366. 10.1021/ja406712c. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Robert M.; Savéant J. M. Concerted Proton-Electron Transfer Reactions in Water. Are the Driving Force and Rate Constant Depending on pH When Water Acts as Proton Donor or Acceptor?. J. Am. Chem. Soc. 2007, 129 (18), 5870–5879. 10.1021/ja067950q. [DOI] [PubMed] [Google Scholar]
- Zhang L. Co-Immobilization of (2,2′-Bipyridyl) (Pentamethylcyclopentadienyl)-Rhodium Complex and NAD-Dependent Dehydrogenases for Enzymatic Electrosynthesis; Ph.D. Dissertation, Université de Lorraine: , Villers-lés-Nancy, France, 2016. [Google Scholar]
- NIST X-Ray Photoelectron Spectroscopy Database, NIST Standard Reference Database Number 20; National Institute of Standards and Technology: Gaithersburg MD, 2000.
- Contour J. X-Ray Photoelectron Spectroscopy and Electron Microscopy of Pt-Rh Gauzes Used for Catalytic Oxidation of Ammonia. J. Catal. 1977, 48 (1–3), 217–228. 10.1016/0021-9517(77)90093-8. [DOI] [Google Scholar]
- Schmickler W.; Santos E.. Interfacial Electrochemistry; Springer: Berlin, 2010, pp 51–65; 145−175 10.1007/978-3-642-04937-8_13 [DOI] [Google Scholar]
- Gileadi E.Physical Electrochemistry, Fundamentals, Techniques and Applications; Wiley-VCH: Weinheim, 2011, pp 44–51; 66–75. [Google Scholar]
- Eberhardt D.; Santos E.; Schmickler W. Hydrogen Evolution on Silver Single Crystal Electrodes—First Results. J. Electroanal. Chem. 1999, 461 (1–2), 76–79. 10.1016/S0022-0728(98)00093-X. [DOI] [Google Scholar]
- Jackson M. N.; Jung O.; Lamotte H. C.; Surendranath Y. Donor-Dependent Promotion of Interfacial Proton-Coupled Electron Transfer in Aqueous Electrocatalysis. ACS Catal. 2019, 9 (4), 3737–3743. 10.1021/acscatal.9b00056. [DOI] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods Fundamentals and Applications, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, 2001; pp. 87–155. [Google Scholar]
- Costentin C.; Robert M.; Savéant J. M. Adiabatic and Non-Adiabatic Concerted Proton-Electron Transfers. Temperature Effects in the Oxidation of Intramolecularly Hydrogen-Bonded Phenols. J. Am. Chem. Soc. 2007, 129 (32), 9953–9963. 10.1021/ja071150d. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Robert M.; Savéant J. M. Electrochemical Concerted Proton and Electron Transfers. Potential-Dependent Rate Constant, Reorganization Factors, Proton Tunneling and Isotope Effects. J. Electroanal. Chem. 2006, 588 (2), 197–206. 10.1016/j.jelechem.2005.12.027. [DOI] [Google Scholar]
- Pitman C. L.; Finster O. N. L.; Miller A. J. M. Cyclopentadiene-Mediated Hydride Transfer from Rhodium Complexes. Chem. Commun. 2016, 52, 9105–9108. [DOI] [PubMed] [Google Scholar]
- Hoffmann R.Solids and Surfaces: A Chemist’s View of Bonding in Extended Structures; VCH Publishers, Inc.: New York, NY, 1988; pp 68–78. [Google Scholar]
- Doubova L. M.; Trasatti S. Effect of the Crystallographic Orientation of Ag Single Crystal Face Electrodes on the Kinetics of Proton Discharge. J. Electroanal. Chem. 1999, 467 (1), 164–176. 10.1016/S0022-0728(99)00060-1. [DOI] [Google Scholar]
- Khanova L. A.; Krishtalik L. I. Kinetics of the Hydrogen Evolution Reaction on Gold Electrode. A New Case of the Barrierless Discharge. J. Electroanal. Chem. 2011, 660 (2), 224–229. 10.1016/j.jelechem.2011.01.016. [DOI] [Google Scholar]
- Jackson M. N.; Pegis M. L.; Surendranath Y. Graphite-Conjugated Acids Reveal a Molecular Framework for Proton-Coupled Electron Transfer at Electrode Surfaces. ACS Cent. Sci. 2019, 5 (5), 831–841. 10.1021/acscentsci.9b00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francke R.; Schille B.; Roemelt M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide-Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118 (9), 4631–4701. 10.1021/acs.chemrev.7b00459. [DOI] [PubMed] [Google Scholar]
- Keith J. A.; Grice K. A.; Kubiak C. P.; Carter E. A. Elucidation of the Selectivity of Proton-Dependent Electrocatalytic CO2 Reduction by Fac -Re(Bpy)(CO)3Cl. J. Am. Chem. Soc. 2013, 135 (42), 15823–15829. 10.1021/ja406456g. [DOI] [PubMed] [Google Scholar]
- Sullivan B. P.; Bolinger C. M.; Conrad D.; Vining W. J.; Meyer T. J. One- and Two-Electron Pathways in the Electrocatalytic Reduction of CO2 by fac-Re(bpy)(CO)3Cl (bpy = 2,2’-Bipyridine). J. Chem. Soc., Chem. Commun. 1985, 985 (20), 1414–1416. 10.1039/C39850001414. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.