

Abstract

Among the >120 modified ribonucleosides in the prokaryotic epitranscriptome, many tRNA modifications are critical to bacterial survival, which makes their synthetic enzymes ideal targets for antibiotic development. Here we performed a structure-based design of inhibitors of tRNA-(N1G37) methyltransferase, TrmD, which is an essential enzyme in many bacterial pathogens. On the basis of crystal structures of TrmDs from Pseudomonas aeruginosa and Mycobacterium tuberculosis, we synthesized a series of thienopyrimidinone derivatives with nanomolar potency against TrmD in vitro and discovered a novel active site conformational change triggered by inhibitor binding. This tyrosine-flipping mechanism is uniquely found in P. aeruginosa TrmD and renders the enzyme inaccessible to the cofactor S-adenosyl-l-methionine (SAM) and probably to the substrate tRNA. Biophysical and biochemical structure–activity relationship studies provided insights into the mechanisms underlying the potency of thienopyrimidinones as TrmD inhibitors, with several derivatives found to be active against Gram-positive and mycobacterial pathogens. These results lay a foundation for further development of TrmD inhibitors as antimicrobial agents.
Introduction
The emergence of antibiotic resistance in bacterial pathogens on a global scale and the lack of new antibiotics represent a crisis with significant societal and economic impact. This is illustrated by >2 million annual cases multidrug resistant bacterial infections, which result in 23000 deaths.1,2Mycobacterium tuberculosis (Mtb) and Pseudomonas aeruginosa (Pa) are examples of major human pathogens for which drug resistance is emerging as a serious public health problem:1 multidrug resistant Mtb and Pa cause >10000 and ∼6700 deaths per year, respectively. Pa is the cause of the most common hospital-acquired infection among the immunocompromised, the elderly, the chronically ill, and patients with in-dwelling medical devices such as catheters, nasogastric tubes, and drains. These two pathogens illustrate the point that new antibiotics, particularly those that avoid resistance mechanisms and are aimed at novel targets, are urgently needed to alleviate the current antibiotic crisis.
Post-transcriptional ribonucleotide modifications of RNA, especially tRNA, play critical roles in translation in all organisms.3−7 In addition to the essentiality of some of the enzymes catalyzing these modifications for growth, studies with bacteria,4−8 yeast,3,5 and parasites9 have demonstrated that many tRNA modifications are critical in the cell stress response by facilitating selective translation of proteins critical to surviving the stress. Loss of the ability to synthesize these tRNA modifications renders bacteria susceptible to killing by the immune response and other environmental stresses.4,8 Given their role in bacterial cell survival, these critical tRNA modification synthesis enzymes constitute attractive targets for antibiotic development.
The bacterial tRNA (guanine37-N1)-methyltransferase (EC2.1.1.228; TrmD) catalyzes methyl transfer from S-adenosyl-l-methionine (SAM) to the guanine N1 at nucleotide position 37 in a subset of bacterial tRNA isoacceptors (Supporting Information, Figure S1) and has proven to be an essential enzyme in most bacterial species.10−14 While of central importance to mammalian cell health,13 Trm5, the functional homologue of TrmD in eukaryotes, has dissimilar active sites and different binding modes of SAM than TrmD.15,16 These observations suggest that TrmD is an attractive antibiotic target, a conclusion supported by the efforts of several groups to develop TrmD inhibitors.15,17,18 We recently developed a radioactivity-free bioluminescence-based high-throughput screening (HTS) assay that identified a series of novel TrmD inhibitors.17 In 2013, Hill et al.15 used X-ray crystallography-guided fragment screening to develop a series of SAM-competitive TrmD inhibitors against a broad range of bacterial isozymes. These inhibitors showed minimal antibacterial activity, presumably due to poor cell permeability.15 Interestingly, one of these inhibitors, compound 51 (AZ51), showed remarkable inhibitory potency toward all the tested TrmD isozymes and subnanomolar activity against P. aeruginosa TrmD (PaTrmD). Despite this potent TrmD inhibitor activity, AZ51 lacked antibacterial activity.
Building on this work and the TrmD inhibitor scaffolds identified in aforementioned HTS,17 we sought here to understand the structural basis for the potent TrmD inhibition caused by the thienopyrimidinone compounds, with the goal of refining the structures for antibiotic activity. Crystal structures of TrmD from Pa and Mtb in complex with AZ51 revealed conformational changes unique to the Gram-negative bacterial TrmD. On the basis of these structures, we then used the thienopyrimidinone scaffold (Figure 1) to design and synthesize a series of 33 derivatives with the goal of improved potency and antibacterial activity. Structure–activity relationship (SAR) studies defined critical features of the thienopyrimidinone that drive enzyme inhibition potency as well as antibacterial activity.
Figure 1.

Structure of TrmD inhibitors based on the thienopyrimidinone scaffold (A) and their O6-derivatives (B).
Results
AZ51 Has Broad-Spectrum TrmD Inhibition Activity
Previously, Hill et al. discovered an interesting inhibition mechanism where one of the thienopyrimidinone derivatives (compound 38)15 ordered the position of the lid domain of Haemophilus influenzae TrmD (HiTrmD) via its terminal imidazole substituent. The lid domain covered the active site of TrmD and was essential for tRNA binding.19,20 AZ51 is another analogue derived from the thienopyrimidinone scaffold (Figure 1A; Table 1) and is generally more potent than other thienopyrimidinone inhibitors across a variety of Gram-negative and Gram-positive isozymes,15 in particular to PaTrmD. Thus, it raised the question about the potential unique inhibition mechanism of AZ51. We confirmed that it inhibited PaTrmD with nanomolar potency (IC50 180 ± 20 nM; Figure 2A, Table 1) using a different, bioluminescence-based assay.17 In addition, we determined the binding affinities of AZ51 to three bacterial TrmDs by surface plasmon resonance (SPR; Table 2). Interestingly, AZ51 has much stronger binding affinity with Gram-negative bacterial TrmDs than mycobacterial and Gram-positive bacterial TrmDs (100- to 300-fold lower Kd; Table 2). For PaTrmD and Staphylococus aureus TrmD (SaTrmD), this relationship paralleled differences in TrmD inhibition potency by AZ51: PaTrmD Kd 91 nM and IC50180 nM here, 39 nM Hill et al.;15 and SaTrmD Kd 27500 nM and IC50 1200 nM.15 Because the basis for these striking differences in AZ51 binding energetics and inhibition potency for the different bacterial classes may lie in different structures of the AZ51–enzyme complexes, we undertook to solve the crystal structures of AZ51 with PaTrmD and MtbTrmD.
Table 1. Structures and IC50 Values for Thienopyrimidinone PaTrmD Inhibitorsa.


Biochemical potency is represented as IC50 value (μM); values are mean ± SD for at least two independent experiments done in duplicate.
The listed pharmacophores replace the group N(R1)(R2).
Figure 2.

Crystal structures of AZ51 with PaTrmD and MtbTrmD. (A) The IC50 values determined for AZ51 at tRNA concentrations of 1.5 μM (●), 4.5 μM (□), and 15 μM (▲), respectively, are 0.18 ± 0.03, 0.33 ± 0.04, and 0.84 ± 0.09 μM. Each data point represents an average of duplicate experiments with error bars indicated as SD. (B,D) Close-up views of catalytic sites of PaTrmD–AZ51 (B; PDB 6JOE) and MtbTrmD–AZ51 (D; PDB 6JOF) showing the binding modes of inhibitor AZ51, respectively. Polypeptide chains are shown as cartoons (chain A in gray and chain B in cyan), whereas key interacting residues are shown as sticks. AZ51 are shown as sticks with an 2Fo −Fc electron density (gray) map contoured at 1.0σ. Potential interactions involved in AZ51 binding are indicated by broken lines (black, hydrogen bonds; green, stacking interactions). (C) Inhibitor AZ51 binding at catalytic site induces wall-loop conformational change and thereafter blocks substrate tRNA (G37) binding. SAM-bound PaTrmD (PDB 5WYQ) and tRNA-bound H. influenzae TrmD (PDB 4YVI) were superimposed onto AZ51-bound PaTrmD, respectively. Polypeptide chains of PaTrmD–AZ51 are shown as surface, while the bound AZ51 (yellow) and side chain flipped residue Tyr120 (gray) are shown as sticks. PaTrmD–SAM structure (light pink) displays SAM and residue Tyr120 in sticks representation. G37of tRNA substrate in HiTrmD–tRNA structure is shown as sticks in orange. Polypeptide chains of PaTrmD–SAM and HiTrmD–tRNA are omitted for clarity. (E) The superposition of catalytic sites of PaTrmD–AZ51 and MtbTrmD–AZ51. The inhibitor AZ51 and residues in different positions from PaTrmD and MtbTrmD are indicated as sticks.
Table 2. Binding Kinetics of TrmD Ligandsa.
| ligand | KD (μM) | kon(M–1 s–1) | koff (s–1) | |
|---|---|---|---|---|
| PaTrmDc | SAHb | 5.9 | 55500 ± 479 | 0.326 ± 0.002 |
| AZ51 | 0.091 | 128000 ± 318 | 0.0117 ± 0.0000 | |
| 15 | 0.0016 | 13500000 ± 87800 | 0.0218 ± 0.0001 | |
| MtbTrmDd | SAH | 10.1 | 7700 ± 14 | 0.0775 ± 0. 0001 |
| AZ51 | 10.4 | 20100 ± 80 | 0.209 ± 0.0004 | |
| 15 | 0.19 | 645000 ± 3030 | 0.120 ± 0.0003 | |
| SaTrmDe | SAH | 3.3 | 279000 ± 36000 | 0.92 ± 0.03 |
| AZ51 | 27.5 | 59900 ± 566 | 1.65 ± 0.01 | |
| 15 | 3.1 | 2940000 ± 241000 | 9.15 ± 0.73 | |
Binding data determined by surface plasmon resonance (SPR) as described in Experimental Procedures.
S-Adenosylhomocysteine;
Gram-negative bacterial TrmD;
Mycobacterial TrmD.
Gram-positive bacterial TrmD.
AZ51 Induces a Unique Conformational Change of a Wall-Loop Residue in PaTrmD
To gain insight into the molecular mechanism used by AZ51 for PaTrmD inhibition, we soaked a PaTrmD–SAM cocrystal with AZ51 and determined the structure at a resolution of 2.21 Å using a crystal form having a complete PaTrmD dimer in the asymmetric unit (Table 3). The electron density of AZ51 is clearly visible at one active site of the PaTrmD, whereas the copurifying SAM cofactor still occupies the other active site (Figure 2B). The observation that cocrystal soaking with the competitive inhibitor led to the displacement of SAM at only one of the two active sites suggests that either the binding affinity to SAM or the rigidity of binding sites is different for the two active sites within a TrmD biological dimer. However, as the difference in AZ51 binding of two active sites are found in the protein crystal by the soaking method where the structural dynamics of the crystal form are restricted, we cannot exclude the possibility that the difference in AZ51 binding is simply due to the nature of the method. Similar to SAM binding (Supporting Information, Figure S2), inhibitor AZ51 is mainly stabilized by three active-site loops in PaTrmD (named the “cover loop”, “bottom loop”, and “wall loop”) (Figure 2B): the thienopyrimidinone ring is tightly bound in the adenine pocket, is hydrogen-bonded to residues Ile138, Tyr141, and Leu143, and also forms stacking interactions with residues Pro94 and Leu143. The phenyl ring is locked in place by stacking interactions with residues Pro94 and Tyr120, while the piperidine ring forms stacking interactions with residue Tyr120 and is potentially hydrogen-bonded to residue Asp182 by its protonated nitrogen. The terminal amine (i.e., the −NH2 hydrogen bond donor incorporated onto the piperidine ring) interacts with the carboxylate of Glu121, a highly conserved residue among TrmD isozymes (Supporting Information, Figure S3).
Table 3. Data Collection and Refinement Statistics for TrmD Atructuresa.
| PaTrmD–AZ51 | PaTrmD–11 | PaTrmD–15 | ApoMtb–TrmD | MtbTrmD–SAH | MtbTrmD–AZ51 | MtbTrmD–12 | MtbTrmD–15 | |
|---|---|---|---|---|---|---|---|---|
| PDB ID | 6JOE | 5ZHM | 5ZHN | 5ZHI | 5ZHJ | 6JOF | 5ZHK | 5ZHL |
| data collection | ||||||||
| space group | P3221 | P3221 | P3221 | P21 | C121 | C121 | C121 | C121 |
| cell dimensions | ||||||||
| a, b, c (Å) | 85.50, 85.50, 147.54 | 84.50, 84.50, 147.27 | 84.67, 84.67, 148.56 | 44.17, 113.07, 44.21 | 72.96, 50.76, 53.31 | 73.07, 51.38, 57.95 | 73.09, 50.80, 58.081 | 73.69, 50.23, 57.94 |
| α, β, γ (deg) | 90.00, 90.00, 120.00 | 90.00, 90.00, 120.00 | 90.00, 90.00, 120.00 | 90.00, 110.75, 90.00 | 90.00, 95.10, 90.00 | 90.00, 90.18, 90.00 | 90.00, 90.56, 90.00 | 90.00, 90.95, 90.00 |
| solvent content (%) | 52 | 51 | 52 | 38 | 35 | 41 | 40 | 40 |
| resolution (Å) | 42.75–2.21 | 49.09–2.76 | 42.33–2.65 | 41.30–2.20 | 53.10–1.75 | 42.03–2.20 | 58.08–2.30 | 41.50–2.25 |
| no. of reflns | 267240 (21374) | 167650 (24471) | 201645 (27032) | 72052 (5534) | 55961 (8132) | 44682 (3655) | 23534 (3432) | 32518 (4380) |
| no. of unique reflns | 32130 (2724) | 16240 (2335) | 18516 (2392) | 19717 (1588) | 18952 (2704) | 10831 (917) | 8972 (1287) | 9936 (1397) |
| Wilson B-factor (Å2) | 56.2 | 70.2 | 82.0 | 16.8 | 16.2 | 20.5 | 19.7 | 24.3 |
| Rmerge (%) | 5.1 (84.3) | 8.8 (99.3) | 5.7 (131.3) | 5.5 (22.6) | 9.0 (40.8) | 4.8 (17.0) | 11.5 (43.8) | 5.8 (23.8) |
| CC(1/2) | 0.999 (0.874) | 0.998 (0.917) | 0.999 (0.865) | 0.997 (0.929) | 0.994 (0.721) | 0.998 (0.945) | 0.989 (0.733) | 0.997 (0.942) |
| I/σI | 21.1 (3.0) | 15.2 (2.7) | 22.0 (2.5) | 16.1 (5.2) | 6.9 (2.5) | 18.9 (6.7) | 6.6 (2.3) | 13.9 (5.8) |
| completeness (%) | 99.8 (98.8) | 99.9 (100.0) | 100.0 (100.0) | 96.1 (91.8) | 96.7 (94.6) | 97.9 (94.8) | 94.2 (92.8) | 97.8 (95.3) |
| multiplicity | 8.3 (7.8) | 10.3 (10.5) | 11.3 (10.9) | 3.7 (3.5) | 3. 0 (3.0) | 4.1 (4.0) | 2.6 (2.7) | 3.3 (3.1) |
| Refinement | ||||||||
| monomers in ASU | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 |
| no. reflns | 31929 | 16202 | 18474 | 18708 | 18930 | 10818 | 8972 | 9900 |
| Rwork/Rfree | 0.2090/0.2455 | 0.2150/0.2751 | 0.2079/0.2543 | 0.1596/0.2001 | 0.1768/0.2021 | 0.2182/0.2619 | 0.1733/0.2265 | 0.1583/0.2110 |
| no. of non-hydrogen atoms | ||||||||
| protein | 3826 | 3786 | 3789 | 3253 | 1634 | 1610 | 1629 | 1633 |
| water | 191 | 123 | 50 | 87 | 145 | 81 | 122 | 159 |
| ligands | 60 | 53 | 60 | NA | 32 | 28 | 30 | 30 |
| average B-factor (Å2) | ||||||||
| protein | 69.20 | 85.03 | 102.90 | 19.95 | 19.79 | 27.09 | 27.85 | 29.05 |
| water | 76.24 | 73.95 | 92.18 | 18.25 | 39.32 | 32.23 | 38.95 | 42.66 |
| ligands | 62.95 | 72.81 | 98.33 | NA | 25.31 | 31.11 | 46.36 | 39.25 |
| inhibitor | 62.82 | 70.75 | 98.33 | NA | 25.32 | 31.11 | 46.36 | 39.25 |
| RMS deviations | ||||||||
| bond lengths (Å) | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| bond angles (deg) | 1.01 | 1.11 | 1.08 | 1.30 | 0.99 | 1.04 | 1.07 | 1.07 |
| Ramachandran plots | ||||||||
| favored (%) | 97.1 | 96.0 | 97.1 | 98.6 | 99.1 | 97.5 | 99.0 | 98.1 |
| allowed (%) | 99.8 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| no. of outliers | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Values in parentheses are for the highest resolution shell; NA, not applicable.
In contrast to the SAM-bound structure, AZ51 binding induces conformational changes of the wall loop, whereupon the side chain of aromatic ring of Tyr120 flips about 180° and forms stacking interactions with both the phenyl and piperidine rings of AZ51 (Figure 2C). This feature appears unique to AZ51 and PaTrmD and was not observed in other reported TrmD inhibitor structures.15,17 In addition, because residue Tyr120 is strictly conserved among TrmD orthologues (Supporting Information, Figure S3), AZ51 can presumably also trigger a comparable side chain flip in other TrmD isozymes. The conformational rearrangement of protein upon ligand binding generally could link to complex binding kinetics.21−23 Compared to SAH binding to PaTrmD, SPR reveals a faster (∼2-fold) on-rate (association rate) and slower (∼30-fold) off-rate (dissociation rate) for AZ51 binding (Table 2).
To further understand the biophysical parameters governing AZ51 binding, the thermal stability of PaTrmD in the presence of AZ51 or the SAM byproduct S-adenosyl-l-homocysteine (SAH) were determined (Figure 3D; Supporting Information, Figure S4). Both active-site ligands decrease the thermal vibration (ΔTm = 4–29 °C), suggesting that the stability of PaTrmD is enhanced by ligand binding. In particular, AZ51 is able to significantly increase the Tm to 79 °C, which is 17 °C higher than thermostabilization conferred by SAH. Therefore, formation of a highly stable complex of PaTrmD with AZ51 binding possibly causes a conformational change at the active site, which locks the structure in a very stable state. We also showed that AZ51 interacts with many other bacterial TrmDs to increase the enzymes’ thermal stability (Supporting Information, Figure S5). This further confirms that AZ51 has broad-spectrum TrmD-binding activity.
Figure 3.

Interactions of compound 15 with TrmDs. (A,B) The polypeptide chains are shown as cartoons, whereas the residues involved in inhibitor binding are shown as sticks. Ligands are shown as sticks with 2Fo – Fc electron density maps (gray) contoured at 1σ. (A) Detailed interactions between 15 and PaTrmD (chain A) (PDB 5ZHN). (B) The interactions between 15 and MtbTrmD (PDB 5ZHL). (C) In PaTrmD, compound 15 induces a conformational change of wall-loop residues, which could block substrate tRNA (G37) binding. SAM-bound PaTrmD and tRNA-bound H. influenzae TrmD (PDB 4YVI) were superimposed onto 15-bound PaTrmD, respectively. (D) The thermal shift of PaTrmD and MtbTrmD in the presence of SAH (2 mM), AZ51 (0.1 mM), or 15 (0.1 mM) was analyzed. Error bars represent mean ± SD. (E) A superposition of PaTrmD–15 (subunit 1 and 2), MtbTrmD–15, and HiTrmD–tRNA (PDB 4YVI) showing the close contacts between 15 and interdomain linker. The interdomain linker of HiTrmD (residues 157–174), and tRNA are shown as cartons, whereas guanosine is shown as stick. (F) Dose-dependent inhibition of PaTrmD at varying tRNA titrations. The IC50 values determined for compound 15 at tRNA concentrations of 1.5 μM (●), 4.5 μM (□), and 15 μM (▲), respectively, are 0.02 ± 0.01, 0.06 ± 0.01, and 0.26 ± 0.02 μM, respectively. Each data point represents an average of duplicate experiments with error bars indicated as SD.
The protein structural changes induced by AZ51 binding to PaTrmD also raised questions about the effect of the changes on tRNA binding. To test this idea, we modeled the tRNA substrate into the AZ51-bound structure by superimposing the structures of PaTrmD–AZ51 (this work) and HiTrmD bound to tRNA (PDB 4YVI) (Figure 2C). Interestingly, we found that the flipped side chain of Tyr120 overlaps with G37 of the tRNA substrate, indicating that the AZ51-bound structure becomes unsuitable for tRNA binding. This is consistent with the results of the tRNA competition assay (Figure 2A). Taken together, we conclude that, in the PaTrmD structure, AZ51 not only mimics substrate SAM and competitively occupies the SAM binding pocket but also induces conformational changes in the active-site wall loop that probably could prevent the binding of tRNA. This hypothesis of “dual functions” of AZ51 may account for the high potency of this molecule in enzyme inhibitory activity for PaTrmD. Given the striking differences in AZ51 binding energetics with PaTrmD and MtbTrmD, these results with PaTrmD raised questions about AZ51-induced structural changes in MtbTrmD.
AZ51 Binds to MtbTrmD without Inducing Conformational Changes
As noted earlier, compared with its binding activity with Gram-negative TrmDs, AZ51 binds with much lower affinity to mycobacterial and Gram-positive15 bacterial TrmDs (Table 2). In addition, AZ51 increases the Tm of PaTrmD by 29 °C, while the corresponding ΔTm values of mycobacterial and Gram-positive bacterial TrmDs range from 1 to 13 °C (Figure 3D; Supporting Information, Figures S4, S5). These observations suggest that AZ51 may use a different mechanism for binding to mycobacterial and Gram-positive bacterial TrmDs in spite of the fact that the overall protein sequence similarity is high among bacterial TrmDs (Supporting Information, Figure S3, Table S1). To gain insight into the inhibitory mechanism on MtbTrmD, we crystallized its free form and determined the structure at 2.20 Å resolution. We then optimized the crystal soaking conditions and successfully soaked SAH and AZ51 into the MtbTrmD crystals, which allowed determination of the structures at high resolution (Table 3).
As shown in Supporting Information, Figure S2D, MtbTrmD forms a biological dimer with the active site located at the dimer interface, as with PaTrmD. Indeed, MtbTrmD and PaTrmD are highly similar in structure with RMS deviations of 1.3 Å for all C-α atoms after superposition. The electron density of SAH is clearly identified at the active site in MtbTrmD–SAH structure, and SAH is positioned by three active-site loops (Supporting Information, Figure S2D). The adenine base is stacked between Pro85 and Leu138 and forms hydrogen bonds with main chains atoms of Ile133, Gly134, Tyr136, and Leu138. The 2′ hydroxyl group of the ribose makes a hydrogen bond to the carbonyl oxygen of Pro83. The amino acid tail interacts with side chain of Thr84 and carbonyl oxygen of Glu112 by forming hydrogen bonds. A protein sequence alignment of multiple bacterial TrmDs shows that the residues involved in adenine base interactions are strictly conserved but more diverse for those interacting with the ribose and tail moiety of SAH (Supporting Information, Figure S3). Compared to MtbTrmD, SAM in PaTrmD has four additional hydrogen bonds with side chains of residues Tyr91, Gln95, Asp182, and His185, where the corresponding residues in MtbTrmD (Val82, Ala86, Glu180, and Ser183) do not interact with SAH (Supporting Information, Figure S2E). Thus, these additional interactions observed at active site in PaTrmD could account for the higher binding affinity for SAM and SAH, which is consistent with SPR data (Table 2).
The crystal structure of MtbTrmD–AZ51 complex was determined at a resolution of 2.20 Å (Table 3). Similar to the AZ51 binding mode in PaTrmD (Figure 2B), AZ51 is positioned at the SAH binding site by interacting with three active-site loops of MtbTrmD (Figure 2D). Residues involved in AZ51 interactions are strictly conserved in other bacterial TrmDs including PaTrmD (Supporting Information, Figure S3). However, the binding of AZ51 does not induce the side chain flip of Tyr111 in MtbTrmD, while the corresponding residue Tyr120 in PaTrmD turned 180° to form stacking interactions with the phenyl ring of the inhibitor (Figure 2E), suggesting a more rigid active site in MtbTrmD compared to PaTrmD. In the absence of the conformational changes at the wall loop, the piperidine ring of AZ51 in MtbTrmD is positioned differently than in PaTrmD, resulting in the loss of interactions between the terminal amine and the side chain of Glu112. Even though the binding mode of the thienopyrimidinone ring is highly similar in MtbTrmD and PaTrmD, the conformational changes induced by AZ51 in PaTrmD lead to additional interactions (hydrophobic and hydrogen-bonding) and eventually higher binding affinity. Thus, the structural flexibility of the wall loop in Gram-negative bacterial TrmDs appears to play a unique role in the binding of AZ51. Exploiting this unique mechanism of AZ51 inhibition of TrmD, we next designed and synthesized a series of derivatives with the goal of improving enzyme inhibition and antibacterial activity.
Design and Synthesis of Thienopyrimidinone Derivatives
The PaTrmD crystal structure was used to design a series of inhibitors based on the thienopyrimidinone molecular scaffold shown in Figure 1. Inspired by the unique mechanism leading to a potential tRNA competition instead of exclusively designing substituents that make contacts with residues Glu121, Asp182, and Tyr120 as found with AZ51, our idea driving the inhibitor design process was that alkyl and aryl substitutions at R1 and R2 would allow full access to the free space outside the SAM binding site, with the hydrophobicity of the substituent and the charge distribution around the centered ammonium ion enhancing enzyme inhibitory activity and thus antibacterial potency.
On the basis of these design principles, we synthesized a series of thienopyrimidinone analogues. The key aldehyde 8 was synthesized using a reaction sequence and procedures similar to those reported by Hill et al.:15 reaction of methyl 2-oxopropanoate (1) and ethyl 2-cyanoacetate (2) in the presence of sulfur in DMF afforded the thiophene amino diester (3), which upon condensation with formamidine acetate in ethanol under reflux yielded thienopyrimidinone (4). Trans amidation of 4 with benzylamine derivative (7), which was synthesized from 4-formylbenzonitrile (5) followed by treatment with trifluoracetic acid, afforded the key aldehyde 8 (Scheme 1). We then modified the procedure of Hill et al.15 for reductive amination of aldehyde 8 with various amines. We found that the reductive amination with titanium isopropoxide (Ti(OiPr)4), sodium triacetoxyborohydride (NaBH(OAc)3) was sluggish and needed long reaction times. Thus, we optimized the same transformation using sodium cyanoborohydride (NaCNBH3) in methanol with catalytic amount of acetic acid at ambient temperature for 18 h, which afforded compounds 9–26. Various Boc-protected diamines were coupled using the above-mentioned procedure to afford compounds 27–30, which, on treating with 4 N HCl in dioxane, afforded compounds 31–34.
Scheme 1. Synthesis of TrmD Inhibitors Based on the Thienopyrimidinone Scaffold; Synthesis of Compounds 9–26 and 31–34 Using Reductive Amination Method.

Conditions: (a) sulfur, triethylamine DMF, 50 °C; (b) formamidine acetate, ethanol, reflux; (c) ethylene glycol, pyridinium-p-toluenesulfonate, Dean–Stark; (d) LiAlH4, THF, 0 °C to room temparature; (e) Et3N, EtOH, reflux; TFA, DCM, 0 °C to ambient temperature; (f) amine, NaCNBH3, cat. acetic acid, MeOH; (g) 4 N HCl in dioxane.
Biochemical Structure–Activity Relationships (SAR) for TrmD Inhibitors
To explore the relationship between inhibitor structure and the potency of TrmD inhibition, we quantified the concentrations of compounds producing 50% inhibition of TrmD methylation of a tRNA substrate. Compared with AZ51, inhibitory activity was significantly reduced (∼6–16-fold) when the R1 and R2 positions are present in a cyclic structure (without the terminal amine) shown in compounds 9 and 10 (Table 1). This observation is consistent with our structural data (explored next), where the terminal amine group as a hydrogen bond donor forms the interaction with PaTrmD. Furthermore, compounds 14, 15, 16, and 17 explore the optimal bulk structure of a single substitution, with benzyl (14), cyclohexyl (16), and adamantyl (17) groups all showing submicromolar activity. However, the n-octyl substitution in 15 confers 5–30-fold more potent inhibitory activity (IC50 ∼ 24 nM), which suggests that the long straight-chain alkyl group could play a role in PaTrmD binding. This observation prompted us to quantify the effect of alkyl chain length on enzyme activity. Accordingly, we synthesized compounds 21–24 with 4-, 6-, 10-, and 12-methylene group alkyl chain lengths. This series of analogues of 15 shows a clear optimal chain length of 8–10 carbons for the most potent TrmD inhibitory activity (Supporting Information, Figure S6). Substitutions at the R2 position, however, generally had a detrimental effect on TrmD inhibitory activity (11–13, 18, 19, and 20) when the substituent at R1 position is also bulky, which suggests steric limitations beyond the SAM binding pocket. For example, when −H at R2 position of compound 15 is substituted with −Et where compound 19 is generated, the enzyme inhibitory activity is significantly decreased from 24 to 370 nM (Table 1). Take compound 20 as another example, when substituents at both R1 and R2 positions are bulk structures (n-octyl and benzyl groups), its enzyme inhibitory activity is nearly abolished.
Thienopyrimidinone Inhibitors Induce the Same Conformational Change at Active Site as AZ51 in PaTrmD
To further investigate the inhibitory mechanism of the thienopyrimidinone compounds, we solved the crystal structures of PaTrmD in complex with 11 and 15 (Table 3). The interactions of PaTrmD with 15 are illustrated in Figure 3A, and with 11 in Figure 4A. Similar to the AZ51 binding mode (Figure 2A), these thienopyrimidinone inhibitors are mainly stabilized by the three active-site loops (cover, bottom, and wall loops). At the cover loop, the thienopyrimidinone ring remains tightly bound in the adenine pocket, with hydrogen bonds to residues Ile138, Tyr141, and Leu143, and stacking interactions with residues Pro94 and Leu143. The phenyl ring of 11 and 15 is locked in place by stacking interactions with residues Pro94 and Tyr120 in the bottom loop. Electron densities for the thienopyrimidinone and phenyl rings are clearly visible in the structures of all thienopyrimidinone inhibitors. However, the electron densities for the flexible tails (substituents) of the thienopyrimidinone inhibitors are poorly visible and may possess multiple conformations. Similar to the AZ51 crystal structure, the binding of thienopyrimidinone inhibitors to PaTrmD induces a conformational change of the wall loop, in which the side chain of residue Tyr120 flips ∼180° and forms stacking interactions with the phenyl ring (Figure 3A; Figure 4A). Compound 15 also highly thermostabilizes PaTrmD (ΔTm = 12 °C) (Figure 3D; Supporting Information, Figure S4) and other bacterial TrmDs (Supporting Information, Figure S5). It is noteworthy that the correlation is not strong between enzyme inhibition IC50 values (Table 1) and thermal stability Tm (Supporting Information, Figure S4), indicating that the substituents play additional roles in enzyme inhibition other than forming a thermally stable complex with the protein, such as tRNA competition. We will elaborate this point using compound 15 as an example next.
Figure 4.
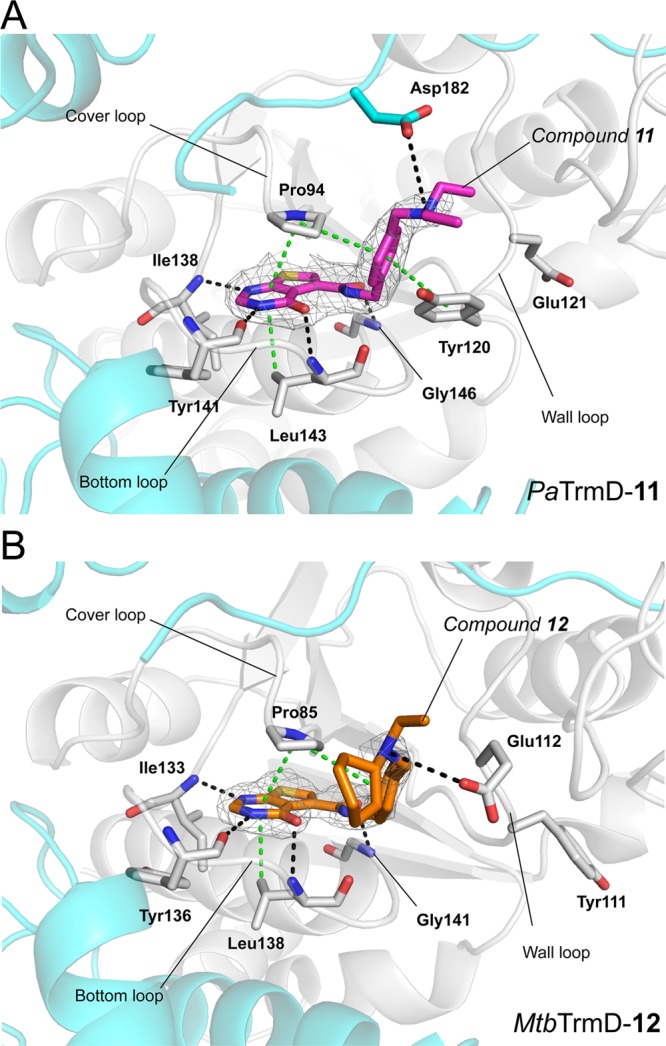
Crystal structures of compound 11 bound to PaTrmD (A; PDB 5ZHM) and compound 12 bound to MtbTrmD (B; PDB 5ZHK), respectively. The polypeptide chains are shown as cartoons, whereas the residues involved in inhibitor binding are shown as sticks. Ligands are shown as sticks with 2Fo – Fc electron density maps (gray) contoured at 1.0σ. Potential interactions involved in inhibitor binding are indicated by broken lines (black, hydrogen bonds; green, hydrophobic contacts including stacking interactions).
To investigate the potential role of thienopyrimidinone inhibitors in tRNA binding, we then modeled the tRNA substrate with the PaTrmD–15 structure by superimposing the HiTrmD–tRNA (PDB 4YVI) structure (Figure 3C). Similar to the mechanism inferred for AZ51, we found that the flipped side chain of Tyr120 also overlapped with G37 of tRNA substrate, indicating that the 15-bound structure was unavailable for tRNA binding. The role of the flexible alkyl chain of 15 was explored below.
Crystal Structures of MtbTrmD in Complex with Thienopyrimidinone Inhibitors
To further define the structural differences between PaTrmD and MtbTrmD, we soaked 12 (Figure 4B) and 15 (Figure 3B) into MtbTrmD crystals and solved the structures at high resolution (Table 3; 2.30 and 2.25 Å, respectively). Similar to the AZ51 binding mode in MtbTrmD, 12 (Figure 4B) and 15 (Figure 3B), are positioned at the SAH binding site by interacting with the three active-site loops of MtbTrmD, but the inhibitors did not induce the side chain flip of Tyr111 (Figure 3A). Inhibitor 15 shows over 100-fold lower affinity for MtbTrmD compared to the PaTrmD binding affinity (Table 2), further indicating that the flip of the Tyr side chain could play an important role in binding to the TrmD protein. Furthermore, similar to the PaTrmD–15 structure, the location of the substituent of 15 is defined by relatively weak electron density in MtbTrmD. Taken together, we found that the thienopyrimidinone-based inhibitors adopt similar binding modes in both MtbTrmD and PaTrmD by interacting with conserved residues, except for the ability to induce a conformational change in the active-site wall loop.
Thienopyrimidinone Substituent Flexibility Is Critical for Potent TrmD Inhibition
The structure–activity studies noted earlier identified optimal R1 and R2 substituents on the thienopyrimidinone scaffold, with a single alkyl chain of 8–10 carbons producing 8–50-fold stronger affinity to TrmDs compared with AZ51 (Table 2). We next explored the mechanistic basis for this potency. In crystal soaking experiments with PaTrmD, most thienopyrimidinone compounds replaced SAM at only one of the two active sites in the dimer, presumably due to the stronger affinity of SAM for binding to the second active site and/or different dynamics of two active sites. Unexpectedly, we were able to identify 15 at both active sites of the dimer (Supporting Information. Figure S7). We assume that the successful replacement of SAM at both active sites resulted from a higher affinity of 15 relative to other compounds. The electron densities of the alkyl substituent in 15 at the two active sites were poorly resolved, indicating flexibility and multiple orientations. Interestingly, the unique side chain flip of residue Tyr120 induced by 15 only occurs at one of the active sites, which indicates that, in the crystal soaking condition, the two active sites have different structural environments that require conformational flexibility in 15 for binding. To further investigate the role of the alkyl chain of 15 in enzyme inhibition, we superimposed the published structure of tRNA-bound HiTrmD20 onto the PaTrmD–15 structure and found a potentially close contact between the flexible 15-alkyl chain and the interdomain linker, which was also observed in MtbTrmD (Figure 3E). Moreover, the hydrophobicity of the alkyl chain probably favors the wall-loop conformational change leading to the formation of a hydrophobic core and facilitating ligand binding. In Table 2, 15 shows much higher binding on-rate (kon) to PaTrmD compared with AZ51 which leads to ∼50-fold higher binding affinity (KD). This observation could be due to the role of the hydrophobic alkyl chain in forming the hydrophobic core at the active site. To test this hypothesis, we replaced the long alkyl group with diethylene glycol to reduce the hydrophobicity of the substituent but still retaining a certain level of structural flexibility (compounds 25 and 26 in Table 1). Clearly, the ethylene glycol substituents reduce the enzyme inhibitory activity by 4–8-fold, compared with the alkyl chain, further suggesting that hydrophobicity or lipophilicity of the substituent facilitate TrmD inhibitory activity.
Taken together, we show that 15 occupies the SAM binding site and induces a Tyr120 side-chain flip. From the crystal structures, we hypothesize that the flipped side chain and the flexible alkyl chain could interfere with tRNA binding. To further investigate this hypothesis, we have performed a tRNA competition assay, in which the IC50 value was observed to increase by 13-fold from 0.02 to 0.26 μM when the tRNA concentration was increased (Figure 3F).
O6-Substituted Thienopyrimidinones
To extend the biochemical SAR study, we assessed the effect of O6-subtituents on the TrmD inhibitory activity of thienopyrimidinones (Figure 1B). We synthesized a series of O6-substituted analogues with alkyl substituents of varying lengths by treating 37, 38, and 10 with alkyl/arylalkyl halides in the presence of K2CO3 in DMF to afford O6-alkylated analogues 39–47 (Scheme 2). PaTrmD IC50 values were then determined for the compounds using the in vitro assay described earlier (Table 4). The result shows that these compounds do not inhibit PaTrmD, with the exception of 47, which is likely to undergo facile degradation in which the aryl substituent is released to form 15. The basis for the lack of activity of the O6-substituted thienopyrimidinone derivatives likely results from the limited space available to accommodate the O6-group in the adenine-base binding site (Figure 2B). In addition to lower enzyme inhibitory activity compared to the other thienopyrimidinone, the O6-substituted analogues also only show a minor impact on protein thermal stabilization (ΔTm = 1–2 °C), suggesting weak interactions with PaTrmD (Supporting Information, Figure S4).
Scheme 2. Synthesis of O6-Substituted Thienopyrimidinones.

Conditions: (a) Et3N, EtOH, reflux; (b) alkyl bromide, K2CO3, DMF; (c) 4 N HCl in dioxane.
Table 4. Structures and IC50 TrmD Inhibition Values for O6-Subsituted Thienopyrimidinone Derivatives.


NA = not active up to 50 μM.
Thienopyrimidinone Analogues with Long Alkyl Substituents Possess Antibacterial Activity
On the basis of the biochemical activity of the thienopyrimidinone analogues as TrmD inhibitors, we next quantified the antibacterial activity of the compounds. The first analysis involved a single-dose (100 μM) growth inhibition assay against a collection of Gram-positive and Gram-negative bacteria. Of the compounds listed in Table 1, only 15, 23, and 24 showed growth inhibitory activity at 100 μM against Gram-positive bacteria and mycobacteria. We then used these compounds to determine minimal inhibitory (MIC) (Table 5) and minimal bactericidal (MBC) concentrations (Table 5). The discovery of the activity of the 15 series analogues against Gram-positive bacteria and mycobacteria is in agreement with the broad-spectrum binding affinity of 15 to three TrmDs (Table 2; Supporting Information, Figure S5). It is worth noting that 15 and 24 also exhibit antibacterial activity against the Gram-negative pathogens Acinetobacter baumannii and Salmonella enteritidis with high MIC50/MIC90 values. Thus, 15, 23, and 24 show signs of broad-spectrum antibacterial activity, possibly due to their multiple TrmD targets. In an attempt to extend and improve the antibacterial activity to Gram-negative bacteria, we either added primary amines24 to 15 and its series analogues (Scheme 1), or conjugated with siderophores25,26 (Supporting Information, Scheme S1), where we synthesized compounds 31–34, 53, and 57, respectively (Table 1). These compounds retained submicromolar TrmD inhibitory activity, although they did not show activity against Gram-negative bacteria and even lost the activity to Gram-positive bacteria (data not shown).
Table 5. Antibacterial Activities (μM) for Selected Thienopyrimidinone Analogues.
|
15 |
23 |
24 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pathogen | MIC50a | MIC90a | MBC99.9b | MIC50 | MIC90 | MBC99.9 | MIC50 | MIC90 | MBC99.9 | |
| Gram-negative | P. aeruginosa | NAc | NA | NA | NA | NA | NA | NA | NA | NA |
| A. baumannii | 62 | 95 | >100 | >100 | >100 | >100 | 75 | 99 | >100 | |
| K. pneumoniae | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
| S. enteritidis | NA | NA | NA | NA | NA | NA | >100 | >100 | >100 | |
| E. coli | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
| Gram-positive | S. aureus | 31 | 36 | 54 | 6 | 8.5 | 34 | 5 | 8.5 | 34 |
| E. faecalis | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
| S. pneumoniae | 60 | 73 | >100 | 34 | 56 | >100 | 62 | 74 | >100 | |
| mycobacteria | M. smegmatis | 58 | 72 | >100 | 13 | 18 | 36 | 17 | 23 | 69 |
| M. tuberculosis | 78 | >100d | >100 | 22 | 43 | 75 | 44 | 88 | nde | |
MIC50 and MIC90 represent hit concentrations inhibiting growth by 50% and 90%, respectively.
MBC99.9 represents hit concentrations kill bacteria by 99.9%.
NA = no bacterial activity.
Denotes detectable growth inhibition or bactericidal activity but less than 50% for MIC50, less than 90% for MIC90, and less than 99.9% for MBC99.9.
nd = not determined.
As with the TrmD inhibitors developed by Hill et al.,15 there is little relationship between the potency of TrmD inhibition by the new thienopyrimidinone compounds developed here and their antibacterial activity. The most likely explanation is poor cell permeability, although it is possible that inhibition of the otherwise essential TrmD by the compounds is incomplete in the cellular environment. That bacterial uptake is responsible for the observed antibacterial activity is supported by the observation of stronger activity against two Gram-positive pathogens (S. aureus and S. pneumoniae) compared to the single Gram-negative pathogen, A. baumannii, with its additional outer cell membrane (Table 5). However, mycobacteria have a complex mycolic acid-rich cell wall that presents a drug permeability problem similar to Gram-negative bacteria, yet M. smegmatis and M. tuberculosis show sensitivity to TrmD inhibitors similar to Gram-positive S. aureus (Table 5). This idiosyncratic activity could result from mechanisms of antibacterial activity other than TrmD inhibition, drug efflux pumps, or compound degradation. The strong SAR for TrmD inhibition by thienopyrimidinone compounds established here provides a foundation for pursuing antibacterial SAR.
Hemolytic Activity of the Thienopyrimidinone Compounds
To further explore the behavior of the thienopyrimidinone analogues, we assessed the ability of the compounds to rupture red blood cells as an index of membrane disrupting potential. The hemolytic activity of all compounds is shown in Supporting Information, Table S2. In general, most of the tested compounds show no or weak hemolytic activity at the highest tested concentration (100 μM).
Discussion and Conclusions
Elaborating on a thienopyrimidinone scaffold, we prepared and analyzed a series of TrmD inhibitors, which revealed a novel SAM-competitive, active site Tyr-flipping inhibition mechanism that distinguished Gram-negative TrmDs from Gram-positive and mycobacterial counterparts. Several of these compounds showed nanomolar TrmD inhibition, tRNA-competitive binding, and micromolar antimicrobial activity against Gram-positive bacteria and, in some instances, Gram-negatives and mycobacteria.
Experimental Section
Protein Expression and Purification
Production of PaTrmD protein was described previously.14 Briefly, a truncated fragment (spanning residues Leu5-Asp250) of PaTrmD was cloned into the vector pNIC28-Bsa4 and transformed into chemically competent Escherichia. coli BL21 (DE3) Rosetta T1R cells. For M. tuberculosis TrmD, S. aureus TrmD, E. faecalis TrmD, and S. pneumoniae TrmD, the full-length genes were inserted into vector pYUB28b-cHIS6 and transformed into chemically competent BL21 (DE3). The following protein expression and purification procedures were applied to all TrmD proteins. E. coli cells harboring a plasmid were cultivated in Luria–Bertani (LB) medium at 37 °C to OD600 0.6–0.8. The proteins were then overexpressed by the addition of 0.5 mM IPTG at 16 °C for 24 h. Cells were pelleted and stored at −80 °C.
All enzyme purification was performed on ÄKTA (GE Healthcare) chromatography system at 4 °C. The thawed pellets from 1 L cell cultures were resuspended in 30 mL of lysis buffer (20 mM Na-HEPES, pH 7.5, 0.3 M NaCl, 5% glycerol, 0.5 mM TCEP), sonicated in the presence of one tablet of EDTA-free protease inhibitors (Roche), and the lysates cleared by centrifugation at 22000 rpm for 45 min in a JA-25.50 rotor. The proteins were purified by immobilized metal affinity chromatography (IMAC) using HisTrap HP IMAC column (GE Healthcare) and by size exclusion on a HiLoad Superdex 200 16/60 column (GE Healthcare). Purified proteins were concentrated to 20 mg/mL by ultrafiltration in 20 mM Na-HEPES, pH 7.5, 0.3 M NaCl, 10% (v/v) glycerol, 0.5 mM TCEP, and were stored at −80 °C.
tRNA in Vitro Synthesis and Purification
The preparation of tRNA substrates has been reported previously.14 A PaTrmD substrate, tRNALeu(CAG), was synthesized using in vitro transcription by MEGAshortscrip T7 Transcription Kit (cat. no. AM1354, Thermo Scientific) according to the manufacturer’s instructions. DNA template and primers (Supporting Information, Table S3) were obtained from Integrated DNA Technologies, Inc. (IDT). The transcribed tRNA was desalted in RNase-free water and purified on an Agilent HPLC 1200 equipped with an Agilent Bio SEC-3 size-exclusion column (300 Å pore size, 3 μm particle size, 7.8 mm i.d.) operated at 60 °C eluted with 100 mM ammonium acetate buffer. The purity of the tRNA product was further assessed on an Agilent 2100 Bioanalyzer with a small RNA chip, and the tRNA was quantified using a Nanodrop spectrophotometer. The final product was desalted in RNase-free water, concentrated to 20 mg/mL, and frozen at −20 °C.
TrmD Inhibitor Assay
The tRNA methyltransferase activity of TrmD was measured by a bioluminescence-based assay using the MTase-Glo Assay Kit (Promega, cat. no. V7601) in a 384-well format. Generation of SAH was measured by its conversion to ADP by the MTase-Glo reagent, with MTase-Glo detection solution applied to convert ADP to ATP, which is then used by luciferase to generate a luminescent signal proportional to the coupled TrmD methyltransferase activity. Luminescence was measured using a plate-reading luminometer (BioTek Synergy 4 microplate reader) and was correlated to SAH concentration using an SAH standard curve generated according to manufacturer’s instructions. The TrmD concentration used in this assay is 8 nM. Thus, the estimated limit of IC50 detection for a substrate competitive inhibitor is about 4 nM, which is half of the molar concentration of the enzyme. The detailed TrmD activity assay and kinetic studies have been described in our recent work.14,17
The TrmD methyltransferase reaction was performed at 37 °C in 5 μL reaction mixtures containing 8 nM TrmD enzyme, 50 mM Tris-HCl buffer, pH 8.0, 80 mM NaCl, 1 mM MgCl2, 0.5 mM DTT, 0.01% (v/v) Triton X-100, 2 μM SAM, and 0.5 μM tRNALeu(CAG), and 1× MTase-Glo reagent. TrmD enzyme in 1× assay buffer (50 mM Tris-HCl buffer, pH 8.0, 80 mM NaCl, 1 mM MgCl2, 0.5 mM DTT, 0.01% v/v Triton X-100) was preincubated with an inhibitor in a serial dilution (0–500 μM) at ambient temperature for 15 min. The negative-control mix was prepared in an identical manner except that 1× assay buffer was used in place of the inhibitor solution. The reaction was initiated by adding the TrmD enzyme mixture to a final concentration of 0.25 μg/mL. The plate was sealed and incubate at 37 °C for 45 min. After reaction, the mixture was mixed with 5 μL of MTase-Glo detection solution and further incubated for 30 min at ambient temperature in the dark. The luminescent signal was read in the microplate luminometer and background subtracted from all sample and control reads before analyzing the net signal intensity in Prism GraphPad. The rate for each activity assay with a series of compound titrations was expressed as a percentage of the control assay and analyzed using nonlinear regression fit in Prism GraphPad to estimate IC50 values for each compound. The assay was done in two independent experiments performed in technical duplicates.
Surface Plasmon Resonance (SPR)
SPR analysis was performed using a BIACORE T200 instrument (GE Healthcare) equipped with a CM5-S sensor chip. The surfaces of all flow cells were activated for 7 min with a 1:1 mixture of 0.1 M NHS and 0.1 M EDC at a flow rate of 10 μL/min. Anti-His antibody was captured on all surfaces to ∼12000 RU. Surfaces were then blocked with a 7 min injection of 1 M ethanolamine, pH 8.0. The protein TrmD, at a concentration of 50 μg/mL in 50 mM Tris-HCl pH8, 150 mM NaCl, 1 mM MgCl2, 1 mM DTT, and 0.05% Tween-20, was captured on the anti-His antibody surface to a density of ∼2200 RU. Flow cell no. 1 was left blank to serve as a reference surface. Different concentrations (2-fold dilutions) of the test analyte were injected from lowest to highest concentration in the same buffer over the flow cell at 30 μL/min for a contact time of 30 s and dissociation time of 240 s. Analysis of inhibitor binding affinity to TrmD was quantified using 2-fold dilutions. Data were collected at a rate of 10 Hz, and at a temperature of 25 °C. The KD value was calculated using steady-state affinity fit.
Crystallization and Data Collection: PaTrmD
The crystal screening and optimization for PaTrmD has been described in our recent work.14 Briefly, Crystals of PaTrmD were grown by mixing equal volumes of 20 mg mL–1 protein and precipitant solution containing 0.1 M Tris-HCl, pH 8.6–8.8, 20% (v/v) MPD, 20% (w/v) PEG 1000, and 5% (w/v) PEG200 and incubating at 20 °C using vapor diffusion method (hanging-drop). The crystals were soaked with 1–5 mM inhibitor in the precipitating solution supplemented with 20% (v/v) glycerol at 20 °C for at least 4 h and rapidly frozen in liquid nitrogen. X-ray diffraction data were collected at the PXIII beamline in SLS (Villigen, Switzerland) for the PaTrmD–AZ51 crystal. For both PaTrmD–11 and PaTrmD–15 crystals, X-ray diffraction data were collected at the MX2 beamline in Australian Synchrotron.
Crystallization and Data Collection: MtbTrmD
Crystallization conditions were screened at 20 °C with protein concentration at 10 mg/mL using the vapor diffusion method and commercial crystallization screens in Intelli 96-3 wells sitting-drop plates. Three precipitant:protein ratios (1:1, 1:2, and 2:1) were tested using the mosquito crystallization robot (Art Robbins Instruments) and drop volumes of 0.2 μL. Optimized crystals of the MtbTrmD were obtained using the vapor diffusion method (sitting drops) in 24-well trays by mixing a volume of 2 μL of protein with 1 μL of a precipitant solution containing 100 mM Bis-Tris propane at pH 6.5, 20% (w/v) PEG3350, and 0.1 M ammonium acetate. Prior to data collection, crystals were soaked with 1–5 mM inhibitor (for obtaining inhibitor-bound structure) or 5 mM SAH in their respective precipitating solution supplemented with 20% (v/v) glycerol and rapidly frozen in liquid nitrogen. X-ray diffraction data were collected at the MX1 beamline in the Australian Synchrotron for apo MtbTrmD crystal, at the PXIII beamline in SLS (Villigen, Switzerland) for the MtbTrmD–SAH and MtbTrmD–12 crystals, and at the MX1 and MX2 beamlines in the Australian Synchrotron for MtbTrmD–AZ51 andMtbTrmD–15 crystals, respectively.
Structure Determination and Refinement: PaTrmD
Diffraction intensities were reduced with XDS,27 scaled, merged, and truncated with SCALA/TRUNCATE.28,29 The structures were determined by molecular replacement using Phaser,30 with the structure from PaTrmD–SAM (PDB 5WYQ) as search probe. Model for the 3D structure of PaTrmD was built iteratively at the computer graphics using COOT31 and refined using Autobuster.32 Waters and ligands were added to the model after a few runs of refinement. The geometrical parameters for inhibitors were generated using program PRODRG.33
Structure Determination and Refinement: MtbTrmD
Data reduction is the same as described above for PaTrmD. The structures were determined by molecular replacement using Phaser,30 with the MtbTrmD structure modeled from I-TASSER server34 as search probe. A model for the 3D structure of MtbTrmD was built using COOT31 and refined using Autobuster,32 except that apo MtbTrmD structure was refined by Refmac.35 The geometrical parameters for inhibitors were generated using program PRODRG.33
The quality of the structures was assessed using the MOLPROBITY server36 (molprobity.biochem.duke.edu), and figures were generated using the Pymol software.37 Data collection and structure refinement parameters are summarized in Table 3.
Thermal Stability Assay
The thermal stability analysis of PaTrmD was performed in a 96-well PCR plate (Bio-Rad) with 50 μL per reaction containing 5× SYPRO Orange dye (Invitrogen), 4 μM test protein, and the test ligand(s) at various concentrations. The assay buffer (50 mM Tris-HCl pH8, 150 mM NaCl, 1 mM MgCl2, 1 mM DTT, and 0.05% Tween-20) was added instead of the test ligand as a negative control. The temperature was increased from 25 to 95 °C in an i-Cycler iQ5 real-time PCR (Bio-Rad). The thermal stability curve and the temperature midpoint Tm for the protein-unfolding transition was analyzed using the Bio-Rad iQ5 software.
Antibacterial Activity Assay
The synthetic compounds were first screened at a fixed concentration of 100 μM against a series of bacteria strains by monitoring the bacterial growth in broth at 96-well format. Bacteria P. aeruginosa PA14 (−), A. baumannii ATCC 17961 (−), K. pneumoniae ATCC 13883 (−), S. enteritidis ATCC 13076 (−), E. coli BW25113 (−), and S. aureus ATCC 43300 (methicillin-resistant) (+) were tested in cation-adjusted Mueller–Hinton (MH) broth at 37 °C. The E. faecalis (+) ATCC 51299 strain was tested in brain heart infusion broth. S. pneumoniae ATCC 49619 (+) was tested in MH medium supplemented with 3% defibrinated sheep blood at 37 °C and 5% CO2. M. smegmatis mc2155 (+) was cultured in the complete Middlebrook 7H9 medium.4 Briefly, four single colonies of each bacterial strain were picked from a freshly streaked plate and grown overnight at 37 °C. The overnight culture was subcultured to fresh medium and further grown at 37 °C to obtain log-phase bacteria. The bacterial concentration was further adjusted to approximately 5 × 105 cells per mL of each well in the presence or absence of 100 μM of the test compound. The total culture volume of each well was 100 μL. Ampicillin and kanamycin were testing in parallel as positive controls. DMSO control and blank control were applied. The culture was allowed to grow for 24 h at 37 °C before cell density was measured by OD600 using a BioTek Synergy 4 microplate reader. The percent bacterial growth (percentage survival) was expressed as the OD600 value of the test well as a percentage of that from the untreated control wells. For S. pneumoniae, the cells were spin down and the supernatant was measured at OD450. The percentage survival was expressed as the OD450 value of the test well as a percentage of that from the untreated control wells.
Minimum inhibitory concentration (MIC) of the hits from the screening were determined by serial broth microdilution method and following the procedures described above, where cell suspensions were incubated with serial dilutions of test compound. The experiment was done in two independent experiments performed in duplicate. MIC50 and MIC90 represent the concentration of compound that inhibit 50% and 90% bacterial growth respectively as compared to drug-free controls. Experiments were performed in biological duplicates. The MBC was determined by CFU enumeration on MH agar plates (except BHI agar plates for E. faecalis and TSA agar plates with 5% sheep blood for S. pneumoniae) after exposure to a given concentration of test compound. Bacterial cultures were grown to mid log phase, adjusted to a final cell density of 5 × 105 CFU/mL, and then treated with concentrations equivalent to 1×, 2×, and 4× MIC90 of test compound for 24 h at 37 °C. Drug-free cultures were plated at the start of the experiment to determine the bacterial load of the inoculum. After incubation for 24 h, the compound-treated cultures were plated to determine CFU. MBC99.9 are defined as the concentration of the test compound that caused at least 1000-fold reduction in CFU as compared to the untreated inoculum at time point zero.
Bacterial culture of M. tuberculosis and its MIC determination was performed in BSL-3 laboratory in NUS Singapore, and the details were described previously.38,39 Briefly, M. tuberculosis (ATCC 27294) was maintained in complete Middlebrook 7H9 medium supplemented with 0.05% (v/v) Tween 80, 0.5% (v/v) glycerol, and 10% (v/v) Middlebrook albumin-dextrose-catalase at 37 °C in PETG sterile square bottles with shaking at 80 rpm in 10 mL volumes. MICs are determined by the broth dilution method as described. Mid log phase cultures (OD600 = 0.3–0.6) are spun down by centrifugation, resuspended in fresh medium, and adjusted to an OD600 = 0.1. Cell suspension (100 μL) was added into wells containing 100 μL of 2-fold serially diluted compound (highest concentration 100 μM) in transparent flat-bottomed 96-well plates, sealed with Breath-Easy membranes. Isoniazid (Sigma-Aldrich) was used as a positive control. The inoculated plates were incubated for 7 d with shaking at 80 rpm at 37 °C. Cultures were manually resuspended, and absorbance at 600 nm was measured using a Tecan Infinite M200 Pro spectrophotometer. MIC50 and MIC90 represent, the concentration of compound that inhibit 50% and 90% bacterial growth, respectively, as compared to drug-free controls. Experiments were performed in biological duplicates. The MBC was determined by CFU enumeration on complete 7H10 agar plates after exposure to a given concentration of test compound. Bacterial (M. tuberculosis H37Rv) cultures were grown to mid log phase, adjusted to a final OD600 = 0.05, and then treated with concentrations equivalent to 1×, 2×, and 4×x MIC90 of the test compounds for 7 d at 37 °C with shaking at 80 rpm. Drug-free cultures were plated at the start of the experiment to determine the bacterial load of the inoculum. After incubation for 7 days, the compound-treated cultures were plated to determine CFU. MBC99.9 are defined as the concentration of the test compound that caused 1000-fold reduction in CFU as compared to the untreated inoculum at time point zero.
Hemolysis Assay
Fresh human red blood cells (RBCs) were washed with PBS until the supernatant was clear after centrifugation. The pellet was resuspended in PBS to an OD600 of 24 and added (100 μL) to each well of a 96-well U-bottom plate. Candidate compounds were serially diluted in PBS and added (100 μL) to the wells. Triton X-100 was used as a positive control. After one hour of incubation at 37 °C without shaking, cells were centrifuged at 1000g for 15 min. The supernatant was diluted and OD450 measured using a BioTek Synergy 4 microplate reader. The experiment was done in triplicate.
Synthetic and Analytical Chemistry
All reagents, starting materials, and solvents (including dry solvents) were obtained from commercial suppliers and used as such without further purification. Reactions were carried out in oven-dried glassware under a positive pressure of argon unless otherwise mentioned. Air-sensitive reagents and solutions were transferred via syringe or cannula and were introduced to the apparatus via rubber septa. Reactions were monitored by thin-layer chromatography (TLC) with 0.25 mm precoated silica gel plates (60 F254). Visualization was accomplished with either UV light, iodine adsorbed on silica gel, or by immersion in an ethanolic solution of phosphomolybdic acid (PMA), p-anisaldehyde, or KMnO4, followed by heating with a heat gun for ∼15 s. Column chromatography was performed on silica gel (100–200 or 230–400 mesh size). All final compounds have purity ≥95 as determined by Shimadzu Prominence UFLC using a reverse phase column [Phenomenex C18 column (5 μm, 250 mm × 4.60 mm)] and a solvent gradient of A (0.05% TFA in H2O) and solvent B (0.039% TFA in 90% ACN and 10% H2O). Method A: Solvent B, 0–100% in 25 min. Method B: Solvent B, 0–60% in 10 min to 60–100% in 10 min. Deuterated solvents for NMR spectroscopic analyses were used as received. All 1H NMR and 13C NMR spectra were obtained using a 400 MHz spectrometer. Coupling constants were measured in hertz. All chemical shifts were quoted in ppm, relative to TMS, using the residual solvent peak as a reference standard. HRMS (ESI) were recorded on a 6520 QTOF mass spectrometer equipped with a dual-spray electrospray ionization source (Agilent Technologies, Santa Clara, CA). Chemical nomenclature was generated using Chem Bio Draw Ultra 14.0.
General Procedure for Reductive Amination
Compound 8 (100 mg, 0.32 mmol) and corresponding amine (0.64 mmol) were suspended in MeOH (5.0 mL), and a catalytic amount of acetic acid was added with stirring at ambient temperature for 30 min. NaCNBH3 (0.96 mmol) was added in portions over 5 min. The reaction mixture was stirred at room temperature for 24 h. Upon completion of the reaction, the solution was diluted with 10% aqueous sodium bicarbonate (5.0 mL) and extracted with ethyl acetate (3 × 5.0 mL) or 10% MeOH in dichloromethane (4 × 10 mL). Organic layer was washed with brine (5.0 mL) and dried over anhydrous Na2SO4. The product was purified using column chromatography (5–10% methanol in chloroform) to get the desired compound as off-white solids.
General Procedure for Transamidation
To a solution of amines 35 and 36 (1.5 equiv) and ester 4 (1.0 equiv) in anhydrous ethanol was added trimethylamine (3.0 equiv), and the resulting reaction mixture was stirred under vigorous reflux for 24 h, and the reaction mixture was cooled, evaporated, and purified by column chromatography using 5–15% MeOH in dichloromethane.
For Boc-Protected Secondary Amines
Products obtained above were dissolved in dry dichloromethane and treated with 20–30 equiv of 4 N HCl in dioxane at 0 °C and stirred at ambient temperature for 4–6 h. After complete consumption of starting material, the reaction mass was evaporated and washed with diethyl ether to afford an off-white solid as their HCl salts.
General Procedure for the Synthesis of O6-Substituted Thienopyrimidinone Analogues
Under argon atmosphere alkyl or aryl alkyl bromide (2.0 equiv) was added to a suspension of compounds (37, 38, and 10, 1.0 equiv) and K2CO3 (3.0 equiv) in anhydrous DMF at 0 °C, with the resulting reaction mixture was stirred at ambient temperature for 3–4 h. The progress of the reaction was monitored by TLC and was eventually quenched with saturated aqueous NH4Cl and extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. The crude material was purified by column chromatography (silica gel 200–400 mesh) using 5–10% MeOH in dichloromethane to afford product as off-white solids. O6-Substituted products obtained above were dissolved in dry dichloromethane and treated with 20–30 equiv of 4 N HCl in dioxane at 0 °C and stirred at ambient temperature for 4–6 h after complete consumption of starting material the reaction mass was evaporated and washed with diethyl ether to afford off-white solid as their HCl salts (39–44, 47).
4-Oxo-N-(4-(piperidin-1-ylmethyl)benzyl)-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (9)
HPLC 98.63% (method B, tR 8.62 min). 1H NMR (400 MHz, DMSO-DMSO-d6) δ 11.48 (bs, 1H), 8.35 (s, 1H), 8.26 (s, 1H), 7.35–7.28 (m, 4H), 4.52 (d, J = 5.4 Hz, 2H), 3.41 (s, 2H), 2.31 (m, 4H), 1.49–1.36 (m, 4H), 1.35 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.7, 160.8, 160.7, 147.1, 138.0, 137.1, 132.9, 131.2, 129.5, 127.6, 119.7, 62.8, 54.2, 43.0, 25.8, 24.3. HRMS (ESI): m/z calculated for C20H22N4O2S [M + H]+ 383.15417, found 383.15414.
N-(4-(Morpholinomethyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (10)
HPLC 99.1% (method A, tR 12.27 min). 1H NMR (400 MHz, DMSO-d6) δ 11.41 (s, 1H), 8.32 (s, 1H), 8.20 (s, 1 H), 7.36–7.12 (m, 4H), 4.49 (d, J = 5.6 Hz, 2H), 3.55–3.48 (m, 4H), 3.38 (s, 2H), 2.29 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 167.5, 160.8, 160.3, 146.5, 137.8, 136.5, 132.5, 131.5, 129.6, 127.6, 119.6, 66.4, 62.4, 53.4, 42.9. HRMS (ESI): m/z calculated for C19H21N4O3S [M + H]+ 385.13344, found 385.13303.
N-(4-((Diethylamino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (11)
HPLC 99.29% (method A, tR 14.68 min). 1H NMR (400 MHz, CDCl3) δ 11.60 (t, J = 5.2 Hz, 1H), 8.37 (s, 1H), 8.14 (bs, 1H), 7.93 (s, 1H), 7.41–7.30 (m, 4H), 4.60 (d, J = 5.4 Hz, 2H), 3.78 (s, 2H), 2.73 (t, J = 7.2 Hz, 4H), 1.14 (d, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ ppm 167.7, 161.4, 161.1, 145.5, 138.6, 132.6, 131.3, 130.1, 127.9, 119.7, 56.3, 46.2, 43.5, 10.2. HRMS (ESI): m/z calculated for C19H23N4O2S [M + H]+ 371.15417, found 371.15325.
N-(4-((Cyclohexyl(ethyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (12)
HPLC 97.98% (method B, tR10.03 min). 1H NMR (400 MHz, DMSO-d6) δ 11.35 (t, J = 5.4 Hz, 1H), 8.38 (s, 1H), 8.28 (s, 1H), 7.39–7.24 (m, 4H), 4.52 (d, J = 5.4 Hz, 2H), 3.64 (s, 2H), 2.46 (q, merged with DMSO peak, 2H), 1.80–1.58 (m, 4H), 1.55–1.49 (m, 1H), 1.24–0.96 (m, 6H), 0.94 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.6, 160.7, 160.3, 146.6, 133.0, 131.6, 128.9, 127.6, 119.8, 59.4, 53.4, 44.1, 43.0, 28.7, 26.2, 26.0, 14.0. HRMS (ESI): m/z calculated for C23H29N4O2S [M + H]+ 425.20112, found 425.20149.
N-(4-((Benzyl(ethyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (13)
HPLC 96.90% (method B, tR 8.09 min). 1H NMR (400 MHz, DMSO-d6) δ 13.05 (s, 1H), 11.30 (t, J = 5.1 Hz, 1H), 8.39 (s, 1H), 8.28 (s, 1H), 7.39–7.21 (m, 9H), 4.53 (d, J = 3.5 Hz, 2H), 3.53 (m, 4H), 2.40–2.48 (q, merged with DMSO, 2H), 1.02 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.6, 160.6, 160.14, 146.4, 140.0, 138.7, 137.8, 132.9, 131.6, 129.0, 128.6, 127.7, 127.2, 119.8, 57.4, 57.1, 46.8, 43.0, 12.0. HRMS (ESI): m/z calculated for C24H25N4O2S [M + H]+ 433.16982, found 433.16908.
N-(4-((Benzylamino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (14)
HPLC 96.21% (method B, tR 10.72 min). 1H NMR (400 MHz, DMSO-d6) δ 11.40 (t, J = 5.24, 1H), 8.35 (s, 1H), 8.25 (s, 1H), 7.24–7.39 (m, 9H), 4.54 (d, J = 5.48 Hz, 2H), 3.82 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 167.3, 160.8, 160.4, 147.1, 139.7, 138.4, 137.5, 132.5, 130.5, 128.3, 128.2, 127.3, 126.8, 119.2, 51.9, 51.7, 42.6. HRMS (ESI): m/z calculated for C22H21N4O2S [M + H]+ 405.13852, found 405.13817.
N-(4-((Octylamino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (15)
HPLC 97.80% (method B, tR 12.36 min). 1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.21 (s, 1H), 8.20 (s, 1H), 7.36 (m, 4H), 4.52 (d, J = 5.40, 2H), 3.91 (s, 2H), 2.69 (t, J = 7.48, 2H), 1.51 (m, 2H), 1.15–1.21 (m, 10H), 0.83 (t, J = 6.24 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.5, 160.8, 160.7, 149.5, 138.9, 134.3, 132.5, 129.1, 128.9, 127.4, 118.8, 50.9, 47.3, 42.4, 31.2, 28.7, 28.5, 27.0, 26.4, 22.1, 13.9. HRMS (ESI): m/z calculated for C23H31N4O2S [M + H]+ 427.21677, found 427.21677.
N-(4-((Cyclohexylamino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (16)
HPLC 99.50% (method A, tR 14.48 min). 1H NMR (400 MHz, CD3OD) δ 11.47 (t, J = 5.52 Hz, 1H), 8.36 (s, 1H), 8.28 (s, 1H), 7.40–7.46 (m, 4H), 4.57 (d, J = 5.44 Hz, 2H), 4.11 (s, 2H), 2.05–2.08 (m, 1H), 1.74–1.76 (m, 3H), 1.58–1.61 (m, 1H), 1.20–1.26 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 167.5, 161.9, 161.1, 146.4, 139.9, 132.0, 130.9, 130.7, 129.7, 128.0, 119.5, 56.9, 42.7, 29.1, 24.8, 24.2, 8.0. HRMS (ESI): m/z calculated for C21H25N4O2S [M + H]+ 397.16982, found 397.16964.
N-(4-((((3s,5s,7s)-Adamantan-1-yl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (17)
HPLC 98.55% (method B, tR 6.13 min). 1H NMR (400 MHz, DMSO-d6) δ 11.35 (s, 1H), 8.14 (m, 2H), 7.34 (d, J = 7.40, 2H), 7.14 (d, J = 7.68, 2H), 4.29 (s, 2H), 3.92 (s, 2H), 2.11 (m, 10H), 1.64 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 167.7, 161.4, 160.2, 145.6, 139.2, 132,4, 131.2, 130.6, 130.5, 128.0, 119.7, 58.6, 43.6, 43.6, 38.5, 35.7, 29.3. HRMS (ESI): m/z calculated for C25H29N4O2S [M + H]+ 449.27269, found 449.27393.
N-(4-((Benzyl(hexyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (18)
HPLC 98.94% (method B, tR 7.83 min). 1H NMR (400 MHz, DMSO-d6) δ 11.30 (t, J = 5.4 Hz, 1H), 8.37 (s, 1H), 8.26 (s, 1H), 7.37–7.15 (m, 9H), 4.51 (d, J = 5.6 Hz, 2H), 3.48 (bs, 4H), 2.31 (t, J = 7.1 Hz, 2H), 1.43 (t, J = 7.0 Hz, 2H), 1.25–1.03 (m, 6H), 0.77 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.6, 160.6, 146.5, 137.8, 131.6, 129.0, 128.9, 128.6, 127.6, 127.2, 119.7, 58.0, 57.7, 53.0, 43.0, 31.4, 26.8, 26.6, 22.5, 14.3. HRMS (ESI): m/z calculated for C28H33N4O2S [M + H]+ 489.23242, found 489.23181.
N-(4-((Ethyl(octyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (19)
HPLC 99.33% (method B, tR 18.02 min). 1H NMR (CDCl3, 400 MHz) δ 11.24 (bs, 1H), 8.47 (s, 1H), 7.85 (s, 1H), 7.41–7.26 (m, 4H), 4.67 (d, J = 5.0 Hz, 2H), 3.67 (s, 2H), 2.60 (q, J = 6.8 Hz, 2H), 2.52 (t, J = 7.12 Hz, 2H), 1.58–1.45 (m, 2H), 1.34–1.20 (m, 10 H), 1.08 (t, J = 7.0 Hz, 3H), 0.88 (t, J = 6.04 Hz, 3H). 13C NMR (CDCl3, 100 MHz) δ 167.8, 161.4, 161.02, 145.2, 137.6, 136.6, 132.7, 131.4, 129.5, 127.7, 119.7, 57.2, 52.7, 46.8, 43.7, 31.8, 29.5, 29.2, 27.4, 26.0, 22.6, 14.1, 10.9. HRMS (ESI): m/z calculated C25H35N4O2S [M + H]+ 455.24807, found 455.24853.
N-(4-((Benzyl(octyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (20)
HPLC 96.76% (method B, tR 13.97 min). 1H NMR (400 MHz, DMSO-d6) δ 11.34 (t, J = 5.4 Hz, 1H), 8.37 (s, 1H), 8.26 (s, 1H), 7.37–7.15 (m, 9H), 4.51 (d, J = 5.6 Hz, 2H), 3.48 (bs, 4H), 2.31 (t, J = 7.1 Hz, 2H), 1.43 (t, J = 7.0 Hz, 2H), 1.25–1.03 (m, 10H), 0.77 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.6, 160.6, 146.5, 137.8, 131.6, 129.0, 128.9, 128.6, 127.6, 127.2, 58.0, 57.7, 53.0, 43.0, 31.4, 31.3, 31.2, 26.8, 26.6, 22.5, 14.3. HRMS (ESI): m/z calculated for C30H37N4O2S [M + H]+ 517.33498, found 517.33460.
N-(4-((Butylamino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (21)
HPLC 97.55% (method B tR 9.21 min). 1H NMR (400 MHz, DMSO-d6) δ 11.34 (t, J = 5.5 Hz, 1 H), 9.39 (br s, 1H), 8.38 (s, 1H), 8.28 (s, 1H), 7.70–7.48 (m, J = 8.2 Hz, 2H), 7.48–7.26 (m, J = 8.1 Hz, 2H), 4.56 (d, J = 5.4 Hz, 2H), 4.07 (t, J = 5.7 Hz, 2H), 2.90–2.75 (m, 2H), 1.63 (td, J = 7.7, 15.6 Hz, 2H), 1.43–1.20 (m, 2H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.1, 160.2, 159.6, 145.9, 139.6, 132.3, 131.2, 130.7, 130.2, 127.5, 119.4, 49.5, 46.1, 42.5, 27.3, 19.3, 13.5. HRMS (ESI): m/z calculated for C19H23N4O2S [M + H]+ 371.15417, found 371.15436.
4-Oxo-N-(4-((pentylamino)methyl)benzyl)-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (22)
HPLC 99.74% (method B, tR 9.94 min). 1H NMR (400 MHz, DMSO-d6) δ 13.25 (br s, 1H), 11.34 (t, J = 5.5 Hz, 1H), 9.34 (br s, 1H), 8.38 (s, 1H), 8.28 (s, 1H), 7.64–7.48 (m, 2H), 7.46–7.33 (m, 2H), 4.56 (d, J = 5.4 Hz, 2H), 4.07 (t, J = 5.7 Hz, 2H), 2.93–2.70 (m, 2H), 1.72–1.58 (m, 2H), 1.33–1.20 (m, 4H), 0.85 (t J = 6.95 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.1, 160.2, 159.6, 145.9, 139.6, 132.3, 131.2, 130.7, 130.2, 127.5, 119.3, 49.5, 46.3, 42.4, 28.1, 24.8, 21.6, 13.7. HRMS (ESI): m/z calculated for C20H25N4O2S [M + H]+ 385.16982, found 385.17037.
N-(4-((Decylamino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (23)
HPLC 97.03% (method B, tR 13.89 min). 1H NMR (400 MHz, DMSO-d6) δ 12.28 (s, 1H), 8.21 (s, 1H), 8.20 (s, 1H), 7.36 (m, 4H), 4.52 (d, J = 5.40, 2H), 3.91 (s, 2H), 2.69 (t, J = 7.48, 2H), 1.51 (m, 2H), 1.15–1.21 (m, 14H), 0.83 (t, J = 6.24 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.5, 160.7, 159.6, 149.5, 138.9, 134.3, 132.5, 129.1, 128.9, 127.4, 118.8, 50.9, 47.3, 42.4, 31.4, 31.3, 31.2, 28.7, 28.5, 27.0, 26.4, 22.1, 13.9. HRMS (ESI): m/z calculated for C25H35N4O2S [M + H]+ 455.31964, found 455.31954.
N-(4-((Dodecylamino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (24)
HPLC 95.24% (method A, tR 21.49 min). 1H NMR (400 MHz, DMSO-d6) δ 13.17 (br s, 1H), 11.34 (t, J = 5.7 Hz, 1H), 9.20 (s, 2H), 8.39 (s, 1H), 8.29 (s, 1H), 7.36–7.20 (m, 4H), 4.55 (d, J = 5.40, 2H), 4.01 (s, 2H), 2.84 (t, J = 7.48, 2H), 1.51 (m, 2H), 1.15–1.21 (m, 18H), 0.83 (t, J = 6.24 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.5, 160.7, 160.1, 149.5, 138.9, 134.3, 132.5, 129.1, 128.9, 118.8, 127.4, 50.9, 47.3, 42.4, 31.4, 31.3, 31.2 (2C), 28.7, 28.5, 27.0, 26.4, 22.1, 13.9. HRMS (ESI): m/z calculated for C27H39N4O2S [M + H]+ 483.35094, found 483.35047.
N-(4-(((2-(2-Hydroxyethoxy)ethyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (25)
HPLC 95.90% (method A, tR 12.26 min). 1H NMR (DMSO-d6, 400 MHz) δ 11.8 (brs, 1H), 8.29 (s, 1H), 8.23 (s, 1H), 7.30 (bs, 4H), 4.52 (d, J = 5.6 Hz, 2H), 3.75 (s, 2H), 3.51–3.47 (m, 4H), 3.42–3.39 (m, 2H), 2.69 (t, J = 5.6 Hz, 2H). 13C NMR (DMSO-d6, 100 MHz) δ 167.7, 161.9, 160.9, 148.2, 138.6, 138.1, 133.0, 130.3, 128.7, 127.7, 119.5, 72.6, 69.7, 60.6, 52.7, 48.2, 42.9. HRMS (ESI): m/z calculated for C19H23N4O4S [M + H]+ 403.14354 found, 403.14400.
N-(4-(((2-(2-Ethoxyethoxy)ethyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (26)
HPLC 95.59% (method A, tR 13.33 min). 1H NMR (DMSO-d6, 400 MHz) δ 11.74 (t, J = 5.2 Hz, 1H), 8.30 (s, 1H), 8.24 (s, 1H), 7.30 (bs, 4H), 4.52 (d, J = 5.6 Hz, 2H), 3.76 (s, 2H), 3.51–3.44 (m, 6H), 3.42 (q, J = 14.0 Hz, 7.0 Hz, 2H), 2.70 (t, J = 5.6 Hz, 2H), 1.07 (t, J = 7.0 Hz, 3H). 13C NMR (DMSO-d6, 100 MHz) δ 167.7, 161.6, 160.8, 147.9, 138.4, 138.1, 132.9, 130.5, 128.7, 127.7, 119.6, 70.1, 69.7, 69.6, 66.0, 52.6, 48.1, 42.9, 15.5. HRMS (ESI): m/z calculated for C21H27N4O4S [M + H]+ 431.17530, found 431.17422.
N-(4-(((2-(2-(2-Aminoethoxy)ethoxy)ethyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide dihydrochloride (31)
HPLC 99.72% (method A, tR 12.14 min). 1H NMR (DMSO-d6, 400 MHz) δ 13.2 (br s, 1H), 11.34 (t, J = 5.5 Hz, 1H), 9.53 (br s, 1H), 8.38 (s, 1H), 8.28 (s, 1H), 8.16 (br s, 2H), 7.57 (d, J = 8.1 Hz, 2H), 7.40 (d, J = 8.1 Hz, 2H), 4.56 (d, J = 5.6 Hz, 2H), 4.17 (t, J = 5.5 Hz, 2H), 3.70 (t, J = 5.3 Hz, 2H), 3.63 (d, J = 5.4 Hz, 2H), 3.06–2.89 (m, 4H). 13C NMR (DMSO-d6, 100 MHz) δ 167.5, 160.7, 160.0, 146.4, 140.1, 132.7, 131.7, 131.1, 130.7, 127.9, 119.8, 70.0, 69.9, 66.9, 66.0, 50.1, 45.8, 42.9, 38.8. HRMS (ESI): m/z calculated for C21H28N5O4S [M + H]+ 446.18620, found 446.18690.
N-(4-(((7-Aminohexyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide dihydrochloride (32)
HPLC 97.21% (method A, tR 12.28 min). 1H NMR (DMSO-d6, 400 MHz) δ 13.17 (br s, 1H), 11.34 (t, J = 5.2 Hz, 1H), 9.29 (br s, 1H), 8.39 (s, 1H), 8.29 (s, 1H), 7.94 (br s, 3H), 7.56–7.52 (s, 2H), 7.43–7.39 (m, 2H), 4.57 (d, J = 5.6 Hz, 2H), 4.12–4.05 (m, 2H), 2.89–2.69 (m, 4H), 1.70–1.49 (m, 4H), 1.35–1.27 (m, 4H). 13C NMR (DMSO-d6, 100 MHz) δ 167.6, 160.7, 160.0, 146.4, 140.2, 132.7, 131.7, 131.1, 130.7, 127.9, 119.8, 50.0, 46.6, 42.9, 39.0, 27.1, 25.9, 25.7, 25.5. HRMS (ESI): m/z calculated for C21H28N5O2S [M + H]+ 414.19637, found 414.19824.
N-(4-(((5-Aminopentyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide dihydrochloride (33)
HPLC 96.66% (method B, tR 7.55 min). 1H NMR (DMSO-d6, 400 MHz) δ 11.42–11.36 (m, 1H), 8.38 (s, 1H), 8.29 (s, 1H), 7.51 (d, J = 7.9 Hz, 2H), 7.40 (d, J = 7.9 Hz, 2H), 4.57 (d, J = 5.6 Hz, 2H), 4.07 (s, 2H), 2.86–2.78 (m, 2H), 2.78–2.72 (m, 2H), 1.68–1.50 (m, 4H), 1.40–1.30 (m, 2H). 13C NMR (DMSO-d6, 100 MHz) δ 167.6, 160.7, 160.1, 146.5, 140.2, 132.7, 131.7, 130.6, 128.0, 119.8, 50.0, 46.5, 42.9, 38.8, 26.8, 25.2, 23.3. HRMS (ESI): m/z calculated for C20H26N5O2S [M + H]+ 400.18072, found 400.18176.
N-(4-(((4-Aminobutyl)amino)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide dihydrochloride (34)
HPLC 96.53% (method A, tR 11.75 min). 1H NMR (DMSO-d6, 400 MHz) δ 11.33 (t, J = 5.5 Hz, 1H), 8.39 (s, 1H), 8.29 (s, 1H), 7.58–7.53 (m, 2H), 7.44–7.38 (m, 2H), 4.57 (d, J = 5.8 Hz, 2H), 4.12–4.06 (m, 2H), 2.93–2.73 (m, 4H), 1.78–1.56 (m, 4H). 13C NMR (DMSO-d6, 100 MHz) δ 167.5, 160.7, 160.0, 146.4, 140.1, 132.7, 131.7, 131.1, 130.7, 127.9, 119.8, 49.9, 46.0, 42.9, 40.6, 24.5, 22.7. HRMS (ESI): m/z calculated for C19H24N5O2S [M + H]+ 386.16507, found 386.16558.
N-(4-((Benzylamino)methyl)benzyl)-4-methoxythieno[2,3-d]pyrimidine-5-carboxamide (39)
HPLC 98.34% (method B, tR 5.87 min). 1H NMR (400 MHz, DMSO-d6) δ 11.22 (t, J = 5.4 Hz, 1H), 8.56 (s, 1H), 8.40 (s, 1H), 7.38–7.20 (m, 9H), 4.54 (d, J = 5.7 Hz, 2H), 3.66 (d, J = 3.5 Hz, 4H), 3.54 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 160.6, 159.8, 149.3, 141.2, 139.9, 137.6, 132.6, 132.3, 128.6, 128.5, 128.4, 127.7, 127.0, 119.0, 52.6, 52.3, 43.0, 34.8. HRMS (ESI): m/z calculated for C23H23N4O2S [M + H]+ 419.15428, found 419.15340.
N-(4-((Benzylamino)methyl)benzyl)-4-propoxythieno[2,3-d]pyrimidine-5-carboxamide (40)
HPLC 96.53% (method B, tR 11.59 min). 1H NMR (400 MHz, DMSO-d6) δ 11.18 (t, J = 5.7 Hz, 1H), 9.75 (br s, 2H), 8.59 (s, 1H), 8.38 (s, 1H), 7.64–7.46 (m, 4H), 7.46–7.32 (m, 5H), 4.57 (d, J = 5.7 Hz, 2H), 4.09 (m, 4H), 3.99 (t, J = 7.3 Hz, 2H), 1.79–1.60 (m, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.5, 160.9, 159.3, 149.0, 140.4, 132.7, 132.3, 130.9, 130.8, 130.6, 129.3, 129.0, 127.9, 119.3, 50.2, 50.0, 48.7, 42.8, 22.2, 11.2. HRMS (ESI): m/z calculated for C25H27N4O2S [M + H]+ 447.18558, found 447.18638.
N-(4-((Benzylamino)methyl)benzyl)-4-(hexyloxy)thieno[2,3-d]pyrimidine-5-carboxamide (41)
HPLC 97.90% (method B, tR 13.75 min). 1H NMR (400 MHz, DMSO-d6) δ 11.17 (t, J = 5.6 Hz, 1H), 8.59 (s, 1H), 8.40 (s, 1H), 7.41–7.20 (m, 9H), 4.54 (d, J = 5.7 Hz, 2H), 4.02 (t, J = 7.4 Hz, 2H), 3.69–3.60 (m, 4H), 1.75–1.61 (m, 2H), 1.32–1.18 (m, 8H), 0.83 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.5, 160.8, 159.2, 148.9, 138.3, 132.8, 132.3, 128.9, 128.8, 128.6, 127.7, 127.5, 119.2, 52.1, 51.9, 47.2, 42.9, 31.2, 28.9, 26.1, 22.4, 14.3. HRMS (ESI): m/z calculated for C28H33N4O2S [M + H]+ 489.23253, found 489.23220.
N-(4-((Octylamino)methyl)benzyl)-4-moxythieno[2,3-d]pyrimidine-5-carboxamide (42)
HPLC 98.25% (method B, tR 7.86 min). 1H NMR (400 MHz, DMSO-d6) δ 11.24 (br s, 1H), 9.24 (br s, 2H), 8.59 (s, 1H), 8.40 (s, 1H), 7.59–7.47 (m, 2H), 7.44–7.35 (m, 2H), 4.58 (d, J = 5.4 Hz, 2H), 4.20–3.99 (m, 4H), 3.55 (s, 3H), 2.82 (br s, 2H), 1.63 (br s, 2H), 1.24 (br s, 10H), 0.85 (t, J = 6.9 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.1, 160.2, 159.3, 148.8, 139.8, 132.0, 131.8, 130.6, 130.1, 130.0, 127.4, 127.3, 118.4, 49.5, 46.3, 42.3, 34.3, 31.1, 28.4, 28.3, 25.9, 25.1, 22.0, 13.9. HRMS (ESI): m/z calculated for C24H32N4O2S [M + H]+ 441.23253, found 441.23220.
N-(4-((Octylamino)methyl)benzyl)-4-propoxythieno[2,3-d]pyrimidine-5-carboxamide (43)
HPLC 98.99% (method A, tR 19.59 min). 1H NMR (400 MHz, DMSO-d6) δ 11.19 (t, J = 5.5 Hz, 1H), 9.29 (br s, 2H), 8.61 (s, 1H), 8.39 (s, 1H), 7.63–7.45 (m, J = 7.8 Hz, 2H), 7.45–7.28 (m, J = 7.8 Hz, 2H), 4.57 (d, J = 5.6 Hz, 2H), 4.15–3.95 (m, 4H), 2.82 (br s, 2H), 1.79–1.66 (m, 2H), 1.63 (br s, 2H), 1.23 (m, 10H), 0.98–0.74 (m, 6H). 13C NMR (100 MHz, DMSO-d6) δ 166.0, 160.3, 158.7, 148.5, 139.8, 132.1, 131.8, 130.6, 130.1, 127.3, 118.7, 49.4, 48.1, 46.2, 42.2, 31.1, 28.4, 28.3, 25.9, 25.1, 22.0, 21.7, 13.9, 10.7. HRMS (ESI): m/z calculated for C26H37N4O2S [M + H]+ 469.26383, found 469.26471.
4-(Hexyloxy)-N-(4-((octylamino)methyl)benzyl)thieno[2,3-d]pyrimidine-5-carboxamide (44)
HPLC 99.53% (method B, tR 9.78 min). 1H NMR (400 MHz, DMSO-d6) δ 11.19 (t, J = 5.6 Hz, 1H), 9.30 (br s, 2H), 8.61 (s, 1H), 8.40 (s, 1H), 7.65–7.45 (m, J = 7.8 Hz, 2H), 7.45–7.29 (m, J = 7.8 Hz, 2H), 4.58 (d, J = 5.6 Hz, 2H), 4.13–3.95 (m, 4H), 2.90–2.70 (m, 2H), 1.66 (dd, J = 6.5, 12.4 Hz, 4H), 1.23 (m, 9H), 1.27 (m, 7H), 0.84 (t, J = 6.4 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 166.5, 160.9, 159.2, 149.0, 140.4, 132.7, 132.4, 131.1, 130.7, 127.9, 119.2, 50.0, 47.2, 46.7, 42.8, 31.6, 31.2, 28.9, 28.9, 26.5, 26.1, 25.7, 22.5, 22.4, 14.4, 14.3. HRMS (ESI): m/z calculated for C29H43N4O2S [M + H]+ 511.31067, found 511.31397.
4-Methoxy-N-(4-(morpholinomethyl)benzyl)thieno[2,3-d]pyrimidine-5-carboxamide (45)
HPLC 99.06% (method A, tR 12.85 min). 1H NMR (400 MHz, DMSO-d6) δ 11.23 (t, J = 5.6 Hz, 1H), 8.57 (s, 1H), 8.40 (s, 1H), 7.28 (m, 4H), 4.54 (d, J = 5.8 Hz, 2H), 3.59–3.53 (m, 7H), 3.43 (s, 2H), 2.38–2.30 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 166.7, 160.7, 159.8, 149.3, 138.2, 138.2, 132.6, 132.3, 129.5, 127.7, 119.0, 66.6, 62.6, 53.6, 42.9, 34.8. HRMS (ESI): m/z calculated for C20H23N4O3S [M + H]+ 399.14920, found 399.14837.
N-(4-(Morpholinomethyl)benzyl)-4-propoxythieno[2,3-d]pyrimidine-5-carboxamide (46)
HPLC 97.34% (method A, tR 14.71 min). 1H NMR (400 MHz, DMSO-d6) δ 11.16 (s, 1H), 8.58 (s, 1H), 8.39 (s, 1H), 7.26 (q, J = 8.1 Hz, 4H), 4.52 (d, J = 5.8 Hz, 2H), 3.99 (t, J = 7.3 Hz, 2H), 3.53 (t, J = 4.5 Hz, 4H), 3.41 (s, 2H), 2.31 (m, 4H), 1.70 (d, J = 7.3 Hz, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.1, 160.3, 158.8, 148.5, 137.8, 136.4, 132.3, 131.9, 129.0, 127.1, 118.8, 66.2, 62.1, 53.1, 48.2, 42.3, 21.8, 10.7. HRMS (ESI): m/z calculated for C22H27N4O3S [M + H]+ 427.18050, found 427.17984.
4-((4-Azidobenzyl)oxy)-N-(4-((octylamino)methyl)benzyl)thieno[2,3-d]pyrimidine-5-carboxamide (47)
HPLC 98.70% (method A, tR 21.09 min). 1H NMR (400 MHz, DMSO-d6) δ 11.00 (t, J = 5.7 Hz, 1H), 9.27 (br s, 2H), 8.82 (s, 1H), 8.40 (s, 1H), 7.52 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.6 Hz, 2H), 7.11 (d, J = 8.5 Hz, 2H), 5.26 (s, 2H), 4.56 (d, J = 5.7 Hz, 2H), 4.08 (t, J = 5.6 Hz, 2H), 2.82 (m, 2H), 1.71–1.58 (m, 2H), 1.35–1.11 (m, 10H), 0.84 (t, J = 6.95 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.0, 160.4, 158.6, 148.4, 139.8, 139.0, 132.9, 132.2, 131.9, 130.6, 130.1, 129.6, 127.3, 119.3, 118.9, 49.5, 48.9, 46.3, 42.3, 31.1, 28.4, 28.4, 25.9, 25.2, 22.0, 13.9. HRMS (ESI): m/z calculated for C30H36N7O2S [M + H]+ 558.26523, found 558.26636.
N-(4-((1,5-Dihydroxy-4-oxo-1,4-dihydropyridine-2-carboxamido)methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (53)
HPLC 97.09% (method B, tR 9.36 min). 1H NMR (DMSO-d6, 400 MHz) δ 13.07 (s, 1H), 12.03 (s, 1H), 11.28 (s, 1H), 8.38 (s, 1H), 8.28 (s, 1H), 7.83 (s, 1H), 7.59 (s, 1H), 7.36–7.25 (m, 4H), 4.54–4.47 (m, 4H). 13C NMR (DMSO-d6, 100 MHz) δ 167.5, 160.6, 160.0, 159.9, 147.5, 146.7, 146.3, 138.0, 137.9, 132.8, 132.6, 131.6, 129.1, 128.1, 128.0, 127.6, 119.8, 112.9, 43.0, 42.6. HRMS (ESI): m/z calculated for C21H18N5O6S [M + H]+ 468.09778, found 468.09756.
N-(4-(((5-(1,5-Dihydroxy-4-oxo-1,4-dihydropyridine-2-carboxamido)pentyl)amino) methyl)benzyl)-4-oxo-3,4-dihydrothieno[2,3-d]pyrimidine-5-carboxamide (57)
HPLC 97.09% (method B, tR 9.36 min). 1H NMR (DMSO-d6, 400 MHz) δ 13.05 (s, 1H), 11.70–11.60 (m, 1H), 11.28 (s, 1H), 8.38 (s, 1H), 8.34–8.21 (m, 2H), 7.80 (s, 1H), 7.56 (s, 1H), 7.32–7.27 (m, 2H), 7.23–7.17 (m, 2H), 4.51 (d, J = 5.2 Hz, 2H), 4.23 (d, J = 5.6 Hz, 2H), 3.03–2.96 (m, 2H), 2.19–2.04 (m, 2H), 1.59–1.45 (m, 4H), 1.41–1.22 (m, 2H). 13C NMR (DMSO-d6, 100 MHz) δ 172.4, 167.5, 160.6, 160.0, 159.8, 146.5, 146.3, 138.8, 137.7, 132.8, 127.8, 119.8, 112.7, 43.0, 42.2, 39.0, 35.8, 29.2, 26.7, 25.5. HRMS (ESI): m/z calculated for C27H29N6O7S [M + H]+ 581.18184, found 581.18358.
Acknowledgments
We are grateful to AstraZeneca for their gift of compound AZ51. We thank the technical assistances from beamline scientists in Australian Synchrotron and Swiss Light Source. We also thank Dr. Ghader Bashiri from The University of Auckland (New Zealand) for the gift of vector pYUB28b. This research was supported by the National Research Foundation of Singapore through the Singapore-MIT-Alliance for Research and Technology (SMART) Infectious Disease and Antimicrobial Resistance Interdisciplinary Research Groups; SMART Innovation Centre grant ING137070-BIO to P.C.D., L.C.F., and J.L.; and AcRF grants Tier1 RG154/14 and MOE2015-T2-2-075 to J.L.
Glossary
Abbreviations Used
- ATCC
American Type Culture Collection
- CFU
colony forming unit
- Hi
Haemophilus influenzae
- MBC
minimum bactericidal concentration
- MIC
minimum inhibitory concentration
- Mtb
Mycobacterium tuberculosis
- Pa
Pseudomonas aeruginosa
- Sa
Staphylococus aureus
- SAH
S-adenosyl-l-homocysteine
- SAM
S-adenosyl-l-methionine
- SPR
surface plasmon resonance
- TrmD
tRNA-(N1G37) methyltransferase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b00582.
Mechanism of action of TrmD enzyme; crystal structures of TrmDs; TrmD sequence alignment and similarity; thermal shift of PaTrmD; thermal shift of other TrmDs; effect of R1 chain length on TrmD inhibitory activity of 15 analogues; compound 15 binding modes in PaTrmD; TrmD sequence identities; hemolytic activity; primers and DNA template for tRNA synthesis; synthesis scheme of pyridone siderophore conjugates; 1H NMR and 13C NMR spectra of synthesized compounds (PDF)
Molecular formula strings (CSV)
Crystal structure of 5ZHIMtbTrmD (PDB)
Crystal structure of 5XHJMtbTrmD–SAH (PDB)
Crystal structure of 5ZHKMtbTrmD–12 (PDB)
Crystal structure of 5ZHLMtbTrmD–15 (PDB)
Crystal structure of 5ZHMPaTrmD–11 (PDB)
Crystal structure of 5ZHNPaTrmD–15 (PDB)
Crystal structure of 6JOFMtbTrmD–AZ51 (PDB)
Crystal structure of 6JOEPaTrmD–AZ51 (PDB)
Accession Codes
Coordinates and structure factors for PaTrmD–AZ51, PaTrmD–11, PaTrmD–15, apo MtbTrmD, MtbTrmD–SAH, MtbTrmD–AZ51, MtbTrmD–12, and MtbTrmD–15 have been deposited in the Protein Data Bank under accession codes 6JOE, 5ZHM, 5ZHN, 5ZHI, 5ZHJ, 6JOF, 5ZHK, and 5ZHL, respectively. The authors will release the atomic coordinates and experimental data upon article publication.
Author Present Address
○ Y.H.C.: Tychan Pte. Ltd., 79 Ayer Rajah Crescent, #05-03, Singapore 139955.
Author Present Address
● P.G.: MSD Translational Medicine Research Centre, Merck Research Laboratories, Singapore.
Author Present Address
◆ E.T.B.: Pfizer Vaccines Research and Development, Pearl River, New York 10965, United States.
Author Present Address
¶ T.D.: Center for Discovery and Innovation, Hackensack Meridian Health, 340 Kingsland Street Building 102, Nutley, New Jersey 07110, United States.
Author Contributions
The manuscript was written through contributions of all authors and all authors have given approval to the final version of the manuscript. W.Z., K.K.P., and S.B. contributed equally. E.T.B., N.O., C.F.L., J.L., and P.C.D. conceived of the studies and managed all experiments. W.Z., K.K.P., S.B., Y.H.W., Y.H.C., M.E., T.D., C.F.L., J.L., and P.C.D. designed experiments. W.Z., K.K.P., S.B., Y.H.W., Q.N., Y.H.C., M.E., and P.G. carried out the experiments. Y.H.W. collected the diffraction data. W.Z. and J.L. analyzed diffraction data and refined structures. K.K.P., S.B., V.G., and S.G. performed the chemical syntheses. P.G. performed the Mtb activity assay. W.Z. and S.H.L. carried out the SPR experiment.
The authors declare no competing financial interest.
Supplementary Material
References
- Antibiotic Resistance Threats in the United States; Centers for Disease Control and Prevention, 2013.
- Vogel G.Meet WHO’s dirty dozen: The 12 bacteria for which new drugs are most urgently needed. Science 2017, 10.1126/science.aal0829. [DOI] [Google Scholar]
- Chan C. T.; Dyavaiah M.; DeMott M. S.; Taghizadeh K.; Dedon P. C.; Begley T. J. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010, 6 (12), e1001247 10.1371/journal.pgen.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chionh Y. H.; McBee M.; Babu I. R.; Hia F.; Lin W.; Zhao W.; Cao J.; Dziergowska A.; Malkiewicz A.; Begley T. J.; Alonso S.; Dedon P. C. tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat. Commun. 2016, 7, 13302. 10.1038/ncomms13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. T.; Pang Y. L.; Deng W.; Babu I. R.; Dyavaiah M.; Begley T. J.; Dedon P. C. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012, 3, 937. 10.1038/ncomms1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netzer N.; Goodenbour J. M.; David A.; Dittmar K. A.; Jones R. B.; Schneider J. R.; Boone D.; Eves E. M.; Rosner M. R.; Gibbs J. S.; Embry A.; Dolan B.; Das S.; Hickman H. D.; Berglund P.; Bennink J. R.; Yewdell J. W.; Pan T. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature 2009, 462 (7272), 522–526. 10.1038/nature08576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley U.; Dyavaiah M.; Patil A.; Rooney J. P.; DiRenzo D.; Young C. M.; Conklin D. S.; Zitomer R. S.; Begley T. J. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol. Cell 2007, 28 (5), 860–870. 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroensuk J.; Atichartpongkul S.; Chionh Y. H.; Wong Y. H.; Liew C. W.; McBee M. E.; Thongdee N.; Prestwich E. G.; DeMott M. S.; Mongkolsuk S.; Dedon P. C.; Lescar J.; Fuangthong M. Methylation at position 32 of tRNA catalyzed by TrmJ alters oxidative stress response in Pseudomonas aeruginosa. Nucleic Acids Res. 2016, 44 (22), 10834–10848. 10.1093/nar/gkw870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng C. S.; Sinha A.; Aniweh Y.; Nah Q.; Babu I. R.; Gu C.; Chionh Y. H.; Dedon P. C.; Preiser P. R. tRNA epitranscriptomics and biased codon are linked to proteome expression in Plasmodium falciparum. Mol. Syst. Biol. 2018, 14, e8009 10.15252/msb.20178009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda I.; Sakaguchi R.; Liu C.; Gamper H.; Hou Y.-M. The temperature sensitivity of a mutation in the essential tRNA modification enzyme tRNA methyltransferase D (TrmD). J. Biol. Chem. 2013, 288 (40), 28987–28996. 10.1074/jbc.M113.485797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dwyer K.; Watts J. M.; Biswas S.; Ambrad J.; Barber M.; Brule H.; Petit C.; Holmes D. J.; Zalacain M.; Holmes W. M. Characterization of Streptococcus pneumoniae TrmD, a tRNA methyltransferase essential for growth. J. Bacteriol. 2004, 186 (8), 2346–2354. 10.1128/JB.186.8.2346-2354.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K.; Ehrlich S. D.; Albertini A.; Amati G.; Andersen K. K.; Arnaud M.; Asai K.; Ashikaga S.; Aymerich S.; Bessieres P.; Boland F.; Brignell S. C.; Bron S.; Bunai K.; Chapuis J.; Christiansen L. C.; Danchin A.; Débarbouillé M.; Dervyn E.; Deuerling E.; Devine K.; Devine S. K.; Dreesen O.; Errington J.; Fillinger S.; Foster S. J.; Fujita Y.; Galizzi A.; Gardan R.; Eschevins C.; Fukushima T.; Haga K.; Harwood C. R.; Hecker M.; Hosoya D.; Hullo M. F.; Kakeshita H.; Karamata D.; Kasahara Y.; Kawamura F.; Koga K.; Koski P.; Kuwana R.; Imamura D.; Ishimaru M.; Ishikawa S.; Ishio I.; Le Coq D.; Masson A.; Mauël C.; Meima R.; Mellado R. P.; Moir A.; Moriya S.; Nagakawa E.; Nanamiya H.; Nakai S.; Nygaard P.; Ogura M.; Ohanan T.; O’Reilly M.; O’Rourke M.; Pragai Z.; Pooley H. M.; Rapoport G.; Rawlins J. P.; Rivas L. A.; Rivolta C.; Sadaie A.; Sadaie Y.; Sarvas M.; Sato T.; Saxild H. H.; Scanlan E.; Schumann W.; Seegers J. F. M. L.; Sekiguchi J.; Sekowska A.; Séror S. J.; Simon M.; Stragier P.; Studer R.; Takamatsu H.; Tanaka T.; Takeuchi M.; Thomaides H. B.; Vagner V.; van Dijl J. M.; Watabe K.; Wipat A.; Yamamoto H.; Yamamoto M.; Yamamoto Y.; Yamane K.; Yata K.; Yoshida K.; Yoshikawa H.; Zuber U.; Ogasawara N. Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (8), 4678–4683. 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk G. R.; Jacobsson K.; Nilsson K.; Johansson M. J. O.; Byström A. S.; Persson O. P. A primordial tRNA modification required for the evolution of life?. EMBO J. 2001, 20 (1–2), 231–239. 10.1093/emboj/20.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaroensuk J.; Wong Y. H.; Zhong W.; Liew C. W.; Maenpuen S.; Sahili A. E.; Atichartpongkul S.; Chionh Y. H.; Nah Q.; Thongdee N.; McBee M. E; Prestwich E. G; DeMott M. S; Chaiyen P.; Mongkolsuk S.; Dedon P.; Lescar J.; Fuangthong M.. Crystal structure and catalytic mechanism of the essential m1G37 tRNA methyltransferase TrmD from Pseudomonas aeruginosa. RNA 2019, 10.1261/rna.066746.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill P. J.; Abibi A.; Albert R.; Andrews B.; Gagnon M. M.; Gao N.; Grebe T.; Hajec L. I.; Huang J.; Livchak S.; Lahiri S. D.; McKinney D. C.; Thresher J.; Wang H.; Olivier N.; Buurman E. T. Selective inhibitors of bacterial t-RNA-(N(1)G37) methyltransferase (TrmD) that demonstrate novel ordering of the lid domain. J. Med. Chem. 2013, 56 (18), 7278–7288. 10.1021/jm400718n. [DOI] [PubMed] [Google Scholar]
- Goto-Ito S.; Ito T.; Kuratani M.; Bessho Y.; Yokoyama S. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat. Struct. Mol. Biol. 2009, 16 (10), 1109–1115. 10.1038/nsmb.1653. [DOI] [PubMed] [Google Scholar]
- Zhong W.; Koay A.; Ngo A.; Li Y.; Nah Q.; Wong Y. H.; Chionh Y. H.; Ng H. Q.; Koh-Stenta X.; Poulsen A.; Foo K.; McBee M.; Choong M. L.; El Sahili A.; Kang C.; Matter A.; Lescar J.; Hill J.; Dedon P. Targeting the bacterial epitranscriptome: Discovery of novel tRNA-(N1G37) methyltransferase (TrmD) inhibitors with antibiotic activity. ACS Infect. Dis. 2019, 5, 326–335. 10.1021/acsinfecdis.8b00275. [DOI] [PubMed] [Google Scholar]
- Elkins P. A., Bonnette W. G., Williams S. P., Madauss K. P., Stuckey J. A.. Crystal structure of TrmD, a M1G37 tRNA methyltransferase with SAM-competitive compounds. Available from: rcsb.org. PDB 4YQD (accessed Nov 11, 2015), 4YQT (accessed March 16, 2016), 4YQS (accessed March 16, 2016), 4YQR (accessed March 16, 2016), 4YQQ (accessed March 16, 2016), 4YQP (accessed March 16, 2016), 4YQO (accessed March 16, 2016), 4YQN (accessed March 16, 2016), 4YQL (accessed March 16, 2016), 4YQK (accessed March 16, 2016), 4YQJ (accessed March 16, 2016), 4YQI (accessed March 16, 2016), 4YQG (accessed March 16, 2016), 4YQC (accessed March 16, 2016), 4YQB (accessed March 16, 2016), 4YQA (accessed March 16, 2016), 4YQ9 (accessed March 16, 2016), 4YQ8 (accessed March 16, 2016), 4YQ7 (accessed March 16, 2016), 4YQ6 (accessed March 16, 2016), 4YQ5 (accessed March 16, 2016), 4YQ4 (accessed March 16, 2016), 4YQ3 (accessed March 16, 2016), 4YQ2 (accessed March 16, 2016), 4YQ1 (accessed March 16, 2016), 4YQ0 (accessed March 16, 2016), 4YPZ (accessed March 16, 2016), 4YPY (accessed March 16, 2016), 4YPX (accessed March 16, 2016), 4YPW (accessed March 16, 2016), 5D9F (accessed Aug 31, 2016).
- Ahn H. J.; Kim H. W.; Yoon H. J.; Lee B. I.; Suh S. W.; Yang J. K. Crystal structure of tRNA(m1G37)methyltransferase: insights into tRNA recognition. EMBO J. 2003, 22 (11), 2593–2603. 10.1093/emboj/cdg269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T.; Masuda I.; Yoshida K-i; Goto-Ito S.; Sekine S-i; Suh S. W.; Hou Y.-M.; Yokoyama S. Structural basis for methyl-donor-dependent and sequence-specific binding to tRNA substrates by knotted methyltransferase TrmD. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (31), E4197–E4205. 10.1073/pnas.1422981112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels K. G.; Suo Y.; Oas T. G. Conformational kinetics reveals affinities of protein conformational states. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (30), 9352–9357. 10.1073/pnas.1502084112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral M.; Kokh D. B.; Bomke J.; Wegener A.; Buchstaller H. P.; Eggenweiler H. M.; Matias P.; Sirrenberg C.; Wade R. C.; Frech M. Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nat. Commun. 2017, 8, 2276. 10.1038/s41467-017-02258-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang X.; Zhou H. X. Rate constants and mechanisms of protein-ligand binding. Annu. Rev. Biophys. 2017, 46, 105–130. 10.1146/annurev-biophys-070816-033639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545 (7654), 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. F.; Mitton-Fry M. J.; Arcari J. T.; Barham R.; Casavant J.; Gerstenberger B. S.; Han S.; Hardink J. R.; Harris T. M.; Hoang T.; Huband M. D.; Lall M. S.; Lemmon M. M.; Li C.; Lin J.; McCurdy S. P.; McElroy E.; McPherson C.; Marr E. S.; Mueller J. P.; Mullins L.; Nikitenko A. A.; Noe M. C.; Penzien J.; Plummer M. S.; Schuff B. P.; Shanmugasundaram V.; Starr J. T.; Sun J.; Tomaras A.; Young J. A.; Zaniewski R. P. Pyridone-conjugated monobactam antibiotics with gram-negative activity. J. Med. Chem. 2013, 56 (13), 5541–5552. 10.1021/jm400560z. [DOI] [PubMed] [Google Scholar]
- Aoki T.; Yoshizawa H.; Yamawaki K.; Yokoo K.; Sato J.; Hisakawa S.; Hasegawa Y.; Kusano H.; Sano M.; Sugimoto H.; Nishitani Y.; Sato T.; Tsuji M.; Nakamura R.; Nishikawa T.; Yamano Y. Cefiderocol (S-649266), A new siderophore cephalosporin exhibiting potent activities against Pseudomonas aeruginosa and other gram-negative pathogens including multi-drug resistant bacteria: Structure activity relationship. Eur. J. Med. Chem. 2018, 155, 847–868. 10.1016/j.ejmech.2018.06.014. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 133–144. 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. IUCr. Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006, 62 (1), 72–82. 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60 (12), 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Smart O. S.; Womack T. O.; Flensburg C.; Keller P.; Paciorek W.; Sharff A.; Vonrhein C.; Bricogne G. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 368–380. 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüttelkopf A. W.; van Aalten D. M. F. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf. 2008, 9, 40. 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67 (4), 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B.; Snoeyink J.; Richardson J. S.; Richardson D. C. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.8; Schroödinger, LLC, 2015.
- Yee M.; Gopal P.; Dick T.. Missense Mutations in the Unfoldase ClpC1 of the Caseinolytic Protease Complex Are Associated with Pyrazinamide Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2016, 61, 10.1128/AAC.02342-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal P.; Tasneen R.; Yee M.; Lanoix J. P.; Sarathy J.; Rasic G.; Li L.; Dartois V.; Nuermberger E.; Dick T. In Vivo-Selected Pyrazinoic Acid-Resistant Mycobacterium tuberculosis Strains Harbor Missense Mutations in the Aspartate Decarboxylase PanD and the Unfoldase ClpC1. ACS Infect. Dis. 2017, 3 (7), 492–501. 10.1021/acsinfecdis.7b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.