

Abstract
Motivation
Optimal growth temperature is a fundamental characteristic of all living organisms. Knowledge of this temperature is central to the study of a prokaryote, the thermal stability and temperature dependent activity of its genes, and the bioprospecting of its genome for thermally adapted proteins. While high throughput sequencing methods have dramatically increased the availability of genomic information, the growth temperatures of the source organisms are often unknown. This limits the study and technological application of these species and their genomes. Here, we present a novel method for the prediction of growth temperatures of prokaryotes using only genomic sequences.
Results
By applying the reverse ecology principle that an organism’s genome includes identifiable adaptations to its native environment, we can predict a species’ optimal growth temperature with an accuracy of 5.17°C root-mean-square error and a coefficient of determination of 0.835. The accuracy can be further improved for specific taxonomic clades or by excluding psychrophiles. This method provides a valuable tool for the rapid calculation of organism growth temperature when only the genome sequence is known.
Availability and implementation
Source code, genomes analyzed and features calculated are available at: https://github.com/DavidBSauer/OGT_prediction.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Growth conditions of an organism are essential to its characterization. However, these values may be unknown in bacteria or archaea that are difficult to culture, ‘unculturable’ or otherwise poorly characterized. Reverse ecology posits that the evolutionary effects of an organism’s native environment is reflected by adaptations in its genome (Li et al., 2008). Therefore, an organism’s native environment can be identified by comparing its genome to the genomes of other organisms from a range of environments. Notably, this is done without experimental manipulation or interrogation of the organism beyond genome sequencing. Such reverse ecology strategies have been successful in studying adaptation to soil conditions (Turner et al., 2010), salinity (Hohenlohe et al., 2010) and temperature (Ellison et al., 2011).
Of these environmental pressures, temperature, being a description of the internal energy of the environment, is a particularly strong driving force for adaptation. Prokaryotes are often viable over a range of temperatures, which varies by species. For a particular organism, increasing temperature beyond it’s growth range, corresponding to increased internal energy, can lead to loss of structure in proteins and nucleic acids. Conversely, a sub-optimal temperature leads to reduced enzyme kinetics and stiffening lipid membranes. Each of these biological consequences may be deleterious to un-adapted organisms. Therefore, it is perhaps not surprising that an organism’s optimal growth temperature (OGT) correlates to quantifiable properties (referred to as ‘features’ in this work) in the organism’s nucleotide and protein sequences. Features correlated with OGT can be identified in the genomic (Kawashima et al., 2000), tRNA (Galtier and Lobry, 1997; Hurst and Merchant, 2001), rRNA (Galtier and Lobry, 1997; Hurst and Merchant, 2001; Khachane et al., 2005), open reading frame (ORF) (Lynn et al., 2002; Singer and Hickey, 2003), and in the proteomic sequences (Lobry and Chessel, 2003; Singer and Hickey, 2003; Tekaia et al., 2002; Zeldovich et al., 2007). Correlations between OGT and tRNA G + C content (Galtier and Lobry, 1997; Hurst and Merchant, 2001) or the charged versus polar amino acid ratios (Suhre and Claverie, 2003) are particularly well known.
Clearly, OGT is a necessary parameter for analyzing physiological processes of an organism or activities of its genes and proteins. (Nguyen et al., 2017; Perl et al., 2000). However, the experimental determination of OGT is laborious (Elliott, 1963; Honglin et al., 1993), and sometimes unattainable (Stewart, 2012) for certain prokaryotes. Also, recorded OGT or environmental temperature may be inconsistently measured, particularly in genetic samples not obtained from pure culture (Kunin et al., 2008). Further, for metagenomic samples the conditions during collection may significantly differ from the originating species' growth environment. This can be due to the organism or its genetic material being found distant from its originating environment (Rose et al., 2014), or the collected genomic material may be from organisms which are inviable (Cangelosi and Meschke, 2014). Even in pure culture in the laboratory, experimental growth conditions can vary greatly (Hearing et al., 1989) and may not be at the source organisms’ OGT (Hashimoto et al., 2004).
While many previous studies have aimed to identify genes and proteins (Wang et al., 2015), mutations (Perl et al., 2000) and mechanisms (Nguyen et al., 2017) that drive thermal adaptation, there is also great value in using these adaptive differences to provide data of an organism’s native environment when it otherwise may not be known or well-described. A number of parameters have been identified which correlate with OGT (Suhre and Claverie, 2003). However, those correlations are often weak and therefore of limited predictive value alone. Here, we aim to predict a prokaryotic species’ OGT only from its genomic sequence. We set out to develop a novel tool for the ecological characterization of a species based solely on its genome, to aid in the study of thermoadaptation and the bioprospecting of thermoadapted genes.
2 Materials and methods
2.1 Species ecological data and taxonomic classification
Experimentally measured OGTs of various species were used as previously published without modification (Sauer et al., 2015). Taxonomic assignments for each species were collected from NCBI (Benson et al., 2017).
2.2 Genomic sequences, gene extraction and feature calculation
All available top level genome sequences for each species with a measured OGT were downloaded from Ensembl Bacteria (Kersey et al., 2016). tRNA, rRNA and ORF sequences were then extracted from these genomes. tRNA sequences were identified and extracted with tRNAScan-SE 2.0 (Lowe and Chan, 2016) with general settings. Ribosomal RNA genes were identified with Barrnap 0.9 (https://github.com/tseemann/barrnap) using superkingdom specific hidden Markov models, and 16S rRNA sequences extracted from the genome using BEDtools 2.27.1 (Quinlan and Hall, 2010). Open reading frame sequences were extracted with Prodigal 2.6.3 (Hyatt et al., 2010) using default settings. The proteome of each genome was generated by in silico translation using Prodigal.
Features were calculated for all contigs in the deposited genomes and derived sequences of tRNA and 16S rRNA genes, ORFs and protein sequences. No attempt was made to identify or exclude exogenous sequences. Only standard nucleotides (ATGC) and amino acids (ACDEFGHIKLMNPQRSTVWY) in each nucleic acid and protein sequence were considered when calculating features. Non-standard and ambiguous nucleotides and amino acids were ignored. All features were calculated on a per genome basis, and then averaged by species. The Pearson correlation coefficient (r) between feature value and species OGT was calculated.
2.3 Dataset generation
The generalizability of the multiple linear regressions was ensured using the holdout method (James et al., 2013; Kohavi, 1995). Species were randomly assigned to training or test datasets prior to multiple linear regression. The training and test dataset consisted of 80 and 20% of the species, respectively. The test dataset was never used for regression. These test species were only used for evaluation of the final multiple linear regressions, comparing the calculated and measured OGTs.
2.4 Multiple linear regression
Optimal growth temperatures were predicted by multiple linear regression against the quantitative genomic, tRNA, rRNA, ORF and proteome features. Only those features with an absolute value of r greater than 0.3 were used as predictor variables for multiple linear regression (Supplementary Table S1). To address multicollinearity of the features and to minimize overfitting of the regression, features were added progressively to the multiple linear regression using a forward stepwise method. The initial predictor variable set consisted of only the feature most correlated with OGT. To this each of the other correlated features were added individually, and multiple linear regressions were calculated. If the adjusted coefficient of determination (2) between measured and predicted OGTs for the training set increased for any regression, the additional feature which most increased 2 was added to the predictor variable set. This new set of predictor variables was then used for additional rounds of feature addition and regression, until 2 did not increase. The regressions were also required to be over-determined, with feature addition stopped if the number of features equaled the number of species in the training dataset. Regressions were carried out using the LinearRegression module of scikit-learn (Pedregosa et al., 2011).
2.5 Multiple linear regression evaluation
To evaluate the accuracy of the multiple linear regressions the predicted and measured OGT of the test dataset species were compared. The coefficient of determination (R2) and root-mean-square error (RMSE) between predicted and reported OGTs of the test set were used to report regression accuracy.
2.6 De novo OGT prediction and validation
All top-level genomes in Ensembl Bacteria were downloaded for each species without a reported OGT in Sauer et al. (2015). Taxonomic assignment and feature calculation were performed as described above. The OGT of each species was predicted using the most taxonomic specific regression available with an R2 greater than 0.5. When available, these predicted OGTs were compared to the species’ reported OGT in BacDive (Söhngen et al., 2016).
2.7 Species distance comparison
Pairwise species distances collected from the All-Species Living Tree project’s SSU rRNA tree (Yarza et al., 2008) (June 2018 release) using the Biopython Phylo library (Talevich et al., 2012) and used without modification. When comparisons were done relative to a single species, a species was chosen at random from those species with an OGT at the median OGT. Pairwise comparisons were evaluated explicitly without assuming commutativity.
2.8 Software
Analyses were carried out with custom scripts in Python 3.6.5 using Biopython 1.72 (Cock et al., 2009), NumPy 1.15.1 (van der Walt et al., 2011), SciPy 1.1.0, Scikit-learn 0.19.2 (Pedregosa et al., 2011) and MatPlotLib 2.2.3 (Hunter, 2007).
3 Results
3.1 Prokaryote genome redundancy is highly skewed
Of the initial 8098 prokaryotic species with a reported OGT, genome sequences were available for 2719 species. These sequenced species were composed of 2549 Bacteria and 170 Archaea, with OGTs ranging from 4 to 103°C. A total of 36, 804 sequenced genomes for these species were downloaded, indicating multiple genomes for each species on average. However, the number of genomes per species was highly skewed, with great redundancy for model organisms and pathogens (Supplementary Fig. S1C). To avoid having these relatively few species dominate the analysis, features were averaged by species and all regressions were done by species rather than by genome.
3.2 Individual genome-derived features correlate with OGT
Based on the reverse genomics principle that an organism’s adaptation to its environment is reflected within its genome, we hypothesize that a species’ OGT could be predicted based on characteristics of its genome and genome-derived sequences. This hypothesis was supported by previous noted correlations between OGT and individual features of the genomic (Galtier and Lobry, 1997; Kawashima et al., 2000; Sabath et al., 2013 ), tRNA (Galtier and Lobry, 1997; Hurst and Merchant, 2001), rRNA (Galtier and Lobry, 1997; Hurst and Merchant, 2001; Khachane et al., 2005), open reading frame (Li et al., 2007; Lynn et al., 2002; Hurst and Merchant, 2001; Singer and Hickey, 2003; Zheng and Wu, 2010) and proteomic or protein sequences (Burra et al., 2010; Cambillau and Claverie, 2000; Haney et al., 1999; Kreil and Ouzounis, 2001; Lobry and Chessel, 2003; Puigbò et al., 2008; Robinson-Rechavi et al., 2006; Sælensminde et al., 2007; Singer and Hickey, 2003; Suhre and Claverie, 2003; Tekaia et al., 2002; Zeldovich et al., 2007). These features are quantifiable properties of the sequence, such as G + C content, length and nucleotide or amino acid fraction. Of the features calculated, 47 were found in this work to be correlated with OGT in the present dataset by the Pearson correlation coefficient with |r| > 0.3 (Fig. 1, Supplementary Table S1). However, these individual correlations to OGT were often weak and therefore insufficient for the calculation of a species’ growth temperature. Furthermore, there was a strong association among many features (Supplementary Fig. S2). We therefore decided to consider them simultaneously, using multiple linear regression. In order to minimize multicollinearity and avoid overfitting, features were added to the regression progressively until the regression no longer improved. Regression generalizability was evaluated with a test dataset, species that were never used in calculating the coefficients of the multiple linear regression. We classified features based on the source sequences (genomic, tRNA, rRNA, open reading frames and proteome). Multiple linear regressions were calculated, progressively increasing the feature classes used in the regression.
Fig. 1.
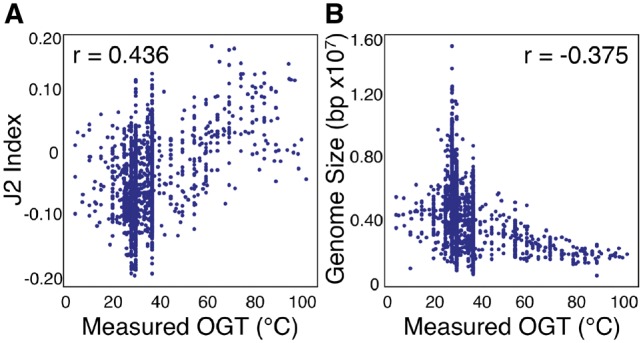
Individual genome-derived features correlate weakly with the originating species’ OGT. Measured optimal growth temperature for each species versus J2 index of genomic dinucleotide fractions (A) and total genome size (B)
3.3 A regression using only the genomic sequence weakly predictive of OGT
The genomic sequence provides information about the nucleotide content, nucleotide order and chromosomal structure of an organism’s hereditable genetic material. In the absence of any other knowledge, this sequence still reflects adaptations to the particular thermal environment of the organism. For example, total genome size has been shown to be negatively correlated with a species’ OGT (Sabath et al., 2013). Accordingly, it has been proposed that the reduced time and energy of genomic replication offers selective advantages at higher temperatures. Additionally, the necessity of maintaining genomic structure with increased temperature is thought to be reflected in a species’ genomic dinucleotide fractions (Amano et al., 1997), which is quantified in the J2 index (Kawashima et al., 2000).
In the present dataset, individual nucleotide and dinucleotide fractions of the genome, the J2 index, the G + C content and total size were calculated for each genome. Of these features, the J2 index, genome size and the CT and AG dinucleotide fractions correlated with OGT, but only weakly. Using these poorly correlated and collinear input features for regression, the resulting multiple linear regression is poor at predicting OGT with a root mean squared error (RMSE) of 10.6°C (R2 = 0.310) (Supplementary Fig. S3).
3.3 tRNA and rRNA sequences improve OGT prediction
tRNA and rRNA are nucleic acids whose structure, and enzymatic activity in the case of rRNA, are essential to cell viability. Therefore, the direct correlation of OGT to G + C content of tRNAs (Galtier and Lobry, 1997; Hurst and Merchant, 2001) and rRNAs (Galtier et al., 1999; Khachane et al., 2005) is thought to reflect the increase in base pair hydrogen bonding necessary to maintain the structure of these nucleic acids at elevated temperatures. While a subset of the previously analyzed genomic sequence, we hypothesized that features derived from these tRNA and rRNA sequences might be more strongly correlated with OGT. To this end, we identified their sequences bioinformatically. tRNA and 16S rRNA sequences were identified in 100% and 99% of the species respectively, reflecting the highly conserved nature of these genes.
Using these identified tRNA and rRNA sequences, nucleotide fractions and G + C content were calculated for each. All calculated features for tRNA and rRNA sequences were correlated with OGT. Calculating a new linear regression with the OGT using tRNA features, in addition to genomic features, improved accuracy (RMSE = 7.90°C, R2 = 0.616) (Fig. 2A). Similarly, a regression calculated with rRNA and genomic features also improved accuracy (RMSE = 7.33°C, R2 = 0.669) (Fig. 2B). By using all available tRNA, rRNA and genomic features, a still more accurate linear regression was calculated (RMSE = 7.12°C, R2 = 0.688) (Fig. 2C).
Fig. 2.

Using genomic and genic sequences improve OGT prediction accuracy. Predicted versus measured OGT for each species, using linear regressions with features derived from genomic and (A) tRNA, (B) rRNA or (C) tRNA and rRNA sequences. Species used for regression and evaluation are shown in dark purple and green, respectively. The dotted line indicates a perfect prediction
3.4 ORF sequences improve OGT prediction
As tRNA and rRNA features clearly improve the ability to predict a species’ OGT, we examined if other gene sequences might also improve the regression. In particular open reading frames, which code for proteins but exclude the non-coding regions of the genome, were considered. We hypothesized that using coding regions alone would increase sensitivity to changes in OGT, possibly due to the sensitivity of protein expression to mRNA structure. Additionally, codon biases have previously been reported to correlate with OGT (Zeldovich et al., 2007), likely reflecting both amino acid differences and the necessity of maintaining proper codon-anticodon pairing in differing thermal environments. Furthermore, the greater number of ORFs in a genome, relative to tRNAs and rRNAs, make the features of ORFs less sensitive to mispredictions or to single gene aberrations. Therefore, ORF derived features were hypothesized to more sensitively and accurately report on the thermal environment than tRNA or rRNA sequences.
We identified ORFs within the genomic sequences bioinformatically. From these ORFs, a number of derived features were calculated including nucleotide and dinucleotide fractions, codon fractions, start and stop codon fractions, the coding ratio and fraction of the genome, the ORF density of the genome, G + C and A + G content, and average ORF length. Of these, nine were found to be correlated with OGT. These include the A + G content, codon and dinucleotide fractions and the fraction of the alternative start codon TTG. These ORF derived features, in addition to the genomic, tRNA and rRNA features, were used to calculate a new multiple linear regression with significantly improved accuracy (RMSE = 6.35°C, R2 = 0.752) (Fig. 3).
Fig. 3.
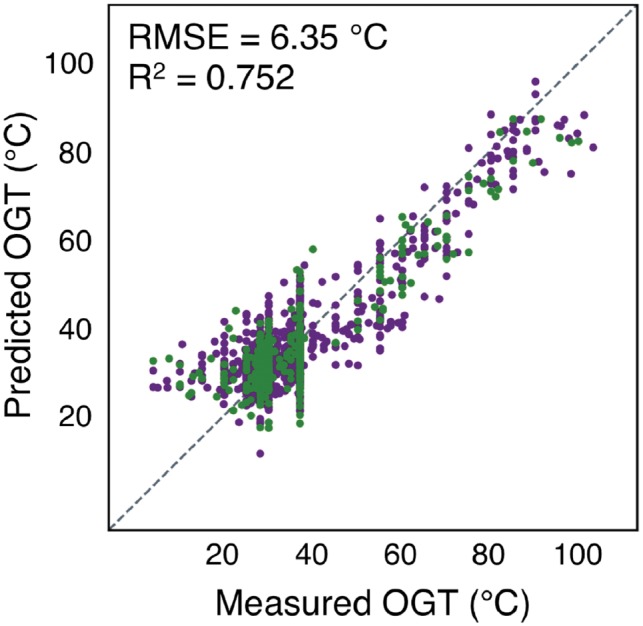
Open reading frame sequences further improve OGT prediction accuracy. Predicted versus measured OGT for each species, using a linear regression with features derived from genomic, tRNA, rRNA and ORF sequences. Species used for regression and evaluation are shown in dark purple and green, respectively. The dotted line indicates a perfect prediction
3.5 Including proteome-wide features significantly improves OGT prediction
While ORF feature correlation to OGT partially reflects the adaptation of the coding regions and mRNAs to the thermodynamic environment, it has been suspected that these correlations also reflected adaptations in each species’ proteome to OGT (Zeldovich et al., 2007). Temperature is known to correlate with protein folding, biochemistry and enzyme kinetics, all of which are essential to organismal viability (Cambillau and Claverie, 2000; Singer and Hickey, 2003; Suhre and Claverie, 2003). Based on these biological consequences, proteome derived features were hypothesized to be especially sensitive to the thermal environment. Therefore, the proteome was translated from each species’ ORFs, and features calculated from the derived proteome. These features included amino acid fractions, the fraction of the proteome that is charged or thermolablile, and the EK/QH, LK/Q, Polar/Charged and Polar/Hydrophobic amino acid ratios.
Supporting the hypothesis that proteins are particularly sensitive to temperature, proteome derived features were found to have the strongest correlation to OGT (Supplementary Table S1), with the greatest correlation being the fraction of the proteome composed of the amino acids ILVWYGERKP (Zeldovich et al., 2007). The linear regression of OGT using proteome features, in addition to previously described features, significantly improved accuracy (RMSE = 5.17°C, R2 = 0.835) (Fig. 4, Supplementary Eq. S1).
Fig. 4.
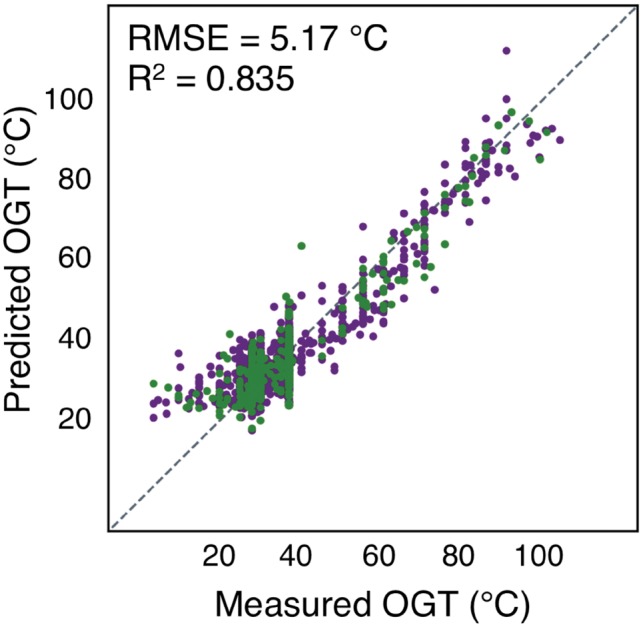
Proteome derived features significantly improve OGT prediction accuracy. Predicted versus measured OGTs for each species, using a linear regression with features derived from genomic, tRNA, rRNA, ORF and proteome sequences. Species used for regression and evaluation are shown in dark purple and green, respectively. The dotted line indicates a perfect prediction
3.6 Taxonomic clade specific regressions are the most accurate
The regressions described up to this point were made using all prokaryotic species. However, we had noted that the number of individual features correlated with OGT was much higher in Archaea than Bacteria (Supplementary Table S1). We therefore tested whether superkingdom specific regressions would be more accurate than the regression of all prokaryotes (Fig. 5).
Fig. 5.

Taxon specific linear regressions are most accurate. Predicted versus measured OGT for each species using superkingdom specific linear regressions for Archaea (A) and Bacteria (B). Species used for regression and evaluation are shown in dark purple and green, respectively. The dotted line indicates a perfect prediction
Using the NCBI taxonomic assignment for each species, an Archaea-only regression dramatically improved accuracy for these species (RMSE = 5.95°C, R2 = 0.938) (Eq S2). However, the Bacteria-only regression was worse (RMSE = 4.93°C, R2 = 0.767) (Eq S3), likely reflecting the greater sequence diversity and narrowed OGT range of this superkingdom. We also examined if the regressions could be further improved when the data is separated into lower taxonomic ranks. OGT regression was limited to those clades with at-least 100 species with a measured OGT to ensure the significance of the regression. Of the individual phyla, the most accurate regressions are found in the Euryarchaeota (RMSE = 6.69°C, R2 = 0.870), Firmicutes (RMSE = 4.82°C, R2 = 0.815) and Actinobacteria (RMSE = 3.18°C, R2 = 0.513) (Supplementary Fig. S4). In contrast, the Proteobacteria (RMSE = 4.87°C, R2 = 0.262) and Bacteroidetes (RMSE = 7.20°C, R2 = 0.324) regressions had more weakly correlated predicted and reported OGTs. Significant predictive regressions could also be calculated on the class, order and family ranks. However, it is worth noting that the current species classification methods have known discrepancies and inconsistencies (Parks et al., 2018), which make comparisons of taxon specific regressions of the same rank difficult.
4 Discussion
Knowing an organism’s optimal growth characteristics is central to addressing basic biological questions about how organisms adapt to a particular environmental niche. Further, the systematic study of adaption often requires the optimal growth conditions of the species of origin for each species and gene or protein examined. Additionally, proteins from organisms adapted to particular environmental niches are often particularly suited for structural biology (Jiang et al., 2002; Karpowich and Wang, 2013; Yernool et al., 2004) and industrial applications (Acharya and Chaudhary, 2012; Koskinen et al., 2008).
However, if the growth characteristics of already sequenced organisms are uncharacterized, the physiochemical properties of these genes that otherwise might be inferred are lost (Kunin et al., 2008). Consequently, this limits the use of these genomes in academic study and mining for biotechnology applications. Exacerbating this issue, high throughput sequencing has enabled rapid growth in the number of available genomic, metagenomic and derived proteomic sequences. This growth in genetic information is likely to outpace the laborious experimental task of characterizing the growth conditions of each species, leading to an increasing number of genomic sequences with unknown growth characteristics. This is already apparent in those organisms which have been ‘unculturable’ to date, but which have been sequenced by metagenomics.
It should be noted that prokaryotes are known to exchange genetic material via a variety of mechanisms. The effect of this material to thermal adaptation is unknown, nor is the subsequent evolution of these sequences after acquisition. Therefore no attempt was made to identify or exclude any exogenous genetic material. As calculated here, genomes containing exogenous genetic material approximate the weighted mean of the OGT of each originating species modified by some unknown amount of post-acquisition adaptation to the current optimal growth conditions. This process has been seen in the adaptation of prokaryotes to temperature (Zhaxybayeva et al., 2009) and other ecological niches (Wiedenbeck and Cohan, 2011), and in the directed evolution of enzymes (Akanuma et al., 1998; Merz et al., 2000).
4.1 OGT can be accurately predicted using only genome-derived features
To satisfy the need for growth condition data when only genomic sequences are available, we demonstrate a novel reverse ecology tool to accurately predict the OGT using solely the genomic sequence as input. Our method can predict the OGT for sequenced Archaea and Bacteria.
Genome classification is clearly essential for the most accurate prediction of OGT. The programs used for tRNA, rRNA and ORF identification all require some level of taxonomic classification. When applying the general prokaryotic regression, this is only requires the relatively simple exclusion of eukaryotic samples prior to sequencing (Venter et al., 2004). However, the most accurate OGT regressions are taxon specific, and therefore genomic samples require further classification. This assignment is routinely addressed in silico, using specialized bioinformatic tools which can easily assign taxonomic clade to genomic material (Kim et al., 2016; Wood and Salzberg, 2014).
As a simple proof-of-concept, the prokaryotic genomes were classified by superkingdom using the best scoring 16S rRNA hidden Markov model in Barrnap (Supplementary Fig. S5). The regressions for Archaea (RMSE = 6.31°C, R2 = 0.931) and Bacteria (RMSE = 4.96°C, R2 = 0.782) were of similar accuracy to the NCBI based regression.
4.2 Excluding genome size does not alter the regression accuracy
While prokaryote genome size is strongly correlated with OGT, it is unique among all features used here in requiring a complete genome for calculation. Therefore, this feature might not be available in metagenomic samples, or otherwise incompletely assembled genomes. Excluding this feature has only a minor impact on the regression for all prokaryotes (RMSE = 5.50°C, R2 = 0.813), or the separate regressions for Bacteria (RMSE = 5.19°C, R2 = 0.741), or Archaea (RMSE = 5.77°C, R2 = 0.941) (Supplementary Fig. S6).
4.3 Psychrophiles are poorly fit
While the final regressions of prokaryotes and Bacteria were generally accurate, species with optimal growth temperatures less than approximately 25°C are clearly poorly fit. This outcome is unsurprising, as few psychrophilic sequences are present in the dataset (Supplementary Fig. S1), and the mechanisms of thermoadaptation to higher and lower temperatures are not equivalent (Yang et al., 2015). Excluding those species with an OGT of less than or equal to 25°C yields a slightly better general prokaryotic regression (RMSE = 4.39°C, R2 = 0.874) (Supplementary Fig. S7). The archaeal regression improved modestly (RMSE = 5.99°C, R2 = 0.934), while the bacterial regression improved significantly (RMSE = 4.29°C, R2 = 0.801), reflecting the known OGT ranges of each superkingdom.
4.4 Accuracy not due to genomic similarity
Considering the increased accuracy of taxon specific regression, we hypothesized that adaptation to temperature may have occurred multiple times throughout evolutionary history, possibly by distinct mechanisms. In such a case, only the adaptations shared by all species would give the strongest correlation feature and OGT. Therefore, examining specific taxa individually, adaptations found only in that particular clade may become apparent. Additionally, by focusing on specific taxa the feature variance due to genetic variation is minimized, increasing the sensitivity of identifying OGT correlated features. Notably, particular taxa often specialize in particular ecological niches (Oren et al., 2009). Therefore, we hypothesized that the taxa specific regressions would minimize any confounding effects of adaptations to other environmental pressures.
Growth temperature can represent an ecological niche preferred by some taxa (Rothschild and Mancinelli, 2001). This presents the possibility that the regressions, and underlying OGT correlated features, are confounded by genomic similarity. We therefore examined if the difference in species’ OGTs or species’ features were correlated to species-species evolutionary distance for all prokaryotic species, based on 16S rRNA sequence distance. To a first order approximation, if OGT were correlated with genomic similarity in the present dataset, then the magnitude of OGT difference between two species should be proportional to the distance between those species (Supplementary Fig. S8A). However, while closely related species have small differences in OGT, there is no general correlation between OGT differences and species-species distance (Supplementary Fig. S8B). Similarly, based on the same hypothesis of species-species similarity driving OGT similarity, species distance from a reference species should have a minimum at that reference species’ OGT (Supplementary Fig. S8C). In contrast, when compared against a single species at the median OGT, there is no apparent trend (Supplementary Fig. S8D). Notably, the most distantly related species were found at or near the median OGT. As a specific example, mesophiles Pontibacillus marinus and Halorubrum lacusprofundi have identical OGTs (30°C) but are only distantly related (distance = 0.985). In contrast, the thermophile Sulfurihydrogenibium azorense is more closely related to Pontibacillus marinus (distance = 0.624) despite having an OGT difference of 38°C.
We next evaluated if feature values are correlated with species-species distance, using the strongly OGT correlated proteomic frequency of the amino acids ILVWYGERKP (r = 0.736) as a prototype. While closely related species have similar feature values, there is no general correlation between the difference in feature value and species-species distance (Supplementary Fig. S9B). For comparison, we also examined feature difference versus OGT difference. As species-species distance is a directionless value, we plotted the absolute OGT difference for consistency, with the results as expected for a linearly correlated feature (Supplementary Fig. S9C).
Finally, we ensure genomic similarity was not underlying regression accuracy by evaluating differences in regression accuracy versus species-species distance. Considering all species pairwise, the regression accuracy does not systematically change with species-species distance (Supplementary Fig. S9D).
These results suggest that features-OGT correlations and regression reflect specific adaptations to temperature, rather than species’ genomic similarity. Further, even if genomic similarity plays an underlying role in the accuracy of the described regressions, calculating features once is likely significantly more computationally efficient than comparing the genome of a query species to the genomes of all other characterized species.
4.5 Improvements over comparable methods
Our method significantly expands and improves upon the individual features previously described to correlate with OGT. By studying a much larger set of genomes, a more precise correlation between each feature and OGT can be calculated. Further, by using multiple features, more accurate and predictive regression models have been calculated. Notably, our method improves on previously reported analyses requiring particular genes being present in the genome, thereby making the method more general in application (Jensen et al., 2012). Also, this method quantitatively predicts an OGT rather than using classification (psychrophile, mesophile, thermophile or hyperthermophile). This improves on methods which predict OGT ranges (Jensen et al., 2012; Li et al., 2010; Lin and Chen, 2011; Taylor and Vaisman, 2010), where classification necessarily limited accuracy.
The most comparable method is reported by Zeldovich et al. (2007) calculating OGT from the proteome as OGT = 937F – 335, where F is the sum of the proteome fraction for the amino acids IVYWREL (Zeldovich et al., 2007). Using the current larger dataset, we calculate a lower correlation (r = 0.726) than previously reported. This is likely a consequence of more genomic sequences being available, and our keeping of individual species separate rather than averaging those with the same OGT. By considering more features derived from the source organism’s genome, the prokaryotic regression presented here clearly advances upon this previous method.
4.6 Application and validation
Applying these regressions, we predicted OGTs for those species with a genomic sequence available, but without a reported OGT in Sauer et al. (2015). In total, 1254 species’ OGTs were predicted (Supplementary Table S2). Of the species with newly predicted OGTs, 528 species had a reported optimal growth temperature in BacDive (Söhngen et al., 2016). The predicted and measured OGTs were strongly correlated (RMSE = 5.18°C, R2 = 0.758), validating the predictive value of this method (Supplementary Fig. S10).
While we focus on growth temperature, the same principle could be readily applied to other quantifiable characteristics of an organism’s optimal growth environment, such as pH, salinity, osmolarity or oxygen concentration. Similarly, eukaryotic OGTs may be predictable from genomic sequences using this method with modifications to account for differences in rRNA genes, ORF identification and mRNA splicing.
Supplementary Material
Acknowledgements
The authors thank Jennifer Marden for discussion and critical review of this manuscript.
Funding
This work was supported by the National Institutes of Health [R01- GM121994, R01-DK099023, R01-GM093825, R01-NS108151 to D-N.W]. D.B.S. was supported in part by a Postdoctoral Fellowship [PF-17-135-01] from the American Cancer Society and by the Office of the Assistant Secretary of Defense for Health Affairs, through the Peer Reviewed Cancer Research Program under Award No. W81XH-16-1-0153. Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
Conflict of Interest: none declared.
References
- Acharya S., Chaudhary A. (2012) Bioprospecting thermophiles for cellulase production: a review. Braz. J. Microbiol. Publ. Braz. Soc. Microbiol., 43, 844–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akanuma S. et al. (1998) Serial increase in the thermal stability of 3-isopropylmalate dehydrogenase from Bacillus subtilis by experimental evolution. Protein Sci., 7, 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano N. et al. (1997) Genomes and DNA conformation. Biol. Chem., 378, 1397–1404. [DOI] [PubMed] [Google Scholar]
- Benson D.A. et al. (2017) GenBank. Nucleic Acids Res., 45, D37–D42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burra P.V. et al. (2010) Reduction in structural disorder and functional complexity in the thermal adaptation of prokaryotes. PloS One, 5, e12069.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambillau C., Claverie J.M. (2000) Structural and genomic correlates of hyperthermostability. J. Biol. Chem., 275, 32383–32386. [DOI] [PubMed] [Google Scholar]
- Cangelosi G.A., Meschke J.S. (2014) Dead or alive: molecular assessment of microbial viability. Appl. Environ. Microbiol., 80, 5884–5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock P.J.A. et al. (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott R.P. (1963) Temperature-gradient incubator for determining the temperature range of growth of microorganisms. J. Bacteriol., 85, 889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison C.E. et al. (2011) Population genomics and local adaptation in wild isolates of a model microbial eukaryote. Proc. Natl. Acad. Sci. USA, 108, 2831–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N. et al. (1999) A nonhyperthermophilic common ancestor to extant life forms. Science, 283, 220–221. [DOI] [PubMed] [Google Scholar]
- Galtier N., Lobry J.R. (1997) Relationships between genomic G+C content, RNA secondary structures, and optimal growth temperature in prokaryotes. J. Mol. Evol., 44, 632–636. [DOI] [PubMed] [Google Scholar]
- Haney P.J. et al. (1999) Thermal adaptation analyzed by comparison of protein sequences from mesophilic and extremely thermophilic Methanococcus species. Proc. Natl. Acad. Sci. USA, 96, 3578–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H. et al. (2004) Comparative study on circadian rhythms of body temperature, heart rate, and locomotor activity in three species hamsters. Exp. Anim., 53, 43–46. [DOI] [PubMed] [Google Scholar]
- Hearing J. et al. (1989) Isolation of Chinese hamster ovary cell lines temperature conditional for the cell-surface expression of integral membrane glycoproteins. J. Cell Biol., 108, 339–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenlohe P.A. et al. (2010) Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet., 6, e1000862.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honglin Z. et al. (1993) Determination of thermograms of bacterial growth and study of optimum growth temperature. Thermochim. Acta, 216, 19–23. [Google Scholar]
- Hunter J.D. (2007) Matplotlib: a 2D graphics environment. Comput. Sci. Eng., 9, 90–95. [Google Scholar]
- Hurst L.D., Merchant A.R. (2001) High guanine-cytosine content is not an adaptation to high temperature: a comparative analysis amongst prokaryotes. Proc. Biol. Sci., 268, 493–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. et al. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James G. et al. (2013) An Introduction to Statistical Learning. Springer, New York. [Google Scholar]
- Jensen D.B. et al. (2012) Bayesian prediction of bacterial growth temperature range based on genome sequences. BMC Genomics, 13, S3.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. et al. (2002) Crystal structure and mechanism of a calcium-gated potassium channel. Nature, 417, 515–522. [DOI] [PubMed] [Google Scholar]
- Karpowich N.K., Wang D.-N. (2013) Assembly and mechanism of a group II ECF transporter. Proc. Natl. Acad. Sci. USA, 110, 2534–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T. et al. (2000) Archaeal adaptation to higher temperatures revealed by genomic sequence of Thermoplasma volcanium. Proc. Natl. Acad. Sci. USA, 97, 14257–14262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey P.J. et al. (2016) Ensembl Genomes 2016: more genomes, more complexity. Nucleic Acids Res., 44, D574–D580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khachane A.N. et al. (2005) Uracil content of 16S rRNA of thermophilic and psychrophilic prokaryotes correlates inversely with their optimal growth temperatures. Nucleic Acids Res., 33, 4016–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. et al. (2016) Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res., 26, 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohavi R. (1995) A study of cross-validation and bootstrap for accuracy estimation and model selection. In Proceedings of the 14th International Joint Conference on Artificial Intelligence - Volume 2, IJCAI’95. Morgan Kaufmann Publishers Inc., San Francisco, CA, USA, pp. 1137–1143.
- Koskinen P.E.P. et al. (2008) Bioprospecting thermophilic microorganisms from icelandic hot springs for hydrogen and ethanol production. Energy Fuels, 22, 134–140. [Google Scholar]
- Kreil D.P., Ouzounis C.A. (2001) Identification of thermophilic species by the amino acid compositions deduced from their genomes. Nucleic Acids Res., 29, 1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V. et al. (2008) A bioinformatician’s guide to metagenomics. Microbiol. Mol. Biol. Rev. MMBR, 72, 557–578. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. et al. (2007) Sequences downstream of the start codon and their relations to G + C content and optimal growth temperature in prokaryotic genomes. Antonie Van Leeuwenhoek, 92, 417–427. [DOI] [PubMed] [Google Scholar]
- Li Y. et al. (2010) A novel scoring function for discriminating hyperthermophilic and mesophilic proteins with application to predicting relative thermostability of protein mutants. BMC Bioinformatics, 11, 62.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.F. et al. (2008) ‘ Reverse ecology’ and the power of population genomics. Evol. Int. J. Org. Evol., 62, 2984–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Chen W. (2011) Prediction of thermophilic proteins using feature selection technique. J. Microbiol. Methods, 84, 67–70. [DOI] [PubMed] [Google Scholar]
- Lobry J.R., Chessel D. (2003) Internal correspondence analysis of codon and amino-acid usage in thermophilic bacteria. J. Appl. Genet., 44, 235–261. [PubMed] [Google Scholar]
- Lowe T.M., Chan P.P. (2016) tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res., 44, W54–W57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn D.J. et al. (2002) Synonymous codon usage is subject to selection in thermophilic bacteria. Nucleic Acids Res., 30, 4272–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz A. et al. (2000) Improving the catalytic activity of a thermophilic enzyme at low temperatures. Biochemistry, 39, 880–889. [DOI] [PubMed] [Google Scholar]
- Nguyen V. et al. (2017) Evolutionary drivers of thermoadaptation in enzyme catalysis. Science, 355, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren A. et al. (2009) Emended descriptions of genera of the family Halobacteriaceae. Int. J. Syst. Evol. Microbiol., 59, 637–642. [DOI] [PubMed] [Google Scholar]
- Parks D.H. et al. (2018) A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol., 36, 996–1004. [DOI] [PubMed] [Google Scholar]
- Pedregosa F. et al. (2011) Scikit-learn: machine learning in Python. J. Mach. Learn. Res., 12, 2825–2830. [Google Scholar]
- Perl D. et al. (2000) Two exposed amino acid residues confer thermostability on a cold shock protein. Nat. Struct. Biol., 7, 380–383. [DOI] [PubMed] [Google Scholar]
- Puigbò P. et al. (2008) Gaining and losing the thermophilic adaptation in prokaryotes. Trends Genet., 24, 10–14. [DOI] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson-Rechavi M. et al. (2006) Contribution of electrostatic interactions, compactness and quaternary structure to protein thermostability: lessons from structural genomics of Thermotoga maritima. J. Mol. Biol., 356, 547–557. [DOI] [PubMed] [Google Scholar]
- Rose M. et al. (2014) Are community environmental surfaces near hospitals reservoirs for gram-negative nosocomial pathogens? Am. J. Infect. Control, 42, 346–348. [DOI] [PubMed] [Google Scholar]
- Rothschild L.J., Mancinelli R.L. (2001) Life in extreme environments. Nature, 409, 1092–1101. [DOI] [PubMed] [Google Scholar]
- Sabath N. et al. (2013) Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol. Evol., 5, 966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sælensminde G. et al. (2007) Structure-dependent relationships between growth temperature of prokaryotes and the amino acid frequency in their proteins. Extremophiles, 11, 585–596. [DOI] [PubMed] [Google Scholar]
- Sauer D.B. et al. (2015) Rapid Bioinformatic Identification of Thermostabilizing Mutations. Biophys. J., 109, 1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer G.A.C., Hickey D.A. (2003) Thermophilic prokaryotes have characteristic patterns of codon usage, amino acid composition and nucleotide content. Gene, 317, 39–47. [DOI] [PubMed] [Google Scholar]
- Söhngen C. et al. (2016) BacDive–The Bacterial Diversity Metadatabase in 2016. Nucleic Acids Res., 44, D581–D585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart E.J. (2012) Growing unculturable bacteria. J. Bacteriol., 194, 4151–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K., Claverie J.-M. (2003) Genomic correlates of hyperthermostability, an update. J. Biol. Chem., 278, 17198–17202. [DOI] [PubMed] [Google Scholar]
- Talevich E. et al. (2012) Bio.Phylo: a unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinformatics, 13, 209.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor T.J., Vaisman I.I. (2010) Discrimination of thermophilic and mesophilic proteins. BMC Struct. Biol., 10, S5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekaia F. et al. (2002) Amino acid composition of genomes, lifestyles of organisms, and evolutionary trends: a global picture with correspondence analysis. Gene, 297, 51–60. [DOI] [PubMed] [Google Scholar]
- Turner T.L. et al. (2010) Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat. Genet., 42, 260–263. [DOI] [PubMed] [Google Scholar]
- Venter J.C. et al. (2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science, 304, 66–74. [DOI] [PubMed] [Google Scholar]
- van der Walt S. et al. (2011) The NumPy Array: a structure for efficient numerical computation. Comput. Sci. Eng., 13, 22–30. [Google Scholar]
- Wang Q. et al. (2015) The survival mechanisms of thermophiles at high temperatures: an angle of omics. Physiology (Bethesda), 30, 97–106. [DOI] [PubMed] [Google Scholar]
- Wiedenbeck J., Cohan F.M. (2011) Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev., 35, 957–976. [DOI] [PubMed] [Google Scholar]
- Wood D.E., Salzberg S.L. (2014) Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol., 15, R46.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.-L. et al. (2015) Low temperature adaptation is not the opposite process of high temperature adaptation in terms of changes in amino acid composition. Genome Biol. Evol., 7, 3426–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarza P. et al. (2008) The All-Species Living Tree project: a 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst. Appl. Microbiol., 31, 241–250. [DOI] [PubMed] [Google Scholar]
- Yernool D. et al. (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature, 431, 811–818. [DOI] [PubMed] [Google Scholar]
- Zeldovich K.B. et al. (2007) Protein and DNA sequence determinants of thermophilic adaptation. PLoS Comput. Biol., 3, e5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaxybayeva O. et al. (2009) On the chimeric nature, thermophilic origin, and phylogenetic placement of the Thermotogales. Proc. Natl. Acad. Sci. USA, 106, 5865–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H., Wu H. (2010) Gene-centric association analysis for the correlation between the guanine-cytosine content levels and temperature range conditions of prokaryotic species. BMC Bioinformatics, 11, S7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.