

Abstract
Cell encapsulation within 3D hydrogels is an attractive approach to develop effective cell-based therapies. However, little is known about how cells respond to the dynamic microenvironment resulting from hydrogel gelation-based cell encapsulation. Here, a tunable biomimetic hydrogel system that possesses alterable gelation kinetics and biologically relevant matrix stiffness is developed to study 3D dynamic cellular responses during encapsulation. Hydrogels are synthesized by cross-linking thiolated hyaluronic acid and thiolated chondroitin sulfate with polyethylene glycol diacrylate under cell-compatible conditions. Hydrogel properties are tailored by altering thiol substitution degrees of glycosaminoglycans or molecular weights of cross-linkers. Encapsulation of human mesenchymal stem cells through hydrogel gelation reveals high cell viability as well as a three-stage gelation-dependent cellular response in real-time focal adhesion kinase (FAK) phosphorylation in live single cells. Furthermore, stiffer hydrogels result in higher equilibrium FAK activity and enhanced actin protrusions. Our results demonstrate the promise of hydrogel-mediated cellular responses during cell encapsulation.
Keywords: glycosaminoglycans, hydrogels, human mesenchymal stem cells, cell encapsulation, focal adhesion kinase
ToC_ABSTRACT
Introduction
Mesenchymal stem cells (MSCs) offer a promising cell source for musculoskeletal regenerative engineering due to their ease of availability, high expansion capacity, and multipotency.[1] Hydrogels composed of water-swollen and cross-linked polymeric networks have been widely investigated as engineered matrices to support cell growth, differentiation, and organization to form regenerated tissue due to extracellular matrix (ECM)-mimicking and cell-interactive properties.[2],[3] Cell encapsulation within 3D hydrogels is an attractive approach for developing effective cell-based therapies by recapitulating the native cellular microenvironment.[4, 5] Precise control of material properties and optimization of cell-material interactions are critical to promote effective regeneration.[6] For example, MSCs could sense the differences in mechanical environments and initiate the mechanotransduction pathways, which ultimately would affect cell fate and distinct lineage differentiation.[7-9] Using hydrogels that stiffen or soften post cell encapsulation, pioneering studies have elucidated the importance of timing and magnitude of matrix stiffness changes in the regulation of cell behaviors and functions.[10-13] However, it remains largely unknown whether and how cells react to temporal hydrogel gelation during 3D encapsulation, which involves a transition of cell microenvironment from a viscoelastic fluid to an elastic solid. Here, we developed a novel tunable bioinspired glycosaminoglycan hydrogel system via thiol-ene click chemistry to study cellular responses during 3D encapsulation of human MSCs (hMSCs).
Natural glycosaminoglycans (GAGs) including hyaluronic acid (HA) and chondroitin sulfate (CS) have attracted significant research interests to develop hydrogels because they offer both a biomimetic microenvironment found in natural ECM and the availability of carboxylic acid and hydroxyl groups necessary for modification.[7] Specifically, HA, a non-sulfated GAG, is composed of alternating D-glucuronic acid and N-acetyl-D-glucosamine, and plays a key role in cellular processes including cell proliferation, morphogenesis, inflammation, and wound repair.[7, 14] CS, a sulfated GAG, is involved in interactions with various extracellular molecules such as growth factors and cytokines/chemokines to support cellular activities.[15, 16] Both HA and CS have good biocompatibility and biodegradability.[7] However, HA- or CS-derived hydrogels alone suffer from limited mechanical properties and uncontrollable degradation rates, which necessitates the need to combine with other polymers. For example, to combine the desirable benefits of both HA and CS into one integrated hydrogel system, collagen or gelatin was introduced to covalently cross-link HA with CS by means of collagen self-assembly,[17] copper(I)-catalyzed azide-alkyne cycloaddition,[18] or photo-cross-linking.[19] However, the application of these hydrogels fabricated from pure natural ECM-derived macromolecules as an engineered cellular matrix is hindered by the lack of appropriate control over the hydrogel molecular structure and cell-hydrogel interactions. Furthermore, slow reaction kinetics[17] and use of copper catalysts[18] present translational challenges for cell-related applications, especially when cells need to be encapsulated directly during the hydrogel formation.
In this study, we developed a biomimetic covalent hydrogel system under mild conditions to investigate 3D cell encapsulation by in situ cross-linking thiolated HA (HA-SH) and thiolated CS (CS-SH) with polyethylene glycol diacrylate (PEGDA) (Figure 1). This HA/CS/PEG (HCP) hydrogel system is unique, because it combines the controllable characteristics of polyethylene glycol (PEG) for efficient modulation of hydrogels’ physical properties[14, 15] with the biological benefits of HA and CS to recapitulate the biological and structural cues of native tissue ECM.[7, 15] Thiol-ene click chemistry was utilized due to its high efficiency and reliability without the need for using external stimuli, as well as absence of free radicals, catalysts, and by-products.[20, 21] To our knowledge, this is the first report that integrated natural/synthetic composite hydrogels of HA, CS, and PEG were fabricated through thiol-ene click chemistry. We demonstrated that various hydrogel properties including gelation kinetics, modulus, swelling, and morphology were efficiently modulated by varying thiol degree of substitution (DS) of GAG or modulating molecular weight (MW) of PEGDA. hMSCs were further encapsulated within the hydrogels and evaluated for cell viability, dynamics of focal adhesion kinase (FAK, an early mechanosensor that responds to changes in matrix stiffness[1, 22]) signalling, morphology and cytoskeleton. Interestingly, using a newly designed FAK peptide sensor, [23] hMSCs were found to exhibit a three-stage gelation-dependent cellular response in real-time FAK phosphorylation during 3D encapsulation (Figure 1). These results provide important new insights into real-time cellular responses during hydrogel-based cell encapsulation, and lay the foundation for fine-tuning hydrogel properties and cell-hydrogel interactions to promote in situ tissue regeneration.
Figure 1.

Schematic of the tunable HCP hydrogel design for cell encapsulation. hMSCs respond to microenvironment changes in a three-stage gelation-dependent manner as polymer phase evolves from a precursor fluid to a polymeric network (gel) via thiol-ene Michael addition click reactions.
Experimental
Thiolation of HA and CS
Carboxyl groups in HA and CS were functionalized to thiol groups by a two-step reaction scheme (Figure 2A). In a typical procedure, HA (1 g, 2.5 mmol) was dissolved in 100 mL 2-(N-morpholino) ethanesulfonic acid (MES) buffer (0.1 M MES, 0.1 M NaCl, pH 6.0) and allowed to dissolve overnight. EDC (2.4 g, 12.5 mmol) and NHS (3.9 g, 34 mmol) were added and allowed to react for 2 h. Then cystamine dihydrochloride (5.65 g, 25 mmol) was added to the mixture and allowed to react overnight while being stirred. The reaction mixture was exhaustively dialyzed (MWCO: 12-14 kDa) against distilled 0.1 M NaCl for 60 h, 25% ethanol for 12 h, and distilled (DI) water for 12 h. After dialysis, the product, HA-conjugated cystamine (HA-Cys), was lyophilized and kept at −20 °C. HA-conjugated cystamine (0.25 g) was dissolved in PBS or DI water with a concentration of 5 mg/mL. Then a calculated amount of DTT, depending on designed thiol DS, was added to the flask and pH was adjusted to the range of 7-8 with 1 M NaOH. After the mixture was stirred for 20 h, NaCl was added (1% w/v). Subsequently, thiolated HA solution was precipitated in 10-fold ethanol three times. The precipitation was dissolved in H2O at a concentration of approximately 5 mg/mL and the purified product was lyophilized and kept at −80 °C. Similar procedures were applied in the synthesis of CS-SH. The details are described in the section of Thiolation of CS, Supporting Information. The structures of functionalized HA and CS were confirmed by 1H NMR spectroscopy (D2O, Bruker ARX 400 MHz). The -Cys DS of HA-Cys and CS-Cys compounds were calculated using Equation S1. The thiol DS of HA-SH and CS-SH was calculated using Equation S2.
Figure 2.
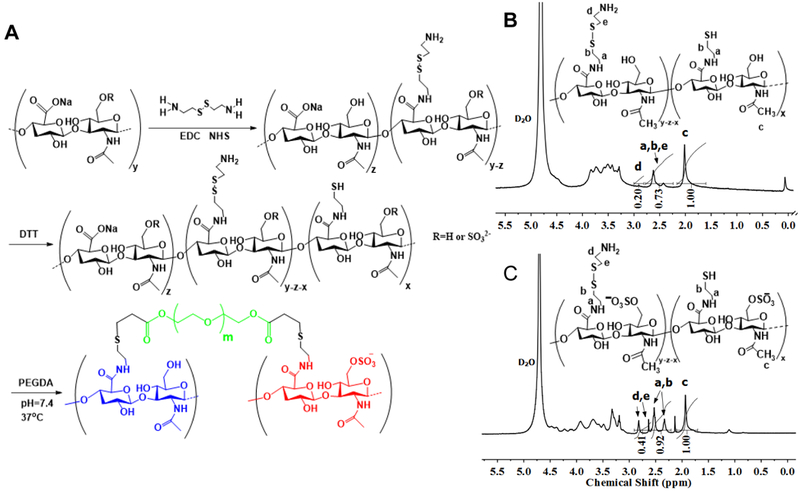
(A) HCP hydrogel formation. Carboxyl groups in HA or CS were reacted with cystamine dihydrochloride by activation with EDC and NHS. Reduction of cystamine by DTT resulted in free thiol groups in the HA or CS backbone. Cross-linking of HA-SH and CS-SH mixtures by conjugate addition using PEGDA. 1H NMR spectra confirmed successful synthesis of (B) HA-SH and (C) CS-SH.
Hydrogel formation
A total of 6 types of hydrogels were prepared by varying MW of PEGDA and thiol DS of HA-SH and CS-SH (Table S3). Under both high and low thiol DS, lyophilized HA-SH and CS-SH were separately dissolved in degassed PBS to prepare 1% and 5% (w/v) stock solutions under a nitrogen atmosphere, respectively. Three stock solutions of PEGDA700 with 5% (w/w), PEGDA3400 with 20% (w/w), and PEGDA8000 with 33% (w/w) were prepared in degassed PBS, respectively. Each of the mixtures of HA-SH and CS-SH in the volume ratio of 1: 1.03 was vortexed gently for about 15 seconds immediately following addition of the PEGDA stock solution. PEGDA stock solutions with varying weight concentrations were utilized for hydrogels with the same thiol DS to maintain the same feeding molar ratio of thiol groups to acrylate groups (1.07) and nearly the same acrylate molar concentrations (0.0089M and 0.0127 M at low and high thiol DS, respectively). The time to form a gel (denoted as gelation time) is defined as the time when the gel, in an inverted state, shows no fluidity for 1 min.[21] The experiment was performed in triplicate.
Swelling tests
Hydrogel samples (~200 μL) were prepared as described above. The swelling ratios of the prepared gels were obtained by the weighing method. In detail, the cylinder-shaped hydrogels were incubated at 37 °C in PBS for 60 h to reach the swelling equilibrium. The excess PBS was aspirated away. Then, the remaining saturated hydrogels were weighed (Ws), washed with distilled water 3 times, and lyophilized. The dry gels were weighed again (Wd). The experiments were performed in triplicate. The swelling ratio of the hydrogels was expressed as: Ws/Wd.[21]
Cryo scanning electron microscope (Cryo-SEM)
Cryo-SEM (Nova NanoSEM) at temperatures in the range of −100 to −140 °C at 3.0 kV was used to analyze the microstructure of hydrogels. Samples were positioned on a vacuum transfer rod, slam-frozen to −190 °C in nitrogen slush, and moved to the cryostat chamber at −130 °C. The top rivet was flicked off to produce a fractured surface that was sublimed at −90 °C for 2 min and coated with palladium.
Rheological characterization
Rheological experiments were carried out with a Discovery Series Hybrid Rheometer (DHR)-3 (TA) using parallel plate (20 mm diameter, 0°) in the oscillatory mode at 37 °C. 450 μL of a gel precursor solution was mixed by vortexing at room temperature for 15 seconds before loading onto the rheometer. Hydrogels were cast between the lower Peltier plate (preheated at 37 °C) and upper parallel plate. The parallel plate geometry was set to a gap of 1000 μm. Each hydrogel sample was used for only one test. A strain sweep from 0.01 to 10% strain was conducted to determine the linear-viscoelastic regime on a formed gel. As the strain was varied up to 10% strain (Figure S4), constant storage modulus (G’) values of HCP700L gels indicated that strains in the range of 0.01% to 10% were in the linear-viscoelastic regime. Consequently, a strain of 0.1% was selected for the subsequent frequency sweep tests. The linear G’ of HCP700L and HCP3400 gels with respect to frequency was determined. At low frequencies from 0.1 to 10 rad/s, G’ does not change, indicating the solid-like nature of the gel. As a result, 0.1% strain and a frequency of 6.28 rad/s were used for the time sweeps until a plateau of G’ was reached. Strain sweeps and frequency sweeps were performed in duplicate. Time sweeps were performed in triplicate.
Cross-linking efficiency
Cross-linking efficiency was assessed by thiol conversions and acrylate conversions. The total number of free thiol groups at the prescribed time was determined using Ellman’s assay. [21] Thermo Nicolet 6700 FTIR/FT-Raman Spectrophotometer was used to obtain Raman spectra. Using the adsorption at around 1640 cm−1 (-C=C-) and 1665 cm−1 (amide bond) as an analysis peak and an internal standard, respectively, the consumption of PEGDA can be calculated according to the Equation (1):
| 1 |
Where x, A0, and At, are acrylate conversions, the peak areas at 0 min, and the peak areas at 60 min, respectively.
Cell encapsulation and culture
hMSCs (Lonza) were cultured in a growth medium containing DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 1% glutamine (Life technologies), and 1% penicillin/streptomycin (Life technologies) at 37 °C with 5% CO2. HA-SH was sterilized by ethanol and was prepared in stock solutions under sterile conditions. CS-SH and PEGDA stock solutions were sterilized by filtration. For cell encapsulation in the hydrogels, cell pellets were obtained by centrifugation, followed by a brief resuspension with a hydrogel precursor solution through pipetting up and down gently to ensure a homogeneous distribution. Then cell suspension with a density of 8 × 105 cells/mL was transferred to an inverted pre-cut 1 mL syringe and incubated at 37 °C until complete gelation. Subsequently, hydrogels were injected to a 24 well tissue culture plate and incubated in growth media. The medium was changed every other day thereafter.
FAK study
The real-time monitoring of FAK phosphorylation of live single cells was conducted to examine cell-hydrogel interactions utilizing a fluorescence lifetime imaging microscopy (FLIM) with FAK phosphorylation biosensors.[23] In brief, ~5000 hMSCs were incubated with a ~20 μM permeable FAK phosphorylation biosensor in growth media for 15 min and washed three times with PBS. hMSCs with FAK peptide biosensors were then suspended in a 120 μL HCP3400 hydrogel precursor solution in a glass bottom dish (#1.5; MatTek Corporation, Ashland, MA) and loaded onto the stage of FLIM. Negative control samples contained a non-reactive PEG (MW: 3000 Da) instead of PEGDA3400 in the polymer precursor fluid. Measurements were repeated over 3-5 cells. Approximate 2-3 min were required for the FLIM test setup. To prepare the samples for real-time probing FAK phosphorylation of live single cells within different hydrogels after hydrogel stiffness reached equilibrium, hMSCs were suspended in 120 μL hydrogel precursor solutions in glass bottom dishes and incubated at 37 °C until complete gelation, following uptake of FAK biosensors. Then, encapsulated hMSCs were filled with a fresh medium and loaded onto the stage for FLIM analysis. The average fluorescence lifetime of FAKSOR was derived from 50 cells for HCP700L and HCP3400L gels, 26 cells for HCP8000L gels, and 20 cells for HCP700 gels. The FAK data are representative of three technical and three independent biological replicates. For FLIM imaging, a Time Correlated Single Photon Counting Fluorescence Lifetime Imaging system (MicroTime 200, Picoquant, and Germany) was utilized. Detailed instrumentation has been reported in previous publications.[24, 25]
Cell viability
The viability of the cells encapsulated in hydrogels after 1 and 5 days culture was investigated by performing a live/dead assay (Life technologies) according to the manufacturer’s instructions.
Immunofluorescent staining
Immunofluorescent staining was performed for cell nuclei and cytoskeletal protein actin. Briefly, cell-laden hydrogels were fixed in 4% paraformaldehyde and blocked with 1% BSA in PBS. Then samples were stained for F-actin with tetramethylrhodamine B isothiocyanate (TRITC)-conjugated phalloidin and for nuclei with 4’,6-diamidino-2-phenylindole (DAPI) according to the manufacturer’s instructions. Imaging was performed with a confocal microscope (Nikon A1R).
Statistical Analysis
Differences among groups were assessed by two-tailed unpaired t tests to identify statistical differences. All experiments were conducted independently at least three times, unless otherwise mentioned. Data were presented as mean ± standard deviation (SD). Single, double, and triple asterisks represent p < 0.05, 0.01, and 0.001, respectively. A p-value of 0.05 was set as the criterion for statistical significance.
Results and Discussion
HCP hydrogel synthesis, gelation, swelling, and morphology
The HCP hydrogels were fabricated by cross-linking of HA-SH and CS-SH using PEGDA via thiol-ene Michael addition under physiological conditions (Figure 2A). We first prepared HA-SH and CS-SH according to a previous method[26] with modified procedures via a two-pot synthesis procedure. First, carboxyl groups in HA and CS were reacted with amine groups of cystamine dihydrochloride via activation with EDC (Figure 2A). The structures of HA- and CS-conjugated Cys were confirmed by 1H NMR spectroscopy (Figure S1). By altering the molar ratios of carboxyl groups in HA to EDC from 2 to 10, DS of -Cys (4.1-43.1%) were obtained (Table S1). The CS/EDC molar ratio of 1:10 led to 69.6% DS of -Cys. Subsequently, reduction of -Cys by DTT gave rise to free thiol groups. The thiolation resulted in a new absorption appearing at 2573 cm−1 in Raman spectra (Figure S2) and the decrease of the Cys disulfide pendant signals at δ=2.32-2.82 ppm in 1H NMR spectra (Figure 2B-C). We also investigated the effects of pH and DTT amounts on the thiol DS of HA-SH and CS-SH. The results of thiol DS determined by Ellman’s assay and 1H NMR were consistent and listed in Table S2. By altering the pH of reaction mixture, the thiol DS of CS-SH could be increased from 18.2% to 38.3%. The pH of 7.2 was selected to offer reactive thiolate and avoid the possible degradation of HA and CS. By increasing the amount of DTT from 8 fold to 15 fold, the thiol DS of HA-SH rose from 5.3% to 16.5%. This two-pot reaction minimized the disulfide formation and allowed for efficient control of thiol DS, which is especially important when high MW of HA was adopted in this study. In addition, the residual -Cys groups allow for additional functionalization of the hydrogels if needed.
Hydrogels were then formed by simple mixing of HA-SH, CS-SH and PEGDA in PBS at 37 oC. The in situ hydrogel formation was confirmed by a tube-inverting method (Figure 3A). A series of HCP hydrogels were obtained by varying MW of PEGDA and thiol DS of GAG while maintaining the CS/HA mass ratio and feeding molar ratio of thiol groups to acrylate groups at 5.1[27] and 1.07, respectively (Table S3). When the MW of PEGDA was at 700, 3400, and 8000 Da, hydrogels were denoted as HCP700L, HCP3400L, and HCP8000L at the low thiol DS (thiol DS of HA-SH and CS-SH were 5.4% and 23.5%, respectively) and accordingly as HCP700, HCP3400, and HCP8000 at the high thiol DS (thiol DS of HA-SH and CS-SH were 11.9% and 34.1%, respectively).
Figure 3.
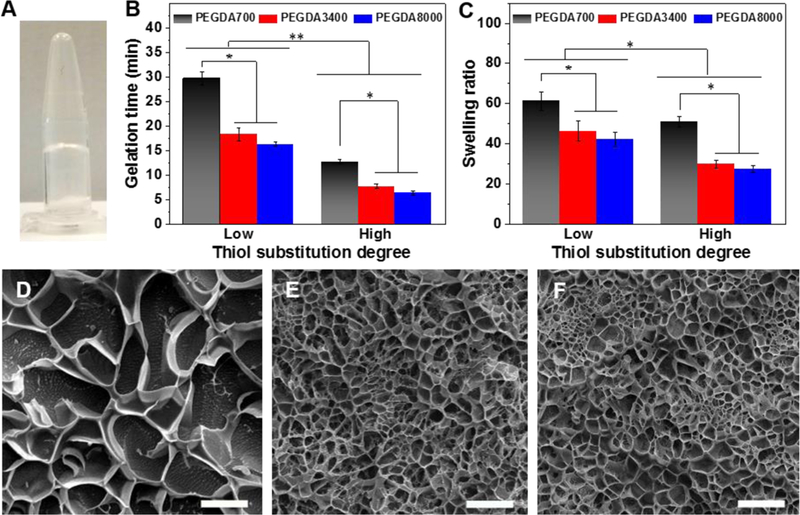
Characterization of gelation, swelling behavior, and morphology of HCP hydrogels. (A) Photograph of hydrogel formation. (B) Gelation times determined by the vial tilting method in PBS at 37 °C. (C) Swelling ratios of hydrogels. Representative Cryo-SEM images of (D) HCP700L, (E) HCP3400 L, and (F) HCP8000L hydrogels. Scale bars: 10 μm. All data are reported as mean ± SD (n = 3). * P < 0.05 and ** P < 0.01.
As shown in Figure 3B, when the thiol DS went up, hydrogel gelation time significantly decreased. HCP700L, HCP3400L, and HCP8000L gels exhibited gelation times of ~29.7, ~18.3, and ~16.3 min, respectively. In contrast, HCP700, HCP3400, and HCP8000 hydrogels exhibited significantly shorter gelation times of ~12.7, ~7.6, and ~6.3 min, respectively. The alterable gelation time within 5-30 min is highly suitable for cell encapsulation and delivery into the targeted site.[28] A too-short gelation time would lead to inefficient mixing, heterogeneous gelation, needle clogging during injection, and inconsistent cell responses,[28] whereas a too-long gelation time would adversely affect the effectiveness of hydrogel/cell retention at targeted injection sites. [4]
The HCP hydrogels are highly swollen in water resulting from the hydrophilicity of PEG, HA and CS molecules and the maintenance of cross-linked HCP networks. As the thiol DS increased, equilibrated swelling ratios of hydrogels cross-linked with PEGDA700, PEGDA3400, and PEGDA8000 decreased ~1.20 fold (61.3 ± 4.6 vs 51.0 ± 2.6), ~1.55 fold (46.4 ± 4.9 vs 29.9 ± 2.0), and ~1.53 fold (42.1 ± 3.7 vs 27.5 ± 1.8) fold, respectively (Figure 3C). At a higher thiol DS, the significantly lower equilibrated swelling ratios indicated the formation of denser cross-linked networks.
Cryo-SEM was used for relative comparison of hydrogel morphology and structure.4 Cryo-SEM images of hydrogels showed porous microstructures. HCP700L hydrogels possessed relatively larger pores than that of HCP3400L and HCP8000L hydrogels (Figure 3D-F).
We further characterized the ability of HCP hydrogels to support transport of macromolecules. Fluorescein isothiocyanate labelled dextran (FITC-dextran) was used as a model macromolecule. Figure S3 shows that the increase of PEGDA MW with constant feeding molar concentrations led to a slower release of FITC-dextran. The cumulative percentage release of FITC-dextran after 58 hours was approximately 84.3% for HCP700L, 77.6% for HCP3400L, and 72.2% for HCP8000L.
Rheological characterization of hydrogels
Both strain and frequency sweeps were performed in duplicate to verify that the shear modulus measurements were taken within the linear viscoelastic regime (Figure S4). Then, to monitor the evolution of polymeric network formation, we conducted time sweep rheological experiments in triplicate (representive results were showed in Figure 4A-C). Initially, G' (elastic response, an indication of the gel stiffness) was smaller than G'', indicating a viscoelastic fluid feature of polymer precursors. Then, G' and G'' increased rapidly as the gelation proceeds. Taking the HCP700 hydrogels (Figure 4A) for example, a higher buildup rate of G' than G'' led to the crossover of G' and G'' at the gel point of ~7.2 min (t = tgel), indicating the formation of elastic 3D networks. After the gel point, G' continued to increase until ~47.2 min (tgel ≤ t ≤ tplateau), resulting from the establishment of denser networks. Finally, G' reached a plateau (G'plateau) when interactions among HA-SH, CH-SH, and PEGDA approached completion. Given the fact that ~2-3 min were required for rheological test setup, tgel values determined by rheology were in agreement with those obtained using the vial-inverting method. The G' was well correlated with the thiol conversions to explain changes in the rheological properties of the thiol-ene Michael hydrogel system. During the initial stage of the reaction (t < ~10 min), thiol conversions in HCP700 gels increased quickly resulting in a rapid increase in G' (Figure S5, Figure 4A). As the thiol conversion increased from 57.4% to 78.8%, the G' increased by 2 orders of magnitude. Finally, both the thiol conversion and G' of HCP700 gels reached steady states within ~50 min. The results were consistent with previous work in a UV curing thiol-ene hydrogel system,[29] confirming that hydrogel stiffening is driven by thiol-Michael addition.
Figure 4.
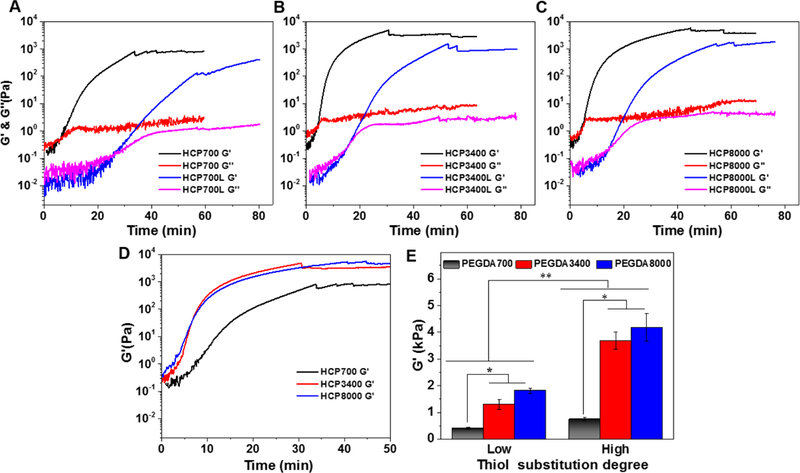
Rheological properties of HCP hydrogels. G’ and G“ as a function of time for (A) HCP700L and HCP700 hydrogels, (B) HCP3400L and HCP3400 hydrogels, and (C) HCP8000L and HCP8000 hydrogels. (D) Time dependence of G’ of HCP700, HCP3400, and HCP8000 hydrogels. (E) G’Plateau of gels as a function of thiol DS. Data are reported as mean ± SD (n = 3). * P < 0.05 and ** P < 0.01.
We next studied how the MW of PEGDA influenced G'plateau and gelation kinetics. In order to evaluate the reactivity of cross-linkers with different MWs, the same theoretical chemical cross-linking densities (i.e., the same feeding molar concentrations of thiol and PEGDA) were used in this study.[30] As shown in Figure 4D, HCP3400 and HCP8000 gels exhibited similar but significantly faster gelation kinetics than those of HCP700 gels. Accordingly, as shown in Figure 4D-E, HCP3400 and HCP8000 gels were comparably stronger, with similar G'plateau of 3168 ± 189 Pa and 3831 ± 390 Pa, respectively, than HCP700 hydrogels with a lower G'plateau of 771 ± 31 Pa. At low thiol DS, PEGDA MW showed similar effects on G'plateau and gelation kinetics. Adjusting MW of PEGDA at the same theoretical chemical cross-linking densities allows for control over gelation kinetics and stiffness in a broad range, while maintaining the GAG concentration constant.
To investigate the underlying mechanism, we further assessed the effective cross-linking density by determining acrylate conversions with Raman spectroscopy. Figure 5A shows Raman spectra of HCP700 hydrogel upon mixing (at 0 min) and after 60 min reaction. At 0 min, absorption bands at 2573 cm−1, 1640 cm−1, and 1716 cm−1, which correspond to -SH, -C=C-, and carbonyl group (-C=C-C=O), respectively, were clearly observed. In contrast, after 60 min, the -SH absorption decreased to a non-detectable level; absorption bands of -C=C- and carbonyl groups were significantly weakened; and a new absorption band that is assigned to -S-C-C-C=O appeared at 1738 cm−1, resulting from formation of a carbon-sulfur bond. Figure 5B shows Raman spectra of HCP700, HCP3400, and HCP8000 hydrogels at 60 min. The absorption band at ~1665 cm−1 ascribed to the amide bonds of HA-SH and CS-SH (Figure 5A) was used as the internal reference to characterize unreacted acrylate groups, because this absorption was constant and equal among different hydrogels during the course of the reactions. Acrylate conversions in HCP700, HCP3400, and HCP8000 hydrogels were found to be 79.3%, 95.6%, and 91.8%, respectively. The longer chain length PEGDA conjugated at one end presents greater probability to react with free thiol groups to form cross-links, thereby resulting in higher effective cross-linking densities in HCP3400 and HCP8000 hydrogels.[31] On the other hand, PEGDA700 would need a higher concentration to form a continuous network[32] and favour the formation of small loops and intramolecular cross-linking,[33] which together largely led to the lower gelation rates and G'plateau of HCP700. These results were in line with previous works in alginate-[33] based and thiolated HA-[31] based two-component hydrogel systems, which demonstrated that the increase in the chain length of cross-linkers (bi-functional PEG, MW ≤ 3400) caused the increase of G'plateau. When the PEGDA MW increased from 3400 to 8000 Da at the same theoretical chemical cross-linking densities, insignificant changes in gelation kinetics and stiffness were due to a lower effective cross-linking density resulting from enhanced steric effects of PEGDA8000, despite a higher polymer weight percentage.[30]
Figure 5.
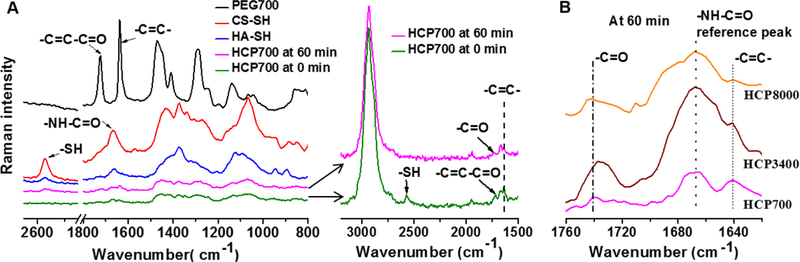
Raman spectra of (A) lyophilized HCP700 hydrogel at 0 min and 60 min, HA-SH, CS-SH, and PEGDA700. Raman spectra of (B) lyophilized HCP700, HCP3400, and HCP8000 hydrogels at 60 min.
The gelation kinetics and stiffness of HCP hydrogels could also be tuned by varying the thiol DS of GAG. With other conditions nearly identical, faster stiffening rates and higher G'plateau values were observed in the high thiol DS group, presumably attributable to their higher cross-linking densities. For example, as shown in Figure 4A-C, the tgel values of HCP700, HCP3400, and HCP8000 hydrogels are ~7.2, ~4.2, and ~3.9 min, respectively, whereas HCP700L, HCP3400L, and HCP8000L took significantly longer to reach the gel point with tgel values of ~25.4, ~15.2, and ~14.0 min, respectively. As the thiol DS decreased, the tplateau values for hydrogels using PEGDA700, PEGDA3400, and PEGDA8000 as cross-linkers increased ~1.7 fold (47.2 ± 5.2 vs 78.1 ± 0.0 min), ~1.6 fold (32.3 ± 1.8 vs 50.4 ± 3.4 min), and ~1.5 fold (35.4 ± 2.7 4 vs 51.6 ± 4.4 min), respectively. Accordingly, as the thiol DS increased, the G'plateau values for hydrogels cross-linked with PEGDA700, PEGDA3400, and PEGDA8000 increased ~1.9 fold (410 ± 30 Pa vs 771 ± 31 Pa), ~2.4 fold (1300 ± 190 Pa vs 3168 ± 189 Pa), and ~2.1 fold (1810 ± 106 Pa vs 3831 ± 390 Pa), respectively (Figure 4E). The G'plateau span from approximately 0.4 kPa to 4 kPa is in the range of stiffness for soft tissues.[7-8] The ability to independently tailor hydrogels with gelation kinetics over the timescale of minutes-to-hours and with biologically relevant stiffness in the kPa range [7-8] is desirable for investigating real-time cellular responses during cell encapsulation.
hMSC responses to HCP hydrogels
To measure how cells respond to the HCP hydrogels, in vitro studies were performed by encapsulating hMSCs during gelation of HCP hydrogels. Real-time hMSC responses in response to microenvironment changes during hydrogel gelation were characterized by monitoring dynamics of FAK signalling in live single cells utilizing FLIM with a newly designed FAK peptide sensor (FAKSOR) (Figure 6A).[23-25] FAK, a cytoplasmic non-receptor tyrosine kinase, is sensitive to changes in matrix stiffness and its activation plays a key role in transmitting extracellular cues to intracellular targets.[1, 22] As illustrated in Figure 6A, FAKSOR was first incubated with hMSCs to enable cellular uptake using an optimized protocol.[23] Then, hMSCs loaded with FAKSOR were suspended in HCP3400 hydrogel precursor solutions for FLIM measurements. The HCP3400 platform was utilized because 1) its stiffening rate (~30 min) offered an appropriate time window to characterize FAK signaling dynamics in live cells and 2) its stiffening range (0.01-3.2 kPa) spanned that of elasticity changes during MSC lineage commitment.[34] Gelation-mediated hMSC responses were observed during the cell encapsulation, which involved three sequential stages as the polymer precursor fluid evolved into a solid gel. Initially and immediately after being mixed with polymer precursors, hMSCs were essentially suspended in a viscoelastic fluid (Stage I, 0-5 min). Then, hydrogels began to transition from a viscoelastic fluid to an elastic solid followed by progressive increases in matrix stiffness (Stage II). During this period, single cell FLIM images showed that fluorescence lifetimes of FAKSOR increased along with time (Figure 6B). Normalized average fluorescence lifetimes of FAKSOR increased steadily up to ~14%, denoting that hMSCs were able to sense and respond to the gradual stiffening of their local hydrogel microenvironment (Figure 6C, red line). In comparison, average fluorescence lifetime of FAKSOR remained nearly unchanged in the polymer precursor fluid with a non reactive PEG, which suggested the absence of hydrogel formation and hydrogel-induced cellular response (Figure 6C, black line). Once G’ reached the plateau (Stage III, 30-60 min), the fluorescence lifetime of FAKSOR leveled off over 30 min, indicating formation of a stabilized cell-laden gel network (Figure 6D). After G’ reached the plateau, we also measured the real-time FAK phosphorylation of hMSCs that were encapsulated within different hydrogels. Representative FLIM images demonstrated that the fluorescence lifetimes of FAKSOR increased as the hydrogel G’plateau increased (Figure 7A). Quantitative analysis (Figure 7B) revealed that the average fluorescence lifetime values of FAKSOR were significantly greater in HCP3400L (3.64 ns) and HCP 8000L (3.76 ns) than in HCP700L (3.37 ns). As shown in Figure 7C, we also found that the average fluorescence lifetime of FAKSOR was significantly greater in HCP700 than in HCP700L gels (3.48 vs 3.37 ns), which further manifested the regulation of cellular FAK phosphorylation in response to matrix stiffness. Our real-time observations are in line with the relationship between matrix stiffness and FAK activation in 2D models[35, 36], and extend those findings to single cells of 3D encapsulation process.
Figure 6.
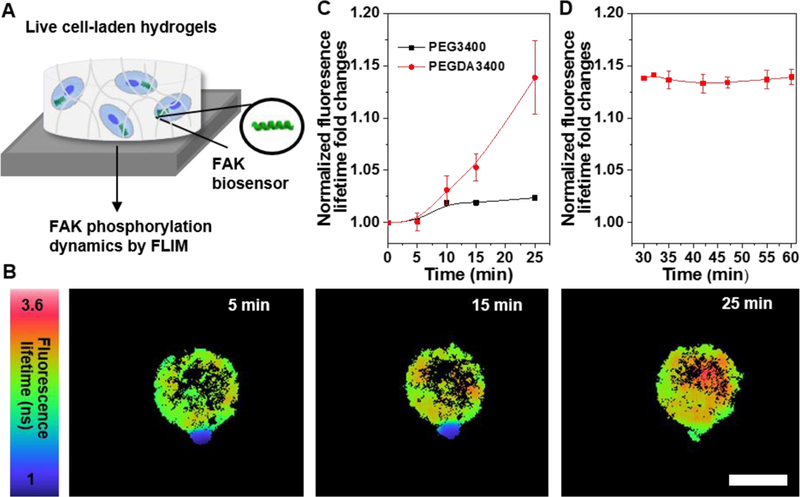
Dynamic FAK phosphorylation is dependent on hydrogel gelation during encapsulation. (A) Schematic of monitoring FAK phosphorylation of live hMSCs containing FAKSOR within hydrogels by FLIM. (B) Fluorescence lifetime images of hMSCs encapsulated in HCP3400 hydrogels as a function of time. Scale bar 20 μm. (C) Quantitative analysis showed a single hMSC gradually increased FAK phosphorylation over time during cell encapsulation in HCP3400 hydrogels(n=3). (D) FAKSOR average lifetime in hMSC-laden HCP3400 hydrogels remained constant over 30 minutes right after G’ reached plateau (n=5). All data are reported as mean ± SD.
Figure 7.

Dynamic FAK phosphorylation is dependent on hydrogel stiffness after encapsulation. (A) Fluorescence lifetime images showed hMSCs exhibited stiffness-dependent FAK phosphorylation. Scale bar 20 μm. (B) Quantitative analysis showed hMSCs increased FAK phosphorylation as the hydrogel stiffness increased (26 ≤ n ≤ 50). (C) Quantitative analysis showed a significant difference in FAKSOR average lifetime in hMSC-laden HCP700L and HCP700 hydrogels (20 ≤ n ≤ 26). All data are reported as mean ± SD. * P < 0.05 and *** P < 0.001.
Confocal images using a live/dead assay demonstrated that HCP hydrogels supported cell encapsulation and cell culture with high viability (> 95%) (Figure 8A-C and S6). Encapsulated hMSCs displayed an overall spherical morphology, which observation was in line with previous reports on HA/PEG and CS/PEG hydrogel cell cuture.[15, 37, 38] hMSCs responded to the stiffness differences of hydrogels as evidenced from the changes in actin cytoskeleton, an indicator of cell-material interactions. Specifically, hMSCs encapsulated in HCP700L hydrogels showed fewer defined actin fibers as compared to those in HCP3400L and HCP8000L hydrogels (Figure 8D-F). Furthermore, cortical actin protrusions of hMSCs into the surrounding matrix were only observed in HCP3400L and HCP8000L hydrogels. The protrusions play a critical role in 3D cell motility.[39] This finding was consistent with the FLIM results on FAK phosphorylation. Our data relating extracellular hydrogel properties to intracellular real-time FAK phosphorylation and cytoskeleton provide important new insights into cell-hydrogel interactions during cell encapsulation.
Figure 8.
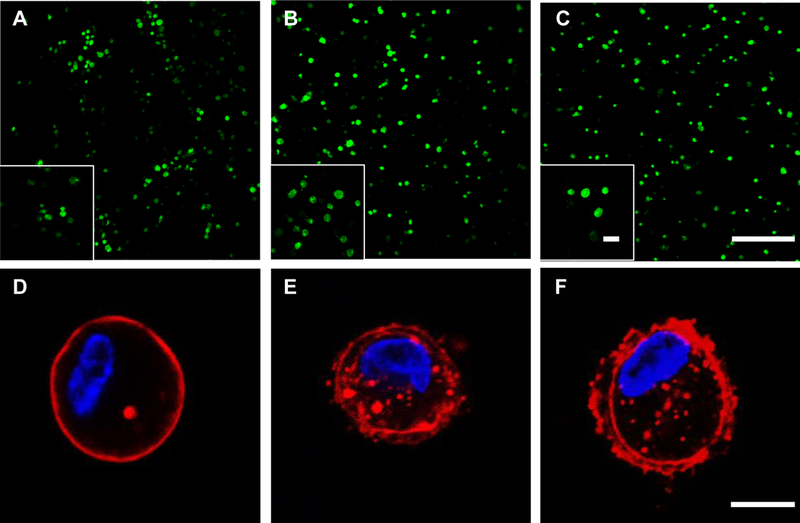
HCP hydrogels support high cell viability and stiffer hydrogels result in greater actin protrusion formation of encapsulated hMSCs. Live/Dead assay of hMSCs encapsulated in (A) HCP700L (B) HCP3400L (C) HCP8000L hydrogels after 1 day of culture; Scale bar: 200 μm, inset scale bar: 50 μm. Green: live cells. Red: dead cells. Immunofluorescent staining of an encapsulated hMSC within (D) HCP700L (E) HCP3400L (F) HCP8000L hydrogels after 1 day of culture. Red: actin. Blue: DAPI. Scale bar: 10 μm.
Conclusions
We have developed a tunable biomimetic HCP hydrogel system consisting of thiolated HA and thiolated CS cross-linked with PEGDA through thiol-ene Michael addition click reactions. This system synergistically combined the controllable characteristics of PEG with the ECM-mimicking properties of HA and CS. Variations in cross-linking by altering thiol DS or modulating PEGDA MW allowed for effectively tuning hydrogel physical properties, including the swelling, morphology, gelation kinetics, and stiffness. In vitro studies with hMSCs demonstrated the ability of the HCP hydrogels to support 3D cell encapsulation with high viability. Using FLIM with a newly designed FAK peptide sensor, we have determined for the first time a three-stage gelation-dependent hMSC response in FAK phosphorylation in live single cells as the cell microenvironment transitioned from a liquid state to an elastic solid and equilibrated when hydrogel stiffness reached the plateau. We have also shown that stiffer hydrogels promoted higher equilibrium FAK phosphorylation and more protrusion formation. These results highlighted the promise to tune hydrogel properties for achieving optimal cellular responses during cell encapsulation, and paved the way for further development of cell-instructive biomaterials for musculoskeletal tissue regeneration.
Supplementary Material
Acknowledgements
Funding support from the National Institutes of Health grant R03AR068108 and Purdue Start-up Package is greatly appreciated.
(Additional Supporting Information may be found in the online version of this article.)
Abbreviations
- hMSCs
human mesenchymal stem cells
- FAK
focal adhesion kinase
- MSCs
mesenchymal stem cells
- ECM
extracellular matrix
- GAGs
glycosaminoglycans
- HA
hyaluronic acid
- CS
chondroitin sulfate
- HA-SH
thiolated HA
- CS-SH
thiolated CS
- PEGDA
polyethylene glycol diacrylate
- HCP
HA/CS/PEG hydrogels
- PEG
polyethylene glycol
- DS
degree of substitution
- MW
molecular weight
- FAK
focal adhesion kinase
- EDC
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- NHS
N-hydroxysuccinimide Ellman s Reagent, 5, 5’-dithiobis (2-nitrobenzoic acid)
- DTT
DL-Dithiotreitol
- MES
2-(N-morpholino) ethanesulfonic acid
- PBS
phosphate buffer saline
- HA-Cys
HA-conjugated cystamine
- CS-Cys
CS-conjugated cystamine
- Cryo-SEM
Cryo scanning electron microscope
- FLIM
fluorescence lifetime imaging microscopy
- TRITC
tetramethylrhodamine B isothiocyanate
- DAPI
4’,6-diamidino-2-phenylindole
- SD
standard deviation
- FITC-dextran
Fluorescein isothiocyanate labelled dextran
- FAKSOR
focal adhesion kinase peptide sensor.
Footnotes
The table of contents entry should be 50-60 words long, and the first phrase should be bold. The entry should be written in the present tense and third person. The text should be different from the abstract text.
A new biomimetic hydrogel composed of hyaluronic acid, chondroitin sulfate, and polyethylene glycol is developed through thiol-Michael addition for 3D dynamic cell encapsulation. The hydrogel possesses tunable gelation kinetics and biologically-relevant stiffness. Encapsulated mesenchymal stem cells show three-stage gelation-dependent responses as cell microenvironment evolves from precursor fluids to polymeric networks. Stiffer hydrogels promote higher FAK phosphorylation and enhanced actin protrusions
REFERENCES
- [1].Lv Q, Deng M, Ulery BD, Nair LS, Laurencin CT, Clinical Orthopaedics and Related Research® 2013, 471, 2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Deng M, James R, Laurencin CT, Kumbar SG, NanoBioscience IE EE Transactions on 2012, 11, 3. [DOI] [PubMed] [Google Scholar]
- [3].Kumbar S, Laurencin C, Deng M, “Natural and synthetic biomedical polymers”, Burlington, San Diego: Elsevier, 2014. [Google Scholar]
- [4].Nicodemus GD, Bryant SJ, Tissue Engineering Part B: Reviews 2008, 14, 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Orive G, Hernandez RM, Gascon AR, Calafiore R, Chang TMS, Vos PD, Hortelano G, Hunkeler D, Lacik I, Shapiro AMJ, Pedraz JL, Nat Med 2003, 9, 104. [DOI] [PubMed] [Google Scholar]
- [6].Narayanan N, Jiang C, Uzunalli G, Thankappan SK, Laurencin CT, Deng M, Regenerative Engineering and Translational Medicine 2016, 2, 69. [Google Scholar]
- [7].Julian T, Yujie M, Bruekers SMC, Shaohua M, Huck WTS, Advanced Materials 2014, 26, 125. [DOI] [PubMed] [Google Scholar]
- [8].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- [9].Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera-Feliciano J, Mooney DJ, Nat Mater 2010, 9, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guvendiren M, Burdick JA, Nature Communications 2012, 3, 272. [DOI] [PubMed] [Google Scholar]
- [11].Yang C, Tibbitt MW, Basta L, Anseth KS, Nat Mater 2014, 13, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Stowers RS, Allen SC, Suggs LJ, Proceedings of the National Academy of Sciences 2015, 112, 1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tamate R, Ueki T, Kitazawa Y, Kuzunuki M, Watanabe M, Akimoto AM, Yoshida R, Chemistry of Materials 2016, 28, 6401. [Google Scholar]
- [14].Lam J, Truong NF, Segura T, Acta Biomater 2014, 10, 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Varghese S, Hwang NS, Canver AC, Theprungsirikul P, Lin DW, Elisseeff J, Matrix Biology 2008, 27, 12. [DOI] [PubMed] [Google Scholar]
- [16].Freudenberg U; Liang Y; Kiick KL; Werner C, Advanced Materials 2016, 28, 8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhang L, Li K, Xiao W, Zheng L, Xiao Y, Fan H, Zhang X, Carbohydrate Polymers 2011, 84, 118. [Google Scholar]
- [18].Hu X, Li D, Zhou F, Gao C, Acta Biomaterialia 2011, 7, 1618. [DOI] [PubMed] [Google Scholar]
- [19].Levett PA, Melchels FPW, Schrobback K, Hutmacher DW, Malda J, Klein TJ, Acta Biomaterialia 2014, 10, 214. [DOI] [PubMed] [Google Scholar]
- [20].Kharkar PM, Rehmann MS, Skeens KM, Maverakis E, Kloxin AM, ACS Biomaterials Science & Engineering 2016, 2, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zheng Shu X, Liu Y, Palumbo FS, Luo Y, Prestwich GD, Biomaterials 2004, 25, 1339. [DOI] [PubMed] [Google Scholar]
- [22].Mitra SK, Hanson DA, Schlaepfer DD, Nat Rev Mol Cell Biol 2005, 6, 56. [DOI] [PubMed] [Google Scholar]
- [23].Damayanti NP, Buno K, Narayanan N, Voytik Harbin SL, Deng M, Irudayaraj JMK, Analyst 2017, 142, 2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Damayanti N, PaulaáCraig A, Irudayaraj J, Analyst 2013, 138, 7127. [DOI] [PubMed] [Google Scholar]
- [25].Damayanti NP, Parker LL, Irudayaraj JM, Angew Chem Int Ed Engl 2013, 52, 3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shu XZ, Liu Y, Luo Y, Roberts MC, Prestwich GD, Biomacromolecules 2002, 3, 1304. [DOI] [PubMed] [Google Scholar]
- [27].Muzzarelli RAA, Greco F, Busilacchi A, Sollazzo V, Gigante A, Carbohydrate Polymers 2012, 89, 723. [DOI] [PubMed] [Google Scholar]
- [28].Darling NJ, Hung Y-S, Sharma S, Segura T, Biomaterials 2016, 101, 199. [DOI] [PubMed] [Google Scholar]
- [29].Chiou B-S, Khan SA, Macromolecules 1997, 30, 7322. [Google Scholar]
- [30].Jeon O, Song SJ, Lee K-J, Park MH, Lee S-H, Hahn SK, Kim S, Kim B-S, Carbohydrate Polymers 2007, 70, 251. [Google Scholar]
- [31].Young JL, Engler AJ, Biomaterials 2011, 32, 1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Padmavathi NC, Chatterji PR, Macromolecules 1996, 29, 1976. [Google Scholar]
- [33].Eiselt P, Lee KY, Mooney DJ, Macromolecules 1999, 32, 5561. [Google Scholar]
- [34].Parekh SH, Chatterjee K, Lin-Gibson S, Moore NM, Cicerone MT, Young MF, Simon CG, Biomaterials 2011, 32, 2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cukierman E, Pankov R, Stevens DR, Yamada KM, Science 2001, 294, 1708. [DOI] [PubMed] [Google Scholar]
- [36].Miron-Mendoza M, Seemann J, Grinnell F, Biomaterials 2010, 31, 6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nguyen LH, Kudva AK, Saxena NS, Roy K, Biomaterials 2011, 32, 6946. [DOI] [PubMed] [Google Scholar]
- [38].Kim J, Kim IS, Cho TH, Lee KB, Hwang SJ, Tae G, Noh I, Lee SH, Park Y, Sun K, Biomaterials 2007, 28, 1830. [DOI] [PubMed] [Google Scholar]
- [39].Fraley SI, Feng Y, Krishnamurthy R, Kim D-H, Celedon A, Longmore GD, Wirtz D, Nat Cell Biol 2010, 12, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.