

Huntington's disease (HD) is traditionally considered as a triad of movement, cognitive, and emotional disorders.1, 2, 3, 4 According to current clinical practice, “manifest HD” is diagnosed primarily according to motor criteria, that is, when a clinician has 99% confidence of “an otherwise unexplained extrapyramidal movement disorder” in someone with a family history of HD. It is desirable to incorporate other features of the triad into a new classification system for clinical care and research. Large observational studies, including TRACK‐HD, PREDICT‐HD, COHORT‐HD, and PHAROS, have greatly expanded our understanding of HD natural history. Subtle motor, cognitive, and emotional changes begin years before motor‐manifest HD, and brain changes likely begin even earlier, motivating the consideration of consistent definitions across a wide range including prior to the appearance of manifest HD.5, 6, 7, 8, 9, 10, 11, 12, 13, 14
In a previous publication, 3 of us (C.R., B.L., and R.R.) suggested modifying current diagnostic criteria to more broadly incorporate clinical features of HD.15 That article provided background about the natural history of HD, the current diagnostic criteria, and our proposed new diagnostic categories.
The Movement Disorder Society commissioned a task force to consider the issues in research and clinical definitions of HD and to develop a lexicon. Christopher Ross was selected as chair with cochairs Francisco Cardoso and Ralf Reilmann. The charge was to “select and convene a committee of HD experts, with involvement of patient and family representatives, to discuss diagnostic categories for Huntington's disease” based on the recent studies of natural history and biomarkers for HD and to “produce a set of recommendations for diagnostic classifications of HD.” The task force met in person on February 4 and 5, 2017, followed by several teleconferences. The task force proposed 3 categories, presymptomatic HD, prodromal HD, and manifest HD, with presymptomatic and prodromal together comprising the premanifest HD period (Fig. 1, Table 1). The criteria for these classifications were developed using an informal consensus approach and include both cognitive and motor components.
Figure 1.
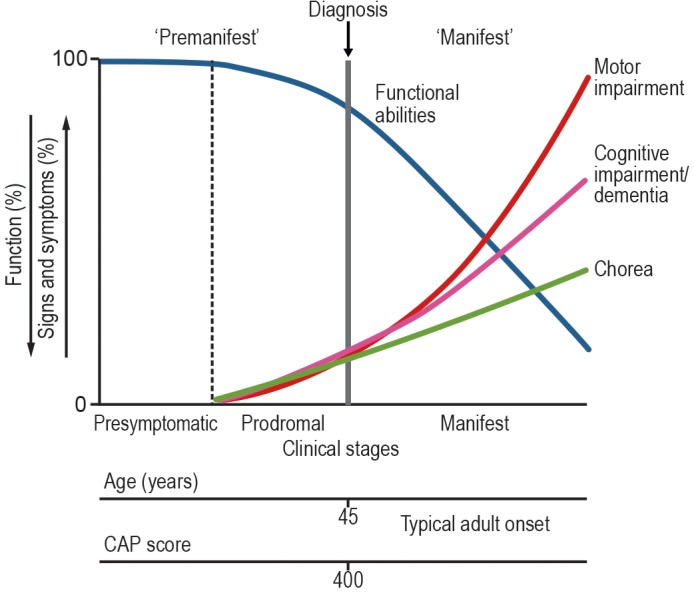
Diagnostic categories defined in this paper in the context of Huntington's disease (HD) natural history. The Cytosine, Adenine, Guanine (CAG) expansion in the Huntingtin gene is present at birth, and the mRNA and protein are widely expressed during development and adulthood, so the biological processes underlying the clinical syndrome are continuously active during the individual's lifetime. The extent of exposure to the effects of the CAG expansion can be quantified using the CAG age product (CAP) score (see text). The “premanifest HD” period before diagnosable onset according to the criteria proposed includes both the “presymptomatic HD” period, when there are no detectable clinical features, and the prodromal HD period, when there are subtle changes in motor and cognitive (and often emotional) function, with consequent subtle changes in functional abilities. During the manifest HD period, the motor and cognitive features progress, and functional abilities decline.
Table 1.
Criteria for diagnoses in individuals with a CAG‐repeat expansion in Huntingtin
| Diagnosis | Motor | Cognitive | Potential Treatment |
|---|---|---|---|
| (1) Presymptomatic HD | Dx conf 0–2 | Normal | (1) Disease modifying |
| (2) Prodromal HD (either A or B) |
A) Dx conf 2 B) Dx conf 3 |
(A) + Minor or major neurocognitive changes (B) With normal (unchanged) cognition |
(2A or B) Symptomatic or disease modifying |
| (3) Manifest HD (either A or B) |
A) Dx conf 3 B) Dx conf 4 |
(A) + Minor or major neurocognitive changes (B) With normal (unchanged) cognition |
(3A or B) Symptomatic or disease modifying |
Potential treatments apply to each of the 3 diagnoses regardless of the criteria for meeting the diagnosis. It is expected that the ability to define signs and symptoms would be enhanced by longitudinal follow‐up and assessments.
Dx conf, Diagnostic Confidence; HD, Huntington's disease.
Manifest HD
We propose adding nonmotor signs, particularly cognitive signs, to current motor diagnostic criteria15 (see Table 1). HD is a clinical diagnosis made on the basis of family history, personal history, neurological and psychiatric examinations, and genetic and any other appropriate testing. Extrapyramidal movement disorder is diagnosed based on the neurological examination, and the severity can be quantified using the Unified HD Rating Scale (UHDRS) motor examination,16 which yields a “total motor score” ranging from 0 to 124. Motor abnormalities in individuals tested positive for the Cytosine, Adenine, Guanine (CAG) expansion that causes HD or individuals with a family history of HD are rated on a 0 to 4 “diagnostic confidence” scale. When defining manifest HD, normal is rated 0, nonspecific abnormalities are rated 1, possible HD (“50% probability” of onset) is scored 2, probable onset (“90% confidence”) is rated 3, and definite (“99% probability”) onset is rated 4. Although the diagnostic confidence scale has the disadvantage of implying a “pseudo‐precision” via the somewhat arbitrary probability thresholds, it has a long history of use by HD clinicians and researchers. HD is a clinical diagnosis, and so a Total Motor Score (TMS) threshold for diagnosis is not suggested or implied. In the PREDICT‐HD study, which followed 225 premanifest cases through motor diagnosis, the mean TMS score at diagnosis was around 15, with a range from about 8 to 35. For individuals not followed longitudinally but seen for the first time, a higher motor score may be appropriate to make a diagnosis of definite HD.
In contrast, cognitive disorder is more difficult to diagnose because cognitive abilities vary considerably in the general population. It is optimal to confirm cognitive changes with longitudinal detailed neuropsychological testing, although this is not always feasible or affordable and should not be required. Information from family members or coworkers can provide crucial data about whether there appears to be cognitive impairment with change over time.
The task force proposes that cognitive disorder in manifest HD should be diagnosed according to the criteria of the Diagnostic and Statistical Manual, Fifth Edition (DSM)17 for either major neurocognitive disorder or minor neurocognitive disorder. A summary of how major neurocognitive disorder is defined in the DSM is as follows: first, evidence is provided of significant cognitive decline from a previous level of performance in 1 or more cognitive domains (complex attention, executive function, learning and memory, language, perceptual–motor, or social cognition). This is based on concern of the individual, a knowledgeable informant, or the clinician that there has been a significant decline in cognitive function and a substantial impairment in cognitive function, preferably documented by standardized neuropsychological testing or, in its absence, another quantified clinical assessment. Second, the cognitive deficits interfere with the independence in everyday activities (ie, at a minimum, requiring assistance with complex instrumental activities of daily living such as paying bills or managing medications). An important aspect of the DSM criteria is that the cognitive difficulties represent a decline from a previous level of functioning.
The tests listed in the National Institute of Neurological Disorders and Stroke Common Data Elements for Huntington's disease, cognitive domain (https://www.commondataelements.ninds.nih.gov/HD.aspx#tab=Data_Standards) provide a useful resource, with the Montreal Neuro Cognitive Assessment (MoCA) perhaps the simplest and most widely used screening test. The Symbol Digit Modalities Test, which is part of the UHDRS cognitive battery, is another well‐validated test for HD. Several executive function tests are listed, with the Trail‐Making Test perhaps the simplest to use for screening (an abbreviated trail‐making test is part of the MoCA).
A summary of how mild neurocognitive disorder is defined is as follows: first, evidence is provided of modest cognitive decline from a previous level of performance in 1 or more cognitive domains (complex attention, executive function, learning and memory, language, perceptual–motor, or social cognition), based on concern of the individual, a knowledgeable informant, or the clinician. Second, there is a modest impairment in cognitive performance. In this case, the cognitive impairments do not interfere with independence in everyday activities, but greater effort, compensatory strategies, or accommodation may be required.
It is important to exclude cognitive disorder secondary to depression, which can appear clinically similar to the cognitive disorder of HD, both sometimes described as having "subcortical" features. Depression and its associated cognitive disorder are not infrequent in all individuals and can appear many years before the onset of other features of HD and subsequently resolve,2, 14 and depression and other mood disorders are treatable, so the task force agreed that depression by itself should not be used to diagnose HD.
Other emotional disorders (irritability, apathy, and personality change) are more variable in their presentation, can occur at any time during the course of HD, and are common in individuals who do not have a CAG repeat expansion, thus making emotional changes challenging for establishing a diagnosis of HD.18, 19
One important complication for the diagnosis of HD is that individuals with HD often have reduced or absent awareness of their impairments, or anosognosia. It is vital that clinicians engage with family members or other informants who often can provide critical longitudinal information, beginning with the initial presentation and continuing throughout the course of HD.
Manifest HD causes functional disability. This might be relatively subtle in the earlier phases, for example, greater expenditure of time or effort on adequate work performance, but there is then progressive decline in social and occupational function, and finally difficulty with basic activities of daily living. The division of HD into 5 clinical “stages” based only on functional capacity scores, and not on any biological criteria, appeared arbitrary to the committee, and 3 broad clinical periods such as “early,” “moderate,” and “severe” were favored.
We propose that the combination of a motor diagnostic confidence of 3 (notionally “90%”), plus minor neurocognitive disorder, be sufficient for a diagnosis of manifest HD. Some individuals may already have major neurocognitive disorder when seen for diagnosis. Manifest HD could still be diagnosed by motor criteria alone, which would still require a diagnostic confidence of 4.
Prodromal HD
We propose a new diagnostic category of “prodromal HD.” Motor and cognitive changes appear subtly and progress slowly over years during this period until they are prominent enough for a diagnosis of manifest HD. Cognitive changes during the prodromal period often consist of "executive dysfunction," with difficulty recalling and sequencing multiple tasks.20, 21, 22, 23 This corresponds to the DSM category of “Minor Neurocognitive Disorder.”
We propose that a motor diagnostic confidence of 2 plus clear (although possibly subtle) cognitive changes, that is, “minor neurocognitive disorder,” be sufficient for a diagnosis of prodromal HD. TMS scores for individuals followed longitudinally might typically be in the range of 5 to 15, although as for manifest HD, a motor score threshold is not suggested and not implied, and higher scores may be appropriate for patients being seen for the first time.
Prodromal HD may have relatively minor impact on functional capacity, but treatment of motor (eg, tetrabenazine and related compounds or dopamine receptor blockers), cognitive (behavioral strategies), or emotional features (eg antidepressants, other pharmacological treatments for irritability or behavioral interventions) may be warranted. Counseling regarding family and social issues and strategies for coping with the developing illness is also important.24 The potential benefit of earlier access to treatment (both symptomatic and disease modifying—see later) is a key motivator for defining this category.
Presymptomatic HD
We believe that it is important to establish a category for individuals who have the CAG expansion but as yet have no signs or symptoms related to HD, that is, prior to prodromal HD. These individuals would have no significant motor signs on exam (diagnostic confidence 0 or 1, nonspecific findings) and no cognitive changes. Individuals in this group may be candidates for future disease‐modifying treatment to delay or prevent the onset.
Premanifest HD
The term premanifest is defined as the period prior to manifest HD, that is, inclusive of both the presymptomatic period and the prodromal period.
Issues for Discussion
Approach to Diagnosis in Individuals Who Have Not Had Genetic Testing
There was an agreement that similar categories would be applicable in the absence of genetically confirmed HD. The following terms are suggested: at risk for HD, but nonmanifest (ie, no signs or symptoms); clinically prodromal HD, and clinically manifest HD.15 The diagnostician would likely need more definitive clinical evidence, especially longitudinal data, in those at risk but without genetic testing when compared with individuals with confirmed CAG repeat expansion. There are numerous HD‐like syndromes that need to be considered in the differential diagnosis, especially in those without a clear family history.3, 25, 26 Thus, the criteria described in Table 1 are to be reserved for individuals with a positive genetic test for the HD CAG repeat expansion.
Role of Genetic Testing and Biomarkers for Diagnosis
Task force members generally agreed that the diagnostic categories should currently refer to the clinical status of the patient rather than to genetic or diagnostic testing or biomarker determination. HD is a clinical diagnosis. A combination of CAG repeat length and age, usefully summarized as the CAG age product or CAP score,27, 28 roughly predicts onset in groups of patients and serves as a useful longitudinal index of exposure to the effects of the CAG repeat expansion. However, CAG length by itself only explains about 50% in the variance of onset age. Genetic modifiers provide additional information.29 Quantitative motor examination (eg, Q‐Motor instrumentation30) is useful in research, but it has not been evaluated within the diagnostic setting.
Many years of study have consistently shown that progressive changes in structural magnetic resonance imaging (and likely functional magnetic resonance imaging and magnetic resonance spectroscopy) begin well before manifest HD,5, 7, 9, 31, 32, 33 and perhaps even before the period of prodromal HD. Blood and cerebrospinal fluid biomarkers, especially mutant Huntingtin (Htt) and neurofilament light‐chain levels,34, 35 are becoming increasingly relevant. Imaging and other tests should be used to rule out other conditions (especially in older individuals) and to determine whether other conditions may be contributing to the clinical presentation. However, biomarkers, although very useful for studying groups of patients and likely for clinical trials, at present are currently insufficiently precise and well validated for the diagnosis of individual patients.
“Cognitive Onset” of HD?
It is increasingly clear that cognitive dysfunction is important in causing functional disability and that prominent cognitive impairment can occur with relatively less noticeable motor changes. It is less clear whether substantial cognitive impairment occurs frequently in the absence of any detectable change on motor exam.23, 36, 37 The use of a multidimensional diagnosis including the cognitive, motor, behavioral, and functional aspects of the UHDRS (question 80) resulted in a slightly earlier diagnosis of HD than when based on motor exam alone (question 17),38 although the difference may have reflected functional as well as cognitive changes. Furthermore, the subgroup described as “predominantly cognitively impaired” in that study actually had higher motor scores than the other subgroups. Longitudinal neuropsychological testing (although time consuming and expensive) can be useful for cognitive assessment, contributing to the diagnosis of both manifest HD and prodromal HD. Individuals with early cognitive and behavioral changes may have greater anosognosia, and thus may not present in a timely fashion for motor examination. Even in the same family and with the same repeat expansion size, HD does not have an identical‐appearing onset and progression. Thus we retain motor criteria for the diagnosis of HD, but highlight the importance of cognition.
Emotional Aspects of HD
There was general agreement about the importance of emotional alterations in HD. Personality changes, especially apathy and irritability, are increasingly appreciated as important contributors to functional disability. The TRACK‐HD study7 has shown that apathy can begin quite early in what we would now term the prodromal phase, consistent with clinical experience. However, great caution should be taken before considering emotional changes exclusively for diagnosis of HD: emotional changes are quite common in individuals without the CAG expansion and may be even more common in individuals at risk for HD because of the disruptive effects of illness on family life.2 Furthermore, depression may be mistaken for apathy, and a readily treatable diagnosis should be prioritized. Nonetheless, emotional changes in someone at risk or with a known CAG expansion should trigger close follow‐up of both motor and cognitive features and additional testing as appropriate.
Possible Subtypes of HD
The committee discussed whether the diagnostic categories would apply equally well to all individuals with HD. Juvenile‐onset patients often have more bradykinesia, dystonia, and rigidity, and less (sometimes essential no) chorea, than adult‐onset cases, whereas late‐onset patients often have chorea‐predominant HD,39, 40 but both variants are well described by the current criteria. Cognitive onset has been proposed to be the most relevant for HD subtypes such as juvenile‐onset HD.39
Application to Research and Clinical Practice
We believe that ultimately the same categories should be used in both clinical and research settings to facilitate the transition from clinical trials to clinical practice. The set of classification criteria proposed are the result of an informal consensus process. The criteria are the result of holistic considerations of a selective group of publications and expert opinion. For these criteria to be refined both in clinical practice and research, it is desirable to validate this classification for accuracy, representativity, and usability in future studies. The currently proposed criteria are primarily designed for research because they use the research‐based examination, the UHDRS; however, as clinical trials of disease‐modifying therapy are advancing rapidly and early intervention may be most beneficial, we believe that clinical application should come soon, and that it is especially timely to include the prodromal HD diagnosis. We urge diagnostic and clinical practices to continue to be based on research and evidence‐based criteria.
The composition of the task force was designed to be international, and we hope that our ideas will have wide international application, but we also are aware of the importance of regional and cultural issues. In some countries, such as the United States, the diagnosis and treatment of HD is often restricted by insurance rules to neurologists. The task force recommends that any physician with relevant training and experience should be considered qualified to diagnose and treat HD, including psychiatrists, especially given the increasing awareness of the importance of the cognitive and emotional aspects of the disease. We also highlight that sensitive clinical judgment is of course paramount in discussing diagnostic and prognostic issues with patients and families.
Topics for the Future
Basic and clinical research in HD is moving rapidly, with clinical trials in progress (eg, clinicaltrials.gov, NCT02519036) or in planning stages for several strategies for huntingtin lowering. These disease‐modifying strategies have the potential to be applied in the prodromal or presymptomatic periods, making the availability of these diagnostic categories especially relevant for clinical practice.41, 42 In addition, biomarker research is advancing rapidly.28, 34, 35 The proposed categories are based on clinical examination and have the limitation of the use of the diagnostic confidence scale with its disadvantage of implying a “pseudo‐precision” via the “probability” thresholds. For many reasons, therefore, we propose a frequent reexamination of these diagnostic categories and a reassessment in 2 to 3 years, especially in relation to biomarkers.
We also urge further research on the topics discussed previously. Some topics for further study include the nature of early cognitive and emotional changes and their correlation with imaging and other biomarkers and the question of which signs and symptoms are most important in causing functional disability. The combination of datasets from the large observational studies will facilitate these studies, for instance, the identification of early cognitive changes.43 An important question for the timing of treatment is the point at which neuronal cell death or irreversible neuronal changes begin, as treatment, if safe, should ideally begin before that point. Given the limited access to different brain regions of some of the large molecules currently under study, understanding of the regional differences (and possible spread within the brain) of pathology at different points in the natural history will be especially important. All of these questions could use the CAP score more systematically to help order and sequence clinical and neurobiological changes and help in designing clinical trials.44 It would be especially valuable to design measures of the earliest changes in functional abilities. In keeping with the international nature of the task force, it would also be useful to know the extent of regional differences in the impact of definitions and clinical diagnoses. Perhaps most important will be research to demonstrate the efficacy of Htt‐lowering or other disease‐modifying therapies given during the premanifest period to delay or even conceivably prevent the onset of manifest HD.
Comments and Suggestions
We welcome comments and suggestions. They should be sent to the chair (caross@jhu.edu).
Author Roles
1) Research project: A. Conception, B. Organization, C. Execution; 2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3) Manuscript: A. Writing of the first draft, B. Review and Critique.
C.A.R.: 1A, 1B, 1C, 3A, 3B
R.R.: 1A, 1B, 1C, 3B
F.C.: 1B, 1C, 3B
E.M.: 1C, 3B
C.T.: 1C, 3B
J.S.: 1C, 3B
B.L.: 1C, 3B
Z.P.: 1C, 3B
B.L.: 1C, 3B
A.M.: 1C, 3B
J.L.: 1C, 3B
T.S.: 1C, 3B
J.B.: 1C, 3B
S.T.: 1C, 3B
Disclosures
Ethical Compliance Statement
The authors confirm that, since there were no subjects or patients involved, patient consent or the approval of an institutional review board were not relevant nor required for this work. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest
No relevant funding sources or conflicts of interest, given the nature of the article. No off‐label uses of medications or other treatments discussed. Participants received financial support for travel to the meeting February 4 and 5, 2017, from the Movement Disorder Society. Costs for phone conferences were paid by the Movement Disorder Society.
Financial Disclosure for previous 12 months
C.M.T reports employment with Virginia Commonwealth University and Virginia Commonwealth University Health Systems. She received the following honoraria: Huntington Study Group for scientific advisory consulting to uniQure; Rosenthal Research, contracted by unnamed sponsor, for structured interview on Huntington disease treatment; Tremor Research Group for service as the elected Vice President of Tremor Research Group; and Virginia Neurological Society for serving as faculty lecturer at the 2019 annual Virginia Neurological Society meeting. Dr. Testa is the Virginia Commonwealth University site principal investigator for the Enroll‐HD study sponsored by the nonprofit Cure HD Initiative (CHDI) Foundation and is the site principal investigator for the SIGNAL study, contracted through the Huntington Study Group and sponsored by Vaccinex. Dr. Testa is a site principal investigator for the Dystonia Coalition study; the full study is funded under National Institutes of Health/National Organization for Rare Diseases (NORD) Grant 5 U54 NS065701. The other authors declare that there are no additional disclosures to report.
Acknowledgments
We thank the International Parkinson and Movement Disorder Society for support and Kristy L. Moeller for coordination. We thank Huntington's Disease Society of America and International Huntington Association for comments. We also thank the anonymous peer reviewers for helpful critiques of our initial submission.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Huntington G. On chorea. Med Surg Rep 1872;26:317–321. [Google Scholar]
- 2. Folstein SE. Huntington's Disease: A Disorder of Families. Baltimore, MD: Johns Hopkins University Press; 1989. [Google Scholar]
- 3. Cardoso F. Huntington disease and other choreas. Neurol Clin 2009;27(3):719–736, vi. [DOI] [PubMed] [Google Scholar]
- 4. Donaldson I, Marsden CD, Schneider S. Marsden's Book of Movement Disorders. Oxford, UK: Oxford University Press; 2012. [Google Scholar]
- 5. Tabrizi SJ, Scahill RI, Durr A, et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK‐HD study: the 12‐month longitudinal analysis. Lancet Neurol 2011;10(1):31–42. [DOI] [PubMed] [Google Scholar]
- 6. Huntington Study Group COHORT Investigators , Dorsey ER. Characterization of a large group of individuals with Huntington disease and their relatives enrolled in the COHORT study. PloS One 2012;7(2):e29522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early‐stage Huntington's disease in the TRACK‐HD study: analysis of 36‐month observational data. Lancet Neurol 2013;12(7):637–649. [DOI] [PubMed] [Google Scholar]
- 8. Paulsen JS, Long JD, Johnson HJ, et al. Clinical and biomarker changes in premanifest Huntington disease show trial feasibility: a decade of the PREDICT‐HD study. Front Aging Neurosci 2014;6:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Paulsen JS, Long JD, Ross CA, et al. Prediction of manifest Huntington's disease with clinical and imaging measures: a prospective observational study. Lancet Neurol 2014;13(12):1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Duijn E, Reedeker N, Giltay EJ, Eindhoven D, Roos RAC, van der Mast RC. Course of irritability, depression and apathy in Huntington's disease in relation to motor symptoms during a two‐year follow‐up period. Neurodegener Dis 2014;13(1):9–16. [DOI] [PubMed] [Google Scholar]
- 11. Zielonka D, Mielcarek M, Landwehrmeyer GB. Update on Huntington's disease: advances in care and emerging therapeutic options. Parkinsonism Relat Disord 2015;21(3):169–178. [DOI] [PubMed] [Google Scholar]
- 12. Huntington Study Group PHAROS Investigators , Biglan KM, Shoulson I, et al. Clinical‐Genetic Associations in the Prospective Huntington at Risk Observational Study (PHAROS): implications for clinical trials. JAMA Neurol 2016;73(1):102–110. [DOI] [PubMed] [Google Scholar]
- 13. Jacobs M, Hart EP, van Zwet EW, et al. Progression of motor subtypes in Huntington's disease: a 6‐year follow‐up study. J Neurol 2016;263(10):2080–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Epping EA, Kim J‐I, Craufurd D, et al. Longitudinal psychiatric symptoms in prodromal Huntington's disease: a decade of data. Am J Psychiatry 2016;173(2):184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reilmann R, Leavitt BR, Ross CA. Diagnostic criteria for Huntington's disease based on natural history. Mov Disord 2014;29(11):1335–1341. [DOI] [PubMed] [Google Scholar]
- 16. Huntington Study Group . Unified Huntington's Disease Rating Scale: reliability and consistency. Mov Disord 1996;11(2):136–142. [DOI] [PubMed] [Google Scholar]
- 17. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition Washington, DC: American Psychiatric Association; 2013. [Google Scholar]
- 18. Zappacosta B, Monza D, Meoni C, et al. Psychiatric symptoms do not correlate with cognitive decline, motor symptoms, or CAG repeat length in Huntington's disease. Arch Neurol 1996;53(6):493–497. [DOI] [PubMed] [Google Scholar]
- 19. Smith MM, Mills JA, Epping EA, Westervelt HJ, Paulsen JS, PREDICT‐HD Investigators of the Huntington Study Group. Depressive symptom severity is related to poorer cognitive performance in prodromal Huntington disease. Neuropsychology 2012;26(5):664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington's disease decades before diagnosis: the Predict‐HD study. J Neurol Neurosurg Psychiatry 2008;79(8):874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stout JC, Paulsen JS, Queller S, et al. Neurocognitive signs in prodromal Huntington disease. Neuropsychology 2011;25(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Rourke JJF, Beglinger LJ, Smith MM, et al. The Trail Making Test in prodromal Huntington disease: contributions of disease progression to test performance. J Clin Exp Neuropsychol 2011;33(5):567–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harrington DL, Smith MM, Zhang Y, Carlozzi NE, Paulsen JS, PREDICT‐HD Investigators of the Huntington Study Group. Cognitive domains that predict time to diagnosis in prodromal Huntington disease. J Neurol Neurosurg Psychiatry 2012;83(6):612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McCusker EA, Loy CT. Huntington disease: the complexities of making and disclosing a clinical diagnosis after premanifest genetic testing. Tremor Hyperkinetic Mov N Y N 2017;7:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Margolis RL, Ross CA. Diagnosis of Huntington disease. Clin Chem 2003;49(10):1726–1732. [DOI] [PubMed] [Google Scholar]
- 26. Schneider SA, Bhatia KP. Huntington's disease look‐alikes. Handb Clin Neurol 2011;100:101–112. [DOI] [PubMed] [Google Scholar]
- 27. Zhang Y, Long JD, Mills JA, et al. Indexing disease progression at study entry with individuals at‐risk for Huntington disease. Am J Med Genet Part B Neuropsychiatr Genet 2011;156B(7):751–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol 2014;10(4):204–216. [DOI] [PubMed] [Google Scholar]
- 29. Genetic Modifiers of Huntington's Disease (GeM‐HD) Consortium . Identification of genetic factors that modify clinical onset of Huntington's disease. Cell 2015;162(3):516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Reilmann R, Bohlen S, Kirsten F, Ringelstein EB, Lange HW. Assessment of involuntary choreatic movements in Huntington's disease—toward objective and quantitative measures. Mov Disord 2011;26(12):2267–2273. [DOI] [PubMed] [Google Scholar]
- 31. Aylward EH, Sparks BF, Field KM, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 2004;63(1):66–72. [DOI] [PubMed] [Google Scholar]
- 32. Harrington DL, Long JD, Durgerian S, et al. Cross‐sectional and longitudinal multimodal structural imaging in prodromal Huntington's disease. Mov Disord 2016;31(11):1664–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sturrock A, Laule C, Wyper K, et al. A longitudinal study of magnetic resonance spectroscopy Huntington's disease biomarkers. Mov Disord 2015;30(3):393–401. [DOI] [PubMed] [Google Scholar]
- 34. Wild EJ, Boggio R, Langbehn D, et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington's disease patients. J Clin Invest 2015;125(5):1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Byrne LM, Rodrigues FB, Blennow K, et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington's disease: a retrospective cohort analysis. Lancet Neurol 2017;16(8):601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Loy CT, McCusker EA. Is a motor criterion essential for the diagnosis of clinical huntington disease? PLoS Curr 2013;5 10.1371/currents.hd.f4c66bd51e8db11f55e1701af937a419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ross CA, Pantelyat A, Kogan J, Brandt J. Determinants of functional disability in Huntington's disease: role of cognitive and motor dysfunction. Mov Disord 2014;29(11):1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Biglan KM, Zhang Y, Long JD, et al. Refining the diagnosis of Huntington disease: the PREDICT‐HD study. Front Aging Neurosci 2013;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ribaï P, Nguyen K, Hahn‐Barma V, et al. Psychiatric and cognitive difficulties as indicators of juvenile huntington disease onset in 29 patients. Arch Neurol 2007;64(6):813–819. [DOI] [PubMed] [Google Scholar]
- 40. Chaganti SS, McCusker EA, Loy CT. What do we know about Late Onset Huntington's Disease? J Huntingt Dis 2017;6(2):95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wild EJ, Tabrizi SJ. Therapies targeting DNA and RNA in Huntington's disease. Lancet Neurol 2017;16(10):837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Deb A, Frank S, Testa CM. New symptomatic therapies for Huntington disease. Handb Clin Neurol 2017;144:199–207. 10.1016/B978-0-12-801893-4.00017-1 [DOI] [PubMed] [Google Scholar]
- 43. Garcia TP, Wang Y, Shoulson I, Paulsen JP, Marder K. Disease progression in Huntington disease: an analysis of multiple longitudinal outcomes. J Huntingtons Dis 2018;7(4):337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Long JD, Landwehrmeyer B, Palermo G, Schobel S, Tabrizi SJ. Enrichment strategy in early‐to‐moderate manifest Huntington's disease (HD) based on CAG‐age product (CAP) ≥400 threshold. Paper presented at: Huntington Study Group 2018; November 8–10, 2018; Houston, TX.