

ABSTRACT
Background
The phenotypic spectrum of adenylyl cyclase 5 (ADCY5)‐related disease has expanded considerably since the first description of the disorder in 1978 as familial essential chorea in a multiplex family.
Objective
To examine recent advances in the understanding of ADCY5‐related dyskinesia and outline a diagnostic approach to enhance clinical detection.
Methods
A pragmatic review of the ADCY5 literature was undertaken to examine unique genetic and pathophysiological features as well as distinguishing clinical features.
Results
With over 70 cases reported to date, the phenotype is recognized to be broad, although distinctive features include prominent facial dyskinesia, motor exacerbations during drowsiness or sleep arousal, episodic painful dystonic posturing increased with stress or illness, and axial hypotonia with delayed developmental milestones. Uncommon phenotypes include childhood‐onset chorea, myoclonus‐dystonia, isolated nongeneralized dystonia, and alternating hemiplegia.
Conclusion
The ongoing expansion in clinical features suggests that ADCY5 remains underdiagnosed and may account for a proportion of “idiopathic” hyperkinetic movement disorders. Enhanced understanding of its clinical features may help clinicians improve the detection of complex or uncommon cases.
Keywords: episodic movement disorders, ADCY5 mutation, phenotypic spectrum
The advent of next‐generation massively parallel sequencing technologies has given rise to rapid discoveries in underlying genetic causes of previously undiagnosed rare clinical syndromes. Descriptions of unifying clinical features, however, remains a crucial aspect in this evolving process. An example is the recognition of adenylyl cyclase 5 (ADCY5) gene mutations as a novel cause for a range of “dyskinetic” phenotypes. Detailed clinical descriptions of a curious familial disorder began in 1978 with the recognition that sporadic paroxysmal choreoathetosis in a young woman1 had been transmitted to her daughter.2 That same year, a family with “dyskinesia and facial myokymia,” in which the movements were both dystonic and choreic, was reported.3 A combination of further clinical descriptions4 and familial linkage and exome sequencing analysis in this multigenerational family subsequently led to the discovery of a missense variant in the ADCY5 gene.5 There has since been a rapid expansion of the reported phenotypes and pathogenic variants, including the one responsible for disease in the family first published in 1967.6 These genetic studies, in combination with functional studies of the effect of variants in vitro, have shed light on possible underlying disease mechanisms.7, 8 Here, we review some of these advances with the aim of highlighting features of this novel disorder for clinicians to help improve detection.
Methods
References for this review were identified by searches of PubMed and Google Scholar between 1965 and February of 2019 and from references of relevant articles and book chapters. Search terms included “Adenylyl cyclase,” “ADCY5,” “paroxysmal dyskinesias,” “episodic movement disorders,” and “sleep related movement disorders.” No language restrictions were imposed.
Results
From Molecule to Disease
Adenylyl cyclases (ACs) comprise a family of molecules involved in the conversion of adenosine triphosphate (ATP) to cyclic adenosine‐3’, 5’‐monophosphate (cAMP), a second messenger in a broad range of cellular activities. Six sequence dissimilar AC classes exist with eukaryotic groups belonging to class 3. Class 3 isoforms are membrane bound and divided into four subgroups based on their regulatory properties. Of these, group 3 comprises the highly homologous ADCY5 and ADCY6 proteins.9, 10
ADCY5 consists of 1,261 amino acids encoded by a 21‐exon gene on chromosome 3. It contains an N‐terminus and two six‐unit membrane‐spanning helices (M1 and M2) which flank intracellular catalytic cyclase domains (C1 and C2). ADCY5 is partly regulated by dopamine receptors (D): D1 stimulating and D2 inhibiting.10, 11, 12, 13, 14 Stimulation results in catalytic domains (C1a and C2a) interacting to form an ATP‐binding heterodimer pocket.15 Bound ATP is converted into cAMP and pyrophosphate, which propagate downstream activity (Fig. 1). Inhibitory proteins conversely bind C1 and impair this interaction.10, 11, 12, 13
Figure 1.
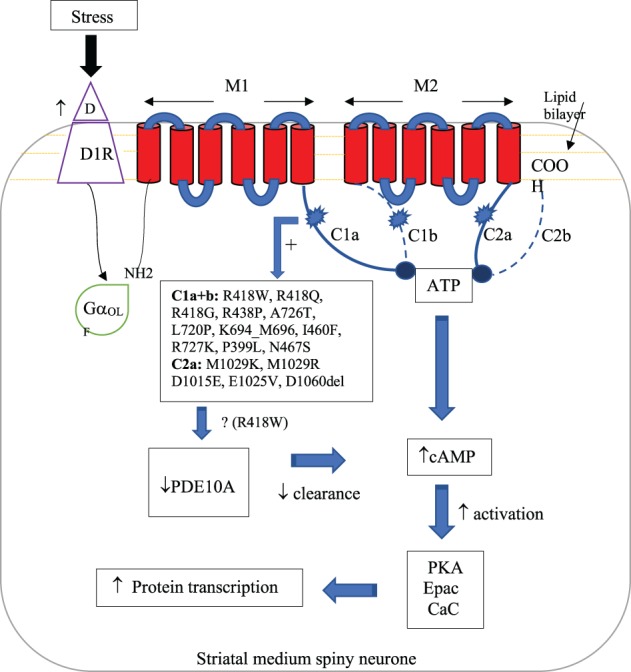
Proposed disease mechanism. Stress increases striatal dopamine synthesis and release, heightening receptor sensitivity while activating ADCY5 through GαOLF. ADCY5 mutations result in a gain‐of‐function response to this stimulus with increased binding of ATP by the catalytic pocket (C1a and C2a) as well as downstream decreased PDE10A expression. Both of these increase levels of cAMP and subsequent downstream cellular activity. Abbreviations: D, dopamine; D1R, dopamine receptor 1; GαOLF, stimulatory G protein; ATP, adenosine triphosphate; PKA, protein kinase A; Epac, exchange proteins activated by cAMP; CaC, calcium channels; PDE10A, phosphodiesterase 10A.
Chen and colleagues first identified a familial ADCY5 mutation of neurological consequence—c.2176G>A, p.A726T—in 20125 before further describing c.1252C>T, p.R418W in 2 sporadic cases.7 Mutational outcomes were somewhat different despite affecting the same domain (C1) of the protein. The p.A726T variant was associated with a milder phenotype, and this genotype‐phenotype correlation continues to hold as additional families with this variant have been found.6 Further mutations continue to be described (Fig. 1),15, 16, 17, 18, 19, 20, 21, 22, 23 although the 418 residue appears to be a hotspot (~50% of cases). C1a and C2a mutations possibly result in moderate‐to‐severe presentations, whereas C1b mutations and somatic mosaicism in milder disease are lacking axial symptomatology.6, 16
Gender‐balanced, autosomal dominance predominates in families, although recessive inheritance is noted in two families with biallelic pathogenic variants.17, 24 With the exception of c.g2176a;p.A726T,6 however, the majority of newly identified cases result from de novo mutations. The relatively common occurrence of somatic mosaicism in this disorder and the incomplete data on parents of seemingly sporadic cases make it difficult to be more precise. In one study, 6 of 13 sporadic cases and the progenitors in two multigenerational families were mosaic.6 There are other reports of suspected mosaicism in a more mildly affected parent in 1 of 4 cases23 and 1 of 2 cases.19 The read depth at the site of the variant in diagnostic exomes may not be sufficient to rule out mosaicism, and, in most cases, parents of sporadic cases are not evaluated.
Disease pathophysiology is increasingly, though, not entirely understood. A hypothetical disease mechanism is summarized in Figure 1 and detailed as follows. Functional studies suggest increased intracellular cAMP in response to chemical stress stimulation, indicating mutational gain of function.7, 8 Amino acid substitutions conceivably alter the binding‐pocket conformation, resulting in increased affinity for stimulatory proteins.5 ADCY5 is highly expressed in the nucleus accumbens and striatum, structures involved in sleep‐wake cycle arousal and coordination of movements, respectively.25 given that these structures subserve the mesolimbic dopaminergic system, their responses are typically stress sensitive.26 Stress can increase striatal dopamine synthesis and release, while heightening receptor sensitivity.26, 27 This typical response could be potentiated by the gain‐of‐function mutation and decreased expression of phosphodiesterase 10A (noted in some ADCY5‐dyskinesia cases), an enzyme implicated in cAMP hydrolysis.28 A combination of these molecular and brain regional features may explain the stress‐sensitive, sleep‐associated hyperkinetic phenotypes.25, 29
Although knockout mouse models manifest hypokinetic features,30, 31 loss of function (c.2088+1G>A) has been implicated in human hyperkinetic movements in one family.18 The mechanism by which contrasting haploinsufficiency and missense mutations result in similar presentations is unclear. A recent imaging study, however, suggests some complexity to mutational effects, with p.R418W patients suffering decreased striatal dopamine transporter expression, nigral neuron and microstructural loss,28 and possible prodromal nigral dysfunction.28 Long‐term follow‐up of ADCY5 cases will be crucial in understanding the clinical relevance of these findings.
Less explored aspects include ADCY5‐mediated neural developmental in cerebellar granular cells through the Hedgehog pathway. Although ADCY5 overexpression represses the pathway,32 the clinical implications are uncertain. Cardiac and pancreatic expression are also poorly detailed in the neurological literature. Overexpression in mice results in a cardiomyopathy whereas disruption confers protection.33, 34 Single‐nucleotide polymorphisms (rs11708067, rs2877716) are associated with an increased risk of type 2 diabetes by virtue of lowering ADCY5 pancreatic islet cell expression with resultant glucose hypometabolism.35
On balance, cellular pathology and brain regional findings to date largely reconcile core clinical features. The biological basis for clinical heterogeneity, however, remains unexplained and requires further exploration.
“Core” Clinical Features
As mentioned above, documentation of the range of clinical manifestations of the disease began in 1978 in two American families of European ancestry. At the time, one of these families was postulated to be suffering from a form of “essential” (benign) chorea.3 In 2001, Fernandez and colleagues further described features from five generations while proposing an alternate diagnosis, “familial dyskinesia and facial myokymia” (FDFM),4 to acknowledge a seemingly novel and unique syndrome. The progenitor in the second family was described as having sporadic paroxysmal choreoathetosis with hemiplegia,1 but the disorder was much more severe and constant in her daughter and included many of the features later found to be part of the ADCY5‐dyskinesia phenotypic spectrum.2 We will now consider some of these clinical aspects. Cases shown in Videos 1 to 4 provide examples of the phenotypic manifestations of ADCY5‐related dyskinesia.
Facial “Myokymia”
FDFM was characterised by the onset of generalized choreic dyskinesia in conjunction with perioral and ‐orbital movements resembling myokymia in either childhood or adolescence. Although myokymia was reportedly demonstrated on electromyography (EMG) in some family members,4 this finding has not been replicated in all subsequent cases, in which the facial movements represent a mixture of myoclonus and chorea.6, 36 Further case descriptions led to the adoption of the more universally accepted terminology of ADCY5‐related dyskinesia.6
The dyskinetic movements have been variably described as chorea, athetosis, dystonia, or myoclonus or a combination thereof. Limb EMG studies, in some cases, suggest a combination of myoclonus and chorea.5 Both clinical and electrophysiological distinctions can, at times, be challenging.37 Whereas these movements can affect all limbs, they are typically more prominent in the upper limbs.
“Ballistic Bouts” Worsened by Sleep
Exacerbations of hyperkinetic movements in a paroxysmal manner is a hallmark feature of the disease, although some prefer the term episodic in view of a lack of predictable stereotyped triggers.16 These episodes can last for minutes to hours and gradually worsen in frequency and severity into the third decade of life before becoming either constant or completely resolving. Some describe them as “ballistic bouts” and they can manifest with a characteristic painful (although not always), dystonic truncal flexion‐extension, which progresses to retrocollis and upper limb extension. A noteworthy exacerbation, either in drowsiness or during sleep, has been reported.38 Polysomnographic studies suggest preceding hypopnea before the movements in some cases. Poor sleep efficacy has also been noted, though the implicated underlying cause seems to vary. Although some suggest this to be a result of prolonged latency,16, 39 others have found latency to be unimpaired while implicating sleep disruption from movements in the N2, rapid eye movement (REM), and awakening stages.40 Other precipitating factors include emotional stress or laughter and intercurrent illness (with or without pyrexia), during which time the movements can occur in clusters and with increased frequency.6, 16, 38 Whereas alcohol has no discernible impact, sneezing has been found to worsen dyskinesia in some.16 Caffeine exacerbated dyskinesia in some cases16; however, a dramatic amelioration of the paroxysmal dyskinesias was noted in 1 patient.41 The episodes can also wax and wane over weeks to months in the absence of triggers. Whereas ballistic bouts are nonepileptic, focal epilepsy can rarely co‐occur in ADCY5. Semiology is of dissociation and rhythmic head movements, with EEG recordings implicating a bifrontocentral focus.4, 16
Axial Hypotonia
Interepisodic neurological abnormalities do occur albeit variably. The most characteristic of these is axial hypotonia. Onset is typically in infancy with impact on motor milestones. Severity ranges from difficulties sitting up to an inability to walk independently into teenage years. An adaptive distinctive method of mobilization, termed “frog‐like,” has been noted in severe cases.16 Axial hypotonia generally improves with age, although it can persist into adulthood.
Less Certain and Variable Features
Dystonic posturing in all limbs and writer's cramp have been noted variably. Close to half have recordable signs of spasticity (increased tone, hyper‐reflexia, and extensor plantar responses) at all disease stages, whereas spastic paraparesis has been documented in an adult.42 Detailed eye movement assessments suggest saccadic initiation and execution impairment particularly in the vertical plane. This, with associated hypometria, is suggestive of oculomotor apraxia.16, 39 Although slow, uncoordinated gait abnormalities have been described, convincing cerebellar gait features have not been well documented.6, 16
Language development delay may potentially be related to abnormal tongue and facial movements. Whereas a majority progress to normal language, some experience ongoing dysarthria. Cognitive impairment has been documented variably, with deficits in learning and problem solving noted on neuropsychological testing. Whereas mild nonspecific impairments in IQ and cognition have been observed, specific deficits in spatial working and episodic memory are evident on detailed assessments.6, 16, 39, 43 These abnormalities occur across ages, and it remains unclear why they are variably observed. Their true frequency is uncertain given the lack of detailed cognitive assessments in reported cases. Psychiatric symptoms can occur intermittently throughout life and include depression, psychosis with delusions and auditory hallucinations, and obsessive‐compulsive (OC) symptoms.6, 16, 24, 43 Although OC symptoms and autistic‐like behavior are related to haploinsufficiency,44 their occurrence with gain‐of‐function mutations once again highlights the complexity of genotype‐phenotype correlation. Cardiac failure from dilated cardiomyopathy has been reported as has cardiac death of unclear aetiology.4, 17 Although this corresponds with ADCY5 expression, it remains unclear why this finding is not universal. The current movement disorder focus of ascertainment and reporting may be partly to blame.
Rare Phenotypes and Diagnostic Considerations
The core features described above are considered pathognomonic and should prompt genetic testing (conventional testing, such as cerebrospinal fluid analysis and brain imaging, is typically normal). A broad range of diagnostic possibilities should, however, be considered when confronted with these mixed phenotypes. Among these are paroxysmal dyskinesias. Although conventionally understood to be triggered by clear precipitants (kinesigenic, exercise, and hypnogenic), and characterized by normal interepisodic examinations, these conditions have an array of noted genetic causes (PRRT2, MR‐1, SLC2A1, etc.). It is becoming increasingly understood, however, that different mutations in established genes can modify phenotypes with cases manifesting a mixture of additional interepisodic features while lacking clear triggers. ADCY5 has been implicated in some established syndromes and should therefore be considered a diagnostic possibility in these cases, despite a lack of core features. Further consideration should also be given to patients presenting with prominent sleep‐related movements during the diagnostic process. Here, we detail some of these considerations (summarized in Table 1).
Table 1.
Distinguishing features of disorders that may mimic ADCY5
| Predominant Phenotype | Differential Diagnosis | Distinguishing Features (From ADCY5) | Useful Diagnostic testing (Abnormalities) |
|---|---|---|---|
| Sleep‐related motor and behavioral disorders | Sleep‐related hypermotor epilepsy |
|
|
| Myoclonus at sleep onset, non‐REM parasomnias, sleep‐related rhythmic movement disorder, benign nocturnal alternating hemiplegia of childhood |
|
Video‐EEG (unique semiology) | |
| Childhood onset chorea | Athetoid cerebral palsy |
|
MRI brain (focal dysplasia) |
| Sydenham disease |
|
ASO, anti‐DNAse B titers | |
| NKX2‐1–related benign hereditary chorea |
|
|
|
| PDE10A and 2A mutations |
|
MRI brain (striatal lesions) | |
| Myoclonus‐ dystonia | SGCE mutations |
|
Genetic testing |
| Focal dystonia + tremor | ANO3 mutations | Absence of ADCY5 core features | Genetic testing |
| Alternating hemiplegia | ATP1A3 mutations | Absence of interepisodic dyskinesia | Genetic testing |
Sleep Predominance
The prominence of dyskinetic episodes in relation to sleep warrants the consideration of a number of sleep‐related motor and behavioral disorders as alternate diagnostic possibilities. Chief among these is sleep‐related hypermotor epilepsy (SHE), although clear distinguishing features from ADCY5 exist.45 Whereas SHE patients can suffer from episodes beginning in childhood, they tend to occur at any time during sleep, which is atypical for ADCY5. SHE episodes can also occur in clusters and are stereotypical in individuals, although manifest with a wide range of phenomenology (vocalization, cycling, episodic nocturnal wandering, etc.) across cases, unlike ADCY5.46 The unique phenotype of asymmetric tonic/dystonic seizures (previously mistaken as nocturnal paroxysmal dystonia) can, however, resemble ADCY5 semiology.46 Secondary generalized seizures are not universal and should not be relied upon to exclude SHE. Furthermore, the epilepsy observed in ADCY5 cases have different clinical and electrophysiological semiology, as previously detailed.4, 6 Efforts should therefore be made to capture events on video‐EEG. Although several SHE genes have been identified (CHRNA4, KCNT1, and DEPDC5), no ADCY5 cases have coexistent SHE.46 A less compelling possibility would be myoclonus at sleep onset.47, 48 Although the semiology (nonrhythmic myoclonic jerks with flexion of trunk, hips, and knees occurring exclusively during the transition between wakefulness and sleep with disruption of sleep) somewhat resembles ballistic bouts, coexistent core ADCY5 features would distinguish the conditions. Less pertinent considerations include non‐REM parasomnias,49, 50 sleep‐related rhythmic movement disorder,51 and benign nocturnal alternating hemiplegia of childhood.52 Although some of these conditions occur in sleep transition, they lack core ADCY5 features and present with distinct phenomenology.
Chorea Predominance
Infantile‐onset chorea presents unique diagnostic considerations. Although many of these cases have been previously diagnosed as dyskinetic (athetoid) cerebral palsy, this diagnosis should be avoided if the neurological syndrome is progressive and the history lacks an intrauterine or perinatal insult or accompanying imaging abnormalities. Cases lacking these features have been demonstrated to have ADCY5 mutations.16 Sydenham chorea is another consideration whereby movements occur continuously from an onset in childhood with a gradual resolution in later stages. Although this can be observed in ADCY5 cases, a more typically presentation is of movement onset in infancy, with episodic worsening occurring later in childhood. Benign hereditary chorea (BHC) is another differential consideration in these cases. A well‐recognized cause is mutations in NKX2‐1. ADCY5 mutations have, however, been implicated in BHC cases that are NKX2‐1–mutation negative.19 The increased expression of ADCY5 in the striatum longitudinally in contrast to NKX2‐1 results in some noteworthy clinical differences. First, ADCY5 cases suffer more severe dystonic posturing and progression of symptoms into adulthood, unlike the relatively mild course with improvements after childhood noted with NKX2‐1. The exacerbation of dyskinesia by stress, action, and excitement and the sleep‐related ballistic bouts observed in ADCY5 also distinguish these disorders. Finally, mutations in these genes result in different extraneural features—cardiomyopathy as a rare feature of ADCY5 and hypothyroidism, respiratory distress, and ligamentous laxity in NKX2‐1 patients.53
In ADCY5‐negative cases of childhood onset chorea with variable associated core features, testing for PDE10A 47 and PDE2A 49 mutations should be considered. PDE genes encode an enzyme that hydrolyses cAMP and cyclic guanosine monophosphate downstream from ADCY5. Mutations severely decrease respective enzymatic activity and therefore clearance. Cases of largely de novo dominant (p.Phe300Leu and p.Phe334Leu) and inherited recessive homozygous mutations (p.Tyr107Cys and p.Ala116Pro) in PDE10A and an isolated case with a de novo homozygous missense mutation (c.1439A>G; p.Asp480Gly) in PDE2A have been described.54, 55, 56 Phenotypes seem to vary with the types of mutations. Dominant cases manifest with a milder disease marked by diurnal fluctuations of childhood‐onset chorea that variably progresses into adulthood. Characteristic striatal T2‐hyperintense changes on MRI lasting years is a unique diagnostic feature. Recessive PDE10A cases demonstrate a more severe phenotype with axial hypotonia, delayed developmental milestones, and cognitive abnormality, but without MRI changes.57, 58
Myoclonus‐Dystonia Predominance
Presentations with both early‐onset generalized and focal dystonia have been noted. In cases of generalized dystonia, patients typically present with dystonia in combination with myoclonus (myoclonus‐dystonia phenotype).20 The location of dystonia can be widespread and involve the face (blepharospasm and orolingual), cervical, and axial regions while relatively sparing the lower body, although dystonic gait abnormalities have been described. Onset is typically in childhood and associated with developmental delay. The commonest cause of myoclonus‐dystonia is mutations in the epsilon sarcoglycan gene (SGCE).59 Some cases that are negative for this abnormality have ADCY5 mutations. Potential distinguishing features include a lack of alcohol response, more frequent facial involvement, the presence of axial hypotonia rather than dystonia, and the less frequent presence of psychiatric symptoms in ADCY5 cases.20, 43, 59 Although cervical dystonia can occur in conjunction with other ADCY5 features,21 this was the sole presentation in two members of a single family.21 These patients suffered exclusively from cervical dystonia and head tremor in their late twenties. Another cause for this presentation is mutations in anoctamin 3 (ANO3). Reliable clinical features to distinguish between cases of ANO3 and ADCY5 presenting with isolated craniocervical dystonia with prominent tremor and myoclonus‐dystonia59, 60, 61 are currently lacking.
Alternating Hemiplegia Predominance
A number of ADCY5 patients have displayed features of alternating hemiplegia in childhood (AHC).1, 22 Core ADCY5 features occur in conjunction with brief hemiplegic attacks beginning in childhood and worsening in duration over time. The episodes can variably affect different body parts (including speech) whereas consciousness is preserved. Attacks can follow periods of rest, tiredness, or occur while eating and are not related to typical ADCY5 triggers nor demonstrate improvements after sleep as observed with other causes of AHC, particularly mutations is the ATP1A3 gene.22, 62 The presence of interepisodic dyskinesia is a distinguishing feature of ADCY5 from ATP1A3, although more definitive characteristics are currently lacking.
Cases of these isolated phenotypes have arisen from exploratory testing of ADCY5 in these populations after exclusion of more familiar genetic causes. Available evidence suggests that ADCY5 is a rare cause of these presentations, and the detection rates of these approaches are low in populations studied (e.g., 11% of pediatric undiagnosed hyperkinetic movements).23 This is compounded further by contrasting reports, with some groups finding no cases of ADCY5 mutations in their populations (e.g., myoclonus‐dystonia),22 despite others reporting substantial proportions with similar phenotypes.20 More clarity in distinguishing clinical features in cases lacking core ADCY5 features is therefore necessary before widespread testing of these phenotypes can be recommended in clinical practice.
Treatment Considerations
A range of medications have been trialed to manage symptoms with variable success.4, 6, 16, 17, 18, 19, 42 Acetazolamide (up to 30 mg/kg) appears to be the most efficacious option in the control of chorea and dyskinesia. Other beneficial agents for dyskinesia include trihexyphenidyl (up to 30 mg/day), tetrabenazine (50 mg/day), clonazepam, propranolol, levocarnitine, melatonin, chlordiazepoxide, and methylphenidate,63 although these medications have also been reported by some to worsen symptoms. Clonazepam has been found to improve nocturnal dystonic episodes and axial hypotonia in some cases. Variable responses have been noted with antiepileptics; low‐dose gabapentin (20 mg/kg) may improve coexisting epilepsy, although higher doses worsen dyskinesias4; levetiracetam has provided moderate movement improvements in some cases4, 6, 16; carbamazepine, topiramate, ethosuximide, primidone, and phenobarbital do not seem to have any impact. Multiple other agents, including antihistamines, levodopa, antidepressants, and CoQ10, either have no impact or worsen symptoms. Botulinum neurotoxin has been used with good success for cervical dystonia21 whereas pallidal DBS seems to provide modest improvements for the hyperkinetic movements, but not for axial hypotonia.64
Conclusion
The spectrum of phenotypes associated with ADCY5 mutations has dramatically expanded since its relatively recent description in the family originally described as having FDFM. The unique core features of facial myoclonus, axial hypotonia, and ballistic bouts in relation to drowsiness and sleep are worthy of recognition by clinicians to further improve detection. Although mutations have been implicated less frequently in other well‐recognized phenotypes—BHC, AHC, myoclonus‐dystonia, and isolated dystonia—more case descriptions are required to establish whether or not signature ADCY5 features occur in these cases. The field has advanced a great deal in a short period. Improved clinical detection will help to further advance this fast‐maturing genetic disorder.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
N.V.: 1A, 1B, 1C, 2A, 2B
K.P.B.: 2B
A.E.L.: 2B
W.H.R.: 2B
A.J.E.: 1A, 1B, 2B
Disclosures
Ethical Compliance Statement: The authors confirm that the approval of an institutional review board was not required for this work. Written informed patient consent was obtained in person and documented in accord with Journal guidelines on individual hospital proforma for the videos published. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: This work was supported by grants 1R01NS069719 from the National Institutes of Health and Merit Review Award Number 101 CX001702 from the Clinical Sciences R&D (CSRD) Service of the United States Department of Veterans Affairs to W.H.R. The authors report no conflicts of interest.
Financial Disclosures for previous 12 months: Nirosen Vijiaratnam has received educational support from AbbVie, Stada, Ipsen, and Merc; speaker's honorarium from AbbVie and Stada; and advisory boards for AbbVie. Kailash P. Bhatia has received consulting fees/honoraria for speaking at meetings from Ipsen, Cavion, Retrophin, Merz, Allergan, Teva, and Ultragenyx pharma. He has grant support from a Horizon 2020 EU grant. He receives royalty for books published by OUP and Cambridge Publishers. He has received a stipend for editorial work for the Movement Disorders Clinical Prqctice journal and has received honoraria from the International Parkinson and Movement Disorder Society (MDS) to teach at international MDS meetings and summer/winter schools. Anthony E. Lang has served as an advisor for AbbVie, Acorda, Biogen, Bristol‐Myers Squibb, Janssen, Sun Pharma, Merck, and Corticobasal Degeneration Solutions; received honoraria from Sun Pharma, Medichem, Medtronic, AbbVie, and Sunovion; received grants from Brain Canada, Canadian Institutes of Health Research, Corticobasal Degeneration Solutions, Edmond J Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, National Parkinson Foundation, Parkinson Society Canada, and W. Garfield Weston Foundation; and received publishing royalties from Elsevier, Saunders, Wiley‐Blackwell, Johns Hopkins Press, and Cambridge University Press. Wendy H. Raskind receives grant support from the NIH and the Department of Veterans Affairs. She receives royalties for patent 10,036,067: Mutations in PKCγ are a cause for spinocerebellar ataxia. Alberto J. Espay has received grant support from the NIH, Great Lakes Neurotechnologies, and the Michael J Fox Foundation; personal compensation as a consultant/scientific advisory board member for AbbVie, Adamas, Acadia, Acorda, Neuroderm, Impax, Sunovion, Lundbeck, Osmotica Pharmaceutical, and USWorldMeds; publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer; and honoraria from USWorldMeds, Lundbeck, Acadia, Sunovion, the American Academy of Neurology, and the MDS.
Supporting information
Case 1, Video S1. This is a 27‐year‐old woman with a history of autism, delayed motor milestones, and special education needs in school for speech and language difficulties. She had a 1‐year history of gait abnormalities. The video demonstrates an unusual dystonic gait particularly with left leg, pelvic, and trunk involvement as well as flexion of the upper limbs (right more than left) and slowness and loss of dexterity on finger movements in both hands.
Case 2. This 16‐year‐old boy experienced motor regression at the age of 12 months losing his ability to roll over and balance himself while sitting. At 18 months, he could pull himself up to stand but not walk; at the age of 4 to 5 years, he could walk with a walker and at age 6 he began using a wheelchair. He had no cognitive disturbances. Two and a half years before he was first seen, he began having painful spasms beginning in his hands and subsequently spreading to involve his trunk and legs. These would occur at least weekly, lasting for 2 to 3 minutes at a time followed by rest and then recurrence of the movements on and off sometimes for 3 to 4 hours at a time. The movements would occur particularly at night when lying down, but would not wake him from sleep. Shortly before he was seen, he also developed more complex semirhythmic movements of the left hand. Video S2. The patient provides a history describing the nature of the paroxysmal events.
Video S3. demonstrates the interictal clinical examination. He has dysarthria with slow tongue movements, and his mouth is held open most of the time. He demonstrates a variety of abnormal involuntary movements, including myoclonic jerks (particularly the face), axial tremor, choreoathetotic movements of the limbs, axial hypotonia, lower limb hypertonia, hyperactive deep tendon reflexes, ankle clonus and extensor plantar responses, and the tendency to sit with his legs crossed over each other.
Case 3, Video S4. This 24‐year‐old man of Pakistani ancestry demonstrates generalized chorea, with prominent facial and tongue involvement. Axial hypotonia is noted (Video courtesy: Dr. Bettina Balint).
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Perez‐Borja C, Tassinari AC, Swanson AG. Paroxysmal choreoathetosis and seizures induced by movement (reflex epilepsy). Epilepsia 1967;8:260–270. [DOI] [PubMed] [Google Scholar]
- 2. Bird T, Carlson C, Hornung M. Ten year follow‐up of paroxysmal choreoathetosis: a sporadic case becomes familial. Epilepsia 1978;19:129–132. [DOI] [PubMed] [Google Scholar]
- 3. Bird TD, Hall JG. Additional information on familial essential (benign) chorea. Clin Genet 1978;14:271–272. [DOI] [PubMed] [Google Scholar]
- 4. Fernandez M, Raskind W, Wolff J, et al. Familial dyskinesia and facial myokymia (FDFM): a novel movement disorder. Ann Neurol 2001;49:486–492. [PubMed] [Google Scholar]
- 5. Chen YZ, Matsushita MM, Robertson P, et al. Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol 2012;69:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen DH, Meneret A, Friedman JR, et al. ADCY5‐related dyskinesia: broader spectrum and genotype‐phenotype correlations. Neurology 2015;85:2026–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen YZ, Friedman JR, Chen DH, et al. Gain‐of‐function ADCY5 mutations in familial dyskinesia with facial myokymia. Ann Neurol 2014;75:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doyle TB, Hayes MP, Chen DH, et al. Functional characterization of AC5 gain‐of‐function variants: impact on the molecular basis of ADCY5‐related dyskinesia. Biochem Pharmacol 2019;163:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Linder JU. Class 3 adenylyl cyclases: molecular mechanism of catalysis and regulation. Cell Mol Life Sci 2006;63:1736–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buck J, Sinclair ML, Schapal L, et al. Cytosolic adenylyl cyclase defines a unique signalling molecule in mammals. Proc Natl Acad Sci U S A 1999;96:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Onda T, Hashimoto Y, Nagai M, et al. Type‐specific regulation of adenylate cyclases. Selective pharmacological stimulation and inhibition of adenylyl cyclase isoforms. J Biol Chem 2001;276:47785–47793. [DOI] [PubMed] [Google Scholar]
- 12. Cooper DM. Molecular and cellular requirements for the regulation of adenylate cyclases by calcium. Biochem Soc Trans 2003;31(Pt 5):912–915. [DOI] [PubMed] [Google Scholar]
- 13. Beazely MA, Watts VJ. Regulatory properties of adenylate cyclases type 5 and 6: a progress report. Eur J Pharmacol 2006;535:1–12. [DOI] [PubMed] [Google Scholar]
- 14. Girault JA, Greengard P. The neurobiology of dopamine signalling. Arch Neurol 2004;61:641–644. [DOI] [PubMed] [Google Scholar]
- 15. Tesmer JJ, Sunahara RK, Gilman AG, et al. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgamma S. Science 1997;278:1907–1916. [DOI] [PubMed] [Google Scholar]
- 16. Chang FC, Westenberger A, Dale RC, et al. Phenotypic insights into ADCY5‐associated disease. Mov Disord 2016;31:1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barrett MJ, Williams ES, Dhamija R. Autosomal recessive inheritance of ADCY5‐related generalized dystonia and myoclonus. Neurol Genet 2017;3:e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carapito R, Paul N, Untrau M, et al. A de novo ADCY5 mutation causes early‐onset autosomal dominant chorea and dystonia. Mov Disord 2015;30:423–427. [DOI] [PubMed] [Google Scholar]
- 19. Mencacci NE, Erro R, Wiethoff S, et al. ADCY5 mutations are another cause of benign hereditary chorea. Neurology 2015;85:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Douglas AG, Andreoletti G, Talbot K, et al. ADCY5‐related dyskinesia presenting as familial myoclonus‐dystonia. Neurogenetics 2017;18:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zech M, Boesch S, Jochim A, et al. Clinical exome sequencing in early‐onset generalised dystonia and large‐scale resequencing follow‐up. Mov Disord 2017;32:549–559. [DOI] [PubMed] [Google Scholar]
- 22. Westenberger A, Max C, Bruggemann N, et al. Alternating hemiplegia of childhood as a new presentation of adenylate cyclase 5‐mutation‐associated disease: a report of two cases. J Pediatr 2017;181:306–308. [DOI] [PubMed] [Google Scholar]
- 23. Carecchio M, Mencacci NE, Iodice A, et al. ADCY5‐related movement disorders: frequency, disease course and phenotypic variability in a cohort of paediatric patients. Parkinsonism Relat Disord 2017;41:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bohlega SA, Abou‐Al‐Shaar H, AlDakheel A, et al. Autosomal recessive ADCY5‐Related dystonia and myoclonus: expanding the genetic spectrum of ADCY5‐related movement disorders. Parkinsonism Relat Disord 2019. Feb 28. pii: S1353‐8020(19)30084‐7. 10.1016/j.parkreldis.2019.02.039. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 25. Matsuoka I, Suzuki Y, Defer N, et al. Differential expression of type 1, 2 and 5 adenylyl cyclase gene in the postnatal developing rat brain. J Neurochem 1997;68:498–506. [DOI] [PubMed] [Google Scholar]
- 26. Finlay JM, Zigmond MJ. The effects of stress on central dopaminergic neurones: possible clinical implications. Neurochem Res 1997;22:1387–1394. [DOI] [PubMed] [Google Scholar]
- 27. Howes OD, McCutcheon R, Owen MJ, et al. The role of genes, stress and dopamine in the development of schizophrenia. Biol Psychiatry 2017;81:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Niccolini F, Mencacci NE, Yousaf T, et al. PDE10A and ADCY5 mutations linked to molecular and microstructural basal ganglia pathology. Mov Disord 2018;33:1961–1965. [DOI] [PubMed] [Google Scholar]
- 29. Callaway CW, Henriksen SJ. Neuronal firing in the nucleus accumbens is associated with the level of cortical arousal. Neuroscience 1992;51:547–553. [DOI] [PubMed] [Google Scholar]
- 30. Iwamoto T, Okumura S, Iwatsubo K, et al. Motor dysfunction in type 5 adenylyl cyclase‐null mice. J Biol Chem 2003;278:16936–16940. [DOI] [PubMed] [Google Scholar]
- 31. Lee KW, Hong JH, Choi IY, et al. Impaired D2 dopamine receptor function in mice lacking type 5 adenylyl cyclase. J Neurosci 2002;22:7931–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vuolo L, Herrera A, Torroba B, et al. Ciliary adenylyl cyclases control the Hedgehog pathway. J Cell Sci 2015;128:2928–2937. [DOI] [PubMed] [Google Scholar]
- 33. Okumura S, Takagi G, Kawabe J, et al. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci U S A 2003;100:9986–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ho D, Yan L, Iwatsubo K, et al. Modulation of beta‐adrenergic receptor signalling in heart failure and longevity: targeting adenylyl cyclase type 5. Heart Fail Rev 2010;15:495–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hodson DJ, Mitchell RK, Marselli L, et al. ADCY5 couples glucose to insulin secretion in human islets. Diabetes 2014;63:3009–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tunc S, Bruggemann N, Baaske MK, et al. Facial twitches in ADCY5‐associated disease‐Myokymia or myoclonus? An electromyography study. Parkinsonism Relat Disord 2017;40:73–75. [DOI] [PubMed] [Google Scholar]
- 37. Hallett M. Analysis of abnormal voluntary and involuntary movements with surface electromyography In: Desmedt JE, ed. Motor Control Mechanism in Health and Disease. New York, NY: Raven; 1983:907–914. [PubMed] [Google Scholar]
- 38. Friedman JR, Meneret A, Chen DH, et al. ADCY5 mutation carriers display pleiotropic paroxysmal day and night time dyskinesias. Mov Disord 2016;31:147–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Balint B, Antelmi E, Mencacci NE, et al. Occulomotor apraxia and disrupted sleep with nocturnal ballistic bouts in ADCY5‐related disease. Parkinsonism Relat Disord 2018;54:103–106. [DOI] [PubMed] [Google Scholar]
- 40. Méneret A, Roze E, Maranci JB, et al. Sleep in ADCY5‐related dyskinesia: prolonged awakenings caused by abnormal movements. J Clin Sleep Med 2019;15:1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Méneret A, Gras D, McGovern E, et al. Caffeine and the dyskinesia related to mutations in the ADCY5 gene. Ann Intern Med 2019. Jun 11. 10.7326/L19-0038. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 42. Waalkens AJE, Vansenne F, van der Hout AH, et al. Expanding the ADCY5 phenotype toward spastic paraparesis. Neurol Genet 2018;4:e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vijiaratnam N, Newby R, Kempster PA. Depression and psychosis in ADCY5‐related dyskinesia‐part of the phenotypic spectrum? J Clin Neurosci 2018;57:167–168. [DOI] [PubMed] [Google Scholar]
- 44. Kim H, Lee Y, Park JY, et al. Loss of adenylyl cyclase type‐5 in the dorsal striatum produces autistic‐like behaviours. Mol Neurobiol 2017;54:7994–8008. [DOI] [PubMed] [Google Scholar]
- 45. Provini F, Plazzi G, Tinuper P, et al. Nocturnal frontal lobe epilepsy. A clinical and polygraphic overview of 100 consecutive cases. Brain 1999;122(Pt 6):1017–1031. [DOI] [PubMed] [Google Scholar]
- 46. Tinuper P, Bisulli F, Cross JH, et al. Definition and diagnostic criteria of sleep‐related hypermotor epilepsy. Neurology 2016;86:1834–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brown P, Thompson PD, Rothwell JC, et al. Axial myoclonus of propriospinal origin. Brain 1991;114(Pt 1A):197–214. [PubMed] [Google Scholar]
- 48. Montagna P, Provini F, Plazzi G, et al. Propriospinal myoclonus upon relaxation and drowsiness: a cause of severe insomnia. Mov Disord 1997;12:66–72. [DOI] [PubMed] [Google Scholar]
- 49. Breen DP, Hogl B, Fasano A, et al. Sleep‐related motor and behavioural disorders: recent advances and new entities. Mov Disord 2018;33:1042–1055. [DOI] [PubMed] [Google Scholar]
- 50. Ohayon MM, Guilleminault C, Priest RG. Night terrors, sleepwalking, and confusional arousals in the general population: their frequency and relationship to other sleep and mental disorders. J Clin Psychiatry 1999;60:268–276; quiz, 277. [DOI] [PubMed] [Google Scholar]
- 51. Khan A, Auger RR, Kushida CA, et al. Rhythmic movement disorder. Sleep Med 2008;9:329–330. [DOI] [PubMed] [Google Scholar]
- 52. Wagener‐Schimmel LJ, Nicolai J. Child neurology: benign nocturnal alternating hemiplegia of childhood. Neurology 2012;79:e161–e163. [DOI] [PubMed] [Google Scholar]
- 53. Parnes M, Bashir H, Jankovic J. Is Benign Hereditary Chorea Really Benign? Brain‐Lung Thyroid Syndrome Caused by NKX2‐1 Mutations. Mov Disord Clin Pract 2018;6:34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mencacci NE, Kamsteeg EJ, Nakashima K, et al. De novo mutations in PDE10A cause childhood‐onset chorea with bilateral striatal lesions. Am J Hum Gen 2016;98:763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Salpietro V, Perez‐Dueñas B, Nakashima K, et al. A homozygous loss‐of‐function mutation in PDE2A associated to early‐onset hereditary chorea. Mov Disord 2018;33:482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Esposito S, Carecchio M, Tonduti D, et al. A PDE10A de novo mutation causes childhood onset chorea with diurnal fluctuations. Mov Disord 2017;32:1646–1647. [DOI] [PubMed] [Google Scholar]
- 57. Diggle CP, Sukoff Rizzo SJ, Popiolek M, et al. Biallelic mutations in PDE10A lead to loss of striatal PDE10A and a hyperkinetic movement disorder with onset in infancy. Am J Hum Genet 2016;98:735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Miyatake S, Koshimizu E, Shirai I, et al. A familial case of PDE10A‐associated childhood‐onset chorea with bilateral striatal lesions. Mov Disord 2018;33:177–179. [DOI] [PubMed] [Google Scholar]
- 59. Peall KJ, Kurian MA, Wardle M, et al. SCGE and myoclonus dystonia: motor characteristics, diagnostic criteria and clinical predictors of genotype. J Neurol 2014;261:2296–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miltgen M, Blanchard A, Mathieu H, et al. Novel heterozygous mutation in ANO3 responsible for craniocervical dystonia. Mov Disord 2016;31:1251–1252. [DOI] [PubMed] [Google Scholar]
- 61. Stamelou M, Charlesworth G, Cordivari C, et al. The phenotypic spectrum of DYT24 due to ANO3 mutations. Mov Disord 2014;29:928–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sweney MT, Newcomb TM, Swoboda KJ, et al. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, alternating hemiplegia of childhood, rapid‐onset dystonia‐Parkinsonism, CAPOS and beyond. Pediatr Neurol 2015;52:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tübing J, Bohnenpoll J, Spiegler J, et al. Methylphenidate can improve chorea in NKX2.1 and ADCY5 mutation‐positive patients—a report of two children. Mov Disord Clin Pract 2018;5:343–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dy ME, Chang FC, Jesus SD, et al. J Child Neurol. Treatment of ADCY5‐associated dystonia, chorea, and hyperkinetic disorders with deep brain stimulation: a multicentre case series. J Child Neurol 2016;31:1027–1035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Case 1, Video S1. This is a 27‐year‐old woman with a history of autism, delayed motor milestones, and special education needs in school for speech and language difficulties. She had a 1‐year history of gait abnormalities. The video demonstrates an unusual dystonic gait particularly with left leg, pelvic, and trunk involvement as well as flexion of the upper limbs (right more than left) and slowness and loss of dexterity on finger movements in both hands.
Case 2. This 16‐year‐old boy experienced motor regression at the age of 12 months losing his ability to roll over and balance himself while sitting. At 18 months, he could pull himself up to stand but not walk; at the age of 4 to 5 years, he could walk with a walker and at age 6 he began using a wheelchair. He had no cognitive disturbances. Two and a half years before he was first seen, he began having painful spasms beginning in his hands and subsequently spreading to involve his trunk and legs. These would occur at least weekly, lasting for 2 to 3 minutes at a time followed by rest and then recurrence of the movements on and off sometimes for 3 to 4 hours at a time. The movements would occur particularly at night when lying down, but would not wake him from sleep. Shortly before he was seen, he also developed more complex semirhythmic movements of the left hand. Video S2. The patient provides a history describing the nature of the paroxysmal events.
Video S3. demonstrates the interictal clinical examination. He has dysarthria with slow tongue movements, and his mouth is held open most of the time. He demonstrates a variety of abnormal involuntary movements, including myoclonic jerks (particularly the face), axial tremor, choreoathetotic movements of the limbs, axial hypotonia, lower limb hypertonia, hyperactive deep tendon reflexes, ankle clonus and extensor plantar responses, and the tendency to sit with his legs crossed over each other.
Case 3, Video S4. This 24‐year‐old man of Pakistani ancestry demonstrates generalized chorea, with prominent facial and tongue involvement. Axial hypotonia is noted (Video courtesy: Dr. Bettina Balint).