

Abstract
Peroxiredoxins (Prx) are enzymes that efficiently reduce hydroperoxides through active participation of cysteine residues (CP, CR). The first step in catalysis, the reduction of peroxide substrate, is fast, 107 − 108 M−1s−1 for human Prx2. In addition, the high intracellular concentration of Prx positions them not only as good antioxidants but also as central players in redox signaling pathways. These biological functions can be affected by post-translational modifications that could alter the peroxidase activity and/or interaction with other proteins. In particular, inactivation by hyperoxidation of CP, which occurs when a second molecule of peroxide reacts with the CP in the sulfenic acid form, modulates their participation in redox signaling pathways. The higher sensitivity to hyperoxidation of some Prx has been related to the presence of structural motifs that disfavor disulfide formation at the active site, making the CP sulfenic acid more available for hyperoxidation or interaction with a redox protein target. We previously reported that treatment of human Prx2 with peroxynitrite results in tyrosine nitration, a post-translational modification on non-catalytic residues, yielding a more active peroxidase with higher resistance to hyperoxidation. In this work, studies on different mutants of hPrx2 confirm that the presence of the tyrosyl side-chain of Y193, belonging to the C-terminal YF motif of eukaryotic Prx, is necessary to observe the increase in Prx2 resistance to hyperoxidation. Moreover, our results underline the critical role of this structural motif on the rate of disulfide formation that determines the differential participation of Prx in redox signaling pathways.
Keywords: Peroxiredoxin, post-translational modification (PTM), tyrosine nitration, peroxynitrite, hyperoxidation, hydrogen peroxide, redox signaling, oxidative stress
Graphical Abstract
Introduction
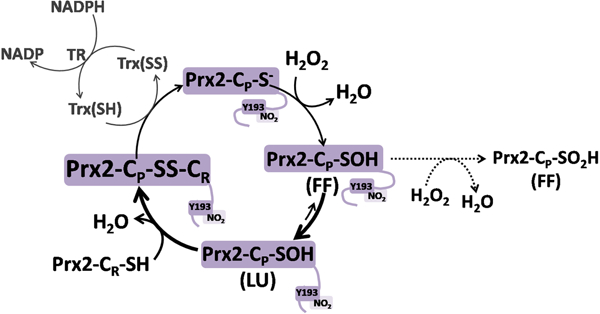
Peroxiredoxins (Prx) are enzymes that efficiently reduce hydroperoxides through active participation of one or two cysteine residues (1). The catalytic cycle can be divided into three steps: 1) the reduction of peroxide via oxidation of the peroxidatic cysteine (CP-SH) to sulfenic acid (CP-SOH), 2) the resolution step, that for typical 2-Cys Prx involves the condensation of the sulfenic acid with the resolving cysteine (CR-SH) to form an intersubunit disulfide and 3) the reduction of the disulfide at the expense of thioredoxin or other reductant. The resolution step demands a conformational change to bring both cysteine residues together in order to make the disulfide bond, the so-called FF (fully-folded) to LU (locally-unfolded) transition. The CP-SOH has a second potential fate; it can either react with the resolving cysteine CR-SH (which requires the FF to LU transition) or (in FF) with a second peroxide molecule forming cysteine sulfinic acid CP-SO2H (hyperoxidation).
The first step in catalysis, the reduction of peroxide substrate, is fast. In particular, the rate constant for the reduction of hydrogen peroxide (H2O2) by the peroxidatic cysteine of human Prx2 (a typical 2-Cys Prx) is 1 x 108 M−1 s−1 (2), more than a million times faster than the reaction of H2O2 with a low molecular weight thiol (3). Structural and computational studies have helped uncover the particular active site architecture that explains this extraordinary acceleration (4–7). In addition, the intracellular concentration of Prx is high (10 – 400 µM), positioning them not only as good antioxidants but also as central players in redox signaling pathways (8,9).
These biological functions can be affected by post-translational modifications (PTM) that could alter aspects of the peroxidase activity and/or the interaction with other proteins. The latter would be extremely important in the transduction of the signal in a redox-relay mechanism and in the reduction step of the catalytic cycle (10,11). Any covalent modification of the CP will inactivate the enzyme, as observed before for nitrosation or glutathionylation (12–14). Hyperoxidation of CP also leads to inactivation of Prx2, at least during a period of time before the slow sulfiredoxin-dependent reduction of CP-SO2H takes place (15). It was noted that the bacterial Prx were more resistant to hyperoxidation than the eukaryotic counterparts which led to a classification into robust vs sensitive Prx. Early comparative structural analysis revealed that sensitive Prx present YF and GGLG motifs close to the active site that are largely absent in prokaryotes (16,17). The YF sequence motif in the C-terminal helix of one subunit covers helix α2 containing the CP of the adjacent subunit, hindering the unfolding of the CP-containing α2 to reach CR and form the disulfide. More recently, additional molecular determinants were discovered, motifs A and B, that even discriminate distinct levels of sensitivity to hyperoxidation amongst the sensitive Prx (18).
We have previously reported that treatment of human Prx2 with peroxynitrite results in tyrosine nitration, a PTM on non-catalytic residues (19). Even though Prx2 is a peroxynitrite reductase, the enzyme gets nitrated (on tyrosine residues) as well as hyperoxidized (on its peroxidatic cysteine) in turnover (2,20). Under conditions of high oxidant flux, most of intracellular Prx is found in the disulfide-oxidized form, and in this form, it is more easily modified on tyrosine residues by peroxynitrite. Nitration of tyrosines after peroxynitrite treatment of the disulfide-containing Prx2 surprisingly resulted in an enzyme more active as a peroxidase, with higher resistance to hyperoxidation (19).
Protein tyrosine nitration is a common PTM occurring under conditions of nitroxidative stress, which alters the structure and function of the modified protein. The biological mechanism of protein nitration is a radical mechanism involving the one-electron oxidation of a tyrosine residue to form the tyrosyl radical followed by addition of nitrogen dioxide (NO2•) to yield 3-nitrotyrosine (21,22). The radicals derived from peroxynitrite homolysis represent a major source of tyrosine nitration in vivo (23). Although non-enzymatically catalyzed, specificity in protein nitration is achieved in vivo (24). Proteomic analysis revealed the presence of 3-nitrotyrosine on select proteins as well as specific tyrosine residues. PrxII E from Arabidopsis thaliana was found nitrated after inducing oxidative stress in plants (25) and Prx1 was found nitrated (on Y194) on Jurkat cells exposed to peroxynitrite (26). Nitrated proteins have been detected in plasma and tissues of patients with oxidative stress-associated pathologies and aging. In particular, nitrated Prx2 was found in patients with early Alzheimer’s disease (27) and mitochondrial Prx3 was found nitrated in diabetic mice hearts (28).
Human Prx2 is a decamer composed of five head-to-tail homodimers and each monomer contains seven tyrosine residues, all of them at least somewhat exposed to the solvent (Figure 1). Mass spectrometry analysis of disulfide-containing Prx2 treated with peroxynitrite revealed that tyrosine nitration was the main modification on the protein and nitration of three residues (Y33, Y126 and Y193) was confirmed (19). Although peroxynitrite treatments could be adjusted to limit modification of the Prx2 protein molecules to 1–1.5 sites of nitration on average, all preparations were undoubtedly complex mixtures with up to two sites modified, potentially also on different tyrosine residues within the mixture. Despite this, the peroxynitrite-treated Prx2 always behaved as a more robust Prx compared with the non-treated enzyme.
Figure 1.

Representation of the human Prx2 dimer. The 7 tyrosine residues (magenta) from chain A (light gray) are shown, as well as the cysteine residues of both active sites of the dimer: CP and CR of chain A (light gray); and CP’ and CR’ of chain B (dark gray); CP is C51, CR is C172. Figure constructed using Pymol from pdb 1QMV.
Herein, mutants of human Prx2 were produced in order to unravel the effects of tyrosine nitration on the activity of this protein, which could impact on its role as a H2O2 sensor/transducer. In particular, specific co-translationally nitrated variants of the protein on different tyrosine residues were generated using genetic code expansion technology to incorporate unnatural amino acids. Together, our results confirm that the presence of Y193 is necessary to observe an increase in peroxidase activity and a more robust Prx2 after nitration by peroxynitrite. The results underline the critical role of this C-terminal residue on the rate of disulfide formation that affects the fate of Prx2 in redox signaling pathways.
Materials and methods
Chemicals
Dithiothreitol (DTT), N-ethylmaleimide (NEM) and reduced nicotinamide adenine dinucleotide phosphate (NADPH) were purchased from AppliChem (Germany). Hydrogen peroxide (H2O2), 3-nitro-L-tyrosine and diethylenetriaminepentaacetic acid (DTPA) were purchased from Sigma-Aldrich (USA).4,4’-Dithiodipyridine (DTDPy) was from Acros Organics (Fisher Scientific, USA). Peroxynitrite was synthesized as in (29). All other reagents were of analytical grade and used as received.
Methods
Peroxide and protein quantification
The concentration of H2O2 was measured at 240 nm (ɛ240 = 39.4 M−1 cm−1) (30), peroxynitrite concentration was determined at alkaline pH at 302 nm (ɛ302 = 1670 M−1 cm−1) (31) and E.coli thioredoxin reductase (TR) concentration was measured at 280 nm (ɛ280 = 51700 M−1cm−1) (32). Prx2 wild-type, mutants and E. coli thioredoxin 1 (Trx) were quantified by the absorption at 280 nm, using the corresponding ɛ determined with https://web.expasy.org/protparam/: ɛ280 = 21555 M−1 cm−1 for (Prx2)SS, (Prx2 Y193F)SS, and (Prx2 Y193M)SS,ɛ280 = 21430 M−1 cm−1 for (Prx2)SH, (Prx2 Y193F)SH, (Prx2 Y93G)SH, and ɛ280 (EcTrx)SH = 14060 M−1 cm−1. The co-translationally nitrated variants of Prx2 (NO2Y33, NO2Y43 and NO2Y126 Prx2) were quantified using the Bradford method and BSA for the calibration curve.
Protein expression and purification
- Wild-type Prx2, Y193F, Y193G and Y193M Prx2 mutants
Recombinant proteins were expressed with two different constructs; non-tagged recombinant wild-type Prx2 and the Y193F mutant were expressed and purified as in (33) using a pET17 vector in BL21-Gold(DE3) with the modification that lactose autoinduction media, ZYM-5052, containing 100 µg ml−1 ampicillin, was used for expression overnight at 25 °C (34). The QuikChange mutagenesis protocol was used to create the Y193F mutation in the wild-type vector. On the other hand, Y193G and Y193M Prx2 were expressed and purified as reported for PRDX1 in (35) using a pET28a plasmid containing the coding sequence of wild type HsPrx2 fused to a His-tag and a cleavage site for the protease TEV at the N-terminus (synthesized and codon-optimized by Genscript). Mutagenesis was performed using a QuikChangeTM site-directed mutagenesis kit (Stratagene). All of the site-directed mutations were confirmed by DNA sequencing.
Purified proteins were concentrated and stored at −20 °C in buffer A (50 mM sodium phosphate and 150 mM NaCl, 0.1 mM DTPA, pH 7.4). Purity was evaluated by SDS-PAGE.
- Co-translationally nitrated Prx2 variants
The pET17 clone containing the gene for wild-type Prx2 was mutated to convert the native amber TAG stop codon to TAA, an ochre stop codon. Individual Tyr codons at positions 33, 43, 126 and 193 were then mutated to a TAG amber codon. BL21-ai (arabinose inducible) competent cells were co-transformed with one of these mutated plasmids and pDule2–3nY-5B, a plasmid expressing mutated tRNA and aminoacyl-tRNA synthetase genes which direct nitrotyrosine incorporation at TAG codons (36). Protein expression was carried out in the arabinose induction media described by Hammill et al (37) modified to include a final concentration of 1 mM 3-nitrotyrosine from a 50 mg ml−1 stock dissolved in 1 M HCl. Overnight growth at 37 °C was found to be optimal for expression of Prx2 containing nitrotyrosine. Purification of nitrotyrosine-containing Prx2 was carried out as described above for non-tagged, wild-type Prx2. Attempted expression of Prx2 with nitrotyrosine incorporation at codon 193 consistently gave a mix of truncated as well as successfully extended protein, which could not be separated during the purification steps. Re-engineering of the expression vector to include cleavable fusion proteins at the C-terminus was also unsuccessful in generating the nitrated protein at Y193.
- Proteins for the coupled activity assay
E. coli thioredoxin 1 (EcTrx) was expressed and purified according to (38) and E. coli thioredoxin reductase (EcTR) as reported in (39). Protein purity was evaluated by SDS-PAGE.
Prx2 thiol reduction and oxidation
For reduction of purified Prx2, the enzyme was incubated with 1 mM DTT for 30 min at room temperature immediately before the experiment, and the mixture was passed twice through a Vivaspin ultrafiltration spin column (5 kDa MWCO, GE Healthcare) pre-equilibrated with the assay buffer. Thiol concentration was determined immediately after recovery of the protein. Controlled oxidation of reduced Prx2 to its disulfide form was achieved with the addition of 0.6 equivalents of H2O2 per thiol, probed to be enough oxidant to form the disulfide without hyperoxidation to sulfinic acid (19).
Nitration of Prx2 with peroxynitrite
Peroxynitrite has been shown to react with Prx2 CP, hyperoxidizing it to its sulfinic and even sulfonic acid form (2). To prevent these modifications of CP, treatment with peroxynitrite was performed on the disulfide-oxidized enzyme. Disulfide-oxidized Prx2 (130 µM) was treated with a five-fold excess peroxynitrite in a flux-like addition, as described in (19). This protocol had to be followed rigorously in order to obtain reproducible results after nitration, considering that nitration yields depend on the rate of radicals production from peroxynitrite (21,40). As a control, peroxynitrite was previously decomposed in the assay buffer and then added to the protein (reverse-order addition, ROA). After treatment with peroxynitrite or its decomposition products, Prx2 thiols were reduced with DTT and oxidized to disulfide when needed, as described above.
NADPH-linked peroxidase activity (coupled assay)
Peroxidase activity was measured spectrophotometrically through a coupled assay following the NADPH consumption at 340 nm (ɛ340 = 6.2 mM−1 cm−1) in a cuvette containing 200 µM NADPH, 1 µM EcTR, 8 µM EcTrx1 and 0.5 µM Prx in buffer A. The reaction was started by adding the specified amount of H2O2. All spectrophotometric measurements were made with a Cary 50 spectrophotometer (Varian, Australia).
Determination of Chyp1% in turnover
Using the NADPH-linked peroxidase activity assay, Chyp1% (as the peroxide concentration at which 1 out of every 100 Prx molecules would be inactivated per turnover) was determined for wild type and Prx2 mutants according to (41).
Kinetics followed by intrinsic fluorescence
Reaction of pre-reduced enzymes with H2O2 was detected by following the change in their intrinsic fluorescence as described in (42) in an Applied Photophysics SX20 stopped-flow fluorimeter with a mixing time of less than 2 ms, with λex = 280 nm, measuring the total emission above 320 nm. Prx (0.25 µM) in buffer A was reacted with different concentrations of H2O2 diluted in buffer A. At low oxidant concentrations (<10 µM) the time courses were biphasic (a fast drop, followed by a slow increase in the signal) and fit to a double exponential function. The fast phase, rapid decrease in fluorescence was associated to the reaction Prx-CP-SH + H2O2 → Prx-CP-SOH (kfast=kH2O2). When working at higher H2O2 concentrations, the downward phase became faster than the dead time of mixing (thus hard to register), and the slower phase was fit to a monoexponential decay (kslow). This slower recovery of fluorescence, kslow, was associated with the two processes competing for CP-SOH: disulfide formation (kres) and hyperoxidation to CP-SO2H (khyp).
| equation 1 |
From the curve kslow vs [H2O2], the bimolecular rate constant of hyperoxidation, khyp (slope), and the first-order rate constant of resolution, kres (y-axis intercept) were determined for each protein assayed.
Direct determination of Chyp1%
Knowing the rate constants of the two competing processes, kres and khyp, the fraction of inactive sulfinylated Prx, finact, at specific concentrations of H2O2 can be estimated as:
| equation 2 |
The value of Chyp1% as the concentration of H2O2 that yields 1% inactivated/hyperoxidized Prx can be calculated from finact= 0.01, thus:
| equation 3 |
Prx2 circular dichroism (CD) spectra analysis
Spectra of Prx2 wild-type, mutants and nitro variants were acquired at 20 °C using a Jasco 810 spectropolarimeter (with a Jasco CDF-4265/15 Peltier-effect device). Near-UV measurements were carried out in 1 cm cells containing 40 µM Prx2 in buffer A. For far-UV measurements, 0.1 cm cells were used, and protein samples were diluted to 4 µM in the same buffer. Scans of buffer were properly smoothed and subtracted from the corresponding averaged sample spectra.
Bioinformatics and conformational stability predictions
The contribution of a given side chain to the global stability of the Prx2 was explored by means of two different computational methods: FOLDX (43,44) and Dynamut (45). The former is based on an empirical physic force field, whereas the latter is based on a statistical force field which additionally introduces the dynamics component to mutation analysis. In both cases the calculation of the free energy contribution was carried out using the high-resolution 3D structure of a Prx2 (PDB ID: 1QMV) in which the hyperoxidized Cys 51 residue was first converted to Cys and then, the routine ‘repair’ tool from FOLDX was run in order to minimize the energy of the model (side-chains with wrong torsion angles or Van der Waals’ clashes were identified and corrected by changing the side-chain rotamers). In addition, the calculations were performed using the dimer AB and the mutation was introduced only in chain A. The was considered for the interpretation of the calculations.
The dimer AB from Prx2 structure PDB ID: 1QMV was examined using Frustratometer (46) in order to infer the localization of energetic frustration in the native protein structure. Frustration is a measure of how favorable a particular contact is relative to sets of possible interactions. The individual contacts can be classified regarding their frustration index values in contacts minimally, neutral, or highly frustrated. Mutational frustration was evaluated, that is: how favorable the native residues are relative to other possible residues in that location. Frustatometer evaluates every possible mutation of the amino acid pair that forms a particular contact in a fixed structure. Importantly, the energy change observed comes from the particular contact probed but also from interactions of each residue with other residues of the protein.
Molecular dynamics (MD) simulations
The coordinates corresponding to Prx2 structure (PDB ID: 1QMV) were used as starting structure for classical molecular dynamics simulation. Given that CP is oxidized to sulfinic acid, it was in silico transformed to Cys (thiolate state). Additionally, Y193G and Y193F mutants were prepared. Mutations were performed in every chain of the decamer. Each system was solvated with TIP3P water molecules (47) and a standard minimization protocol was applied. The systems were heated up from 100 K to 300 K, using the Berendsen thermostat and subsequently switched to a constant isotropic pressure to allow the density to equilibrate. The SHAKE algorithm was applied and a 2 fs time step was settled. Unrestrained MD simulations were performed with the Amber16 software (48) and all used residue parameters corresponding to the ff14SB force field (49). Production MDs were run at 300K for 100 ns. Analysis was performed using Amber Tools package.
Thermal-induced denaturation
Heat-induced denaturation of Prx2 wild-type and mutants was carried out in a Chronos FD fluorimeter (ISS, USA) equipped with a Peltier-type temperature controller. Thermal denaturation was recorded following changes in fluorescence spectra (excitation at 280 nm, emission from 310 nm to 450 nm) from 25 °C to 65 °C at 5 °C intervals and allowing 10 min for equilibration at each temperature. The protein concentration was 2 µM in 50 mM phosphate buffer, pH 7.4, 150 mM NaCl, 0.1 mM DTPA, 1 mM DTT and 1 M urea (to reduce temperature of melting and avoid aggregation).
Results
Our previous studies on nitration of human Prx2 were performed on the enzyme purified from red blood cells, thus, the first step was to obtain the human recombinant protein in order to generate different mutations on its tyrosine residues. Importantly, the same peroxynitrite-dependent effects were observed on the recombinant protein as on protein purified from red blood cells, i.e. the peroxidase activity increased with a decrease in hyperoxidation sensitivity (Figure 2A and 2B). The analysis of steady-state kinetics (coupled assay) at different H2O2 concentrations allows in a simple graphical way, easy comparison of the sensitivity of these enzymes to oxidative inactivation. Given the low apparent Km of Prx2 for H2O2 and the catalytic mechanism of this protein (2,8), it is expected that, at low peroxide concentrations, all the substrate is consumed at maximum velocity as observed when 10 µM H2O2 is used (Figure 2A). However, for a sensitive enzyme like Prx2, the peroxide consumption rate at a concentration as low as 20 µM already significantly decreases during turnover (Figure 2A), which has been associated with inactivation by hyperoxidation of its peroxidatic cysteine (41). Nitration of Prx2 by peroxynitrite yields an enzyme able to work at maximal activity at higher H2O2 concentrations over many turnovers, i.e. more resistant to hyperoxidation (Figure 2B).
Figure 2.

Peroxidase activity of recombinant wild-type Prx2, with and without peroxynitrite treatment, and specific nitro variants. (A) Coupled assay of peroxidase activity of wild type Prx2. For activity measurements, 200 µM NADPH, 1 µM EcTR, 8 µM EcTrx1 and 0.5 µM Prx2 were used, and reaction was started with 10 µM H2O2 (black), 20 µM H2O2 (dark gray) or 50 µM H2O2 (light gray). Consumption of NADPH was followed at 340 nm. (B) Coupled assay peroxidase activity of nitrated wild-type Prx2 in the same conditions used in A. Inset. Coupled assay peroxidase activity of wild-type Prx2 non-treated (black) or treated with a 5-fold excess peroxynitrite (gray) using 10 µM H2O2. (C) Coupled assay peroxidase activity of nitro-variant NO2Y33 Prx2 and (D) NO2Y126 Prx2, in the same conditions used in A
Co-translational incorporation and kinetic analysis of specific nitro-tyrosine residues
Treatment of native Prx2 with peroxynitrite resulted in detectable nitration of three of the seven tyrosine residues of the protein (Y33, Y126 and Y193, Figure 1), but this nitration was shown to be non-stoichiometric and complex (19,50). One approach to study the role of tyrosine nitration on Prx2 functionality was to use the elegant method developed by R. Mehĺs group that uses genetic code expansion to site-specifically incorporate non-canonical amino acids into proteins (51,52). We succeeded in producing homogeneous, high-quality nitrated forms of Prx2 on residues Y33 and Y126, as confirmed by ESI-TOF MS (Figure 1S) and UV-vis absorption spectra (Figure 2S). Far-UV CD spectra (Figure 3S) confirmed that the nitro variants displayed essentially the same secondary structure as the wild type in the reduced as well as the oxidized form (50). The near-UV CD spectra showed a notable decrease of the signal around 280 nm after disulfide formation as observed before for the wild type enzyme (50).
Peroxidase activity of these nitro variants was measured using the coupled assay following NADPH consumption at 340 nm, as shown in Figures 2C and 2D. Interestingly, both nitro variants were active peroxidases in the conditions of our assay, although differences in the susceptibility to hyperoxidation were observed. Again, the activity using the coupled assay was measured at three different H2O2 concentrations, giving a sense of the differential susceptibility to hyperoxidation. While NO2Y33 showed a decrease in peroxidase activity at a H2O2 concentration as low as 20 µM (Figure 2C), NO2Y126 Prx2 was more resistant to hyperoxidation, not showing loss of activity until 50 µM H2O2 (Figure 2D). Importantly, none of the mononitrated variants assayed were as robust as the peroxynitrite-treated enzyme (Figure 2B).
In addition, NO2Y43 Prx2 was expressed and successfully purified. Although Y43 was not detected as nitrated by MS after peroxynitrite treatment of Prx2 (19), it is located in a critical position in proximity to the interface between dimers and accessible to the solvent (Figure 1). As seen for the other nitrovariants, the mononitrated variant on Y43 did not behave as the nitrated wtPrx2 (Figure 4S).
Unfortunately, homogenous NO2Y193 Prx2 could not be adequately expressed since during synthesis in E. coli this variant was prone to becoming prematurely truncated, a result observed by other authors when trying to incorporate modified amino acid residues at the C-terminus of a protein (53). The obtained mixture of full-length protein presumably containing 3-nitrotyrosine along with protein truncated at the C-terminus could not be resolved during purification, suggesting that dimerization of the Prx2 is not significantly affected by the truncation. As described in more detail below, however, the characterization of a panel of site-directed mutants at position Y193 did prove to be useful.
Characterization of Prx2 Y193 mutants
Since the NO2Y193 variant could not be obtained, point mutations were designed to test the important structural/functional role of this residue at position 193. The first approach was to delete the tyrosine residue at that position and the mutant Y193F was generated. Although the mutant Y193F Prx2 showed higher peroxidase activity than the wild type enzyme in the NADPH-linked coupled assay (Figure 3A), (probably due to the fact that the C-terminus was modified, which has repeatedly been related to changes in activity for typical 2-Cys Prx (54–57)), the effect of peroxynitrite treatment on this mutant was totally different than for the wild type enzyme (Figure 3A). Disulfide-containing Y193F Prx2 treated with peroxynitrite yielded a nitrated protein on tyrosine residues other than Y193, as shown in Figure 3C. However, the nitrated Y193F Prx2 was a less active peroxidase, still sensitive to inactivation by hyperoxidation (Figure 3A and 3B). It is worth noting that peroxynitrite is capable of nitrating tyrosine and tryptophan residues but not phenylalanine (58).
Figure 3.

Effect of nitration on NADPH-linked peroxidase activity of wild type vs Y193F Prx2. (A) Non-treated Y193F Prx2 is shown in black while protein treated with a 5-fold excess of peroxynitrite is shown in dark gray. Wild type Prx2 activity is shown in light gray. To start the reactions, 10 µM H2O2 was added and consumption of NADPH was followed at 340 nm. (B) The same treatments and assays were done with nitrated Y193F Prx2 using increasing concentrations of H2O2 [10 µM H2O2 (black), 20 µM H2O2 (dark gray) or 50 µM H2O2 (light gray)]. (C) Western blot of wild-type and Y193F Prx2, non-treated or treated with 5-fold excess peroxynitrite were resolved by a 12% reducing SDS-PAGE. A polyclonal antibody against nitrated tyrosine residues (59) was used in a 1/5,000 dilution and a secondary antibody conjugated to HRP was used in a 1/100,000 dilution.
Steady-state kinetics (coupled assay) at different H2O2 concentrations showed that Y193F Prx2 was more sensitive to hyperoxidation than the wild type (Figure 4A). In addition, a separate kinetic analysis was performed by stopped-flow spectrofluorimetry following changes in intrinsic fluorescence after mixture of the reduced enzyme with various concentrations of H2O2 (Figure 4B). As reported previously for Prx oxidation by peroxides (6,20,41,60,61), two phases of fluorescence changes were observed, a fast decrease in emission associated with the oxidation step, followed by a slower recovery of fluorescence associated with the resolution step (Figure 4B). For Y193F Prx2, from the fast peroxide-dependent phase, a second-order rate constant of 1.3 x 108 M−1 s−1 was determined for the reaction of CP-SH with H2O2 (Figure 4C), a value similar to the previously reported for different Prx isoforms (2,20,42). However, the slow phase, associated with the competition between the two pathways the sulfenic form of the enzyme could follow, hyperoxidation or disulfide formation (resolution step), behaved differently than the wild type enzyme with a higher kres (0.64 vs 0.2 s−1) but the same khyp = 1.2 x 103 M−1 s−1 (Figure 4D, Table 1). The faster resolution rate constant obtained for the variant Y193F is in accordance with a slightly faster peroxidase activity and a modestly more robust enzyme. These results suggest that removing a hydroxyl group in the C-terminal region of Prx2 is sufficient to lead to dynamic or conformational changes in the enzyme that can alter its catalytic activity.
Figure 4.

Kinetic analysis of the reaction of H2O2 with Y193F Prx2. (A) NADPH-linked peroxidase activity of Y193F Prx2. Reaction was started with the addition of 10 µM H2O2 (black), 20 µM H2O2 (dark gray) or 50 µM H2O2 (light gray). (B) Intrinsic fluorescence profile of the reaction of 0.5 µM Y193F Prx2 with 1 µM H2O2. (C) Fast phase kinetic constant determined using different H2O2 concentrations. (D) Dependence of the kinetic constant of the second (slow) phase on H2O2 concentration. λex = 295 nm.
Table 1.
Kinetic constants for disulfide formation and hyperoxidation and determination of Chyp1% value of different Prx.
| kres (s−1) | khyp (M−1 s−1) | direct Chyp1% (µM) | Chyp1% (µM) | |
|---|---|---|---|---|
| Prx2RBC | 0.419 ± 0.004 a 2.0 b |
(2.6 ± 0.4) × 103 a 1.2 × 104 b |
1.61 1.67 |
|
| wt Prx2 | 0.197 ± 0.001 0.20 c |
(1.73 ± 0.06) × 103 2.0 × 103 c |
1.14 1.00 |
5 d |
| Nitrated wt Prx2 | ¥ | ¥ | 136 | |
| Y193F Prx2 | 0.627 ± 0.002 | (2.31 ± 0.09) × 103 | 2.71 | |
| Y193G Prx2 | 76 ± 1 | (1.3 ± 0.2) × 103 | 585 | |
| AhpC | 77 ± 1 | (0.41 ± 0.03) × 103 | 1880 | > 5000 d |
| Prx1 | 11 c | 1.7 × 103 c | 65 | 50 d 62 e |
| Prx3 | 20 b 2 f |
1.2 × 104 b 1.1 × 103 f |
17 18 |
127 e |
Randall et al, unpublished results
Peskin et al (64)
Dalla Rizza et al (20)
Bolduc et al (63)
Nelson et al (41)
De Armas et al 2019 (61)
could not be determined by intrinsic fluorescence kinetics Chyp1% refers to the parameter defined in Nelson et al. (41) direct Chyp1% was calculated using equation 3
Values shown in black were obtained in this work. Values shown in gray were collected from the literature and used to calculate direct Chyp1% for Prx2RBC, wt Prx2, Prx1 and Prx3.
The kres and khyp values from this work were obtained following changes in intrinsic fluorescence in a SF spectrophotometer. All experiments were performed in buffer A (pH 7.4).
Computational analysis regarding the contribution of different side-chains in position 193 to the global stability of Prx2 suggested that a glycine residue in that position would significantly destabilize the protein, while a phenylalanine would rigidize the region (Figure 5A). Mutation of position 193 to Met was predicted as neutral or modestly destabilizing, with a small enhancement in flexibility. In addition, the energetics of Prx2 dimer structure were explored by means of the Frustratometer tool (Figure 5B). Briefly, for a contact between two residues defined as highly frustrated, it means that most other amino acid pairs at that location would be more favorable for folding than those found in Prx2 sequence (the native ones). Even though the core of the dimer seems to be unfrustrated, some regions of the protein present clusters of highly frustrated contacts. Among them, the C-terminal region is shown to be highly frustrated. In particular, residue Tyr 193 showed the highest frustration index value (Figure 5B). This fact suggests that this residue may not necessarily provide stability to the protein but would potentially contribute to the Prx2 function. Thus, a Gly residue at position 193 is expected to increase the local backbone flexibility, whereas a Phe residue would rigidify this α-helical stretch, as judged by the Dynamut analysis (Figure 5A). In fact, preliminary molecular dynamics simulations show that residues at the C-terminal of the mutant Y193G display more fluctuations than the rest of the backbone chain compared to the wt Prx2 (Figure 5S), suggesting a higher flexibility of this loop when glycine (and not phenylalanine) is in position 193, as predicted.
Figure 5.

Energetics and dynamics of Prx2 and mutants. (A) Predictions of the contribution of residue 193 to the conformational stability of Prx2: FOLDX and Dynamut were used to test the effect of point mutations at position 193 in chain A (light and dark gray, respectively). In addition, Dynamut was used to evaluate the effect of these mutations on dynamics (cyan). (B) Frustration by residue for chains A and B (cyan and black, respectively). Positive (> 0.55) values indicate residues minimally frustrated and negative (˂ −1) values indicate residues highly frustrated. The arrow indicates residue Y193 All the calculations were carried out using the dimer AB from PDB ID: 1QMV, in which hyperoxidized Cys was previously mutated to Cys using FOLDX.
Following up on these computational results, the mutants Y193G and Y193M were generated, since the first one would theoretically yield a much more destabilized protein than the latter (Figure 5). As shown in Figure 6, insertion of a Met residue in position 193 gave an inactive Prx2, while Y193G was a more active and robust variant of the enzyme.
Figure 6.

Kinetic analysis of the reduction of H2O2 by wild type, Y193G and Y193M Prx2. (A) NADPH-linked peroxidase activity of wild-type Prx2 is shown in black, Y193G Prx2 in dark gray and Y193M Prx2 in light gray. Ten µM H2O2 was added to start the reactions and consumption of NADPH was followed at 340 nm. (B) NADPH-linked peroxidase activity was followed for the Y193G Prx2 mutant in the same conditions, using 10 µM H2O2 (black), 20 µM H2O2 (dark gray) or 50 µM H2O2 (light gray). (C) Oxidation of Y193G Prx2 followed by intrinsic fluorescence. Dependence of the kinetic constant of the slow phase on H2O2 concentration.
Interestingly, the mutant Y193G Prx2, displaying a more unstable C-terminal region (Figure 5C, Figure 5S) behaved very similarly to the nitrated enzyme, showing higher peroxidase activity and higher robustness than the wild type enzyme (Figure 6B). Availability of this pure, single site variant also enabled evaluation of rate constants by intrinsic fluorescence kinetics (Figure 6C), revealing a faster resolution step (kres = 76 s−1) and a similar hyperoxidation rate constant, khyp = 1.3 × 103 M−1 s−1 (Table 1).
Analysis of hyperoxidation sensitivity and structural features of Y193F and Y193G
The parameter Chyp1% is the concentration of H2O2 that yields 1% of inactivated Prx per turnover, previously described as a quantitative measure of Prx susceptibility toward peroxide-mediated hyperoxidation (41). Herein, a straightforward way to calculate this parameter is described using the rate constants of the two competing reactions for CP-SOH, disulfide formation or hyperoxidation, as determined during the stopped flow kinetic analysis (equation 3). For Y193G Prx2 we obtained a value of Chyp1% higher than 500 µM to achieve 1% inactivation per turnover, compared to ~1.6 µM for the wild type enzyme (Table 1) supporting the higher robustness of this mutant to H2O2-induced inactivation. For Y193F Prx2 analyzed by this approach, Chyp1% was only slightly increased compared to wild type enzyme, ~3 µM (Table 1). Rate constants for the nitrated wild type Prx2 could not be determined using stopped flow fluorimetry due to heterogeneity of the sample and quenched Trp fluorescence, but using the coupled assay and previous approach (41) Chyp1% was determined to be ~136 µM (Table 1).
In addition, as observed before for the peroxynitrite-treated Prx2 (50), the near-UV CD spectra of the reduced form of Y193G resembles its disulfide-oxidized form (Figures 7A and 7B), while Y193F only modestly exhibited this perturbation. The fluorescence spectra (Figure 7C) show a red shift of the maximum emission and decreased in intensity for Y193G mutant (center mass of 352 nm compared to 349 nm for the wild type protein), indicating a shift of tryptophan residues to a more polar environment. Prx2 has two tryptophan residues, W86 close to CP, and W176 at the C-terminal region; W176 is probably the one being more exposed in the Y193G Prx2 structure. Global protein stability of the Prx2 wild type and mutant proteins was also assessed by monitoring fluorescence changes during thermal denaturation. A lower apparent melting temperature of denaturation was obtained for the Y193G mutant compared to wild type (Figure 7D) indicating a decrease in global stability by insertion of a glycine at position 193, while Y193F mutant displayed an intermediate stability.
Figure 7.

Spectroscopic analysis of Prx2 mutants Y193F and Y193G. (A) Near UV-CD spectra of the reduced form of wild type Prx2 (black), Y193F (dark gray) and Y193G (light gray). (B) Near UV-CD spectra of disulfide-oxidized wild type Prx2 (black), Y193F (dark gray) and Y193G (light gray). (C) Emission spectra of wild type Prx2 (black), Y193F (dark gray) and Y193G (light gray) mutants in phosphate buffer pH 7.4, 1 mM DTT, 1 M urea, λex = 280 nm. (D) Thermal-induced denaturation followed by intrinsic fluorescence (λex = 280 nm, λem = 340 nm). Apparent melting temperatures of 47 °C, 44 °C and 40 °C were obtained for wt Prx2 (black), Y193F (dark gray), and Y193G (light gray), respectively.
Nitration of AhpC, a model robust Prx
AhpC, a very well characterized robust Prx, lacks the YF motif present in the sensitive enzymes. As previously reported (62), the kinetics of H2O2 reaction with AhpC from Salmonella typhimurium followed by intrinsic fluorescence showed similar behavior to other 2-Cys Prx. More specifically, AhpC behaved most like mutant Y193G Prx2, displaying a slow recovery of fluorescence after the initial fast drop, from which kres and khyp, as well as a Chyp1% value of ~1.9 mM, were obtained herein (Table 1, Figure 6S). The same described peroxynitrite treatment of human Prx2 (using a 5-fold excess of peroxynitrite) was performed on AhpC, protecting its cysteine residues by first forming the disulfide bond in the active site. After treatment, nitration on tyrosine residues was confirmed by western blot analysis (not shown), and the peroxidase activity was assessed using the coupled assay (Figure 7S). Interestingly, nitration of AhpC, a Prx lacking a tyrosine residue at the C-terminus, did not increase the enzyme activity; on the contrary, it rendered AhpC moderately less active as a peroxidase, which is the same effect as observed for the nitrated Y193F Prx2 mutant, also lacking Y193 at the C-terminal. Nitration of AhpC was never reported before and revealed a different behavior for this pathogen-derived Prx compared to the mammalian host counterpart Prx2.
Discussion
We previously reported that nitration of human Prx2 transforms this sensitive enzyme into a more robust peroxidase, less sensitive to hyperoxidation, and our results pointed to Y193 nitration as likely responsible for that effect (19). In order to unravel the molecular mechanism for this modulation, Prx2 variants with a co-translationally incorporated nitrotyrosine residue were envisioned. In practice, three proteins with a unique nitration site per monomer were obtained, NO2Y33, NO2Y43 and NO2Y126 (Figures 1S, 2S, 3S). This elegant approach enabled studies with homogeneous samples, which cannot be achieved by protein treatment with peroxynitrite, however, none of these nitro variants behaved as the nitrated wild type (Figures 2 and 4S) (22). Unfortunately, it was not possible to effectively incorporate a nitrotyrosine into position 193 belonging to the YF motif linked to eukaryotic Prx sensitivity to hyperoxidation. On the other hand, the effects on conformation and function of the chemical alteration of side-chain 193 were explored by site-directed mutagenesis. Different mutants for this position were prepared and studied. Surprisingly, a neutral substitution of Y193 by phenylalanine resulted in a variant slightly more resistant to hyperoxidation than the wild type (Figure 3A), underlying the importance of this structural motif for Prx2 functional modulation. Given this first interesting result, other mutants at this position that would stabilize or destabilize the protein were designed based on theoretical calculations (Figure 5). Preliminary molecular dynamics simulations suggest that incorporation of glycine at position 193 increases the fluctuations of the protein backbone in the C-terminal region more than observed for the wild type Prx2 (Figure 5S). As expected, this mutant with a more flexible C-terminal loop that favors the transition to the LU conformation, displayed a higher rate of disulfide formation, thus, more resistance to hyperoxidation (Figure 6, Table 1). Interestingly, while none of the nitro variants produced herein behaved like the nitrated wild type enzyme, the incorporation of a glycine in place of tyrosine at position 193 very much resembled it. In addition, peroxynitrite treatment of Y193F Prx2 (lacking a tyrosine residue at the C-terminal) yielded a protein nitrated on tyrosine residues but less active and still sensitive to hyperoxidation, contrary to what was observed for the wild type enzyme. Altogether, these results confirm that, although in vitro, Prx2 can get nitrated on other Tyr residues, the tyrosyl side-chain at position 193 is necessary for the observed increase in peroxidase activity and hyperoxidation resistance after peroxynitrite treatment. This effect is likely achieved by increased flexibility of the C-terminal loop so that the CP sulfenic acid can more easily reach the resolving cysteine to form the disulfide bond and protect the enzyme from hyperoxidation.
The results obtained for the Prx2 mutants have allowed a direct correlation between time courses of the coupled assay at increasing H2O2 concentrations and stopped-flow kinetics analysis (Figures 3, 4 and 6). With the rate constants obtained by intrinsic fluorescence, a value of Chyp1% was calculated as a measure of inactivation sensitivity, which mostly agree with the reported ones obtained in turnover (Table 1) (41,63). The faster the disulfide is formed (higher kres), the more resistant the enzyme is to hyperoxidation as long as the reactivity of the CP-SOH with H2O2 does not change significantly, and this is reflected in a higher concentration of H2O2 needed to inactivate 1% of the protein (Chyp1%). For instance, Prx2 showed a decrease in peroxidase activity during turnover with H2O2 concentrations as low as 20 µM, while Prx1 needs higher H2O2 concentrations to become inactivated (20). This correlates well with lower kres and Chyp1% values for Prx2 compared to Prx1 (Table 1). Unfortunately, peroxynitrite-treated Prx2 could not be reasonably analyzed by changes in intrinsic fluorescence, due to the heterogeneity of the sample combined with quenching of Trp fluorescence emission (19,50). However, Chyp1% in turnover was determined, yielding a value of 136 µM H2O2 (Figure 8S, Table 1), consistent with the desensitization of the enzyme by nitration as shown directly by the coupled assay (Figure 2).
These functional studies correlate with the near-UV CD spectra results, in which the conformational change occurring during oxidation of the active site can be detected (Figure 7). As previously reported, proteins less sensitive to oxidative inactivation by H2O2 also have a lower CD signal around 280 nm, already resembling the disulfide oxidized form even in their reduced state (50), which relates to the kres values discussed in this work. That is, with a more readily displaced C-terminus, the FF to LU transition of CP-SOH is facilitated and the disulfide can more readily form.
As previously pointed out (9,20), the kinetic pause at the resolution step opens different pathways for Prx to participate in redox signaling. The longer the lifetime of the sulfenic acid, higher the chance to get hyperoxidized but also to react with other protein thiols to translate the signal through a redox relay. On the other hand, a faster resolution step favors disulfide Prx formation, which can also function in a redox relay, probably interacting with a different set of targets.
In conclusion, our results not only confirm Y193 as a critical residue in modulation of Prx2 activity by peroxynitrite, but also highlight the importance of the YF motif at the C-terminus as a sensitive structural point that determines the fate of Prx2 in redox signaling pathways.
Supplementary Material
Highlights.
Tyrosine nitration of hPrx2 renders a peroxidase more resistant to hyperoxidation
Nitro Y193 at C-term YF motif increases the rate of disulfide formation
Side-chain at position 193 affects the rate of the resolution step
Chyp1% calculated from rate constants correlates well with resolution kinetics
Rate of resolution step impacts on the fate of Prx2 in redox signaling
Acknowledgements
Financial support was provided by Universidad de la República (CSIC C632-348) to AD, Agencia Nacional de Investigación e Innovación (FCE 2013_100581) to LMR and R01 GM119227 from the National Institutes of Health to LBP. LMR and JDR were partially supported by CAP Universidad de la República and PEDECIBA, Uruguay. We thank Drs. Cristina Furdui and Tom Forshaw for mass spectrometry analysis of purified Prx2 nitro variants, with support for instrumentation provided by the Kimbrell family. We thank Dr. Ari Zeida for running MD simulations, Drs. Beatriz Alvarez and Gerardo Ferrer for helpful discussions, Dr. Bruno Manta for critical reading of the manuscript and Lucía Blixen for graphical work assistance.
Footnotes
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wood ZA, Schroder E, Harris RJ, and Poole LB (2003) Structure, mechanism and regulation of peroxiredoxins. Trends in biochemical sciences 28, 32–40 [DOI] [PubMed] [Google Scholar]
- 2.Manta B, Hugo M, Ortiz C, Ferrer-Sueta G, Trujillo M, and Denicola A (2009) The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Archives of biochemistry and biophysics 484, 146–154 [DOI] [PubMed] [Google Scholar]
- 3.Winterbourn CC, and Metodiewa D (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free radical biology & medicine 27, 322–328 [DOI] [PubMed] [Google Scholar]
- 4.Hall A, Parsonage D, Poole LB, and Karplus PA (2010) Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. Journal of molecular biology 402, 194–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagy P, Karton A, Betz A, Peskin AV, Pace P, O’Reilly RJ, Hampton MB, Radom L, and Winterbourn CC (2011) Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. J Biol Chem 286, 18048–18055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Portillo-Ledesma S, Sardi F, Manta B, Tourn MV, Clippe A, Knoops B, Alvarez B, Coitino EL, and Ferrer-Sueta G (2014) Deconstructing the catalytic efficiency of peroxiredoxin-5 peroxidatic cysteine. Biochemistry 53, 6113–6125 [DOI] [PubMed] [Google Scholar]
- 7.Zeida A, Reyes AM, Lebrero MC, Radi R, Trujillo M, and Estrin DA (2014) The extraordinary catalytic ability of peroxiredoxins: a combined experimental and QM/MM study on the fast thiol oxidation step. Chemical communications 50, 10070–10073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferrer-Sueta G, Manta B, Botti H, Radi R, Trujillo M, and Denicola A (2011) Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chemical research in toxicology 24, 434–450 [DOI] [PubMed] [Google Scholar]
- 9.Randall LM, Ferrer-Sueta G, and Denicola A (2013) Peroxiredoxins as preferential targets in H2O2-induced signaling. Methods Enzymol 527, 41–63 [DOI] [PubMed] [Google Scholar]
- 10.Pace PE, Peskin AV, Konigstorfer A, Jasoni CJ, Winterbourn CC, and Hampton MB (2018) Peroxiredoxin interaction with the cytoskeletal-regulatory protein CRMP2: Investigation of a putative redox relay. Free radical biology & medicine 129, 383–393 [DOI] [PubMed] [Google Scholar]
- 11.Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T, Scharf AN, and Dick TP (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nature chemical biology 11, 64–70 [DOI] [PubMed] [Google Scholar]
- 12.Engelman R, Weisman-Shomer P, Ziv T, Xu J, Arner ES, and Benhar M (2013) Multilevel regulation of 2-Cys peroxiredoxin reaction cycle by S-nitrosylation. J Biol Chem 288, 11312–11324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fang J, Nakamura T, Cho DH, Gu Z, and Lipton SA (2007) S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc Natl Acad Sci U S A 104, 18742–18747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park JW, Piszczek G, Rhee SG, and Chock PB (2011) Glutathionylation of peroxiredoxin I induces decamer to dimers dissociation with concomitant loss of chaperone activity. Biochemistry 50, 3204–3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowther WT, and Haynes AC (2011) Reduction of cysteine sulfinic acid in eukaryotic, typical 2-Cys peroxiredoxins by sulfiredoxin. Antioxidants & redox signaling 15, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall A, Karplus PA, and Poole LB (2009) Typical 2-Cys peroxiredoxins--structures, mechanisms and functions. The FEBS journal 276, 2469–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perkins A, Nelson KJ, Parsonage D, Poole LB, and Karplus PA (2015) Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends in biochemical sciences 40, 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolduc JA, Nelson KJ, Haynes AC, Lee J, Reisz JA, Graff AH, Clodfelter JE, Parsonage D, Poole LB, Furdui CM, and Lowther WT (2018) Novel hyperoxidation resistance motifs in 2-Cys peroxiredoxins. J Biol Chem 293, 11901–11912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Randall LM, Manta B, Hugo M, Gil M, Batthyany C, Trujillo M, Poole LB, and Denicola A (2014) Nitration transforms a sensitive peroxiredoxin 2 into a more active and robust peroxidase. J Biol Chem 289, 15536–15543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dalla Rizza J, Randall LM, Santos J, Ferrer-Sueta G, and Denicola A (2019) Differential parameters between cytosolic 2-Cys peroxiredoxins, PRDX1 and PRDX2. Protein science : a publication of the Protein Society 28, 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radi R (2004) Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A 101, 4003–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trujillo M, Ferrer-Sueta G, and Radi R (2008) Peroxynitrite detoxification and its biologic implications. Antioxidants & redox signaling 10, 1607–1620 [DOI] [PubMed] [Google Scholar]
- 23.Trujillo M, Alvarez B, Souza JM, Romero N, Castro L, Thomson L and Radi R (2010) Nitric Oxide and Pathobiology, Academic Press, Orlando, FL [Google Scholar]
- 24.Denicola A, Alvarez B, and Thomson L (2008) 3-Nitrotyrosine, a post-translational modification associated with nitroxidative stress, Transworlk Research Network, India [Google Scholar]
- 25.Takahashi M, Shigeto J, Sakamoto A, Izumi S, Asada K, and Morikawa H (2015) Dual selective nitration in Arabidopsis: Almost exclusive nitration of PsbO and PsbP, and highly susceptible nitration of four non-PSII proteins, including peroxiredoxin II E. Electrophoresis 36, 2569–2578 [DOI] [PubMed] [Google Scholar]
- 26.Ghesquiere B, Colaert N, Helsens K, Dejager L, Vanhaute C, Verleysen K, Kas K, Timmerman E, Goethals M, Libert C, Vandekerckhove J, and Gevaert K (2009) In vitro and in vivo protein-bound tyrosine nitration characterized by diagonal chromatography. Molecular & cellular proteomics : MCP 8, 2642–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reed TT, Pierce WM Jr., Turner DM, Markesbery WR, and Butterfield DA (2009) Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. Journal of cellular and molecular medicine 13, 2019–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turko IV, Li L, Aulak KS, Stuehr DJ, Chang JY, and Murad F (2003) Protein tyrosine nitration in the mitochondria from diabetic mouse heart. Implications to dysfunctional mitochondria in diabetes. J Biol Chem 278, 33972–33977 [DOI] [PubMed] [Google Scholar]
- 29.Romero N, Radi R, Linares E, Augusto O, Detweiler CD, Mason RP, and Denicola A (2003) Reaction of human hemoglobin with peroxynitrite. Isomerization to nitrate and secondary formation of protein radicals. J Biol Chem 278, 44049–44057 [DOI] [PubMed] [Google Scholar]
- 30.Nelson DP, and Kiesow LA (1972) Enthalpy of decomposition of hydrogen peroxide by catalase at 25 degrees C (with molar extinction coefficients of H 2 O 2 solutions in the UV). Analytical biochemistry 49, 474–478 [DOI] [PubMed] [Google Scholar]
- 31.Radi R, Beckman JS, Bush KM, and Freeman BA (1991) Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J Biol Chem 266, 4244–4250 [PubMed] [Google Scholar]
- 32.Wang PF, Veine DM, Ahn SH, and Williams CH Jr. (1996) A stable mixed disulfide between thioredoxin reductase and its substrate, thioredoxin: preparation and characterization. Biochemistry 35, 4812–4819 [DOI] [PubMed] [Google Scholar]
- 33.Haynes AC, Qian J, Reisz JA, Furdui CM, and Lowther WT (2013) Molecular basis for the resistance of human mitochondrial 2-Cys peroxiredoxin 3 to hyperoxidation. J Biol Chem 288, 29714–29723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Studier FW (2005) Protein production by auto-induction in high density shaking cultures. Protein expression and purification 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 35.Portillo-Ledesma S, Randall LM, Parsonage D, Dalla Rizza J, Karplus PA, Poole LB, Denicola A, and Ferrer-Sueta G (2018) Differential Kinetics of Two-Cysteine Peroxiredoxin Disulfide Formation Reveal a Novel Model for Peroxide Sensing. Biochemistry 57, 3416–3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooley RB, Feldman JL, Driggers CM, Bundy TA, Stokes AL, Karplus PA, and Mehl RA (2014) Structural basis of improved second-generation 3-nitro-tyrosine tRNA synthetases. Biochemistry 53, 1916–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammill JT, Miyake-Stoner S, Hazen JL, Jackson JC, and Mehl RA (2007) Preparation of site-specifically labeled fluorinated proteins for 19F-NMR structural characterization. Nature protocols 2, 2601–2607 [DOI] [PubMed] [Google Scholar]
- 38.Santos J, Marino-Buslje C, Kleinman C, Ermacora MR, and Delfino JM (2007) Consolidation of the thioredoxin fold by peptide recognition: interaction between E. coli thioredoxin fragments 1–93 and 94–108. Biochemistry 46, 5148–5159 [DOI] [PubMed] [Google Scholar]
- 39.Mulrooney SB (1997) Application of a single-plasmid vector for mutagenesis and high-level expression of thioredoxin reductase and its use to examine flavin cofactor incorporation. Protein expression and purification 9, 372–378 [DOI] [PubMed] [Google Scholar]
- 40.Batthyany C, Souza JM, Duran R, Cassina A, Cervenansky C, and Radi R (2005) Time course and site(s) of cytochrome c tyrosine nitration by peroxynitrite. Biochemistry 44, 8038–8046 [DOI] [PubMed] [Google Scholar]
- 41.Nelson KJ, Parsonage D, Karplus PA, and Poole LB (2013) Evaluating peroxiredoxin sensitivity toward inactivation by peroxide substrates. Methods Enzymol 527, 21–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Portillo-Ledesma S, Randall LM, Parsonage D, Dalla Rizza J, Karplus PA, Poole LB, Denicola A, and Ferrer-Sueta G (2018) Differential Kinetics of Two-Cysteine Peroxiredoxin Disulfide Formation Reveal a Novel Model for Peroxide Sensing. Biochemistry [DOI] [PMC free article] [PubMed]
- 43.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, and Serrano L (2005) The FoldX web server: an online force field. Nucleic acids research 33, W382–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Durme J, Delgado J, Stricher F, Serrano L, Schymkowitz J, and Rousseau F (2011) A graphical interface for the FoldX forcefield. Bioinformatics 27, 1711–1712 [DOI] [PubMed] [Google Scholar]
- 45.Rodrigues CH, Pires DE, and Ascher DB (2018) DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic acids research 46, W350–W355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parra RG, Schafer NP, Radusky LG, Tsai MY, Guzovsky AB, Wolynes PG, and Ferreiro DU (2016) Protein Frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics. Nucleic acids research 44, W356–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jorgensen WL, J. C., Madura JD, Impey RW, Klein ML. (1983) Comparison of simple potential functions for simulating liquid water. The Journal of Chemical Physics 79, 926–935 [Google Scholar]
- 48.Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, and Walker RC (2013) Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. Journal of chemical theory and computation 9, 3878–3888 [DOI] [PubMed] [Google Scholar]
- 49.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015) ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of chemical theory and computation 11, 3696–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Randall L, Manta B, Nelson KJ, Santos J, Poole LB, and Denicola A (2016) Structural changes upon peroxynitrite-mediated nitration of peroxiredoxin 2; nitrated Prx2 resembles its disulfide-oxidized form. Archives of biochemistry and biophysics 590, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neumann H, Hazen JL, Weinstein J, Mehl RA, and Chin JW (2008) Genetically encoding protein oxidative damage. Journal of the American Chemical Society 130, 4028–4033 [DOI] [PubMed] [Google Scholar]
- 52.Porter JJ, and Mehl RA (2018) Genetic Code Expansion: A Powerful Tool for Understanding the Physiological Consequences of Oxidative Stress Protein Modifications. Oxidative medicine and cellular longevity 2018, 7607463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mukai T, Hoshi H, Ohtake K, Takahashi M, Yamaguchi A, Hayashi A, Yokoyama S, and Sakamoto K (2015) Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Scientific reports 5, 9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chang TS, Jeong W, Choi SY, Yu S, Kang SW, and Rhee SG (2002) Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J Biol Chem 277, 25370–25376 [DOI] [PubMed] [Google Scholar]
- 55.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, and Marks PA (2008) HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc Natl Acad Sci U S A 105, 9633–9638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woo HA, Yim SH, Shin DH, Kang D, Yu DY, and Rhee SG (2010) Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 140, 517–528 [DOI] [PubMed] [Google Scholar]
- 57.Zykova TA, Zhu F, Vakorina TI, Zhang J, Higgins LA, Urusova DV, Bode AM, and Dong Z (2010) T-LAK cell-originated protein kinase (TOPK) phosphorylation of Prx1 at Ser-32 prevents UVB-induced apoptosis in RPMI7951 melanoma cells through the regulation of Prx1 peroxidase activity. J Biol Chem 285, 29138–29146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alvarez B, Rubbo H, Kirk M, Barnes S, Freeman BA, and Radi R (1996) Peroxynitrite-dependent tryptophan nitration. Chemical research in toxicology 9, 390–396 [DOI] [PubMed] [Google Scholar]
- 59.Brito C, Naviliat M, Tiscornia AC, Vuillier F, Gualco G, Dighiero G, Radi R, and Cayota AM (1999) Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. Journal of immunology 162, 3356–3366 [PubMed] [Google Scholar]
- 60.Carvalho LAC, Truzzi DR, Fallani TS, Alves SV, Toledo JC Jr., Augusto O, Netto LES, and Meotti FC (2017) Urate hydroperoxide oxidizes human peroxiredoxin 1 and peroxiredoxin 2. J Biol Chem 292, 8705–8715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Armas MI, Esteves R, Viera N, Reyes AM, Mastrogiovanni M, Alegria TGP, Netto LES, Tortora V, Radi R, and Trujillo M (2019) Rapid peroxynitrite reduction by human peroxiredoxin 3: Implications for the fate of oxidants in mitochondria. Free radical biology & medicine 130, 369–378 [DOI] [PubMed] [Google Scholar]
- 62.Parsonage D, Nelson KJ, Ferrer-Sueta G, Alley S, Karplus PA, Furdui CM, and Poole LB (2015) Dissecting peroxiredoxin catalysis: separating binding, peroxidation, and resolution for a bacterial AhpC. Biochemistry 54, 1567–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bolduc JA, Nelson KJ, Haynes AC, Lee J, Reisz JA, Graff AH, Clodfelter JE, Parsonage D, Poole LB, Furdui CM, and Lowther WT (2018) Novel hyperoxidation resistance motifs in 2-Cys peroxiredoxins. J Biol Chem [DOI] [PMC free article] [PubMed]
- 64.Peskin AV, Dickerhof N, Poynton RA, Paton LN, Pace PE, Hampton MB, and Winterbourn CC (2013) Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J Biol Chem 288, 14170–14177 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.