

Abstract
Lung cancer is a major global health problem, as it is the leading cause of cancer-related deaths worldwide. Major advances in the identification of key mutational alterations have led to the development of molecularly targeted therapies, whose efficacy has been limited by emergence of resistance mechanisms. US Food and Drug Administration (FDA)-approved therapies targeting angiogenesis and more recently immune checkpoints have reinvigorated enthusiasm in elucidating the prognostic and pathophysiological roles of the tumour microenvironment in lung cancer. In this Review, we highlight recent advances and emerging concepts for how the tumour-reprogrammed lung microenvironment promotes both primary lung tumours and lung metastasis from extrapulmonary neoplasms by contributing to inflammation, angiogenesis, immune modulation and response to therapies. We also discuss the potential of understanding tumour microenvironmental processes to identify biomarkers of clinical utility and to develop novel targeted therapies against lung cancer.
Lung cancer is the leading cause of cancer-related mortality, with non-small-cell lung cancer (NSCLC) as the most prevalent form with a poor 5-year survival of~15%1. Despite advances in treatment options including surgery, radiation, chemotherapy and targeted therapies, prognosis remains poor owing to the presence oflocally advanced or widely metastatic tumours in the majority of patients at the time of diagnosis2. However, extensive genomic characterization of NSCLC has led to the identification of molecular subtypes of NSCLC that are oncogene addicted and exquisitely sensitive to targeted therapies3. These include activating mutations in epidermal growth factor receptor (EGFR) and BRAF or echinoderm microtubule-associated protein-like 4 (EML4)-anaplastic lymphoma kinase (ALK) fusions and ROS1 receptor tyrosine kinase fusions. Drugs that target the tyrosine kinase domain of these driver oncogenes have resulted in improved response rates and survival in patients with metastatic disease4. Unfortunately, this represents only 15–20% of patients and, while these interventions are effective initially, efficacy in the majority of the patients is limited by emergence of resistance mechanisms4. Therefore, further molecular characterization of the tumour landscape has the potential to identify novel biomarkers and molecular targets that impact disease progression and enable the design of novel therapeutic strategies5.
In the past decade, the central role of the tumour microenvironment (TME) in the initiation and progression of primary de novo lung carcinoma has been recognized3,6,7. In addition, extrathoracic malignancies including breast and colon cancer and melanoma systemically reprogramme the lung microenvironment to support the colonization and outgrowth of disseminated tumour cells (DTCs) to generate secondary lung tumours8. The TME in both primary and secondary lung tumours is recognized as a target-rich environment for the development of novel anticancer agents. In fact, drugs targeting various components of the TME including vascular endothelial growth factor (VEGF), aromatase and immune checkpoints have already been approved for use in the clinic2.
In this Review, we summarize recent advances showing how the specialized lung TME supports both primary lung cancer and metastasis from extrapulmonary carcinoma and discuss how the mechanistic understanding of aberrant molecular signalling networks stimulated by tumour-stromal interactions has the potential to provide novel diagnostic, prognostic and therapeutic opportunities. We also highlight technological advances in live imaging, multiscale profiling and deconvolution of bulk gene expression data for mapping the microenvironmental landscape.
The altered TME landscape
The anatomical and cellular features of the normal lung serve as a defensive barrier against foreign pathogens and particulates. However, in inflammatory states such as chronic obstructive pulmonary disease (COPD)9, the lung microenvironment displays features that may support carcinogenesis (FIG. 1). Human lung adenocarcinomas encompass unique lung cancer subtypes with distinct cellular and mutational heterogeneity3. Importantly, this heterogeneity is not only limited to tumour epithelial cells but also spans the TME, which includes vasculature, cancer-associated fibroblasts (CAFs), extracellular matrix (ECM) and infiltrating immune cells. In human NSCLC, stage-dependent immune cell infiltration10,11 suggests that the TME contributes to lung carcinogenesis and may have prognostic utility. As such, specific TME states are being considered as potential biomarkers to determine the stage and/or type of disease, clinical outcome and therapeutic responses (BOX 1).
Fig. 1 |. The heterogeneous microenvironment of the lung.
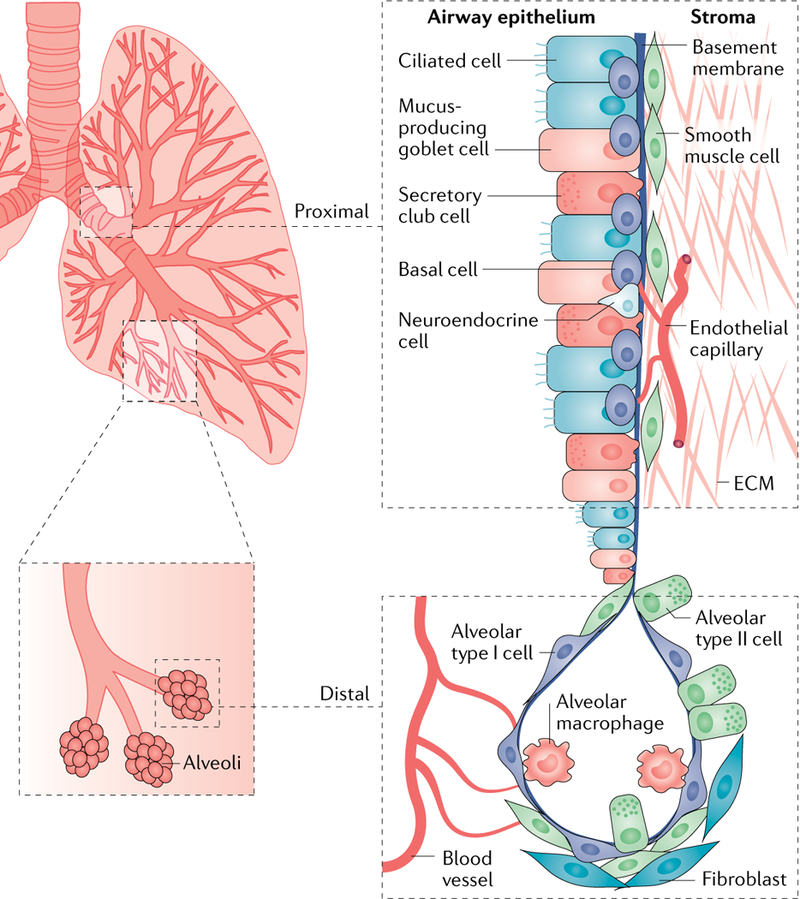
A schematic of the normal lung showing anatomic regions encompassing the proximal and distal airways is shown. The proximal airways are composed of ciliated cells, secretory club cells, undifferentiated basal cells, mucus-producing goblet cells and neuroendocrine cells; the distal airways are composed of alveolar type I and type II cells256. Other cell types in the lung microenvironment include smooth muscle cells, fibroblasts, endothelial cells and immune cells, including resident alveolar macrophages and dendritic cells. Vascular capillaries line the alveolar walls to facilitate gaseous exchange and infiltration of circulating immune cells257. Endothelium-derived angiocrine signalling induces and sustains regenerative lung alveolarization258,259. Resident alveolar macrophages maintain immunological homeostasis; however, they can also contribute to inflammation and the development of pre-malignant lung lesions in mice260. Patients with chronic obstructive pulmonary disease (COPD) are at a higher risk of developing lung cancer9, and their lungs show remodelling of the airway epithelium and alterations in lung inflammatory cells including neutrophils, monocytes and macrophages, which express elevated levels of pro-inflammatory mediators. A population of tissue-resident memory T cells resides in the lung airways, and they are generated by a diverse set of pathogens penetrating mucosal barriers and are believed to confer protective immunity to secondary infections261. ECM, extracellular matrix.
Box 1 |. The tumour microenvironment as a prognostic or predictive biomarker in lung cancer.
The exploration of the tumour microenvironment (TME) as a prognostic, diagnostic or predictive biomarker is an area of active research. A summary of the promising biomarkers is provided below.
Cancer-associated fibroblasts (CAFs) are indicators of poor prognosis associated with nodal metastases and risk of recurrence215, and CAF gene signatures stratify patients with NSCLC into low-risk and high-risk groups216 and predict patient survival217.
An extracellular matrix signature with prognostic and predictive value for response to adjuvant chemotherapy has been identified218.
Increased tumour-infiltrating lymphocytes and a higher CD4+:CD8+ T cell ratio are associated with a favourable prognosis and survival219,220. Forkhead box P3 (FOXP3)+ regulatory T (Treg) cells and intratumoural cyclooxygenase 2 (COX2) expression were associated with worse recurrence-free survival in node-negative non-small-cell lung cancer (NSCLC)221.
An international study in patients with colon cancer has recommended the implementation of an immunoscore as a predictor of risk of recurrence independently of the tumour-node-metastasis (TNM) staging in the clinic222. In lung cancer, the immunoscore has not yet been implemented; however, given the success in colon cancer, an accelerated development is expected223–225.
Trials with programmed cell death protein 1 (PD1) and PD1 ligand 1 (PDL1) inhibitors rely on PDL1 expression as a biomarker to predict clinical benefit. However, clinical efficacy has been reported in only ~10% of patients with PDL1-expressing tumours144,145,150, with many patients with PDL1+ tumours showing no benefit149. The heterogeneity of PDL1 expression within tumours226,227, the dynamic nature of PDL1 expression and the use of different PDL1 antibody platforms with different cut-off levels call for further standardization152,228,229.
Superior immune checkpoint inhibition (ICI) responses in smokers reflects the higher number of non-synonymous mutations in smokers230. Higher non-synonymous tumour mutational burden (TMB) is associated with improved objective response rates to ICI231 and constitutes a predictive biomarker in randomized trials of immune checkpoint inhibitors with other agents158,232. TMB was predictive of clinical efficacy independent of PDL1 expression233, suggesting that both TMB and PDL1 can be used as biomarkers for patient selection.
Amongst myeloid cells, a neutrophil transcription signature predicted mortality in NSCLC186, myeloid-derived suppressor cells (MDSCs) were associated with negative clinical outcomes234, and the polarization status (M1 versus M2) of tumour-associated macrophages (TAMs), which are abundant in NSCLC51, is associated with improved outcome in NSCLC235. However, the prognostic role of TAMs remains unresolved, as these findings have been debated54. Future efforts are aimed towards understanding differences in the local TAM phenotypes and identification of markers for reliable detection.
A high density of lysosomal membrane-associated protein 3 (LAMP3)-positive mature dendritic cells (DCs) in tertiary lymphoid structures is associated with cytotoxic T cell infiltration and predicts better outcome81,236,237. Calreticulin associated with an increased infiltration of mature DCs and effector memory T cells predicted favourable outcome in NSCLC238,239.
Despite these advances, a prognostic role of many TME biomarkers has not been adopted in clinical practice, and future studies encompassing larger patient cohorts, standardized platforms of detection and multi-institutional studies are warranted to achieve higher sensitivity and specificity of these potential biomarkers.
In the lung TME, malignant cancer cells can reprogramme the tumour-infiltrating stromal cells, which in turn contribute to carcinogenesis6,7. A growing appreciation of the role of the TME in promoting lung carcinogenesis has driven the development of anticancer therapies that target the TME. Therefore, understanding the heterotypic reciprocal crosstalk between cancer cells, various stromal cells and ECM described below is an area of active research (FIG. 2).
Fig. 2 |. The heterogeneous microenvironment of lung cancer.
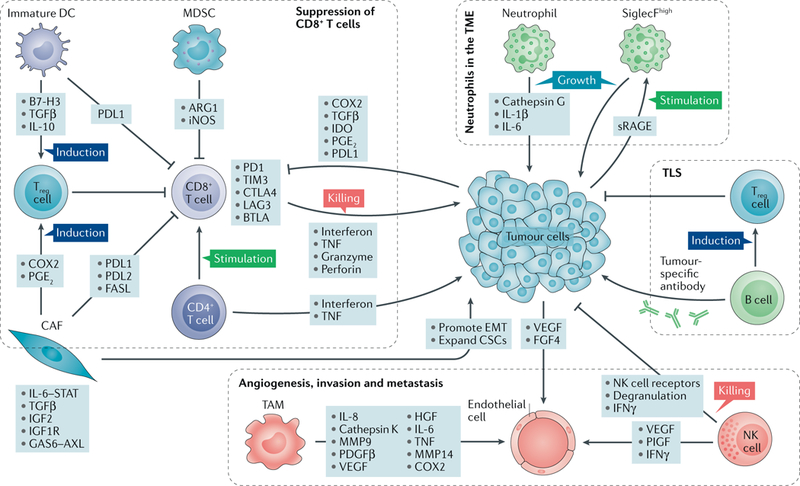
The lung tumour microenvironment (TME) includes a variety of tumour-reprogrammed stromal cells. A major hallmark of immunosuppression in the TME is the inactivation of cytotoxic CD8+T cells, which is achieved through diverse pathways. Immature dendritic cells (DCs) produce transforming growth factor-β (TGFβ), which expands the population of immunosuppressive forkhead box P3 (FOXP3)+ regulatory T (Treg) cells, which in turn inhibit CD8+T cells. DCs also express programmed cell death protein 1 (PD1) ligand 1 (PDL1) to directly suppress CD8+ T cells. Myeloid-derived suppressor cells (MDSCs) express arginase 1 (ARG1) and inducible nitric oxide synthase (iNOS), which contribute to T cell inhibition. Cancer-associated fibroblasts (CAFs) can also suppress the activity of CD8+ T cells by inducing Treg cells or by expressing PDL1, PDL2 and FAS ligand (FASL). Furthermore, tumour cells can directly produce inhibitory molecules including cyclooxygenase 2 (COX2), prostaglandin E2 (PGE2), PDL1 and indoleamine 2,3-dioxygenase (IDO), which can blunt the activity of CD8+ tumour-infiltrating lymphocytes. Neutrophils are major players in the lung TME, as they impact tumour progression through secretion of cytokines (interleukin-1β (IL-1β) and IL-6). Tumours secrete soluble receptor for advanced glycosylation end product (sRAGE), which systemically activates osteoblasts in the bone marrow to produce tumour-infiltrating sialic acid-binding immunoglobulin-like lectin F (SiglecF) neutrophils, which promote tumour growth by increasing angiogenesis, myeloid cell differentiation, T cell suppression and tumour cell proliferation. Endothelial cells respond to vascular endothelial growth factor (VEGF) from various sources including tumour cells, natural killer (NK) cells and tumour-associated macrophages (TAMs) to induce angiogenesis. In addition to promoting angiogenesis, TAMs may also activate other pathways to promote invasion and metastasis through secretion of various factors including IL-6, COX2, matrix metalloproteinase 9 (MMP9) and MMP14. Besides inducing angiogenesis, NK cells also mediate immune suppression by undergoing dysfunctional phenotypes characterized by downregulation of NK receptors, loss of degranulation potential and reduced interferon-γ (IFNγ) expression. B cells in tertiary lymphoid structures (TLS) can either generate tumour-specific antibodies or undergo a dysfunctional state that induces Treg cells. BTLA, B and T lymphocyte attenuator; CSCs, cancer stem cells; CTLA4, cytotoxic T lymphocyte-associated antigen 4; EMT, epithelial-to-mesenchymal transition; FGF4, fibroblast growth factor 4; GAS6, growth arrest-specific protein 6; HGF, hepatocyte growth factor; IGF1R, insulin-like growth factor 1 receptor; IGF2, insulin-like growth factor 2; LAG3, lymphocyte activation gene 3; PDGFβ, platelet-derived growth factor-β; PlGF, placental growth factor; STAT, signal transducer and activator of transcription; TIM3, T cell membrane protein 3; TNF, tumour necrosis factor.
Cancer-associated fibroblasts
Human NSCLC often exhibits desmoplasia, which is characterized by the presence of CAFs12. In solid tumours, CAFs mediate cancer cell proliferation, angiogenesis, invasion, metastasis and drug resistance13. CAFs isolated from human lung cancer tissues secrete interleukin-6 (IL-6), which stimulates Janus kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3) signalling in human lung cancer cells to increase metastasis in vivo14. CAFs also express insulin-like growth factor 2 (IGF2), which through ligation of the IGF1 receptor (IGF1R) on cancer stem cells (CSCs) induces expression of the stemness marker NANOG, leading to CSC enrichment15. CAFs also drive expression of α11β1 integrin, leading to increased ECM stiffness that contributes to increased growth and metastasis of patient-derived xenografts16,17. Intriguingly, one study showed that CAFs from human NSCLC exhibit increased autophagy and glycolytic metabolism18; however, the consequences of this finding in carcinogenesis remain unknown.
CAFs also modulate immune responses in the TME. For example, CAFs isolated from a subset of human NSCLC expressing the ligands of the programmed cell death protein 1 (PD1) receptor, PD1 ligand 1 (PDL1) and PDL2, were shown to suppress T cell function in coculture experiments19. Similarly, in a genetic mouse model of lung cancer, CAFs were able to cross-present antigens complexed with major histocompatibility complex class I (MHC I) to antigen-specific CD8+ T cells, leading to antigen-specific upregulation of PDL2 and FAS ligand (FASL) within these T cells and resulting in their elimination20.
The response to chemotherapy can also be modulated by CAFs, as cisplatin-induced expression of the receptor tyrosine kinase AXL (also known as UFO) ligand growth arrest-specific protein 6 (GAS6) in CAFs promoted migration of AXL-expressing lung cancer cells in mice21. Consistent with these findings in mice, the 5-year disease-free survival was significantly worse in patients with AXL+ tumour cells and GAS6+ stroma, suggesting the potential of combining chemotherapy with AXL or GAS6 inhibition22. Given the multifaceted nature of CAFs, it will be important for future studies to identify factors that drive the transcriptional reprogramming of CAFs in the lung TME, as this may aid the development of CAF-targeting therapies.
Extracellular matrix
The ECM, composed of collagens, proteoglycans and glycosaminoglycans, is a major constituent of the TME and mediates interactions between cancer cells and stromal cells to promote carcinogenesis23. In the KRAS-driven NSCLC mouse model (KrasG12D;Trp53fl/fl), matrix metallo-proteinase 14 (MMP14) expressed on the membranes of both cancer cells and myeloid cells mediated proteolytic processing of heparin-binding EGF-like growth factor (HB-EGF), leading to its activation; activated EGF stimulated the EGFR signalling pathway to promote tumour growth24. In another study, the receptor for the glycosaminoglycan hyaluronan, known as hyaluronan-mediated motility receptor (HMMR), enhanced ECM-mediated signalling and promoted tumour out-growth in the lung following intratracheal administration of human tumour cells in mice25. Loss-of-function mutations in the tumour suppressor LKB1 (also known as STK11), which are observed in ~30% of KRAS-mutated human lung adenocarcinomas, contribute to multiple cellular functions including cellular metabolism, cell polarity, invasion and metastasis26. In mice, LKB1 deficiency upregulated the ECM protein lysyl oxidase through the mTOR-hypoxia-inducible factor 1a (HIF1α) signalling axis, which in turn mediated collagen crosslinking and β1 integrin signalling activation27. Together, these studies suggest that targeting the ECM network including MMP14, hyaluronan and lysyl oxidase has potential therapeutic value. ECM density has also been shown to function as a major barrier to T cell infiltration into tumour beds in human NSCLC28. Therefore, targeting the ECM network may facilitate T cell infiltration into tumour beds for effective antitumour efficacy.
Endothelial cells
Angiogenesis, the formation of new blood vessels from pre-existing vasculature, is a key hallmark of cancers29. Tumour-derived angiogenic factors contribute to endothelial cell migration and proliferation, leading to formation of new blood capillaries that support tumour progression, invasion and metastasis. Several angiogenesis inhibitors including bevacizumab have either been approved or are in clinical trials for the treatment of NSCLC; however, it is striking that mechanistic insights into angiogenesis in lung carcinogenesis are limited. In the KRAS-driven mouse model, endothelial expression of the microRNAs (miRNAs) miR-143 and miR-145 promoted angiogenesis-mediated tumour growth30, suggesting the therapeutic potential of endothelial-specific inhibition of these miRNAs. The NAD-dependent protein deacetylase sirtuin 1, a key regulator of angiogenesis, was shown to increase vessel density and promote the growth of Lewis lung carcinoma (LLC) xenografts by downregulating delta-like ligand 4 (DLL4)-Notch signalling and through deacetylation of the NOTCH1 intracellular domain in lung cancer-derived endothelial cells31.
However, the prevailing concept that tumour growth is strictly dependent on neo-angiogenesis has been challenged, as a subset of patients with NSCLC exhibit non-angiogenic growth patterns. These tumours appear to exploit pre-existing blood vessels rather than induce angiogenesis and importantly have a worse prognosis than their angiogenic counterparts32. Hence, it is unlikely that antiangiogenic treatments would be effective for all patients with lung cancer, and this may explain the modest clinical benefits associated with antiangiogenic therapies in NSCLC. Therefore, understanding the complex biology of the tumour vasculature and angiogenic signalling in NSCLC constitutes an important area of research. Indeed, analysis of 180 human NSCLC specimens captured multiple unique and dominant angiogenic phenotypes33. Given that angiogenesis is dependent on a complex network of different cellular compartments regulated by a balance of angiogenic and antiangiogenic factors, a greater understanding of tumour-specific angiogenesis has the potential to aid the rational design of antiangiogenic interventions.
The immune TME
The innate and adaptive immune cells in the lung TME harbour both tumour-promoting and tumour-suppressing activities, which may also predict clinical outcome34,35. Genome-wide expression analysis has begun to provide molecular insights into the tumour-induced reprogramming of infiltrating lymphoid and myeloid cells in both mouse and human NSCLC36,37.
Myeloid cell populations
Myeloid cells of granulocytic and monocytic lineages are components of innate immunity and contribute to carcinogenesis in various ways38,39. Myeloid cells can also regulate adaptive immunity by controlling tumour-infiltrating lymphocyte (TIL) composition, activation and antitumour function40. Major efforts in the field are now directed towards developing a comprehensive atlas of the immune TME. Indeed, recent analysis of myeloid cells from tumours and matched adjacent non-neoplastic lung tissue of patients with NSCLC identified differentially regulated genes36,37, and a comprehensive analysis in the KRAS-driven mouse model unravelled myeloid-tumour crosstalk networks41. Insights from such analyses have the potential to enable the design of therapeutic strategies that target tumour-stroma crosstalk pathways.
Dendritic cells (DCs) are myeloid derived and recognized as central orchestrators of antitumoural immunity by virtue of their ability to cross-present tumour antigens to prime T lymphocytes in the draining lymph nodes42. However, tumours can suppress DC function or alter the TME to recruit immune-suppressive DCs. In patients with NSCLC, DCs upregulate the co-inhibitory molecule B7-H3 (also known as CD276) and therefore fail to stimulate T cells43. Human DCs also produce transforming growth factor-β (TGFβ), which induces the differentiation of CD4+ T cells into CD4+CD25+forkhead box P3 (FOXP3)+ regulatory T (Treg) cells that suppress T cell proliferation44. A preclinical study showed that administration of DCs transduced with the chemoattractant CC-chemokine ligand 21 (CCL21) led to increased infiltration of DCs and CD4+ and CD8+ T cells in the lung TME, resulting in reduced tumour burden45. These findings led to a phase I trial being conducted in patients with advanced NSCLC in which intratumoural vaccination with autologous DCs expressing CCL21 showed an increase in CD8+ T cell infiltration into tumours, as well as increased PDL1 expression on tumour cells46. Given that the efficacy of DC vaccines as a monotherapy is limited by immunosuppressive mechanisms in the TME, these results provide a rationale for combining immunotherapy with in situ vaccination. More recently, DC vaccine platforms have begun to incorporate innovative approaches for improving DC maturation, proliferation, antigen presentation and selection and have been used in combination with other therapies including immune checkpoint inhibitors and standard of care therapies in solid tumours including NSCLC47.
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous cell population that can suppress T cell proliferation and cytokine production38. Currently, MDSCs can be further divided into two major subsets: poly-morphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs). PMN-MDSCs share many morphological and phenotypic characteristics of neutrophils, whereas M-MDSCs are more similar to immature monocytes. However, they also have different molecular and biochemical characteristics, which have not been completely characterized48,49. In both mice and human cancers, PMN-MDSCs that phenotypically resemble neutrophils represent the most abundant population of MDSCs49. A study in patients with NSCLC showed that endoplasmic reticulum stress-driven inositol-requiring protein 1 (IRE1; also known as ERN1)-X-box binding protein 1 (XBP1) signalling converts regular neutrophils into highly immunosuppressive lectin-type oxidized low-density lipoprotein receptor 1 (LOX1)+ PMN-MDSCs50. Given the marked increase of LOX1+ MDSCs in patients with NSCLC, future studies are likely to assess the potential of LOX1+ cells as a prognostic biomarker in NSCLC treatment. Additionally, either eliminating LOX1+PNM-MDSCs or targeting the IRE1–XBP1 pathway that contributes to the generation of these cells may have potential therapeutic application in NSCLC.
Tumour-associated macrophages (TAMs) are abundant components of the NSCLC immune infiltrate51, are highly plastic and display a variety of phenotypes, including Ml (classically activated, pro-inflammatory with antitumour activity) and M2 (alternatively activated, proangiogenic and immunosuppressive with protumour activity), which are governed by the nature of the local microenvironments51,52. The current view is that the different macrophage phenotypes present within the TME is a continuum and therefore cannot be captured solely with the M1–M2 dichotomy53. Given this heterogeneity of phenotypes, the prognostic relevance of TAMs in NSCLC remains unresolved54, and future studies aimed towards a more comprehensive analysis of the clinical importance of TAMs in NSCLC are needed. In the KRAS-driven mouse model, TAMs expressing IL-6 interact with IL-6 receptors on cancer cells, leading to the activation of tumour-promoting STAT3 signalling41. Lungs also harbour resident alveolar macrophages, which promote carcinogenesis in mice55,56. While macrophage depletion impacts tumour growth in a carcinogen-induced model of lung adenocarcinoma56, TAM targeting with inhibitors of colony-stimulating factor 1 receptor (CSF1R) have exhibited limited benefit in clinical trials57. In one pre-clinical study, as expected, CSF1R inhibition effectively eliminated TAMs; however, an unexpected accumulation of immunosuppressive PMN-MDSCs triggered by CXC-chemokine ligand 1 (CXCL1) released from CAFs was observed58. Notably, combined inhibition of CSF1R and CXC-chemokine receptor 2 (CXCR2) (the receptor for CXCL1) decreased PMN-MDSC recruitment and abrogated tumour growth58. The existence of this mechanism in human NSCLC has not been investigated.
Neutrophils infiltrating mouse tumours can either promote carcinogenesis by supporting tumour-related inflammation, angiogenesis and metastasis or restrict tumour growth through the expression of antitumour and cytotoxic mediators59,60. However, such functional diversity has not been evaluated in human lung cancer. In human NSCLC, neutrophils dominate the immune landscape11. A recent study identified a unique subset of tumour-infiltrating neutrophils that expressed markers of both neutrophils and antigen-presenting cells (APCs), enabling this cell type to cross-present antigens, as well as augment antitumour T cell responses61. Neutrophils also express IL-1β, which mediates resistance to nuclear factor-κB (NF-κB) inhibitors in the KRAS-driven mouse model62. Furthermore, neutrophil depletion was associated with a pronounced reduction in lung carcinogenesis in LLC xenograft and carcinogen (methylcholanthrene)-induced mouse models of lung cancer37,63. A recent study showed that secretion of soluble receptor for advanced glycosylation end product (sRAGE) by KRAS-driven lung adenocarcinomas systemically activated osteocalcin-positive osteoblasts in the bone marrow of mice64. Bone marrow activation led to the production of sialic acid-binding immunoglobulin-like lectin F (SiglecF; also known as Siglec5) neutrophils that homed to the lungs to promote tumour growth by promoting angiogenesis, myeloid cell differentiation, T cell suppression and tumour cell proliferation. While the relevance of this type of neutrophils in humans remains unknown, the authors did identify a SiglecFhigh neutrophil signature that was associated with poor patient survival in patients with lung cancer. Future studies characterizing tumour-infiltrating neutrophil phenotypes during human NSCLC progression may allow precise targeting to eliminate specific pro-tumorigenic subsets or increase the activity of antitumorigenic subsets.
Natural killer (NK) cells are lymphocytes of the innate immune system that play a key role in antitumour immunity by virtue of directly recognizing tumour cells as targets65. As cancer cells have evolved multiple strategies to escape CD8+ T cell recognition, the importance of NK cells in directly mediating tumour cell destruction has recently gained prominence66. Several studies have characterized the frequency, localization, phenotype and functional status of NK cells infiltrating the lung TME in mice and humans67. Factors that regulate NK cell response in the lung TME include TGFβ, which mediates polarization of NK cells towards a proangiogenic phenotype, as determined by expression of VEGF, placental growth factor (P1GF) and interferon-γ (IFNγ)68. A dysfunctional state of NK cells was associated with downregulation of NK cell receptors, decreased degranulation and loss of IFNγ expression in human NSCLC67. Despite these findings, NK cells from pleural effusions of patients with lung cancer expressed MHC I and released cytokines upon exposure to both allogeneic and autologous tumour cells in vitro69. While insights into NK cell biology have increased in recent years, NK cell subset identification and function in lung carcinogenesis remain to be elucidated.
Lymphoid cell populations
T lymphocytes infiltrating tumours, particularly the CD4+ and CD8+ T cells, and their immunoregulatory cytokines represent adaptive immunity, execute key effector cytotoxic functions in the TME and mediate responses to immune checkpoint inhibition (ICI). In the KRAS-driven mouse model, PD1 inhibition resulted in increased proliferation of all T cell subsets and effector cytokine production by CD4+ T helper 1 (TH1) cells, while the depletion of CD4+ and/or CD8+ T cells in combination with a PD1 antibody resulted in reduced therapeutic efficacy of the PD1 blockade70.
Immunosuppressive FOXP3+ Treg cells are induced by cyclooxygenase 2 (COX2; also known as PTGS2)-mediated upregulation of prostaglandin E2 (PGE2) in tumour cells71. As expected, Treg cell depletion reduced lung tumour burden in mice72 and was associated with an increased expression of granzyme A, granzyme B, perforin and IFNγ in CD8+ TILs73. In the KRAS-driven mouse model, PD1 inhibition was associated with upregulation of PD1 and IFNγ in a proportion of Treg cells, suggesting loss of immunosuppressive activity70. Within the CD4+ T cell populations, a fine balance between the immunosuppressive Treg cells and pro-inflammatory TH17 cells is associated with effective adaptive immune responses in lung carcinogenesis74. However, given the context-dependent pro-tumorigenic and antitumorigenic roles of TH17 cells, further investigations are needed to characterize the mechanistic and prognostic value of these two populations in NSCLC75,76.
CD8+ cytotoxic T cells elicit tumour clearance by targeting tumour cells for destruction through production of IFNγ, tumour necrosis factor (TNF) and granzyme B after ligation of their T cell receptors (TCRs)77. However, progressing tumours employ various means of inhibiting CD8+ T cells. For example, Treg cells can directly suppress the antitumour function of CD8+ T cells73. Furthermore, persistent antigen exposure can lead to T cell dysfunction or exhaustion characterized by loss of effector and memory functions, a phenomenon first recognized in the setting of chronic viral infections78. In patients with NSCLC, disease progression is correlated with increased expression of markers of T cell exhaustion including PD1, T cell membrane protein 3 (TIM3; also known as HAVCR2), cytotoxic T lymphocyte-associated antigen 4 (CTLA4), lymphocyte activation gene 3 (LAG3) and B and T lymphocyte attenuator (BTLA) on CD8+ TILs79. Indeed, treatment with anti-PD1 therapy increased the number of highly proliferative (Ki67+) PD1+ CD8+ T cells in the circulation of patients with NSCLC80. Despite the central role of CD8+ T cells in antitumour immunity, an efficient antitumour immune response requires the cooperation of both CD8+ and CD4+ T cells, as the therapeutic efficacy of PD1 inhibition was only partially reversed by depletion of CD8+ T cells but was completely abolished by the additional depletion of CD4+ T cells in the KRAS-driven mouse model70.
Tumour-infiltrating B cells have emerged as key players in the TME. In patients with NSCLC, tumour-infiltrating B cells and CD4+ T cells reside within the tertiary lymphoid structures (TLS), and these structures correlate with better prognosis81. It was demonstrated that tumour-infiltrating B cells harvested from patients can present endogenous tumour antigens to CD4+ TILs and alter the CD4+ TIL phenotype in vitro82. Interestingly, activated tumour-infiltrating B cells were associated with activated IFNγ+CD4+ T cells, and exhausted tumour-infiltrating B cells were associated with immunosuppressive Treg cells82. These studies suggest that in the TME, the ability of tumour-infiltrating B cells to influence CD4+ TILs constitutes a potential novel therapeutic target in NSCLC immunotherapy.
Immunosuppressive mechanisms
Improving the efficacy of immunotherapy requires a better understanding of the immunomodulatory roles of the immune cells within the TME83. Therefore, understanding cancer cell-intrinsic and cell-extrinsic pathways that influence the immune microenvironment has emerged as an active area of investigation.
In the context of cancer cell-intrinsic pathways of immunosuppression, a major effort is directed towards understanding how cancer cells have devised means to suppress the tumoricidal capabilities of the immune cells. Emerging studies have begun to show that oncogenic driver mutations in KRAS, p53 and EGFR are associated with discrete immune phenotypes in both mice and human lung adenocarcinomas84. Accordingly, there is marked interest in understanding exactly how these cancer cell-intrinsic pathways alter immune cells in the TME85, and some of these mechanisms are displayed in FIG. 3.
Fig. 3 |. Cancer cell-intrinsic pathways mediate immunosuppression in lung cancer.
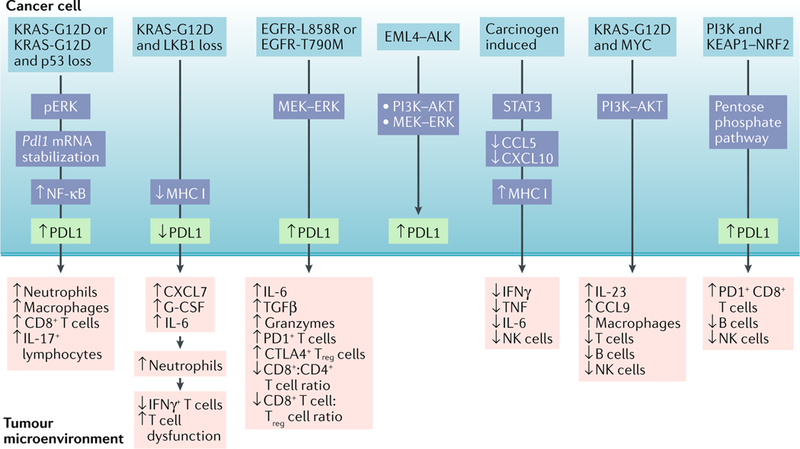
Lung adenocarcinomas with specific driver mutations exhibit discrete immune phenotypes. The observation that programmed cell death protein 1 (PD1) ligand 1 (PDL1) expression was increased in patients with KRAS-mutant lung cancer led to the finding that mutant KRAS in mouse tumours induces PDL1 expression either through phosphorylated ERK (pERK) signalling or Pdl1 mRNA stabilization to mediate immune escape262,263. These studies advocate the use of either ERK inhibitors or inhibition of the AU-rich element-binding protein tristetraprolin (TTP), which is responsible for PDL1 stabilization, for the treatment of KRAS tumours. Loss of the tumour suppressor LKB1 in KRAS-mutant mouse tumours is associated with increased accumulation of immunosuppressive neutrophils, exhausted T cells, increased pro-inflammatory cytokines including interleukin-6 (IL-6) and decreased PDL1 expression. While PD1 monotherapy was ineffective in this setting, its therapeutic efficacy can be restored with either neutrophil depletion or an IL-6-neutralizing antibody166. Similarly, immune suppression in KRAS-mutant mouse tumours may be a result of MYC co-activation leading to upregulation of IL-23 and CC-chemokine ligand 9 (CCL9), which mediates the exclusion of B cells, T cells and natural killer (NK) cells, as well as the recruitment of proangiogenic macrophages264. As expected, inhibition of CCL9 and IL-23 can decrease MYC-induced tumour progression. Clinical observations that epithelial growth factor receptor (EGFR)-mutant tumours are associated with high PDL1 expression265 are consistent with the observation that EGFR-driven mouse tumours suppress T cell function by activating PD1 and PDL1 (REF266). Patients with echinoderm microtubule-associated protein-like 4 (EML4)-anaplastic lymphoma kinase (ALK) fusion oncogene-driven lung cancer also show increased PDL1 expression and, in mice, it was found that EML4-ALK-mediated activation of the PI3K-AKT and MEK-ERK pathways upregulates PDL1 expression267. In carcinogen-induced lung tumorigenesis, signal transducer and activator of transcription 3 (STAT3) activation in cancer cells inhibits pro-inflammatory chemokines, increases major histocompatibility complex class I (MHC I) expression, decreases effector cytokines and blunts NK cell-mediated toxicity268. Despite these findings, the link between oncogenic drivers and PDL1 expression remains controversial, as one study demonstrated an association of PDL1 expression with mutant EGFR but not KRAS or ALK in patients with non-small-cell lung cancer (NSCLC)269, while another study failed to correlate PDL1 expression with mutant EGFR in patients147. In yet another study, PDL1 expression was associated with mTOR activation in patients with NSCLC driven by a wide spectrum of driver mutations, and the combination of mTOR and PD1 inhibitors could reduce tumour growth, increase tumour-infiltrating lymphocytes and decrease regulatory T (Treg) cells in mouse lung cancers270. An emerging concept lends support for oncogene cooperativity in driving immune evasion. Co-activation of KRAS and MYC or PI3K and Kelch-like ECH-associated protein 1 (KEAP1)-nuclear factor erythroid-2-related factor 2 (NRF2) reprogrammes the tumour microenvironment (TME) to promote lung adenocarcinoma264,271. Indeed, genetic deficiency of Keap1 and Pten can increase the efficacy of immune checkpoint inhibition271. As cancer cell-intrinsic pathways alter immunity in unique ways, this calls for studies involving large cohorts of patients with NSCLC to establish how specific genomic alterations impact the TME composition and function; this would be valuable for designing combination treatments for patients who have been selected on the basis of their tumour genomic alterations and immune contexture. CTLA4, cytotoxic T lymphocyte-associated antigen 4; CXCL, CXC-chemokine ligand; G-CSF, granulocyte colony-stimulating factor; IFNγ, interferon-γ; NF-κB, nuclear factor-κB; TGFβ, transforming growth factor-β; TNF, tumour necrosis factor.
With respect to tumour-extrinsic mechanisms mediating immunosuppression, a major focus is to understand the contributions of the reprogrammed tumour-infiltrating immune cells. MDSCs express L-arginase and inducible nitric oxide synthase (iNOS), which deplete L-arginine in the TME; loss of L-arginine downregulates TCR expression, leading to T cell dysfunction, and confers poor survival in patients with NSCLC86,87. The tryptophan-catabolizing enzyme indoleamine 2,3-dioxygenase (IDO), which is expressed by a variety of cells including tumour cells, macrophages and DCs, has emerged as a potent mediator contributing to the immune escape of tumours, as tryptophan deficiency results in MDSC expansion, decreased cytotoxic T lymphocyte response and Treg cell accumulation. In KRAS-driven mouse models, IDO increased metastasis through IL-6-dependent inflammation and MDSC-driven immune escape88. IDO can meta-bolically reprogramme immune cells, as IDO deficiency in MDSCs was shown to downregulate nutrient-sensing AMP-activated protein kinase (AMPK) activity, leading to their inactivation, whereas in CD8+ T cells, IDO deficiency increased AMPK activation associated with an enhanced effector function in an LLC xenograft mouse model89. Thus, IDO inhibition may represent a two-pronged therapeutic approach for inhibiting MDSCs while simultaneously eliciting effector CD8+ T cells. Unfortunately, a recent phase III trial (ECHO-301)90 of IDO inhibition in combination with ICI failed in patients with melanoma, resulting in a major set-back for IDO-based immunotherapy regimens. As the ECHO-301 trial90 did not measure IDO expression to stratify patients, future studies will need to identify biomarkers that better predict responders to IDO-based therapy combinations.
The lung as a site of metastasis
The lung is also a frequent site of metastases formation from a variety of extrathoracic malignancies91,92. Malignancies including breast, skin and colon cancer secrete factors and extracellular vesicles, which systemically reprogramme the lung microenvironment to generate pre-metastatic niches. Studies in mouse models have shown that these pre-metastatic niches, which are characterized by host immune and inflammatory cells, organ-specific chemoattractants, growth factors and ECM-modifying proteins, provide permissive microenvironments that support extravasation, colonization and metastatic outgrowth of DTCs8,93 (FIG. 4).
Fig. 4 |. The lung metastatic niche.
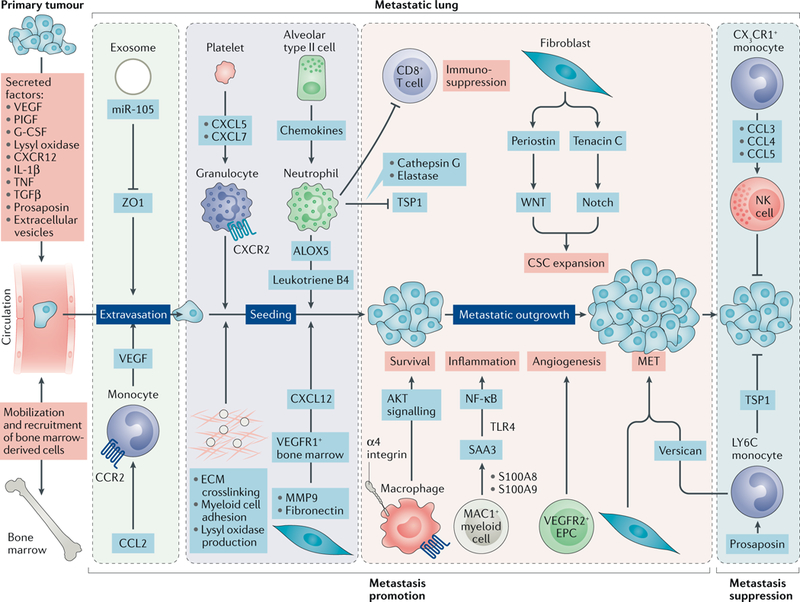
Primary tumours secrete various growth factors, cytokines and enzymes (vascular endothelial growth factor A (VEGFA), placental growth factor (P1GF), transforming growth factor-β (TGFP), tumour necrosis factor (TNF) and lysyl oxidase) and shed extracellular vesicles into the circulation that systemically impact the lung microenvironment. Tumour-derived factors can also activate bone marrow compartments, resulting in the mobilization and recruitment of bone marrow-derived cells into the lungs. Together, these activities contribute to the generation of pre-metastatic niches in the lung. Extravasation of disseminated tumour cells (DTCs) into the lung parenchyma is facilitated by microRNA miR-105 delivered by breast cancer exosomes, which silence the tight junction protein zona occludens 1 (ZO1) to allow DTCs to breach vascular barriers. In addition, CC-chemokine ligand 2 (CCL2) gradients recruit CC-chemokine receptor 2 (CCR2)+ inflammatory monocytes, which produce VEGF to increase vascular permeability. Following extravasation, the pre-metastatic niche contributes to the seeding and colonization of the DTCs in the lungs. Hypoxia in primary breast tumours upregulates lysyl oxidase, which systemically oxidizes lysine residues in collagen and elastin, resulting in covalent crosslinking of these molecules in the lungs. Collagen crosslinking increases the adhesion of myeloid cells to generate microenvironments conducive to DTC colonization. Platelets secrete CXC-chemokine ligand 5 (CXCL5) and CXCL7, which recruit granulocytes to facilitate metastatic seeding170. Exosome-derived small nuclear RNAs (snRNAs) activate Toll-like receptor 3 (TLR3) in lung alveolar type II cells to induce chemokine secretion, which in turn mediates neutrophil recruitment. Neutrophils, via arachidonate 5-lipoxygenase (ALOX5)-dependent leukotriene synthesis, stimulate adhesion, chemotaxis and vascular permeability to support colonization of leukotriene receptor-positive DTCs. In addition, the extracellular matrix (ECM) component fibronectin enables clustering of bone marrow-derived VEGF receptor 1 (VEGFR1)+ haematopoietic cells, leading to increased production of CXCL12, which recruits CXC-chemokine receptor 4 (CXCR4)+ DTCs to the pre-metastatic niche272. Following seeding of DTCs, α4 integrin-expressing macrophages interact with vascular cell adhesion protein 1 (VCAM1)+ DTCs to induce pro-survival AKT signalling. The polarized neutrophils in the pre-metastatic lungs may suppress activation of CD8+ T cells to dampen anti-metastatic immunity. Subsequently, metastatic outgrowth is supported by various pathways. Neutrophils may secrete cathepsin G and neutrophil elastase to proteolytically degrade the antitumorigenic factor thrombospondin 1 (TSP1) to promote the metastatic outgrowth. Tumour-secreted factors induce expression of chemoattractants, such as S100A8 and S100A9, by lung endothelial cells and MAC1 (also known as CD11b)+ myeloid cells, which facilitate the homing of tumour cells to the pre-metastatic niche, through the induction of serum amyloid A3 (SAA3)273. Notably, SAA3 stimulates nuclear factor-κβ (NF-κβ) signalling in a TLR4-dependent manner to facilitate inflammation-mediated metastasis. DTCs can induce expression of periostin or tenacin C in lung fibroblasts to support cancer stem cell (CSC) maintenance and expansion through WNT and Notch signalling, respectively. The colonized mesenchymal tumour cells may undergo an epithelial conversion that is supported by paracrine interactions with reprogrammed monocytic cells. The myeloid-derived proteoglycan versican mediates inhibition of the TGFβ-SMAD2 or SMAD3 pathway in DTCs that induces mesenchymal-to-epithelial transition (MET) and promotes metastatic outgrowth. Through another mechanism, mesenchymal DTCs can also express TSP2 to activate local fibroblasts, which in turn promote MET of cancer cells106. Lastly, metastatic outgrowth can be facilitated by infiltrating bone marrow-derived endothelial progenitor cells (EPCs) and endothelial cells, which contribute to the switch to angiogenesis. The pre-metastatic niche can exhibit features of metastasis suppression, as lung endothelial cells expressing CX3C-chemokine ligand 1 (CX3CL1) attract CX3C-chemokine receptor 1 (CX3CR1)+ patrolling monocytes, which through expression of CCL3, CCL4 and CCL5 recruit cytotoxic natural killer (NK) cells to inhibit metastasis. Similarly, metastatic incompetent tumours express the glycoprotein prosaposin, which systemically reprogrammes myeloid cells in the lungs to express TSP1, which inhibits metastatic outgrowth. G-CSF, granulocyte colony-stimulating factor; IL-1β, inter1eukin-1β; LY6C, lymphocyte antigen 6C; MMP9, matrix metalloproteinase 9.
Following dissemination into the circulation, DTCs extravasate into the lung parenchyma by breaching lung endothelial barriers94,95. Following extravasation, several mechanisms within the premetastatic niche facilitate DTC colonization. Hypoxia in the primary breast tumour induced by elevated levels of HIF1α triggers expression of lysyl oxidases, which systemically crosslinks collagen in the lungs, increasing the adhesion of myeloid cells to generate niches that support colonization96. In another mechanism, lung alveolar type II cells induce chemokine secretion that recruits neutrophils97. Neutrophils, via arachidonate 5-lipoxygenase (ALOX5)-dependent leukotriene synthesis, support proliferation and colonization of leukotriene B4 receptor 2 (LTB4R2)-positive DTCs98. Polarized neutrophils in the pre-metastatic lungs can also suppress activation of CD8+ T cells to dampen anti-metastatic immunity99. Furthermore, neutrophils can secrete cathepsin G and neutrophil elastase to degrade the antitumorigenic factor thrombospondin 1 (TSP1), thereby promoting metastases in the lung100. A recent study showed that atypical chemokine receptor 2 (ACKR2) expressed on haematopoietic progenitor cells restrains myeloid differentiation and function, and targeting host ACKR2 increased the ability of neutrophils to target to lung metastases in a breast cancer mouse model101.
Following colonization, it is important for DTCs to be able to survive in the foreign microenvironment of the metastatic lung, and this is known to be facilitated by α4 integrin-expressing macrophages, which induce pro-survival AKT signalling in DTCs102. DTCs also exploit fibroblasts to produce periostin and tenascin C, which induce WNT and Notch signalling, respectively, in DTCs to promote expansion103,104. Mesenchymal tumour cells may undergo an epithelial conversion (mesenchymal-to-epithelial transition (MET)), which is supported by the myeloid-derived proteoglycan versican105 or through activated, local fibroblasts generated by tumour-derived TSP2 (REF106). In addition, metastatic outgrowth is supported by angiogenesis, which is mediated by infiltrating bone marrow-derived endothelial progenitor cells107 or VEGF-induced VEGF receptor 2 (VEGFR2) phosphorylation that in turn triggers calcineurin-nuclear factor of activated T cells (NFAT) signalling to transactivate the angiogenic growth factor angiopoietin 2 (ANG2) in the lung endothelium108.
NK cells have been recognized to play an important role in tumour immunosurveillance and metastasis control109. The discovery that CD96 is a negative regulator of NK cell activity led to the use of CD96-neutralizing antibodies in preclinical models. These antibodies enhanced NK cell IFNγ-dependent effector function to inhibit lung metastasis in mice110,111. However, in the context of the pre-metastatic niche, hypoxic tumour cell-derived CCL2, by virtue of recruiting granulocytic myeloid cells from the bone marrow to the lungs, creates an immune-suppressive pre-metastatic niche through suppression of NK cell cytotoxicity and maturity112. Importantly, a recent study identified an NK cell checkpoint IL-1 receptor 8 (IL-1R8; also known as SIGIRR), which restrains NK cell maturation and effector function; inhibition of IL-1R8 unleashed NK cells to impair metastasis in the lungs of mice113.
Intriguingly, some studies have also described metastasis-suppressive niches. For instance, certain metastatic incompetent tumours express the glycoprotein prosaposin, which systemically reprogrammes myeloid cells in the lungs to express TSP1, generating metastasis-suppressive niches in mice114. Similarly, lung endothelial cells expressing CX3-chemokine ligand 1 (CX3CL1; also known as fractalkine) attract -CX3C-chemokine receptor 1 (CX3CR1)+ patrolling monocytes, which through expression of CCL3, CCL4 and CCL5 recruit cytotoxic NK cells to prevent metastasis to the lung in mice115.
Local pre-existing inflammation in the lung caused by exposure to cigarette smoke may contribute to increased metastasis. For example, cigarette smoke-induced inflammation is correlated with increased metastasis to the lungs from primary breast cancers in mice and humans116,117. The mechanisms governing metastasis-promoting effects of inflammation include the ubiquitin-CXCR4 signalling axis118, elevated levels of E-selectin expression in lung tissue119 and NF-κB activation in airway epithelial cells120 or increased CCL2 expression and macrophage infiltration in the lung microenvironment121. Recently, it was shown that T cell-intrinsic expression of oxygen-sensing prolyl hydroxylase domain (PHD) proteins restrained lung inflammation by decreasing TH1 cell differentiation, inducing Treg cells and suppressing CD8+ T cell effector function122. DTCs can exploit these immunologically tolerant metastatic niches for metastatic progression122.
In many patients, DTCs that colonize the lungs may undergo dormancy, and targeting dormancy constitutes an attractive therapeutic strategy for minimizing the risk of cancer recurrence123,124. While mechanisms underlying cancer cell-intrinsic pathways of dormancy regulation are beginning to be identified123,124, emerging evidence also implicates the contribution of the local microenvironment125,126 (BOX 2). Primary lung adenocarcinomas also exhibit metastasis, with the common metastatic sites being the bone, brain, liver and adrenal glands. The contribution of specialized microenvironments that support colonization and progression of metastases is now being elucidated127 (BOX 3).
Box 2 |. The lung microenvironment contributes to metastatic dormancy.
Dormancy is generally defined as the process by which cancer cells survive in a quiescent state and evade therapy. The tumour microenvironment at the metastatic site contributes to the initiation and maintenance of metastatic dormancy125. A detailed understanding of the mechanisms of metastatic dormancy and colonization is important for the development of novel therapeutics. In the lung, endothelial tip cells in the perivascular niche express thrombospondin 1 (TSP1), which induces dormancy in disseminated breast cancer cells240. Activation of the endothelium through expression of periostin and transforming growth factor-β1 (TGFβ1) can induce disseminated tumour cell (DTC) proliferation and mediate possible escape from dormancy. An endogenous inhibitor of bone morphogenetic protein 4 (BMP4; a TGFβ family member), COCO (also known as DAND5), and non-canonical discoidin domain receptor 1 (DDR1) signalling was shown to reactivate dormant metastatic breast cancer cells in the lung241,242. In addition, targeting the cytoskeleton through inhibition of myosin light chain kinase (MLCK)243 or through inhibition of type I collagen244 induced dormancy and inhibited metastatic outgrowth in breast cancer. Inflammation in the lungs led to the awakening of dormant DTCs through the expression of ZEB1, a well-known regulator of epithelial-to-mesenchymal transition (EMT)245.
Maintaining DTCs in a quiescent state has the potential to transform cancer into a chronic manageable disease; however, at some point, the dormant cells may proliferate under the influence of a new stimulus. Therefore, approaches to directly eliminate dormant tumour cells are considered the preferred anti-metastatic strategy.
Box 3 |. The lung tumour microenvironment contributes to lung cancer metastasis to other organs.
Approximately 20–40% of patients with non-small-cell lung cancer (NSCLC) develop brain metastases during their lifetime, with poor overall survival246,247. Astrocytes are involved in metastasis from the lung to the brain through expression of specific inflammatory cytokines such as interleukin-8 (IL-8), IL-1β, IL-6 and tumour necrosis factor (TNF) and their subsequent secretion into the microenvironment of the brain248. Astrocytes also secrete matrix metalloproteinase 2 (MMP2) and MMP9, which contributes to tumour cell invasion and formation of metastases249. Placental growth factor (PlGF) secreted by small-cell lung cancer cells can trigger a vascular endothelial growth factor receptor 1 (VEGFR1)-RHO-associated kinase (ROCK) signalling axis to breach tight junctions in brain endothelial cells to promote cancer cell transendothelial migration250. Serpin expression by lung cancer cells can facilitate vascular co-option and protect these cells from death signals in the reactive brain microenvironment251.
In addition to the brain, primary lung adenocarcinomas also metastasize to the bone. In preclinical models of lung cancer, hyperactivation of the WNT-T cell factor (TCF) pathway led to activation of the target genes of WNT and TCF, lymphoid enhancer-binding factor 1 (LEF1) and homeobox B9 (HOXB9) to promote invasion and outgrowth of bone metastases252. Osteoclast-derived CXC-chemokine ligand 12 (CXCL12; also known as SDF1a) signalling through CXC-chemokine receptor 4 (CXCR4) on lung cancer cells activated the ERK-nuclear factor-κB (NF-κB) pathway, resulting in increased MMP9 secretion, thereby mediating lung cancer cell invasion and subsequent bone metastasis253. Analysis of the immune cell infiltration in matched primary and metastatic lesions from patients with NSCLC revealed lower CD8+:CD4+ T cell and CD8+:CD68+ T cell ratios in metastases (lung, brain, liver, distant lymph nodes, kidney and bone), which were indicative of a tolerogenic and tumour-promoting microenvironment at the metastatic site254. In another study using a lung adenocarcinoma mouse model, lung cancer cells were shown to produce IL-6 to mobilize galectin 3-expressing inflammatory myeloid cells into the circulation; the subsequent interaction of these metastatic cancer cells, through cell surface expression of an oncofetal galectin 3 carbohydrate ligand (T antigen), with the myeloid cells within the metastatic niche resulted in metastatic colonization255.
The lung TME as a target for therapy
The identification of critical cellular and molecular pathways in the TME has yielded potential strategies to target key cancer vulnerabilities6,128 (TABLE 1), and drugs targeting TME components are being investigated in clinical trials (Supplementary Table 1).
Table 1 |.
Therapeutic targets in the lung tumour microenvironment
| Target | Function | Genetic perturbation |
Pharmacological agent |
In vivo validation models | Refs |
|---|---|---|---|---|---|
| Cytokines and chemokines | |||||
| CCL2 | Chemokine for monocytes that increases tumour cell extravasation and angiogenesis | NA | Monoclonal antibody | Metastasis to lung: tail vein implantation; mammary fat pad implantation | 95,121,171 |
| CCL9 | Chemokine that recruits macrophages, excludes B and T cells from the tumour mass and increases angiogenesis | NA | Monoclonal antibody | NSCLC: LSL-KrasG12D;Rosa26–1ox-STOP-lox MycERT2 GEMM | 264 |
| IL-1β | Cytokine that binds to the IL-1R to promote epithelial cell proliferation and induces IL-17 in γδT cells; also the target in the trial of canakinumab | NA | Monoclonal antibody | Metastasis to lung: subcutaneous implantation NSCLC: lkkbfl/fl;LysM-Cre GEMM treated with urethane; Ccsp-rtTA (tet-O)-KrasG12D GEMM; K14-Cre; Cdhfl/fl;Trp53fl/fl GEMM |
62,99,141 |
| IL-6 | Cytokine that promotes antiinflammatory responses, proliferation, angiogenesis, EMT and drug resistance | shRNA and siRNA | Monoclonal antibody and receptor neutralizing antibody | NSCLC: Ccsp-Cre;LSL-KrasG12D GEMM with nontypeable Haemophilus influenzae strain 12 lysate exposure; subcutaneous implantation; KrasG12D; Trp53fl/fl GEMM | 274–278 |
| IL-17 A | Cytokine released from TH17 and yδT cells, resulting in neutrophilmediated CD8+T cell suppression | NA | Monoclonal antibody | Metastasis to lung: tail vein implantation; K14-Cre;Cdhfl/fl;Trp53fl/fl GEMM | 76,99 |
| IL-23 | Cytokine that induces immune suppression via exclusion of B cells, T cells and NK cells from tumours | NA | Monoclonal antibody | NSCLC: LSL-KrasG12D;Rosa26-LSL-MycERT2 GEMM | 264 |
| TGFβ1 | Cytokine that promotes EMT, inhibits antitumour immunity and promotes resistance to targeted therapies and chemotherapies | shRNA | SB431542 (TGFβR inhibitor) | NSCLC: subcutaneous implantation | 275–277,279,280 |
| TGFβ3 | Tumour-derived cytokine that induces stromal periostin and supports CSC maintenance and expansion | TGFβR2ΔTM soluble decoy receptor | NA | Metastasis to lung: MMTV-PyMT GEMM; mammary fat pad implantation | 103 |
| Other secreted factors | |||||
| HGF | Growth factor leading to c-MET signalling and responsible for resistance to EGFR tyrosine kinase inhibitors | NA | Monoclonal antibody and antagonist (NK4) | NSCLC: subcutaneous implantation | 281–283 |
| IGF2 | Enriches CSCs via IGF1R signalling | shRNA | NA | NSCLC: subcutaneous implantation | 15,17,264 |
| Periostin | Fibroblast-derived ECM protein that supports CSC maintenance and expansion | Postn−/− mice | Monoclonal antibodies | Metastasis to lung: MMTV-PyMT GEMM; mammary fat pad implantation | 103 |
| PGE2 | Promotes FOXP3 expression and Treg cell activities to suppress antitumour immunity | Ptger2−/− mice | Monoclonal antibody | NSCLC: subcutaneous implantation | 71 |
| sRAGE | Produced by lung tumours to stimulate tumour-promoting neutrophil release from bone | Recombinant protein | NA | NSCLC: KrasLSL-G12D/WT;Trp53fl/fl GEMM; tail vein implantation | 64 |
| Tenascin C | Tumour-derived ECM protein that regulates CSC fitness in the metastatic niche | shRNA | NA | Metastasis to lung: mammary fat pad implantation; tail vein implantation | 104 |
| TIMP1 | NF-κB target that increases FAK-ERK signalling through CD63 to increase tumour proliferation | shRNA | NA | NSCLC: subcutaneous implantation; LSL-KrasG12D;CA2-Cre-shp53 GEMM | 284 |
| VEGFA | Pro-angiogenic factor that also contributes to CSC phenotypes, invasion and chemoresistance | NA | Monoclonal antibody and VEGFR inhibitors | Metastasis to lung: tail vein implantation; subcutaneous implantation; subcapsular renal implantation | 108,285 |
| Versican | ECM proteoglycan stimulating MET, proliferation and metastasis; also suppresses inflammation | shRNA | NA | Metastasis to lung: tail vein implantation; MMTV-PyMT GEMM | 105,121 |
| Cytokine and chemokine receptors | |||||
| CXCR2 | Chemokine receptor on granulocytes that enables recruitment to increase metastasis | NA | Monoclonal antibody | Metastasis to lung: tail vein implantation | 170,286 |
| CXCR4 | Chemokine receptor responding to extracellular ubiquitin after injury to increase lung metastasis | NA | Monoclonal antibody (AMD3100) | Metastasis to lung: tail vein implantation with intratracheal LPS or live Escherichia coli DH5α administration; mammary fat pad implantation with intratracheal DH5a administration | 118 |
| IL-1R | Cytokine receptor that responds to IL-1β to promote epithelial cell proliferation and induces IL-17 in γδ T cells | NA | Soluble receptor antagonist (IL-1RA) | Metastasis to lung: subcutaneous implantation NSCLC: lkkbfl/fl;LysM-Cre GEMM treated with urethane; Ccsp-rtTA (tet-O)-KrasG12D GEMM; K14-Cre; Cdhfl/fl;Trp53fl/fl GEMM |
62,99 |
| IL-1R8 | Cytokine co-receptor, negative regulator of IL receptor and TLR signalling and immune checkpoint for NK cell maturation and effector function | Il1r8−/− mice | NA | Metastasis to lung: intramuscular implantation | 113 |
| Surface receptors | |||||
| CD96 | Negative regulator of NK cell function via interaction with CD155 | Cd96−/− mice | Monoclonal antibody | Metastasis to lung: tail vein implantation; mammary fat pad implantation | 111 |
| E-selectin | Inflammation-induced adhesion molecule on endothelial cells mediating tumour adhesion | NA | Monoclonal antibody | Metastasis to lung: mammary fat pad implantation with LPS treatment | 119 |
| HMMR | Hyaluronan receptor associated with inflammation, cell survival and expansion of micrometastases | shRNA | Hyaluronidase | NSCLC: intratracheal implantation with bleomycin treatment; intraarterial implantation | 25 |
| α6β4 and α6β1 integrins | Exosome adhesion molecules regulating vascular leakiness, expression of lung-specific S100 genes and SRC signalling | ITGB4 shRNA | NA | Metastasis to lung: tail vein or cardiac implantation following exosome administration via tail vein, cardiac or retroorbital injection | 287 |
| α11β1 integrin | Adhesion molecule that interacts with collagen, induces IGF2 and increases tumour growth and metastasis | Itga11−/− | NA | Metastasis to lung: subcutaneous implantation; endobronchial implantation | 16,17 |
| LGR5 | WNT signal transduction protein increasing stem cell fitness in the metastatic niche | shRNA | NA | Metastasis to lung: mammary fat pad implantation; tail vein implantation | 104 |
| PD1 | Inhibitory receptor and negative regulator of T cell function | NA | Monoclonal antibody | NSCLC: tail vein implantation | 70,80,288 |
| TIM3 | Inhibitory receptor of T cell function and adaptive resistance mechanism to PD1 and PDL1 blockade | NA | Monoclonal antibody | NSCLC: Tet-op-EgfrT790M/L858R;Cc10-rtTA GEMM; LSL-KrasG12D GEMM | 194 |
| TLR3 | Alveolar cell pattern recognition receptor that induces metastatic niche formation via production of chemokines, pro-metastatic gene expression and neutrophil recruitment | Tlr3−/− mice | NA | Metastasis to lung: subcutaneous implantation; tail vein implantation following exosome administration via tail vein injection | 97 |
| VCAM1 | Adhesion molecule resulting in pro-survival AKT signalling upon interaction with the a4 integrin subunit | shRNA | Antibody blocking the a4 integrin subunit | Metastasis to lung: mammary fat pad implantation; tail vein implantation | 102 |
| Enzymes | |||||
| ALOX5 | Neutrophil leukotriene-generating enzyme aiding metastatic colonization | Alox5−/−mice | Zileuton | Metastasis to lung: MMTV-PyMT GEMM; tail vein implantation; mammary fat pad implantation | 98 |
| Cathepsin G | Degrades TSP1, increasing inflammation and angiogenesis | Ctsg−/− mice | Sivelestat | NSCLC: tail vein implantation with or without intranasal LPS administration; subcutaneous implantation with intranasal LPS administration | 100 |
| COX2 | Promotes FOXP3 expression and Treg cell activities to suppress antitumour immunity | Cox2−/−mice | SC58236 and celecoxib | NSCLC: subcutaneous implantation; CC10 TAg GEMM | 71 |
| HDAC6 | A HDAC that promotes Treg cell recruitment and represses activation machinery on T cells and dendritic cells | NA | ACY-1215 (ricolinostat) | NSCLC: KrasLSL-G12D/WT; Trp53fl/fl GEMM; EgfrLSL-T790M/L858R GEMM | 163 |
| IDO1 | Tryptophan-catabolizing enzyme that expands myeloid-derived suppressor cells and Treg cells and impairs T cell responses | Ido1−/− mice and siRNA | Methylthiohydantoin-DL-tryptophan and 1-methyl-D-tryptophan | NSCLC: tail vein implantation; cardiac implantation; LSL-KrasG12D GEMM | 88,89 |
| LysyL oxidase, LysyL oxidase Like 2 and LysyL oxidase Like 4 | Crosslinks basement membrane collagen IV in pre-metastatic niches for myeloid cell accumulation | shRNA and Loxl2fl/fl mice | PXS-S2B | Metastasis to lung: MMTV-PyMT+/T;MMTV-Cre+/T GEMM; tail vein implantation; mammary fat pad implantation with or without intraperitoneal tumour cell-conditioned media administration | 96,289 |
| MMP14 | Processes HB-EGF, leading to EGFR signalling and tumour growth | Dominant negative MMP14 | NSC405020 | NSCLC: tail vein implantation | 24 |
| Neutrophil eLastase | Degrades TSP1, increasing inflammation and angiogenesis | Elane−/− mice | Sivelestat | NSCLC: tail vein implantation with or without intranasal LPS administration; subcutaneous implantation with intranasal LPS administration | 100 |
| RNA binding factors | |||||
| miR-105 | Exosomal miRNA targeting ZO1; disrupts endothelial monolayers and increases metastasis | Anti-miR-105 | NA | Metastasis to lung: cardiac implantation following exosome administration via tail vein injection; mammary fat pad implantation | 94 |
| miR-122 | A vesicle miRNA that suppresses glucose uptake and pyruvate kinase, increasing metastasis | Anti-miR-122 | NA | Metastasis to lung: cardiac implantation with pre-implantation and post-implantation exosome administration via tail vein injection; mammary fat pad implantation | 290 |
| miR-143 and miR-145 | Stromal miRNAs targeting CAMK1D, leading to endothelial proliferation and angiogenesis | miR-143– miR145−/− mice |
NA | NSCLC: KrasLSL-G12D/+; Trp53fl/fl GEMM; KrasLSL-G12D/+; Trp53fl/fl; Ccsp-rtTA+ GEMM; tail vein implantation; subcutaneous implantation | 30 |
| MSI1 | RNA binding protein blocking translation of the Notch inhibitor NUMB in the metastatic niche | shRNA | NA | Metastasis to lung: mammary fat pad implantation; tail vein implantation | 104 |
ALOX5, arachidonate 5-lipoxygenase; CAMK1D, calcium/calmodulin-dependent protein kinase type 1D; CCL, CC-chemokine ligand; COX2, cyclooxygenase 2; CSC, cancer stem cell; CXCR, CXC-chemokine receptor; ECM, extracellular matrix; EGFR, epithelial growth factor receptor; EMT, epithelial-to-mesenchymal transition; FAK, focal adhesion kinase; FOXP3, forkhead box P3; GEMM, genetically engineered mouse model; HB-EGF, heparin-binding EGF-like growth factor; HDAC6, histone deacetylase 6; HGF, hepatocyte growth factor; HMMR, hyaluronan-mediated motility receptor; IDO1, indoleamine 2,3-dioxygenase 1; IGF1R, insulin-like growth factor 1 receptor; IGF2, insulin-like growth factor 2; IL, interleukin; IL-1R, IL-1 receptor; LGR5, leucine-rich repeat-containing G proteincoupled receptor 5; LPS, lipopolysaccharide; MET, mesenchymal-to-epithelial transition; miRNA, microRNA; MMP14, matrix metalloproteinase 14; MMTV, mouse mammary tumour virus; MSI1, Musashi 1; NA, not applicable; NF-κB, nuclear factor-κB; NK cell, natural killer cell; NSCLC, non-small-cell lung cancer; PD1, programmed cell death protein 1; PDL1, PD1 ligand 1; PGE2, prostaglandin E2; PyMT, polyoma middle T antigen; shRNA, short hairpin RNA; siRNA, small interfering RNA; sRAGE, soluble receptor for advanced glycosylation end product; TAg, SV40 large T antigen; TGFβ, transforming growth factor-β; TGFβR, TGFβ receptor; TGFβR2ΔTM, soluble form of TGFβR2 lacking the transmembrane domain; TH17 cell, T helper 17 cell; TIM3, T cell membrane protein 3; TIMP1, tissue inhibitors of metalloproteinase 1; TLR, Toll-like receptor; Treg cell, regulatory T cell; TSP1, thrombospondin 1; VCAM1, vascular cell adhesion protein 1; VEGFA, vascular endothelial growth factor A; VEGFR, VEGF receptor; ZO1, zona occludens 1.
Antiangiogenic therapies
The VEGF pathway is a key mediator of tumour angiogenesis, and angiogenesis inhibitors targeting various components of this pathway including VEGF ligands, VEGFRs and downstream signalling components have been developed. These agents have been extensively tested in advanced NSCLC, some of which have been approved by the US Food and Drug Administration (FDA)129. The VEGF antibody bevacizumab was FDA-approved in 2006 for use in combination with carboplatin and paclitaxel chemotherapy as a first-line treatment for advanced, non-squamous NSCLC130. Additionally, ramucirumab, a VEGFR2-targeting antibody, has been approved in combination with docetaxel chemotherapy for the treatment of patients with metastatic NSCLC who have progressed on or after platinum-based chemo-therapy131. Finally, nintedanib, an antibody targeting the tyrosine kinases VEGFR1, VEGFR2, VEGFR3, platelet-derived growth factor receptor-α (PDGFRα), PDGFRβ, fibroblast growth factor receptor 1 (FGFR1), FGFR2 and FGFR3, has been approved by the European Union for use in combination with docetaxel for the treatment of patients with advanced NSCLC with disease progression after front-line platinum-based chemotherapy132. Currently, a plethora of antiangiogenic agents including aflibercept (known as a ‘VEGF trap’, as it binds to circulating VEGF ligands) and the receptor tyrosine kinase inhibitors sunitinib, sorafenib, motesanib, cediranib and vandetanib are being evaluated in clinical trials133.
Despite documented benefits, results from phase III clinical trials have shown that treatment with anti-angiogenic therapies is often associated with somewhat modest increases in median overall survival (OS) and progression free survival (PFS) (Supplementary Table 1). With a growing number of antiangiogenic agents being approved or considered for approval, the requirement for biomarkers is critical for determining drug efficacy and safety, as patient selection may be key to improving these modest clinical benefits seen to date.
Anti-inflammatory therapies
Epidemiological evidence and clinical trial results have indicated that long-term use of aspirin, which is primarily designed to prevent cardiovascular disease, is associated with a reduction in cancer deaths after long-term follow-up134,135. By contrast, a large population-based study, showed a lack of association between aspirin use before diagnosis and lung cancer-specific mortality136. A meta-analysis of several studies related to aspirin use revealed heterogeneous responses, suggesting that it may be important to identify particular patient populations that could benefit from aspirin use137. The COX2 inhibitor celecoxib initially displayed clinical benefit in patients with high COX2-expressing NSCLC138; however, a follow-up, randomized, double-blind, phase III trial failed to demonstrate improved survival139. Concern over toxicity risks with nonsteroidal anti-inflammatory drugs (NSAIDs), which include COX2 inhibitors, has limited their utility140. Recently, a large randomized trial of canakinumab, an antibody targeting IL-1β in patients with atherosclerosis, showed that patients receiving canakinumab had a statistically significant reduction in new lung cancer incidence and mortality141. A phase III trial to evaluate the efficacy and safety of canakinumab as adjuvant therapy in patients with NSCLC is underway142 (Supplementary Table 1).
Immunotherapies
Immunotherapy aims to either improve immune cell function or inhibit immunosuppressive activity143. Inhibition of immunosuppressive signalling networks with ICI, which restores T cell-mediated antitumour immunity, has changed practice guidelines in multiple solid tumours including lung cancer. In 2015, nivolumab, a PD1 antibody targeting the PD1–PDL1 axis, became the first immune checkpoint inhibitor to be approved for treatment of NSCLC. Since then, other immune checkpoint inhibitors have been granted approval as either monotherapy or in combination with other agents (Supplementary Table 1). In the following subsections, we discuss the seminal trials that led to such approvals, as well as the current thinking on biomarkers predictive of response or resistance to ICI.
ICI monotherapy for metastatic NSCLC.
There are currently three PD1 and PDL1 antibodies approved for the treatment of metastatic NSCLC in the second or third line. The approval of the PD1 antibody nivolumab was based on the results of two phase III trials — CheckMate 017, which enrolled patients with tumours with squamous histology, and CheckMate 057, which enrolled patients with tumours with non-squamous histology. In these studies, patients with advanced NSCLC who had progressed during or after prior therapy were randomly allocated nivolumab or docetaxel, a standard second-line chemotherapeutic agent. In both trials, the primary end point of OS was superior with nivolumab treatment144,145. A 3-year follow-up report from both trials showed that both OS and PFS significantly favoured nivolumab (OS: 17% versus 8%; PFS: 10% versus < 1%)146.
Subsequently, another PD1 antibody, pembroli-zumab, was approved for previously treated patients with NSCLC (regardless of histology) whose tumours expressed PDL1 by immunohistochemistry. Initial approval was based on results from the large multicohort phase Ib KEYNOTE-001 trial, where pembrolizumab was used for the treatment of patients with metastatic NSCLC, some of whom had received prior therapy147. Subsequently, the KEYNOTE-010 study showed that pembrolizumab exhibited superior OS compared with docetaxel in patients with previously treated metastatic NSCLC whose tumours had at least 1% total PDL1 expression148. KEYNOTE-001 also found that response rates and survival were highest in patients expressing PDL1 in at least 50% of tumour cells. This finding was corroborated in a landmark phase III trial (KEYNOTE-024), where patients with untreated, metastatic NSCLC with tumours expressing PDL1 in ≥ 50% of cancer cells and with no activating EGFR or ALK mutations were randomized to receive pembrolizumab or standard platinum-doublet chemotherapy. In this study, OS, PFS and response rates were all significantly higher with pembrolizumab149. As a result, pembrolizumab alone without chemotherapy has become a current standard of care for this patient population.
Atezolizumab is currently the only PDL1 antibody approved for patients with previously treated metastatic NSCLC. Similar to the PD1 trials described above, the OAK trial showed superior survival with atezolizumab over docetaxel. This outcome was observed regardless of PDL1 expression levels on tumour cells or tumour-infiltrating immune cells150. Trials are in progress to evaluate the efficacy of other PDL1 antibodies, such as durvalumab and avelumab for the treatment of metastatic NSCLC. However, durvalumab has also been evaluated in patients with inoperable, locally advanced, nonmetastatic (stage III) NSCLC in a randomized trial151. In this study, patients completing the standard treatment of combined chemotherapy and radiation were assigned to receive either durvalumab or placebo for 12 months. Remarkably, median PFS was threefold higher, and time to recurrence or death was nearly doubled with durvalumab. Furthermore, improvement was observed in both smokers and never smokers and at all levels of PDL1 expression in tumour cells. This has led to a rapid breakthrough designation by the FDA of durvalumab for locally advanced, stage III NSCLC with no progression after chemoradiation.
Collectively, ICI trials are rapidly changing the treatment landscape of patients with metastatic and locally advanced NSCLC. Importantly, second-line immunotherapy is becoming a standard for patients who have progressed on first-line chemotherapy alone using Eastern Cooperative Oncology Group (ECOG) performance status. In addition, while the approval of the PD1 antibody pembrolizumab for first-line therapy in patients with advanced NSCLC with tumours expressing PDL1 in ≥ 50% of cancer cells is revolutionary, only 25–50% of all patients with NSCLC express PDL1, with less than half of these expressing PDL1 at the regulatory mandated level of expression152. Further improvements in clinical outcomes require exploring the efficacy of various combination therapeutic strategies.
Combination therapy.
To increase the response rates of current immunotherapies, a major focus is to find appropriate combinations to act synergistically to mount potent immunological responses153–155. Cytotoxic therapy such as chemotherapy and radiation may alter the TME to increase the efficacy of ICI. For example, cisplatin, some anthracyclines and radiation can induce immunogenic cell death, which increases tumour antigen load and increases MHC expression and antigen cross-presentation by activated DCs, leading to an influx of TILs2,153. On the basis of evidence from pre-clinical studies, the landmark trial KEYNOTE-189 randomized untreated patients with advanced non-squamous NSCLC regardless of PDL1 expression to standard chemotherapy plus pembrolizumab or standard chemotherapy alone156. All clinical outcome end points such as response rates, PFS and OS were higher with the combined therapy across all levels of PDL1 expression, including tumours with < 1% PDL1 expression. Another strategy currently being tested is the combination of two immune checkpoint inhibitors that target non-redundant pathways of T cell inactivation. The combination of the CTLA4 antibody ipilimumab and the PD1 antibody nivolumab showed promise in a phase I trial157. A larger phase III multipart trial (CheckMate 227) included a study randomizing patients with untreated, metastatic tumours with some level of PDL1 expression to nivolumab alone, nivolumab plus ipilimumab or standard chemotherapy158. There were two co-primary end points: PFS in all patients and PFS in patients with non-synonymous high tumour mutational burden (TMB) defined as ten or more mutations per megabase. PFS in all patients at 1 year was significantly higher with the combination of nivolumab and ipilimumab than with chemotherapy (31% versus 17%). Of the 44% of patients with high TMB, 1-year PFS with the ICI combination was 42% compared with 13% with chemotherapy alone and 29% with nivolumab monotherapy; yet, no significant difference between the treatment regimens in patients with low TMB was observed. Although this trial has not yet resulted in FDA approval for dual ICI therapy, it is notable for being the first trial to assess the value of TMB as an efficacious predictive biomarker when used as a predefined end point in a randomized lung cancer trial.
Other combination strategies are being explored in ongoing early-phase or more advanced clinical trials. These strategies include combining immune checkpoint inhibitors with epigenetic modifiers, oncolytic viruses or VEGF antibodies. The combination of atezolizumab with bevacizumab plus platinum-doublet chemotherapy has already been shown to elicit a survival advantage over chemotherapy plus bevacizumab alone159.
Given the successes above, the potential for other agents to synergize with ICI is being investigated in preclinical models. For example, extensive preclinical data suggest that radiation may not only contribute to local tumour control but may also augment a systemic antitumour immune response, a phenomenon known as the abscopal effect160. Radiation administered to a liver lesion in combination with anti-CTLA4 therapy improved metastatic disease in other organs in a patient with NSCLC161. However, several factors including the fractionation schedule, dosing, sequence, timing and safety need optimization. There is also increasing evidence suggesting that epigenetic drugs may improve the therapeutic responses of ICI162. For example, a histone deacetylase 6 (HDAC6) inhibitor increased T cell activation and improved the function of APCs, whereas the bromodomain inhibitor JQ1 attenuated CD4+FOXP3+ Treg cells in peripheral blood and single cell suspensions generated from human NSCLCs. Notably, in vivo validation was demonstrated, as combined treatment reduced tumour growth and improved survival in spontaneous mutant KRAS and EGFR-driven mouse models of lung adenocarcinoma163. Consistent with these findings, patients with NSCLC first primed with the DNA methyltransferase inhibitor 5-azacytidine showed improved responses to PD1 or PDL1 therapy164.
Resistance to immunotherapies.
Undoubtedly, the past 5 years have ushered in a transformation in our treatment paradigms for lung cancer. However, despite these advances, most patients with NSCLC either do not respond to ICI or develop resistance after initial response with subsequent disease progression. This has generated an unmet clinical need for biomarkers to identify patients who may benefit from immunotherapies (BOX 1) and a better understanding of the mechanisms of tumour escape.
Tumour immune evasion eventually develops through a variety of pathways including the emergence of antigen loss variants, defects in antigen presentation (human leukocyte antigen (HLA) loss, downregulation of MHC expression and mutations in β2 microglobulin, a component of MHC I molecules), defects in IFNγ signalling and/or activation of alternative inhibitory pathways such as other immune check-points or IDO. A recent study also identified LKB1 alterations as the most prevalent genomic driver of primary resistance to PD1 inhibitors in KRAS-mutant lung adenocarcinoma165,166. A deeper understanding of the complexities of immune response and evasion will further enable the design of more effective and durable immunotherapy regimens. Consequently, we advocate for preoperative or window of opportunity trials in earlier stages of disease, where tissue and blood is obtained before and during treatment and after surgical resection of the tumour mass. For example, 45% of patients had a major pathological response in a recent trial of preoperative anti-PD1 inhibition in early-stage operable disease167. Access to human material obtained before, during and after treatment and at the time of relapse for translational studies will provide great insight into the evolution of an effective immune response.
Pre-metastatic niche targeting
The pathways driving the establishment of a pre-metastatic niche constitute potential therapeutic targets for the prevention and early treatment of metastases. In preclinical models, an inhibitor of ALOX5 (known as zileuton and used as an asthma inhibitor) reduced lung metastasis by decreasing leukotriene production in neutrophils; notably, neutrophil counts remained unaffected98. In addition, targeting the neutrophil proteases cathepsin G and neutrophil elastase with the small molecule inhibitor sivelestat decreased lung metastases100, suggesting potential for clinical translation. While these studies support therapeutic targeting of the pro-metastatic role of neutrophils in cancer, importantly, such approaches must be applied with caution, as neutrophils also exert anti-metastatic activity in other experimental mouse models168. Lysyl oxidase-neutralizing antibodies have shown efficacy in preclinical studies169, and efforts are underway to develop antibodies for use in humans. Several studies have proposed therapies that inhibit the recruitment of immune cells to the pre-metastatic niche, including CXCR2 targeting to decrease platelet-mediated granulocyte recruitment170. Similarly, blockade of CCL2-CC-chemokine receptor 2 (CCR2) signalling reduced metastasis by decreasing mobilization ofmonocytes95. However, paradoxically, discontinuation of CCL2 inhibition led to a larger release of monocytes into the blood as well as increased angiogenesis and cancer cell proliferation in the lungs, resulting in reduced survival of the mice171. Harnessing potential targets from metastasis-suppressive niches is also being considered for therapy. For example, a peptide of five amino acids in length derived from prosaposin with TSP1-inducing activity reduced metastases formation in the lungs114.
Challenges and future directions
Despite major advancements in basic and clinical research pertaining to the role of the TME in lung cancer, several key questions remain to be addressed to create effective treatment opportunities so that optimal clinical outcomes are achieved.
Improving standard therapies
Antiangiogenic therapies have produced some favourable outcomes despite key limitations pertaining to dose-limiting toxicities, emergence of primary or acquired resistance and the lack of reliable validated biomarkers to guide selection of patients with NSCLC172,173. Current studies have focused on combining antiangiogenic agents with therapies targeting the cancer cell. For example, the combination of bevacizumab with the EGFR inhibitor erlotinib received approval as first-line treatment for patients with EGFR-mutant NSCLC in Europe in 2016 and is now being tested in the United States174.
In the clinical space of immunotherapy, the challenge is to increase response rates in patients with NSCLC, and a major effort towards finding predictive biomarkers for patient selection and combination agents to increase treatment efficacy is underway. For example, an important barrier constitutes the lack of biomarkers to assess early responses to immunotherapy, and recently, circulating free DNA was used to assess immunotherapy response in lung cancer175. Following the success of ipilimumab in combination with nivolumab in patients with NSCLC with high TMB158, other modalities including radiation, chemotherapy and antiangiogenic therapies are being tested176,177. Recently, the combination of the VEGFR2 inhibitor ramucirumab and pembrolizumab showed a disease control rate of 85% in a phase I trial of patients with advanced NSCLC178. Considerable enthusiasm has developed for the use of ICI in early-stage and locally advanced cancer179,180. The timing is pertinent, as implementation of low-dose computed tomography (CT) screening programmes in populations at risk has already begun to increase the number of patients with NSCLC diagnosed at an early stage181. While such trials carry the promise of improving systemic immunity against occult metastases, several substantial challenges in this approach exist including defining the optimal treatment schema and appropriate drug combinations, measuring treatment responses as determined by the evaluation of tumour volume or immune-related responses and quantifiable end points (PFS and OS) and, importantly, the selection of patients who are likely to benefit.
Another challenge with immune checkpoint inhibitors is their ability to activate host immune responses, causing undesired adverse effects. Recently, immune checkpoint antibodies conjugated to a matrix-binding peptide, when administered directly into tumours, showed maximal retention in the tumour ECM and generated effective antitumour responses with minimal adverse effects182.
Technological advancements
Technological barriers need to be overcome to unravel the TME landscape and the intricate network of signalling pathways, as they may lead to the identification of prognostic and diagnostic biomarkers and targets for therapy development. Emerging studies have begun to interrogate the complex tumour-stroma crosstalk in preclinical models41, which has led to the development of the computational software CCCExplorer, which assesses cell-cell communication pathways from multicellular genomic data sets41. Recently, an innovative approach unravelled TME transcriptomes in human lung tumours at single cell resolution183. Other technological advancements in multiplexed immuno-histochemical staining, multiparametric flow cytometry, cytometry by time of flight (CyTOF) and single cell sequencing have also resolved TME features directly in clinical specimens10,36,184,185. Regression-based deconvolution of bulk gene expression data is being used to quantify specific cellular compositions of human tumours to predict clinical outcomes and for biomarker discovery and therapeutic target identification186. This approach has the potential to be developed into a clinical test, as it does not rely on a predetermined panel of markers and can determine cellular heterogeneity from limited amounts of fresh, frozen and fixed clinical specimens.
The emerging area of radiomics has the potential to directly utilize medical images (positron emission tomography (PET), magnetic resonance imaging (MRI) and CT) to better define tumour spatial complexity and heterogeneity187. In preclinical models, real-time intravital optical imaging is beginning to be used to visualize dynamic cellular interactions during tumour progression, specifically between disseminated cancer cells and immune cells188 (Supplementary Movie 1). Although imaging of the intact lung presents challenges owing to local respiratory motion within the lungs, this has now been overcome by using a vacuum stabilized imaging window188,189. In addition, whole lung organ imaging was used to estimate 3D TAM composition and their response to CSF1R blockade in a KRAS-mutant xenograft mouse model of lung cancer190.
Developing better lung cancer models
Genetically engineered mouse models (GEMMs) carrying activating mutations in EGFR or KRAS, or EML4-ALK translocations, continue to provide key mechanistic insights related to TME reprogramming and function191–193. However, GEMMs are unresponsive to single-agent immune checkpoint inhibitors194,195, which has been attributed to a low mutational burden and reduced lymphocytic infiltration196–198. Whether these GEMMs can be used to provide mechanistic insights into non-responsiveness to ICI remains to be determined. Recently, immunocompetent mutant KRAS orthotopic mouse models with mutational burdens comparable to non-smoking early-stage patients with NSCLC have shown responses to single-agent immune checkpoint inhibitors70,199. There is also an increased interest in using patient-derived xenograft (PDX) models, as they recapitulate aspects of individual patient tumours and have been predictive of clinical outcomes and precision medicine strategies200,201. However, as immunodeficient mice used for propagating human tumours have a deficiency of key adaptive immune cells, efforts are underway to ‘humanize’ these models by engineering in human immune effectors and engrafting human peripheral blood mononuclear cells202. However, additional barriers continue to limit the use of PDX models, such as potential mouse-specific evolution of the human tumours203 and mouse H2 but not HLA-restricted T cell education in the mouse thymus204.
A major effort in the field is to develop ex vivo human cancer models that can address the contribution of the microenvironment to tumour progression as a function of time. Ex vivo human NSCLC explant models, which have the advantage of retaining the TME, are being explored to evaluate response to therapy205. Recently, a human NSCLC model was engineered on a chip that mimics organ microenvironment-specific cancer growth, tumour dormancy and responses to therapy over time206.
The pre-metastatic niche in the clinic
The pre-metastatic niche has potential clinical utility to identify patients with breast cancer who are at an increased risk of metastasis at the time of diagnosis of the primary tumour. However, studies so far describing the pre-metastatic niche have been mainly performed in mouse models. The observation that MDSCs secrete S100A8 and S100A9 in the pre-metastatic niche was used to develop S100A9-specific single-photon emission computed tomography (SPECT) whole body imaging to visualize pre-metastatic lungs of a preclinical model of breast cancer metastasis207. Similarly, high-affinity PET radiopharmaceutical targeting of the α4β1 integrin was used to identify the pre-metastatic niche208. The clinical relevance of such an approach in identifying patients with breast cancer who are at increased risk of metastasis is awaited. Deconstructing the complexity of the cellular and molecular interactions in the pre-metastatic niche in the human lung has proved challenging owing to a dearth of clinical specimens; however, the forthcoming implementation of rapid autopsy programmes is likely to yield such specimens. Moreover, determining how long the pre-metastatic niche remains primed following surgery in patients with cancer is important because lung metastasis may develop months or years after surgical resection of the primary tumour. Future investigations are also likely to focus on determining similarities and differences between the reprogrammed TME associated with the primary lung carcinoma and the pre-metastatic niche, as this would allow development of therapies that can selectively target either the primary or the secondary tumours in the lung.
The TME in precision medicine
Currently, personalized medicine approaches are focused on the evolution of mutant cancer genes to assess therapeutic responses209, and the utility of the TME has remained largely unexplored. The role of the TME in influencing the treatment responses of cancer cells suggests opportunities for rational, precise and individualized treatments210. Given that the TME landscape is continually changing as a function of tumour evolution and treatment responses, molecular and functional imaging has the potential to match treatment regimens to the specific landscape in individual patients211. Recent studies have also begun to include the TME as a predictive biomarker to identify patients who may benefit from immunotherapies149,212. However, other parameters including engagement of additional immune checkpoints, deficient T cell priming and the inflammatory state of the tumour (cold versus hot tumours) are being considered to guide precision immunotherapy approaches. Indeed, recently, an immunogenomic analysis of >10,000 tumours including lung cancer identified 6 immune subtypes that are likely to impact future prognostic and therapeutic strategies213 (BOX 1). It is expected that such multiscale profiling approaches will lead to the generation of a patient-specific TME atlas, with prognostic and therapeutic implications. Recently, the generation of autologous tumour organoids cocultured with peripheral blood lymphocytes from patients with NSCLC has the potential to assess the sensitivity of tumour cells to T cell-mediated killing in individual patients214.
Conclusion
The complex microenvironment of the lung supports the development of both primary lung carcinomas and metastasis from extrapulmonary neoplasms and offers an untapped resource of targets for therapy development. Mechanistic insights into the tumour-reprogrammed microenvironmental landscape in lung cancer together with the development of drugs that specifically inhibit the components of the landscape have ushered in a new era of cancer medicine. Particularly, certain immunotherapies have performed exceptionally well compared with standard chemotherapy and have the potential to become the new standard of care in a subset of advanced patients with NSCLC. Identifying predictive biomarkers to optimize the benefit of these therapies remains a clinical challenge. Although both PDL1 and TMB are being used as predictive biomarkers to select for patients for therapy, many patients will not qualify just based on these two markers, and there is a requirement to develop additional biomarkers to ensure that larger patient populations benefit. The concept that one size fits all is now obsolete, and this has ushered in the era of precision medicine in lung cancer, which is currently focused on individual molecular testing and cancer genomic analysis to help guide physicians to identify appropriate therapies. However, to maximize patient outcomes, the stage is set to combine the genetic information with the TME landscape, which may allow development of rationally designed combination therapies. Importantly, the success of targeting angiogenesis and immune cells has reinvigorated enthusiasm in elucidating the prognostic and pathophysiological roles of other components of the TME in lung cancer to enable clinical evaluation.
Supplementary Material
Aromatase
An enzyme responsible for the biosynthesis of oestrogen.
Chronic obstructive pulmonary disease
(COPD). A type of obstructive lung disease that includes chronic bronchitis and emphysema and is characterized by long-term breathing problems and poor airflow.
Desmoplasia
Excessive growth of fibrous or connective tissue around an invasive tumour.
Angiocrine signalling
Signalling that involves secreted and membrane-bound inhibitory and stimulatory growth factors, trophogens, chemokines, cytokines, extracellular matrix components, exosomes and other cellular products that are supplied by tissue-specific endothelial cells to help regulate homeostatic and regenerative processes in a paracrine or juxtacrine manner
Degranulation
The process by which a cell releases the contents of its membrane-bound vesicles, called granules.
Tertiary lymphoid structures
(TLS). Transient ectopic lymphoid organizations that may develop after birth in nonlymphoid tissues in situations of chronic inflammation and cancer.
Doublet chemotherapy
A regimen of platinum compounds including cisplatin or carboplatin used as a standard of care in the management of advanced patients with non-small-cell lung cancer.
Immunogenic cell death
A form of cell death caused by, for example, cytostatic agents and radiation therapy that induces an effective antitumour immune response through activation of dendritic cells, which increase T cell priming.
Vascular co-option
A mechanism by which tumours obtain a blood supply from pre-existing vessels without undergoing angiogenesis. This is commonly observed in highly vascularized organs including the lungs and brain.
Cytometry by time of flight
(CyTOF). A variation of flow cytometry in which antibodies are labelled with heavy metal ion tags rather than fluorochromes. Readout is by time-of-flight mass spectrometry. This allows measurement of several cellular parameters in single samples without spectral overlap.
Radiomics
High-throughput mining of quantitative image features from standard of care medical imaging that enables data to be extracted and applied within clinical decision support systems to improve diagnostic, prognostic and predictive accuracy.
Radiopharmaceutical
The combination of a radioactive molecule, a radionuclide, that permits external detection and a biologically active molecule or drug that acts as a carrier and determines localization and biodistribution.
Rapid autopsy programmes
Programmes to arrange and perform autopsies on an urgent basis after death to collect tumour samples and other tissues for research purposes and clinical assessments.
Acknowledgements
The authors thank B. Sleckman, G. Koretzky and J. Cubillos-Ruiz for reading the manuscript and providing insights. They also thank S. Lee for editing this manuscript and Y. Ban and R. Shaykhiev for help with figures. The authors acknowledge funding support from US National Institutes of Health (U01 CA188388), US Department of Defense and Meyer Cancer Center Pilot grants. The authors apologize for being unable to include several excellent studies owing to space limitations.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41568-018-0081-9.
References
- 1.Siegel RL, Miller KD & Jemal A Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Herbst RS, Morgensztern D & Boshoff C The biology and management of non-small cell lung cancer. Nature 553, 446–454 (2018).This review highlights recent progress in lung cancer biology and therapeutic strategies that are impacting outcomes for patients with advanced-stage NSCLC.
- 3.Chen Z, Fillmore CM, Hammerman PS, Kim CF & Wong KK Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev Cancer 14, 535–546 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayekar MK & Bivona TG Current landscape of targeted therapy in lung cancer. Clin. Pharmacol. Ther. 102, 757–764 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Jamal-Hanjani M et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D & Coussens LM Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322 (2012).This review highlights the TME as a key hallmark of carcinogenesis.
- 7.Quail DF & Joyce JA Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peinado H et al. Pre-metastatic niches: organ-specific homes for metastases. Nat. Rev. Cancer 17, 302–317 (2017).This is a comprehensive review on the pre-metastatic niche in cancer.
- 9.Houghton AM Mechanistic links between COPD and lung cancer. Nat. Rev. Cancer 13, 233–245 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Banat GA et al. Immune and inflammatory cell composition of human lung cancer stroma. PLOS ONE 10, e0139073 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kargl J et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat. Commun. 8, 14381 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bremnes RM et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac Oncol. 6, 209–217 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Kalluri R The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Wang L et al. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through 1L-6/STAT3 signaling pathway. Oncotarget 8, 76116–76128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen WJ et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 5, 3472 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Navab R et al. Integrin a 11 (31 regulates cancer stromal stiffness and promotes tumorigenicity and metastasis in non-small cell lung cancer. Oncogene 35, 1899–1908 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu CQ et al. Integrin alpha 11 regulates 1GF2 expression in fibroblasts to enhance tumorigenicity of human non-small-cell lung cancer cells. Proc. Natl Acad. Sci. USA 104, 11754–11759 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaudhri VK et al. Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol. Cancer Res. 11, 579–592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nazareth MR et al. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J. Immunol. 178, 5552–5562 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Lakins MA, Ghorani E, Munir H, Martins CP & Shields JD Cancer-associated fibroblasts induce antigen-specific deletion of CD8. Nat. Commun. 9, 948 (2018).References 19 and 20 show that CAFs modulate immune responses in the TME, as they express PDL1 to blunt activation of T cells and cross-present antigens to eliminate antigen-specific CD8+ T cells via PDL2 and FASL.
- 21.Kanzaki R et al. Gas6 derived from cancer-associated fibroblasts promotes migration of Axl-expressing lung cancer cells during chemotherapy. Sci. Rep. 7, 10613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye X et al. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene 29, 5254–5264 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Lu P, Weaver VM & Werb Z The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395–406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stawowczyk M et al. Matrix metalloproteinase 14 promotes lung cancer by cleavage of heparin-binding EGF-like growth factor. Neoplasia 19, 55–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevens LE et al. Extracellular matrix receptor expression in subtypes of lung adenocarcinoma potentiates outgrowth of micrometastases. Cancer Res. 77, 1905–1917 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marcus AI & Zhou W LKB1 regulated pathways in lung cancer invasion and metastasis. J. Thorac Oncol. 5, 1883–1886 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao Y et al. LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proc. Natl Acad. Sci. USA 107, 18892–18897 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salmon H et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 122, 899–910 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanahan D & Weinberg RA The hallmarks of cancer. Cell 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Dimitrova N et al. Stromal expression of miR-143/145 promotes neoangiogenesis in lung cancer development. CancerDiscov. 6, 188–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie M, Liu M & He CS S1RT1 regulates endothelial Notch signaling in lung cancer. PLOS ONE 7, e45331 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pezzella F et al. Non-small-cell lung carcinoma tumor growth without morphological evidence of neo-angiogenesis. Am. J. Pathol. 151, 1417–1423 (1997). [PMC free article] [PubMed] [Google Scholar]
- 33.McClelland MR et al. Diversity of the angiogenic phenotype in non-small cell lung cancer. Am. J. Respir. Cell. Mo!. Biol. 36, 343–350 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fridman WH, Zitvogel L, Sautes-Fridman C & Kroemer G The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 14, 717–734 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Gajewski TF, Schreiber H & Fu YX Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lavin Y et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 169, 750–765 (2017).This is the first study to perform multiscale immune profiling using CyTOF and single-cell sequencing in patients with early-stage lung cancer.
- 37.Durrans A et al. Identification of reprogrammed myeloid cell transcriptomes in NSCLC. PLOS ONE 10, e0129123 (2015).This is the first study to sort individual myeloid cell subsets from tumour tissue and matched adjacent normal tissue from freshly harvested early-stage lung cancer specimens to identify tumour reprogrammed, differentially regulated genes.
- 38.Veglia F, Perego M & Gabrilovich D Myeloid-derived suppressor cells coming of age. Nat. Immunol. 19, 108–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engblom C, Pfirschke C & Pittet MJ The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 16, 447–462 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Schmid MC & Varner JA Myeloid cells in the tumor microenvironment: modulation of tumor angiogenesis and tumor inflammation. J. Oncol. 2010, 201026 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi H et al. Transcriptome analysis of individual stromal cell populations identifies stroma-tumor crosstalk in mouse lung cancer model. Cell Rep. 10, 1187–1201 (2015).This study interrogates the complex tumour-stroma crosstalk in a mutant KRAS-driven lung cancer mouse model, which led to the development of the computational software CCCExplorer, which assesses cell-cell communication pathways from multicellular genomic data sets.
- 42.Kastenmuller W, Kastenmuller K, Kurts C & Seder RA Dendritic cell-targeted vaccines—hope or hype? Nat. Rev. Immunol. 14, 705–711 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Schneider T et al. Non-small cell lung cancer induces an immunosuppressive phenotype of dendritic cells in tumor microenvironment by upregulating B7-H3. J. Thorac Oncol. 6, 1162–1168 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Dumitriu IE, Dunbar DR, Howie SE, Sethi T & Gregory CD Human dendritic cells produce TGF-β1 under the influence of lung carcinoma cells and prime the differentiation of CD4+CD25+Foxp3+ regulatory T cells. J. Immunol. 182, 2795–2807 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Yang SC et al. Intrapulmonary administration of CCL21 gene-modified dendritic cells reduces tumor burden in spontaneous murine bronchoalveolar cell carcinoma. Cancer Res. 66, 3205–3213 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Lee JM et al. Phase I trial of intratumoral injection of CCL21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and CD8+ T-cell infiltration. Clin. Cancer Res. 23, 4556–4568 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saxena M & Bhardwaj N Re-emergence of dendritic cell vaccines for cancer treatment. Trends Cancer 4, 119–137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabrilovich DI Myeloid-derived suppressor cells. Cancer Immunol. Res. 5, 3–8 (2017).This is a comprehensive review on MDSCs in cancer.
- 49.Coffelt SB, Wellenstein MD & de Visser KE Neutrophils in cancer: neutral no more. Nat. Rev. Cancer 16, 431–446 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Condamine T et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 1, aaf8943 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Almatroodi SA, McDonald CF, Darby IA & Pouniotis DS Characterization of M1/M2 tumour-associated macrophages (TAMs) and Th1/Th2 cytokine profiles in patients with NSCLC. Cancer Microenviron. 9, 1–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohri CM, Shikotra A, Green RH, Waller DA & Bradding P Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur. Respir. J. 33, 118–126 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Ginhoux F, Schultze JL, Murray PJ, Ochando J & Biswas SK New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 17, 34–40 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Conway EM et al. Macrophages, inflammation, and lung cancer. Am. J. Respir. Crit. Care Med. 193, 116–130 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Wang DH et al. Progression of EGFR-mutant lung adenocarcinoma is driven by alveolar macrophages. Clin. Cancer Res. 23, 778–788 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Fritz JM et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front. Immunol. 5, 587 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cannarile MA et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 5, 53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar V et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell 32, 654–668 (2017).This study provides a new mechanism of resistance to CSF1R inhibitors and suggests the implementation of a rationally designed combination therapy.
- 59.Sionov RV, Fridlender ZG & Granot Z The multifaceted roles neutrophils play in the tumor microenvironment. CancerMicroenviron. 8, 125–158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mantovani A, Cassatella MA, Costantini C & Jaillon S Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 11, 519–531 (2011).This is a comprehensive review describing how neutrophils regulate immunity.
- 61.Singhal S et al. Origin and role of a subset of tumor-associated neutrophils with antigen-presenting cell features in early-stage human lung cancer. Cancer Cell 30, 120–135 (2016).This study shows that APC-like ‘hybrid neutrophils’ cross-present antigens and trigger antitumour T cell responses in human lung cancer.
- 62.McLoed AG et al. Neutrophil-derived IL-1 p impairs the efficacy of NF-κB inhibitors against lung cancer. Cell Rep. 16, 120–132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vikis HG et al. Neutrophils are required for 3-methylcholanthrene-initiated, butylated hydroxytoluene-promoted lung carcinogenesis. Mol. Carcinog. 51, 993–1002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Engblom C et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF. Science 358, eaa15081 (2017).This study highlights a new mechanism by which primary lung tumours mediate crosstalk with the bone marrow to generate a new class of tumour-promoting neutrophils.
- 65.Spits H & Cupedo T Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu. Rev. Immunol. 30, 647–675 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Cerwenka A & Lanier LL Natural killers join the fight against cancer. Science 359, 1460–1461 (2018). [DOI] [PubMed] [Google Scholar]
- 67.Carrega P & Ferlazzo G Natural killers are made not born: how to exploit NK cells in lung malignancies. Front. Immunol. 8, 277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bruno A et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 15, 133–142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vacca P et al. NK cells from malignant pleural effusions are not anergic but produce cytokines and display strong antitumor activity on short-term IL-2 activation. Eur. J. Immunol. 43, 550–561 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Markowitz GJ et al. Immune reprogramming via PD-1 inhibition enhances early-stage lung cancer survival. JCI Insight 3, e96836 (2018).This study demonstrates the efficacy of anti-PD1 monotherapy and its impact on the immune microenvironment in an orthotopic mutant KRAS-driven model of NSCLC.
- 71.Sharma S et al. Tumor cyclooxygenase-2/prostaglandin E2-dependent promotion of FOXP3 expression and CD4+CD25+ T regulatory cell activities in lung cancer. Cancer Res. 65, 5211–5220 (2005).This study shows that CAFs express COX2, which promotes secretion of PGE2 to induce immune-suppressive FOXP3+ Treg cells.
- 72.Granville CA et al. A central role for Foxp3+regulatory T cells in K-Ras-driven lung tumorigenesis. PLOS ONE 4, e5061 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ganesan AP et al. Tumor-infiltrating regulatory T cells inhibit endogenous cytotoxic T cell responses to lung adenocarcinoma. J. Immunol. 191, 2009–2017 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marshall EA et al. Emerging roles of T helper 17 and regulatory T cells in lung cancer progression and metastasis. Mol. Cancer 15, 67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petersen RP et al. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer 107, 2866–2872 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Reppert S et al. A role for T-bet-mediated tumour immune surveillance in anti-IL-17A treatment of lung cancer. Nat. Commun. 2, 600 (2011). [DOI] [PubMed] [Google Scholar]
- 77.Dunn GP, Old LJ & Schreiber RD The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Wherry EJ & Kurachi M Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 15, 486–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thommen DS et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol. Res. 3, 1344–1355 (2015). [DOI] [PubMed] [Google Scholar]
- 80.Kamphorst AO et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl Acad. Sci. USA 114, 4993–4998 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Germain C et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am. J. Respir. Crit. Care Med. 189, 832–844 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Bruno TC et al. Antigen-presenting intratumoral B cells affect CD4. Cancer Immunol. Res. 5, 898–907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Turley SJ, Cremasco V & Astarita JL Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 15, 669–682 (2015). [DOI] [PubMed] [Google Scholar]
- 84.Busch SE et al. Lung cancer subtypes generate unique immune responses. J. Immunol. 197, 4493–4503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Spranger S & Gajewski TF Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 18, 139–147 (2018).This is a comprehensive review on the impact of cancer cell-intrinsic pathways on immunomodulation.
- 86.Liu CY et al. Population alterations of L-arginase-and inducible nitric oxide synthase-expressed CD11b+/CD14−/CD15+/CD33+myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 136, 35–45 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Feng PH et al. CD14+S100A9+ monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. Am. J. Respir. Crit. Care Med. 186, 1025–1036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smith C et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. 2, 722–735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schafer CC et al. Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget 7, 75407–75424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02752074?term=NCT02752074&rank=1 (2018). [DOI] [PubMed]
- 91.Woodard PK, Dehdashti F & Putman CE Radiologic diagnosis of extrathoracic metastases to the lung. Oncol. (WillistonPark) 12, 431–438; discussion 441–442 (1998). [PubMed] [Google Scholar]
- 92.Francia G, Cruz-Munoz W, Man S, Xu P & Kerbel RS Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat. Rev. Cancer 11, 135–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu Y & Cao X Characteristics and significance of the pre-metastatic niche. Cancer Cell 30, 668–681 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Zhou W et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 25, 501–515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Qian BZ et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Erler JT et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44 (2009).This study demonstrates that hypoxia in the primary breast tumour triggers expression of lysyl oxidase, which systemically crosslinks collagen in the lungs, to increase adhesion of myeloid cells, generating niches that support metastatic colonization.
- 97.Liu Y et al. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell 30, 243–256 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Wculek SK & Malanchi I Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coffelt SB et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.El Rayes T et al. Lung inflammation promotes metastasis through neutrophil protease-mediated degradation of Tsp-1. Proc. Natl Acad. Sci. USA 112, 16000–16005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Massara M et al. ACKR2 in hematopoietic precursors as a checkpoint of neutrophil release and anti-metastatic activity. Nat. Commun. 9, 676 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen Q, Zhang XH & Massague J Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 20, 538–549 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Malanchi I et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481, 85–89 (2012). [DOI] [PubMed] [Google Scholar]
- 104.Oskarsson T et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 17, 867–874 (2011).References 103 and 104 show that tumour cell-fibroblast crosstalk promotes metastasis.
- 105.Gao D et al. Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition. Cancer Res. 15, 1384–1394 (2012).This study shows that reprogrammed myeloid cells express the proteoglycan versican to promote MET of DTCs in the lungs.
- 106.Del Pozo Martin Y et al. Mesenchymal cancer cell-stroma crosstalk promotes niche activation, epithelial reversion, and metastatic colonization. Cell Rep. 13, 2456–2469 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gao D et al. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science 319, 195–198 (2008). [DOI] [PubMed] [Google Scholar]
- 108.Minami T et al. The calcineurin-NFAT-angiopoietin-2 signaling axis in lung endothelium is critical for the establishment of lung metastases. Cell Rep. 4, 709–723 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lopez-Soto A, Gonzalez S, Smyth MJ & Galluzzi L Control of metastasis by NK cells. Cancer Cell 32, 135–154 (2017). [DOI] [PubMed] [Google Scholar]
- 110.Carrega P et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56brightCD16- cells and display an impaired capability to kill tumor cells. Cancer 112, 863–875 (2008). [DOI] [PubMed] [Google Scholar]
- 111.Blake SJ et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. CancerDiscov. 6, 446–459 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Sceneay J et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 72, 3906–3911 (2012). [DOI] [PubMed] [Google Scholar]
- 113.Molgora M et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 551, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Catena R et al. Bone marrow-derived gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discov. 3, 578–589 (2013).This study shows that metastatic incompetent tumours express the glycoprotein prosaposin to reprogramme myeloid cells in the lungs to express TSP1 to generate metastasis-suppressive niches. A peptide of five amino acids in length derived from prosaposin with TSP1-inducing activity has anti-metastatic activity in preclinical models.
- 115.Hanna RN et al. Patrolling monocytes control tumor metastasis to the lung. Science 350, 985–990 (2015).This study shows that lung endothelial cells attract CX3CR1 + patrolling monocytes, which through expression of CCL3, CCL4 and CCL5 recruit cytotoxic NK cells to prevent metastasis to the lung in mice.
- 116.Murin S, Pinkerton KE, Hubbard NE & Erickson K The effect of cigarette smoke exposure on pulmonary metastatic disease in a murine model of metastatic breast cancer. Chest 125, 1467–1471 (2004). [DOI] [PubMed] [Google Scholar]
- 117.Murin S & Inciardi J Cigarette smoking and the risk of pulmonary metastasis from breast cancer. Chest 119, 1635–1640 (2001). [DOI] [PubMed] [Google Scholar]
- 118.Yan L, Cai Q & Xu Y The ubiquitin-CXCR4 axis plays an important role in acute lung infection-enhanced lung tumor metastasis. Clin. Cancer Res. 19, 4706–4716 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jiang M et al. Systemic inflammation promotes lung metastasis via E-selectin upregulation in mouse breast cancer model. Cancer Biol. Ther. 15, 789–796 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stathopoulos GT et al. Host nuclear factor-kappaB activation potentiates lung cancer metastasis. Mol. Cancer Res. 6, 364–371 (2008). [DOI] [PubMed] [Google Scholar]
- 121.Said N, Sanchez-Carbayo M, Smith SC & Theodorescu D RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J. Clin. Invest. 122, 1503–1518 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Clever D et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell 166, 1117–1131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ghajar CM Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 15, 238–247 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Aguirre-Ghiso JA, Bragado P & Sosa MS Metastasis awakening: targeting dormant cancer. Nat. Med. 19, 276–277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Linde N, Fluegen G & Aguirre-Ghiso JA The relationship between dormant cancer cells and their microenvironment. Adv. Cancer Res. 132, 45–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Romero I, Garrido F & Garcia-Lora AM Metastases in immune-mediated dormancy: a new opportunity for targeting cancer. Cancer Res. 74, 6750–6757 (2014). [DOI] [PubMed] [Google Scholar]
- 127.Wood SL, Pernemalm M, Crosbie PA & Whetton AD The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat. Rev. 40, 558–566 (2014). [DOI] [PubMed] [Google Scholar]
- 128.Pitt JM et al. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 27, 1482–1492 (2016). [DOI] [PubMed] [Google Scholar]
- 129.Pilotto S et al. Anti-angiogenic drugs and biomarkers in non-small-cell lung cancer: a ‘hard days night’. Curr. Pharm. Des. 20, 3958–3972 (2014). [DOI] [PubMed] [Google Scholar]
- 130.Sandler A et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 355, 2542–2550 (2006).This study is responsible for the FDA approval of bevacizumab for patients with advanced NSCLC.
- 131.Garon EB et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet 384, 665–673 (2014).This study led to the approval of ramucirumab, a VEGFR2-targeting antibody in combination with docetaxel chemotherapy for the treatment of patients with metastatic NSCLC who have progressed on or after platinum-based chemotherapy.
- 132.Reck M et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 15, 143–155 (2014).This study led to the approval in the European Union of nintedanib in combination with docetaxel for the treatment of patients with advanced pretreated NSCLC.
- 133.Tabchi S & Blais N Antiangiogenesis for advanced non-small-cell lung cancer in the era of immunotherapy and personalized medicine. Front. Oncol. 7, 52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang D & Dubois RN Prostaglandins and cancer. Gut 55, 115–122 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cuzick J et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann. Oncol. 26, 47–57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mc Menamin U, Cardwell CR, Hughes CM & Murray LM Low-dose aspirin and survival from lung cancer: a population-based cohort study. BMC Cancer 15, 911 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hochmuth F, Jochem M & Schlattmann P Meta-analysis of aspirin use and risk of lung cancer shows notable results. Eur. J. Cancer Prev. 25, 259–268 (2016). [DOI] [PubMed] [Google Scholar]
- 138.Edelman MJ, Hodgson L, Wang X, Kratzke RA & Vokes EE Cyclooxygenase-2 (COX-2) as a predictive marker for the use of COX-2 inhibitors in advanced non-small-cell lung cancer. J. Clin. Oncol. 30, 2019–2020; author reply 2020 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Edelman MJ et al. Phase III randomized, placebo-controlled, double-blind trial of celecoxib in addition to standard chemotherapy for advanced non-small-cell lung cancer with cyclooxygenase-2 overexpression: CALGB 30801 (Alliance). J. Clin. Oncol. 35, 2184–2192 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Solomon DH et al. The risk of major NSAID toxicity with celecoxib, ibuprofen, or naproxen: a secondary analysis of the PRECISION trial. Am. J. Med. 130, 1415–1422 (2017). [DOI] [PubMed] [Google Scholar]
- 141.Ridker PM et al. Effect of interleukin-1 (3 inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017). [DOI] [PubMed] [Google Scholar]
- 142.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03447769?term=NCT03447769&rank=1 (2018). [DOI] [PubMed]
- 143.Zavala VA & Kalergis AM New clinical advances in immunotherapy for the treatment of solid tumours. Immunology 145, 182–201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Brahmer J et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 373, 123–135 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Borghaei H et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).References 144 and 145 are responsible for the approval of the PD1 antibody nivolumab.
- 146.Vokes EE et al. Nivolumab versus docetaxel in previously treated advanced non-small-cell lung cancer (CheckMate 017 and CheckMate 057): 3-year update and outcomes in patients with liver metastases. Ann. Oncol. 29, 959–965 (2018). [DOI] [PubMed] [Google Scholar]
- 147.Garon EB et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 372, 2018–2028 (2015).This study is responsible for the approval of the PD1 antibody pembrolizumab for previously treated patients with NSCLC (regardless of the tumour histology) whose tumours expressed PDL1.
- 148.Herbst RS et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387, 1540–1550 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Reck M et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N. Engl. J. Med. 375, 1823–1833 (2016).This study reports on a randomized phase III trial, which led to the FDA approval of pembrolizumab for patients with advanced NSCLC expressing PDL1 in > 50% of tumour cells.
- 150.Rittmeyer A et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389, 255–265 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Antonia SJ et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N. Engl. J. Med. 377, 1919–1929 (2017).This study is responsible for the rapid breakthrough designation by the FDA of durvalumab for locally advanced, stage III NSCLC with no progression after chemoradiation.
- 152.Rimm DL et al. A prospective, multi-institutional, pathologist-based assessment of 4 immunohistochemistry assays for PD-L1 expression in non-small cell lung cancer. JAMA Oncol. 3, 1051–1058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sharma P & Allison JP Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Patel SA & Minn AJ Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity 48, 417–433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zappasodi R, Merghoub T & Wolchok JD Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell 33, 581–598 (2018).References 153, 154 and 155 discuss the functional roles and determinants of immune checkpoint blockade, immunotherapy combinations and biomarkers for treatment optimization.
- 156.Gandhi L et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 378, 2078–2092 (2018).This paper reports on a double-blind phase III trial confirming that the addition of pembrolizumab to standard chemotherapy of pemetrexed and a platinum drug results in significantly longer OS and PFS than chemotherapy alone irrespective of PDL1 levels in previously untreated metastatic non-squamous NSCLC without EGFR or ALK mutations.
- 157.Hellmann MD et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 18, 31–41(2017).This paper reports a phase I trial showing that in patients with high TMB (at least 10 mutations per megabase), nivolumab plus ipilimumab significantly improve PFS compared with chemotherapy irrespective of PDL1 levels.
- 158.Hellmann MD et al. Nivolumab plus Ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 378, 2093–2104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02366143?term=NCT02366143&rank=1 (2018). [DOI] [PubMed]
- 160.Ngwa W et al. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 18, 313–322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Golden EB, Demaria S, Schiff PB, Chachoua A & Formenti SC An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol. Res. 1, 365–372 (2013).This study is the first to demonstrate an abscopal response to radiation and ipilimumab in a patient with metastatic NSCLC.
- 162.Jones PA, Issa JP & Baylin S Targeting the cancer epigenome for therapy. Nat. Rev Genet. 17, 630–641 (2016). [DOI] [PubMed] [Google Scholar]
- 163.Adeegbe DO et al. Synergistic immunostimulatory effects and therapeutic benefit of combined histone deacetylase and bromodomain inhibition in non-small cell lung cancer. Cancer Discov. 7, 852–867 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Juergens RA et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 1, 598–607 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Skoulidis F et al. Mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. CancerDiscov. 8, 822–835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Koyama S et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 76, 999–1008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Forde PM et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N. Engl. J. Med. 378, 1976–1986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Granot Z et al. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 20, 300–314 (2011).This study describes a metastasis-suppressive role for neutrophils.
- 169.Cox TR, Gartland A & Erler JT Lysyl oxidase, a targetable secreted molecule involved in cancer metastasis. Cancer Res. 76, 188–192 (2016). [DOI] [PubMed] [Google Scholar]
- 170.Labelle M, Begum S & Hynes RO Platelets guide the formation of early metastatic niches. Proc. Natl Acad. Sci. USA 111, E3053–E3061 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Bonapace L et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515, 130–133 (2014). [DOI] [PubMed] [Google Scholar]
- 172.Kurzrock R & Stewart DJ Exploring the benefit/risk associated with antiangiogenic agents for the treatment of non-small cell lung cancer patients. Clin. Cancer Res. 23, 1137–114 (2017). [DOI] [PubMed] [Google Scholar]
- 173.Moserle L, Jimenez-Valerio G & Casanovas O Antiangiogenic therapies: going beyond their limits. Cancer Discov. 4, 31–41 (2014). [DOI] [PubMed] [Google Scholar]
- 174.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01532089?term=NCT01532089&rank=1 (2018). [DOI] [PubMed]
- 175.Goldberg SB et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin Cancer Res. 24, 1872–1880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fukumura D, Kloepper J, Amoozgar Z, Duda DG & Jain RK Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat. Rev. Clin. Oncol. 15, 325–340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Khan KA & Kerbel RS Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev Clin. Oncol. 15, 310–324 (2018).References 176 and 177 discuss the rationale for combining antiangiogenic therapies with immunotherapy.
- 178.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02443324?term=NCT02443324&rank=1 (2018). [DOI] [PubMed]
- 179.Berman AT & Simone CB Immunotherapy in locally-advanced non-small cell lung cancer: releasing the brakes on consolidation? Transl Lung Cancer Res. 5, 138–142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Taunk NK et al. Immunotherapy and radiation therapy for operable early stage and locally advanced non-small cell lung cancer. Transl Lung Cancer Res. 6, 178–185 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Aberle DR et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 365, 395–409 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ishihara J et al. Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl Med. 9, eaan0401 (2017). [DOI] [PubMed] [Google Scholar]
- 183.Lambrechts D et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 184.Remark R et al. In-depth tissue profiling using multiplexed immunohistochemical consecutive staining on single slide. Sci. Immunol. 1, aaf6925 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lizotte PH et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight 1, e89014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gentles AJ et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 21, 938–945 (2015).This study demonstrates that deconvolution of bulk gene expression data can be used to estimate cellular compositions of human tumours.
- 187.Lambin P et al. Radiomics: the bridge between medical imaging and personalized medicine. Nat. Rev. Clin. Oncol. 14, 749–762 (2017). [DOI] [PubMed] [Google Scholar]
- 188.Entenberg D et al. A permanent window for the murine lung enables high-resolution imaging of cancer metastasis. Nat. Methods 15, 73–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Looney MR et al. Stabilized imaging of immune surveillance in the mouse lung. Nat. Methods 8, 91–96 (2011).References 188 and 189 show real-time dynamic interactions in the lung microenvironment.
- 190.Cuccarese MF et al. Heterogeneity of macrophage infiltration and therapeutic response in lung carcinoma revealed by 3D organ imaging. Nat. Commun. 8, 14293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Politi K et al. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 20, 1496–1510 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.DuPage M, Dooley AL & Jacks T Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc. 4, 1064–1072 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Maddalo D et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516, 423–427 (2014).References 191, 192 and 193 describe mouse models of lung cancer with prominent oncogenic drivers including mutant EGFR and KRAS and the EML-ALK4 translocation.
- 194.Koyama S et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 7, 0501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Pfirschke C et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 44, 343–354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Westcott PM et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 517, 489–492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.McFadden DG et al. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc. Natl Acad. Sci. USA 113, E6409–E6417 (2016).This study shows that oncogene-driven lung adenocarcinomas in GEMMs harbour few somatic mutations compared with human lung adenocarcinomas. By contrast, reference 70 shows that an orthotopic mutant KRAS-driven mouse model of NSCLC harbours a mutational burden comparable to non-smoker early-stage human NSCLC.
- 198.DuPage M et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 19, 72–85 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Li HY et al. The tumor microenvironment regulates sensitivity of murine lung tumors to PD-1/PD-L1 antibody blockade. Cancer Immunol. Res. 5, 767–777(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Aparicio S, Hidalgo M & Kung AL Examining the utility of patient-derived xenograft mouse models. Nat. Rev. Cancer 15, 311–316 (2015). [DOI] [PubMed] [Google Scholar]
- 201.Morgan KM, Riedlinger GM, Rosenfeld J, Ganesan S & Pine SR Patient-derived xenograft models of non-small cell lung cancer and their potential utility in personalized medicine. Front. Oncol. 7, 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zitvogel L, Pitt JM, Daillére R, Smyth MJ & Kroemer G Mouse models in oncoimmunology. Nat. Rev. Cancer 16, 759–773 (2016). [DOI] [PubMed] [Google Scholar]
- 203.Ben-David U et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 49, 1567–1575 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Walsh NC et al. Humanized mouse models of clinical disease. Annu. Rev Pathol. 12, 187–215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Karekla E et al. Ex vivo explant cultures of non-small cell lung carcinoma enable evaluation of primary tumor responses to anticancer therapy. Cancer Res. 2, 2029–2039 (2017). [DOI] [PubMed] [Google Scholar]
- 206.Hassell BA et al. Human organ chip models recapitulate orthotopic lung cancer growth, therapeutic responses, and tumor dormancy in vitro. Cell Rep. 21, 508–516 (2017). [DOI] [PubMed] [Google Scholar]
- 207.Eisenblaetter M et al. Visualization of tumor-immune interaction - target-specific imaging of S100A8/A9 reveals pre-metastatic niche establishment. Theranostics 7, 2392–2401 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Shokeen M et al. Molecular imaging of very late antigen-4 (α4β1 integrin) in the premetastatic niche. J. Nucl. Med. 53, 779–786 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kim C & Giaccone G Precision oncology in non-small-cell lung cancer: opportunities and challenges. Nat. Rev. Clin. Oncol. 15, 348–349 (2018). [DOI] [PubMed] [Google Scholar]
- 210.Ostman A The tumor microenvironment controls drug sensitivity. Nat. Med. 18, 1332–1334 (2012). [DOI] [PubMed] [Google Scholar]
- 211.Penet MF, Krishnamachary B, Chen Z, Jin J & Bhujwalla ZM Molecular imaging of the tumor microenvironment for precision medicine and theranostics. Adv. Cancer Res. 124, 235–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Herbst RS et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Thorsson V et al. The immune landscape of cancer. Immunity 48, 812–830 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Dijkstra KK et al. Generation of tumor-reactive T cells by co-culture of peripheral blood lymphocytes and tumor organoids. Cell 174, 1586–1598 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ito M et al. Prognostic impact of cancer-associated stromal cells in stage I lung adenocarcinoma patients. Chest 142, 151–158 (2012). [DOI] [PubMed] [Google Scholar]
- 216.Navab R et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl Acad. Sci. USA 108, 7160–7165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Gocheva V et al. Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proc. Natl Acad. Sci. USA 114, E5625–E5634 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lim SB, Tan SJ, Lim WT & Lim CT An extracellular matrix-related prognostic and predictive indicator for early-stage non-small cell lung cancer. Nat. Commun. 8, 1734 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Brambilla E et al. Prognostic effect of tumor lymphocytic infiltration in resectable non-small-cell lung cancer. J. Clin. Oncol. 34, 1223–1230 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Reynders K & De Ruysscher D Tumor infiltrating lymphocytes in lung cancer: a new prognostic parameter. J. ThoracDis. 8, E833–E835 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Shimizu K et al. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J. Thorac Oncol. 5, 585–590 (2010). [DOI] [PubMed] [Google Scholar]
- 222.Pagés F et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391, 2128–2139 (2018). [DOI] [PubMed] [Google Scholar]
- 223.Donnem T et al. Strategies for clinical implementation of TNM-Immunoscore in resected nonsmall-cell lung cancer. Ann. Oncol. 27, 225–232 (2016).References 222 and 223 discuss the implementation of the immunoscore for predicting risk in colon cancer and strategies to apply a similar immunoscore in the clinic for lung cancer, respectively.
- 224.Paulsen EE et al. Assessing PDL-1 and PD-1 in non-small cell lung cancer: a novel immunoscore approach. Clin. Lung Cancer 18, 220–233 (2017). [DOI] [PubMed] [Google Scholar]
- 225.Remark R et al. The non-small cell lung cancer immune contexture. A major determinant of tumor characteristics and patient outcome. Am. J. Respir. Crit. Care Med. 191, 377–390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Rehman JA et al. Quantitative and pathologist-read comparison of the heterogeneity of programmed death-ligand 1 (PD-L1) expression in non-small cell lung cancer. Mod. Pathol. 30, 340–349 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Mansfield AS et al. Temporal and spatial discordance of programmed cell death-ligand 1 expression and lymphocyte tumor infiltration between paired primary lesions and brain metastases in lung cancer. Ann. Oncol. 27, 1953–1958 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hirsch FR et al. PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the blueprint PD-L1 IHC assay comparison project. J. Thorac Oncol. 12, 208–222 (2017). [DOI] [PubMed] [Google Scholar]
- 229.Ratcliffe MJ et al. Agreement between programmed cell death ligand-1 diagnostic assays across multiple protein expression cutoffs in non-small cell lung cancer. Clin. Cancer Res. 23, 3585–3591 (2017). [DOI] [PubMed] [Google Scholar]
- 230.Govindan R et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 150, 1121–1134 (2012).This study uses genomic analysis of lung cancer to show that the average mutation frequency is more than tenfold higher in smokers than in non-smokers.
- 231.Rizvi NA et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).This study shows that higher TMB in patients with NSCLC is associated with improved objective response rates, durable clinical benefit and PFS.
- 232.Carbone DP et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N. Engl. J. Med. 376, 2415–2426 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hellmann MD et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 33, 853–861 (2018).This study shows that TMB is predictive of clinical efficacy independent of PDL1 expression.
- 234.Messmer MN, Netherby CS, Banik D & Abrams SI Tumor-induced myeloid dysfunction and its implications for cancer immunotherapy. Cancer Immunol. Immunother. 64, 1–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Jackute J et al. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. 19, 3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Dieu-Nosjean MC et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J. Clin. Oncol. 26, 4410–4417 (2008). [DOI] [PubMed] [Google Scholar]
- 237.Goc J et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8 + T cells. Cancer Res. 74, 705–715 (2014). [DOI] [PubMed] [Google Scholar]
- 238.Fucikova J et al. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res. 76, 1746–1756 (2016). [DOI] [PubMed] [Google Scholar]
- 239.Stoll G et al. Calreticulin expression: Interaction with the immune infiltrate and impact on survival in patients with ovarian and non-small cell lung cancer. Oncoimmunology 5, e1177692 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ghajar CM et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 15, 807–817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Gao H et al. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 150, 764–779 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Gao H et al. Multi-organ site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell 166, 47–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Barkan D et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 68, 6241–6250 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Barkan D et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 70, 5706–5716 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.De Cock JM et al. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res. 76, 6778–6784 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Villano JL et al. Incidence of brain metastasis at initial presentation of lung cancer. Neuro Oncol. 17, 122–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Ali A, Goffin JR, Arnold A & Ellis PM Survival of patients with non-small-cell lung cancer after a diagnosis of brain metastases. Curr. Oncol. 20, e300–e306 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Seike T et al. Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin. Exp. Metastasis 28, 13–25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Wang L et al. Astrocytes directly influence tumor cell invasion and metastasis in vivo. PLOS ONE 8, e80933 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Li B et al. Elevated PLGF contributes to small-cell lung cancer brain metastasis. Oncogene 32, 2952–2962 (2013). [DOI] [PubMed] [Google Scholar]
- 251.Valiente M et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 156, 1002–1016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Nguyen DX et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 138, 51–62 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Tang CH, Tan TW, Fu WM & Yang RS Involvement of matrix metalloproteinase-9 in stromal cell-derived factor-1/CXCR4 pathway of lung cancer metastasis. Carcinogenesis 29, 35–43 (2008). [DOI] [PubMed] [Google Scholar]
- 254.Müller P et al. Metastatic spread in patients with non-small cell lung cancer is associated with a reduced density of tumor-infiltrating T cells. Cancer Immunol. Immunother. 65, 1–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Reticker-Flynn NE & Bhatia SN Aberrant glycosylation promotes lung cancer metastasis through adhesion to galectins in the metastatic niche. CancerDiscov. 5, 168–181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hogan BL et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15, 123–138 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Stevens T et al. Lung vascular cell heterogeneity: endothelium, smooth muscle, and fibroblasts. Proc. Am. Thorac Soc. 5, 783–791 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Ding BS et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 147, 539–553 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lee JH et al. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 156, 440–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Morales-Nebreda L, Misharin AV, Perlman H & Budinger GR The heterogeneity of lung macrophages in the susceptibility to disease. Eur. Respir. Rev. 24, 505–509 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Takamura S Niches for the long-term maintenance of tissue-resident memory T cells. Front. Immunol. 9, 1214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Chen N et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 66, 1175–1187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Coelho MA et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity 47, 1083–1099 (2017).References 262 and 263 provide mechanistic insights into how activated KRAS upregulates PDL1.
- 264.Kortlever RM et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 171, 1301–1315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Azuma K et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 25, 1935–1940 (2014). [DOI] [PubMed] [Google Scholar]
- 266.Akbay EA et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 3, 135520161363 (2013).References 265 and 266 show that mutant EGFR induces PDL1 expression to mediate immune escape in human and mouse lung tumours, respectively.
- 267.Ota K et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signaling pathways in non-small cell lung cancer. Clin. Cancer Res. 21, 4014–4021 (2015).References 262 and 267 show that mutant KRAS and the EML-ALK4 oncoprotein induce PDL1 expression, respectively.
- 268.Ihara S et al. Inhibitory roles of signal transducer and activator of transcription 3 in antitumor immunity during carcinogen-induced lung tumorigenesis. Cancer Res. 72, 2990–2999 (2012). [DOI] [PubMed] [Google Scholar]
- 269.D’Incecco A et al. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br. J. Cancer 112, 95–102 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Lastwika KJ et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 76, 227–238 (2016).This study provides mechanistic insights into how activation of the AKT-mTOR pathway upregulates PDL1.
- 271.Best SA et al. Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. CellMetab. 27, 935–943 (2018).References 264 and 271 demonstrate the contribution of oncogene cooperativity in mediating immune evasion in lung cancer.
- 272.Kaplan RN et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 (2005).This is the first study to demonstrate that tumour-induced bone marrow-derived progenitor cells generate a pre-metastatic niche that supports colonization of metastatic cells.
- 273.Hiratsuka S et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 10, 1349–1355 (2008). [DOI] [PubMed] [Google Scholar]
- 274.Caetano MS et al. IL6 blockade reprograms the lung tumor microenvironment to limit the development and progression of K-ras-mutant lung cancer. Cancer Res. 76, 3189–3199 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Abulaiti A et al. Interaction between non-small-cell lung cancer cells and fibroblasts via enhancement of TGF-β signaling by IL-6. Lung Cancer 82, 204–213 (2013). [DOI] [PubMed] [Google Scholar]
- 276.Gao SP et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Invest. 117, 3846–3856 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Yao Z et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl Acad. Sci. USA 107, 15535–15540 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Serresi M et al. Polycomb repressive complex 2 is a barrier to KRAS-driven inflammation and epithelialmesenchymal transition in non-small-cell lung cancer. Cancer Cell 29, 17–31 (2016). [DOI] [PubMed] [Google Scholar]
- 279.Goldstraw P et al. Non-small-cell lung cancer. Lancet 378, 1727–1740 (2011). [DOI] [PubMed] [Google Scholar]
- 280.Hasegawa Y et al. Transforming growth factor-beta1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer 91, 964–971 (2001). [PubMed] [Google Scholar]
- 281.Wang W et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin. Cancer Res. 15, 6630–6638 (2009). [DOI] [PubMed] [Google Scholar]
- 282.Owusu BY et al. Targeting the tumor-promoting microenvironment in MET-amplified NSCLC cells with a novel inhibitor of pro-HGF activation. Oncotarget 8, 63014–63025 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Yano S et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J. Thorac Oncol. 6, 2011–2017 (2011). [DOI] [PubMed] [Google Scholar]
- 284.Xia Y et al. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat. Cell Biol. 14, 257–265 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Butler JM, Kobayashi H & Rafii S Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 10, 138–146 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Labelle M, Begum S & Hynes RO Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20, 576–590 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Hoshino A et al. Tumour exosome integrins determine organotropic metastasis. Nature 527, 329–335 (2015).This study demonstrates organotropic metastasis by showing that tumour exosomal α6β4 and α6β1 integrins interact with specific resident cells in the lung to achieve lung tropism.
- 288.Zhang Y, Huang S, Gong D, Qin Y & Shen Q Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell. Mol. Immunol. 7, 389–395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Salvador F et al. Lysyl oxidase-like protein LOXL2 promotes lung metastasis of breast cancer. Cancer Res. 77, 5846–5859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Fong MY et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat. Cell Biol. 17, 183–194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.