

Abstract
Magnetic microbeads exhibit rapid separation characteristics and are widely employed for biomolecule and cell isolations in research laboratories, clinical diagnostics assays and cell therapy manufacturing. However, micrometer particle diameters compromise biomarker recognition, which leads to long incubation times and significant reagent demands. Here, a stimuli-responsive binary reagent system is presented that combines the nanoscale benefits of efficient biomarker recognition and the microscale benefits of rapid magnetic separation. This system comprises magnetic nanoparticles and polymer-antibody (Ab) conjugates that transition from hydrophilic nanoscale reagents to microscale aggregates in response to temperature stimuli. The binary reagent system was benchmarked against Ab-labeled Dynabeads® in terms of biomarker isolation kinetics, assay speed and reagent needs. Surface plasmon resonance (SPR) measurements showed that polymer conjugation did not significantly alter the Ab’s binding affinity or kinetics. ELISA analysis showed that the unconjugated Ab, polymer-Ab conjugates and Ab-labeled Dynabeads exhibited similar equilibrium dissociation constants (Kd), ~ 2 nM. However, the binary reagent system isolated HIV p24 antigen from spiked serum specimens (150 pg/mL) much more quickly than Dynabeads, which resulted in shorter binding times by 10’s of minutes, or about 30-50% shorter overall assay times. The binary reagent system showed improved performance because the Ab molecules were not conjugated to large, solid microparticle surfaces. This stimuli-responsive binary reagent system illustrates the potential advantages of nanoscale reagents in molecule and cell isolations for both research and clinical applications.
Keywords: Stimuli-responsive, polymer-protein conjugates, immunoassays, SPR, ELISA
Graphical Abstract
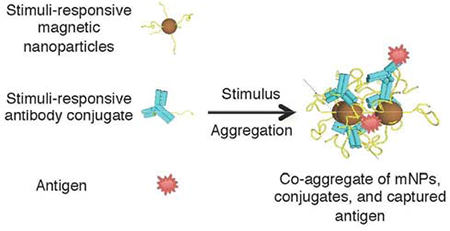
INTRODUCTION
Bioseparations capture molecules of interest for identification and quantification, or to clean up samples before downstream processing. Magnetic microbeads are employed as the solid supports in separations of cells[1],[2],[3], proteins[4],[5], oligonucleotides[6] and pathogens[7] because they provide accessible surfaces for immobilizing affinity reagents, and they respond sharply to a modest magnetic field for efficient separations[8]. These magnetic microbeads are standard reagents in clinical immunoanalyzer platforms such as the Beckman Coulter Dxl and Access® systems and the Abbott Laboratories ARCHITECT system. Magnetic microbeads provide separation advantages due to their large diameters (≥ 1 μm), but their use also imposes a paradox. The micrometer particle size imparts poor diffusion characteristics, which leads to long incubation times for antigen binding. Also, there are inefficiencies in Ab loading onto microparticle surfaces that lead to significant losses of Ab affinity. Thus, the properties of magnetic microbeads limit assay speeds, binding efficiencies and detection sensitivity. On the other hand, magnetic nanoparticles (< 50 nm) have many potential advantages related to their diffusive properties and binding capacity. However, most magnetic nanoparticles (mNPs) suffer from low magnetophoretic mobility, limiting their utility for separations using modest magnetic fields.
We previously reported stimuli-responsive, polymer-coated mNPs that maintain the diffusive advantages of small nanoparticles while introducing switchable transitions to larger, aggregated mNPs with excellent magnetophoretic mobilities[9],[10]. Here, a binary reagent system is presented that comprises two stimuli-responsive reagents: mNPs and polymer-antibody (Ab) conjugates (Figure 1). Stimuli-responsive polymers, such as the temperature-responsive polymer poly(N-isopropylacrylamide), or pNIPAM, respond sharply and reversibly to physical or chemical stimuli by changing their conformation and physicochemical properties (e.g from a hydrophilic state to a more hydrophobic state). This polymer displays lower critical solution temperature (LCST) behavior at solution temperatures of about 32°C[11], and this is salt-controllable[12]. Stimuli-responsive polymers like pNIPAM have been grafted onto surfaces[13],[14], membranes[15],[16] and gold nanoparticles[17] to capture, enrich and detect target biomolecules. They also have been used to synthesize micelles[18], hydrogels[19],[20], drug conjugates[21] and protein conjugates[22] for potential use as drug delivery devices. By decoupling the recognition component (polymer-Ab conjugate) from the separation component (mNPs), the diffusive and binding advantages of small nanoscale reagents are exploited. This system comes with the additional advantage of controllable transitions from a stable colloidal suspension to micron-sized aggregates with high magnetophoretic mobility.
Figure 1.
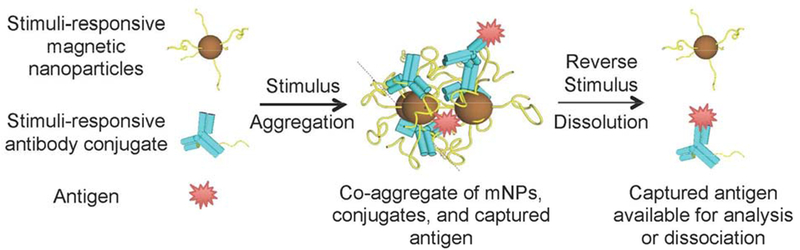
The stimuli-responsive binary reagent system, comprising stimuli-responsive magnetic nanoparticles (mNPs) and polymer-Ab conjugates. After application of an appropriate stimulus, the mNPs and conjugates co-aggregate with captured antigen to form magnetically-separable species. Reversing the stimulus provides the captured antigen free of magnetic particles.
There is a need for more rapid and higher sensitivity p24 immunoassays. The HIV core protein p24 is a biomarker used to diagnose HIV infection[23]. Higher sensitivity p24 diagnostic tests would allow earlier detection of HIV to narrow the window period[24] and minimize the possibility of false negative readings. Faster p24 diagnostic tests would improve the throughput of clinical immunoanalyzers and have additional benefits for point-of-care assays in low resource settings[25]. Many laboratories have innovated technologies that could improve immunoassay performance, ranging from microfluidic platforms[5],[7] to two-phase systems[26]. However, conventional magnetic microbeads are used in most clinical immunoanalyzers. For example, magnetic microbeads (e.g., Dynabeads) are used to capture p24 in 4th generation HIV immunoassays such as the Abbott ARCHITECT HIV Ag/Ab Combo Test[4]. We hypothesized that the binding and capture of p24 would be faster with the nanoscale binary reagent system than with microscale magnetic beads. So, p24 was used as a model biomarker to benchmark the binding affinities and kinetics of the stimuli-responsive binary reagent system against Dynabeads.
EXPERIMENTAL SECTION
Materials and Reagents.
Information for materials and reagents is included in Supporting Information.
PNIPAM Synthesis and Modification.
Reversible addition-fragmentation chain transfer (RAFT) polymerization produced two classes of pNIPAM: one with a dodecyl (C12) tail and one with an ethyl (C2) tail. To produce pNIPAM with a C12 tail (C12-pNIPAM), NIP AM (9.38 g, 83 mmol), DMP (816 mg, 1.69 mmol), ACVA (47.4 mg, 0.160 mmol) and dioxane (23.5 g) were sealed in a round bottom flask, purged with N2 for 20 minutes and held at 70°C for 2 hours. To produce pNIPAM with a C2 tail (C2-PNIPAM), NIPAM (4.97 g, 43.9 mmol), ECT (32.6 mg, 0.0120 mmol), ACVA (3.5 mg, 0.001 mmol) and dioxane (12.5 g) were sealed in a round bottom flask, purged with N2 for 20 minutes and held at 70°C for 4 hours. The polymers were collected by precipitation into 90% pentane/10% ether followed by vacuum drying. To produce an amine-reactive NHS ester (pNIPAM-NHS), C2-PNIPAM (2.5 g) and NHS (0.016 g) were dissolved in 10 mL DCM. DCC (0.029 g) was added to this solution, and the reaction was mixed at 20°C for 24 hours. The polymer was collected by precipitation into pentane and dried in vacuo. The polymer characterizations using SEC and NMR are included in the Supporting Information.
Polymer-Antibody Conjugate Preparation.
Polymer-Ab conjugates were prepared at a polymer:antibody molar ratio of 100:1. Anti-HIV-1 p24 monoclonal antibody was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAED, NEH:HIV-1 p24 Hybridoma (183-H12-5C) from Dr. Bruce Chesebro[27]. The Ab was diluted into 25 mM Na2CO3/NaHCO3, pH 9.5 and cooled. The pNIPAM-NHS was injected into the Ab solution and mixed for 18 hours at 4°C. Free polymer was removed by ultrafiltration with 100 kDa MWCO centrifugal filter units (Millipore) or by aqueous SEC (Supporting Information). Characterization and functional assays of the polymer-Ab conjugates are given in the Supporting Information.
Magnetic Nanoparticle (mNP) Synthesis and Purification.
Magnetic nanoparticles coated with pNIPAM were synthesized with modifications of previous protocols[9]. C12-pNIPAM (0.9 g, 0.138 mmol) was solubilized in 50 mL tetraglyme at 100°C for 30 minutes. Next, Fe(CO)5 (200 μL, 1.48 mmol) was injected into the polymer solution and the temperature was increased to 190°C. The reaction was held at 190°C for 6 hours. The polymer-coated mNPs were collected by precipitation into pentane and then dried in vacuo. Then, the mNPs were subjected to ultrafiltration in DH2O with a 100 kDa MWCO membrane and lyophilized.
Surface Plasmon Resonance (SPR) Biosensor Experiments.
The binding affinities of the Ab and polymer-Ab conjugates were measured by SPR. Running conditions and instrument details are given in the Supporting Information. Seven p24 (analyte) samples, from 50.0 nM to 68.6 pM, were prepared in HBS-EP, and each was run in a mixed order, in triplicate. This approach assessed the reproducibility of binding and managed potential systematic bias to the order of injection. Multiple blank (buffer) injections were used to assess and subtract system artifacts. The association and dissociation phases for all analyte concentrations were monitored for 240 seconds (s) and 600 s, respectively, at a flow rate of 75 μL/min. Longer dissociation phase experiment of 5400 s also was performed with the 16.7 nM p24 sample at a flow rate of 75 μL/min. Between experiments, the surfaces were regenerated with 10 mM glycine, pH 1.5 for 30 s, at a flow rate of 50 μL/min. SPR analyses are given in the Supporting Information.
ELISA-Based Binding of anti-p24, polymer-Ab conjugates and Dynabeads-anti-p24.
A competitive ELISA (developed in-house) measured the apparent binding affinities of unconjugated Ab, polymer-Ab conjugates and Dynabeads-anti-p24 for p24 antigen. The monoclonal anti-p24 IgG used in this ELISA was from the NIH AIDS Reagent Program[27], and the ELISA protocol has been published elsewhere[28]. ELISA analyses via BIOEQS to determine the Kd values for unconjugated Ab, polymer-Ab conjugates and Dynabeads-anti-p24 are given in the Supporting Information.
HIV-1 p24 – Antibody Reaction Kinetics.
The p24 binding kinetics of the binary reagent system and the Dynabeads-anti-p24 were compared. Samples in 10 mM PBS with final concentrations of 150 pg/mL p24, 2 mg/mL mNPs, 5% human serum, 350 mM NaCl and 1.938 μg/mL polymer-Ab conjugate were mixed for 0, 1, 2.5, 5, 10 or 15 minutes at 20°C. After incubating, the mNPs and conjugates were co-aggregated by warming at 37°C for 2 minutes and isolated with a magnetic field for 2 minutes at 37°C. The supemate was analyzed for free p24 via ELISA[28]. For the Dynabeads-anti-p24, samples in 10 mM PBS (350 mM NaCl) with final concentrations of 150 pg/mL p24, 5% human serum and 242 μg/mL Dynabeads-anti-p24 (1.938 μg/mL Ab equivalent) were prepared. The incubation, heating and magnetic separation steps were identical to that of the binary reagent system experiments. The anti-p24 IgG:p24 antibody:antigen molar ratio for both the binary reagent system and the Dynabeads-anti-p24 was 2000:1, creating a 4000:1 ratio of antibody binding sites:p24. This ratio was chosen from ELISA-based binding experiments above. Simulations of the reaction kinetics, as binding progress curves, were also performed, as described in the Supporting Information.
RESULTS AND DISCUSSION
The binary reagent assay system.
The binary reagent system comprises stimuli-responsive mNPs and polymer-Ab conjugates. The C12-pNIPAM was synthesized via RAFT polymerization[29]. The physical and chemical properties of C12-pNIPAM were shown in Supporting Figure 1. The C12-pNIPAM was then used to synthesize stimuli-responsive mNPs, which comprised iron oxide cores coated with pNIPAM[9–10]. All experiments were performed with two different batches of mNPs (20.0 ± 0.45 nm diameter, measured by dynamic light scattering). The thermoresponsive character of the mNPs was shown by the LCST (n=2) of 36.4 ± 0.085°C in PBS (Figure 2A). The mNP LCST was ~6°C greater than literature values for pNIPAM[11]. The mNPs zeta potentials were ~0 to −10 mV (data not shown), so we hypothesize that the higher LCST values for the mNPs was due to electrostatic repulsion of the negatively charged particle surfaces. The pNIPAM polymer was also salt-responsive[12]. The addition of 200 mM NaCl to mNPs in PBS decreased the LCST (n=2) from 36.4 ± 0.085°C to 32.3 ± 0.233°C (Figure 2A). Therefore, all experiments were performed in solutions with 350 mM final NaCl concentration (150 mM NaCl in PBS, 200 mM NaCl additional), in which a temperature stimulus of 37°C aggregated mNPs for separation. In fact, after warming mNPs to 37°C for 2 minutes, 95% of the mNPs were magnetically separated within 30 seconds (Figure 2B). However, the mNPs are stable colloids, so they can only be separated after they are aggregated by a heat/salt stimulus. For example, in non-aggregated state (Figure 2D, 0 seconds), <10% of the mNPs are separated. The rapid aggregation and separation kinetics of mNPs suggested that these reagents could be used as platforms to quickly isolate target molecules from biological samples.
Figure 2.
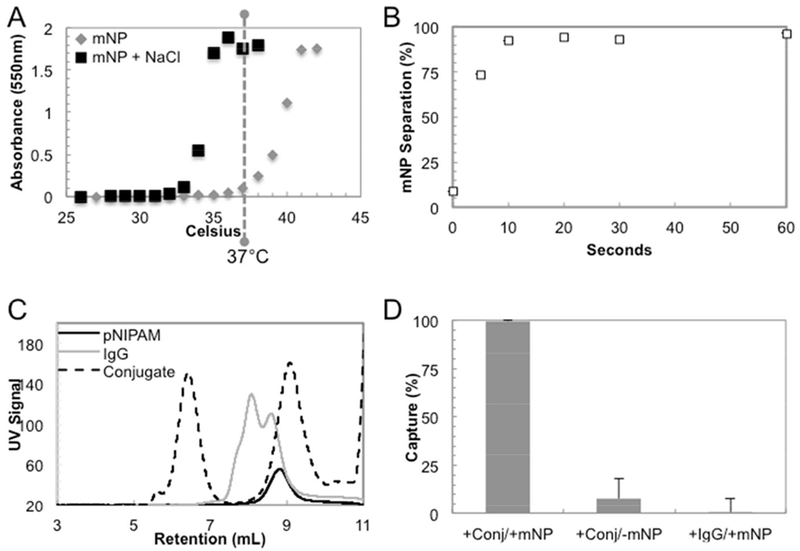
Functional characteristics of the binary reagent system. The LCST of mNPs was 36-37°C and salt-controllable (A). The mNPs can be separated with ~97% efficiency in less than 1 minute (n=2) after a temperature stimulus of 37°C (B). Standard deviations were <1.5% at each data point. SEC chromatograms shows high molecular weight polymer-Ab conjugates resolved from unreacted polymer and Ab. (C) Polymer-Ab conjugates (conj) were only captured in the presence of mNPs, and there is no non-specific capture of the native Ab (IgG) by mNPs (D).
The second component of the binary reagent system is the polymer-Ab conjugates. RAFT was used to synthesize C2-pNIPAM with a terminal carboxylate. The physical and chemical properties of C2-pNIPAM were shown in Supporting Figure 1. After polymerization, the carboxylate was converted to an N-hydroxysuccinimide (NHS) ester with carbodiimide chemistry. The NHS modified pNIPAM was used for covalent conjugation of anti-p24 Ab via amine groups. This ‘grafting-to’ approach has been used to produce many types of thermoresponsive polymer-protein conjugates for affinity precipitations[14],[30],[31],[32]. Aqueous SEC resolved the conjugates at 6.5 minutes, unmodified Ab at 8 minutes and unreacted pNIPAM at 9 minutes (Figure 2C). The polymer-Ab conjugates eluted first because one or more pNIPAM chains were covalently conjugated to the Ab, resulting in high molecular weight conjugates. The polymer-Ab conjugates were purified from both unmodified IgG and unreacted pNIPAM by collecting the fraction from 5.5-7 minutes. Biophysical and binding characterization of the polymer-Ab conjugate was measured by SPR and described below.
The binary reagent system combines the mNPs and polymer-Ab conjugates (Figure 1). The capture efficiency of polymer-Ab conjugate by mNPs was determined by quantifying the remaining (uncaptured) conjugates in supemates after aggregation and magnetic separation (Supporting Information). Approximately 99% of polymer-Ab conjugates were captured by mNPs after a 2-minute, 37°C stimulus. The polymer-Ab conjugates were not captured in the absence of mNPs, and there was little (<1%) non-specific capture of unmodified Ab by the mNPs (Figure 2D). Since target molecules (e.g., p24) are bound to polymer-Ab conjugates, the p24 separation is specific. Therefore, pNIPAM was required on both mNPs and the Ab for rapid and effective co-aggregation and magnetic capture of the polymer-Ab conjugate. Thus, the two components of the binary reagent system have similar stimuli-responsive behavior.
Other work[33],[34],[35] described stimuli-responsive, streptavidin-coated mNPs coated with biotinylated Abs to capture biomolecules. In those studies, the recognition and separation reagents were coupled, which limited the amount of capture Ab and target antigen that was immobilized on mNP surfaces. Our binary reagent system decoupled biomarker recognition (by polymer-Ab conjugates) from the separation reagent (the mNPs) until the aggregation stimulus was applied. Therefore, biomarker recognition efficiency can be maximized by increasing the concentration of polymer-Ab conjugates independently of the mNPs. Nagaoka and colleagues reported a related binary reagent system that quantified thyroid stimulating hormone with stimuli-responsive mNPs and polymer-Ab conjugates[36]. Their approach only allowed detection of target molecules. By contrast, our binary reagent system first isolates target molecules after co-aggregation of mNPs and polymer-Ab conjugates. Then, by reversing the stimulus (e.g., cooling the solution), the aggregates re-solubilize into mNPs and polymer-Ab conjugates with bound target molecules. This re-solubilization allows direct detection of the target biomolecules or their release using standard dissociation protocols, depending on the downstream application.
Binding affinities of anti-p24 Ab and polymer-Ab conjugates by SPR.
SPR biosensor analysis[37] measured the apparent binding affinities (Kd), kinetics (kon and koff) and overall binding capacity for the unconjugated Ab and the polymer-Ab conjugates (Supporting Information). These analyses allowed assessment of the impact of polymer conjugation on the Ab binding properties. The association, dissociation and long dissociation kinetic “sensorgrams” were overlaid (Supporting Figure 2) with fitted curves[38] for the unconjugated Ab and polymer-Ab conjugates. Both the association and dissociation phases fit the 1:1 binding model, which indicated that each Ab sample was homogeneous in its p24 binding properties. This was particularly noteworthy for the polymer-Ab conjugates. These polymer-Ab conjugates were heterogeneous in the number of polymers bound to each Ab, but this sample was homogeneous in binding properties. The Kd, kon and koff for the Ab and polymer-Ab conjugate were derived from the fits (Table 1). The three-fold difference in Kd between the Ab (0.165 nM) and polymer-Ab conjugate (0.501 nM) was largely due to a faster koff for the p24:polymer-Ab conjugate complex. In applications where the target needs to be removed from the Ab for downstream processing, a higher koff could be advantageous. However, it was a concern for binding assays, so the Rvalues were considered as dissociation half-life constants (τ1/2). Both the Ab and polymer-Ab conjugates showed τ1/2 values greater than one hour (Table 1). This timeframe was significantly longer than the minutes-long incubations in typical capture assays, so the practical signficance of the koff difference was minor. In this study, the Ab:p24 and polymer-Ab:p24 complexes remained largely intact throughout the time-critical steps in the binding assays.
Table 1.
Biophysical and kinetic properties of unconjugated anti-p24 antibody (Ab), stimuli-responsive polymer-Ab conjugates and antibody-labeled Dynabeads® measured by SPR, ELISA and binding progress analyses.
| Method | Sample | Kd (nM) | kon (M−1s−1) | koff (s−1) | τ1/2 (h) | Rmax (R.U.) Fitted | Rmax (R.U.) Theoretical | |
|---|---|---|---|---|---|---|---|---|
| SPR | Ab | 0.165 | 2.42×l05 | 4.00×10−5 | 4.8 | 11.6 | 24.0 | 48.3 % |
| Polymer-Ab | 0.501 | 2.06×l05 | 10.3×l0−5 | 1.9 | 7.6 | 21.7 | 35.1% | |
| ELISA | Ab | 1.25 | - | - | - | - | - | - |
| Polymer-Ab | 2.27 | - | - | - | - | - | - | |
| Dynabeads®-anti-p24 | 2.45 | - | - | - | - | - | - | |
| Binding Progress | Polymer-Ab | 1.7 | 3.6×105 | 61.4×l0−5 | 0.32 | - | - | - |
| Dynabeads®-anti-p24 | 2.4 | 0.37×l05 | 8.8×l0−5 | 2.2 | - | - | - |
In SPR experiments, artifacts and biases can complicate head-to-head comparisons of different Abs or forms of Abs[39]. The goodness of fit to the 1:1 binding model for both Ab forms suggested that the Abs were not significantly nor selectively impacted by the potentially heterogeneous chip surface[40]. Another possible bias could occur if a heterogeneous sample (i.e., polymer-Ab conjugates) appeared homogeneous in binding properties because only a subset of the molecules were captured or active on the SPR chip. We used a goat anti-mouse Fc “capture” Ab to hold the unconjugated Abs or polymer-Ab conjugates on the SPR chip, which provided a more reproducible and cleaner analysis of the two Ab forms than directly attaching the Ab forms to the SPR chip (data not shown)[41]. Since the rates of capture to the chip surface for the unconjugated Ab and polymer-Ab conjugates were similar (data not shown), it was unlikely that the capture Ab was unable to bind or retain subsets of the heterogeneous polymer-Ab conjugates. This result also suggested that polymer conjugation did not negatively impact the Fc interaction with the capture Ab. By comparing the fitted and theoretical Rmax values, the percentage of active, competent Abs captured on the chip surface was determined. The results (Table 1) suggested that the polymer-Ab conjugates and unconjugated Abs had similar activities, with values of 35.1% and 48.3%, respectively. The ~13% difference in activities may be due to a loss of activity of some Abs due to polymer conjugation, a difference in extinction coefficients or the poly-carboxylated SPR chip surface[40]. Regardless, the SPR results suggested that conjugating multiple pNIPAM chains to the Ab had nominal impact on the binding affinity, kinetics and capacity of the Ab.
Binding affinities of anti-p24 Ab, polymer-Ab conjugates and Dynabeads-anti-p24.
The binding affinity and kinetic properties of the binary reagent system were benchmarked against antibody-labeled Dynabeads through an ELISA (Supporting Information). Here, the Ab forms were the assay titrants. Since the bivalent Ab molecule has two independent but identical binding sites, the typical multi-parameter dose-response algorithm was not appropriate to account for changing distributions of unbound, singly-bound, and doubly-bound p24:Ab species during the titration[42],[43],[44]. So, we applied the binding analysis program BIOEQS to fit the raw data and recover a Kd value for the intrinsic binding interaction (Figure 3 and Supporting Information). Compared to SPR results[30], ELISA showed that the unconjugated Ab and polymer-Ab conjugates had ~10x and ~5x apparent lower affinity (Kd) for p24, respectively (Table 1). These differences were not unexpected. Comparable affinity differences have been recovered for identical molecules with different brands of SPR surfaces[40],[41],[46]. So, differences in the SPR chip and the ELISA plate surfaces could contribute systematic and random uncertainties to the recovered Kd values. SPR and ELISA also have different uncertainties, and their analytical rigor has been debated[47],[48],[49],[50]. Additionally, activity differences in the titrant stocks (p24 vs. Ab) likely contributed systematic uncertainty to the recovered Kd values. Nonetheless, the apparent affinities recovered from SPR and ELISAs were similar (Table 1). Therefore, the intrinsic binding properties for the unconjugated Ab (Kd = 1.25 nM), polymer-Ab conjugates (Kd = 2.27 nM) and Dynabeads®-anti-p24 (Kd = 2.45 nM) were only slightly different based on the molecular compositions of these Ab forms.
Figure 3.
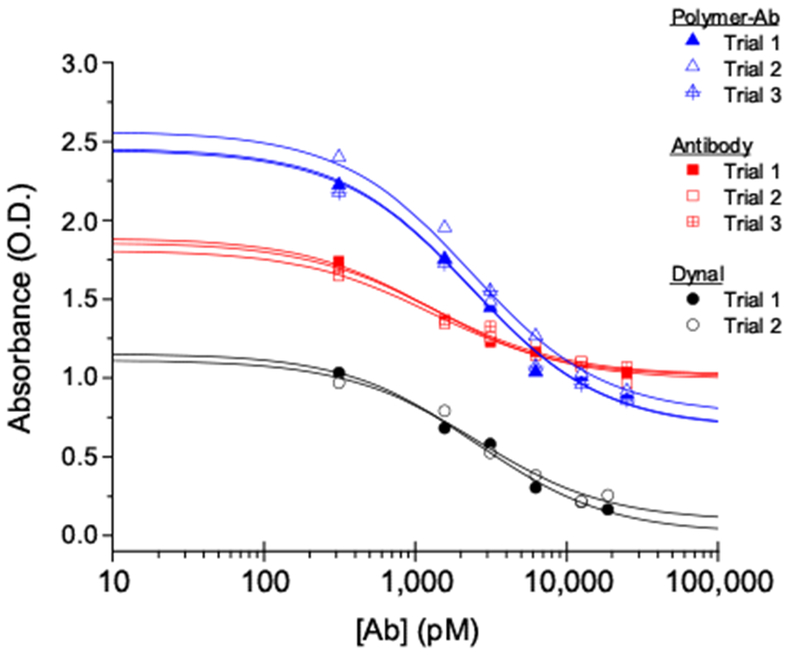
Binding isotherms for replicate competitive ELISA trials for unconjugated Ab (squares), polymer-Ab conjugates (triangles) and Dynabeads®-anti-p24, or Dynal (circles). Lines through the data correspond to the results from BIOEQS analyses.
In developing the binary reagent system, we hypothesized that decoupling the Ab from magnetic particle surfaces would elicit numerous advantages compared to other particle systems with covalently-coupled Ab (e.g., Dynabeads, Miltenyi Biotec MACS® MicroBeads). For example, polymer-Ab conjugates should be able to freely diffuse and bind target molecules without hindrance from a bulky particle, leading to rapid antigen capture and shorter assay times. So, it was surprising to see the similar ELISA-derived binding affinities (Kd) for the binary reagent and Dynabeads systems, which suggested that the two system had similar capture and separation performance. However, reactions with similar affinity constants can be composed of very different kinetic properties (kon and koff). Of the two rate constants, the second-order kon primarily determines the time for binding reactions to approach and reach their equilibrium states. The binding rate constants for the Dynabeads-anti-p24 could not be assessed by SPR for direct comparison to the binary reagent system, so we obtained apparent binding kinetic parameters of both systems by evaluating the reaction progress curves[51].
Binding kinetics of polymer-Ab conjugates and Dynabeads-anti-p24.
The apparent binding kinetic parameters of the polymer-Ab conjugates and Dynabeads-anti-p24 were measured with identical Ab concentrations. These experiments served to benchmark the performance of the binary reagent and Dynabeads-anti-p24. The binding progress curves (Figure 4) were transformed into the percent of p24 captured over time, with corresponding fits in the same units (Supporting Information). Under these conditions, the binary reagent captured ~90% of the p24 (the reaction equilibrium state) in approximately 8 minutes. The Dynabeads-anti-p24 required nearly 50 minutes to reach equilibrium (~90% bound), so the binary reagent saves 10’s of minutes in this p24 assay, or about 30-50% of overall assay time. The apparent kon and koff were recovered from the progress curve analyses (Table 1 and Supporting Information). The subsequently derived Kd values for the polymer-Ab conjugates (1.7 nM) and Dynabeads-anti-p24 (2.4 nM) were comparable to the values recovered from the ELISA (Figure 4 and Table 1). It is interesting that the progress curve rate differences between the binary reagent and Dynabeads systems was due to a nearly lOx lower association rate constant (kon) for the Dynabeads-anti-p24. The significant difference in the progress curve rates between the binary reagent and Dynabeads-anti-p24 likely reflected some combination of differences in their diffusion properties, hydrodynamic factors, sizes and integrity of the anti-p24 Ab.
Figure 4.
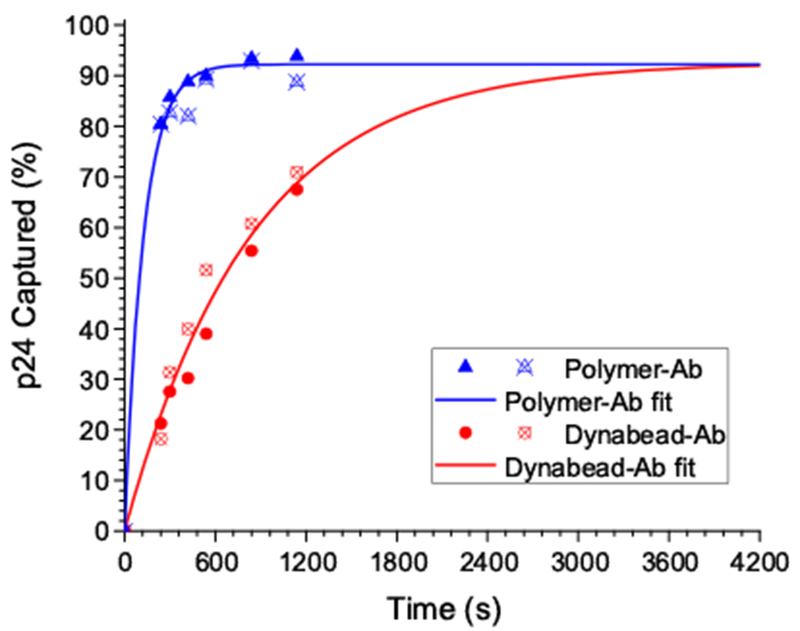
ELISA-based binding progress curve as the percent of p24 captured over time and overlaid fits (to recover kon and koff values). Replicate data points were included for the Dynabead-anti-p24(circles) and polymer-Ab conjugates (triangles).
Assuming that the microbeads (diameter = 2.8 μm), polymer-Ab conjugates (diameter ~ 20 nm) and mNPs (diameter ~ 20 nm) behaved like spherical particles in water at 40°C, the diffusion coefficient of microbeads (1.25× 10−9 cm2/s) was 100x lower than the binary reagent system (1.4×10−7 cm2/s). Additionally, at any Ab concentration, there were many more “particles” of polymer-Ab conjugate diffusing in solution than particles of Dynabeads-anti-p24. This was because a single Dynabead contained numerous Ab molecules confined to its surface, which limited the Ab’s effective “particle” concentration. It consequently limited the diffusional degrees of freedom and the mass action-based molecular collisional rate. One implication of having Abs sequestered into fewer numbers of slower-diffusing particles was that the smaller polymer-Ab conjugates encountered p24 molecules more frequently. This resulted in more rapid binding than the larger Dynabeads-anti-p24 particles. The dissociation process also was impacted by size-dependent diffusion differences, and the binary reagent system showed a slightly faster p24 dissociation rate than the Dynabeads-anti-p24 system (Table 1). We hypothesized that, after an Ab:p24 dissociation event, the slow diffusing Dynabeads traversed a relatively small volume of solution space away from the dissociated p24 molecule. So, the p24 molecules did not readily mix with the bulk of solution after dissociation. This allowed rapid primary geminate recombination of a significant portion of the p24 to the slowly moving Dynabeads-anti-24 particles before those Dynabeads dispersed appreciably from the dissociated p24. These particles also exhibited very high local Ab concentrations because many Abs were bound to the particle surfaces. This recombination phenomenon manifested as a slower overall koff (i.e. the rate process of p24 dissociating and mixing with the bulk of solution)[52]. As a consequence, the slower association and dissociation rates for the Dynabeads system balanced. This generated an apparent affinity value similar to the binary reagent system even though the two capture systems had vastly different kinetic rates comprising their similar affinity values.
Simulations of binding progress curves (Supporting Figure 3) showed that, regardless of the starting Ab concentration (either IX or 9X), the binary reagent system reached equilibrium significantly faster than the Dynabeads. This also revealed the significantly greater reagent needs for the Dynabeads system to achieve a comparable kinetic performance to the binary reagent system. For example, to reach equilibrium at approximately 8 minutes, about 9X more Dynabeads-Ab were required than binary reagents. These binding reaction simulations also benchmarked parameters for assay performance. The simulations showed that increasing the Ab reagent concentrations by 9X increased mass action effects. This raised the amount of p24 captured at the equilibrium state to 99%, but it also predicted the incubation times necessary to acquire that level of binding. Assuming that there are no deleterious Ab:Ab interactions at high Ab concentrations, assay developers can determine the relative benefits of achieving faster assay times with higher Ab concentrations.
To our knowledge, this was the first direct comparison of antigen binding rates between nanoscale polymer-affinity reagent conjugates and commercial magnetic microbeads. The comparison against Dynabeads was appropriate for several reasons. First, most mNPs (~10 nm diameter) that behave as stable colloids in aqueous environments exhibit low magnetophoretic mobility and slow (≥ hours) magnetic separation with typical magnetic separation devices. Other mNPs with better magnetophoretic mobilities have limited colloidal stability. Also, the binary reagent system has potential benefits for biomolecule separations like immunoprecipitations and in vitro diagnostics, and most of these separations utilize Dynabeads. Although Dynabeads were used as model microbeads for benchmarking purposes, conclusions from this work likely apply to other Ab-labeled particle systems. It was shown that the magnetic microbeads had inherently slower reaction kinetics compared to those of the binary reagent system. The slower kinetic parameters were due to their large size and their sequestration of Ab to the particle surfaces. These diffusion and binding consequences could be general issues with the microbeads, such that no matter the intrinsic binding kinetics of a given Ab, the Ab’s attachment to a bead will decrease the kon for its antigen. Second, polymer conjugation to the Ab had nominal impact on the affinity and binding properties of the Ab. So, the assay kinetics and binding performance of the Ab in the binary reagent system were not impacted significantly, but the effects of polymer conjugation on other Abs should be tested. ELISAs provided a bridge from a solution based assay analysis to more biophysically rigorous SPR assays that provided unique kinetic parameters. SPR and ELISAs may not be appropriate for antibody systems with substantially higher affinities (i.e., Kd values in the mid nM to low pM), but more sensitive analytical and biophysical based solution methods could be considered[49],[52]. Finally, the ability to titrate separately the mNP and polymer-Ab reagents in order to optimize assay speeds and binding efficiencies makes the binary reagent system attractive in terms of cost-effectiveness.
CONCLUSIONS
This report shows that the stimuli-responsive binary reagent system achieves biomolecule separation with significantly faster kinetics and higher efficiency than magnetic microbeads. The binary reagent system decouples the molecular recognition events from the magnetic separation: it enables rapid recognition of target molecules by the polymer-Ab conjugate in the soluble, hydrophilic state and rapid magnetic separation (< 30 seconds) of the captured molecules after co-aggregation of the conjugates with mNPs. It also allows titration of the affinity reagent independent of the mNPs, thus maintaining or even enhancing binding performance while using fewer reagents. Biophysical and ELISA characterization elucidated the advantages of the binary reagent system and potential limitations of larger magnetic microbeads in terms of Ab: antigen binding kinetics. The binary reagent system is a modular platform that is easily customized for different affinity reagents, offering potential advantages in biomarker diagnostics, sample cleanup, bioprocessing and cell isolations.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Anthony Convertine for providing the chain transfer agent ECT. Financial support was provided by NIH GM100558 and CA174581. Additional funding was provided by the Life Sciences Discovery Fund (LSDF) 2008 Innovative Research Projects, 2010 Second Round Commercialization, and 2012 PreCede grant.
Footnotes
Supporting Information
Detailed experimental methods. Polymer characterization. SPR sensorgrams. Simulation data of p24 binding kinetics. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Casavant BP; Mosher R; Warrick JW; Maccoux LJ; Berry SMF; Becker JT; Chen V; Lang JM; McNeel DG; Beebe DJ Methods 2013, 64, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kalos M; Levine BL; Porter DL; Katz S; Grupp SA; Bagg A; June CH Sci Transl Med 2011, 3, 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chen A; Byvank T; Chang WJ; Bharde A; Vieira G; Miller BL; Chalmers JJ; Bashir R; Sooryakumar R Lab Chip 2013, 13, 1172–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chavez P; Wesolowski L; Patel P; Delaney K; Owen SM J. Clin. Virol 2011, 52, S51–S55. [DOI] [PubMed] [Google Scholar]
- (5).Berry SM; Regehr KJ; Casavant BP; Beebe DJ JALA-J. Lab. Autom 2013, 18, 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Adams NM; Bordelon H; Wang KKA; Albert LE; Wright DW; Haselton FR ACS Appl. Mater. Inter 2015, 7, 6062–6069. [DOI] [PubMed] [Google Scholar]
- (7).Hung LY; Huang TB; Tsai YC; Yeh CS; Lei HY; Lee GB Biomed. Microdevices 2013, 15, 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Borlido L; Azevedo AM; Roque ACA; Aires-Barros MR Biotechnol. Adv 2013, 31, 1374–1385. [DOI] [PubMed] [Google Scholar]
- (9).Lai JJ; Hoffman JM; Ebara M; Hoffman AS; Estournes C; Wattiaux A; Stayton PS Langmuir 2007, 23, 7385–7391. [DOI] [PubMed] [Google Scholar]
- (10).Lai JJ; Nelson KE; Nash MA; Hoffman AS; Yager P; Stayton PS Lab Chip 2009, 9, 1997–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Schild HG Prog. Polym. Sci 1992, 17, 163–249. [Google Scholar]
- (12).Freitag R; Garret-Flaudy F Langmuir 2002, 18, 3434–3440. [Google Scholar]
- (13).Hyun J; Lee WK; Nath N; Chilkoti A; Zauscher SJ Am. Chem. Soc 2004, 126, 7330–7335. [DOI] [PubMed] [Google Scholar]
- (14).Hoffman JM; Ebara M; Lai JJ; Hoffman AS; Folch A; Stayton PS Lab Chip 2010, 10, 3130–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schacher F; Rudolph T; Wieberger F; Ulbricht M; Muller AHE ACS Appl. Mater. Inter 2009, 1, 1492–1503. [DOI] [PubMed] [Google Scholar]
- (16).Golden AL; Battrell CF; Pennell S; Hoffman AS; Lai JJ; Stayton PS Bioconjugate Chem. 2010, 21, 1820–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Nash MA; Waitumbi JN; Hoffman AS; Yager P; Stayton PS ACS Nano 2012, 6, 6776–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lee YJ; Kang HC; Hu J; Nichols JW; Jeon YS; Bae YH Biomacromolecules 2012, 13, 2945–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hoare T; Young S; Lawlor MW; Kohane DS Acta Biomater. 2012, 8, 3596–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Betancourt T; Pardo J; Soo K; Peppas NA J. Biomed. Mater. Res. A 2010, 93a, 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lavignac N; Lazenby M; Franchini J; Ferruti P; Duncan R Int. J. Pharm 2005, 300, 102–112. [DOI] [PubMed] [Google Scholar]
- (22).Matsumoto NM; Buchman GW; Rome LH; Maynard HD Eur. Polym. J 2015, 69, 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Collins ML; Irvine B; Tyner D; Fine E; Zayati C; Chang CA; Horn T; Ahle D; Detmer J; Shen LP; Kolberg J; Bushnell S; Urdea MS; Ho DD Nucleic Acids Res. 1997, 25, 2979–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Branson BM; Stekler JD J. Infect. Dis 2012, 205 (4), 521–524. [DOI] [PubMed] [Google Scholar]
- (25).Parpia ZA; Elghanian R; Nabatiyan A; Hardie DR; Kelso DM JAIDS-J. Acq. Imm. Def 2010, 55, 413–419. [DOI] [PubMed] [Google Scholar]
- (26).Chiu RY; Nguyen PT; Wang J; Jue E; Wu BM; Kamei DT Ann. Biomed. Eng 2014, 42, 2322–32. [DOI] [PubMed] [Google Scholar]
- (27).Chesebro B; Wehrly K; Nishio J; Perryman S J. Virol. 1992, 66, 6547–6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Roy D; Nehilla BJ; Lai JJ; Stayton PS ACS Macro Lett. 2013, 2, 132–136. [DOI] [PubMed] [Google Scholar]
- (29).Chiefari J; Chong YK; Ercole F; Krstina J; Jeffery J; Le TPT; Mayadunne RTA; Meijs GF; Moad CL; Moad G; Rizzardo E; Thang SH Macromolecules 1998, 31, 5559–5562. [Google Scholar]
- (30).Li HM; Bapat AP; Li M; Sumerlin BS Polym. Chem.-UK 2011, 2, 323–327. [Google Scholar]
- (31).Magoshi T; Ziani-Cherif H; Ohya S; Nakayama Y; Matsuda T Langmuir 2002, 18, 4862–4872. [Google Scholar]
- (32).Pan LC; Chien CC J. Biochem. Bioph. Meth 2003, 55, 87–94. [DOI] [PubMed] [Google Scholar]
- (33).Ohnishi NF,H; Hideyuki J; Wang J-M; An CI; Fukusaki E; Kataoka K; Ueno K; Kondo A NanoBiotechnology 2006, 2, 43–49. [Google Scholar]
- (34).Xie XM; Ohnishi N; Takahashi Y; Kondo A J. Magn. Magn. Mater 2009, 321, 1686–1688. [Google Scholar]
- (35).Hoshino A; Ohnishi N; Yasuhara M; Yamamoto K; Kondo A Biotechnol. Progr 2007, 23, 1513–1516. [DOI] [PubMed] [Google Scholar]
- (36).Nagaoka H; Sato Y; Xie XM; Hata H; Eguchi M; Sakurai N; Watanabe T; Saitoh H; Kondo A; Sugita S; Ohnishi N Anal. Chem 2011, 83, 9197–9200. [DOI] [PubMed] [Google Scholar]
- (37).Schuck P Annu. Rev. Bioph. Biom 1997, 26, 541–566. [DOI] [PubMed] [Google Scholar]
- (38).Rich RL; Myszka DG J. Mol. Recognit 2000, 13, 388–407. [DOI] [PubMed] [Google Scholar]
- (39).Gorshkova II; Svitel J; Razjouyan F; Schuck P Langmuir 2008, 24, 11577–11586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Gertig ET In Handbook of Surface Plasmon Resonance, Schasfoort RBM, Tudos AJ Eds.; Royal Society of Chemistry: Cambridge, UK, 2008; pp 173–220. [Google Scholar]
- (41).Lu B; Smyth MR; O’Kennedy R Analyst 1996, 121, R29–R32. [Google Scholar]
- (42).Bobrovnik SA J. Biochem. Bioph. Meth 2003, 57, 213–236. [DOI] [PubMed] [Google Scholar]
- (43).Stevens FJ Mol. Immunol 1987, 24, 1055–1060. [DOI] [PubMed] [Google Scholar]
- (44).Stevens FJ; Bobrovnik SA J. Immunol. Methods 2007, 328, 53–58. [DOI] [PubMed] [Google Scholar]
- (45).Rosales T; Royer CA Anal. Biochem 2008, 381, 270–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Abdiche Y; Malashock D; Pinkerton A; Pons J Anal. Biochem 2008, 377, 209–217. [DOI] [PubMed] [Google Scholar]
- (47).Campbell K; Huet AC; Charlier C; Higgins C; Delahaut P; Elliott CT J. Chromatogr. B 2009, 877, 4079–4089. [DOI] [PubMed] [Google Scholar]
- (48).Heinrich L; Tissot N; Hartmann DJ; Cohen R J. Immunol. Methods 2010, 352, 13–22. [DOI] [PubMed] [Google Scholar]
- (49).Nechansky A J. Pharmaceut. Biomed 2010, 51, 252–254. [DOI] [PubMed] [Google Scholar]
- (50).Vaisocherova H; Faca VM; Taylor AD; Hanash S; Jiang SY Biosens. Bioelectron 2009, 24, 2143–2148. [DOI] [PubMed] [Google Scholar]
- (51).Hardy F; Djavadi-Ohaniance L; Goldberg ME J Immunol Methods 1997, 200, 155–159. [DOI] [PubMed] [Google Scholar]
- (52).Huppert D; Pines E; Agmon N J Opt Soc Am B 1990, 7, 1545–1550. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.