

Abstract
Two pairs of enantiomers, (−) and (+)-securidanes A (1 and 2) and B (3 and 4) featuring unprecedented triarylmethane (TAM) skeletons, were isolated from Securidaca inappendiculata. Their structures were established by spectroscopic data, X-ray crystallography, and CD analysis. A plausible biosynthetic pathway for 1−4 based on the co-isolated precursors was proposed. Bioinspired total synthesis of 1−4 was completed in high yield, which in turn corroborated the biosynthetic hypothesis. Compounds 1−4 showed good inhibition against protein tyrosine phosphatase 1B (PTP1B). The molecular docking demonstrated that the strongest inhibitor 3 (IC50 = 7.52 μM) reaches deeper into the binding pocket and has an additional H-bond.
1. Introduction
Compounds incorporating the triarylmethane (TAM) motif are well recognized in materials science, such as fluorescent probes, organic dyes, and metal ion sensors [1–5]. It is particularly interesting that TAM derivatives have also demonstrated a broad spectrum of biological significance including anticancer, K+ channel blocking, histidine protein kinase inhibitory, and antiparasitic, antiviral, and antitubercular activities [6–10]. Given the important roles of TAMs in materials science and medicinal chemistry, this compound class has attracted widespread attention in the area of organic chemistry and a number of synthetic methods have been developed by involving the key steps of Friedel−Crafts reaction, transition-metal-catalytic cross-coupling reaction, and reductive dehydroxylation of triarymethanol derivatives, including a few approaches of asymmetric catalysis [11–25], which has led to the synthesis of a large array of TAMs. However, only a very limited number of flavonoid derivatives with a TAM motif have been identified from natural products hitherto [26–29].
The plant Securidaca inappendiculata Hassk. (Polygalaceae) is mainly distributed in the south of China and the tropical regions of Asia. The whole plants have been applied in the remedies of traditional Chinese medicine [30], from which a number of xanthone, benzophenone, and sterol derivatives were identified with anti-inflammatory, anti-HIV, and MAO inhibitory activities [31–34]. In this study, two enantiomeric pairs, (−)- and (+)-securidanes A (1 and 2) and B (3 and 4) featuring new TAM skeletons, were obtained as optically pure compounds by chiral separation from the stems of S. inappendiculata (Figure 1). Compounds 1−4 biosynthetically descended from a diphenylmethane and a diphenyl derivative are unprecedented, and a plausible biosynthetic pathway based on the coisolated precursor 5 was proposed. Bioinspired total synthesis of 1−4 was completed in high yield, which in turn corroborated the biosynthetic hypothesis. PTP1B plays a profound role in cell regulation, growth, and the onset of human diseases. Its overexpression causes persistent dephosphorylation of insulin receptor, stimulating the insulin-resistant phenotype in type 2 diabetes and obesity [35]. PTP1B has thus been considered as a potential therapeutic target for type 2 diabetes and obesity [36, 37]. Compounds 1−4 showed PTP1B inhibitions with IC50 values ranging from 7.5 to 15.6 μm. The molecular docking showed that the strongest inhibitor 3 reaches deeper into the binding pocket and has an additional H-bond. Herein, we present the isolation, chiral separation, structural elucidation, biological evaluation, and bioinspired total synthesis of 1–4.
Figure 1.
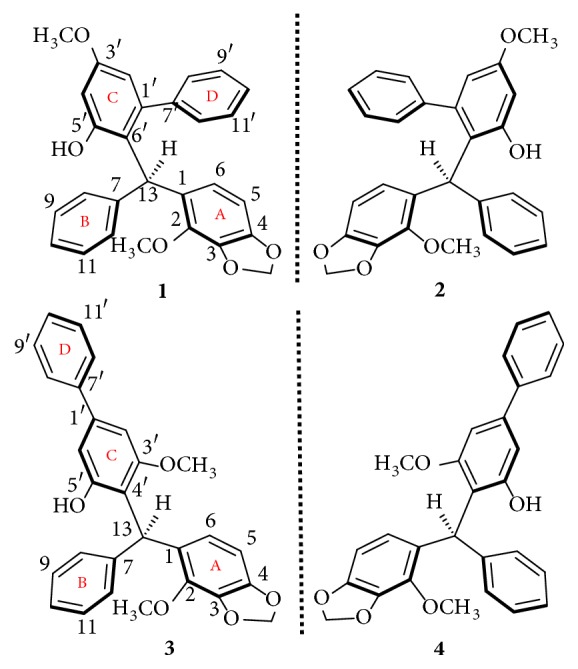
Structures of compounds 1–4.
2. Results
(−)-Securidane A (1) was obtained as colorless crystals (in MeOH) with [α]22D −76.9 (c 0.42, MeOH). The molecular formula, C28H24O5 with 17 double-bond equivalents (DBEs), was determined by the HRESIMS ion at m/z 441.1700 [M + H]+ (calcd 441.1697) and the NMR data. Its IR absorption bands showed the presence of hydroxy (3487 cm−1) and aromatic (1613 and 1576 cm−1) functionalities. The 1H NMR data () displayed the diagnostic resonances of two methoxy and one methylenedioxy groups. The 13C NMR data () with the aid of DEPT experiments revealed the existence of two methyls, one methylene, 15 methines (14 sp2 and one sp3), and 10 sp2 quaternary carbons. Comprehensive analysis of 1H and 13C NMR data indicated the presence of four phenyl groups (two mono- and two tetrasubstituted), which accounted for 16 out of the 17 DBEs, and the remaining one DBE required one more ring in the molecule. A singlet proton signal at δH 5.40 that did not show correlations with any carbons in the HSQC spectrum () was assigned to a hydroxy group. Particularly, the diagnostic sp3 methane group (δH 5.58, s; δC 45.0) suggested that it is a TAM derivative.
The planar structure of 1 was established by 2D NMR analysis (). Three proton-bearing coupling fragments as drawn in bold bonds were revealed by 1H-1H COSY spectrum. In the HMBC spectrum, the key correlations from H-13 to C-1, C-7, and C-6′ attached three phenyls to C-13. The HMBC correlations of H-12′/C-1′ and H-2′/C-7′ placed the remaining phenyl unit at C-1′ to form a diphenyl motif via the C-1′−C-7′ bond. Two methoxyls were located at C-2 and C-3′ by the HMBCs of 2-OCH3/C-2 and 3′-OCH3/C-3′, respectively. The only hydroxy group was assigned to C-5′ by the key HMBC correlation of 5′-OH/C-5′. The downfield shifted methylene signals (δH 5.93, 5.95; δC 101.2) was assigned to a 3,4-methylenedioxy group by the chemical shifts and the HMBCs from the two protons to both C-3 and C-4. The planar structure of 1 was thus delineated. The absolute configuration of 1 was unambiguously determined as 13R by a single crystal X-ray diffraction study (Figure 2), in which the anomalous dispersion of Cu Kα radiation was applied and the absolute structure parameter of −0.13(7) was acquired.
Figure 2.

(a) X-ray structure of 1. (b) Molecule assembly in crystals.
(+)-Securidane A (2) shared the same molecular formula and identical NMR data with 1 (, Figures and ), but had an opposite specific rotation [α]22D +72.3 (c 0.48, MeOH) and CD curve to that of 1 (Figure 3), indicating that it is the enantiomer of 1 and 13S-configured. Crystallization of 2 from methanol allowed for a successful performance of X-ray diffraction analysis (Figure 4), which not only confirmed its absolute configuration [absolute structure parameter 0.04(10)], but also provided solid evidence to understand the conformation and molecular assembling patterns of enantiomeric pair of triarylmethane-type compounds 1 and 2 in the solid state.
Figure 3.
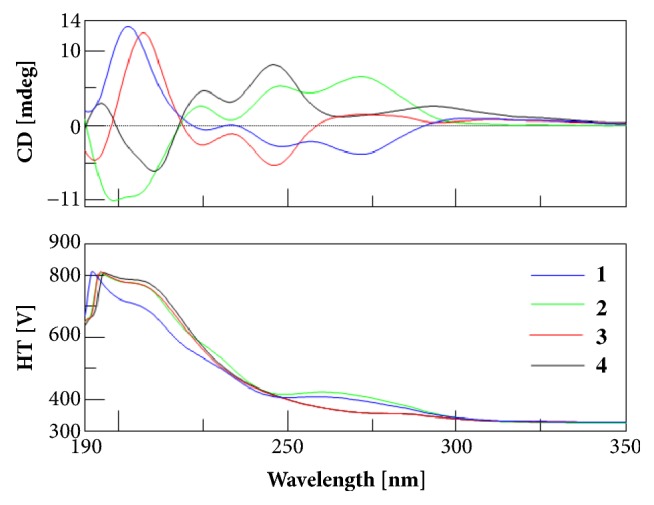
CD spectra of 1–4.
Figure 4.

(a) X-ray structure of 2. (b) Molecule assembly in crystals.
(−)-Securidane B (3), [α]22D −43.5 (c 0.37, MeOH), had a molecular formula of C28H24O5 as determined by the HRESIMS ion at m/z 441.1707 [M + H]+ (calcd 441.1697). The NMR data () showing great similarities to those of 1 suggested that it is also a TAM analogue. The major differences observed in its 13C NMR spectrum () were the C-4′ and C-6′. The quaternary C-4′ (δC 116.7) and the methine C-6′(δC 102.4) resonated downfield (ΔδC +14.4) and upfield (ΔδC −17.0), respectively, as compared to those of 1, indicative of an alternative conjugation between the diphenyl and diphenylmethane motifs. The planar structure of 3 was finally constructed by 2D NMR spectra (Figures and –), especially HMBC data. The linkage between two motifs via a C-13−C-4′ bond was confirmed by the key HMBC correlation from H-13 (δH 6.30, s) to C-4′. The substituted patterns in two motifs were assigned to be identical to those of 1 by the HMBC correlations. The tendency of its CD curve (Figure 3) is compatible to that of 1, suggesting that 3 had a 13R-configuration. This was supported by its negative specific rotation as compared to 1.
(+)-Securidane B (4) possessed the same molecular formula C28H24O5 and the identical NMR data as those of 3 (, Figures and ), while showing opposite CD curve (Figure 3) and specific rotation of [α]22D = +42.3 to those of 3. It was thus assigned as the enantiomer of 3 to be 13S-configured.
A direct LC-ESIMS analysis of the fresh ethanolic extract of the plant stems showed the presence of the diagnostic ion peaks at m/z 441 [M + H]+ and 439 [M − H]− for 1−4 (), indicating that they are not artifacts produced in the separation.
A possible biosynthetic pathway for 1−4 was proposed (Scheme 1). The co-isolate 5 [9] and a natural product 6 [38] were served as the biosynthetic precursors. Although 6 has not been isolated in this study, it is presumed to exist in the plant either in a low concentration or with a very short lifespan after production. Reduction of 5 by NADPH would produce the key intermediate 7, which was readily transformed to a very stable carbocation 7i. Nucleophilic attack of C-4′ or C-6′ of 6 on to 7i via electrophilic aromatic substitution reaction would produce (−)- and (+)-securidanes A (1 and 2) (route A in pink) and B (3 and 4) (route B in green).
Scheme 1.

Plausible biosynthetic pathway of compounds 1–4.
To confirm the biosynthetic hypothesis, we carried out a bioinspired total synthesis of 1−4. The retrosynthetic analysis (Scheme 2) involves a biomimetic assembling of 6a and 7 via Kim's protocol [25] as the key step to furnish the targeted TAM frameworks of 1−4, in which 7 is the key biosynthetic intermediate, and 6a is the MOM ether of the other biosynthetic precursor 6. Synthesis of fragment 7 in turn was envisioned to arise from aldehyde 10 by a Grignard reaction. While the aldehyde 10 could be made by the treatment of 11 under formylation condition, biaryl compound 6a could be readily prepared from 8 in two steps.
Scheme 2.

Bioinspired retrosynthetic analysis of 1−4.
2.1. Synthesis of 7
Compound 11 was prepared in 87% yield by alkylation of 12 [39]. Formylation of 11 then produced two isomeric aldehydes 10 and 10a in a ratio of 2:1 [40, 41]. Addition of Grignard reagents formed from bromobenzene (9) to the aldehyde 10 afforded the desired alcohol 7 in a good yield of 99% (Scheme 3) [42].
Scheme 3.

Synthesis of 7, (i) NaH, HMPA, CH2I2, rt, 87%; (ii) DMF, POCl3, 100°C, 7 h, 52%; (iii) Mg, I2, bromobenzene, THF, rt, 99%.
2.2. Synthesis of 6a
Biaryl 6a was synthesized from the known starting material 8 (Scheme 4). Enolization of 8 under acidic condition in methanol at room temperature afforded 8a, which was then converted into 6 by refluxing with Hg(OAc)2 in AcOH for 7 h [43]. 6a was finally obtained by protection of the hydroxyl of 6 with MOM ether [44].
Scheme 4.

Synthesis of 6a, (i) H2SO4, MeOH, rt, 92%; (ii) AcOH, Hg(OAc)2, reflux, 7 h, 60%; (iii) CH3OCH2Cl, NaH, THF, 0°C to rt, 98%.
2.3. Synthesis of 1−4
With the key fragments 6a and 7 in hand, we next focused on their assembling in the presence of Fe (III) [24] (Scheme 5). The desired coupling products were obtained as a mixture of two racemic pairs 1a−4a, which were dominated by the racemates 3a and 4a (ca. 95%, 1:1) with the minor racemic products 1a and 2a (ca. 5%, 1:1) (determined by HPLC analysis). Deprotection of the MOM ethers [45] afforded a mixture of two pair racemates in 79% total yield, which was further separated into four optically pure compounds 1−4 by chiral HPLC preparation. The products of chemical synthesis were dominated by (−)- and (+)-securidane B (3 and 4, 95%) due largely to the different steric hindrance in two coupling models, while the natural isolates 1−4 from the plant were accounted for approximately 25% each (Figures –). It is suggested that the transition state in the key step of chemical coupling is different from that of the biosynthesis in the plant, and the MOM ether protection group of chemical synthesis is likely an influencing factor for the ratios of the products.
Scheme 5.

Synthesis of 1−4, (i) Fe (ClO4)3•xH2O, CH3CN, rt, 57%; (ii) HCl, MeOH, reflux, 79%.
2.4. PTP1B Inhibitory Evaluation
Compounds 1−4 were tested for the inhibitory effects on PTP1B enzyme by using an in vitro assay [46], and a well-recognized natural PTP1B inhibitor oleanolic acid was used as the positive control (IC50 = 4.14 ± 0.59 μM). Compounds 1−4 showed remarkable inhibition with IC50 values of 15.6 ± 1.37, 12.6 ± 3.68, 7.5 ± 0.74, and 10.5 ± 2.86 μM, respectively. This is the first report of TAMs as PTP1B inhibitors.
2.5. Molecular Docking
The main structural modules furnishing the ligand binding pocket of PTP1B have been demonstrated as the catalytic loop, the YRD motif, and the WPD loop [47]. The molecular docking results (Figures 5, S7, and S8) revealed that the best scoring docking conformations of compounds 1−4 at the binding pocket of PTP1B and their interacting patterns are similar. Taking compound 3 as an example (Figure 5), ring A participates in hydrophobic interactions with Arg47, Asp48, and Val49 at the YRD motif and forms a hydrogen bond with the main-chain of Arg47. Rings C and D bury into a hydrophobic pocket mostly contributed by Try46 at the YRD motif, Phe182 at the WPD loop, and residues at the catalytic site including Ala217, Ile219, and Gly220. Four compounds interact with the same binding pocket, while 3 and 4, a pair of enantiomers, reach deeper into this hydrophobic pocket as compared to 1 and 2. This may contribute to the higher potency of 3 and 4 to PTP1B. The additional hydrogen bond between ring A of 3 and PTP1B, which is devoid in the complex of 4 and PTP1B, makes 3 to be the most potent inhibitor among those four compounds.
Figure 5.

Docking pose of 3 showing key interactions between the compound and PTP1B. (a) The protein is shown as light grey cartoon, while the catalytic loop, the YRD motif, and the WPD loop are colored green, yellow, and cyan, respectively; 3 is represented as a ball-and-stick model with carbon and oxygen colored magenta and red, respectively. (b) The protein is shown by molecular surface and 3 is represented as sticks. (c) Key interactions between 3 and PTP1B are analyzed by Ligplot+.
3. Discussion
In conclusion, we have identified optically pure (−)- and (+)-securidanes A and B (1−4) featuring unprecedented TAM skeletons from a Chinese medicinal plant S. inappendiculata. A plausible biosynthetic pathway for 1−4 based on the coisolated precursor 5 was proposed. Bioinspired total synthesis of 1−4 was achieved in high yield, which in turn corroborated the biosynthetic hypothesis. Compounds 1−4 showed good inhibition against PTP1B. Our efforts provide for the first time the chemophysical data for the optically pure TAM analogues, a bioinspired total synthesis of TAMs, and TAMs as PTP1B inhibitors. This finding is of great importance for understanding the biosynthesis of natural TAMs and exploration of their medicinal potency.
4. Materials and Methods
4.1. General Experimental Procedures
The general experiments were completed according to the reported general procedures with minor modification (Experimental Section, Supporting Information) [48].
4.1.1. Plant Material
The detail information of the plant of S. inappendiculata was included in the Experimental Section, Supporting Information.
4.1.2. PTP1B Inhibition Assay
A colorimetric assay for the measurement of PTP1B inhibition was performed according to the reported protocols (Experimental Section, Supporting Information) [46, 49].
4.1.3. Molecular Docking
The crystal structure of PTP1B in complex with one of benzotriazole inhibitors (PDB ID: 1Q6P) was used to prepare the receptor structure, and the centroid of the inhibitor was selected as the center of grid boxes [50]. Water molecules, ions, and the inhibitor were deleted before docking performance. The receptor was then prepared using Protein Preparation and Grid Preparation tools in the Schrödinger Maestro interface. As for ligands, the 3D structures of compounds 1–4 were optimized with B3LYP/6-31G∗ using GAUSSIAN 09 [51, 52]. Molecular docking were performed using the Glide extraprecision mode with default settings [53, 54]. The OPLS-2005 force field was used for minimization and grid generation, while OPLS-2001 was used for docking. Key interactions between compound 3 and PTP1B were analyzed by Ligplot+ [55].
Acknowledgments
The authors thank Professor S. Q. Tang of Guangxi Normal University for the identification of the plant material. The National Natural Science Foundation (nos. 21532007, U1302222, 81321092) and the Foundation from the MOST (2012CB721105) of China are gratefully acknowledged.
Data Availability
All data are available in the manuscript or supplementary materials.
Conflicts of Interest
The authors declare no competing financial interests.
Authors' Contributions
All authors designed the research. B. Zhou performed compounds isolation and structure identification. D. X. Liu and Y. Li contributed to the synthesis work. J. Y. Li and J. Li finished PTP1B inhibitory assay. X. J. Yuan and Y. C. Xu performed the molecular docking. B. Zhou, D. X. Liu, and X. J. Yuan wrote the manuscript. J. M. Yue supervised the project and provided comments and revisions on the manuscript. B. Zhou and D. X. Liu contributed equally to this work.
Supplementary Materials
Table S1. Figures S1~S43: general experimental procedures, X-ray crystal data of 1 and 2, biomimetic total synthesis of 1–4, PTP1B inhibitory assay, and molecular docking of 1–4.
References
- 1.Nolan E. M., Lippard S. J. Tools and tactics for the optical detection of mercuric ion. Chemical Reviews . 2008;108(9):3443–3480. doi: 10.1021/cr068000q. [DOI] [PubMed] [Google Scholar]
- 2.Nair V., Thomas S., Mathew S. C., Abhilash K. G. Recent advances in the chemistry of triaryl- and triheteroarylmethanes. Tetrahedron . 2006;62(29):6731–6747. doi: 10.1016/j.tet.2006.04.081. [DOI] [Google Scholar]
- 3.Miura T., Urano Y., Tanaka K., Nagano T., Ohkubo K., Fukuzumi S. Rational design principle for modulating fluorescence properties of fluorescein-based probes by photoinduced electron transfer. Journal of the American Chemical Society . 2003;125(28):8666–8671. doi: 10.1021/ja035282s. [DOI] [PubMed] [Google Scholar]
- 4.Muramatsu H., Okumura A., Shibata K., Matsui M. Synthesis, absorption spectra, and photostability of triarylmethane dye ethynylogues containing trifluoromethyl group(s) Chemische Berichte . 1994;127(9):1627–1632. doi: 10.1002/cber.19941270914. [DOI] [Google Scholar]
- 5.Duxbury D. F. The photochemistry and photophysics of triphenylmethane dyes in solid and liquid media. Chemical Reviews . 1993;93(1):381–433. doi: 10.1021/cr00017a018. [DOI] [Google Scholar]
- 6.Palchaudhuri R., Nesterenko V., Hergenrother P. J. The complex role of the triphenylmethyl motif in anticancer compounds. Journal of the American Chemical Society . 2008;130(31):10274–10281. doi: 10.1021/ja8020999. [DOI] [PubMed] [Google Scholar]
- 7.Wulff H., Zhorov B. S. K+ channel modulators for the treatment of neurological disorders and autoimmune diseases. Chemical Reviews . 2008;108(5):1744–1773. doi: 10.1021/cr078234p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S., Malik V., Kaur N., Kaur K. A simple synthesis of di(uracilyl)aryl methanes and 1,ω-bis[di(uracilyl)methyl]benzenes. Tetrahedron Letters . 2006;47(48):8483–8487. doi: 10.1016/j.tetlet.2006.09.158. [DOI] [Google Scholar]
- 9.Mibu N., Yokomizo K., Uyeda M., Sumoto K. Synthesis and antiviral activities of some 4,4′- and 2,2′-dihydroxytriphenylmethanes. Chemical & Pharmaceutical Bulletin . 2005;53(9):1171–1174. doi: 10.1248/cpb.53.1171. [DOI] [PubMed] [Google Scholar]
- 10.Parai M. K., Panda G., Chaturvedi V., Manju Y. K., Sinha S. Thiophene containing triarylmethanes as antitubercular agents. Bioorganic & Medicinal Chemistry Letters . 2008;18(1):289–292. doi: 10.1016/j.bmcl.2007.10.083. [DOI] [PubMed] [Google Scholar]
- 11.Huang Y., Hayashi T. Asymmetric synthesis of triarylmethanes by rhodium-catalyzed enantioselective arylation of diarylmethylamines with arylboroxines. Journal of the American Chemical Society . 2015;137(24):7556–7559. doi: 10.1021/jacs.5b03277. [DOI] [PubMed] [Google Scholar]
- 12.Nambo M., Crudden C. M. Modular synthesis of triarylmethanes through palladium-catalyzed sequential arylation of methyl phenyl sulfone. Angewandte Chemie International Edition . 2014;53(3):742–746. doi: 10.1002/anie.201307019. Angew. Chem. 126, 761–765 (2014) [DOI] [PubMed] [Google Scholar]
- 13.Matthew S. C., Glasspoole B. W., Eisenberger P., Crudden C. M. Synthesis of enantiomerically enriched triarylmethanes by enantiospecific suzuki-miyaura cross-coupling reactions. Journal of the American Chemical Society . 2014;136(16):5828–5831. doi: 10.1021/ja412159g. [DOI] [PubMed] [Google Scholar]
- 14.Matsuo K., Saito S., Yamaguchi S. Photodissociation of B-N Lewis adducts: A partially fused trinaphthylborane with dual fluorescence. Journal of the American Chemical Society . 2014;136(36):12580–12583. doi: 10.1021/ja506980p. [DOI] [PubMed] [Google Scholar]
- 15.Creutz S. E., Peters J. C. Catalytic reduction of N2 to NH3 by an Fe-N 2 complex featuring a C-atom anchor. Journal of the American Chemical Society . 2014;136(3):1105–1115. doi: 10.1021/ja4114962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Q., Srinivas H. D., Dasgupta S., Watson M. P. Nickel-catalyzed cross-couplings of benzylic pivalates with arylboroxines: Stereospecific formation of diarylalkanes and triarylmethanes. Journal of the American Chemical Society . 2013;135(9):3307–3310. doi: 10.1021/ja312087x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris M. R., Hanna L. E., Greene M. A., Moore C. E., Jarvo E. R. Retention or inversion in stereospecific nickel-catalyzed cross-coupling of benzylic carbamates with arylboronic esters: Control of absolute stereochemistry with an achiral catalyst. Journal of the American Chemical Society . 2013;135(9):3303–3306. doi: 10.1021/ja311783k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J., Bellomo A., Creamer A. D., Dreher S. D., Walsh P. J. Palladium-catalyzed C(sp3)-H arylation of diarylmethanes at room temperature: Synthesis of triarylmethanes via deprotonative-cross-coupling processes. Journal of the American Chemical Society . 2012;134(33):13765–13772. doi: 10.1021/ja3047816. [DOI] [PubMed] [Google Scholar]
- 19.Taylor B. L., Harris M. R., Jarvo E. R. Synthesis of Enantioenriched Triarylmethanes by Stereospecific Cross-Coupling Reactions. Angewandte Chemie International Edition . 2012;51(31):7790–7793. doi: 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]
- 20.Yuan F.-Q., Gao L.-X., Han F.-S. PdCl2-catalyzed efficient allylation and benzylation of heteroarenes under ligand, base/acid, and additive-free conditions. Chemical Communications . 2011;47(18):5289–5291. doi: 10.1039/c1cc10953g. [DOI] [PubMed] [Google Scholar]
- 21.Taylor B. L. H., Swift E. C., Waetzig J. D., Jarvo E. R. Stereospecific nickel-catalyzed cross-coupling reactions of alkyl ethers: Enantioselective synthesis of diarylethanes. Journal of the American Chemical Society . 2011;133(3):389–391. doi: 10.1021/ja108547u. [DOI] [PubMed] [Google Scholar]
- 22.McGrew G., Temaismithi J., Carroll P., Walsh P. Synthesis of Polyarylated methanes through cross-coupling of tricarbonylchromium-activated benzyllithiums. Angewandte Chemie International Edition . 2010;49:p. 5541. doi: 10.1002/ange.201000957. Angew. Chem. 122, 5673–5676 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bing Y., Selassie D., Paradise R. H., et al. Circular dichroism tensor of a triarylmethyl propeller in sodium chlorate crystals. Journal of the American Chemical Society . 2010;132(21):7454–7465. doi: 10.1021/ja1018892. [DOI] [PubMed] [Google Scholar]
- 24.Thirupathi P., Kim S. S. Fe(ClO4)3·×H2O-Catalyzed direct C-C bond forming reactions between secondary benzylic alcohols with different types of nucleophiles. Tetrahedron . 2010;66(16):2995–3003. doi: 10.1016/j.tet.2010.02.063. [DOI] [Google Scholar]
- 25.Thirupathi P., Sung S. K. InBr3: A versatile catalyst for the different types of Friedel-Crafts reactions. The Journal of Organic Chemistry . 2009;74(20):7755–7761. doi: 10.1021/jo9014613. [DOI] [PubMed] [Google Scholar]
- 26.Dibwe D. F., Awale S., Kadota S., Morita H., Tezuka Y. Muchimangins G-J, fully substituted xanthones with a diphenylmethyl substituent, from securidaca longepedunculata. Journal of Natural Products . 2014;77(5):1241–1244. doi: 10.1021/np5000445. [DOI] [PubMed] [Google Scholar]
- 27.Dibwe D. F., Awale S., Kadota S., Morita H., Tezuka Y. Muchimangins e and F: Novel diphenylmethyl-substituted xanthones from Securidaca longepedunculata. Tetrahedron Letters . 2014;55(11):1916–1919. doi: 10.1016/j.tetlet.2014.01.149. [DOI] [Google Scholar]
- 28.Jin C., Michetich R. G., Daneshtalab M. Flavonoids from Stellera chamaejasme. Phytochemistry . 1998;50(3):505–508. doi: 10.1016/S0031-9422(98)00588-3. [DOI] [Google Scholar]
- 29.Seligmann O., Wagner H. Structure determination of melanervin, the first naturally occurring flavonoid of the triphenylmethane family. Tetrahedron . 1981;37(15):2601–2606. doi: 10.1016/S0040-4020(01)98963-X. [DOI] [Google Scholar]
- 30.Chen S. K., Chen B. Y., Li H. In Zhongguo Zhiwu Zhi . Vol. 43. Beijing: Science Press; 1997. [Google Scholar]
- 31.Zuo J., Mao K.-J., Yuan F., Li X., Chen J.-W. Xanthones with anti-tumor activity isolated from Securidaca inappendiculata. Medicinal Chemistry Research . 2014;23(11):4865–4871. doi: 10.1007/s00044-014-1051-8. [DOI] [Google Scholar]
- 32.Zhang L., Yang X., Xu L., Yang S. Three New Xanthones from the Roots of Securidaca inappendiculata. Heterocycles . 2005;65(7):p. 1685. doi: 10.3987/COM-05-10392. [DOI] [Google Scholar]
- 33.Kang W., Ji Z., Wang J. A new xanthone from the roots of Securidaca inappendiculata. Chemical Papers . 2009;63(1) doi: 10.2478/s11696-008-0087-y. [DOI] [Google Scholar]
- 34.Yang X.-D., Xu L.-Z., Yang S.-L. Xanthones from the stems of Securidaca inappendiculata. Phytochemistry . 2001;58(8):1245–1249. doi: 10.1016/S0031-9422(01)00356-9. [DOI] [PubMed] [Google Scholar]
- 35.Haftchenary S., Jouk A. O., Aubry I., et al. Identification of bidentate salicylic acid inhibitors of PTP1B. ACS Medicinal Chemistry Letters . 2015;6(9):982–986. doi: 10.1021/acsmedchemlett.5b00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Comeau A. B., Critton D. A., Page R., Seto C. T. A focused library of protein tyrosine phosphatase inhibitors. Journal of Medicinal Chemistry . 2010;53(18):6768–6772. doi: 10.1021/jm100528p. [DOI] [PubMed] [Google Scholar]
- 37.Marx J. Unraveling the causes of diabetes. Science . 2002;296(5568):686–689. doi: 10.1126/science.296.5568.686. [DOI] [PubMed] [Google Scholar]
- 38.Song M.-C., Nigussie F., Jeong T.-S., et al. Phenolic compounds from the roots of Lindera fruticosa. Journal of Natural Products . 2006;69(5):853–855. doi: 10.1021/np060048b. [DOI] [PubMed] [Google Scholar]
- 39.McKittrick B. A., Stevenson R. Synthesis of the yeast antioxidant benzofuran and analogues. Journal of the Chemical Society, Perkin Transactions 1 . 1984:709–712. doi: 10.1039/p19840000709. [DOI] [Google Scholar]
- 40.Benington F., Morin R. D. Contribution to the Structure of Falcatine. Synthesis of Isoanhydrofalcatine Lactam. The Journal of Organic Chemistry . 1962;27(1):142–146. doi: 10.1021/jo01048a035. [DOI] [Google Scholar]
- 41.Wagner A. F., Walton E., Wilson A. N., et al. Synthesis of Degradation Products of 2-(6-Hydroxy-2-methoxy-3, 4-methylenedioxyphenyl)-benzofuran. Journal of the American Chemical Society . 1959;81(18):4983–4989. doi: 10.1021/ja01527a053. [DOI] [Google Scholar]
- 42.Tabuchi S., Hirano K., Satoh T., Miura M. Synthesis of triarylmethanes by palladium-catalyzed c-h/c-o coupling of oxazoles and diarylmethanol derivatives. The Journal of Organic Chemistry . 2014;79(12):5401–5411. doi: 10.1021/jo5010636. [DOI] [PubMed] [Google Scholar]
- 43.Lebeuf R., Robert F., Landais Y. Regioselectivity of birch reductive alkylation of biaryls. Organic Letters . 2005;7(21):4557–4560. doi: 10.1021/ol051377i. [DOI] [PubMed] [Google Scholar]
- 44.Castello L. M., Hornillos V., Vila C., Giannerini M., Fananas-Mastral M., Feringa B. L. Pd-Catalyzed Cross-Coupling of Aryllithium Reagents with 2-Alkoxy-Substituted Aryl Chlorides: Mild and Efficient Synthesis of 3, 3′-Diaryl BINOLs. Organic Letters . 2014;17:62–65. doi: 10.1021/ol5032409. [DOI] [PubMed] [Google Scholar]
- 45.Ramesh C., Ravindranath N., Das B. Simple, efficient, and selective deprotection of phenolic methoxymethyl ethers using silica-supported sodium hydrogen sulfate as a heterogeneous catalyst. The Journal of Organic Chemistry . 2003;68(18):7101–7103. doi: 10.1021/jo030088+. [DOI] [PubMed] [Google Scholar]
- 46.Zhang W., Hong D., Zhou Y., et al. Ursolic acid and its derivative inhibit protein tyrosine phosphatase 1B, enhancing insulin receptor phosphorylation and stimulating glucose uptake. Biochimica et Biophysica Acta (BBA) - General Subjects . 2006;1760(10):1505–1512. doi: 10.1016/j.bbagen.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 47.Johnson T. O., Ermolieff J., Jirousek M. R. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nature Reviews Drug Discovery . 2002;1(9):696–709. doi: 10.1038/nrd895. [DOI] [PubMed] [Google Scholar]
- 48.Zhou B., Shen Y., Wu Y., Leng Y., Yue J.-M. Limonoids with 11β-Hydroxysteroid Dehydrogenase Type 1 Inhibitory Activities from Dysoxylum mollissimum. Journal of Natural Products . 2015;78(8):2116–2122. doi: 10.1021/acs.jnatprod.5b00442. [DOI] [PubMed] [Google Scholar]
- 49.Yu J.-H., Liu Q.-F., Sheng L., Wang G.-C., Li J., Yue J.-M. Cipacinoids A-D, four limonoids with spirocyclic skeletons from cipadessa cinerascens. Organic Letters . 2016;18(3):444–447. doi: 10.1021/acs.orglett.5b03487. [DOI] [PubMed] [Google Scholar]
- 50.Scapin G., Patel S. B., Becker J. W., et al. The structural basis for the selectivity of benzotriazole inhibitors of PTP1B. Biochemistry . 2003;42(39):11451–11459. doi: 10.1021/bi035098j. [DOI] [PubMed] [Google Scholar]
- 51.Frisch M., Trucks G., Schlegel H. B., et al. Gaussian 09, Revision A. 02, Gaussian. Inc., Wallingford, CT. 2009, 200.
- 52.Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. The Journal of Chemical Physics . 1993;98(7):5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 53.Friesner R. A., Murphy R. B., Repasky M. P., et al. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. Journal of Medicinal Chemistry . 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 54.Friesner R. A., Banks J. L., Murphy R. B., et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. Journal of Medicinal Chemistry . 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 55.Laskowski R. A., Swindells M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. Journal of Chemical Information and Modeling . 2011;51(10):2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Figures S1~S43: general experimental procedures, X-ray crystal data of 1 and 2, biomimetic total synthesis of 1–4, PTP1B inhibitory assay, and molecular docking of 1–4.
Data Availability Statement
All data are available in the manuscript or supplementary materials.