

The technical genesis and practice of 8-aminoquinoline therapy of latent malaria offer singular scientific, clinical, and public health insights. The 8-aminoquinolines brought revolutionary scientific discoveries, dogmatic practices, benign neglect, and, finally, enduring promise against endemic malaria.
KEYWORDS: 8-aminoquinolines, CYP2D6, G6PD deficiency, Plasmodium vivax, hemolytic toxicity, latency, plasmochin, primaquine, tafenoquine, therapy
SUMMARY
The technical genesis and practice of 8-aminoquinoline therapy of latent malaria offer singular scientific, clinical, and public health insights. The 8-aminoquinolines brought revolutionary scientific discoveries, dogmatic practices, benign neglect, and, finally, enduring promise against endemic malaria. The clinical use of plasmochin—the first rationally synthesized blood schizontocide and the first gametocytocide, tissue schizontocide, and hypnozoitocide of any kind—commenced in 1926. Plasmochin became known to sometimes provoke fatal hemolytic crises. World War II delivered a newer 8-aminoquinoline, primaquine, and the discovery of glucose-6-phosphate dehydrogenase (G6PD) deficiency as the basis of its hemolytic toxicity came in 1956. Primaquine nonetheless became the sole therapeutic option against latent malaria. After 40 years of fitful development, in 2018 the U.S. Food and Drug Administration registered the 8-aminoquinoline called tafenoquine for the prevention of all malarias and the treatment of those that relapse. Tafenoquine also cannot be used in G6PD-unknown or -deficient patients. The hemolytic toxicity of the 8-aminoquinolines impedes their great potential, but this problem has not been a research priority. This review explores the complex technical dimensions of the history of 8-aminoquinolines. The therapeutic principles thus examined may be leveraged in improved practice and in understanding the bright prospect of discovery of newer drugs that cannot harm G6PD-deficient patients.
INTRODUCTION
Latency in malaria may be described as the asymptomatic persistence of infection by quiescent forms inherently not susceptible to therapies applied to treat the acute attack. Among the human malarias routinely exhibiting latency—those caused by Plasmodium vivax, Plasmodium ovale, and Plasmodium malariae—acute vivax malaria dominates as a global cause of morbidity and mortality, occurring in tens of millions in tropical and temperate Asia, Africa, and the Americas (1). The hepatic latency of endemic P. vivax endows it with a great resilience against conventional methods of malaria control (2). Primary and secondary acute attacks alike often provoke an incapacitating febrile illness that may progress to severe and life-threatening complications, like severe anemia or thrombocytopenia, hemorrhage, acute respiratory distress, hepatic or renal dysfunction, seizures or coma, multiple organ dysfunction, circulatory collapse, and splenic rupture or infarct (3–6). Prompt diagnosis and effective blood schizontocidal therapy arrest the acute attack, with nearly complete recovery occurring within several days (7), but when treatment excludes a hypnozoitocidal therapy aimed at the latent stages of P. vivax, recurrent clinical attacks called relapses occur in the weeks, months, or several years following the primary attack.
Not all patients with acute P. vivax infection left untreated with a hypnozoitocide will relapse, but most will relapse at least several times at rates and periods varying with the geographic origin of infection (8, 9). Scientific understanding of this phenomenon dates to 1901, when physician P. T. Manson reported his relapse in Britain 9 months following quinine therapy of a mosquito-induced primary attack arranged by his famous father, Sir Patrick Manson (10). A single infective inoculation of P. vivax may be thought of as two biologically distinct infections: (i) a single sporozoite-borne primary attack occurring within about a week or two of inoculation and (ii) one to five or more hypnozoite-borne secondary attacks occurring later without further involvement of a mosquito. This review considers the appropriate therapy of both types of infections after the onset of the primary attack, with particular attention given to hypnozoitocidal treatment against latency. The combined therapies are often called radical cure.
The century-long use of 8-aminoquinoline drugs against relapse provides essential historical context and technical detail that underpin a practical understanding of this complex class of therapeutics of enduring great importance in combating the problem of the human malarias. In response to militarily consequential quinine shortages during World War I, the German pharmaceutical company Bayer mobilized a groundbreaking scientific effort to discover synthetic antimalarials. In 1926, it introduced an 8-aminoquinoline, called plasmochin, for the treatment of acute malaria (11), unaware of the impact of this drug on the delayed attacks of latent vivax malaria. Practitioners sought to mitigate the immediately recognized dangerous hemolytic toxicity of plasmochin by administering lower daily doses of it augmented with daily quinine (12), only then recognizing the remarkable ability of that combination to prevent delayed attacks. Radical cure of vivax malaria employing a combination of complementary therapies, blood schizontocidal and hypnozoitocidal, against acute and delayed attacks was thus unwittingly invented. Nonetheless, the unsolved hemolytic toxicity problem greatly impeded the clinical and public health utility of plasmochin.
The loss of the island of Java in the East Indies (modern Indonesia) to an occupying enemy in early 1942 during World War II denied the Allied powers access to the Dutch colonial cinchona plantations that produced almost the entire world quinine supply (13). The event forced the Americans, at war with Germany, to seize Bayer’s trove of synthetic antimalarial intellectual property for dealing with a serious military malaria problem (13). This initially meant using plasmochin combined with another Bayer drug, a blood schizontocide called atabrine, for radical cure. However, in 1943 an unexpected drug-drug interaction disqualified the combined therapy and the Allies became defenseless against relapsing malaria (14). A war-spurred search for a newer 8-aminoquinoline was thus hurriedly set into motion and was expediently focused solely on that chemical class (15).
The animal model for relapsing human vivax malaria, rhesus macaques infected by Plasmodium cynomolgi, was not available for the early preclinical screening of candidate 8-aminoquinolines (16). Winnowing of the many dozens of candidate compounds was limited to those with activity against avian malarias lacking latency and toxicity assessments in birds, rodents, dogs, and monkeys, without any understanding of glucose-6-phosphate dehydrogenase (G6PD) deficiency as the key toxicity problem for this class of compounds (17). Several dozen advanced to clinical development in trials empowered by experimental mosquito-borne P. vivax sporozoite challenge of inmates at two American prisons during the middle to late 1940s and early 1950s (18). Understanding of the role of G6PD deficiency in hemolytic toxicity would come only in 1956, 4 years after the registration of primaquine with the U.S. Food and Drug Administration (FDA). The early preclinical screening of the 8-aminoquinolines occurred in systems unable to recognize or optimize either efficacy against hypnozoites or the crucial hemolytic toxicity problem. The war-spurred development of primaquine for soldiers by those systems delivered a drug seemingly suited to them, but it proved deeply flawed for practical public health use in the impoverished rural tropics where malaria is endemic (19).
Through the second half of the 20th century, the inadequacy of primaquine to an endemic hypnozoite reservoir problem seemed to not register as a priority with the communities of science, medicine, and public health. Until the 2018 introduction of a new 8-aminoquinoline called tafenoquine, primaquine had been the sole therapeutic option for dealing with that reservoir. These drugs invariably provoke acute hemolytic anemia in patients having G6PD deficiency, the most common human genetic abnormality, affecting about 8% of residents in countries where malaria is endemic (20). The extent of harm depends on a complex array of defining factors: the drug and its dosing, the G6PD variant involved, whether the individual is hemi-, homo-, or heterozygous for the X-linked trait, the extent of lyonization among heterozygotes, and the health of the patient at treatment (21). Providers unable to discriminate G6PD-normal from G6PD-deficient patients must often choose between risking harm in the unseen G6PD-deficient minority by prescribing primaquine or withholding it and inviting harm in all by repeated clinical attacks and onward transmission. The prolonged 14 days of primaquine dosing mitigates the potential for toxic harm but imposes adherence difficulties and does not relieve the necessity of clinical monitoring. In practice, where most malaria patients live—very often beyond the reach of clinical services capable of reliably assessing the risk or onset of serious harm and performing rescue of those in hemolytic crisis—few are or should be prescribed primaquine, and fewer still receive or complete the full course (22).
Primaquine thus remains fraught with risk and uncertainty in clinical and public health practice (23, 24). Likewise, the single dose of tafenoquine imposes the strict requirement of ensuring >70% of normal G6PD activity (25). The very substantial threat of the hypnozoite reservoir in endemic vivax malaria may be neutralized by the safe and effective application of these 8-aminoquinolines, which are indeed highly efficacious against it (26–29). Safe access to this singularly complex, highly beneficial, and quite dangerous class of drugs is deeply challenging where clinical care may be distant and limited. Assurance of availability, along with good adherence and safety, requires innovative technical, clinical, and public health solutions guided by appropriate evidence and a clarity of understanding of the practical limitations imposed where most malaria occurs.
Most of the core principles of 8-aminoquinoline therapeutics were accurately described nearly a century ago, but some became forgotten or disregarded as untowardly complex. This carries important repercussions today in the form of therapies not fully optimized or adequate to broadly effective practice involving most malaria patients. This review strives to consolidate varied lines of historic and contemporary evidence in expressing 8-aminoquinoline therapeutic principles in the radical cure of the equally complex latency of P. vivax. Those principles, in turn, identify specific gaps in understanding for research aimed at optimizing current practices involving available 8-aminoquinolines, along with the bright prospects for the discovery of much better ones.
ESSENTIAL TECHNICAL BACKGROUND
Parasites and Infection
In the minutes following natural inoculation of infectious sporozoites into a human by an anopheline mosquito, each parasite invades a single hepatocyte and commences a development process (hepatic schizogony) in which it is destined to end about a week or two later as a mature hepatic schizont containing thousands of merozoites equipped to invade red blood cells (30). The erythrocytic asexual reproduction of merozoites every 48 h (blood schizogony) provokes the typically daily febrile paroxysm of an attack of acute malaria (a 72-h cycle in patients infected with P. malariae). In the instance of P. vivax, but not Plasmodium falciparum, a distinct developmental process leads to an intracellular hepatic form called a hypnozoite. Sporozoites that directly develop into hepatic schizonts and initiate primary patency may be called tachysporozoites, whereas bradysporozoites become hypnozoites with variable periods of latency before activating to provoke delayed attacks called relapses (Fig. 1). Lysenko and colleagues first posited (31) and tested (32) this hypothesis of intrinsic sporozoite polymorphism and genetically defined discrete periods of hypnozoite dormancy in 1977. In 2007, Chen et al. (33) put forth essentially the same “intrinsic clock” hypothesis to explain the timing and singular genetic character of relapses in repatriated Australian soldiers infected at Timor Leste.
FIG 1.

Contemporary schematic representing the biology of Plasmodium vivax primary attack and relapses according to the polymorphic intrinsic latency period hypothesis proposed by Lysenko et al. in 1977 (31). Tachysporozoites launch immediate hepatic schizogony and then a single primary attack. Bradysporozoites develop to latent hypnozoites that later awaken to hepatic schizogony and patent relapses according to intrinsic latency period phenotypes.
The time from hepatic invasion to the emergence of tachysporozoite-borne merozoites into the bloodstream to cause the onset of illness in a primary attack is called prepatency (or incubation period), which is about 8 to 16 days for vivax malaria. Bradysporozoite-borne merozoites and consequent clinical attacks also derive from hepatic schizogony but commence only after a highly variable period of latency: the primary latency period represents the time between a primary tachysporozoite-borne attack and a bradysporozoite-borne first relapse. The period between subsequent relapses may be called the relapse interval. Primary latency varies anywhere from about 3 weeks to 3 years, whereas the relapse interval is usually, but not uniformly, about 2 months. The discrete intrinsic latency period character of any given brood of bradysporozoites and their numbers defines emergent relapse behavior phenotypes. Extrinsic triggers of relapse may occur, but the regularity of relapse behaviors in both natural and controlled environments points to intrinsic phenomena as being dominant. The model of relapse mechanics by underlying parasite genetics of Lysenko et al. (31) is considered here a best fit and most probable hypothesis underpinning distinct relapse behavior phenotypes.
The existence of prepatent or latent forms in some tissue other than the blood had been deduced and supposed by the 1900s, but implication of the liver as that tissue came only in 1948 (34), and the identification of hypnozoites in that organ came in 1982 (35). These had been referred to with necessary ambiguity as “exoerythrocytic” or “tissue” forms. Shannon’s 1946 review of the wartime U.S. government effort at antimalarial drug discovery applied this ambiguous but wholly accurate terminology and understanding of the P. vivax life cycle as it pertained to therapeutic objectives (15). Today we refer to these collectively and accurately as hepatic forms, with either actively developing prepatent schizonts or latent hypnozoites later yielding the same forms.
This review does not include consideration of the sparse evidence regarding presumptive antirelapse therapy (PART) for the other cause of relapsing human malaria, P. ovale, but it does examine relevant data from the animal model for relapse in P. vivax, rhesus macaques infected by P. cynomolgi. That model has been extensively used and is of proven relevance to therapy of latent P. vivax in humans (36, 37). As for P. ovale, recommendations for its therapy essentially mirror those for P. vivax, but with little supporting evidence due to the relative rarity of it, despite a broad geographic range through much of Africa, Asia, and Oceania where malaria is endemic (38, 39). The infection occurs at an extremely low prevalence, being rarely detected in mass blood surveys by microscopy, but it is often seen along with the similarly infrequent human malaria parasite P. malariae when more sensitive molecular diagnostic methods are applied (40, 41).
The Question of Latency in Plasmodium malariae
Although there is little doubt that P. malariae exhibits latency, sometimes for decades, no biological evidence explains it, and hypnozoites are today presumed to not be involved (42). However, the report of a board of experts on malaria chemotherapy convened by the WHO in 1960 (43) expressed the following regarding 8-aminoquinolines: “The main value of these drugs derives from their ability to destroy the exoerythrocytic forms of P. vivax and P. malariae, a property not common to any other chemical group so far tested.” A standard search of the literature using PubMed yielded no clinical trials of primaquine therapy against recurrent P. malariae, but numerous examples of it being used in this manner were reported. Gilles and Hendrickse (44), for example, described using primaquine for “radical cure” of P. malariae in their nephrotic patients. The question of the biological character of this latency is particularly important, given the association of chronic P. malariae infection with a threatening and presumably autoimmune injury to the kidneys (45). A recent study linked infection by P. malariae with a significant risk of morbidity due to anemia and of mortality comparable to that from both P. falciparum and P. vivax infection among hospitalized patients in Indonesia (46). Authoritative treatment advice for P. malariae today does not mention primaquine therapy because its latent stages are supposed to be persisting asexual blood stages and therefore susceptible to blood schizontocidal therapy (42). However, recurrent infections following what appears to be effective blood schizontocidal therapy have been reported; in one case report (47) and others reviewed therein, recurrent parasitemia differed genetically from that cleared 2 months earlier without a likelihood of reinfection. In the absence of definitive evidence to the contrary, the possibility of latent stages of P. malariae (in whatever form or tissue in which they may occur) not being susceptible to effective blood schizontocides should be considered. If broadly effective primaquine indeed terminates latency in this infection and narrowly effective blood schizontocides do not, 8-aminoquinolines may serve a role in the treatment and elimination of P. malariae.
Chemotherapy
8-Aminoquinolines have broad activity against asexual and sexual blood stages, hepatic schizonts and hypnozoites, and sporogonic stages developing in the mosquito host (sporontocidal activity; Fig. 2). This review focuses on the treatment of the hypnozoites responsible for latency in P. vivax. The killing of these hepatic forms by 8-aminoquinolines combined with blood schizontocidal agents, like quinine or chloroquine for arresting the acute attack, has long been called radical cure, i.e., clearing of all parasites regardless of stage or tissue. Technically, though not in practice, the same term could be applied to acute P. falciparum infection treated with both blood schizontocidal therapy and a single gametocytocidal dose (0.25 mg/kg of body weight) of primaquine. Although the term is antiquated and somewhat ambiguous, radical cure remains in common use to describe combined hypnozoitocidal and blood schizontocidal therapy for latency with the relapsing malarias.
FIG 2.

Antimalarial drug classes (red lettering), preventive strategies (bold black), and the life cycle of the plasmodia.
Another antiquated term, terminal prophylaxis, carries more ambiguity and creates confusion. Travelers using a suppressive chemoprophylactic antimalarial (i.e., a blood schizontocide) during exposure to infection must consume a curative regimen of primaquine at the end of travel in order to prevent relapses by hypnozoite-bearing species. This has been called terminal prophylaxis, and it is precisely the rationale for primaquine therapy following a diagnosis of those malarias, i.e., prevention of relapses in a patient presumed to harbor hypnozoites. No diagnostic technology reveals the presence of hypnozoites, and any treatment against them—be it in a healthy traveler or a patient with acute vivax malaria—is therefore presumptive. The term presumptive antirelapse therapy (PART) simplifies and clarifies the terminology of therapeutic strategies and tactics aimed at preventing relapse following either travel (posttravel PART) or a diagnosis of vivax malaria (postpatency PART) (48).
The term schizontocidal requires qualification as hepatic or blood to avoid ambiguity. Blood schizontocides are sometimes referred to simply as schizontocides, but that broader class includes hepatic schizontocides, and the distinction is important in practice. Hepatic schizontocides may sometimes also be blood schizontocidal and hypnozoitocidal, e.g., primaquine against P. vivax, but the dose and regimen for each activity are distinct and of varying clinical context and relevance (Table 1). Primaquine is a proven blood schizontocide in vivax malaria (49, 50) but is weakly so and is not used as such in practice. Its hepatic schizontocidal activity comes into play only when prescribed as daily causal prophylaxis (51, 52), where a single 30-mg adult dose given within 48 h of sporozoite inoculation but not later (53) suffices to prevent vivax or falciparum malaria. The killing of formed hypnozoites, in contrast, requires a total dose of 210 to 420 mg delivered over 7 days to 8 weeks (54).
TABLE 1.
Therapeutic/prophylactic indications and dosing of available 8-aminoquinolines in vivax and falciparum malarias
| Compartment and type of activity | Therapeutic activity or dose |
|||
|---|---|---|---|---|
| Primaquine |
Tafenoquine |
|||
| P. vivax | P. falciparum | P. vivax | P. falciparum | |
| Blood | ||||
| Schizontocidal | Good activity but not prescribed | No activity and not prescribed | Good activity but not prescribed | Good activity but not prescribed |
| Gametocytocidal | Good activity but not prescribed | 15-mg single adult dose | Good activity but not prescribed | Good activity but not prescribed |
| Hepatic | ||||
| Schizontocidal | 30-mg single adult daily dose as causal prophylaxis, off label | 30-mg single adult daily dose as causal prophylaxis, off label | Single weekly 200-mg adult dose as causal prophylaxis, labeled | Single weekly 200-mg adult dose as causal prophylaxis, labeled |
| Hypnozoitocidal | 210–420-mg total adult dose over 7 days to 8 wk | No hypnozoite, not prescribed | Single 300-mg adult dose | No hypnozoite, not prescribed |
Synonymous common names for drugs haunt the nomenclature of antimalarials. Table 2 provides a useful guide to those. Priority in this review is given to the name offered by the discoverers of any given compound, but with the necessary exception of the 4-aminoquinoline compound discovered by Johann Andersag at Bayer’s Elberfeld laboratory in 1934, where it was named resochin. A decade later, the Americans at war obtained this drug and put it forward through clinical development, naming it chloroquine at registration in 1946 (55). Andersag’s resochin became so widely known as chloroquine that putting forth any alternative now is impractical. Andersag’s colleagues at Elberfeld discovered and commercially distributed the 8-aminoquinoline that they called beprochin and pamachin in development but later marketed as plasmochin (13). That drug was widely used and reported on by precisely that name (or plasmoquine) between 1927 and 1952, but the Americans developing primaquine later referred to plasmochin most often as pamaquine. Plasmochin is central to the subject of this review, and it is referred to by that name in acknowledgement of the discoverers of it. The Bayer scientists also discovered and marketed an 9-aminoacridine (a structural precursor of their 4-aminoquinolines) that they called atabrine but that others referred to as mepacrine or quinacrine.
TABLE 2.
Synonymous common names among quinoline antimalarials
| Drug | Chemical class | Experimental designation(s) | Synonymous name(s) | Trade name(s) |
|---|---|---|---|---|
| Hypnozoitocides | ||||
| Plasmochin | 8-Aminoquinoline | SN-971 | Plasmoquine, beprochin, pamaquine, pamaquin, praequine | Plasmochin, Gamefar, Quipenyl |
| Quinocide | 8-Aminoquinoline | SN-15333 | Quinocid, chinocide | |
| Plasmocid | 8-Aminoquinoline | SN-3115 | Rhodaquine, plasmocide | Fourneau-710 |
| Pentaquine | 8-Aminoquinoline | SN-13276s | ||
| Isopentaquine | 8-Aminoquinoline | SN-13274 | ||
| Primaquine | 8-Aminoquinoline | SN-13272, WR2975 | Primax, Primal, Primacip, Malirid | |
| Tafenoquine | 8-Aminoquinoline | WR238605, SB-252263 | Etaquine | Krintafel, Arakoda |
| Blood schizontocides | ||||
| Atabrine | 9-Aminoacridine | SN-11437 | Quinacrine, mepacrine, atebrin, acrinamine | Erion, Quinactine, Akrichin |
| Chloroquine | 4-Aminoquinoline | SN-7618, WR1544 | Resochin | Resochin, Aralen, Nivaquine |
| Sontochin | 4-Aminoquinoline | SN-183 | Sontoquine |
The historic literature considered here applied what may be considered an archaic and sometimes imprecise expression of drug doses. Virtually all of the 8-aminoquinoline literature before 1990 refers to adult daily doses with a presumption of 14 days’ duration rather than daily milligram-per-kilogram amounts; i.e., standard daily doses of 90 mg plasmochin or 30 mg primaquine over a fixed 2-week period would yield total doses of 1.25 g or 420 mg, respectively, or 21 mg/kg and 7 mg/kg, respectively. In almost all instances, the daily dose administered referred to the base exclusive of the weight of varied salts, with the notable exception of quinine therapy (doses often included the weight of the common sulfate salt formulation). These conventions are followed in this review inclusive of older literature, but only up to about 2000, when, for example, 0.5 mg/kg for daily primaquine (for the 2-week standard) or a 7.0-mg/kg total dose (regardless of the duration of dosing) became the preferred standard expression of primaquine treatment.
Total Dose Effect
Wide arrays of dosing strategies appear in the histories of both plasmochin and primaquine. Although each is rapidly eliminated and extensively metabolized, the schedule and duration of administration of an effective total dose vary from days to months. The familiar standard of a single daily dose over 2 weeks evolved with early plasmochin use as a means of mitigating toxicity. The developers of primaquine later followed that strategy with the same dosing regimen and intent and also developed a less hazardous single weekly dosing regimen for 8 weeks. Schmidt and colleagues (56) later demonstrated that prevention of relapse of P. cynomolgi infection in rhesus macaques could be achieved with the same total dose of 8-aminoquinolines administered once, divided daily over a wide range of days, or weekly over several months. Those findings validated what Schmidt et al. (56) termed the “total dose effect”; i.e., the schedule of administering 8-aminoquinoline hypnozoitocidal therapy had almost no impact on efficacy, provided that the same efficacious total dose was delivered. The specific injury that 8-aminoquinolines could inflict on hypnozoites, whatever that may be, thus appears to be irreversible and cumulative in effect.
This fact provides wide flexibility in dosing strategies often driven by consideration of hemolytic toxicity in patients of unknown G6PD status. The developers of primaquine variably considered the maximal tolerated daily dose of primaquine to be 120 or 240 mg in primaquine-insensitive subjects but deemed a single 15-mg daily dose to be relatively safe (albeit still hemolytic) in primaquine-sensitive patients. At first unaware of G6PD deficiency and unable to screen against it, the regimen of 15 mg daily for 14 days thus emerged as the recommended regimen and was widely practiced and perceived as safe without sensitivity screening or clinical monitoring. Decades would pass before realization of the lack of safety for any regimen of 8-aminoquinoline hypnozoitocidal therapy without clinical monitoring or G6PD screening. Likewise, leveraging the total dose effect in patients of known G6PD-normal status to compress the duration of primaquine dosing in order to improve effectiveness would also take the same decades: a recent trial of 7 versus 14 days for delivering a total dose of 7.0 mg/kg demonstrated the similar efficacies of the shorter and longer regimens against relapse in G6PD-screened P. vivax-infected patients (57, 342). G6PD-normal patients may safely consume relatively very large single or daily doses of 8-aminoquinolines that are unacceptably dangerous to G6PD-deficient patients.
Toxicity
The 8-aminoquinoline literature often refers to the well-known toxicities of the 8-aminoquinolines but especially the specific problem of their hemolytic toxicity in G6PD-deficient patients. It may be useful to conceptually separate that toxicity problem from chemical toxicity in all humans without regard to G6PD status, i.e., neural, hepatic, and hematologic toxicity in the broader sense (58). The screening of 8-aminoquinolines during the 1920s and the 1940s included the winnowing of compounds exhibiting conspicuous toxicity in birds, rats, dogs, and monkeys. These delivered compounds of generally good tolerance and safety in most people but not in those hemolytically sensitive to them by the inherited G6PD deficiency abnormality. All 8-aminoquinolines evaluated have proven hemolytic toxicity in those patients; it appears to be a class-wide defect of presumably variable degrees among them.
The terms toxicity or safety and tolerability applied to experimental 8-aminoquinolines in the broader context were almost always exclusive of the special case of hemolytic toxicity in G6PD-deficient subjects. When Clayman and colleagues compared the “maximum tolerated daily [adult] dose” of primaquine versus plasmochin at 120 mg and 63 mg, for example, they referred only to G6PD-normal subjects (59). The same dose ranging in African-American G6PD-deficient subjects found that whereas the hemolysis occurring with primaquine at 15 mg daily could be tolerated, that occurring with primaquine at 30 mg daily could not (60). The minimum effective daily adult dose of primaquine and plasmochin (administered for 14 days) against P. vivax, 22.5 mg and 90 mg, respectively (61), alone supported the assertion of relative safety in the broadest sense, inclusive of the hemolytic sensitivity problem. In other words, greater safety presumed lower hemolytic toxicity with the lower dosing of primaquine. Assumed molar equity in hemolytic toxicity between the two compounds translated as therapeutic primaquine being less hemolytic than therapeutic plasmochin. However, the two drugs were not compared directly with regard to relative hemolytic toxicity in sensitive subjects of any G6PD deficiency variant. No evidence supports the presumption of molar equity in hemolytic toxicity among the 8-aminoquinolines, and variable relative hemolytic toxicities may well be clinically significant. This key characteristic of the 8-aminoquinolines—degrees of variation in relative hemolytic toxicity with molecular character—remains an unknown of great practical and theoretical importance, as will be explained and illustrated later in the context of the development of tafenoquine.
A constant feature of 8-aminoquinoline toxicity in both G6PD-normal and -deficient patients is a usually mild and reversible methemoglobinemia at therapeutic dosing. Some workers accurately refer to this as a phenomenon of hemotoxicity, but methemoglobinemia has not been causally linked with the more specific problem of hemolytic toxicity. The direct involvement of methemoglobinemia in the drug-induced destruction of red blood cells is not established, and at least some early workers considered it to be a phenomenon unrelated to acute hemolytic anemia (60, 62). 8-Aminoquinolines also have known toxicity to formed elements of blood, like reversible granulocytopenia (63). Thus, “hemotoxicity” should be considered a general feature of broad toxicity not specific to or necessarily mechanistically linked to the very specific problem of hemolytic toxicity.
The hemolytic toxicity problem of the 8-aminoquinolines defines almost every feature of their use in practice in striving to mitigate the real risks thus engaged, and yet the problem itself evades precise definition: the mechanism of hemolysis is not known, and the degree of hemolytic toxicity is thus not predictable without direct observation in sensitive human subjects or patients. Quantitative measurement of hemolytic toxicity in anyone given an 8-aminoquinoline applies that crudest and most impractical means possible: the extent of hemolysis (typically assessed by measures of hemoglobin in whole blood) observed in exposed and vulnerable humans. Up to the present day, no technology has permitted preclinical winnowing of 8-aminoquinolines by relative hemolytic toxicity, though many investigators have tried (64–68). Recent work with zebrafish or humanized mice having G6PD-deficient red blood cells represents the latest attempts to do so (69, 70). The first reliable assessment of the relative (to primaquine) hemolytic toxicity of tafenoquine came from a phase 2a trial (71) undertaken more than 25 years after its registration as an investigational new drug.
Metabolism
The complex metabolism of the 8-aminoquinolines in at least several mammalian systems has been studied over the decades (72–75). This review will not fully explore that rich literature but instead focuses on aspects of it of apparent direct relevance to therapy and toxicity. Among the many metabolic derivatives known, the 5-hydroxylated species (with perhaps variable modifications to the 8-amino alkyl chain) emerge as probably the most relevant (76, 77). These highly redox-reactive derivatives appear to be generated by the 2D6 isozyme of cytochrome P-450 (CYP2D6), and their relatively minute amounts and instability impose technical challenges in studying them (78–80). The most widely studied metabolite, a carboxylated species of the parent drug (carboxyprimaquine) produced by reaction with monoamine oxidase, also happens to be the most abundant, stable, slowly eliminated, and easily detected, but it is without apparent therapeutic or toxic activity (81, 82).
In 2016, Marcsisin et al. (83) reviewed the large body of evidence pointing to the involvement of CYP2D6 in generating the therapeutic and toxic metabolic derivatives of primaquine. That evidence included the 2013 report of Bennett et al. (84) describing the therapeutic failure of primaquine against relapse in 2 of 25 experimentally challenged subjects in the United States employing P. vivax-infected mosquitoes from Thailand. Both subjects possessed impaired CYP2D6 genotypes, whereas most successfully treated subjects did not. In 2018, Baird et al. (85) examined the CYP2D6 genotypes and dextromethorphan metabolism phenotypes of subjects in a case-control study nested within a randomized trial of 177 subjects treated with directly observed high-dose primaquine and blood schizontocidal partners for radical cure of acute vivax malaria (27). These infections were acquired in eastern Indonesia, but diagnosis, enrollment, treatment, and prolonged follow-up (12 months) occurred on the Indonesian island of Java, where reinfection was highly improbable. Recurrent infections in these subjects represented nearly unambiguous evidence of relapse and, therefore, therapeutic failure. The case-control study enrolled 21 of the 26 observed therapeutic failures along with 36 controls randomly selected from among the 151 subjects not relapsing and therefore eligible as such. Twenty of the 21 subjects experiencing relapse but relatively few controls had impaired CYP2D6 metabolism (odds ratio [OR] = 18; 95% confidence interval [CI] = 3 to 140). CYP2D6 appears to generate the primaquine metabolite(s) responsible for killing hypnozoites.
Available evidence for plasmochin, primaquine, and tafenoquine in animals and humans suggests that these are prodrugs metabolized to therapeutically active and toxic derivatives, although the evidence for tafenoquine remains incomplete and unresolved (86–88). This exposes both of those features of these drugs to potentially important impacts of pharmacogenetic and pharmacokinetic or pharmacodynamic factors. Evidence reviewed below certainly implies the efficacy- and toxicity-defining importance of each occurring in practice. Understanding the effects of 8-aminoquinolines on the plasmodia and their human hosts first requires understanding how that host and other therapies in them impact the metabolic processing of active and toxic 8-aminoquinoline metabolites. No evidence firmly demonstrates those to be the same derivatives, but some observations later reviewed here suggests that that may be the case.
RELAPSE BIOLOGY AND CHEMOTHERAPY
Antimalarial drug classes have defined specific activities against a particular stage in the complex development cycle of the plasmodia (Fig. 2). Most therapeutically applied drugs belong to the blood schizontocidal class, aimed at arresting the acute attack of malaria provoked by blood schizogony. Among the other classes—hypnozoitocidal, hepatic schizontocidal, gametocytocidal, and sporontocidal—there are only the 8-aminoquinolines available, and, remarkably, they each exert activity across those therapeutic classes. Some of that activity may be narrowly poor, e.g., in the specific instance of primaquine against asexual blood stages, particularly those of P. falciparum but much less so those of P. vivax. Some blood schizontocides exert what is often a species- or stage-specific activity outside of their class, e.g., artemisinin against immature gametocytes of P. falciparum, chloroquine or quinine against mature gametocytes of P. vivax, or atovaquone against tissue schizonts of P. falciparum but not P. vivax (89–91). In general, however, the blood schizontocides are narrowly class specific, whereas 8-aminoquinolines exhibit activity across antimalarial classes. Except for the clinically useful causal prophylactic (hepatic schizontocidal) and gametocytocidal activities of 8-aminoquinolines against all plasmodia, these drugs are optimized for and directed against formed latent hypnozoites seeding the livers of patients infected by P. vivax or P. ovale. Clinically speaking, the blood schizontocidal and sporontocidal class activities of the 8-aminoquinolines are incidental and typically unimportant, although the former may play a role in tafenoquine for chemoprophylaxis (see below).
Understanding the effects of any given chemotherapeutic agent on a plasmodial infection thus requires careful consideration of the underlying biology. An anopheline mosquito-borne infective inoculation may typically contain about a dozen sporozoites (92–95). Ratios of the specific types of them tend to vary with the geographic origin of the strain, with tachysporozoites dominating tropical strains and bradysporozoites dominating those from temperate latitudes (96–98). Unless overwhelming numbers of sporozoites of the North Korean strain of P. vivax are inoculated, for example, no primary attack occurs (96). The ratio of brady- to tachysporozoites must be one in hundreds or thousands in that strain. About 70% to 85% of Indian strains of P. vivax, in contrast, will not relapse (99, 100), and those inocula must be dominated by relatively high ratios of tachysporozoites. The relapse-prone tropical strains of P. vivax usually include relatively balanced ratios of both types (101, 102), as hypothetically represented in Fig. 1.
According to the original hypothesis of polymorphic sporozoites in vivax malaria (31), each bradysporozoite-borne hypnozoite possesses a discrete intrinsic dormancy period ranging from about 3 weeks to several years. Each relapse represents the activation of coinoculated hypnozoites sharing that latency periodicity phenotype, or, more typically, given the spare numbers involved, a discrete hypnozoite seems to usually be involved (33), but not always (103). The timing, frequency, and numbers of relapses occurring in patients (relapse behavior) are thus very likely defined by the ratio of tachy- to bradysporozoites, the total numbers of them inoculated within a single infectious bite, the discrete latency period phenotypes among them, and the number of inoculations occurring within the span of any given latency period. Repeatedly exposed patients may harbor multiple broods of hypnozoites yielding 30 or more distinct relapses (104). One study estimated five to be the average number of hypnozoites harbored by P. vivax-infected subjects living in Thailand (105). Another study estimated that only about 20% (i.e., 1 in 5) of cases of incident patent vivax malaria observed in residents of Papua New Guinea, where malaria is endemic, were mosquito-borne primary attacks (106).
This biology has important ramifications with regard to assessing the therapeutic efficacy of 8-aminoquinolines against hypnozoites. A patient in an area of endemicity with a recurrent parasitemia following radical cure may be experiencing therapeutic failure of the blood schizontocide (a recrudescence), failure of the hypnozoitocide (a relapse), or a new mosquito-borne primary attack (a reinfection). No available molecular technology distinguishes a relapse from a reinfection, and renewed malaria attacks are referred to with correct and necessary ambiguity as recurrences. Unless the subjects of hypnozoitocidal trials are removed from areas where transmission is endemic, the confounding of reinfection among recurrences will diminish estimations of efficacy. Moreover, in terms of the clinical benefits delivered by hypnozoitocides, few trials have assessed the multiplicity of relapses following inadequately treated or untreated infections. The clinical and public health benefits of PART with 8-aminoquinolines may thus be substantially underestimated in conventional clinical trials of them. On the other hand, if local P. vivax parasites tend to relapse late (>6 months) or relatively infrequently, as in India or the Korean Peninsula, the seemingly good efficacy of 8-aminoquinolines may be an illusion revealed only with a relapse control arm (i.e., no hypnozoitocide) and prolonged follow-up (>6 months). In India, for example, the inadequacy of a 5-day primaquine regimen (evaluated in uncontrolled trials and practiced for decades) became apparent only after conducting trials of this appropriate design (107–109).
There must be a very substantial hypnozoite reservoir standing in most communities where malaria is endemic, streaming new attacks and onward transmission therein. Models suggest that eliminating that reservoir would relatively quickly lead to the arrest of transmission in areas of endemicity (110, 111), but that has not occurred in most zones where malaria is endemic, despite a full century of access to 8-aminoquinoline therapies having highly efficacious hypnozoite-killing activities. The hemolytic properties of that class in G6PD-deficient patients, together with the long neglect of P. vivax as a clinical and public health problem (112–115), largely explain the conspicuously poor access to 8-aminoquinolines where they are most needed. The solution to the problem of relapse, so intimately tied to also solving the problem of 8-aminoquinoline hemolytic toxicity, promises extraordinary gains against endemic vivax malaria. The complex technical history that follows here aims to explain the failure to fully exploit 8-aminoquinoline therapeutic potentials in coping with a hemolytic toxicity problem that events of the past century demonstrate to be disabling (19, 116).
TECHNICAL HISTORY OF 8-AMINOQUINOLINES
Before Hypnozoitocides
In 1856, an audacious British teenager named William Henry Perkin birthed the aniline dye industry when he stumbled upon purple mauveine dye in coal tar in an academic effort aimed at synthesizing quinine in his laboratory at home (117). A German silk merchant at Elberfeld, near Dusseldorf, Germany, Friedrich Bayer, and textile dyer Friedrich Weskott formed Friedrich Bayer & Co. in 1863 to commercially exploit the new anilines (118). Perkin’s aniline compounds later enabled Paul Ehrlich’s development of dyes for the precise differential staining of pathological specimens, including fuchsine and methylene blue (119). Ehrlich reasoned that the observed affinity of methylene blue for malaria parasites in fixed blood films could translate into therapeutic activity in living systems, and in 1891, he and Guttman (120) reported the curative effects of methylene blue in two patients with acute vivax malaria. Some consider this report the birth of medicinal chemistry—validation of Ehrlich’s revolutionary idea of bespoke chemical “magic bullets” against microbes and disease—but 3 decades would pass before serious follow-up efforts to synthesize optimized antimalarial compounds could occur (Fig. 3A) (121, 122). Without animal models for toxicity or malaria and necessarily sophisticated chemical synthesis methods, systematic chemical experimentation optimizing safety and efficacy was not yet possible. Methylene blue did nonetheless find limited use as malaria therapy where P. falciparum was considered refractory to quinine, e.g., in Brazil up to 1910 (123). It was not until 1907 that Ehrlich put forth the term “chemotherapy” for the treatment of illness with rationally derived chemicals (13).
FIG 3.

(A) The discoverers of aniline dyes and methylene blue therapy of malaria: on the left, audacious teenager William Henry Perkin (a self-portrait in 1852, aged just 14 years, 4 years before he discovers mauvine in coal tars), and on the right, physician-scientist Paul Ehrlich (in Berlin, 1910; by Alfred Krauth, courtesy of the Wellcome Collection), who cured acute vivax malaria in two patients in 1891. (B) Werner Schulemann (top left, in 1924) and his colleagues August Wingler (top right, in 1934) and Fritz Schoenhoefer (bottom left, in 1939) at Bayer’s Elberfeld facility synthesized plasmochin in 1925. (Bottom right) The Bayer company logo in ca. 1925. Photos courtesy of Bayer AG, Corporate History & Archives, with permission. (C) (Top) Wilhelm Roehl and an unidentified colleague at the Bayer Elberfeld laboratory dosing Javanese rice finches infected by Plasmodium relictum with experimental antimalarial compounds in 1926. Roehl identified plasmochin to be 60 times more effective than quinine against that asexual blood-stage infection. (Bottom left) Portrait of Wilhelm Roehl in the same year. These photos courtesy of Bayer AG, Corporate History & Archives, with permission. (Bottom right) John A. Sinton (in ca. 1938), the prodigious British military malariologist who defined radical cure of Plasmodium vivax in British soldiers in India with optimized therapy with plasmochin combined with quinine and, later, atabrine (photo courtesy of the Wellcome Collection, London, United Kingdom, with permission).
Between 1913 and 1923, Dutch industrialists managed to consolidate a global monopoly on the production and sale of quinine by means of their privately owned and managed Cinchona Bureau (124). This followed decades of deliberate and sophisticated horticultural efforts by Franz Wilhelm Junghuhn during the 1800s in the East Indies (modern Indonesia). His methods allowed the development of productive cinchona plantations on the high volcanic ridges of western Java where uniquely high-yield bark for quinine production were cultivated (125, 126). In part due to the loss of German influence within that cartel and that nation suffering militarily consequential quinine shortages near the end of World War I, the German pharmaceutical firm Bayer vitalized efforts to develop synthetic antimalarials in this era (11).
Shortly after the enabling description of the studies of quinine therapy against Plasmodium relictum in canaries by the Sergent brothers (127) in 1921, Bayer gathered specifically skilled scientists at its Elberfeld laboratory of IG Farbenindustrie (Interessen Gemeinshaft Farberwerke) with the intent of synthetic antimalarial drug discovery (Fig. 3B) (13, 122). By 1924, Wilhelm Roehl, who had worked on therapies for trypanosomiasis in rodents under Ehrlich, mastered and standardized the P. relictum model in Javanese rice finches (Padda oryzivora) for the industrial-scale screening of experimental compounds (128). Roehl and his colleagues undertook a revolutionary task: identify new compounds effective against avian malarias that may also be effective against human malaria.
Those compounds derived from Ehrlich’s methylene blue but were derived with a deliberate eye on the quinoline nucleus of quinine (13) (Fig. 4). At first employing quinine as the lead molecule, Roehl’s efforts were punctuated by repeated failures to discover compounds of any potential clinical interest (13). Turning to methylene blue—then the only other chemical having proven plasmodium-killing activity—he quickly identified dozens of compounds of great potential. Other histories of this work offer visual representations of the rational evolution of the structures of those compounds (13, 122). Roehl’s screening included useful therapeutic indices measuring blood schizontocidal therapeutic activity relative to toxicity, an innovative legacy of Ehrlich’s (122). Importantly, however, his bird model, as with all other models involving birds and their natural malarias, lacked latent exoerythrocytic forms comparable to those responsible for the delayed attacks of vivax malaria (129). The discovery of 8-aminoquinolines involved no intent or ability to target latency in vivax malaria, but a century later, no other class of compounds has been identified to be hypnozoitocidal. As with the discovery of quinine centuries earlier, the discovery of hypnozoitocides was wholly serendipitous.
FIG 4.

Methylene blue as the progenitor of modern synthetic 4- and 8-aminoquinoline antimalarial drugs beginning in 1921 at the Bayer laboratory at Elberfeld near Dusseldorf, Germany, and continuing up to the present day (with tafenoquine). Mefloquine (bottom right) is not a 4-aminoquinoline but a quinoline methanol structurally related to halofantrine and lumefantrine.
The scientists providing Roehl with test compounds were Werner Schulemann, Fritz Schoenhoefer, and August Wingler (Fig. 3B), also at Elberfeld. As they closed in on 8-aminoquinolines with the structure-activity relationships becoming clearer, they narrowed these many prochin derivatives (A-prochin, B-prochin, C-prochin, etc.) in Roehl’s model, finally adding a 6-methoxy after previously discovering its necessity for activity in the quinoline ring of quinine (13). All effective 8-aminoquinoline antimalarials, past and present, possess that 6-methoxy group (Fig. 4). In 1924, they synthesized an 8-aminoquinoline that they called beprochin (the B-prochin from the series, which they later called pamachin and finally marketed as plasmochin) and that proved to be 60 times more active than quinine against asexual blood stages of P. relictum in Roehl’s finches (Fig. 3C) (130). Promising compounds from Elberfeld, like plasmochin in 1925, were evaluated by Franz Sioli in dozens of patients given induced vivax malaria for therapy of neurosyphilis at a psychiatric hospital nearby in Dusseldorf, Germany, and then at a seaman’s hospital in Hamburg, Germany, seeing imported malaria patients (11, 122). The complex stepwise means of molecular therapeutics discovery had been effectively assembled at Bayer’s Elberfeld facilities and accessible hospitals.
That extraordinary work heralded the dawn of molecular discovery in chemotherapeutics. Analogs of quinine and methylene blue and the sophisticated synthetic chemistry producing them, shrewdly coupled to Roehl’s malarious finches and then to induced and imported malaria in human subjects, ultimately provided humanity with chloroquine and primaquine, the mainstay antimalarials through the second half of the 20th century. Countless millions of lives have been spared from malaria alone in the course of the past century by the groundbreaking basic science done at Elberfeld between 1921 and 1939. Those achievements represented an event of signal importance to modern medicine that became overshadowed by a catastrophic global war and are today often not accounted for or acknowledged.
The massive wartime antimalarial development effort in the United States that later occurred (see below)—a multipartner public-private endeavor under precise bureaucratic control by empowered committees of that government—would be held up as the model of discovery in modern biomedical research (13). In fact, however, the core discoveries of 4- and 8-aminoquinolines had already been made by a small group of scientists working in a single laboratory supported by a pharmaceutical firm working in commercial isolation and motivation. The American effort would validate Bayer’s resochin as chloroquine and improve its plasmochin as primaquine, but discoveries of other classes of antimalarial compounds by the Americans (and the wartime development of proguanil by the British) carried relatively minor lasting impacts. In 1955, Schmidt and Coatney (131) (each played direct and important roles in the American development efforts) expressed, “with the possible exception of the work associated with the discovery of pyrimethamine, none of these efforts [from 1946 to 1954] was of great weight nor did they produce results which now command attention either at the practical or theoretical level.” Events of the 6 decades that followed largely vindicate that early modest view of the American antimalarial discovery program. The 4- and 8-aminoquinolines discovered at Elberfeld stand today as singularly relevant and vitally important in combating an onerous and stubborn malaria problem. The extraordinarily novel development of plasmochin, atabrine, resochin, and sontochin at that laboratory heralded a new and brighter scientific age in that struggle.
Plasmochin
The 1927 commercial debut of plasmochin was hailed as a scientific, medical, and economic breakthrough. Its merits included effectively terminating acute vivax malaria attacks, preventing those unexplained delayed attacks, killing otherwise refractory gametocytes of P. falciparum, and possibly breaking the Dutch monopoly on expensive quinine for the therapy of acute malaria (132, 133). In 1938, Field (132) expressed, “The synthesis of plasmoquine [plasmochin] marked an important advance in malarial chemotherapy, the most important advance, perhaps, since the discovery of cinchona bark.” Table 3 summarizes what was immediately understood of the therapeutic properties of plasmochin, explaining much of the contemporary enthusiasm for it. This first rationally designed antimalarial indeed appeared to fully live up to Ehrlich’s envisioned magic bullet. In 1927, Vad and Mohile in India exclaimed, “The days of quinine are numbered” (134).
TABLE 3.
Known therapeutic properties of plasmochin in 1927
| Type of activity | Plasmochin activity against: |
Quinine activity against either species | |
|---|---|---|---|
| Plasmodium vivax | Plasmodium falciparum | ||
| Blood schizontocidal | Yes | No | Yes |
| Efficacy against delayed attacks | Yes | No delayed attacks | No |
| Gametocytocidal | Yes | Yes | No |
| Suppressive prophylactic | Yes | No | Impractical |
| Causal prophylactic | Yes | Yes | No |
Despite its very useful and then unprecedented ability to kill P. falciparum gametocytes, plasmochin had almost no activity against asexual blood stages of that infection (135). Plasmochin was thus advanced as primarily a blood schizontocidal therapy for acute vivax malaria. Early investigators recognized the phenomenon of delayed attacks being peculiar to vivax malaria and that these were somehow biologically distinct from primary attacks. They also understood that quinine had no effect on delayed attacks and quickly realized that plasmochin did. Activity against primary attacks, a given with either quinine or plasmochin in vivax malaria, yielded to efficacy against delayed attacks as the therapeutic endpoint of greatest interest in striving to optimize plasmochin for the therapy of vivax malaria.
In the earliest forays into practice, plasmochin earned a reputation as a drug capable of provoking threatening and sometimes fatal side effects. Sinton and Bird (136) in 1928 noted 3 confirmed deaths due to plasmochin treatment after fewer than a thousand treatments in humans. The authors noted that those deaths occurred despite the close clinical monitoring that came with its tentative initial use. Plasmochin caused severe anemia, jaundice, cyanosis, and methemoglobinemia, typically occurring in a minority of non-Caucasian patients (less than 10%). In a 1927 report from the United Fruit Company (in the United States), for example, Wilhelm Cordes in Cuba described the treatment of 250 predominantly male African-Haitian patients given daily doses of plasmochin ranging downward from 80 to 40 mg (with 1.8 g quinine daily): six of those patients (four at the 80-mg dose) had serious adverse reactions consistent with acute hemolytic anemia. Two of those ended in death within 48 h of the sudden onset of symptoms; cyanosis, jaundice, and hematuria after the 4th or 5th dose (137). Brosius in Panama reported 5 similar deaths with plasmochin therapy in a series of 385 treatments (138). Investigators complained of the very sudden onset of serious complications after several days of dosing, considering serious harm as having already been done by the time of the delayed onset of complications. Plasmochin became rightly regarded as a dangerous drug.
A dose of 100 mg plasmochin daily for 17 days had initially been recommended by Bayer for monotherapy to terminate the acute attack, but that soon evolved to a complex schedule imposing pauses that drew treatment out to about 40 days in attempting to mitigate the serious danger of that regimen (Fig. 5A). In addition to tablets containing 20 mg plasmochin for that indication, Bayer quickly introduced tablets that it called “plasmochin compound” containing 10 mg plasmochin and 125 mg quinine sulfate, a formulation intended for daily dosing (10 tablets for an adult) for up to 4 weeks (136). The combining of plasmochin with quinine also allowed good efficacy against P. falciparum (135). The coformulated tablets delivered the same dangerously high daily dose of plasmochin, then viewed as minimally effective by Bayer. Indeed, the minimally effective 14-day regimen of plasmochin against a New Guinea strain of P. vivax was later proven to be 90 mg daily (139, 140).
FIG 5.

Studies of plasmochin monotherapy versus plasmochin combined with quinine. (A) Complex dosing regimens of 100 mg plasmochin (orange boxes) or 100 mg plasmochin with 1.25 g coformulated quinine (orange and blue boxes), as recommended by the manufacturer, with days of rest (white boxes) or on consecutive days without rest (right), listing therapeutic outcomes among 63 patients thus treated and followed for at least 2 months after the initiation of therapy (based on data from Sinton and Bird [136]). (B) Based on data from Craige et al. (139), the impact of 15 to 63 mg daily plasmochin (for 14 days) on delayed attacks when administered separately or concurrently with quinine (2 g daily for 8 days) in a total of 38 subjects challenged with the Chesson strain of P. vivax and followed for about 1 year. (C) Based on data from Berliner et al. (140), therapeutic outcomes when plasmochin (90 mg daily for 14 days) was administered before or during quinine therapy (2 g daily for 8 days).
The thin margin of efficacy versus toxicity in 8-aminoquinolines remains a constant in their clinical use. Today it may be useful to understand the Bayer strategy underpinning the seemingly illogical strategic pivot with coformulated plasmochin-quinine, i.e., striving to improve plasmochin safety without compromising on its dose. Toxicity findings in uncontrolled trials led Bayer to believe that quinine mitigated the toxic effects of plasmochin, cyanosis (due to methemoglobinemia) specifically (135, 138). The pairing of plasmochin and quinine in the therapy of vivax malaria thus occurred not with consideration of a strategy of lower less toxic dosing but as a deliberate and ultimately failed strategy to mitigate the toxic effects of plasmochin by exploiting an unknown interaction with quinine that seemed to do so. Ultimately, it would be the still poorly understood or appreciated potentiation of 8-aminoquinoline therapeutic activity by partner blood schizontocides (lacking any direct activity against latency) that would later prove defining in optimized practice with plasmochin.
Potentiating Effects of Blood Schizontocides on 8-Aminoquinoline Efficacy
In view of the data summarized in Fig. 5A, where 15 of 50 subjects not given quinine but just 2 of 30 given the coformulated quinine-plasmochin tablets relapsed, Sinton et al. (141) expressed, “The combination of quinine with plasmoquine [plasmochin] seemed to be more effective in the production of both clinical and radical cures [prevention of delayed attacks] of the disease than plasmoquine alone.” Those investigators understood that this presented an opportunity to mitigate plasmochin toxicity by simply lowering the dose of it. They explained, “The results of the continuous course of treatment with plasmoquine and quinine combined, made it seem probable that a high percentage of radical cures might be obtained by continuous treatment with smaller doses of plasmoquine, if combined with the same doses of quinine as before, and at the same time the toxic manifestations might be reduced, if not entirely abolished, by such treatment.” They also considered the possibility of quinine mitigating the toxic effects of plasmochin but found no compelling evidence of it. They instead focused on the possibility of smaller less toxic doses by the observed potentiation of efficacy with quinine coadministration.
In summarizing their experimental findings on this possibility, they concluded, “The combination of quinine and plasmoquine is clearly indicated. It can be seen from our results that when these two drugs are combined, a higher percentage of radical cures is obtained by smaller doses than when either of the drugs is given separately in larger doses.” The report from Sinton et al. (141) explicitly recommended against clinical use of the coformulated plasmochin-quinine tablets as they delivered either too large of a dose of plasmochin or too small of a dose of quinine. They also pointed to the great economy of purchasing the drugs separately. The coformulated plasmochin-quinine tablets thus fell out of favor as quickly as they had been introduced. Investigators instead applied various lower doses of plasmochin coadministered with each generally fixed minimal effective dose of quinine sulfate (1.2 to 2.0 g daily for 8 to 14 days).
Two decades later, during the late 1940s, the developers of primaquine would affirm the findings of Sinton et al. (141) on the synergistic effects of quinine on plasmochin efficacy (Fig. 5B and C). In an experiment designed to optimize plasmochin therapy as the standard against which to compare newer 8-aminoquinolines, Craige et al. (139) combined 15, 31, or 63 mg (daily for 14 days) plasmochin with fixed doses of quinine (2 g daily for 8 days) administered consecutively or concurrently with plasmochin. Relapse occurred in every subject at every consecutive dose (18/18) but only 12/20 subjects concurrently dosed. The highest dose was considered inadequate, so Berliner et al. (140) applied plasmochin at 90 mg daily for 14 days and again compared the consecutive versus concurrent dosing of quinine, with 2 of 5 and 0 of 9 subjects relapsing, respectively. This study confirmed 90 mg plasmochin as the minimally effective dose and the necessary practice of concurrent quinine therapy. When Alving et al. (142) treated 47 subjects with 60 mg daily (for 14 days) of the experimental 8-aminoquinoline called pentaquine with concurrent quinine, 8 (17%) relapsed, whereas 3 of 5 (60%) subjects given the same therapy without concurrent quinine did so. Quinine enhancement of plasmochin therapeutic activity against delayed attacks seemed to also occur among other 8-aminoquinolines.
The developers of primaquine confirmed the phenomenon of hypnozoitocidal synergy of it when combined with quinine. Edgcomb et al. (61) (Fig. 6A) evaluated multiple daily doses of primaquine, settling on 22.5 mg daily for 14 days coadministered with 1.64 mg quinine on each of those days (no relapses occurred among 23 subjects) as being minimally effective. When the same dose was administered without quinine, 4 of 5 subjects had recurrent attacks. Alving et al. (143) pointed to the possibility of partner blood schizontocides in radical cure having “a potentiating effect on the curative action of primaquine” but considered the available data inconclusive. Their follow-up experiment (Fig. 6B) was designed explicitly to settle that question: “The investigations reported in this communication were set up under rigidly controlled experimental conditions in order to establish firmly whether quinine or chloroquine potentiates the action of primaquine against the late tissue stages of vivax malaria” (143).
FIG 6.

Studies of primaquine monotherapy against delayed attacks of vivax malaria. (A) Relapse findings (right) after daily dosing of primaquine and quinine, each administered alone or concurrently, as reported by Edgcomb et al. (61). (B) Relapse findings after daily dosing of primaquine and quinine consecutively (top) or currently (middle) or of primaquine with 1 g chloroquine administered over 24 h (bottom), as reported by Alving et al. (143).
The relatively simple experiment employed a New Guinea strain of P. vivax in 57 inmate volunteers challenged by the bites of anopheline mosquitos scored from 15 to 33 (on a scale of from 10 to 40) by sporozoite load. Upon patency, the subjects were randomized to 3 treatment groups (n = 19 each), with all receiving 15 mg primaquine daily for 14 days with 2 g quinine sulfate for 14 days administered consecutively prior to the commencement of primaquine 2 days after the completion of quinine therapy (group 1), quinine concurrently with primaquine therapy (group 2), or 1.0 g chloroquine administered in two doses over 24 h (group 3). Subjects not relapsing were followed for 114 to 373 days, and relapses occurred between days 17 and 184 (median, 52 days), with 15, 1, and 5 relapses occurring among the individuals in groups 1, 2, and 3, respectively. Alving et al. (143) summarized their findings (Fig. 6B): “We consider the results to indicate that both of these suppressive agents [quinine and chloroquine] potentiate the effect of primaquine against late fixed-tissue stages [hypnozoites] of vivax malaria.”
This conclusion reflects that expressed by Phelps (144) in 1927: “I have reached the conclusion that plasmochin in one course, without the addition of suitable doses of quinine, is of little value in the treatment of any variety of malaria.” Sinton and colleagues (141) had long before considered the phenomenon defining in optimized plasmochin therapy, and Alving et al. (143) confirmed the same with primaquine whether it was administered with quinine or chloroquine. In 2011, Dow et al. (145) demonstrated that the addition of quinine, artemether-lumefantrine, or mefloquine to the treatment of P. cynomolgi infections in rhesus macaques reduced the effective hypnozoitocidal dose of tafenoquine 10-fold. Evidence drawn from at least four different 8-aminoquinolines over nearly a century of experience points to the fact that some blood schizontocidal drugs independently lacking any hypnozoitocidal activity powerfully and consistently potentiate that activity by 8-aminoquinolines.
Schmidt explored a similar phenomenon with mirincamycin with primaquine against P. cynomolgi hypnozoites over 30 years ago; the adjunct more than halved the required curative dose (146). He expressed that “the current study serves to focus attention on a somewhat novel approach to the development of more effective and better-tolerated regimens for radical cure.” Myint et al. (147), reviewing the phenomenon in 2011, hypothesized complementary therapies for radical cure of lower toxicities, summarizing with the following: “it may be possible to increase the therapeutic index of primaquine, and ultimately tafenoquine, with the co-administration of other clinical agents, and thus may optimize the treatment of malaria with 8-aminoquinolines.” No other contemporary work has explored the extent to which other compounds may potentiate 8-aminoquinoline activity against hypnozoites as a means of reducing dose and toxicity. The original idea of Sinton et al. (141) expressed and demonstrated nearly a century ago has yet to be explored, explained, or exploited. Optimizing the potentiation of 8-aminoquinolines may offer a means of greatly mitigating the harmful and deeply constraining hemolytic toxicity of that class. The same may be expressed concerning the observed effects of blood schizontocides on the manifest hemotoxicity of 8-aminoquinolines.
Blood Schizontocide Impacts on 8-Aminoquinoline Toxicity
Early investigators noted that quinine seemed to mitigate the side effects of plasmochin but did so anecdotally in a relatively few uncontrolled trials. MacPhail (148) expressed in 1927, “There appears to be little doubt that plasmochin should be administered in combination with quinine, which seems to diminish its toxic tendencies.” Sinton and Bird (136) expressed more doubtful views on this question: “Although the addition of quinine to plasmochin seems to have some effect in diminishing the toxicity of this drug, it does not appear to be the very marked action which some work would lead one to believe.” Severe plasmochin hemolytic toxicity, seemingly idiosyncratic but linked to unrecognized G6PD deficiency, occurred with or without quinine coadministration. Whatever mitigating effects that quinine may have exerted, these did not significantly diminish the untoward effects or caution necessary with plasmochin imposed by the specific hemolytic toxicity issue. Nonetheless, quinine and other blood schizontocides exerted measurable impacts on the plasma levels and broader toxicities of the 8-aminoquinolines, either exacerbating or mitigating them.
Plasmochin virtually disappears from plasma within 8 h of dosing, and according to Zubrod et al. (149), this is not due to marked localization or excretion but is due to very rapid metabolic conversion. Those investigators also described marked interindividual variation in the means of the minimal (24 h postdosing) and maximal (3 h postdosing) daily plasma concentrations of plasmochin without exposure to any other drug. Mean maximal daily plasma concentrations with 60-mg doses varied between 54 and 2,924 μg/liter on the third day and 89 and 1,400 μg/liter on the sixth day, with the lower and higher values representing the values for the same subjects on each day. This interindividual variability in plasma levels appeared with all of the experimental 8-aminoquinolines evaluated (17) and may reflect the then unknown importance of inherent variability in the catalytic activity of the cytochrome P-450 2D6 isozyme in the metabolism of 8-aminoquinolines (79–81). In 1981, Carson et al. (17) pointed out the seemingly paradoxical inverse relationship between the plasma levels of the parent drug and therapeutic activity, perhaps explained by the extent of conversion of the parent drug to the active metabolite(s). Other investigators declared that no correlation existed between plasmochin plasma levels and therapeutic activity (140). The 8-aminoquinolines appear to be prodrugs delivering metabolites with therapeutic and toxic activities in a manner likely to be impacted by other therapies that could interfere with the normal metabolism of them.
The Bayer blood schizontocide atabrine (quinacrine, mepacrine; Fig. 4), introduced commercially in 1932, provoked 10-fold increases in plasma plasmochin levels (149, 150). Zubrod et al. (149) administered nine daily 30-mg plasmochin doses that showed average plasma concentrations of about 40 μg/liter; the subsequent seven daily doses included a subtherapeutic 30-mg atabrine dose, and plasma plasmochin concentrations rose to about 450 μg/liter after 3 daily doses. Both drugs were then stopped, with the rapid disappearance of plasma plasmochin. Atabrine is very slowly excreted (similar to chloroquine), and when daily plasmochin alone was resumed after a 35-day pause, the plasma concentrations of it again reached greatly elevated levels (250 to 350 μg/liter). Even relatively very low levels of atabrine disturbed the normal metabolism of plasmochin.
Earle et al. (151) compared the toxic effects of plasmochin given with or without atabrine. Table 4 summarizes their findings with regard to the methemoglobinemia induced by plasmochin. Whereas diminishing the dose of plasmochin from 90 mg daily to 60 mg and 30 mg daily brought methemoglobin levels from 12.2% to 8.9% and 4.4%, respectively, adding atabrine to the lowest plasmochin dose (30 mg) increased the methemoglobin level to the same level achieved with the highest dose (90 mg), i.e., 12%. In contrast, Clayman et al. (59) observed no impact of coadministered quinine on the plasmochin-induced methemoglobinemia in the dozens of subjects evaluated (Table 4), and quinine had no impact on plasmochin plasma levels. Regardless of the dose or individual steady-state plasma plasmochin concentration, introduction of atabrine into subjects after achieving that concentration provoked profound alteration of it. Earle et al. (151) also measured the impact of atabrine on the well-known plasmochin-induced granulocytopenia. Table 5 summarizes their findings, which tend to reflect the same trends observed with methemoglobinemia in Table 4: a plasmochin dose-dependent effect of decreasing neutrophil counts, with the lowest dose administered with atabrine approximating the effect of the highest plasmochin dose, i.e., 69% versus 64% reductions in neutrophil counts.
TABLE 4.
Atabrine exacerbates plasmochin-induced methemoglobinemia, whereas quinine fails to impact plasmochin-induced methemoglobinemia
| Treatment | No. of subjects | Mean % of MetHba | SD (range) of % of MetHb |
|---|---|---|---|
| Plasmochin or plasmochin-atabrineb | |||
| 30 mg | 29 | 4.4 | 2.4 (0.7–10.0) |
| 60 mg | 13 | 8.9 | 4.5 (1.9–20.8) |
| 90 mg | 24 | 12.2 | 7.0 (0.9–28.7) |
| 30 mg + atabrine (dose not specified) | 6 | 12.2 | 4.6 (6.0–19.3) |
| Plasmochin-quininec | |||
| 31.5 mg | 12 | 4.4 | 3.7 (2.1–15.8) |
| 63 mg | 6 | 12.8 | 4.1 (7.2–21.4) |
| 15 mg + quinine | 10 | 2.5 | 0.7 (1.3–3.6) |
| 31.5 mg + quinine | 27 | 4.5 | 2.1 (1.7–9.2) |
| 63 mg + quinine | 76 | 10.7 | 4.9 (2.9–24.0) |
TABLE 5.
Effect of atabrine on the neutrophil counts in the blood of subjects dosed with plasmochin with or without atabrinea
| Treatment | No. of subjects | Avg no. of neutrophils/ml |
% decrease in neutrophils | |
|---|---|---|---|---|
| Prior to dosing | After 14 days of dosing | |||
| Plasmochin | ||||
| 30 mg | 11 | 4,650 | 2,890 | 38 |
| 60 mg | 4 | 3,878 | 2,920 | 25 |
| 90 mg | 12 | 4,762 | 1,714 | 64 |
| 30 mg plasmochin + atabrine | 6 | 5,093 | 1,493 | 69 |
Adapted from reference 151 with permission of the American Society for Clinical Investigation.
Although quinine did not impact the plasma levels or the methemoglobinemia of plasmochin, it did exert those effects with other 8-aminoquinolines. In those instances, however, quinine very significantly mitigated methemoglobinemia and other signs of toxicity. In 1948, Craige et al. (152) administered 120-mg daily doses of pentaquine alone or with quinine to 5 subjects each for 6 to 14 days: in addition to better efficacy (0/5 versus 2/5 subjects relapsed), coadministration almost completely prevented otherwise frequent and severe weakness or prostration and syncope or postural hypotension. The mean level of methemoglobinemia in these groups was 5.6 g% and 11.3 g%, respectively. In 1950, Clayman et al. (59) similarly reported the mitigating effects of quinine on methemoglobinemia and the impacts on the plasma levels of primaquine administered as 120 mg daily for 14 days (Table 6). Whereas quinine seemed to exacerbate the poor gastrointestinal tolerance of primaquine, it roughly halved the methemoglobinemia and plasma levels of primaquine. Clayman et al. (59) reported a series of experiments where daily doses of primaquine of between 15 and 120 mg were administered to G6PD-normal Caucasians either alone or with concurrent quinine or methylene blue (Fig. 7) daily for 2 weeks, and the impact on primaquine-induced methemoglobinemia was evaluated (the average for the last 5 days of dosing was reported). Quinine or methylene blue more than halved the peak methemoglobin level of 21% with 120 mg primaquine alone to 10% and 8%, respectively.
TABLE 6.
Effect of 120 mg primaquine daily for 14 days administered without or with concurrent 1.64 g quinine on tolerability, methemoglobinemia, and plasma primaquine levelsa
| Treatment | No. of subjects | Age range (yr) | Abdominal discomfort | Symptoms | Mean (range) % of MetHbb | Mean (range) primaquine concn (μg/liter) in plasma |
|---|---|---|---|---|---|---|
| 120 mg primaquine daily for 14 days | 5 | 21–43 | Mild in 3, moderate in 2 | Nausea in 1 | 20.2 (18.3–21.7) | 210 (144–308) |
| 120 mg daily primaquine concurrently with 1.64 mg quinine | 6 | 22–39 | Mild in 2, moderate in 1, severe in 3 | Nausea on 1, vomiting in 1, anorexia in 2 | 10.0 (5.5–15.5) | 99 (74–132) |
Based on data from Edgcomb and colleagues (61).
MetHb, methemoglobin.
FIG 7.

Effects of quinine (QN) and methylene blue (MB) coadministered with increasing daily doses of primaquine (PQ) on methemoglobinemia in Caucasian subjects, showing the average value for the last 5 days of dosing. The figure is based on data from Clayman et al. (59).
Although the relevance of methemoglobinemia to the hemolytic toxicity of the 8-aminoquinolines is unknown and perhaps doubtful (58, 60), the contrasted toxic effects of atabrine on plasmochin (exacerbation), quinine on plasmochin (no effect), and quinine on primaquine (mitigation) point to the fine chemical specificities and consequences of interactions between blood schizontocides and 8-aminoquinolines and the great diversity of blood schizontocides that interact with them. Proguanil, for example, increased the plasma levels of the experimental 8-aminoquinoline called pentaquine (17). Likewise, Lysenko (153) described the frequency of acute methemoglobinemia occurring among patients receiving a daily dose of 23 mg of the Russian 8-aminoquinoline called quinocide as monotherapy (n = 70) or quinocide coadministered with bigumal (proguanil; n = 35), cycloquine (haloquine; n = 8), or chloridine (pyrimethamine; n = 12) at 0.07%, 26%, 100%, and 100%, respectively. Although drugs of diverse classes impact the methemoglobinemia and plasma pharmacokinetics of 8-aminoquinolines, the relevance of these effects to the specific problem of 8-aminoquinoline hemolytic toxicities in G6PD-deficient patients was not specifically reported. Incidentally, however, no known combination of available blood schizontocides and 8-aminoquinolines in practice has proven to be free of hemolytic effects.
In the specific instance of a methylene blue adjunct to radical cure possibly impacting 8-aminoquinoline hemolytic toxicity, Alving and colleagues reported but did not publish the outcome of a single-subject series of experiments examining this possibility (154). After observing that methylene blue apparently potentiated the hypnozoitocidal activity of pentaquine—0 of 3 subjects relapsed when receiving methylene blue with pentaquine and quinine, whereas 4 of 10 subjects receiving the same therapies without methylene blue relapsed—they examined its effect on pentaquine hemolytic toxicity in a sensitive subject. The report reads as follows: “a drug which aids in the reduction of methemoglobin to hemoglobin may be useful in preventing hemolytic anemia due to the presence of oxidants (? quinone imines) within the red cells. This theory has been tested in one volunteer who developed severe acute hemolytic anemia after several days treatment with 60 mgm. of pentaquine base and two grams of quinine sulfate administered concurrently. This man again developed a moderately severe acute hemolytic crisis later in the course of treatment with 30 mgm. of pamaquin [plasmochin] base and two grams of quinine sulfate during the next relapse. He has subsequently been treated with the concurrent administration of 60 mgm. pentaquine base, two grams of quinine sulfate and 0.5 g of methylene blue hydrochloride for fourteen days without the development of intravascular hemolysis. These studies suggest that the administration of methylene blue may be of value in the prevention of acute intravascular hemolysis.”
Regarding this extraordinary observation—the apparently complete abrogation of 8-aminoquinoline hemolytic toxicity in a demonstrably sensitive human subject by a therapeutically potentiating methylene blue adjunct—no record of a follow-up investigation could be located. The development of primaquine (see below) would follow a path to registration with the earnest belief that its hemolytic toxicity did not constitute a serious, much less disqualifying, problem. Abrogating that toxicity would not have registered as a development priority and apparently did not. A nearly 70-year history of extremely poor primaquine effectiveness almost wholly attributable to its hemolytic toxicity would have to unfold before the significance of a means of detoxifying hemolytic 8-aminoquinolines could rise to laboratory and clinical investigation of great merit (see “Future Perspectives” below).
Collectively, the findings summarized here offer compelling evidence of specific drug-drug interactions across most possible combinations of blood schizontocides with 8-aminoquinolines. Although the findings are not detailed here, modern studies affirm that chloroquine, dihydroartemisinin-piperaquine, and pyronaridine-artesunate also affect the pharmacokinetics of primaquine (155–158). The converse effect—8-aminoquinolines impacting blood schizontocide pharmacokinetics—has been consistently negative with both older and modern blood schizontocides (155–157). Fasinu et al. (159) examined the metabolic pathway-specific effects of chloroquine and quinine on primaquine metabolism and suggested shifts favoring therapeutic activity with diminished toxicity. The marked interindividual variation in the plasma levels of 8-aminoquinolines in the face of stable and consistent individual levels, taken with their rapid extensive metabolism to therapeutically active and toxic forms, points to likely perturbation of that metabolism by partner blood schizontocides as the most probable basis of their sometimes profound impacts on 8-aminoquinoline therapeutics.
The modern view of blood schizontocides in radical cure tends to segregate them from the hypnozoitocidal compartment as if they were independent and freely interchangeable partner therapies. Few authorities or experts, for example, express caution over or regard for selection of the partner blood schizontocide for 8-aminoquinoline radical cure, including the U.S. FDA in its 2018 label instructions for the uses of tafenoquine for chemoprevention or radical cure (160, 161). This was not the case nearly a century ago, when Sinton and colleagues (141) outlined quinine-optimized plasmochin therapy and the principles of applying it.
Optimized Plasmochin Therapy
In 1930, Sinton (Fig. 3C) and colleagues lamented the highly variable and seemingly haphazard dosing with plasmochin after its introduction, but they ultimately defined what they considered an optimized standard of 40 mg plasmochin coadministered with 1.25 g quinine daily for 2 weeks (141). Their patients had been British soldiers infected in India (162). American investigations applying Chesson strain P. vivax in American inmate volunteers (effectively 100% of whom experienced relapse following heavy experimental challenge with an isolate recovered from an American soldier infected in New Guinea in 1944 [163]) showed that 63-mg daily uninterrupted dosing for 2 weeks with concurrent daily quinine (also for 2 weeks) was inadequate (Fig. 5B) (139). Berliner et al. (140) applied 90-mg plasmochin uninterrupted daily dosing with concurrent quinine and achieved complete efficacy against Chesson strain P. vivax (Fig. 5C). Nonetheless, when later applied against Korean strains of P. vivax (after essential correction for a relapse rate of 28%), just 27.5-mg daily dosing of plasmochin achieved a cure rate of 97% (164). As would later prove true with primaquine, P. vivax susceptibility to hypnozoitocidal therapies varies by the geographic origin of the strain, with the Chesson and Korean P. vivax strains representing strains with the known extremes of apparently naturally variable hypnozoite susceptibility to 8-aminoquinolines (165). It may thus be apparent that optimized plasmochin therapy (administered for 14 days) was indeed 40 mg daily for the Indian P. vivax strain, 90 mg for the New Guinea P. vivax strain, and 30 mg for the Korean P. vivax strain, with all of these being in the key efficacy-defining context of uninterrupted concurrent daily quinine therapy (>1.25 g base).
Sinton et al. (141) laid out the fundamental therapeutic principles of applying quinine and plasmochin for achieving optimized safety, tolerability, and efficacy against primary and delayed attacks of vivax malaria. Their eight points regarding clinical practice with these drugs reflect the same for contemporary radical cure applying primaquine or tafenoquine with quinine, chloroquine, or other blood schizontocidal therapies: “1) Plasmochin and quinine in combination works better than either drug alone for permanent cure of vivax malaria; 2) Plasmochin should not be used as monotherapy but only in combination with quinine; 3) The dose of quinine must be sufficient to terminate the acute attack; 4) Smaller doses of plasmochin administered over a longer period of time [are] more safe and efficacious than giving larger doses interrupted by pauses; 5) The maximal tolerated daily dose of plasmochin is less than 40 mg; 6) Smaller doses of plasmochin over longer periods [are] preferred over larger doses over shorter periods; 7) Plasmochin therapy should be halted with onset of signs of toxicity; 8) Plasmochin should not be administered to people with co-morbidities like anemia or hepatic or renal dysfunction without compelling cause and very close clinical monitoring.”
The related points 4 and 6 bear mention here as perhaps not reflecting currently accepted therapeutic principles for 8-aminoquinolines. As will be detailed later for primaquine, larger doses administered over shorter or longer interrupted periods, e.g., 7 daily or 8 weekly doses, appear to offer advantages with regard to compliance or safety, respectively, without apparent compromises in efficacy. Furthermore, unlike in the time of Sinton and colleagues, the defining toxicity problem of G6PD deficiency is today understood and thus clinically manageable by screening prior to administering otherwise dangerously hemolytic 8-aminoquinoline dosing regimens.
Sinton et al. (141) offered a prescient synopsis of the toxicity problem with plasmochin of enduring relevance to PART with 8-aminoquinolines: “it does not seem advisable that the drug should be issued for general use by the lay public, but that it should be used only under medical supervision. It may be that smaller non-toxic doses will require a longer period of treatment to effect a radical cure, but this would not be of such importance if a dosage could be determined which would be safe for general use, for one must remember that a large proportion of the population in many areas of the tropics are not within reach of medical aid. The occurrence of many cases of toxaemia among the lay public would tend to bring into disrepute what is undoubtedly a very valuable drug in the treatment of certain forms of malaria.”
According to sources cited by Field (132) in 1938 attributed to the Malaria Commission of the League of Nations Health Organization, plasmochin with quinine or atabrine was not recommended for use outside of the hospital setting and was deemed unsuitable for safe public health use (lay administration without medical supervision) at doses required for the lasting cure of vivax malaria. Plasmochin thus never saw broad use against endemic malaria; no balance between practicality (duration of dosing) and safety (daily dose) was ever achieved. Field (132) remarked that the development of plasmochin “was significant not so much because of the special merits of the drug itself or because any sensational progress in malarial treatment can be recorded from its use but because its synthesis was a pioneer step which led after half a century of stalemate to a fresh advance in our knowledge of malarial chemotherapy and paved the way to further developments in synthetic quinoline derivatives of a more important character.” Plasmochin, he supposed, would be improved, but 80 years later we struggle with the same problem of clinical and public health balance with modern 8-aminoquinolines. The fact that most patients needing radical cure do not live “within the reach of medical aid” remains as true and as great of an impediment to clinical and public health utility.
How we came to accept these dangerous treatments not safely accessible by most malaria patients as suitable and then to become wholly reliant on them for killing hypnozoites for nearly a century is a fraught question that is as difficult and complex as it is important. Key to addressing that question is the historic and technical context of antirelapse drug development during the 1940s aimed specifically at malaria as a serious military problem.
War-Spurred 8-Aminoquinoline Research
The German suite of innovative synthetic antimalarials failed to commercially challenge the primacy of quinine for the therapy of acute malaria during the 1920s and 1930s. The toxicity of plasmochin and the unpleasant side effects of atabrine, along with their relatively high cost, rendered them less competitive, albeit commercially viable (13), but Bayer had superior 4-aminoquinonline products coming: resochin (chloroquine) and sontochin (a methylated and presumed less toxic derivative of chloroquine) (Fig. 4). Walter Kikuth at Elberfeld had replaced Wilhelm Roehl, who died prematurely in 1929. Kikuth set resochin aside in favor of sontochin after Sioli had tried it in just four patients at the Dusseldorf asylum in 1936 (13, 55). Sontochin fared better in Sioli’s hands and awaited final clinical development by the Germans at the outset of war in Europe in September 1939. The Allies liberating Tunis, Algeria, in 1943 captured experimental sontochin tablets from Vichy French investigators. Thus alerted, American authorities then put sontochin and resochin to clinical trials in the United States and in 1946 registered resochin for the treatment of acute falciparum or vivax malaria but then dubbed it chloroquine (55, 166).
The American authorities effectively seized German Bayer’s intellectual property through its American corporate partner, Winthrop Pharmaceuticals in New York City. There is almost no record of American antimalarial drug discovery work during the 1920s and 1930s. Tentative initial efforts were launched only in November 1939 after the outbreak of war in Europe (13). The American wartime raid on Bayer’s trove of antimalarial properties derived from the Elberfeld laboratory during the prewar period spared them the 2 decades of the revolutionary discovery work invested in those molecules. Global war had provoked the Americans into urgent efforts of antimalarial development unfettered by the legitimate Bayer patents held on plasmochin, atabrine, resochin, and sontochin molecules already registered both in Germany and in the United States by 1941.
World War II for the Americans began only in December 1941 with the nearly simultaneous Japanese attacks on their military facilities in Hawaii and the Philippines. Just a few months later, in March 1942, Imperial Japan seized the Netherlands East Indies (modern Indonesia) and the global supplies of quinine that it cultivated. The Allied powers understood the threatening military strategic implications of that loss and rallied vast fiscal and technical resources to meet that challenge, including the expropriation of domestic stocks of quinine, plasmochin, and atabrine. Those proved grossly inadequate to the need, and the authorities then commandeered pharmaceutical factories for the emergency manufacture of plasmochin and atabrine (13, 167).
As events occurred, the Americans could not content themselves with the commandeered stocks or emergency manufacturing of plasmochin and atabrine. An unanticipated interaction between these drugs, discovered only in 1942, seemed to exacerbate an already serious toxicity problem with plasmochin (14). In early 1943, the Surgeon General of the United States warned against the use of that combination for radical cure (168). This blow left millions of soldiers exposed to the risk of repeated debilitating attacks of vivax malaria, including after repatriation to the U.S. mainland, which was on the verge of malaria elimination success. All of these factors motivated the U.S. government to mount a deliberate effort to discover a safer means of radical cure. In 1946, James A. Shannon (Fig. 8) (15)—who would go on to head and indelibly shape the U.S. National Institutes of Health during the 1950s and 1960s—recalled the situation like this: “The direction of the early clinical work (1942-1943) was conditioned largely by the early loss to the United Nations [the Allies] of their normal sources of supply of quinine and by the lack of an adequate stockpile. Those who were intimately concerned with the malaria problem during the first year of the war will recall the gravity of the situation. The worry which was incidental to the lack of an adequate supply of quinine was enhanced by the preliminary reports from the field which carried the suggestion that quinacrine [atabrine] was of little use in the suppression and treatment of the malarias and that pamaquin [plasmochin] as then used had little value in the cure of vivax malaria.”
FIG 8.

Key figures in the development of primaquine from 1943 to 1952 included wartime antimalarial development project leader James A. Shannon (top left, ca. 1968, from the U.S. NIH Almanac); chemist Robert C. Elderfield (top right; in about 1970, with permission of The National Academies Press), who first synthesized primaquine in 1945; physician Alf Alving (bottom left, in about 1968; photo by L. J. Bruce-Chwatt, courtesy of the Wellcome Collection), who oversaw clinical trials of many 8-aminoquinolines for the University of Chicago at Stateville Penitentiary, Joliet, IL; and biologist Clay Huff (bottom right; in 1967, with permission of Elsevier [341]), who experimentally challenged those human subjects at the Stateville Penitentiary facilities with P. vivax-infected Anopheles stephensi mosquitoes.
The U.S. government body orchestrating the research response to the wartime malaria threat was the Board for the Coordination of Malarial Studies, established in early 1941 and seated at the National Academy of Sciences in Washington, DC (15, 55). The people managing that enterprise focused solely on military medical imperatives. As expressed by Shannon, head of clinical development on that board (15), “The primary purpose of the investigations has been, at all times, to satisfy the specific needs of the armed services.” This emphasis would have profound and lasting implications insofar as the therapeutic objective of radical cure is concerned and the problem of the hemolytic toxicity of 8-aminoquinolines bound to it.
Wartime urgency led them to restrict the search for a safer and more effective plasmochin to other 8-aminoquinolines, the only class of drugs (up to the present) known to have activity against the delayed attacks of vivax malaria. According to Carson et al. (17), several hundred 8-aminoquinolines were synthesized and screened for therapeutic activity relative to quinine for the cure of blood-stage Plasmodium gallinaceum in chickens and Plasmodium lophurae in ducks. The investigators understood the inadequacy of these avian malaria models to vivax malaria, knowing that those lacked the persistent tissue stages responsible for delayed attacks. They further understood that this activity of plasmochin had been serendipitously discovered rather than systematically selected and optimized in the quinine-plasmochin radical cure. They carefully studied the data from the handful of plasmochin mimics or frank copies by the French and Russians (e.g., praechin, plasmocid, rhodaquine, and a class of several derived compounds called plasmocids [132, 169]), discouragingly noting that “pamaquin [plasmochin] and many of its analogs possess seriously toxic effects when administered at a dosage well below that which is generally curative” (15). They nonetheless elected to conduct a systematic search of 8-aminoquinolines for a less toxic compound, believing “that previous exploration of the 8-aminoquinolines was inadequate to be certain that pamaquin is the best drug to be derived from that series” (15). The stage was thus set for a discovery effort aimed at a superior drug for the killing of the persistent tissue stage of P. vivax, and concern for the poorly understood but certain dangerous toxicities of 8-aminoquinolines, primarily their hemotoxic and neurotoxic properties, dominated that effort.
Preclinical Screening of 8-Aminoquinolines
As already explained, it may be helpful to conceptualize two distinct problems in considering the toxicity of 8-aminoquinolines: (i) general toxicity to humans broadly and (ii) the special case of hemolytic toxicity restricted to G6PD-deficient humans. Until the 1950s, investigators would have no understanding of that inherited abnormality as the dominating feature of plasmochin toxicity. They could not make the distinction expressed above, considering hemolytic toxicity a seemingly idiosyncratic feature of a broader hemotoxicity problem. Preclinical screening efforts of many dozens of 8-aminoquinolines in the mid-1940s were thus effectively limited to classical toxicity and pharmacology studies in birds, rats, and dogs (16), none of which would predict hemolytic toxicity in G6PD-deficient patients.
These investigations narrowed the field of hundreds of 8-aminoquinolines to just 85, which were then screened for toxicity in rhesus monkeys (17). Among these, two distinct toxicities emerged in accord with specific variation in the aliphatic side chain: (i) severe, irreversible neurotoxicity highly selective for brain stem nuclei, especially III, IV, VI, and VIII (17, 170), and (ii) lethal overdoses involving multiple organ systems but reversible at sublethal doses. Schmidt held up two 8-aminoquinolines as representative of these two toxicity subclasses: SN-3115 and SN-971, plasmocid and plasmochin (Fig. 4), respectively (171). The French synthesized plasmocid (or rhodoquine) in 1931, and it saw clinical use in French colonial territories (132). A number of related compounds (in which the secondary or tertiary terminal amino group was separated from the 8-amino nitrogen by a chain of 2 or 3 methylene groups) were synthesized in Russia (the plasmocids), but by 1952 they had settled on quinocide, differing from primaquine by placement of the methyl group on the otherwise identical side chain (153) (Fig. 4). Plasmochin and related compounds (by having a primary, secondary, or tertiary terminal amino group separated from the 8-amino nitrogen by at least four methylenes) constituted the focus of American clinical development efforts. The dangerously neurotoxic plasmocid subclass of 8-aminoquinolines had been recognized and abandoned in the American preclinical development effort. Notably, however, SN-191 and SN-1452 (Fig. 9) appear as exceptions to this rule; the additional methyl at the 8-amino in the former may have been seen as mitigating, and the latter was evaluated for safety only at one-half the tolerated dose in macaques, without apparently advancing to therapeutic trials in humans (142).
FIG 9.

Experimental 8-aminoquinolines advanced to human trials in the late 1940s.
According to Carson et al. (17) in 1981, more than 50 8-aminoquinolines had been tested in humans since 1944. Since that report predates the renewed clinical work leading to tafenoquine registration (and that amounted to tafenoquine alone), they presumably referred specifically to the U.S. Army experience with these drugs during the 1940s and 1950s. Published records of that era show toxicity and efficacy findings against P. vivax in humans for 23 8-aminoquinolines (18 are illustrated in Fig. 9). The initial 1948 report of these compounds in clinical trials excluded primaquine and SN-3883 [8-(4-aminobutyl)-6-methoxyquinoline] (Fig. 10) (142). All of these compounds went to clinical trials in humans on the basis of standard safety assessments through an ascending series of vertebrate animals ending with rhesus macaques.
FIG 10.

The most active and least toxic 8-aminoquinolines: plasmochin, primaquine, pentaquine, isopentaquine, and SN-3883.
Today it is uncertain how asexual blood-stage antimalarial activity measured in infected ducks and chickens served in selecting the early 8-aminoquinoline candidates. The accounting of Carson et al. (17) of that process seems rationally dismissive of the therapeutic activity for 8-aminoquinolines against avian plasmodia lacking latency. However, Jones et al. (172) described the selection of plasmochin and three experimental 8-aminoquinolines for clinical testing: “These four compounds were selected for human testing because they had high antimalarial activity in avian infections and because their toxicity was low and of the pamaquine type, which, in contradistinction to the plasmacid [sic] type of toxicity, is reversible.” Blood schizontocidal activity against the avian plasmodia apparently played some role, as it serendipitously did in the discovery of plasmochin. Schmidt and Coatney (131) explained in 1955 that the use of P. cynomolgi in rhesus macaques achieved validation only in 1948, then permitting immediate, albeit limited, assessments among the most promising 8-aminoquinolines. It was this late P. cynomolgi work that finally brought primaquine and SN-3883 to compete with pentaquine and isopentaquine in clinical development (Fig. 10). The preclinical screening effort up to 1948 may be judged in retrospect as inadequate to the therapeutic problem of the 8-aminoquinolines; neither hypnozoitocidal activity nor specific hemolytic toxicity composing meaningful therapeutic indices could have been derived.
The selected compounds nonetheless represented relative safety regarding broader toxicity issues and were safely advanced to clinical trials. There is no record of any of the hundreds of research subjects suffering irreversible harm attributable to exposure to those dozens of compounds. The clinical research team headed by Alf Alving (Fig. 8) in the Department of Medicine at the University of Chicago conducted the bulk of those trials using the vigorously relapsing Chesson strain P. vivax, a colony of Anopheles quadrimaculatus mosquitos managed by the laboratory of Clay Huff (Fig. 8) at the University of Chicago, and inmate volunteers at the penitentiary in Joliet, IL (173). Robert Coatney and colleagues at the U.S. National Institutes of Health conducted similar human trials from March 1944 to November 1946 at the United States Penitentiary at Atlanta, GA (174). Both groups would later experiment with an abundant supply of new research subjects after 1950: American soldiers infected by P. vivax on the Korean Peninsula at war, the findings for 3,531 of whom were described in just three consecutive 1953 reports, with the chloroquine treatment-only relapse rate being 44% (n = 738) or 18% (n = 331) (131, 164, 175, 176). As the Germans had done at Elberfeld 2 decades earlier, the Americans assembled an essentially similar stepwise means of antimalarial drug discovery and development but employed a vastly larger and more complex web of partners (13).
Primaquine
Robert Elderfield (Fig. 8) at Columbia University first synthesized 8-(4-amino-1-methyl butylamino)-6-methoxy quinoline in 1945 under a U.S. government wartime contract. The efficient production of pure primaquine was made possible by Elderfield’s innovation of the catalytic reductive condensation of 6-methoxy-8-aminoquinoline with 1-diethylaminopentan-4-one (177, 178). The new compound was designated SN-13272 by the Board for the Coordination of Malarial Studies and ultimately named primaquine by Alving’s research group, which first put the drug into humans in 1948. The name primaquine derived from its defining primary terminal amine of the aliphatic side chain at the 8 position of the quinoline nucleus. SN-3883 proved as active as primaquine against P. vivax Chesson relapse but showed a slightly higher incidence of “annoying side actions” and was not further considered (179).
The scientific debut of primaquine may be considered the December 1950 paper of Edgcomb et al. in the Journal of the National Malaria Society (61). Therein may be found the culmination of a broader series of trials comparing primaquine to the standard of care plasmochin and competing isopentaquine applied against Chesson strain P. vivax. The report summarizes the controls for the challenge trials, 38 subjects treated with blood schizontocides only (quinine, atabrine, chlorguanide, and chloroquine), and all experienced a relapse after those therapies. Table 3 in that report represents the findings of primaquine dose-finding experiments in treating primary attacks of sporozoite-induced infections by Chesson strain P. vivax also treated with 1.64 g quinine daily for 14 days (concurrent dosing with 14 days of daily primaquine). The dose of 15 mg primaquine proved inadequate (4 of 5 subjects relapsed), and doses of 30 mg or 60 mg were completely efficacious (in 5 and 4 subjects, respectively). Follow-up experiments employing the same quinine treatment and concurrent 22.5 mg primaquine daily prevented a first relapse in all 23 subjects challenged.
The investigators also treated subjects with primaquine alone, administering either 22.5 or 45 mg daily for 14 days in 5 subjects each; 4 of 5 and 1 of 5 relapsed, respectively. The authors suggested that most of those relapses were probably recrudescences on the weight of the relatively rapid recurrence of parasitemia (days 12 to 18 after the cessation of therapy). However, other studies with Chesson strain P. vivax determined day 22 postpatency to be the median day of first relapse (180); the recurrences after primaquine alone occurred on days 26 to 32 postpatency, well within the normal range for relapse by P. vivax Chesson. The follow-up investigation by Alving et al. (143) affirming the potentiation of primaquine activity against hypnozoites by quinine or chloroquine, rather than simple recrudescence, has been described (Fig. 6B). A clinical trial of primaquine reported in 2015 included an arm of consecutive primaquine 2 days after rapidly eliminated artesunate monotherapy of the acute attack (27). Unlike the 1955 experiment of Alving et al. (143), where 15 mg daily was applied, the recent trial applied 30 mg daily, and it resulted in good efficacy (92%). The relatively poor efficacy of primaquine alone may be overcome by coadministered blood schizontocide or an increased dose.
Among the field of 21 8-aminoquinolines evaluated against the plasmochin standard in clinical trials, primaquine emerged as superior. In the 1950 report (61), Edgcomb et al. assessed the chemotherapeutic indices (the ratio of the largest tolerated dose divided by the smallest dose capable of preventing “nearly all” relapses) of pamaquine (1.0), isopentaquine (2.5), and primaquine (10), with the estimated doses for 100% cure of Chesson strain P. vivax being 90 to 120 mg, 90 mg, and 22.5 mg, respectively. The maximum tolerated doses for these compounds (in subjects not sensitive to them) were considered to be 90 mg, 120 mg, and 240 mg, respectively. In this light, the primary objective of the mission to discover a therapy less toxic and more effective than plasmochin for delayed attacks of vivax malaria succeeded spectacularly. In April 1954, Alving addressed an audience at the Walter Reed Army Medical Center: “we had in primaquine a much more effective drug for Caucasians. . . . Unfortunately, in 10% of Negroes or dark-skinned races a serious hemolytic defect occurred. . . . This was important because about 10% of our troops in Korea were Negroes” (181).
According to Beutler in 1959 (62), Alving and his colleagues dosed 1,852 Caucasians and 141 African-Americans with 8-aminoquinolines, with just 4 and 14 proving hemolytically sensitive, respectively (OR = 51, 95% CI = 17 to 160, P < 0.0001). The relatively good tolerability and safety of primaquine were thus affirmed in many hundreds of normal subjects but apparently in no more than 18 sensitive subjects. The later military experience with primaquine during the Korean War occurred without knowledge of G6PD deficiency or the ability to discern who may or may not be sensitive, other than understanding that “dark-skinned races” were somehow more prone to hemolytic sensitivity to 8-aminoquinolines. Screening for primaquine sensitivity amounted to dosing and monitoring for the onset of acute hemolytic anemia.
Garrison et al. (182) described supervised treatment with 15 mg daily primaquine for 14 days administered to a total of 864 soldiers repatriated from the Korean Peninsula and suffering late attacks of vivax malaria at military bases in the United States during the summer of 1951. They described no serious adverse reactions and characterized the therapy as “well tolerated.” Soon thereafter a published trial of posttravel PART was undertaken aboard two troop ships repatriating American soldiers from the Korean Peninsula during September 1951 (183). The trial involved 3,536 soldiers, 426 of whom were African-Americans. During the approximately 2-week voyage from Sasebo, Japan, to Seattle, WA, USA, soldiers were randomized to 15 mg primaquine daily (n = 2,060) or a placebo of primaquine (n = 1,476), and those passengers seeking medical attention were evaluated. According to the investigators, “There was no detectable evidence of toxicity among the 2,060 officers and men who received primaquine.”
Archambeault (184) described the mass primaquine treatment (15 mg daily for 14 days) of 415,340 healthy American soldiers also being repatriated from duties on the Korean Peninsula between August 1952 and December 1953. The report provides details regarding the compulsory supervised treatment regimen. His retrospective analysis of shipboard medical records is also reported but largely focused on the successful prevention of late malaria attacks, with slight mention of side effects. The earlier shipboard trial (183) is cited as the evidence of safety and tolerability allowing the operational mass therapy applied. He assessed his own evidence of safety and tolerability succinctly: “There were surprisingly few toxic reactions. . . . in two men methemoglobinemia developed and in one man hemolytic anemia developed. There were a few reports of mild to moderate dusky cyanosis that did not require discontinuance of primaquine therapy” (184). If one accepts “few” as mentioned here as approximately 10, the incidence of recorded toxic events in these operations would have been 1 in 50,000 treatments.
If, as Alving (181) described, about 1% of troops in Korea were G6PD deficient, at least several thousand would have been among Archambeault’s assessed population (184). Hemolytic anemia invariably occurs with administration of 15 mg daily to African-Americans with even moderate G6PD deficiency variants (see “G6PD Deficiency” below); there is no record of a closely monitored research subject known to be G6PD deficient not experiencing acute hemolytic anemia following exposure to daily primaquine at any therapeutic dose. The implicit near absence of hemolytic reactions among Archambeault’s (184) likely thousands of G6PD-deficient soldiers thus seems improbable, although presumably none constituted a clinical emergency.
The armed forces medical people seemed satisfied that the shipboard trials and operations demonstrated that primaquine could be safely administered without testing for sensitivity or clinical supervision. Alving (181) put it like this on 28 April 1954 in a lecture presented to the Course on Recent Advances in Medicine and Surgery at the Army Medical Service Graduate School, Walter Reed Army Medical Center, in Washington, DC: “It also gives us a great deal of reassurance to know that even though primaquine potentially can produce hemolytic anemia, the hemolytic anemia which may occur after 15 mg is apt to be mild.” The U.S. Army researchers and their academic partners thus considered the hemolytic toxicity problem to be safely manageable without clinical supervision. The broader medical and public health communities in the 1950s and 1960s adopted primaquine therapy as such based almost entirely on what amounted to evidence of safety, tolerability, and efficacy derived from the unique U.S. Army experience with the drug. That experience was in exceedingly small numbers of closely observed G6PD-deficient American prisoner volunteers (all of whom hemolyzed with daily primaquine exposure) or far larger numbers of inadequately observed soldiers in the Korean War (in almost none of whom were hemolytic events recorded).
In a retrospective view over 60 years later inclusive of wide and important diversity in G6PD deficiency genotypes and primaquine sensitivity phenotypes, the American military experience and perspective on primaquine toxicity now appears to be overly simplistic and optimistic. As with plasmochin, primaquine would never see broad effective use beyond the sphere of clinical supervision for precisely the same reason: an earned reputation as a sometimes dangerous drug (see “G6PD Deficiency” below). Two decades after the registration of primaquine, the U.S. Army decided to try improving it.
Tafenoquine
A 1948 report from Pullman et al. (185) details a clinical trial of a peculiar aminoquinoline, SN-10275, having 1-phenyl and 3-methoxypyridine, along with 6,8-chloro, substitutions. It suffered toxic intolerance and poor therapeutic efficacy, but its extremely slow excretion (plasma half-life, approximately 24 days) and effective prophylactic suppression of parasitemia fired the imaginations of the investigators. They closed their report with, “However, further investigation of constitutionally related compounds is indicated because a non-toxic drug which retained the antimalarial activity of SN10275 and remained in the body for long periods of time, would have great value in the chronic suppression of malaria.” In 1979, a slowly eliminated 5-phenoxy 8-aminoquinoline called WR238605 (and, much later, tafenoquine) was first synthesized (186) and advanced to clinical development in 1991 (187). Although developed in a program aimed at improved hypnozoitocidal therapy, its promise as a chemoprophylactic agent weighed heavily on its advancement. In 2018, exactly 70 years after Pullman et al. (185) expressed the vision of it for chemoprophylaxis, tafenoquine was registered with the U.S. FDA for the separate indications of prevention of all malarias and the treatment of those that relapse (160, 161). Examining the genesis of tafenoquine and its development pathway provides relevant insights regarding a modern vision of the versatile 8-aminoquinolines for a global malaria problem and the fettered issue of their hemolytic toxicity.
The 1981 report by Davidson et al. (188) details the U.S. Army effort during the late 1960s and through the 1970s to discover and develop an improved hypnozoitocidal 8-aminoquinoline in response to the military operational problems encountered with primaquine against relapse in several million American soldiers deployed to Vietnam during that era. Unsupervised primaquine therapy in repatriated soldiers too often failed to prevent relapse (189, 190), as did the chemoprophylactic regimen of chloroquine combined with primaquine in a single weekly tablet (300 mg and 45 mg, respectively, called the “CP pill”) developed by Alving and colleagues in the early 1960s (191, 192). Davidson et al. (188) expressed, “The toxicity of primaquine limits its clinical usefulness in both prophylactic and therapeutic applications.” The U.S. Army recognized the key problem with its primaquine product long before the inheritors of it in the broader communities of science, medicine, and public health. As it had done with Bayer’s plasmochin, the U.S. Army set about the search for a less toxic and more easily administered primaquine.
The U.S. Army program developing tafenoquine occurred in the Division of Experimental Therapeutics, Walter Reed Army Institute of Research (ET-WRAIR), in Building 500 of the Walter Reed Army Medical Center Annex at Forest Glen, MD. During the early 1980s, this laboratory stood virtually alone as a site of active antimalarial drug discovery and development. In this era, a nadir of awareness of and research on malaria as a global health problem stood as a consequence of both the great gains and eventual collapse of the Global Malaria Eradication Program in 1969. The more important consequence, a global resurgence of malaria morbidity and mortality, was already well under way and would soon greatly worsen, but it was not yet broadly recognized or acknowledged (193). Chloroquine-resistant P. falciparum was known as a serious threat in those years, and most of the efforts at ET-WRAIR focused on developing new blood schizontocides, like halofantrine and mefloquine, to replace that failing drug (194). The development of a safer and more effective radical cure of vivax malaria was a project of peripheral importance, and it moved slowly with relatively few resources.
That program screened approximately 4,000 compounds first for causal prophylactic (hepatic schizontocidal) activity in the rodent Plasmodium yoelii or Plasmodium berghei models and later for hypnozoitocidal activity against P. cynomolgi in rhesus macaques (37, 188). Over 700 compounds representing 10 chemical classes exhibited causal prophylactic activity in the rodent models, with most of them being 8-aminoquinolines. Further, among those chemically diverse hundreds of causally active compounds, only the 6- and 8-aminoquinolines also exhibited hypnozoitocidal activity. The 6-aminoquinolines were uniformly less active than primaquine against P. cynomolgi hypnozoites, while among the 8-aminoquinolines the compound WR225448 (a trifluoromethyl-phenoxy compound identical to tafenoquine but lacking its 2-methoxy group) was about 5 times more potent than primaquine as a hypnozoitocide. This also proved true for other 4-methyl-5-phenoxy-8-aminoquinolines (WR233195, WR233078, WR232584, and WR232956; Fig. 11). Tafenoquine (WR238605) later proved more active than WR225448 against relapse of P. cynomolgi (showing 7-fold greater activity than primaquine) (188). In contrast to the limited preclinical therapeutic screening systems that resulted in both plasmochin and then primaquine (from a broad field of 8-aminoquinoline clinical development candidates that had been assessed against blood stages of avian malarias), tafenoquine had been optimized for hypnozoitocidal therapeutic efficacy in preclinical assessments heavily leveraging the model of rhesus macaque infected by P. cynomolgi. It would later enter into clinical development without competing compounds.
FIG 11.

The 8-aminoquinolines exhibiting >4-fold greater therapeutic activity relative to primaquine against relapse of P. cynomolgi in macaques up to 1981, where WR225448 emerged as the most active, being 4.8-fold more active than the other 5-phenoxy derivatives. WR225448 would later compete with WR242511 (10-fold more active) and WR238605 (tafenoquine, 7-fold more active) for selection as the sole candidate for clinical development.
The U.S. Army’s awareness of the 8-aminoquinoline hemolytic toxicity problem and its G6PD deficiency basis did not necessarily equip it to effectively deal with it in preclinical screening. By 1981, the greatly narrowed pool of candidate compounds all had good preclinical toxicity and efficacy profiles, but no technology could yet predict the relative order and degree of hemolytic toxicity in G6PD-deficient patients among them. In that year, the author took a junior technical position (as a graduate student) at ET-WRAIR. There, under the supervision of David Davidson, Jr., and Joan Decker-Jackson, he strived to develop an in vitro means of measuring relative hemolytic toxicity among 8-aminoquinolines (66, 67, 195, 196). The author left that post in 1984 without having achieved that technical objective and with a lasting impression of having been the sole meager resource committed to it before or after his tenure in that era.
Screening for hemotoxicity in animals did occur, and a study from ET-WRAIR in 1988 reported methemoglobinemia in beagles with tafenoquine and its closest preclinical peers at that late date, WR225448 and WR242511 (Fig. 11) (197). WR242511 stood alone as a 5-arlyoxy derivative among 5-phenoxy peers in late preclinical development. The 5-aryloxy series, first reported in 1982 (198), offered WR242511 as having conspicuously superior blood and hepatic schizontocidal as well as hypnozoitocidal activities relative to primaquine, other 5-aryloxy derivatives (199), and the 5-phenoxy derivatives (200). However, the peak methemoglobinemia levels in beagles were 2.7-, 4.0-, or 7.6-fold higher for tafenoquine, WR225448, and WR242511, respectively, than the level induced by primaquine in the same model (197). Despite having 10- and 18-fold greater hypnozoitocidal and causal prophylactic activities than primaquine against P. cynomolgi in rhesus macaques (201) and ex vivo hepatocytes (202), WR242511 was dropped from the development effort. The greater propensity of WR242511 to generate methemoglobin than primaquine (in beagles) appears to have disqualified WR242511 on the dubious surmise of greater potential for hemolytic toxicity. Another factor, however, could have also played a role; i.e., this compound lacked the long elimination half-life of the 5-phenoxy candidates (about 24 h in macaques [203] versus several weeks) and would have lost appeal as an agent of chemoprophylaxis against P. falciparum, a dominating U.S. Army interest relatively late in the 8-aminoquinoline program (204).
The seemingly potent generation of methemoglobin by WR242511 led to its transition to another U.S. Army group developing therapies for cyanide poisoning. It was evaluated in rhesus macaques, where it generated almost no methemoglobin (205). Sublethal and lethal toxicity with WR242511 occurred at daily doses of 7 mg/kg (still without methemoglobinemia), but the effective dose against relapse of P. cynomolgi in the same animal was just 0.1 mg/kg (versus 0.316 mg/kg for tafenoquine) daily for 7 days (199, 201). The rejection of therapeutically powerful WR242511 from the U.S. Army antimalarial development program, presumably on the basis of high methemoglobinemia in dogs, may have been unfounded. That specific hemotoxicity metric (and animal model) was not validated as a correlate of hemolytic toxicity and, in any event, later proved irrelevant in treated macaques.
The lack of understanding of the mechanism by which 8-aminoquinolines destroy G6PD-deficient erythrocytes disallowed a robust in vitro screening system by which to rationally select a least hemolytic 8-aminoquinoline. Preclinical optimizing of therapeutic activity with minimal hemolytic toxicity, i.e., a therapeutic index of relevance to that specific toxicity problem, was not achieved. The hemolytic toxicity of WR242511 or any other candidate 8-aminoquinoline relative to that of primaquine could not have been known. Perhaps more than any other 8-aminoquinoline in the later U.S. Army preclinical screening program, WR242511 exemplifies the promise of clinical hypnozoitocidal efficacy, with possible hemolytic safety left effectively unexamined.
In the late 1980s, tafenoquine advanced as the lone candidate for clinical development without certainty regarding its hemolytic toxicity relative to primaquine or any other 8-aminoquinoline. The Investigational New Drug (IND) application to the U.S. FDA was filed for tafenoquine in 1991, and the first trials in humans commenced in 1992 (187). Although the U.S. Army developed tafenoquine expressly for the purpose of replacing primaquine in PART, it had recognized 5- to 15-fold greater activity against blood-stage asexual P. falciparum than primaquine, and the idea of using it as a very slowly eliminated chemoprophylactic soon dominated interest and effort. Clinical trials in the late 1990s and early 2000s of weekly tafenoquine for prophylaxis—along with trials of 1- to 3-dose radical cure—were undertaken by the U.S. Army and its GlaxoSmithKline (GSK; UK) drug development partners. These trials uniformly showed good safety, tolerability, and efficacy in nonpregnant G6PD-normal adult subjects (206–215).
Nonetheless, the U.S. Army in that era indefinitely paused its development of tafenoquine for prophylaxis. This was in part due to an unexpected but mild and reversible vortex keratopathy among most subjects taking 200 mg tafenoquine weekly for 6 months, later shown to be inconsequential (216). More importantly, the Fifth Amendment to the Declaration of Helsinki during this period played a pivotal role in discouraging the development of tafenoquine for chemoprophylaxis: ethical dilemmas had been identified for clinical trials of chemoprophylactics in heavily exposed semi-immune subjects unlikely to benefit from chemoprophylaxis (217). The essential use of placebo (or relapse) controls in vulnerable nonimmune subjects also carried both ethical and practical barriers to trials of chemoprophylaxis. The perceived need for at least one additional phase 3 pivotal trial of tafenoquine for chemoprophylaxis—which could not be executed—swayed the U.S. Army to pause on that indication.
Faced with loss of the drug for any indication, in 2006 the U.S. Army/GSK steering committee on tafenoquine chose to finally pursue the less challenging development of it for radical cure of vivax malaria. They approached the Medicines for Malaria Venture in Geneva, Switzerland, with a proposal to do so. In 2008, those public and private partners signed a formal agreement to develop tafenoquine for that indication. At that early stage, the developers reasonably coupled their experimental hypnozoitocide to the chloroquine standard-of-care blood schizontocide for the radical cure of acute vivax malaria. That strategic decision was not made in view of an optimized potentiation of tafenoquine against hypnozoites or mitigated toxicity but in view of optimized regulatory ease of approval relative to pairing it with an artemisinin combined therapy. The developers reasonably presumed the envisioned drug label would be for a single pair of partner drugs for radical cure, i.e., chloroquine and tafenoquine. The already widespread and acknowledged problem of chloroquine-resistant P. vivax could await postregistration trials paired with other blood schizontocides. No other partner blood schizontocides with possible activity-potentiating or toxicity-mitigating effects would be evaluated with tafenoquine along its clinical development pathway to registration.
Unlike primaquine, the pivotal phase 3 clinical trials of tafenoquine as a hypnozoitocide did not occur in naturally infected soldiers or experimentally challenged volunteers but in residents of areas of endemicity (218–220). This imposed the necessity of accepting the confounding of posttreatment recurrences by reinfection during a minimal 6 months of follow-up. The temporal mingling of relapses following the possible therapeutic failure of tafenoquine with reinfections could not be sorted by any known technology. Therefore, estimates of the efficacy of tafenoquine against relapse by P. vivax necessarily include recurrences due to reinfection and may not be directly compared with the much higher and more accurate estimates of primaquine efficacy derived from subjects isolated from the risk of reinfection.
The proportion of patients not having a recurrence after therapy with chloroquine combined with tafenoquine (300-mg single adult dose), primaquine (0.25 mg/kg daily for 14 days), or no hypnozoitocide was 89%, 77%, and 38%, respectively, in one trial (217) and 62%, 70%, and 28%, respectively, in another (219). In a third trial lacking a placebo control (220), 67% and 73% taking tafenoquine or primaquine, respectively (each with chloroquine), remained recurrence free after 6 months. The proportions of posttherapy recurrences due to reinfections rather than treatment failures in these trials cannot be known. There is as yet no unambiguous estimate of the efficacy of single adult dose of 300 mg tafenoquine (combined with a defining blood schizontocide) against relapse. In 2019, such a trial with dihydroartemisinin-piperaquine and tafenoquine radical cure without confounding by reinfection is in progress in Indonesia.
The current availability of tafenoquine for a labeled indication of chemoprophylaxis merits explanation here. In about 2010, the U.S. Army Medical Material Development Activity revisited its earlier decision putting a hold on the development of tafenoquine for a prophylactic indication. It compiled the available data and contracted a pharmaceutical consulting firm to assess the status of the data accumulated over the past 20 years for the development of tafenoquine for chemoprophylaxis. The firm advised that the data in hand were no more or less complete than the data that had been presented to the U.S. FDA by GSK in the chemoprophylaxis dossier for atovaquone-proguanil (Malarone), which 10 years earlier resulted in approval. With that advice and effectively salvaging a trial in Kenya that had been shelved with flawed on-site diagnostic microscopy (a solved problem of false positives) (221), the U.S. Army undertook several small trials and launched a single large safety trial focused on ophthalmologic toxicity in 600 healthy volunteers (216). In 2013, GSK formally expressed to the U.S. Army its decision to not develop tafenoquine for a chemoprophylaxis indication. The U.S. Army then accepted proposals from several pharmaceutical firms for registering tafenoquine for chemoprophylaxis in the United States, and 60 Degrees Pharmaceutical LLC was selected as a U.S. Department of Defense commercial partner for that enterprise. In 2018, that firm successfully registered tafenoquine, labeled as Arakoda, with the U.S. FDA with the sole indication of chemoprophylaxis against malaria (160).
For reasons already discussed, tafenoquine proceeded through this protracted and complex clinical development path without competing 8-aminoquinolines or partner blood schizontocides and without specific knowledge of its hemolytic toxicity relative to primaquine. The first meaningful assessment of that toxicity came only in 2017 with the report by Rueangweerayut and colleagues (71) in Thailand. Those authors concluded that a single 300-mg dose of tafenoquine was approximately as hemolytic as 14 days of 15 mg primaquine daily (210 mg) in otherwise healthy women heterozygous for the moderate (WHO class III) Mahidol variant of G6PD deficiency (and having between a 40% and a 60% of normal enzymatic activity phenotype). Some 30 years after needing this specific knowledge—when the U.S. Army faced having to select a single molecule from among several promising 8-aminoquinolines for advancement to clinical development—tafenoquine was finally determined to be as hemolytic as primaquine.
Tafenoquine greatly exceeds the original chemoprophylactic vision of it expressed by Pullman et al. (185) in 1948. It is safe and well-tolerated in nonpregnant G6PD-normal adult subjects, exerts efficacious blood and hepatic schizontocidal activities over a greatly prolonged presence in plasma, and has shown good efficacy as weekly chemoprophylaxis against all malarias or as a single-dose hypnozoitocidal therapy against relapsing malarias. GSK successfully registered tafenoquine as Krintafel with the U.S. FDA for the indication of the radical cure of vivax malaria in 2018 (161). Tafenoquine is a very significant and hard-won advance in malaria chemotherapeutics. It may well transform the practice of travel medicine against malaria, both as weekly causal prophylaxis and as single-dose PART, posttravel or -patency (222), and yet, this time, the U.S. Army failed its expressed objective of discovering a less toxic primaquine.
The problem of 8-aminoquinoline hemolytic toxicity in G6PD-deficient patients remains the impediment to public health utility first imposed on plasmochin and then primaquine. Tafenoquine, however, comes with greatly improved and rapidly improving G6PD diagnostics for managing the problem (see “Clinical and Public Health Implications” below). The greatest advantage of tafenoquine in therapy—broad coverage of all stages of parasites and the very slow elimination of a single dose—also imposes its greatest safety concern: there is no clinical retreat with the onset of hemolytic anemia. Patients having a confirmed G6PD status of <70% of normal must not receive this treatment (160, 161). Accidental dosing of patients successfully screened as G6PD deficient occurs (208), so access to rescue transfusion and hemodialysis services may prove necessary with clinical use if tafenoquine in hemizygotes or homozygotes incites a threatening hemolytic anemia. Precisely as Sinton and colleagues (141) expressed almost a century ago for plasmochin, any serious harm done by tafenoquine “would tend to bring into disrepute what is undoubtedly a very valuable drug in the treatment of certain forms of malaria.” Tafenoquine is certainly very valuable, but it is also potentially quite dangerous. This is, concisely, the lasting legacy and unsolved dilemma of the extraordinary 8-aminoquinolines.
The vital role of the hemolytic toxicity of the 8-aminoquinolines in G6PD-deficient patients and the intractable neglect of it over many decades (19) emerge as being of clear core importance in dealing with this class of drugs. That neglect was neither irrational nor irresponsible but was driven by both an inability to predict hemolytic toxicity in preclinical screening and a technical misunderstanding of the character and complexity of the problem and its practical consequences for patients lacking adequate access to minimally sufficient clinical services. A technical exploration of that misunderstanding serves the important purpose of rationally addressing it because humanity remains in need of a hypnozoitocide that cannot seriously harm G6PD-deficient patients. The potential for 8-aminoquinolines to meet that need, as evidenced by this technical history of the preclinical development of tafenoquine, has yet to be deliberately explored. Exploiting optimized efficacy and minimized and mitigated toxicity by rationally selected compounds and partner drugs may be informed by a clarity of understanding of the cellular and molecular mechanics of those effects enabling rational and effective preclinical screening (see “Future Perspectives” below).
G6PD DEFICIENCY
Biochemistry and Genetics
G6PD catalyzes the conversion of glucose-6-phosphate to 6-phospho-glucono-δ-lactone in the rate-limiting reaction of the hexose monophosphate shunt (or pentose phosphate pathway). Nicotinamide phosphate (NADP+) cofactor is reduced in the reaction to NADPH, the source electrons for the reduction of glutathione (GSH) (glutathione disulfide [GSSG] → 2GSH) via glutathione reductase and the reduction of methemoglobin to hemoglobin by methemoglobin reductase. G6PD activity maintains a healthy reducing erythrocyte cytosol. When G6PD activity is impaired, the erythrocyte is labile to creation of an oxidizing cytosol by introduced oxidants. That equilibrium favors oxidized species of specific compounds introduced into this system, and in sufficient quantity, these yield the Heinz bodies associated with clinical hemolytic crisis (Fig. 12). Heinz bodies are irregular aggregates of degraded hemoglobin and its heme moiety in oxidatively distressed red blood cells, but their formation is quite specific to chemical poisons like phenylhydrazine or primaquine (223). The simple oxidant sodium nitrite, for example, does not induce Heinz bodies or hemolytic anemia, even when fatal poisoning occurs (224).
FIG 12.
The photomicrograph illustrates Heinz bodies (the dark blue bodies) within red blood cells stained with crystal violet. The thin smear slide derived from a suspension of red blood cells prepared from a sample of venous blood taken from a G6PD-deficient Karen woman living in Thailand incubated for 1 h with acetyl-phenylhydrazine. Germana Bancone at the Shoklo Malaria Research Unit at Mae Sot, Thailand, provided the image, and it is published here with her permission.
G6PD deficiency is the most common and diverse inherited disorder of humans, affecting about 400 million people at a prevalence of about 8% in nations where malaria is endemic (225). The gene expressing G6PD occurs on the X chromosome, and people may thus be either G6PD normal, hemizygous (males only), homozygous (females only), or heterozygous (females only) (Table 7). Over 20,000 base pairs along 13 exons compose the gene, and the many dozens of known single nucleotide polymorphisms associated with specific variants of G6PD deficiency occur all along the length of the gene. Variants may be typed by classes according to WHO criteria based upon the severity of impairment of enzymatic activity and clinical consequences; classes I to V range from severe and potentially dangerous to mild and relatively inconsequential (226). The most common variants are classes II and III, separated by a residual G6PD activity threshold of less than and greater than 10%, respectively. Historically, these have been represented by the Mediterranean variant and the African A− variant, respectively, and primaquine sensitivity phenotypes of severe and moderate sensitivity, respectively. Although the residual enzymatic activity of defective enzyme dominates as a clinically relevant and intuitively simple phenotype, other more complex factors, like the stability of the active G6PD enzyme dimer, may also be relevant (227).
TABLE 7.
Summary of G6PD status, genotype, enzymatic activity phenotype, primaquine safety threshold at a percentage of normal activity and value of qualitative screening for G6PD deficiency
| G6PD status | X allelesa | G6PD activity phenotype (%) | Primaquine safety threshold (%) | Qualitative screening |
|---|---|---|---|---|
| Normal male | XY | >80 | >70 | Safely included |
| Normal female | XX | >80 | >70 | Safely included |
| Hemizygous male | X*Y | <20 | None | Safely excluded |
| Homozygous female | X*X* | <20 | None | Safely excluded |
| Heterozygous female | XX* | 0–100 | >70 | Dangerously included at 30–70% of normal |
X, wild-type (normal) G6PD gene; X*, mutant (deficient) G6PD gene.
The X-linked character of G6PD deficiency imposes sex as a core determinant of its expression and consequent vulnerability to hemolytic agents. Variation in hemolytic sensitivity among hemizygous males and homozygous females must be wholly attributable to the variable degrees of impairment to G6PD enzyme function, i.e., variant-specific determinants of residual enzymatic activity. The highly variable vulnerability of heterozygous females, on the other hand, is dominated by the degree to which the lone abnormal X chromosome is inactivated relative to the normal X chromosome. Mary Lyon hypothesized a random inactivation event during early embryonic development where one of the two X chromosomes in each cell becomes permanently inactive (228). The progeny cells of each retain the inactive chromosome, resulting in females having a fixed ratio of G6PD-normal or -deficient cells, i.e., lyonization and mosaicism for the trait in red blood cell populations randomly ranging from 0% to 100% normal or deficient. Population surveys of the G6PD activity phenotype often find females dominating the 30% to 80% of normal activity range (229–231). The term “intermediate” G6PD deficiency thus refers to these heterozygotes rather than the residual enzyme activity phenotypes per se, with these tending to be dichotomized as wholly normal (>70% of normal) or deficient (<30% of normal) among hemizygous males and homozygous females.
None of these fundamentally important characteristics of G6PD deficiency were understood during the clinical development of primaquine during late 1940s and early 1950s. So-called primaquine sensitivity mirrored the already known and characterized sensitivity to plasmochin in a minority of non-Caucasian males and, more rarely, Caucasians or females (232, 233). That lack of understanding bears directly on the question of how primaquine came to be seen as a suitable solution to the problem of hypnozoitocidal therapy throughout the latter half of the 20th century.
Discovery
Before the U.S. Army undertook its development of 8-aminoquinolines against delayed attacks of vivax malaria in 1944, idiosyncratic allergic reactions, like those of black water fever in patients consuming quinine against malaria, were suspected but not confirmed. The little evidence available tended to argue against that hypothesis; e.g., blood autoagglutinins inconsistently appeared in patients in a postplasmochin hemolytic crisis (151). Also, patterns of risk by race and sex—non-Caucasian males seemed predisposed—were noted but cautiously weighed in light of the risk of the need of plasmochin therapy; i.e., non-Caucasian males were more often exposed to malaria and the necessity of therapy. In 1959, Beutler (62) listed the racial incidence of 8-aminoquinoline-induced hemolytic anemia among 30,590 people exposed to those therapies: 1,314 were races other than Caucasian (predominantly Asian and African), with 86 (6.6%) suffering hemolytic anemia; 29,276 Caucasians were exposed, with 47 (0.16%) suffering hemolytic events (OR = 44, 95% CI = 30 to 62, P < 0.0001). Among people needing and receiving 8-aminoquinoline therapies, those of non-European races were certainly somehow predisposed, and this was understood to point to an inherited abnormality.
The discovery of an inherited defect in glucose metabolism as the primary lesion in primaquine-sensitive subjects during the early to mid-1950s—an unprecedented and wholly unexpected finding at that time—derived from a remarkable series of experiments comparing human subjects of the primaquine-sensitive phenotype to those of the primaquine-insensitive phenotype identified in the development of that drug. The work was conducted on the same prisoner volunteers at the Illinois State Penitentiary in Joliet used by the primaquine development team at the U.S. Army Medical Research Unit embedded within that prison and largely staffed and operated by the University of Chicago (13).
The discoverers of G6PD deficiency first confirmed that plasmochin-sensitive subjects were also sensitive to primaquine (60). Then they isolated that defect to the red blood cells of affected persons, leveraging the technology of 51Cr labeling introduced in 1950 (234). In brief, they transfused erythrocytes of sensitive subjects into insensitive recipients treated with primaquine and observed the destruction of the infused erythrocytes. Conversely, insensitive erythrocytes transfused into sensitive subjects dosed with primaquine appeared to be unaffected (235). Further work demonstrated that older erythrocytes were the most sensitive to destruction (236). At about the same time, work describing the appearance of Heinz bodies in sensitive but not insensitive erythrocytes incubated with phenylhydrazine provided a means to identify sensitive subjects in the laboratory and to assess the phenomenon in vitro (237). Sulfhydryl-interfering poisons like arsenite caused insensitive erythrocytes treated with phenylhydrazine to produce Heinz bodies as if the erythrocytes were from a sensitive subject (238), bringing focus to the glutathione reservoir of erythrocytes in the search for the primary metabolic lesion. When Carson et al. (239) in 1956 reported that hemolysates from sensitive subjects deprived of NADPH could not reduce glutathione, despite added glucose-6-phosphate, impaired G6PD enzyme activity had finally been implicated as the underlying cause of hemolytic sensitivity to 8-aminoquinolines.
8-Aminoquinoline-Induced Hemolytic Anemia
In 1959, Beutler (62) characterized the primaquine-induced hemolytic anemia in G6PD-deficient patients broadly, having yet no notion of the enormous diversity of the disorder or the key clinical and public health implications embedded therein. His experience up to that point had been with a very few African-American subjects, most of them otherwise healthy men. In 2008, Beutler (240) recalled, “In the 1950s it was not generally appreciated that many different mutations could strike a gene. Thus, it was initially assumed that G6PD deficiency was a single disorder.” This is crucially important context today in reading Beutler’s 1959 description of the clinical course of the primaquine-induced hemolytic reaction. Emphasis had been given to the relatively mild and self-limiting nature of hemolysis observed in his research subjects: “Finally, the peripheral blood picture returns entirely to normal and patient is entirely asymptomatic in spite of continued administration of primaquine” (62). The first description of clinically relevant G6PD deficiency variability came only in that year, from subjects of Mediterranean descent in direct comparison to those having the African A− variant (241). Subsequent clinical studies through the 1960s affirmed the relatively extreme primaquine sensitivity of those with Mediterranean G6PD deficiency compared to those with African A− G6PD deficiency; i.e., it was neither mild nor self-limiting in character and appeared potentially threatening to life. In 1970, Zalusky (242) expressed this regarding the Mediterranean variant: “Because even the young red cells have little enzyme activity, hemolysis may continue unabated in the presence of the incitant and may lead to death if blood transfusion is not given early.”
Consideration of the clinical course and consequences of 8-aminoquinoline poisoning of G6PD-deficient patients thus requires acknowledging those two phenotypes (represented by two discrete genotypes) and the certain possibility of many other primaquine sensitivity phenotypes among the many dozens of other specific genotypes (225). No work has systematically surveyed the variants for the primaquine sensitivity phenotype, but the presumptive marker of that phenotype—the percentage of normal G6PD enzymatic activity—is often measured and reported. Indeed, the WHO classification scheme for G6PD variants (226) depends on that phenotype. The rationale for separation of severe class II from moderate class III at the 10% of normal activity threshold was in part dependent on the primaquine sensitivity phenotypes in Mediterranean and African A− variants, i.e., severe versus mild. Nonetheless, no broader correlation has been validated, and some severe hemolytic events occur in patients having a supposedly moderate sensitivity phenotype or a seemingly inadequate exposure to primaquine (243–245). Such deviation from expectation perhaps reflects the broader biochemical (227) and clinical complexity of the abnormality and event (21) to include a likely exacerbation of oxidative liability by the recent acute malaria context of the therapy in practice. Infections may induce a hemolytic crisis in G6PD-deficient patients without the involvement of hemolytic drugs.
G6PD-deficient patients are sensitive to a variety of drugs and chemicals, and the relative hemolytic toxicity among those varies a great deal (62). Few drugs exceed the 8-aminoquinoline archetype primaquine in terms of both the clinically significant hemolytic potential and the numbers of people potentially exposed to harm by indicated therapeutic use (20). Important distinctions in the degree of hemolytic toxicity in G6PD-deficient patients of the same variant may also occur among 8-aminoquinolines, but direct examination of this has been exceedingly rare. In 1960, Alving et al. (246) compared equal daily doses (30 mg base) of primaquine to the 8-aminoquinoline called quinocide (Fig. 4 and 13) in the same African A− subject: quinocide provoked a more rapid, deeper, and more prolonged hemolytic event than primaquine, and mole for mole (each drug has the same molecular weight), quinocide appeared to be substantially more hemolytic than primaquine. The relatively slight molecular distinction between primaquine and quinocide (placement of the methyl group on the aliphatic side chain) produced a clinically significant distinction in hemolytic toxicity. Structure-hemolytic toxicity relationships of the 8-aminoquinolines almost certainly exist but have not been explored or even crudely characterized. We cannot now know if only slightly hemolytic or, indeed, nonhemolytic 8-aminoquinolines exist.
FIG 13.
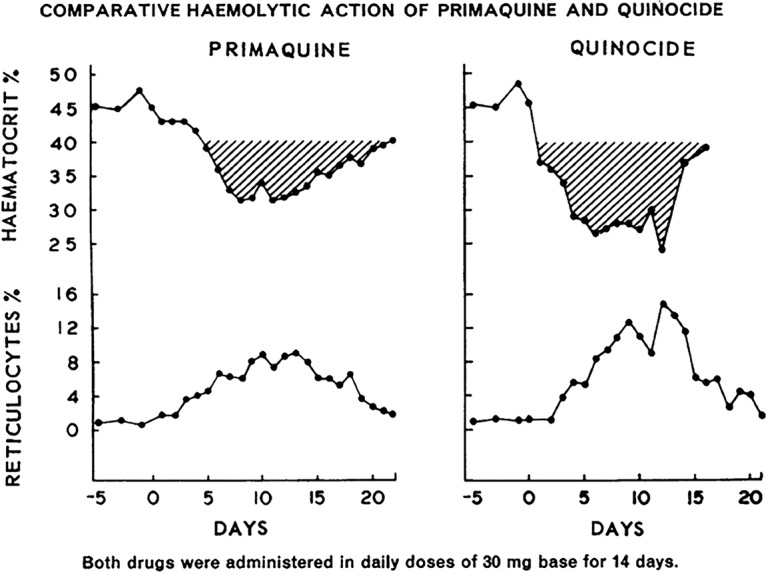
The acute hemolytic anemias induced by closely related 8-aminoquinolines in a single healthy volunteer having the African A− variant of G6PD deficiency exhibiting different degrees of hemolytic toxicity. From Alving et al. (246), reproduced with permission of the Bulletin of the World Health Organization.
More recently, Rueangweerayut et al. (71) showed that a single dose of 200 mg of tafenoquine caused less hemolysis than a total dose of 210 mg primaquine (administered over 14 days) to otherwise healthy heterozygous female volunteers with the Mahidol variant of G6PD deficiency. Although this means of quantifying hemolytic potential may be considered relatively crude and prone to many potential confounders, it is nonetheless direct. Gram for gram, primaquine may be slightly more hemolytic than tafenoquine. On the other hand, mole for mole—tafenoquine is nearly twice the molecular weight of primaquine (464 versus 259 g/mol)—primaquine would appear to be less hemolytic; i.e., 210 mg primaquine contains nearly twice as many molecules as 200 mg of tafenoquine.
The hemolysis provoked by 8-aminoquinolines may thus be appreciated as being profoundly dependent upon the drug administered, its dose and dosing schedule, its metabolism to hemolytic derivatives, the G6PD variant and the alleles involved (hemi-, homo-, or heterozygous), the specific degree of lyonization of the trait in heterozygotes, and the health of the patient at exposure to the hemolytic agent. This wide diversity of important determinants imposes limitations and specificity of the condition regarding the character of 8-aminoquinoline hemolytic toxicities in vulnerable patients. Nonetheless, cautious, appropriately controlled experimental consideration of the event offers a key understanding of the underlying lesion and its dominating determinants.
Mechanism of Hemolytic Toxicity
The extensive literature exploring the molecular and cellular basis of 8-aminoquinoline hemolytic toxicity broadly points to the conspicuous oxidative lability of G6PD-deficient red blood cells. The seminal work using arsenite sulfhydryl poisoning of glutathione, mimicking G6PD-deficient red blood cells, and most of what followed affirmed the lost reducing capacity as the core lesion of hemolytic sensitivity. Many workers concluded that primaquine-induced hemolytic toxicity must therefore be a consequence of a broad oxidative degradation of the red blood cell cytosol, typically pointing to the peroxides and superoxide generated by redox cycling of highly reactive primaquine derivatives (76, 77, 247–251). Those compounds are thus seen as agents generating oxidizing moieties, principally, hydrogen peroxide, that poison G6PD-deficient red blood cells in the manner of, for example, the oxidant poison sodium nitrite. While both primaquine and nitrite certainly introduce an oxidizing stress and methemoglobinemia (252, 253), even fatal acute nitrite poisoning with overwhelming methemoglobinemia is not accompanied by a hemolytic crisis or the Heinz bodies invariably occurring with primaquine poisoning (254). Simple oxidative degradation seems to be an inadequate explanation for how primaquine destroys G6PD-deficient red blood cells. The drug itself rather than the products of its redox cycling must be the agent of red blood cell destruction. This view has long been held by some workers (65, 196, 223, 255), but no direct evidence yet affirms either hypothesis.
The hallmark of some chemically induced acute hemolytic anemias is the Heinz body, first identified in 1928 in connection with plasmochin toxicity (62). The poisons inducing Heinz bodies tend to be relatively reactive redox compounds, and among those, the resonating aromatic compounds tended to be the most hemolytic (223). Further, such compounds may be converted by oxygen to quinone-like substances (quinhydrone and hydroquinone analogs) to oxidize ferrous heme or globin sulfhydryls (223). In vitro, reversible methemoglobin formation precedes the irreversible formation of brown and green pigments that go on to form micellar aggregates of up to 2 to 3 μm presumed to be Heinz bodies (255). It is likely that these reactions, and not the superoxide and peroxides that they generate, explain their specific Heinz body hemolytic character, as Jandl and colleagues hypothesized nearly 60 years ago (223). Itano and colleagues (256–259) later established that the ability of phenylhydrazine analogs to induce Heinz body hemolytic anemias in rats correlated with their ability to act as ligands to ferrihemoglobin in forming an irreversible ferrihemochrome. The phenyl moiety of those poisons covalently bonded to the porphyrin ring in that ferrihemochrome. That specific molecular event was responsible for the Heinz body formation that ultimately destroys erythrocytes.
Figure 14 illustrates the irreversible CYP2D6-catalyzed generation of the primaquine metabolite 5-hyrdoxyprimaquine, followed by the reversible redox reactions to the primaquine semiquinonimine radical and primaquine quinonimine. Hypothetically, the latter compound would dominate a redox equilibrium deprived of reducing equivalents and thus generate irreversible ferrihemochromes, leading to Heinz body formation. Indeed, synthetic 5-hydroxyprimaquine in glutathione-depleted rats caused Heinz body hemolytic anemia (260). An oxidizing red blood cell cytosol depleted of reduced glutathione readily occurs in G6PD-deficient patients (261); an equilibrium favoring dominance of the 5-hydroxylated primaquine quinonimine derivative may thus occur. That species may react as the arylhydrazine ligand of Itano and colleagues (256–259) with ferrihemoglobin to form an irreversible ferrihemochrome. Thus, destabilized hemoglobin denatures and aggregates as Heinz bodies (223, 259, 261). This hypothesis accords with the chemical character of known highly redox-reactive 8-aminoquinoline metabolites and the physiochemical fate and consequences of them in G6PD-deficient patients. These compounds appear to directly provoke Heinz bodies and the acute hemolytic anemia that follows their appearance. The hypothesized irreversible accumulation of ferrihemochromes with relatively small daily doses of primaquine metabolized to redox-reactive 5-hydroxyl derivatives in an oxidative equilibrium state that favors toxic quinonimine species accords with the sudden onset of hemolytic crisis after at least several days.
FIG 14.

Hypothesized metabolism of 8-aminoquinolines, as represented by primaquine, to 5-hydroxylated species. In a G6PD-normal individual, who therefore has a reducing redox equilibrium in the cytosol (larger green arrows, above), the presumably harmless 5-hydroxyprimaquine metabolite dominates. In a G6PD-deficient individual, who therefore has an oxidizing redox equilibrium in the cytosol, the highly reactive primaquine quinonimine species dominates and forms irreversible hemochromes that precipitate as Heinz bodies, causing acute hemolytic anemia. Hb, hemoglobin; MetHb, methemoglobin; GS−, oxidized glutathione.
No current consensus around appropriate evidence affirms the molecular events leading to acute hemolytic anemia, beyond impaired G6PD function being the primary lesion (83). Many factors help explain that gap in understanding. The highly reactive and labile derivatives of primaquine that likely cause hemolytic toxicity have been exceedingly difficult to study, for example, but the dominant factor may be the failure to assign a high priority to answering the question. This may be appreciated by the long technical history of development of the 8-aminoquinoline antimalarials, where the hemolytic toxicity of candidate 8-aminoquiniolines relative to plasmochin or primaquine was not assessed and the lead of the complete abrogation of pentaquine-induced hemolysis by methylene blue adjunct was not followed.
The distinct possibility of a regimen of 8-aminoquinoline therapy that cannot cause harm in G6PD-deficient patients—with or without rationally selected activity-enhancing/toxicity-mitigating partner blood schizontocides or detoxifying adjuncts—has yet to be purposefully pursued in research aimed specifically at that bright prospect. The enabling preclinical optimizing of hemolytic toxicity and possibly mitigating adjuncts will likely require near certainty regarding the cellular and molecular events culminating in Heinz body formation, specific knowledge that would open multiple avenues of promising investigation (see “Future Perspectives” below).
Mild and Self-Limiting Primaquine Sensitivity
Though “mild and self-limiting” is often expressed to describe the character of hypnozoitocidal primaquine-induced hemolytic anemia in G6PD-deficient patients of the African A− variant, that terminology may misrepresent what typically occurs in practice. In most instances of this event during the era of plasmochin therapy and the later development of primaquine, the sudden onset of clinical signs of hemolysis would have prompted the cessation of therapy, typically after the third to fifth dose. Instances of G6PD-deficient patients completing the prescribed 14-day regimen must have been rare, and almost none were reported from practice. The extent of harm possible in large numbers of acutely ill G6PD-deficient patients has not been directly assessed in randomized controlled trials or by rigorous pharmacovigilance of practice. Available reported evidence suggests a very low frequency of serious harm (262), much like Archambeault’s nearly half million shipboard American soldiers (184). Nonetheless, the absence of evidence of harm should not be construed as evidence of the absence of harm. Confidence in safety, especially for a drug of demonstrated toxicity in patients invariably sensitive to it, requires direct evidence gathered in randomized controlled trials or rigorous pharmacovigilance systems specifically designed to gather that evidence.
Experimental full dosing of primaquine under close clinical observation occurred in a series of experiments reported by Alving et al. (246) involving daily dosing with 15 or 30 mg daily for 14 days, 30 mg daily for 120 days, or a weekly dose of 45 mg for 8 weeks. Each of these treatments involved what appears to have been the same three otherwise healthy African-American men having the A− variant of G6PD deficiency, with months of pauses between the experiments. The top panel of Fig. 15 illustrates the course of hemolysis, toleration, and recovery, all occurring despite uninterrupted daily 30-mg primaquine dosing for 120 days. The initial 30% drop in hematocrit (with the onset of hemoglobinuria on day 3 and a hematocrit nadir at about day 10 of dosing) returned to normal after about 28 days. Days 8 to 28 saw a sharp rise in peripheral reticulocytes, and then from days 28 to 120 of dosing, an essentially normal peripheral blood picture was seen. The damage done by primaquine had indeed been clinically inconsequential and self-limiting in those three healthy African-American men under close observation.
FIG 15.

Primaquine-induced hemolytic crises expressed as percent of hematocrit in patients having the African A− or Mediterranean variant of G6PD deficiency, based on data from Alving et al. (246) (top) and Pannacciulli et al. (271) (bottom). In the patient with the African A− variant, there is tolerance to continued primaquine exposure as the vulnerable older red blood cells destroyed by drug are replaced by more G6PD-robust and drug-tolerant reticulocytes and younger normocytes and dosing is extended to 120 days without a hemolytic event. In the patient with the Mediterranean variant, hemolysis deepens with each primaquine exposure, and after the 7th dose, dosing must cease due to danger to the patient (red X). The resumption of dosing 2 weeks later renews the hemolytic crisis, again with the clinical necessity of cessation of therapy. There is no acquired tolerance to primaquine exposure in the patient with the Mediterranean variant.
Alving et al. (246) rationally explained these findings on the basis of the marked age dependency of G6PD activity in erythrocytes originally described by Beutler et al. (236) in 1954. They reasoned that tolerance of primaquine derived from the clearance of vulnerable older erythrocytes (the initial hemolytic event) and replacement by primaquine-insensitive reticulocytes. Multiple lines of evidence later affirmed that deduction (Fig. 15) (263). G6PD variant A− reticulocytes exhibit nearly normal G6PD activity levels, but as those erythrocytes age, the slope of enzymatic activity declines more steeply and deeply than that for G6PD-normal red blood cells. Continuous primaquine dosing would maintain a relatively young erythrocyte population by destroying those aging as they become oxidatively labile and vulnerable to destruction. Indeed, pausing primaquine dosing for a few weeks—permitting A− erythrocytes to normally age—incites another hemolytic event (236).
These findings offered reassurance regarding the clinical use of primaquine without close supervision; i.e., there appeared to be little potential for serious harm even with extraordinarily prolonged high daily dosing. Other studies in populations in areas where malaria is endemic in that era seemed to support the notion of a relatively harmless toxicity problem (264–266). In 1960, an expert group convened by WHO expressed (43), “The group devoted special attention to the side effects of 8-aminoquinolines (primaquine and quinocide) because these may cause an acute intravascular hemolysis in certain primaquine-sensitive individuals, particularly among the dark-skinned peoples of the world. There is ample evidence that when given at recommended dosage, 8-aminoquinolines seldom give rise to symptoms of toxicity.” Such was the sanguine view of the primaquine toxicity problem in that era.
A retrospective critical review of the problem that today includes known key determinants of hemolytic toxicity provides context informing less optimism and much greater caution. The population-based studies in the Americas and temperate Asia, for example, occurred among indigenous people who either did not carry G6PD deficiency or in whom it was exceedingly rare (267–269). Moreover, the population-based interventions were not designed to demonstrate safety in a randomized fashion, although active surveillance for adverse events was often performed (266). Many of the apparently safe mass primaquine administration campaigns were carried out by historically authoritarian governments (e.g., those of the Soviet Union and the Democratic People’s Republic of Korea [266]) socially given to being perhaps overly inclined to consider the greater good over harm to a few. There may well be populations where the unrestrained administration of daily primaquine therapy poses relatively very little danger, e.g., on the Korean Peninsula (269), but in other populations, evidence and reason point to an unacceptable risk of significant harm in doing so (20).
Mild and self-limiting primaquine-induced hemolysis may be considered demonstrated by firm clinical evidence but derived from exceedingly small numbers of closely observed healthy subjects or very large numbers of subjects in inadequately observed or at-risk populations. The likely importance of those constraints may perhaps be evident with the confirmed report of fatal primaquine poisoning of a Brazilian man with primaquine therapy for vivax malaria (30 mg daily for 7 days) having the so-called mild African A− G6PD deficiency variant (243). Mild and self-limiting primaquine sensitivity did not occur in that instance, and no current understanding of the phenomenon may explain the fatal hemolytic event discovered only at postmortem examination of a patient that had been presumed to have not survived acute P. vivax malaria.
Severe Primaquine Sensitivity
The 1959 report of Marks and Gross (241) identified G6PD-deficient subjects of European descent to exhibit far lower levels of residual enzymatic activity than those of African descent. Whereas 500 G6PD-normal subjects had a mean activity of 16.1 U/g hemoglobin, that of 59 G6PD-deficient black males was 2.7 U/g hemoglobin (17% of normal) and that among 22 white males was 0.4 U/g hemoglobin (2.5% of normal). Moreover, they observed nearly normal G6PD activity levels in reticulocytes among deficient black subjects but not in those among deficient white subjects. Bonsignore et al. observed uniformly very low G6PD activity levels among red blood cells from Sardinian subjects of all ages with a history of hemolytic sensitivity to fava beans (favism) (270). Even the reticulocytes of G6PD-deficient Europeans appeared to be potentially vulnerable to destruction by introduced oxidants.
In their 1965 investigation, Pannacciulli et al. (271) expressed, “In mutant Negroes the self-limited course of the drug-induced hemolytic crisis is linked to a relative insensitivity to the drug of the younger red cells which have higher levels of G6PD activity. It seems therefore predictable that in mutant Caucasian males, whose younger red blood cells are almost completely devoid of G6PD activity, the course of the hemolytic crisis may have a somewhat different pattern.” They go on to describe daily primaquine (30 mg) challenge experiments in a G6PD-deficient Sardinian male (Mediterranean variant). In each instance of challenge, a severe hemolytic crisis (approximately 50% hemolysis) occurred, necessitating cession of dosing on the 7th day (Fig. 15, bottom). This investigation demonstrated reticulocytes in the subject to be as vulnerable to hemolysis as older red blood cells, in stark contrast to the well-established tolerance of primaquine dosing in African subjects. Pannacciulli et al. (271) summarized: “From our experiments it is evident that mutant Sardinian males may develop extremely severe hemolytic crises. We received the impression that a continuous administration of primaquine to our patient would have induced a severe anemia, leading to a poor prognosis.” Salvidio et al. (272) pointed to the absence of dangerous favism among blacks, along with differing mechanisms of damaged red blood cell removal (i.e., intravascular alone versus that along with extravascular processes among Sardinians). The graphs of Piomelli et al. (263) from that era clearly illustrated the primaquine sensitivity phenotype distinction with regard to relative G6PD activity levels across ages of red blood cells (Fig. 16).
FIG 16.

Data illustrating the centrifugal gradient separation of erythrocytes by age (C.D.F. is the cumulative distribution function) among G6PD-normal, African A− variant, and Mediterranean variant donors (A, B, and C, respectively). The extent to which G6PD activity declines with the age of the red blood cells in the same gradient and from the same donors is also shown (G, H, and I, respectively). G6PD activity in normal donors (G) decreases slightly with age, whereas in African A− variant donors (H), young red cells exhibit almost normal G6PD activity, but it declines more sharply with age. Mediterranean variant donor red blood cells (I) have very low levels of activity among the youngest reticulocytes and undetectable activity among older cells. Reproduced from Piomelli et al. (263) with permission of the Journal of Clinical Investigation.
The careful laboratory and clinical work on extreme primaquine sensitivity by the Italians was also reported by others (273, 274). Moreover, reports from the field appeared to verify the extreme sensitivity of some to primaquine therapy. In 1968, Abeyaratne and Halpe (275) in Sri Lanka reported four deaths among 21 children admitted to hospital in states of hemolytic crisis attributed to undocumented exposures to primaquine. In 1970, Aung-Than-Batu et al. (276) in Burma (Myanmar) reported 34 to 48% hemolysis in G6PD-deficient patients exposed to daily 15-mg primaquine dosing. Chareonlarp et al. (277) in 1972 examined the effect of a single 45-mg dose of primaquine (having little impact on African A− subjects [246]) in seven Thai subjects of the Mahidol G6PD− variant: 3 patients experienced >15% hemolysis, 2 experienced greater than 5%, and 2 had no hemolytic reaction. In 1976, Chan et al. (278) described severe enzyme deficiency phenotypes among three Chinese G6PD deficiency variants (Canton, B− Chinese, and Hong Kong-Pokfulam). In his 1981 review of the clinical problems associated with primaquine, Clyde (58) expressed, “In variant A− individuals, a standard course of primaquine may elicit a moderate and self-limiting anemia due to the hemolysis of older erythrocytes. In variant B− [Mediterranean] and relative Asian variant individuals, even young erythrocytes are deficient in G6PD and their hemolysis results in progressive hemoglobinemia and hemoglobinuria with fatal outcome unless blood is transfused promptly.” Direct evidence and clinical experience informed that assessment, and no further evidence or experience invalidates it. Primaquine is an unquestionably dangerous drug capable of lethal poisoning when used as indicated in some G6PD-deficient patients.
The most physiologically and clinically relevant lesion in extreme sensitivity to primaquine (and presumably other compounds) appears to be low residual G6PD activity in reticulocytes and younger normocytes. In such patients, hematological recovery in the face of continued daily primaquine dosing would not occur but instead hemolysis would progressively deepen and threaten severe hemolytic anemia. Conversely, sufficient G6PD activity in young erythrocytes predicts recovery and tolerance of continued primaquine exposure. Recent work by Bancone and colleagues (279) demonstrated nearly normal G6PD activity levels among reticulocytes of G6PD-deficient heterozygous Thai women of the class III Mahidol variant. This infers an ability for hematologic recovery despite full daily primaquine dosing, as indeed occurred in patients with that variant (71) and in those with the class II Viangchan variant in a recent trial in Cambodia with eight weekly doses of 45 mg primaquine (280).
The classification of extreme vulnerability to potentially lethal 8-aminoquinoline exposures may hinge upon the reticulocyte G6PD activity phenotype. Except for the African A−, Mahidol, and Mediterranean variants, that phenotype is almost wholly unknown among the dozens of relatively common variants. No work has established a correlation between the residual G6PD activity of reticulocytes and that of whole blood. Although that correlation may be reasonably presumed (Fig. 16), variability in the residual G6PD activity of reticulocytes or their specific vulnerability to particular compounds could occur among the enzymatic variants and be of fundamental clinical importance.
CLINICAL AND PUBLIC HEALTH IMPLICATIONS
Persistent Notion of Safe Primaquine
The 1960 WHO view of harmless primaquine expressed above derived from an expectation of homogeneity in G6PD deficiency and a broad application of an incomplete American understanding and experience. By 1980, although direct evidence by then informed both greater heterogeneity and deeper vulnerability, as detailed above, the WHO view became only slightly tempered: “Primaquine administration in light-skinned subjects with G6PD deficiency may result in a greater degree of hemolysis than in dark-skinned subjects” (281). The same source details three phases of primaquine-induced hemolysis for both African A− and Mediterranean variants explicitly and expressed the third phase in either variant as “an equilibrium phase when, in spite of drug administration, no further hemolysis takes place but there is a shortened survival of erythrocytes and compensation through increased erythropoiesis. The prognosis of this condition is good.” This guidance did not acknowledge the already known inability of patients having severe G6PD variants to compensate and recover with continued drug administration.
More decades would pass before WHO and other authorities would acknowledge that primaquine toxicity in patients with G6PD deficiency is not always “mild and self-limiting” but instead is sometimes severe and potentially progresses to life-threatening complications. In a 2010 publication (282), WHO warned, “In patients with the African variant of G6PD deficiency, the standard course of primaquine generally produces a benign mild and self-limiting anemia. In the Mediterranean and Asian variants, hemolysis may be much more severe.” Finally, in 2014, WHO (283) firmly warned, “Thus, the classification of variants of G6PD deficiency as ‘mild’ and ‘severe’ for the purpose of guiding clinical decisions should be abandoned. Daily primaquine hypnozoitocidal therapy (0.5 mg/kg daily for 14 days) induces potentially life-threatening AHA [acute hemolytic anemia] in patients with all of the known G6PD variants, including the A− variant.”
Among people at risk of infection by P. vivax, Asians dominate in both numbers and the risk of carrying relatively severely impaired G6PD enzymatic activity phenotypes. The Mediterranean variant dominates not only that basin but all of Arabia, Persia, Afghanistan, Pakistan, and western India. East of the center of the Indian subcontinent, a great profusion of variant diversity appears, with most of the variants being WHO class II (20). The hemolytic toxicity of primaquine or tafenoquine—as concluded for plasmochin nearly 90 years ago (132, 141)—prohibits use in people of unknown G6PD status lacking access to clinical services (2). Absent an ability to decisively segregate G6PD-normal from -deficient patients or to notice and rescue those accidentally dosed, the 8-aminoquinolines are simply too dangerous for “public health” use, i.e., too dangerous to be prescribed by paramedics at the periphery of care and beyond the practical reach of clinical monitoring and hemolytic crisis rescue by transfusion or hemodialysis.
The American experience with G6PD deficiency and primaquine through the 1950s unwittingly trivialized a serious problem, and a half century passed before the error was understood, acknowledged, and addressed. Even today the legacy of “mild and self-limiting” hemolytic toxicity in G6PD-deficient patients lingers with a reluctance to accept the threat imposed by the 8-aminoquinolines or the necessity of addressing it scientifically or clinically. A recent survey of primaquine treatment policies revealed that many nations do not express a need for G6PD screening or even caution regarding its use (284). The perceived or imagined good safety and tolerability of primaquine—as in Archambeault’s nearly one-half million soldiers at sea (184)—apparently stands firm in the face of the limited but compelling direct scientific and clinical evidence to the contrary summarized here.
The stubborn reluctance to reject the hopeful but inapt 1960 view of primaquine—as a drug well suited to application without clinical supervision—has probably carried heavy consequences, direct and indirect alike. Directly, patients have suffered serious or fatal hemolytic events with primaquine therapy, in turn leading to rational fear of the drug and a great reluctance to use it. Untreated patients then go on to suffer repeated clinical attacks, some ending in serious illness and death. Indirectly, misplaced satisfaction with primaquine as a solution to the relapse problem stymied research aimed at improving the safety of this drug, either by replacing it with a nonhemolytic therapy or making it more safe with simple and affordable G6PD screening. Finally, in the instance of the development of tafenoquine, the neglect of hemolytic toxicity as a problem to be measured and ranked among preclinical compounds may have dismissed 8-aminoquinolines with good activity and relatively low hemolytic toxicity, but we cannot know because this was not examined. Rational preclinical ranking of 8-aminoquinoline toxicity required an understanding of the precise mechanism of hemolytic toxicity that has never been realized, largely because inadequate priority has been assigned to doing so. No technological barrier separates us from that key knowledge, but the perception of it as unimportant does.
Current practice against latency in vivax and ovale malarias—and against infection by all malarias in the instance of chemoprophylaxis—must cope with the danger of 8-aminoquinoline hemolytic toxicity. Primaquine and tafenoquine may indeed be nearly completely safe with acknowledgment of and adherence to the complex and stringent principles of clinical use summarized below.
Safety of 8-Aminoquinolines in G6PD-Unknown Patients
The unsupervised administration of 8-aminoquinolines as hypnozoitocidal therapy to patients of unknown G6PD status presents a wide range of possible consequences. That encounter may safely relieve the patient from multiple attacks of relapsing malaria, cause a moderate and transient acute hemolytic anemia with the same benefit, or end in severe complications or death shortly after the sudden onset of a severe hemolytic crisis. G6PD deficiency and its many genetic variants and forms of expression dominate as a determinant of those outcomes. The great diversity of G6PD deficiency includes variants of relatively extreme and lesser sensitivity to 8-aminoquinolines, perhaps defined by the residual enzymatic activity of reticulocytes. However, the ratio of those phenotypes among them is largely unexplored and unknown. No reliable evidence affirms the harmlessness of these drugs when administered daily at therapeutic doses in any G6PD-deficient patient (probably excepting female heterozygotes having >70% of normal enzymatic activity in whole blood). In virtually all of them thus exposed, acute hemolytic anemia invariably occurs to degrees varying with the 8-aminoquinoline applied, the dosing regimen, the G6PD variant involved, the alleles present (hemi-, homo-, or heterozygous), and the extent of lyonization of the mutant chromosome among heterozygotes. The clinical consequences of that hemolytic event also depend on the condition of the patient at onset to include those related to acute malaria or other infectious processes, as well as possible exposure to dietary risk, like fava beans. Accessibility to clinical services for rescue from hemolytic crisis ultimately defines those consequences to the patient.
The complexity of this assessment of risks—inclusive of fatal complications—precludes the simplicity of advice that would safely guide unsupervised daily therapy in patients of unknown G6PD status. As Field (132) explained of plasmochin in 1938, currently available 8-aminoquinolines are not suited to use beyond the sphere of access to clinical services. Then, as now, firm evidence of the possible severity of consequences and the great complexity of the determinants of those reasonably imposes that limitation. A possible exception may be populations where G6PD deficiency is exceedingly rare or absent, as in most purely Native Americans or some northern Asia populations.
At least several million patients are diagnosed with acute vivax malaria in clinical settings each year. The proper treatment of that infection in eligible patients must include an 8-aminoquinoline against latency and, therefore, absent ascertainment of G6PD-normal status in that patient, compulsory clinical monitoring over the several days following initiation of daily primaquine therapy (2, 7, 285). Clinical guidelines for that monitoring have not been optimized or validated, but reason may guide providers managing these patients, i.e., by following treatment guidelines from WHO (285) or by recognizing the often sudden and conspicuous signs of onset of acute hemolytic anemia: shortness of breath, cyanosis, or hematuria, in particular. Presentation of these should prompt hospitalization and assessment for transfusion or hemodialysis. Absent any signs the day following the 3rd or 4th dose of primaquine, patients may reasonably be released from monitoring to complete that regimen. Exposure to nonthreatening doses of primaquine as a means to trigger a mild but readily detectable hemolytic event as a means of identifying vulnerable patients without G6PD screening would require clinical optimization and validation in resource-limited settings.
Safety of 8-Aminoquinolines in Patients Screened for G6PD Status
Ascertaining G6PD status in a patient may be accomplished by the quantitative or qualitative approaches summarized in Table 8 and recommended by WHO (286). The quantitative approach relies on the spectrophotometric determination of the conversion of NADP+ to NADPH by G6PD reaction with glucose-6-phosphate in sample hemolysate and is then calculated as the number of units of G6PD activity per gram of hemoglobin. This assay requires a properly equipped, cold chain-supplied laboratory having staff certified to be competent in conducting this relatively sophisticated measurement. Many referral hospitals in areas where malaria is endemic, much less the health care facilities at the periphery of reach of these services, lack this capacity (287). The vast majority of malaria patients do not have access to standard quantitative G6PD testing.
TABLE 8.
Summary of standard and emergent G6PD diagnostic technologies that may improve access to 8-aminoquinoline therapies
| Parameter | Quantitative approaches | Qualitative approaches | ||
|---|---|---|---|---|
| Device or test | Laboratory spectrophotometer | Handheld portable device | Fluorescent spot test | Fluorochrome reduction test |
| Setting of use | Laboratory | Point of care | Laboratory | Point of care |
| Temp of use (°C) | <25 | <35 | <35 | <40 |
| Intended user | Laboratorian | Paramedic | Laboratorian | Paramedic |
| Cold chain supplies | Yes | No | Yes | No |
| Specialized equipment | Yes | Yes | Yes | No |
| Sensitivity for detection of normal activity of: | ||||
| <30% | Nearly perfect | Nearly perfect | Nearly perfect | Nearly perfect |
| >30% | Nearly perfect | Nearly perfect | Very poor | Very poor |
| Suitable for female testing | Yes | Yes | No | No |
| Cost | Very high | High | High | Low |
The standard qualitative option to ascertaining G6PD status has been the fluorescent spot test (FST), developed by Beutler and Mitchell in the 1960s (288). The FST works much like the quantitative assay, except that NADPH brightly fluoresces under UV light, whereas NADP+ does not. The G6PD-catalyzed conversion of NADP+ to NADPH may be thus visualized. This test nonetheless requires specialized laboratory gear and skills, along with a cold chain, and it is relatively expensive at about $5/test for a completely used kit of 50 tests. This testing is as unavailable as quantitative assessments in most areas where malaria is endemic. Since about 2010, at least several commercial biotechnology companies have taken up the challenge of developing G6PD testing kits suited to use at the point of care at the periphery of health care services in the rural tropics where malaria is endemic (287). These technologies include both quantitative and qualitative platforms, and the distinction matters, in the context of the intended use as devices informing a decision to proceed or withhold 8-aminoquinoline therapy.
Among male hemizygotes and female homozygotes of all but a few rare genotypes, G6PD activity is uniformly less than 30% of normal and the hemolysis is usually in excess of 20% of the baseline hemoglobin levels with exposure to 3 to 5 daily 0.25-mg/kg primaquine doses. As pointed out by the early plasmochin workers managing such patients, hemolysis tended to be an all-or-none phenomenon. The exceptional state of mild hemolysis may be limited to the relatively few females having <70% of normal erythrocytes but >30%. This is due to the genetics of the X-linked inheritance of G6PD deficiency and the G6PD mosaicism of female red blood cell populations already explained above. While the specific variant matters, the overriding determinant of the extent of hemolysis in heterozygous females correlates to the extent to which the involved mutant gene remains actively expressing the impaired G6PD enzyme (21). A female having >30% deficient erythrocytes may be considered at risk of a threatening hemolytic crisis, whereas those having >70% normal erythrocytes may not, although that threshold has not been rigorously validated. The measurement of the percentage of normal G6PD activity effectively captures both variant and mosaic variables; i.e., >70% of normal G6PD activity indicates relative safety, whereas below that threshold there is a risk of clinically consequential hemolysis presumed to be inversely proportional to the measurement.
The sex distinction in G6PD deficiency thus carries important diagnostic considerations when the assessment is made qualitatively rather than quantitatively. Qualitative G6PD screening offers distinct advantages over quantitative assessment: it is less expensive, does not require expensive and delicate laboratory equipment or a high degree of technical expertise in the operator, and may be adaptable to point-of-care or bedside assessment. The principal weakness in qualitative tests is the insensitivity for individuals with G6PD deficiency phenotypes of >30% and <70% of normal activity (289, 290), almost all of whom will be female heterozygotes (283, 286). In other words, females of this phenotype will often qualitatively screen as normal and be cleared for treatment with 8-aminoquinolines. Qualitative screening offers very nearly 100% sensitivity and specificity for male hemizygotes, female homozygotes, and female heterozygotes having <30% of normal activity (291–295), but the normal readout for any given female may not be accepted as ensuring 8-aminoquinoline safety (283). This problem explains the necessity of clinical monitoring of all females thus cleared for primaquine therapy and of quantitative testing in connection with single-dose tafenoquine for radical cure. Several point-of-care quantitative G6PD screening devices have recently been developed and may offer many G6PD-normal malaria patients safe access to any 8-aminoquinoline hypnozoitocidal therapy (295).
Safety of 8-Aminoquinolines in G6PD-Deficient Patients
Despite the persistent view of unsupervised primaquine in patients of unknown G6PD status as safe, no known daily dose of any 8-aminoquinoline in a patient of any variant of G6PD deficiency is free of a hemolytic response. The key question is the margin of safety at any given dose and dosing interval; i.e., can the hemolytic reaction be tempered to be within the bounds of clinical safety? In 1960, Alving and colleagues (246) described that a single weekly dose of 45 mg primaquine for 8 weeks in G6PD-deficient African-American volunteers caused insignificant hemolytic anemia and provided good efficacy against relapse of the Chesson P. vivax strain. Despite that trial being performed with very small numbers of volunteers (who may or may not have been negative for the Duffy factor), the WHO in that era adopted this regimen as recommended therapy for G6PD-deficient patients diagnosed with P. vivax malaria, and it still appears in those treatment guidelines (285). However, reports of unsafe hemolytic reactions in class II variants of G6PD deficiency administered this regimen have appeared over the ensuing decades. As early as 1981, Clyde (58) warned against it in Asian variants, despite evidence of moderate hemolysis and tolerability of the regimen in Thai patients with the class III Mahidol variant of G6PD deficiency (277).
A trial of 19 patients with acute P. vivax malaria and the class II Viangchan variant of G6PD deficiency in western Cambodia evaluated the safety of the single 45-mg adult weekly dose for 8 weeks (280). All patients hemolyzed steeply after the first dose, and one required transfusion (but that patient had been treated with hemolytic ciprofloxacin prior to his diagnosis of malaria). These patients nonetheless completed their eight weekly doses without further evidence of significant hemolysis. That trial offers some evidence of apparent tolerability following a hemolytic single-dose exposure to 45 mg primaquine in individuals with the Viangchan variant, as occurs in individuals with the Mahidol and African A− variants but not those with the Mediterranean variant. The reticulocytes and younger normocytes of African A−, Mahidol, and Viangchan variants thus appear to be insensitive to primaquine-induced hemolytic anemia relative to those of Mediterranean and probably other variants.
The most relevant fact to consider in light of these observations is the conspicuously inadequate understanding of clinically relevant hemolytic responses to primaquine or tafenoquine among G6PD variants. We cannot confidently predict the likelihood of survival without access to transfusion or hemodialysis in most G6PD-deficient patients. A survey of residual G6PD activity among the reticulocytes of the many dozens of common variants may provide useful guidance regarding relatively safe (i.e., survivable) administration of any daily regimen of primaquine. Specific knowledge of variants having reticulocyte G6PD activities in the ranges of those of the African A−, Mahidol, or Viangchan variants versus those in the far lower range of the Mediterranean variant would perhaps inform vulnerability to exposures to 8-aminoquinolines likely to result in serious harm unless treatment is promptly halted or the patient is rescued by transfusion or hemodialysis intervention.
8-Aminoquinolines in Chemoprevention
During the development and optimization of plasmochin, primaquine, and tafenoquine, investigators recognized and sought to exploit the cross-stage therapeutic properties of 8-aminoquinolines by applying regimens that prevent primary infection, latency, and onward transmission. Chemoprevention strategies have always been in development play, and practices for each have indeed emerged (53, 222, 296–298). Hepatic schizontocidal activity (against the early postinvasion forms of tachy- and bradysporozoites alike) and gametocytocidal activity offer great clinical and public health advantages against malaria that have historically been constrained by the hemolytic toxicity problem. Only recent work with relatively low-dose primaquine (a 15-mg versus 45-mg single adult dose gametocytocidal therapy in falciparum malaria) demonstrated safety and efficacy in G6PD-deficient patients (299); this application may be the first of impacts for chemoprevention by 8-aminoquinolines.
Chemoprevention in people temporarily exposed to a significant risk of plasmodial infections by the causal (hepatic schizontocidal) activity of 8-aminoquinolines remains hindered by the relatively higher and hemolytically toxic (in G6PD-deficient patients) exposures required. Travel medicine recommendations and practices have always shown an almost exclusive preference for suppressive blood schizontocides, despite the tremendous advantages of causal prophylaxis, principally by being well suited to short-notice or short-duration exposures and effectively preventing latency of relapsing malarias and attacks months following travel (222). The imposed necessity of G6PD screening, along with the requirement for both daily dosing and off-label use in the case of primaquine, seems to have sufficed as an obstacle to broad application in chemoprophylaxis for travelers. The labeled use of tafenoquine as weekly causal chemoprophylaxis against malaria solves all but the G6PD vulnerability problem. Whether the travel medicine community now confronts the G6PD problem by exploiting the rapidly improving diagnostics for that enzymopathy remains to be seen in the regularly updated authoritative recommendations for chemoprevention practice.
The broader communities of science, medicine, and public health engaging the malaria problem must also consider chemoprevention beyond the conventional travel medicine sphere of tourists and soldiers visiting areas of endemicity. Chemopreventive strategies for specific residents of areas of endemicity either seasonally or physiologically vulnerable to threatening attacks of acute malaria have been exhaustively optimized and validated for P. falciparum in sub-Saharan Africa, where malaria is holoendemic (300–303). The cross-species and -stage activities of the 8-aminoquinolines may also offer chemopreventive strategies and practices with positive impacts wherever malaria occurs, in particular, where elimination of local transmission is nearly or completely achieved (304, 305).
Therapeutic Principles of 8-Aminoquinolines
The 8-aminoquinolines provide an enormous therapeutic benefit when safely prescribed and taken, i.e., freedom from multiple recurrent attacks of acute malaria, with each carrying a risk of a missed or late diagnosis and progression to serious complicated illness. Achieving this essential therapeutic objective with a class of drugs capable of serious harm imposes a necessary complexity of clinical use.
The largely accurate therapeutic principles laid out by Sinton and colleagues in 1930 for plasmochin and quinine (141) may be usefully refined and somewhat expanded for contemporary relevance to the clinical and public health enterprise of killing hypnozoites: (i) most 8-aminoquinolines appear to be prodrugs metabolized to therapeutically active and toxic derivatives by CYP2D6; (ii) the therapeutic activity of 8-aminoquinolines against hypnozoites is often potentiated by partner blood schizontocides in radical cure, and the efficacy of such must be in the defining context of the pair; (iii) the hemotoxicity of 8-aminoquinolines may be exacerbated or mitigated by partner blood schizontocides in radical cure, and the safety of such must be in the defining context of the pair; (iv) the total dose of 8-aminoquinolines defines efficacy whether they are administered at once or over days, weeks, or several months, giving great flexibility in terms of striving to balance efficacy, safety, and practicality; (v) the potential for hemolytic harm caused by 8-aminoquinolines in G6PD-deficient patients depends on the drug and its dosing, the health of the patient at dosing, the specific G6PD variant and alleles involved, and the extent of lyonization in heterozygous females, in which harm ranges anywhere from clinically inconsequential to severe and mortal; (vi) qualitative screening for G6PD deficiency safely excludes from primaquine therapeutic exposure hemizygous males, homozygous females, and female heterozygotes having <30% of normal enzymatic activity, but it dangerously includes female heterozygotes having >30% and <70% of normal activity; (vii) quantitative screening for G6PD deficiency safely excludes all potentially G6PD-vulnerable patients at a presumed threshold of >70% of normal activity; and (viii) primaquine hypnozoitocidal therapy may not be safely administered to patients of unknown G6PD status without clinical monitoring, and tafenoquine cannot be safely administered without ascertaining >70% of normal enzymatic activity.
Considerations of safety in potentially G6PD-deficient patients impose great complexity and difficulty in clinical use and severely impede public health applications distant from clinical services. A far greater simplicity of guidance and accessibility in practice than these essential points express may drive the vision of an 8-aminoquinoline therapy that cannot harm G6PD-deficient patients. That is the enduring promise of these compounds endowed by development histories that have left that bright prospect largely unexplored.
FUTURE PERSPECTIVES
Prioritized Research Agenda
Consideration of the complexity imposed in past and current safe practice with 8-aminoquinolines, as detailed above, explains their limited impacts over the past century. The most important barrier to achieving the very substantial potential benefits of 8-aminoquinolines remains the unsolved problem of dangerous hemolytic toxicity in G6PD-deficient residents of areas of endemicity. G6PD screening technologies, as helpful as those may be in a clinical sense today, have yet to prove practical in the public health applications of chemotherapies involving very large populations widely dispersed across isolated rural expanses. Research to improve G6PD diagnostics is certainly important and offers the immediate health dividends that would come with improved safe access, but it is perhaps secondary to the longer-term agenda aimed at 8-aminoquinoline therapies having hemolytic safety profiles that obviate their need. The most important understanding to be drawn from this review of a century of 8-aminoquinoline research and practice is recognition of the hemolytic toxicity problem as unexplored scientific ground of real promise in a technological sense. The cellular and molecular events culminating in the 8-aminoquinoline-induced acute hemolytic anemia are very specific avenues of scientific exploration conceivably delineating a pathway to therapies that cannot harm G6PD-deficient patients. That endeavor requires a prioritization borne of a refusal to accept a therapeutic status quo that imposes either unequivocally dangerous exposure to 8-aminoquinolines, the elaborate means of applying them safely, or simply withholding therapy and allowing relapses.
Development of 8-Aminoquinolines for Public Health Application
In terms of the technical historic development of hemolytic 8-aminoquinolines reviewed here, the forgotten core therapeutic principle of Sinton et al. (141) looms large: optimized efficacy and safety in view of partner drugs that impact those characteristics of the hypnozoitocide. In 1930, Sinton et al. (141) envisioned of plasmochin a combination of radical cure where “the toxic manifestations might be reduced, if not entirely abolished, by such treatment.” Partner therapies to 8-aminoquinolines often profoundly impact both therapeutic activity and toxicity, but these effects have been only opportunistically observed in limited numbers of clinical trials rather than systematically examined for the purpose of optimized therapeutics. That exploration probably first requires in vitro systems that capture these effects, and technologies are today rapidly evolving to suitability to that purpose.
The rhesus macaque infected by P. cynomolgi remains an extraordinarily useful animal model for antirelapse therapies (306). It will no doubt serve a vital role in late preclinical development of any hypnozoitocidal therapy. However, even just exploring the unknown synergistic character of currently used partner blood schizontocides of primaquine or tafenoquine at recommended doses would greatly benefit from a less costly and laborious bench approach. More importantly, the range of that exploration should extend much further, as there remain nearly limitless possible combinations of compounds for radical cure. These would include already discovered 8-aminoquinolines of known good therapeutic activity and broad safety (like SN-3883 or WR242511), dozens of likely partner blood schizontocides or other potentiating compounds, and possibly detoxifying adjuncts. Optimizing hypnozoitocidal therapies for hemolytic safety across these key variables would likely require high-throughput in vitro systems effectively capturing both the hypnozoite-killing and G6PD-deficient erythrocyte-destroying properties of the appropriately metabolized combinations of drugs assessed.
Tremendous progress in systems that may offer that screening capacity has very recently been made, and such systems provide a means of quantifying the synergy of 8-aminoquinolines by any given partner therapy (Table 9) (101, 102, 307–315). Hepatic cellular systems supporting the infection and development of schizonts and hypnozoites may bring the apparently essential involvement of cytochrome P-450 metabolism of the agents under evaluation. If that system exhibits the known synergism of primaquine by quinine in killing hypnozoites, for example, it may also capture these effects more broadly in the practical chemotherapeutic sense. The greatest possible synergism would logically provide the lowest possible exposure to any given hemolytic hypnozoitocide, a rule of Sinton et al. (141). The theoretical features of that technical terrain—where partner therapies in radical cure provide greater or lesser synergies of specific 8-aminoquinolines of variable hemolytic potential—may offer maximally optimized combined therapies for radical cure at a minimized dose and hemolytic toxicity.
TABLE 9.
Summary of emergent technologies applicable to research aimed at improving 8-aminoquinoline safety and efficacya
| Technology | Platform | Capacity | Primary limitation | Reference(s) |
|---|---|---|---|---|
| In vitro hypnozoite development in primary human hepatocytes | 384-well high-throughput system | Hypnozoitocidal efficacy of metabolized 8AQs and potentiation by partner drugs | Live P. vivax sporozoites, along with confirmed hepatic CYP2D6 phenotype, required | 102, 306–310 |
| In vivo hypnozoite development in humanized murine livers | Living humanized mice | Hypnozoitocidal efficacy of metabolized 8AQs and potentiation by partner drugs | Cost of the test animal, along with confirmed hepatic CYP2D6 phenotype required | 311–313 |
| In vitro metabolism of primaquine to active derivatives | Recombinant CYP2D6 systems, high throughput | Hemolytic toxicity assessment and ranking of 8AQs | Access to 8AQs other than primaquine and tafenoquine | 101, 314, 315 |
| In vivo or in vitro metabolism of primaquine to active metabolites | Ultra-HPLC quadrupole time of flight mass spectroscopy | Identifying relevant metabolites | Cost of instrumentation | 80, 316 |
| In vivo G6PD toxicity of 8AQs | Living humanized mice or zebra fish | Hemolytic toxicity across 8AQs and variants of G6PD deficiency | Cost of the test animal, along with confirmed hepatic CYP2D6 phenotype | 69, 70 |
8AQ, 8-aminoquinoline; HPLC, high-performance liquid chromatography.
Key to that exploratory enterprise is the conspicuously necessary and wholly undone task of ranking 8-aminoquinolines for potential hemolytic toxicity in G6PD-deficient patients. If CYP2D6 indeed generates the hemolytically toxic metabolites of the 8-aminoquinolines, then in vitro systems that quantify that toxicity may be composed with relative ease. Progress in such systems has been made with the availability of recombinant cytochrome P-450s (Table 9) (316–319), but none have yet validated an in vitro correlate of 8-aminoquinoline hemolytic toxicity in G6PD-deficient erythrocytes. The hypothetical mechanism of hemolytic toxicity presented in Fig. 14 points to readily quantifiable ferrihemochromes or Heinz bodies as a testable correlate. The number of moles of any given 8-aminoquinoline yielding a fixed quantity of ferrihemochromes or Heinz bodies may well predict the relative or potential hemolytic toxicity in G6PD-deficient patients. That validated correlate, taken together with optimally synergized 8-aminoquinoline killing of hypnozoites in vitro, would finally offer preclinical therapeutic indices of relevance to the primary toxicity problem of these compounds.
A validated in vitro hemolytic toxicity system may also enable a third and somewhat detached approach to the development of nonhemolytic 8-aminoquinolines: detoxifying adjuncts. As already explained, a daily 500-mg adjunct of methylene blue reportedly prevented the acute hemolytic anemia induced by pentaquine/quinine radical cure in a sensitive subject (154). That observation may seem counterintuitive because methylene blue and some other phenothiazine class dyes seem to provoke acute hemolytic anemia in G6PD-deficient patients (320–323). However, as McDonagh et al. (324) pointed out in their 2013 systematic review of methylene blue therapeutics, “It is therefore difficult to pinpoint whether methylene blue is the cause of hemolysis in G6PD-deficient individuals being treated for acquired methemoglobinemia, rather than the precursor agent [inciting methylene blue therapy].” Large-scale trials of adjunctive methylene blue therapy for acute falciparum malaria in hundreds of G6PD-deficient and -normal African children or dozens of otherwise healthy G6PD-deficient African adults reported clinically insignificant reductions of hemoglobin in these patients or subjects (325–327). At least in patients with the African A− variant, methylene blue therapy does not incite acute hemolytic anemia.
The highly diverse phenothiazine derivatives, like the methylene blue archetype, are particularly amenable to systematic structural optimization of tremendously varied physiological effects (328). High-throughput optimization of 8-aminoquinoline hemolytic activity-detoxifying effects may occur in a validated in vitro model for that toxicity in human red blood cells. Discovery of a safe and well-tolerated detoxifying compound that may be coadministered with an otherwise hemolytic 8-aminoquinoline without negatively impacting its therapeutic activity is both conceivable and technologically feasible to test. That discovery effort, perhaps beginning at the same point of 8-aminoquinoline discovery a century ago, i.e., with methylene blue and its derivatives, may be considered a great irony. As with Ehrlich’s two vivax malaria patients in 1891 pointing to a methylene blue lead therapeutic compound at Elberfeld decades later (120), the protection of Alving’s lone pentaquine-sensitive subject by methylene blue in 1949 (154) today suggests a similarly promising and identical lead molecule.
The availability of an envisioned nonhemolytic 8-aminoquinoline could involve a decade or more of discovery effort boldly undertaken despite the competing vision of endemic malaria being driven to elimination in the Americas and the Asia-Pacific—where P. vivax dominates as an endemic problem—within the same period (329–331). In the meantime, and perhaps well beyond 2030, we have only primaquine and tafenoquine and the clinical necessity of coping with their potential for harm in G6PD-deficient patients, where simple, practical, and affordable G6PD diagnostics must play an essential role.
Development of G6PD Screening Technologies Enabling Access
The WHO and partners have defined a product development profile for the ideal G6PD diagnostic device (282, 286). In brief, it must offer superior sensitivity (>95%) and specificity (negative predictive value, >95%), simplicity of use by a lay operator, and an easily understood output (i.e., therapy go or no-go), it must not require a cold chain or power supply (except for simple batteries), it must be operable at ambient temperatures up to 40°C, and it must cost less than $2.00 per test. The intent is to safely put 8-aminoquinolines in the hands of providers at the periphery of malaria diagnosis and treatment services, which are often distant from clinics and hospitals. If that technology is indeed realized and validated as safe and effective, the G6PD-normal majority of patients infected by P. vivax and P. ovale (and perhaps P. malariae) may enjoy the benefit of unfettered safe access to therapies curing the latency of these parasites. That demonstration is by no means trivial, nor is it a complete solution to 8-aminoquinoline toxicity, but it is nonetheless a necessary undertaking essential to expanding safe access to 8-aminoquinolines in the near term.
Managing Relapse Risk without 8-Aminoquinolines
Complete success in G6PD screening technology would not wholly solve the problem of access to hypnozoitocidal therapy. Approximately 90 million pregnant women in the Asia-Pacific alone live at risk of vivax malaria (332). Neither they nor their young infants (<6 months of age), along with approximately 300 million G6PD-deficient patients and perhaps as many as 35% of Asians who are CYP2D6 impaired (333), may receive or benefit from 8-aminoquinoline therapies. There is a compelling need for optimizing and validating alternative means of suppressing or terminating the latency of the relapsing malarias that do not involve these drugs (116, 332, 333). The extensively studied intermittent presumptive treatment regimens for P. falciparum in vulnerable African populations (300–302) may be exemplary but not translatable to the problem of relapse in vulnerable patients untreatable with 8-aminoquinolines. The relatively extreme vulnerability of pregnant women and their fetuses and young infants to acute vivax malaria in particular (334–340) emphasizes an urgency of need to prevent it by validated antirelapse presumptive therapeutic or chemopreventive options.
Essential Research Questions
This review of a century of 8-aminoquinoline discovery and practice brings to light 12 essential research questions posed here in summary: (i) what explains the potentiation of 8-aminoquinoline activity against hypnozoites by compounds having no known activity against those forms, and can the hemolytic toxicity of 8-aminoquinolines be mitigated by optimized drug coadministration that potentiates therapeutic activity to an extent allowing safe dosing of patients without regard to G6PD status; (ii) what composes in vivo or ex vivo systems that capture the known therapeutic potentiation and toxicity-mitigating impacts of other compounds upon the 8-aminoquinolines; (iii) what is the range and order of hemolytic toxicity among 8-aminoquinolines, with particular attention to compounds of known good hypnozoitocidal activity, such as WR242511, or those already evaluated in humans, like isopentaquine or SN-3883; (iv) does a methylene blue adjunct abrogate the hemolytic toxicity of 8-aminoquinolines in G6PD-deficient erythrocytes, and can that effect be clinically optimized using it or other phenothiazine derivatives; (v) does relatively high residual G6PD activity among reticulocytes reliably predict tolerable hemolytic sensitivity to 8-aminoquinolines, and is that phenotype reliably predicted at some whole-blood G6PD activity threshold; (vi) what commonly occurring G6PD variants exhibit African A−/Mahidol-like residual G6PD activity levels in reticulocytes compared to the activity levels in reticulocytes of the Mediterranean variant; (vii) can a simple and affordable point-of-care diagnostic reliably distinguish patients who may not safely receive 8-aminoquinolines from those who may; (viii) can relapse be distinguished from reinfection or recrudescence and thus permit assessment of both the therapeutic efficacy of 8-aminoquinolines and fundamentally important epidemiological features of the hypnozoite reservoir in populations living in areas where malaria is endemic; (ix) does severe G6PD deficiency in reticulocytes completely protect against the P. vivax parasites infecting only those or younger erythroid cells and thereby prevent the need for dangerous exposure to 8-aminoquinoline therapy; (x) are the CYP2D6-derived reactive metabolites responsible for therapeutic activity the same as those responsible for hemolytic toxicity; (xi) how is the risk of relapse in patients either ineligible to receive 8-aminoquinolines (pregnant women, infants) or unable to benefit from them (CYP2D6-null metabolizers) managed; and (xii) does 8-aminoquinoline therapy terminate latency in P. malariae malaria?
CONCLUSIONS
The discovery of the 8-aminoquinolines a century ago realized Paul Ehrlich’s vision of molecular chemotherapy. Drugs of this unique class remain powerful and versatile therapies against malaria but have the conspicuous liability of dangerous hemolytic toxicity in G6PD-deficient patients. Providers must cope with the complex and rigorous therapeutic principles guiding safe and effective 8-aminoquinoline treatment practices summarized herein. People lacking access to the laboratory and clinical services essential to applying those principles cannot safely obtain the great clinical and public health benefits from the prevention of often multiple attacks of acute malaria, each carrying substantial risks of poor outcomes and onward transmission. The relative invisibility of malaria patients typically residing beyond the reach of clinical services, along with the long-marginalized relapsing species of plasmodia infecting them, largely explains why that problem has gone unsolved. In their respective histories, plasmochin, primaquine, and tafenoquine share successive origins in the exigency of malaria in warfare. Soldiers at war dominated hypnozoitocidal development imperatives, and the hemolytic toxicity of 8-aminoquinolines did not register as the very serious problem of safe access that it is for most malaria patients.
The scientific question of the molecular basis of hemolytic toxicity remains unanswered, not for a lack of technological ability but for a want of adequate priority assigned to this problem. The historic illusion of primaquine as a practically harmless 8-aminoquinoline stymied scientific progress, leaving the hemolytic toxicity of 8-aminoquinolines to languish as an acknowledged but unimportant problem. The abandoned lead of methylene blue detoxification of pentaquine in 1949 and the advancement of tafenoquine through preclinical development without knowledge of its relative hemolytic toxicity speak most directly to that perspective. Only in the past decade has significant progress been made in easing the difficulty of safe access to 8-aminoquinolines and has been almost exclusively within the specific realm of G6PD diagnostics suited for use in the malarious rural tropics, and private biotechnological firms rather than research laboratories have undertaken those efforts. The key evidence of the CYP2D6 isozyme as the likely generator of a hypnozoite-killing metabolite(s) came only serendipitously from treatment failures in clinical trials. The complexity and difficulty of P. vivax chemotherapeutics, along with the misplaced confidence in that parasite being a relatively benign infection safely and effectively treated with available drugs, deeply discouraged research aimed at improved therapies for over 70 years.
The slow incidental progress of many decades in understanding 8-aminoquinoline therapeutics, coupled with the very substantial and rapid recent advancement of basic research tools of P. vivax biology, may today be leveraged in the rationally feasible technological enterprise of discovering 8-aminoquinoline therapies that cannot harm G6PD-deficient patients. Knowledge of the relative hemolytic toxicities of 8-aminoquinolines and their respective therapeutic activities with maximized potentiating and toxicity-mitigating partner drugs may conceivably deliver that success. As Sinton and others expressed nearly a century ago, 8-aminoquinoline hypnozoitocides act most effectively in concert with other drugs that also impact their toxicities. Certain knowledge of the molecular events leading to the destruction of both hypnozoites and G6PD-deficient erythrocytes may rationally guide the discovery of 8-aminoquinolines genuinely harmless to G6PD-deficient patients. We possess the technologies to obtain that knowledge; we need only assign sufficiently high priority to having and using it. The rational exploration of 8-aminoquinoline therapeutics has yet to occur. Success in doing so may at last unleash the enormous potential of this singularly powerful class of drugs against a tenacious and insidiously harmful latent malaria foe.
ACKNOWLEDGMENTS
I am supported by grant 106680/Z/14/Z from the Wellcome Trust.
I have served as a consultant on 8-aminoquinolines to the World Health Organization and GlaxoSmithKline (UK) and have received research support funding from both in connection with clinical trials involving 8-aminoquinolines.
Dennis Shanks in Brisbane, Australia, generously provided a rare copy of the 1927 United Fruit Company (Boston, MA, USA) report of early plasmochin experiences at American plantations under management by that company (135, 136, 142, 146), as well as the transcript of the 1954 speech of Alf Alving at the Walter Reed Army Medical Center (149). Robert Snow in Nairobi, Kenya, generously provided an original print of Field’s 1938 report on malaria chemotherapeutics (132). Georges Snounou in Paris, France, provided key references (including reference 125) regarding the French experience with 8-aminoquinoline research in the early 20th century. Anatoly Kondrashin and Alexei Beljaev in Moscow, Russia, provided advice and documentation on the polymorphic sporozoite theorem of A. J. Lysenko. Guy Thwaites in Ho Chi Minh City, Vietnam, generously provided a critical review of the original manuscript and contributed materially to its improvement, as did Cindy Chu in Mae Sot, Thailand. Germana Bancone, also in Mae Sot, generously provided the photomicrograph of Heinz bodies used in this review (Fig. 12). Moshe Schmuklarsky, Laurence Lightner, Geoff Dow, Jonathan Berman, David Wesche, Jason Bennett, and Scott Miller—all formerly with the Division of Experimental Therapeutics at the Walter Reed Army Institute of Research (WRAIR, Forest Glen, MD) or the U.S. Army Medical Material Development Activity (Ft. Detrick, MD)—generously provided their recollections, documentation, and advice regarding the history of the development of tafenoquine by the U.S. Army. The head of the tafenoquine development team at GlaxoSmithKline in London, United Kingdom, J. P. Kliem, also helped shape the description of that history, as did the chief medical and scientific officers at the Medicines for Malaria Venture in Geneva, Switzerland, Stephan Duparc and Timothy Wells. Discussions with Brandon Pybus and Gregory Riechard at the Division of Experimental Therapeutics, WRAIR, in Forest Glen, MD, USA, regarding the vision of harmless 8-aminoquinolines helped shape the expression of such herein. I am indebted to four anonymous reviewers who provided essential criticisms, suggestions, and corrections that substantially improved this article.
Biography

J. Kevin Baird trained in microbiology, protozoology, parasitology, biochemistry, and epidemiology at the University of Maryland (B.Sc., M.Sc.) and Tulane University (Ph.D.). In 1981 he went to work at the Division of Experimental Therapeutics, Walter Reed Army Institute of Research, while pursuing a master’s degree. There he set about the assigned task of developing a means of estimating the relative hemolytic toxicity of 8-aminoquinolines in G6PD-deficient erythrocytes. He left that work incomplete in 1984 upon taking up active duty in the U.S. Navy Medical Service Corps. The ensuing 22 years and postings in the United States and abroad saw work focused on the control, prevention, diagnosis, and treatment of Plasmodium vivax malaria, including strategies for managing the problem of 8-aminoquinoline hemolytic toxicity in G6PD-deficient patients. That work has continued during his now 11 years with the University of Oxford Nuffield Department of Medicine, in which he is posted as its Head of the Eijkman-Oxford Clinical Research Unit at the Eijkman Institute of Molecular Biology in Jakarta, Indonesia. His laboratory in that unit actively conducts clinical trials of tafenoquine and primaquine in the radical cure of vivax malaria aimed at enabling effective control of its endemic latent reservoir.
Footnotes
[This article was published on 31 July 2019 but required an additional change, now reflected in the Note Added after Publication on p. 59. The change to the article was made on 6 August 2019.]
REFERENCES
- 1.Battle KE, Lucas TCD, Nguyen M, Howes RE, Nandi AK, Twohig KA, Pfeffer DA, Cameron E, Rao PJ, Casey D, Gibson HS, Rozier JA, Dalrymple U, Keddie SH, Collins EL, Harris JR, Guerra CA, Thorn MP, Bisanzio D, Fullman N, Huynh CK, Kulikoff X, Kutz MJ, Lopez AD, Mokdad AH, Naghavi M, Nguyen G, Shackelford KA, Vos T, Wang H, Lim SS, Murray CJL, Price RN, Baird JK, Smith DL, Bhatt S, Weiss DJ, Hay SI, Gething PW. 19 June 2019. Mapping the global endemicity and clinical burden of Plasmodium vivax, 2000-17: a spatial and temporal modelling study. Lancet doi: 10.1016/S0140-6736(19)31096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. 2015. Control and elimination of Plasmodium vivax malaria: a technical brief, p 64 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 3.Anstey NM, Douglas NM, Poespoprodjo JR, Price RN. 2012. Plasmodium vivax: clinical spectrum, risk factors and pathogenesis. Adv Parasitol 80:151–201. doi: 10.1016/B978-0-12-397900-1.00003-7. [DOI] [PubMed] [Google Scholar]
- 4.Baird JK. 2013. Evidence and implications of mortality in acute Plasmodium vivax malaria. Clin Microbiol Rev 26:36–57. doi: 10.1128/CMR.00074-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naing C, Whittaker MA, Nyunt Wai V, Mak JW. 2014. Is Plasmodium vivax a severe malaria?: a systematic review and meta-analysis. PLoS Negl Trop Dis 8:e3071. doi: 10.1371/journal.pntd.0003071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Val F, Machado K, Barbosa L, Salinas JL, Siqueira AM, Costa Alecrim MG, Portillo HD, Bassat Q, Monteiro WM, Guimarães Lacerda MV. 2017. Respiratory complications of Plasmodium vivax malaria: systematic review and meta-analysis. Am J Trop Med Hyg 97:733–743. doi: 10.4269/ajtmh.17-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baird JK, Valecha N, Duparc S, White NJ, Price RN. 2016. Diagnosis and treatment of Plasmodium vivax malaria. Am J Trop Med Hyg 95:35–51. doi: 10.4269/ajtmh.16-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Young MD. 1944. Studies on the periodicity of induced Plasmodium vivax. J Natl Malar Soc 3:237–247. [Google Scholar]
- 9.White NJ, Imwong M. 2012. Relapse. Adv Parasitol 80:113–150. doi: 10.1016/B978-0-12-397900-1.00002-5. [DOI] [PubMed] [Google Scholar]
- 10.Manson PT. 1901. Experimental malaria: recurrence after nine months. Br Med J 2:77. doi: 10.1136/bmj.2.2115.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenwood D. 1995. Conflicts of interest: the genesis of synthetic antimalarial agents in peace and war. J Antimicrob Chemother 36:857–872. doi: 10.1093/jac/36.5.857. [DOI] [PubMed] [Google Scholar]
- 12.Sinton JA. 1930. A suggested standard treatment of malaria based upon the results of the controlled investigation of over 3,700 cases. Ind Med Gaz 65:603–620. [PMC free article] [PubMed] [Google Scholar]
- 13.Slater L. 2013. War and disease: biomedical research on malaria in the twentieth century. Rutgers University Press, New Brunswick, NJ. [Google Scholar]
- 14.Baird JK. 2011. Resistance to chloroquine unhinges vivax malaria therapeutics. Antimicrob Agents Chemother 55:1827–1830. doi: 10.1128/AAC.01296-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shannon JA. 1946. Chemotherapy in malaria. Bull N Y Acad Med 22:345–357. [PubMed] [Google Scholar]
- 16.Wiselogle FY. 1946. A survey of antimalarial drugs 1941–1945. J. W. Edwards, Ann Arbor, MI. [Google Scholar]
- 17.Carson PE, Hohl R, Nora MV, Parkhurst GW, Ahmad T, Scanlan S, Frischer H. 1981. Toxicology of the 8-aminoquinolines and genetic factors associated with their toxicity in man. Bull World Health Organ 59:427–437. [PMC free article] [PubMed] [Google Scholar]
- 18.Miller FG. 2013. The Stateville Penitentiary malaria experiments: a case study in retrospective ethical assessment. Perspect Biol Med 56:548–567. doi: 10.1353/pbm.2013.0035. [DOI] [PubMed] [Google Scholar]
- 19.Baird JK. 2015. Origins and implications of neglect of G6PD deficiency and primaquine toxicity in Plasmodium vivax malaria. Pathog Glob Health 109:93–106. doi: 10.1179/2047773215Y.0000000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howes RE, Piel FB, Patil AP, Nyangiri OA, Gething PW, Dewi M, Hogg MM, Battle KE, Padilla CD, Baird JK, Hay SI. 2012. G6PD deficiency prevalence and estimates of affected populations in malaria endemic countries: a geostatistical model-based map. PLoS Med 9:e1001339. doi: 10.1371/journal.pmed.1001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu CS, Bancone G, Nosten F, White NJ, Luzzatto L. 2018. Primaquine-induced haemolysis in females heterozygous for G6PD deficiency. Malar J 17:101. doi: 10.1186/s12936-018-2248-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Douglas NM, Poespoprodjo JR, Patriani D, Malloy MJ, Kenangalem E, Sugiarto P, Simpson JA, Soenarto Y, Anstey NM, Price RN. 2017. Unsupervised primaquine for treatment of Plasmodium vivax malaria relapses in southern Papua: a hospital-based cohort study. PLoS Med 14:e1002379. doi: 10.1371/journal.pmed.1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John GK, Douglas NM, von Seidlein L, Nosten F, Baird JK, White NJ, Price RN. 2012. Primaquine radical cure of Plasmodium vivax: a critical review of the literature. Malar J 11:280. doi: 10.1186/1475-2875-11-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baird JK. 2015. Still defining optimal primaquine therapy against relapse after 63 years of continuous use. Travel Med Infect Dis 13:215–216. doi: 10.1016/j.tmaid.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 25.U.S. Food and Drug Administration. 2018. Prescribing information for Krintafel (tafenoquine) tablets for oral use. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210795s000lbl.pdf. Accessed 31 December 2018.
- 26.Sutanto I, Tjahjono B, Basri H, Taylor WR, Putri FA, Meilia RA, Setiabudy R, Nurleila S, Ekawati LL, Elyazar I, Farrar J, Sudoyo H, Baird JK. 2013. Randomized open-label trial of primaquine against vivax malaria relapse in Indonesia. Antimicrob Agents Chemother 57:1128–1135. doi: 10.1128/AAC.01879-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelwan EJ, Ekawati LL, Tjahjono B, Setiabudy R, Sutanto I, Chand K, Ekasari T, Djoko D, Basri H, Taylor R, Duparc S, Subekti D, Elyazar I, Noviyanti R, Sudoyo H, Baird JK. 2015. Randomized trial of primaquine hypnozoitocidal efficacy when administered with artemisinin-combined blood schizontocides for radical cure of Plasmodium vivax in Indonesia. BMC Med 13:294. doi: 10.1186/s12916-015-0535-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Longley RJ, Sripoorote P, Chobson P, Saeseu T, Sukasem C, Phuanukoonnon S, Nguitragool W, Mueller I, Sattabongkot J. 2016. High efficacy of primaquine treatment of Plasmodium vivax in western Thailand. Am J Trop Med Hyg 95:1086–1089. doi: 10.4269/ajtmh.16-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu CS, Phyo AP, Lwin KM, Win HH, San T, Aung AA, Raksapraidee R, Carrara VI, Bancone G, Watson J, Moore KA, Wiladpharingern J, Proux S, Sriprawat K, Winterberg M, Cheah PY, Chue AL, Tarning J, Imwong M, Nosten F, White NJ. 2018. Comparison of the cumulative efficacy and safety of chloroquine, artesunate, and chloroquine-primaquine in Plasmodium vivax malaria. Clin Infect Dis 67:1543–1549. doi: 10.1093/cid/ciy319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coatney GR, Collins WE, Warren MCW, Contacos PG, 1971. The primate malarias, p 366 Stock no. 1744-0005. U.S. Government Printing Office, Washington, DC. [Google Scholar]
- 31.Lysenko AJ, Beljaev AE, Rybalka VM. 1977. Population studies of Plasmodium vivax. 1. The theory of polymorphism of sporozoites and epidemiological phenomena of tertian malaria. Bull World Health Organ 55:541–549. [PMC free article] [PubMed] [Google Scholar]
- 32.Rybalka VM, Beljaev AE, Lysenko AJ. 1977. Population studies of Plasmodium vivax. 2. Distribution of manifestations in foci of tertian malaria. Bull World Health Organ 55:551–556. [PMC free article] [PubMed] [Google Scholar]
- 33.Chen N, Auliff A, Rieckmann K, Gatton M, Cheng Q. 2007. Relapses of Plasmodium vivax infection result from clonal hypnozoites activated at predetermined intervals. J Infect Dis 195:934–941. doi: 10.1086/512242. [DOI] [PubMed] [Google Scholar]
- 34.Shortt HE, Garnham P. 1948. The pre-erythrocytic development of Plasmodium cynomolgi and Plasmodium vivax. Trans R Soc Trop Med Hyg 41:785–795. doi: 10.1016/S0035-9203(48)80006-4. [DOI] [PubMed] [Google Scholar]
- 35.Krotoski WA, Garnham PC, Bray RS, Krotoski DM, Killick-Kendrick R, Draper CC, Targett GA, Guy MW. 1982. Observations on early and late post-sporozoite tissue stages in primate malaria. I. Discovery of a new latent form of Plasmodium cynomolgi (the hypnozoite), and failure to detect hepatic forms within the first 24 hours after infection. Am J Trop Med Hyg 31:24–35. doi: 10.4269/ajtmh.1982.31.24. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt LH. 1982. Plasmodium cynomolgi infections in the rhesus monkey. Background of studies. Am J Trop Med Hyg 31:609–611. doi: 10.4269/ajtmh.1982.31.609. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt LH. 1983. Appraisals of compounds of diverse chemical classes for capacities to cure infections with sporozoites of Plasmodium cynomolgi. Am J Trop Med Hyg 32:231–257. doi: 10.4269/ajtmh.1983.32.231. [DOI] [PubMed] [Google Scholar]
- 38.Groger M, Fischer HS, Veletzky L, Lalremruata A, Ramharter M. 2017. A systematic review of the clinical presentation, treatment and relapse characteristics of human Plasmodium ovale malaria. Malaria J 16:112. doi: 10.1186/s12936-017-1759-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collins WE, Jeffery GM. 2005. Plasmodium ovale: parasite and disease. Clin Microbiol Rev 18:570–581. doi: 10.1128/CMR.18.3.570-581.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doritchamou JYA, Akuffo RA, Moussiliou A, Luty AJF, Massougbodji A, Deloron P, Tuikue Ndam NG. 2018. Submicroscopic placental infection by non-falciparum Plasmodium spp. PLoS Negl Trop Dis 12:e0006279. doi: 10.1371/journal.pntd.0006279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li P, Zhao Z, Xing H, Li W, Zhu X, Cao Y, Yang Z, Sattabongkot J, Yan G, Fan Q, Cui L. 2016. Plasmodium malariae and Plasmodium ovale infections in the China-Myanmar border area. Malar J 15:557. doi: 10.1186/s12936-016-1605-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collins WE, Jeffery GM. 2007. Plasmodium malariae: parasite and disease. Clin Microbiol Rev 20:579–592. doi: 10.1128/CMR.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.World Health Organization. 1961. Chemotherapy of malaria: report of a technical meeting, 14-19 November 1960, p 92 Technical report series no. 226 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 44.Gilles HM, Hendrickse RG. 1963. Nephosis in Nigerian children: role of Plasmodium malariae and effect of antimalarial treatment. Br Med J 2:27–31. doi: 10.1136/bmj.2.5348.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hendrickse RG. 1976. The quartan malaria nephrotic syndrome. Adv Nephrol 6:229–233. [PubMed] [Google Scholar]
- 46.Langford S, Douglas NM, Lampah DA, Simpson JA, Kenangalem E, Sugiarto P, Anstey NM, Poespoprodjo JR, Price RN. 2015. Plasmodium malariae infection associated with a high burden of anemia: a hospital-based surveillance study. PLoS Negl Trop Dis 9:e0004195. doi: 10.1371/journal.pntd.0004195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rutledge GG, Marr I, Huang GKL, Auburn S, Marfurt J, Sanders M, White NJ, Berriman M, Newbold CI, Anstey NM, Otto TD, Price RN. 2017. Genomic characterization of recrudescent Plasmodium malariae after treatment with artemether/lumefantrine. Emerg Infect Dis 23:1300–1307. doi: 10.3201/eid2308.161582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hill DR, Baird JK, Parise ME, Lewis LS, Ryan ET, Magill AJ. 2006. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am J Trop Med Hyg 75:402–415. doi: 10.4269/ajtmh.2006.75.402. [DOI] [PubMed] [Google Scholar]
- 49.Arnold J, Alving AS, Clayman CB. 1961. Induced primaquine resistance in vivax malaria. Trans R Soc Trop Med Hyg 55:345–350. doi: 10.1016/0035-9203(61)90103-1. [DOI] [PubMed] [Google Scholar]
- 50.Pukrittayakamee S, Chantra A, Simpson JA, Vanijanonta S, Clemens R, Looareesuwan S, White NJ. 2000. Therapeutic responses to different antimalarial drugs in vivax malaria. Antimicrob Agents Chemother 44:1680–1685. doi: 10.1128/aac.44.6.1680-1685.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baird JK, Fryauff DJ, Hoffman SL. 2003. Primaquine for the prevention of malaria in travelers. Clin Infect Dis 37:1659–1667. doi: 10.1086/379714. [DOI] [PubMed] [Google Scholar]
- 52.Olifarhood G, Raeisi A, Ranjbar M, Haghdoust AA, Schapira A, Hashemi S, Masoumi-Asl H, Mozafar Saadati H, Azimi S, Khosravi N, Kondrashin A. 2017. Prophylactic efficacy of primaquine for preventing Plasmodium falciparum and Plasmodium vivax parasitemia in travelers: a meta-analysis and systematic review. Travel Med Infect Dis 17:5–18. doi: 10.1016/j.tmaid.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 53.Arnold J, Alving AS, Hockwald RS, Clayman CB, Dern RJ, Beutler E, Flanagan CL, Jeffery GM. 1955. The antimalarial action of primaquine against the blood and tissue stages of falciparum malaria (Panama, P-F-6 strain). J Lab Clin Med 46:391–397. [PubMed] [Google Scholar]
- 54.Baird JK, Hoffman SL. 2004. Primaquine therapy for malaria. Clin Infect Dis 39:1336–1345. doi: 10.1086/424663. [DOI] [PubMed] [Google Scholar]
- 55.Coatney GR. 1963. Pitfalls in a discovery: the chronicle of chloroquine. Am J Trop Med Hyg 12:121–128. doi: 10.4269/ajtmh.1963.12.121. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt LH, Fradkin R, Vaughan D, Rasco J. 1977. Radical cure of infections with Plasmodium cynomolgi: a function of total 8-aminoquinoline dose. Am J Trop Med Hyg 26:1116–1128. doi: 10.4269/ajtmh.1977.26.1116. [DOI] [PubMed] [Google Scholar]
- 57.Chu CS, Phyo AP, Turner C, Win HH, Poe NP, Yotyingaphiram W, Thinraow S, Wilairisak P, Raksapraidee R, Carrara VI, Paw MK, Wiladphaingern J, Proux S, Bancone G, Sriprawat K, Lee SJ, Jeeyapant A, Watson J, Tarning J, Imwong M, Nosten F, White NJ. 2019. Chloroquine versus dihydroartemisinin-piperaquine with standard high dose primaquine given either for 7 days or 14 days in Plasmodium vivax malaria. Clin Infect Dis 68:1311–1319. doi: 10.1093/cid/ciy735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clyde DF. 1981. Clinical problems associated with the use of primaquine as a tissue schizontocidal and gametocytocidal drug. Bull World Health Organ 59:391–395. [PMC free article] [PubMed] [Google Scholar]
- 59.Clayman CB, Arnold J, Hockwald RS, Yount EH Jr, Edgcomb JH, Alving AS. 1952. Status of primaquine. III. Toxicity of primaquine in Caucasians. JAMA 149:1563–1568. doi: 10.1001/jama.1952.72930340022010b. [DOI] [PubMed] [Google Scholar]
- 60.Hockwald RS, Arnold J, Clayman CB, Alving AS. 1952. Status of primaquine. IV. Toxicity of primaquine in Negroes. JAMA 149:1568–1570. doi: 10.1001/jama.1952.72930340027010c. [DOI] [PubMed] [Google Scholar]
- 61.Edgcomb JH, Arnold J, Yount EH, Alving AS, Eichelberger L, Jeffery GM, Eyles D, Young MD. 1950. Primaquine, SN 13272, a new curative agent in vivax malaria: a preliminary report. J Natl Malar Soc 9:285–292. [PubMed] [Google Scholar]
- 62.Beutler E. 1959. The hemolytic effect of primaquine and related compounds: a review. J Hematol 24:103–139. [PubMed] [Google Scholar]
- 63.Brennecke FE, Alving AS, Arnold J, Bergenstal DM, DeWind LT. 1951. A preliminary report on the effect of certain 8-aminoquinolines in the treatment of rheumatoid arthritis. J Lab Clin Med 38:795–796. [Google Scholar]
- 64.Fraser IM, Vesell ES. 1968. Effects of drugs and drug metabolites on erythrocytes from normal and glucose-6-phosphate dehydrogenase-deficient individuals. Ann N Y Acad Sci 151:777–794. doi: 10.1111/j.1749-6632.1968.tb11938.x. [DOI] [PubMed] [Google Scholar]
- 65.Hopkins J, Tudhope GR. 1974. The effects of drugs on erythrocytes in vitro: Heinz body formation, glutathione peroxidase inhibition and changes in mechanical fragility. Br J Clin Pharmacol 1:191–195. doi: 10.1111/j.1365-2125.1974.tb00235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baird JK, McCormick GJ, Canfield CJ. 1986. Effects of nine synthetic putative metabolites of primaquine on the activity of the hexose monophosphate shunt in intact human red blood cells in vitro. Biochem Pharmacol 35:1099–1106. doi: 10.1016/0006-2952(86)90145-0. [DOI] [PubMed] [Google Scholar]
- 67.Baird JK, Davidson DE Jr, Decker-Jackson JE. 1986. Oxidative activity of hydroxylated primaquine analogs. Non-toxicity to glucose-6-phosphate dehydrogenase-deficient human red blood cells in vitro. Biochem Pharmacol 35:1091–1098. doi: 10.1016/0006-2952(86)90144-9. [DOI] [PubMed] [Google Scholar]
- 68.Davanco MG, Aguiar AC, Dos Santos LA, Padhila EC, Campos ML, de Andrade CR, da Fonseca CR, da Fonseca LM, Dos Santos JL, Chin CM, Krettli AU, Peccinini RG. 2014. Evaluation of antimalarial activity and toxicity of a new primaquine prodrug. PLoS One 9:e105217. doi: 10.1371/journal.pone.0105217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patrinostro X, Carter ML, Kramer AC, Lund TC. 2013. A model of glucose-6-phosphate dehydrogenase deficiency in zebrafish. Exp Hematol 41:697–710. doi: 10.1016/j.exphem.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rochford R, Ohrt C, Baresel PC, Campo B, Sampath A, Magill AJ, Tekwani BL, Walker LA. 2013. Humanized mouse model of glucose-6-phosphate dehydrogenase deficiency for in vivo assessment of hemolytic toxicity. Proc Natl Acad Sci U S A 110:17486–17491. doi: 10.1073/pnas.1310402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rueangweerayut R, Bancone G, Harrell EJ, Beelen AP, Kongpatanakul S, Möhrle JJ, Rousell V, Mohamed K, Qureshi A, Narayan S, Yubon N, Miller A, Nosten FH, Luzzatto L, Duparc S, Kleim J-P, Green JA. 2017. Hemolytic potential of tafenoquine in female volunteers heterozygous for glucose-6-phosphate dehydrogenase deficiency (G6PD Mahidol variant) versus G6PD-normal volunteers. Am J Trop Med Hyg 97:702–711. doi: 10.4269/ajtmh.16-0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McChesney JD, Baker JK, Clark AM, Hufford CD. 1987. Primaquine: studies of mammalian metabolism, p 3–26. In Wernsdorfer WH, Trigg PI (ed), Primaquine: pharmacokinetics, metabolism, toxicity, and activity. John Wiley & Sons, Inc, New York, NY. [Google Scholar]
- 73.Strother A, Bucholz J, Abu-El-Haj S, Allahyrari R, Fraser IM. 1987. Metabolism of primaquine by various animal species, p 27–48. In Wernsdorfer WH, Trigg PI (ed), Primaquine: pharmacokinetics, metabolism, toxicity, and activity. John Wiley & Sons, Inc, New York, NY. [Google Scholar]
- 74.Fletcher KA, Price AH, Barton PF. 1987. The pharmacokinetics and biochemical pharmacology of primaquine in rhesus monkeys and rats, p 49–64. In Wernsdorfer WH, Trigg PI (ed), Primaquine: pharmacokinetics, metabolism, toxicity, and activity. John Wiley & Sons, Inc, New York, NY. [Google Scholar]
- 75.Baker JK, McChesney JD, Jorge LF. 1986. Characterization of the urinary metabolites of primaquine in rats. Pharm Res 3:132–141. doi: 10.1023/A:1016305822808. [DOI] [PubMed] [Google Scholar]
- 76.Fletcher KA, Barton PF, Kelly JA. 1988. Studies on the mechanisms of oxidation in the erythrocyte by metabolites of primaquine. Biochem Pharmacol 37:2683–2690. doi: 10.1016/0006-2952(88)90263-8. [DOI] [PubMed] [Google Scholar]
- 77.Vasquez-Vivar J, Augusto O. 1992. Hydroxylated metabolites of the antimalarial drug primaquine. Oxidation and redox cycling. J Biol Chem 267:6848–6854. [PubMed] [Google Scholar]
- 78.Pybus BS, Marcsisin SR, Jin X, Deye G, Sousa JC, Li Q, Caridha D, Zeng Q, Reichard GA, Ockenhouse C, Bennett J, Walker LA, Ohrt C, Melendez V. 2013. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar J 12:212. doi: 10.1186/1475-2875-12-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Potter BM, Xie LH, Vuong C, Zhang J, Zhang P, Duan D, Luong TL, Bandara Herath HM, Dhammika Nanayakkara NP, Tekwani BL, Walker LA, Nolan CK, Sciotti RJ, Zottig VE, Smith PL, Paris RM, Read LT, Li Q, Pybus BS, Sousa JC, Reichard GA, Marcsisin SR. 2015. Differential CYP 2D6 metabolism alters primaquine pharmacokinetics. Antimicrob Agents Chemother 59:2380–2387. doi: 10.1128/AAC.00015-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Avula B, Tekwani BL, Chaurasiya ND, Fasinu P, Dhammika Nanayakkara NP, Bhandara Herath HMT, Wang YH, Bae JY, Khan SI, Elsohly MA, McChesney JD, Zimmerman PA, Khan IA, Walker LA. 2018. Metabolism of primaquine in normal human volunteers: investigation of phase I and phase II metabolites from plasma and urine using ultra-high performance liquid chromatography-quadrupole time-of-flight mass spectroscopy. Malar J 17:294. doi: 10.1186/s12936-018-2433-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Constantino L, Paixão P, Moreira R, Portela MJ, Do Rosario VE, Iley J. 1999. Metabolism of primaquine by liver homogenate fractions: evidence for monoamine oxidase and cytochrome P450 involvement in the oxidative deamination of primaquine to carboxyprimaquine. Exp Toxicol Pathol 51:299–303. doi: 10.1016/S0940-2993(99)80010-4. [DOI] [PubMed] [Google Scholar]
- 82.Mihaly GW, Ward SA, Edwards G, Orme ML, Breckenbridge AM. 1984. Pharmacokinetics of primaquine in man: identification of the carboxylic acid derivative as a major plasma metabolite. Br J Clin Pharmacol 14:441–446. doi: 10.1111/j.1365-2125.1984.tb02369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marcsisin SR, Reichard G, Pybus B. 2016. Primaquine pharmacology in the context of CYP 2D6 pharmacogenetics: current state of the art. Pharmacol Ther 161:1–10. doi: 10.1016/j.pharmthera.2016.03.011. [DOI] [PubMed] [Google Scholar]
- 84.Bennett JW, Pybus BS, Yadava A, Tosh D, Sousa JC, McCarthy WF, Deye G, Melendez V, Ockenhouse CF. 2013. Primaquine failure and cytochrome P-450 2D6 in Plasmodium vivax malaria. N Engl J Med 369:1381–1382. doi: 10.1056/NEJMc1301936. [DOI] [PubMed] [Google Scholar]
- 85.Baird JK, Louisa M, Noviyanti R, Ekawati L, Elyazar I, Subekti D, Chand K, Gayatri A, Instiaty, Soebianto S, Crenna-Darusallam C, Djoko D, Hasto BD, Meriyenes D, Wesche D, Nelwan EJ, Sutanto I, Sudoyo H, Setiabudy R. 2018. Association of impaired P450 2D6 activity genotype and phenotype with therapeutic efficacy of primaquine treatment for latent Plasmodium vivax malaria. JAMA Netw Open 1:e181449. doi: 10.1001/jamanetworkopen.2018.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marcsisin SR, Sousa JC, Reichard GA, Caridha D, Zeng Q, Roncal N, McNulty R, Careagararja J, Sciotti RJ, Bennett JW, Zottig VE, Deye G, Li Q, Read L, Hickman M, Dhammika Nanayakkara NP, Walter LA, Smith B, Melendez V, Pybus BS. 2014. Tafenoquine and NPC-1161B require CYP 2D6 metabolism for anti-malarial activity: implications for the 8-aminoquinoline class of anti-malarial compounds. Malar J 13:2. doi: 10.1186/1475-2875-13-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.St Jean PL, Carter N, Koh GC, Duparc S, Taylor M, Beaumont C, Llanos-Cuentas A, Rueangweerayut R, Krudsood S, Green JA, Rubio P. 2016. Tafenoquine treatment of Plasmodium vivax: suggestive evidence that CYP2D6 reduced metabolism is not associated with relapse in the phase 2B DETECTIVE trial. Malar J 15:97. doi: 10.1186/s12936-016-1145-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Milner EE, Berman J, Caridha D, Dickson SP, Hickman M, Lee PJ, Marcsisin SR, Read LT, Roncal N, Vesely BA, Xie LH, Zhang J, Zhang P, Li Q. 2016. Cytochrome P-450 metabolism is not necessary for tafenoquine and primaquine to eradicate the erythrocytic stages of Plasmodium berghei. Malar J 15:588. doi: 10.1186/s12936-016-1632-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pukrittayakamee S, Chotivanich K, Chantra A, Clemens R, Looareesuwan S, White NJ. 2004. Activities of artesunate and primaquine against asexual- and sexual-stage parasites in falciparum malaria. Antimicrob Agents Chemother 48:1329–1334. doi: 10.1128/aac.48.4.1329-1334.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jeffery GM. 1958. Infectivity to mosquitoes of Plasmodium vivax following treatment with chloroquine or other antimalarials. Am J Trop Med Hyg 7:207–211. doi: 10.4269/ajtmh.1958.7.207. [DOI] [PubMed] [Google Scholar]
- 91.Shapiro T, Ranasinha CD, Kumar N, Barditch-Crovo P. 1999. Prophylactic activity of atovaquone against Plasmodium falciparum in humans. Am J Trop Med Hyg 60:831–836. doi: 10.4269/ajtmh.1999.60.831. [DOI] [PubMed] [Google Scholar]
- 92.Beier JC, Onyango FK, Koros JK, Ramadhan M, Ogwang R, Wirtz RA, Koech DK, Roberts CR. 1991. Quantitation of malaria sporozoites transmitted in vitro during salivation by wild Afrotropical Anopheles. Med Vet Entomol 5:71–79. doi: 10.1111/j.1365-2915.1991.tb00523.x. [DOI] [PubMed] [Google Scholar]
- 93.Beier JC, Davis JR, Vaughan JA, Noden BH, Beier MS. 1991. Quantitation of Plasmodium falciparum sporozoites transmitted in vitro by experimentally infected Anopheles gambiae and Anopheles stephensi. Am J Trop Med Hyg 44:564–570. doi: 10.4269/ajtmh.1991.44.564. [DOI] [PubMed] [Google Scholar]
- 94.Ponnudurai T, Lensen AH, van Gemert GJ, Bolmer MG, Meuwissen JH. 1991. Feeding behaviour and sporozoite ejection by infected Anopheles stephensi. Trans R Soc Trop Med Hyg 85:175–180. doi: 10.1016/0035-9203(91)90012-n. [DOI] [PubMed] [Google Scholar]
- 95.Rosenberg R, Wirtz RA, Schneider I, Burge R. 1990. An estimation of the number of malaria sporozoites ejected by a feeding mosquito. Trans R Soc Trop Med Hyg 84:209–212. doi: 10.1016/0035-9203(90)90258-g. [DOI] [PubMed] [Google Scholar]
- 96.Shute PG, Lupascu G, Branzei P, Maryon M, Constantinescu P, Bruce-Chwatt LJ, Draper CC, Killick-Kendrick R, Garnham PC. 1976. A strain of Plasmodium vivax characterized by prolonged incubation: the effect of numbers of sporozoites on the length of the prepatent period. Trans R Soc Trop Med Hyg 70:474–481. doi: 10.1016/0035-9203(76)90132-2. [DOI] [PubMed] [Google Scholar]
- 97.Garnham P, Bray RS, Bruce-Chwatt LJ, Draper CC, Killick-Kendrick R, Sergiev PG, Tiburskaja NA, Shute PG, Maryon M. 1975. A strain of Plasmodium vivax characterized by prolonged incubation: morphological and biological characteristics. Bull World Health Organ 52:21–32. [PMC free article] [PubMed] [Google Scholar]
- 98.Krotoski WA, Garnham PCC, Cogswell FB, Collins WE, Bray RS, Gwadz RW, Killick-Kendrick R, Wolf RH, Sinden R, Hollingdale M, Lowrie RC Jr, Koontz LC, Stanfill PS. 1986. Observations on early and late post-sporozoite tissue stages in primate malaria. IV. Pre-erythrocytic schizonts and/or hypnozoites of Chesson and North Korean strains of Plasmodium vivax in the chimpanzee. Am J Trop Med Hyg 35:263–274. doi: 10.4269/ajtmh.1986.35.263. [DOI] [PubMed] [Google Scholar]
- 99.Rajgor DD, Gogtay NJ, Kadam VS, Kocharekar MM, Parulekar MS, Dalvi SS, Vaidya AB, Kshirsagar NA. 2014. Antirelapse efficacy of various primaquine regimens for Plasmodium vivax. Malar Res Treat 2014:347018. doi: 10.1155/2014/347018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Adak T, Valecha N, Sharma VP. 2001. Plasmodium vivax polymorphism in a clinical drug trial. Clin Diagn Lab Immunol 8:891–894. doi: 10.1128/CDLI.8.5.891-894.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mikolajczak SA, Vaughan AM, Kangwangrangsan N, Roobsoong W, Fishbaugher M, Yimamnuaychok N, Rezakhani N, Lakshmanan V, Singh N, Kaushansky A, Camargo N, Baldwin M, Linder SE, Adams JH, Prachumsri J, Kappe SH. 2015. Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe 17:526–535. doi: 10.1016/j.chom.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gural N, Mancio-Silva L, Miller AB, Galstian A, Butty VL, Levine SS, Patrapuvich R, Desai SP, Mikolajczak SA, Kappe SHI, Fleming HE, March S, Sattabongkot J, Bhatia SN. 2018. In vitro culture, drug sensitivity, and transcriptome of Plasmodium vivax hypnozoites. Cell Host Microbe 23:395–406. doi: 10.1016/j.chom.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bright AT, Manary MJ, Tewhey R, Arango EM, Wang T, Schork NJ, Yanow SK, Winzeler EA. 2014. A high resolution case study of a patient with recurrent Plasmodium vivax infections shows that relapses were caused by meiotic siblings. PLoS Negl Trop Dis 8:e2882. doi: 10.1371/journal.pntd.0002882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hill E, Amatuzio DS. 1949. Southwest Pacific vivax malaria: clinical features and observations concerning duration of clinical activity. J Trop Med Hyg 29:203–214. doi: 10.4269/ajtmh.1949.s1-29.203. [DOI] [PubMed] [Google Scholar]
- 105.Adekunle AI, Pinkevych M, McGready R, Luxemburger C, White LJ, Nosten F, Cromer D, Davenport MP. 2015. Modeling the dynamics of Plasmodium vivax infection and hypnozoite reactivation in vivo. PLoS Negl Trop Dis 9:e0003595. doi: 10.1371/journal.pntd.0003595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Robinson LJ, Wampfler R, Betuela I, Karl S, White MT, Li Wai Suen CSN, Hofmann NE, Kinboro B, Waltmann A, Brewster J, Lorry L, Tarongka N, Samol L, Silkey M, Bassat Q, Siba PM, Schofield L, Felger I, Mueller I. 2015. Strategies for understanding and reducing the Plasmodium vivax and Plasmodium ovale hypnozoite reservoir in Papua New Guinean children: a randomized placebo-controlled trial and mathematical model. PLoS Med 12:e1001891. doi: 10.1371/journal.pmed.1001891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gogtay N, Garg M, Kadam V, Kamtekar K, Kshirsagar NA. 1998. A 5 days primaquine regimen as anti-relapse therapy for Plasmodium vivax. Trans R Soc Trop Med Hyg 92:341. doi: 10.1016/s0035-9203(98)91034-3. [DOI] [PubMed] [Google Scholar]
- 108.Baird JK. 1998. Primaquine as anti-relapse therapy for Plasmodium vivax. Trans R Soc Trop Med Hyg 92:687. doi: 10.1016/s0035-9203(98)90814-8. [DOI] [PubMed] [Google Scholar]
- 109.Gogtay NJ, Desai S, Kamtekar KD, Kadam VS, Dalvi SS, Kshirsagar NA. 1999. Efficacies of 5- and 14-day primaquine regimens in the prevention of relapses in Plasmodium vivax infections. Ann Trop Med Parasitol 93:809–812. doi: 10.1080/00034989957790. [DOI] [PubMed] [Google Scholar]
- 110.Roy M, Bouma MJ, Ionides EL, Dhiman RC, Pascual M. 2013. The potential elimination of Plasmodium vivax malaria by relapse treatment: insights from a transmission model and surveillance data from NW India. PLoS Negl Trop Dis 7:e1979. doi: 10.1371/journal.pntd.0001979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.White MT, Karl S, Battle KE, Hay SI, Mueller I, Ghani AC. 2014. Modelling the contribution of the hypnozoite reservoir to Plasmodium vivax transmission. Elife 3:04692. doi: 10.7554/eLife.04692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mendis K, Sina BJ, Marchesini P, Carter R. 2001. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg 64(1-2 Suppl):97–106. doi: 10.4269/ajtmh.2001.64.97. [DOI] [PubMed] [Google Scholar]
- 113.Baird JK. 2007. Neglect of Plasmodium vivax malaria. Trends Parasitol 23:533–539. doi: 10.1016/j.pt.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 114.Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM. 2007. Vivax malaria: neglected and not benign. Am J Trop Med Hyg 77(6 Suppl):79–87. doi: 10.4269/ajtmh.2007.77.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.de Lacerda MV, Zackiewicz C, Alecrim WD, Alecrim MD. 2007. The neglected Plasmodium vivax: are researchers from endemic areas really concerned about new treatment options? Rev Soc Bras Med Trop 40:489–490. doi: 10.1590/S0037-86822007000400026. [DOI] [PubMed] [Google Scholar]
- 116.Baird JK. 2012. Primaquine toxicity forestalls effective therapeutic management of the endemic malarias. Int J Parasitol 42:1049–1054. doi: 10.1016/j.ijpara.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 117.Brightman R. 1956. Perkin and dyestuffs industry in Britain. Nature 177:805–856. [Google Scholar]
- 118.Ambruster HW. 1947. Treason’s peace: German dyes & American dupes, p 420 The Beechhurst Press, New York, NY. [Google Scholar]
- 119.Ehrlich P. 1880. Methodologische Beiträge zur Physiologie und Pathologie der verschiedenen Formen der Leukocyten. Z Klin Med 1:553–560. [Google Scholar]
- 120.Guttman P, Ehrlich P. 1891. Euber die Wirkung des Methylenblau bei Malaria. Berlin Klin Wochenschr 28:953–956. [Google Scholar]
- 121.Krafts K, Hempelmann E, Skorska-Stania A. 2012. From methylene blue to chloroquine: a brief review of the development of antimalarial therapy. Parasitol Res 111:1–6. doi: 10.1007/s00436-012-2886-x. [DOI] [PubMed] [Google Scholar]
- 122.Dascombe MJ, Drew MGB, Evans PG, Ismail FMD. 2007. Rational design strategies for the development of synthetic quinolone and acridine based antimalarials, p 559–609. In Atta-ur-Rahman, Caldwell GW, Choudhary MI, Player MR (ed), Frontiers in drug design & discovery, vol 3 Bentham Science Publishers, Sharjah, United Arab Emirates. [Google Scholar]
- 123.da Silva AF, Benchimol JL. 2014. Malaria and quinine resistance: a medical and scientific issue between Brazil and Germany (1907-19). Med Hist 58:1–26. doi: 10.1017/mdh.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Van der Hoogte AR, Pieters T. 2015. Quinine, malaria, and the Cinchona Bureau: marketing practices and knowledge circulation in a Dutch transoceanic cinchona-quinine enterprise (1920s-30s). J Hist Med Allied Sci 71:197–225. doi: 10.1093/jhmas/jrv009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Taylor N. 1945. Cinchona in Java, p 87 Greenberg: Publisher, New York, NY. [Google Scholar]
- 126.van Leersum N. 1945. Chapters in the history of cinchona. III. Junghuhn and cinchona cultivation, p 190–196. In Science and scientists in the Netherlands Indies. Board for the Netherlands Indies, Surinam and Curacao, New York, NY, and Riverside Press, Cambridge, MA. [Google Scholar]
- 127.Sergent ED, Sergent ET. 1921. Avantages de la quinisation preventive demontres et precises experimentellement (Paludisme des oiseaux). Ann Inst Pasteur 35:125–141. [Google Scholar]
- 128.Slater LB. 2005. Malarial birds: modeling infectious human disease in animals. Bull Hist Med 79:261–294. doi: 10.1353/bhm.2005.0092. [DOI] [PubMed] [Google Scholar]
- 129.Hewitt R. 1940. Bird malaria, p 228 The American Journal of Hygiene monograph series no. 15. The Johns Hopkins Press, Baltimore, MD. [Google Scholar]
- 130.Roehl W. 1926. Die Wirkung des Plasmochins auf die Vogelmalaria. Arch Schiffs Tropenhyg 30:311–318. [Google Scholar]
- 131.Schmidt LH, Coatney GR. 1955. Review of investigations in malaria chemotherapy (USA) 1946 to 1954. Am J Trop Med Hyg 4:208–216. doi: 10.4269/ajtmh.1955.4.208. [DOI] [PubMed] [Google Scholar]
- 132.Field JW. 1938. Notes on the chemotherapy of malaria. VI. Synthetic gametocides of the plasmoquine type. Bull Inst Medical Res Federated Malay States 2:63–79. [Google Scholar]
- 133.Anonymous. 1928. Medicine: Dutch monopoly. Time Magazine, vol XII, no. 14 http://content.time.com/time/magazine/article/0,9171,731964,00.html. Accessed 3 July 2019.
- 134.Vad BG, Mohile GB. 1927. The place of plasmochin in the treatment of malaria. Ind Med Gaz 62:430–434. [PMC free article] [PubMed] [Google Scholar]
- 135.Manson-Bahr P. 1927. The action of plasmochin on malaria. Proc R Soc Med 20:919–926. [PMC free article] [PubMed] [Google Scholar]
- 136.Sinton JA, Bird W. 1928. Studies in malaria, with special reference to treatment. Part IX. Plasmoquine in the treatment of malaria. Indian J Med Res 16:159–177. [Google Scholar]
- 137.Cordes W. 1927. Observations on the toxic effects of plasmochin, p 62–67. In Sixteenth annual report of the Medical Department of the United Fruit Company. United Fruit Company, Boston, MA. [Google Scholar]
- 138.Brosius OT. 1927. Plasmochin in malaria, p 26–53. In Sixteenth annual report of the Medical Department of the United Fruit Company. United Fruit Company, Boston, MA. [Google Scholar]
- 139.Craige B Jr, Jones R Jr, Whorton CM, Pullman TN, Alving AS, Eichelberger L. 1947. Clinical standardization of pamaquin (plasmochin) in mosquito-induced vivax malaria, Chesson strain. Am J Trop Med 27:309–315. doi: 10.4269/ajtmh.1947.s1-27.309. [DOI] [Google Scholar]
- 140.Berliner RW, Earle DP Jr, Taggart JV, Welch WJ, Zubrod CG, Knowlton P, Atchley JA, Shannon JA. 1948. Studies on the chemotherapy of the human malarias. VII. The antimalarial activity of pamaquine. J Clin Invest 27:108–113. doi: 10.1172/JCI101947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sinton JA, Smith S, Pottinger D. 1930. Studies on malaria with special reference to treatment. XII. Further researches into the treatment of chronic benign tertian malaria with plasmoquine and quinine. Indian J Med Res 17:793–814. [Google Scholar]
- 142.Alving AS, Pullman TN, Craige B Jr, Jones R Jr, Whorton CM, Eichelberger L. 1948. The clinical trial of eighteen analogs of pamaquin (plasmochin) in vivax malaria (Chesson strain). J Clin Invest 27:34–45. doi: 10.1172/JCI101963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Alving AS, Arnold J, Hockwald RS, Clayman CB, Dern RJ, Beutler E, Flanagan CL. 1955. Potentiation of the curative action of primaquine in vivax malaria by quinine and chloroquine. J Lab Clin Med 46:301–306. [PubMed] [Google Scholar]
- 144.Phelps BM. 1927. Clinical results with plasmochin, p 70–76. In Sixteenth annual report of the Medical Department of the United Fruit Company. United Fruit Company, Boston, MA. [Google Scholar]
- 145.Dow GS, Gettayacamin M, Hansukjariya P, Imerbsin R, Komcharoen S, Sattabongkot J, Kyle D, Milhous W, Cozens S, Kenworthy D, Miller A, Veazey J, Ohrt C. 2011. Radical curative efficacy of tafenoquine combination regimens in Plasmodium cynomolgi-infected rhesus monkeys (Macaca mulatta). Malar J 10:212. doi: 10.1186/1475-2875-10-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schmidt LH. 1985. Enhancement of curative activity of primaquine by concomitant administration of mirincamycin. Antimicrob Agents Chemother 27:151–157. doi: 10.1128/aac.27.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Myint HY, Berman J, Walker L, Pybus B, Melendez V, Baird JK, Ohrt C. 2011. Improving the therapeutic index of 8-aminoquinolines by use of drug combinations: review of the literature and proposal for future investigations. Am J Trop Med Hyg 85:1010–1014. doi: 10.4269/ajtmh.2011.11-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.MacPhail NP. 1927. Plasmochin in malaria, p 67–69. In Sixteenth annual report of the Medical Department of the United Fruit Company. United Fruit Company, Boston, MA. [Google Scholar]
- 149.Zubrod CG, Kennedy TJ, Shannon JA. 1948. Studies on the chemotherapy of the human malarias. VIII. The physiological disposition of pamaquine. J Clin Invest 27:114–120. doi: 10.1172/JCI101948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shannon JA. 1945. Study of the curative action of plasmochin in vivax malaria, p 38 In Malaria report #358, parts 1 and II. 16 March Board for the Coordination of Malaria Studies, Washington, DC. [Google Scholar]
- 151.Earle DP Jr, Bigelow FS, Zubrod CH, Kane C. 1948. Studies on the chemotherapy of the human malarias. IX. Effect of pamaquine on the blood cells of man. J Clin Invest 27:121–129. doi: 10.1172/JCI101950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Craige B Jr, Eichelberger L, Jones R Jr, Alving AS, Pullman TN, Whorton CM. 1948. The toxicity of large doses of pentaquine (SN-13276), a new antimalarial drug. J Clin Invest 27:17–24. doi: 10.1172/JCI101955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lysenko AY. 1960. Use of quinocide in the treatment and prophylaxis of vivax malaria. Bull World Health Organ 22:641–662. [PMC free article] [PubMed] [Google Scholar]
- 154.Alving AS, Eichelberger L, Arnold J, Edgcomb J. 1949. The clinical testing of antimalarial drugs at Stateville Penitentiary In Semi-annual report 1948-49. NIH malaria report no. 87. National Academy of Sciences Archive for the Board for Coordination of Malaria Studies, Washington, DC. [Google Scholar]
- 155.Hanboonkunupakarn B, Ashley EA, Jittamala P, Tarning J, Pukrittayakamee S, Hanpithakpong W, Chotsiri P, Wattanakul T, Panapipat S, Lee SJ, Day NP, White NJ. 2014. Open-label crossover study of primaquine and dihydroartemisinin-piperaquine pharmacokinetics in healthy Thai adult subjects. Antimicrob Agents Chemother 58:7340–7346. doi: 10.1128/AAC.03704-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pukrittayakamee S, Tarning J, Jittamala P, Charunwatthana P, Lawpoolsri S, Lee SJ, Hanpithakpong W, Hanboonkunupakarn B, Day NPJ, Ashley EA, White NJ. 2014. Pharmacokinetic interactions between primaquine and chloroquine. Antimicrob Agents Chemother 58:3354–3359. doi: 10.1128/AAC.02794-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jittamala P, Pukrittayakamee S, Ashley EA, Nosten F, Hanboonkunupakarn B, Lee SJ, Thana P, Chairat K, Blessborn D, Panapipat S, White NJ, Day NP, Tarning J. 2015. Pharmacokinetic interactions between primaquine and pyronaridine-artesunate in healthy adult Thai subjects. Antimicrob Agents Chemother 59:505–513. doi: 10.1128/AAC.03829-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chairat K, Jittamala P, Hanboonkunupakarn B, Pukrittayakamee S, Hanpithakpong W, Blessborn D, White NJ, Day NPJ, Tarning J. 2018. Enantiospecific pharmacokinetics and drug-drug-interactions of primaquine and blood-stage antimalarial drugs. J Antimicrob Chemother 73:3102–3113. doi: 10.1093/jac/dky297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fasinu PS, Tekwani BL, Avula B, Chaurasiya ND, Nanayakkara NP, Wang YH, Khan IA, Walker LA. 2016. Pathway-specific inhibition of primaquine metabolism by chloroquine/quinine. Malar J 15:466. doi: 10.1186/s12936-016-1509-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.U.S. Food and Drug Administration. 2018. Package insert for Arakoda™ (tafenoquine) for prevention of malaria. Accessed 20 October 2018 https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210607lbl.pdf.
- 161.U.S. Food and Drug Administration. 2018. Package insert for Krintafel™ (tafenoquine) for the treatment of vivax malaria. Accessed 20 October 2018 https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210795s000lbl.pdf.
- 162.Sinton JA. 1927. Studies in malaria with special reference to treatment. Part VII. The intravenous infection of sodium stovarsol in the treatment of benign tertian malaria. Indian J Med Res 15:287–299. [Google Scholar]
- 163.Ehrman FC, Ellis JM, Young MD. 1945. Plasmodium vivax Chesson strain. Science 101:377. doi: 10.1126/science.101.2624.377. [DOI] [PubMed] [Google Scholar]
- 164.Alving AS, Hankey DD, Coatney GR, Jones R Jr, Coker WG, Garrison PL, Donovan WN. 1953. Korean vivax malaria. II. Curative treatment with pamaquine and primaquine. Am J Trop Med Hyg 2:970–976. doi: 10.4269/ajtmh.1953.2.970. [DOI] [PubMed] [Google Scholar]
- 165.Collins WE, Jeffery GM. 1996. Primaquine resistance in Plasmodium vivax. Am J Trop Med Hyg 55:243–249. doi: 10.4269/ajtmh.1996.55.243. [DOI] [PubMed] [Google Scholar]
- 166.Jensen M, Mehlhorn H. 2009. 75 years of resochin in the fight against malaria, p 102 Dusseldorf University Press, Dusseldorf, Germany. [DOI] [PubMed] [Google Scholar]
- 167.Sweeney T. 2003. Malaria frontline: Australian Army research during World War II, p 398 Melbourne University Press, Melbourne, Australia. [Google Scholar]
- 168.Office of the Surgeon General of the United States. 1943. Circular letter no. 153. The drug treatment of malaria, suppressive and clinical. JAMA 123:205. doi: 10.1001/jama.1943.82840390004007. [DOI] [Google Scholar]
- 169.Butler AM. 1945. Clinical toxicity of plasmocide (SN-3115; rhodoquine): review of the Russian and French literature, p 8 In Malaria report no. 401. 9 May Board for the Coordination of Malarial Studies; Washington, DC. [Google Scholar]
- 170.Schmidt IG, Schmidt LH. 1948. Neurotoxicity of the 8-aminoquinolines; lesions in the central nervous system of the rhesus monkey induced by administration of plasmocid. J Neuropathol Exp Neurol 7:368–398. doi: 10.1097/00005072-194810000-00002. [DOI] [PubMed] [Google Scholar]
- 171.Schmidt LH. 1945. The toxicities of the 8-aminoquinolines: relation of structure to the toxicity of these drugs for the monkey, p 19 In Malaria report no. 469. 19 July Board for the Coordination of Malarial Studies; Washington, DC. [Google Scholar]
- 172.Jones R Jr, Craige B Jr, Alving AS, Whorton MC, Pullman TN, Eichelberger L. 1948. A study of the prophylactic effectiveness of several 8-aminoquinolines in sporozoite-induced vivax malaria (Chesson strain). J Clin Invest 27:6–11. doi: 10.1172/JCI101973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Alving AS, Craige B Jr, Pullman TN, Whorton CM, Jones R Jr, Eichelberger L. 1948. Procedures used at Stateville Penitentiary for the testing of potential antimalarial agents. J Clin Invest 27:2–5. doi: 10.1172/JCI101956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Coatney GR, Cooper WC, Ruhe DS. 1948. Studies in human malaria. VI. The organization of a program for testing potential antimalarial drugs in prisoner volunteers. Am J Epidemiol 47:113–119. doi: 10.1093/oxfordjournals.aje.a119180. [DOI] [PubMed] [Google Scholar]
- 175.Jones R Jr, Jackson LS, Di Lorenzo A, Marx RL, Levy BL, Kenny ED, Gilbert M, Johnston MN, Alving AS. 1953. Korean vivax malaria. III. Curative effect and toxicity of primaquine in doses from 10 to 30 mg. daily. Am J Trop Med Hyg 2:977–982. doi: 10.4269/ajtmh.1953.2.977. [DOI] [PubMed] [Google Scholar]
- 176.Coatney GR, Alving AS, Jones R Jr, Hankey DD, Robinson DH, Garrison PL, Coker WG, Donovan WN, Di Lorenzo A, Marx RL, Simmons IH. 1953. Korean vivax malaria. V. Cure of the infection by primaquine administered during long-term latency. Am J Trop Med Hyg 2:985–988. doi: 10.4269/ajtmh.1953.2.985. [DOI] [PubMed] [Google Scholar]
- 177.Leonard NJ. 2018. Robert Cooley Elderfield; May 30, 1904-December 10, 1979, p 14 In Biographical memoirs, vol 78 The National Academies Press, Washington, DC: Accessed 20 October 2018 https://www.nap.edu/read/9977/chapter/2. [Google Scholar]
- 178.Elderfield RC, Craig LC, Lauer WM, Arnold RT, Gensler WJ, Head JD, Bembry TH, Mighton HR, Tinker J, Galbreath J, Holley AD, Goldman L, Maynard JT, Picus N. 1946. Plasmochin and the occurrence of rearrangements in the preparation of certain plasmochin analogs. J Am Chem Soc 68:1516–1523. doi: 10.1021/ja01212a039. [DOI] [PubMed] [Google Scholar]
- 179.Cooper WC, Myatt AV, Hernandez T, Jeffery GM, Coatney GR. 1953. Studies in human malaria. XXXI. Comparison of primaquine, isopentaquine, SN-3883, and pamaquine as curative agents against Chesson strain vivax malaria. Am J Trop Med Hyg 2:949–957. doi: 10.4269/ajtmh.1953.2.949. [DOI] [PubMed] [Google Scholar]
- 180.Pullman TN, Craige B Jr, Alving AS, Whorton CM, Jones R Jr, Eichelberger L. 1948. Comparison of chloroquine, quinacrine (atabrine), and quinine in the treatment of acute attacks of sporozoite-induced vivax malaria (Chesson strain). J Clin Invest 27:51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Alving AS. 1954. Clinical treatment of malaria, chapter 5, part 2 In Medical science publication no. 4, vol II U.S. Army Medical Department, Office of Medical History; Accessed 21 October 2018 http://history.amedd.army.mil/booksdocs/korea/recad2/ch5-2.html. [Google Scholar]
- 182.Garrison PL, Hankey DD, Coker WG, Donovan WN, Jastremski B, Coatney GR, Alving AS, Jones R Jr. 1952. Cure of Korean vivax malaria with pamaquine and primaquine. JAMA 149:1562–1563. doi: 10.1001/jama.1952.72930340021010a. [DOI] [PubMed] [Google Scholar]
- 183.Alving AS, Arnold J, Robinson DH. 1952. Status of primaquine. I. Mass therapy of subclinical vivax malaria with primaquine. JAMA 149:1558–1562. doi: 10.1001/jama.1952.72930340017010. [DOI] [PubMed] [Google Scholar]
- 184.Archambeault CP. 1954. Mass antimalarial therapy in veterans returning from Korea. JAMA 154:1411–1415. doi: 10.1001/jama.1954.02940510011005. [DOI] [PubMed] [Google Scholar]
- 185.Pullman TN, Eichelberger L, Alving AS, Jones R Jr, Craige B Jr, Whorton CM. 1948. The use of SN-10,275 in the prophylaxis and treatment of sporozoite-induced vivax malaria (Chesson strain). J Clin Invest 27:12–16. doi: 10.1172/JCI101949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Brueckner RP, Ohrt C, Baird JK, Milhous WK. 2001. 8-Aminoquinolines, p 123–151. In Rosenthal PJ. (ed), Antimalarial chemotherapy. Humana Press, Totowa, NJ. [Google Scholar]
- 187.Brueckner RP, Lasseter KC, Lin ET, Schuster BG. 1998. First-time-in-humans safety and pharmacokinetics of WR238605, a new antimalarial. Am J Trop Med Hyg 58:645–649. doi: 10.4269/ajtmh.1998.58.645. [DOI] [PubMed] [Google Scholar]
- 188.Davidson DE Jr, Ager AL, Brown BL, Chapple FE, Whitmire RE, Rossan RN. 1981. New tissue schizontocidal antimalarial drugs. Bull World Health Organ 59:463–479. [PMC free article] [PubMed] [Google Scholar]
- 189.Fisher GU, Gordon MP, Lobel HO, Runcik K. 1970. Malaria in soldiers returning from Vietnam. Epidemiologic, therapeutic, and clinical studies. Am J Trop Med Hyg 19:27–39. doi: 10.4269/ajtmh.1970.19.27. [DOI] [PubMed] [Google Scholar]
- 190.Baird JK, Rieckmann KH. 2003. Can primaquine therapy for vivax malaria be improved? Trends Parasitol 19:115–120. doi: 10.1016/S1471-4922(03)00005-9. [DOI] [PubMed] [Google Scholar]
- 191.Schultz MG. 1974. Imported malaria. Bull World Health Organ 50:329–336. [PMC free article] [PubMed] [Google Scholar]
- 192.Barrett O Jr, Skrzypek G, Datel W, Goldstein JD. 1969. Malaria imported to the United States from Vietnam. Am J Trop Med Hyg 18:495–499. doi: 10.4269/ajtmh.1969.18.495. [DOI] [PubMed] [Google Scholar]
- 193.Cohen JM, Smith DL, Cotter C, Ward A, Yamey G, Sabot OJ, Moonen B. 2012. Malaria resurgence: a systematic review and assessment of its causes. Malar J 11:122. doi: 10.1186/1475-2875-11-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ockenhouse CF, Magill AJ, Smith D, Milhous W. 2005. History of U.S. military contributions to the study of malaria. Mil Med 170(4 Suppl):12–16. doi: 10.7205/milmed.170.4s.12. [DOI] [PubMed] [Google Scholar]
- 195.Baird JK, Decker-Jackson JE, Davidson DE Jr.. 1984. An in vitro micro-volume procedure for the rapid measurement of erythrocytic hexose monophosphate shunt activity. Int J Biochem 16:1049–1052. doi: 10.1016/0020-711X(84)90086-7. [DOI] [PubMed] [Google Scholar]
- 196.Baird JK. 1984. Methylene blue-mediated hexose monophosphate shunt stimulation in human red blood cells in vitro: independence from intracellular oxidative injury. Int J Biochem 16:1053–1058. doi: 10.1016/0020-711X(84)90087-9. [DOI] [PubMed] [Google Scholar]
- 197.Anders JC, Chung H, Theoharides AD. 1988. Methemoglobin formation resulting from administration of candidate 8-aminoquinoline antiparasitic drugs in the dog. Fundam Appl Toxicol 10:270–275. doi: 10.1016/0272-0590(88)90311-9. [DOI] [PubMed] [Google Scholar]
- 198.LaMontagne MP, Blumbergs P, Strube RE. 1982. Antimalarials. 14. 5-(Aryloxy)-4-methylprimaquine analogues. A highly effective series of blood and tissue schizontocidal agents. J Med Chem 25:1094–1097. doi: 10.1021/jm00351a017. [DOI] [PubMed] [Google Scholar]
- 199.Chen EH, Tanabe K, Saggiomo AJ, Nodiff EA. 1987. Modifications of primaquine as antimalarials. 4. 5-Alkyoxy derivatives of primaquine. J Med Chem 30:1193–1199. doi: 10.1021/jm00390a012. [DOI] [PubMed] [Google Scholar]
- 200.Nodiff EA, Tanabe K, Chen EH, Saggiomo AJ. 1982. Modifications of primaquine as antimalarials. 3. 5-Phenoxy derivatives of primaquine. J Med Chem 25:1097–1101. doi: 10.1021/jm00351a018. [DOI] [PubMed] [Google Scholar]
- 201.Dhar MM. 1987. Synthesis and screening of new antimalarial drugs. Final report. 30 October Central Drug Research Institute, Lucknow, India: Accessed 4 January 2019 https://apps.dtic.mil/dtic/tr/fulltext/u2/a206103.pdf. [Google Scholar]
- 202.Fisk TL, Millet P, Collins WE, Nguyen-Dinh P. 1989. In vitro activity of antimalarial compounds on the exoerythrocytic stages of Plasmodium cynomolgi and P. knowlesi. Am J Trop Med Hyg 40:235–239. doi: 10.4269/ajtmh.1989.40.235. [DOI] [PubMed] [Google Scholar]
- 203.Marino MT, Peggins JO, Brown LD, Urquhart MR, Brewer TG. 1994. Pharmacokinetics and kinetic-dynamic modelling of an 8-aminoquinoline candidate anticyanide and antimalarial drug (WR242511). Drug Metab Dispos 22:358–366. [PubMed] [Google Scholar]
- 204.Peters W. 1999. The evolution of tafenoquine—antimalarial for a new millennium? J R Soc Med 92:345–352. doi: 10.1177/014107689909200705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Rockwood GA, Duniho SM, Briscoe CM, Gold MB, Armstrong KR, Moran AV, Kahler DW, Baskin SI. 2008. Toxicity in rhesus monkeys following administration of the 8-aminoquinoline 8-[(4-amino-1-methylbutyl)amino]-5-(1-hexyloxy)-6-methoxy-4-methylquinoline (WR242511). J Med Toxicol 4:157–166. doi: 10.1007/BF03161194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Brueckner RP, Coster T, Wesche DL, Shmuklarsky M, Schuster BG. 1998. Prophylaxis of Plasmodium falciparum infection in a human challenge model with WR238605, a new 8-aminoquinoline antimalarial. Antimicrob Agents Chemother 42:1293–1294. doi: 10.1128/AAC.42.5.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Walsh DS, Looareesuwan S, Wilairatana P, Heppner DG Jr, Tang DB, Brewer TG, Chokejindachai W, Viriyavejakul P, Kyle DE, Milhous WK, Schuster BG, Braitman DJ, Brueckner RP. 1999. Randomized dose-ranging study of the safety and efficacy of WR238605 (tafenoquine) in the prevention of relapse of Plasmodium vivax malaria in Thailand. J Infect Dis 180:1284–1287. [DOI] [PubMed] [Google Scholar]
- 208.Shanks GD, Oloo AJ, Aleman GM, Ohrt C, Klotz FW, Braitman D, Horton J, Brueckner R. 2001. A new primaquine analogue, tafenoquine (WRO238605) for prophylaxis against Plasmodium falciparum malaria. Clin Infect Dis 33:1968–1974. doi: 10.1086/324081. [DOI] [PubMed] [Google Scholar]
- 209.Lell B, Faucher JF, Missinou MA, Borrmann S, Dangelmaier O, Horton J, Kremsner PG. 2000. Malaria chemoprophylaxis with tafenoquine: a randomized study. Lancet 355:2041–2045. doi: 10.1016/S0140-6736(00)02352-7. [DOI] [PubMed] [Google Scholar]
- 210.Edstein MD, Walsh DS, Eamsila C, Sasiprapha T, Nasveld PE, Kitchener S, Reickmann KH. 2001. Malaria prophylaxis/radical cure: experiences of the Australian Defence Force. Med Trop (Mars) 61:56–58. [PubMed] [Google Scholar]
- 211.Hale BR, Owusu-Agyei S, Fryauff DJ, Koram KA, Adjuik M, Oduro AR, Prescott WR, Baird JK, Nkrumah F, Ritchie TL, Franke ED, Binka FN, Horton J, Hoffman SL. 2003. A randomized, double-blind, placebo-controlled, dose-ranging trial of tafenoquine for weekly prophylaxis against Plasmodium falciparum. Clin Infect Dis 36:541–549. doi: 10.1086/367542. [DOI] [PubMed] [Google Scholar]
- 212.Nasveld P, Kitchener S, Edstein M, Rieckmann K. 2002. Comparison of tafenoquine (WR238605) and primaquine in the post-exposure (terminal) prophylaxis of vivax malaria in Australian Defence Force personnel. Trans R Soc Trop Med Hyg 96:683–684. doi: 10.1016/s0035-9203(02)90351-2. [DOI] [PubMed] [Google Scholar]
- 213.Walsh DS, Eamsila C, Sasiprapha T, Sangkharomya S, Khaewsathien P, Supakalin P, Tang DB, Jarasrumgsichol P, Cherdchu C, Edstein MD, Rieckmann KH, Brewer TG. 2004. Efficacy of monthly tafenoquine prophylaxis of Plasmodium vivax and multidrug-resistant P. falciparum malaria. J Infect Dis 190:1456–1463. doi: 10.1086/424468. [DOI] [PubMed] [Google Scholar]
- 214.Walsh DS, Wilairatana P, Tang DB, Heppner DG Jr, Brewer TG, Krudsood S, Silachamroon U, Phumratanaprapin W, Siriyanonda D, Looareesuwan S. 2004. Randomized trial of 3-dose regimens of tafenoquine (WR238605) versus low-dose primaquine for preventing Plasmodium vivax malaria relapse. Clin Infect Dis 39:1095–1103. doi: 10.1086/424508. [DOI] [PubMed] [Google Scholar]
- 215.Nasveld PE, Edstein MD, Reid M, Brennan L, Harris IE, Kitchener SJ, Leggat PA, Pickford P, Kerr C, Ohrt C, Prescott W, Tafenoquine Study Team. 2010. Randomized, double-blind study of the safety, tolerability, and efficacy of tafenoquine versus mefloquine for malaria prophylaxis in nonimmune subjects. Antimicrob Agents Chemother 54:792–798. doi: 10.1128/AAC.00354-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Leary KJ, Riel MA, Roy MJ, Cantilena LR, Bi D, Brater DC, van de Pol C, Pruett K, Kerr C, Leary KJ, Veazey JM, Beboso R, Ohrt C. 2009. A randomized, double-blind, safety and tolerability study to assess ophthalmic and renal effects of tafenoquine 200 mg weekly versus placebo for 6 months in healthy volunteers. Am J Trop Med Hyg 81:356–362. doi: 10.4269/ajtmh.2009.81.356. [DOI] [PubMed] [Google Scholar]
- 217.Dow GS, Magill AJ, Ohrt C. 2008. Clinical development of new prophylactic antimalarial drugs after the 5th Amendment to the Declaration of Helsinki. Ther Clin Risk Manag 4:803–819. doi: 10.2147/TCRM.S1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Llanos-Cuentas A, Lacerda MV, Rueangweerayut R, Krudsood S, Gupta SK, Kochar SK, Arthur P, Chuenchom N, Möhrle JJ, Duparc S, Ugwuegbulam C, Kleim J-P, Carter N, Green JA, Kellam L. 2014. Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): a multicentre, double-blind, randomized, phase 2b dose-selection study. Lancet 383:1049–1058. doi: 10.1016/S0140-6736(13)62568-4. [DOI] [PubMed] [Google Scholar]
- 219.Lacerda MVG, Llanos-Cuentas A, Krudsood S, Lon C, Saunders DL, Mohammed R, Yilma D, Batista Pereira D, Espino FEJ, Mia RZ, Chuquiyauri R, Val F, Casapía M, Monteiro WM, Brito MAM, Costa MRF, Buathong N, Noedl H, Diro E, Getie S, Wubie KM, Abdissa A, Zeynudin A, Abebe C, Tada MS, Brand F, Beck H-P, Angus B, Duparc S, Kleim J-P, Kellam LM, Rousell VM, Jones SW, Hardaker E, Mohamed K, Clover DD, Fletcher K, Breton JJ, Ugwuegbulam CO, Green JA, Koh GCKW. 2019. Single dose tafenoquine to prevent relapse of Plasmodium vivax malaria. N Engl J Med 380:215–228. doi: 10.1056/NEJMoa1710775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Llanos-Cuentas A, Lacerda MVG, Hien TT, Vélez ID, Namaik-Larp C, Chu CS, Villegas MF, Val F, Monteiro WM, Brito MAM, Costa MRF, Chuquiyauri R, Casapía M, Nguyen CH, Aruachan S, Papwijitsil R, Nosten FH, Bancone G, Angus B, Duparc S, Craig G, Rousell VM, Jones SW, Hardaker E, Clover DD, Kendall L, Mohamed K, Koh GCKW, Wilches VM, Breton JJ, Green JA. 2019. Tafenoquine versus primaquine to prevent relapse of Plasmodium vivax malaria. N Engl J Med 380:229–241. doi: 10.1056/NEJMoa1802537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dow GS, Liu J, Hetzell B, Thieling S, McCarthy WF, Tang D, Smith B. 2015. Summary of anti-malarial prophylactic efficacy of tafenoquine from three placebo-controlled studies of residents of malaria-endemic countries. Malar J 14:473. doi: 10.1186/s12936-015-0991-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Baird JK. 2018. Tafenoquine for malaria in travelers: evidence. Rationale and recommendations. J Travel Med 25:tay110. doi: 10.1093/jtm/tay110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Jandl JH, Engle LK, Allen DW. 1960. Oxidative hemolysis and precipitation of hemoglobin. I. Heinz body anemias as an acceleration of red cell aging. J Clin Invest 39:1818–1836. doi: 10.1172/JCI104206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Chui JS, Poon WT, Chan KC, Chan AY, Buckley TA. 2005. Nitrite-induced methaemoglobinemia—aetiology, diagnosis, and treatment. Anaesthesia 60:496–500. doi: 10.1111/j.1365-2044.2004.04076.x. [DOI] [PubMed] [Google Scholar]
- 225.Luzzatto L, Nannelli C, Notaro R. 2016. Glucose-6-phosphate dehydrogenase deficiency. Hematol Oncol Clin North Am 30:373–393. doi: 10.1016/j.hoc.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 226.World Health Organization Working Group. 1989. Glucose-6-phosphate dehydrogenase deficiency. Bull World Health Organ 67:601–611. [PMC free article] [PubMed] [Google Scholar]
- 227.Cappellini MD, Fiorelli G. 2008. Glucose-6-phosphate dehydrogenase deficiency. Lancet 371:64–74. doi: 10.1016/S0140-6736(08)60073-2. [DOI] [PubMed] [Google Scholar]
- 228.Lyon MF. 1999. X-chromosome inactivation. Curr Biol 9:R235–R237. doi: 10.1016/S0960-9822(99)80151-1. [DOI] [PubMed] [Google Scholar]
- 229.Kuwahata M, Wijesinghe R, Ho MF, Pelecanos A, Bobogare A, Landry L, Bugora H, Vallely A, McCarthy J. 2010. Population screening for glucose-6-phosphate dehydrogenase deficiencies in Isabel Province, Solomon Islands, using a modified enzyme assay on filter paper blood spots. Malar J 9:223. doi: 10.1186/1475-2875-9-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Satyagraha AW, Sadhewa A, Baramuli V, Elvira R, Ridenour C, Elyazar I, Noviyanti R, Coutrier FN, Harahap AR, Baird JK. 2015. G6PD deficiency at Sumba in eastern Indonesia is prevalent, diverse, and severe: implications for primaquine therapy against relapsing vivax malaria. PLoS Negl Trop Dis 9:e0003602. doi: 10.1371/journal.pntd.0003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Roh ME, Oyet C, Orikiriza P, Wade M, Mwanga-Amumpaire J, Boum Y II, Kiwanuka GN, Parikh S. 2016. Screening for glucose-6-phosphate dehydrogenase deficiency using three detection methods: a cross-sectional survey in southwestern Uganda. Am J Trop Med Hyg 95:1094–1099. doi: 10.4269/ajtmh.16-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Chopra RN, Chaudhuri RN. 1935. Some observations on the toxicity of synthetic anti-malarial remedies. Ind Med Gaz 70:1–5. [PMC free article] [PubMed] [Google Scholar]
- 233.Alving AS, Kellermeyer RW, Tarlov A, Schrier SL, Carson P. 1958. Biochemical and genetic aspects of primaquine-sensitive hemolytic anemia. Arch Intern Med 49:240–248. [DOI] [PubMed] [Google Scholar]
- 234.Gray SJ, Sterling K. 1950. The tagging of red blood cells and blood proteins with radioactive chromium. J Clin Invest 29:1604–1607. doi: 10.1172/JCI102403. [DOI] [PubMed] [Google Scholar]
- 235.Dern RJ, Weinstein IM, LeRoy GV, Talmage DW, Alving AS. 1954. The hemolytic effect of primaquine. I. The localization of the drug-induced hemolytic defect in primaquine-sensitive individuals. J Lab Clin Med 43:303–309. [PubMed] [Google Scholar]
- 236.Beutler E, Dern RJ, Alving AS. 1954. The hemolytic effect of primaquine. IV. The relationship of cell age to hemolysis. J Lab Clin Med 44:439–442. [PubMed] [Google Scholar]
- 237.Beutler E, Dern RJ, Alving AS. 1955. The hemolytic effect of primaquine. VI. An in vitro test for sensitivity of erythrocytes to primaquine. J Lab Clin Med 45:40–50. [PubMed] [Google Scholar]
- 238.Beutler E, Dern RJ, Flanagan CL, Alving AS. 1955. The hemolytic effect of primaquine. VII. Biochemical studies on drug-sensitive erythrocytes. J Lab Clin Med 45:286–295. [PubMed] [Google Scholar]
- 239.Carson PE, Flanagan CL, Ickes CE, Alving AS. 1956. Enzymatic deficiency in primaquine-sensitive erythrocytes. Science 124:484–485. doi: 10.1126/science.124.3220.484-a. [DOI] [PubMed] [Google Scholar]
- 240.Beutler E. 2008. Glucose-6-phosphate dehydrogenase: a historical perspective. Blood 111:16–24. doi: 10.1182/blood-2007-04-077412. [DOI] [PubMed] [Google Scholar]
- 241.Marks PA, Gross RT. 1959. Erythrocyte glucose-6-phosphate dehydrogenase deficiency: evidence of differences between Negroes and Caucasians with respect to this genetically determined trait. J Clin Invest 38:2253–2262. doi: 10.1172/JCI104006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zalusky R. 1970. Drug-associated hemolytic anemia. Bull N Y Acad Med 46:427–437. [PMC free article] [PubMed] [Google Scholar]
- 243.Monteiro WM, Neto-Moura JP, Recht J, Bassat Q, Lacerda MV. 2016. Fatal primaquine-induced hemolysis in a patient with Plasmodium vivax malaria and G6PD A− variant in the Brazilian Amazon. Clin Infect Dis 62:1188. doi: 10.1093/cid/ciw039. [DOI] [PubMed] [Google Scholar]
- 244.Ziai M, Amirhakimi GH, Reinhold JG, Tabatabee M, Gettner ME, Bowman JE. 1967. Malaria prophylaxis and treatment in G6PD deficiency: an observation on the toxicity of primaquine and chloroquine. Clin Pediatr (Phila) 6:242–243. doi: 10.1177/000992286700600416. [DOI] [PubMed] [Google Scholar]
- 245.Choudhry VP, Ghafary A, Zaher M, Qureshi MA, Fazel I, Ghani R. 1990. Drug-induced haemolysis and renal failure in children with glucose-6-phosphate dehydrogenase deficiency in Afghanistan. Ann Trop Paediatr 10:335–338. doi: 10.1080/02724936.1990.11747454. [DOI] [PubMed] [Google Scholar]
- 246.Alving AS, Johnson CF, Tarlov AR, Brewer GJ, Kellermeyer RW, Carson PE. 1960. Mitigation of the haemolytic effect of primaquine and enhancement of its action against exoerythrocytic forms of the Chesson strain of Plasmodium vivax by intermittent regimens of drug administration: a preliminary report. Bull World Health Organ 73:80–92. [PMC free article] [PubMed] [Google Scholar]
- 247.Mills GC, Randall HP. 1958. Hemoglobin catabolism. II. The protection of hemoglobin from oxidative breakdown in the intact erythrocyte. J Biol Chem 232:589–595. [PubMed] [Google Scholar]
- 248.Cohen G, Hochstein P. 1964. Generation of hydrogen peroxide in erythrocytes by hemolytic agents. Biochemistry 3:895–900. doi: 10.1021/bi00895a006. [DOI] [PubMed] [Google Scholar]
- 249.Goldberg B, Stern A. 1976. Superoxide ion as mediator of drug-induced oxidative hemolysis. J Biol Chem 437:6468–6470. [PubMed] [Google Scholar]
- 250.Goldberg B, Stern A. 1975. The generation of O2− by the interaction of the hemolytic agent phenylhydrazine with human hemoglobin. J Biol Chem 250:2401–2403. [PubMed] [Google Scholar]
- 251.Summerfield M, Tudhope GR. 1978. Studies of primaquine in vitro: superoxide radical formation and oxidation of haemoglobin. Br J Clin Pharmacol 6:319–323. doi: 10.1111/j.1365-2125.1978.tb00858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Cowan WK, Evans DA. 1964. Primaquine and methemoglobin. Clin Pharmacol Ther 5:307–309. doi: 10.1002/cpt196453307. [DOI] [PubMed] [Google Scholar]
- 253.Doyle MP, Pickering RA, DeWeert TM, Hoekstra JW, Pater D. 1981. Kinetics and mechanism of the oxidation of human deoxyhemoglobin by nitrites. J Biol Chem 256:12393–12398. [PubMed] [Google Scholar]
- 254.Gautami S, Rao RN, Raghuram TC, Rajagopalan S, Bhat RV. 1995. Accidental acute fatal sodium nitrite poisoning. J Toxicol Clin Toxicol 33:131–133. doi: 10.3109/15563659509000462. [DOI] [PubMed] [Google Scholar]
- 255.Beaven GH, White JC. 1954. Oxidation of phenylhydrazines in the presence of oxyhemoglobin and the origin of Heinz bodies in erythrocytes. Nature 173:389–392. doi: 10.1038/173389b0. [DOI] [PubMed] [Google Scholar]
- 256.Itano H. 1970. Phenyldiimide, hemoglobin, and Heinz bodies. Proc Natl Acad Sci U S A 67:485–492. doi: 10.1073/pnas.67.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Itano HA, Hirota K, Hosokawa K. 1975. Mechanism of induction of haemolytic anaemia by phenylhydrazine. Nature 256:665–667. doi: 10.1038/256665a0. [DOI] [PubMed] [Google Scholar]
- 258.Itano HA, Hosokawa K, Hirota K. 1976. Induction of haemolytic anaemia by substituted phenylhydrazines. Br J Haematol 32:99–104. doi: 10.1111/j.1365-2141.1976.tb01879.x. [DOI] [PubMed] [Google Scholar]
- 259.Itano HA, Hirota K, Vedvick TS. 1977. Ligands and oxidants in ferrihemochrome formation and oxidative hemolysis. Proc Natl Acad Sci U S A 74:2556–2560. doi: 10.1073/pnas.74.6.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Bowman ZS, Oatis JE Jr, Whelan JL, Jollow DJ, McMillan DC. 2004. Primaquine-induced haemolytic anemia: susceptibility of normal versus glutathione-depleted rat erythrocytes to 5-hydroxyprimaquine. J Pharmacol Exp Ther 309:79–85. doi: 10.1124/jpet.103.062984. [DOI] [PubMed] [Google Scholar]
- 261.Beutler E. 1957. The glutathione instability of drug-sensitive red blood cells: a new method for the in vitro detection of drug sensitivity. J Lab Clin Med 49:84–95. [PubMed] [Google Scholar]
- 262.Recht J, Ashley E, White N. 2014. Safety of 8-aminoquinoline antimalarial medicines, p 222 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 263.Piomelli S, Corash LM, Davenport DD, Miraglia J, Amorosi EL. 1968. In vivo liability of glucose-6-phosphate dehydrogenase in GdA− and GdMediterranean deficiency. J Clin Invest 47:940–948. doi: 10.1172/JCI105786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Thaeler AD, Arnold JA, Alving AS. 1953. A clinical study of primaquine in the treatment of malaria among Miskito Indians of Nicaragua. Am J Trop Med Hyg 2:989–999. doi: 10.4269/ajtmh.1953.2.989. [DOI] [PubMed] [Google Scholar]
- 265.Charles LJ. 1960. Observations on the haemolytic effect of primaquine in 100 Ghanaian children. Ann Trop Med Parasitol 54:460–470. doi: 10.1080/00034983.1960.11686008. [DOI] [PubMed] [Google Scholar]
- 266.Kondrashin A, Baranova AM, Ashley EA, Recht J, White NJ, Sergiev VP. 2014. Mass primaquine treatment to eliminate malaria: lessons from the past. Malar J 13:51. doi: 10.1186/1475-2875-13-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Restrepo MA, Gutierrez E. 1968. The frequency of glucose-phosphate dehydrogenase deficiency in Colombia. Am J Hum Genet 20:82–85. [PMC free article] [PubMed] [Google Scholar]
- 268.Lisker R, Loria A, Cordova MS. 1965. Studies on several genetic hematological traits of the Mexican population. VIII. Hemoglobin S, glucose-6-phosphated dehydrogenase deficiency, and other characteristics in a malarial region. Am J Hum Genet 17:179–187. [PMC free article] [PubMed] [Google Scholar]
- 269.Goo YK, Ji SY, Shin HI, Moon JH, Cho SH, Lee WJ, Kim JY. 2014. First evaluation of glucose-6-phosphate dehydrogenase deficiency in vivax malaria endemic regions in the Republic of Korea. PLoS One 9:e97390. doi: 10.1371/journal.pone.0097390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Bonsignore A, Fornaini G, Fantoni A, Leoncini G, Segni P. 1964. Relationship between age and enzymatic activities in human erythrocytes from normal and Fava-bean sensitive subjects. J Clin Invest 43:834–842. doi: 10.1172/JCI104969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Pannacciulli I, Tizianello A, Ajmar F, Salvidio E. 1965. The course of experimentally induced hemolytic anemia in a primaquine-sensitive Caucasian. A case study. Blood 25:92–95. [PubMed] [Google Scholar]
- 272.Salvidio E, Pannacciulli I, Tizianello A, Ajmar F. 1967. Nature of hemolytic crises and the fate of G6PD deficient, drug-damaged erythrocytes in Sardinians. N Engl J Med 276:1339–1344. doi: 10.1056/NEJM196706152762402. [DOI] [PubMed] [Google Scholar]
- 273.George JN, Sears DA, McCurdy PR, Conrad ME. 1967. Primaquine sensitivity in Caucasians: hemolytic reactions induced by primaquine in G6PD deficient subjects. J Lab Clin Med 70:80–93. [PubMed] [Google Scholar]
- 274.Carson PE, Frischer H. 1966. Glucose-6-phosphate dehydrogenase and related disorders of the pentose phosphate pathway. Am J Med 41:744–753. doi: 10.1016/0002-9343(66)90035-0. [DOI] [PubMed] [Google Scholar]
- 275.Abeyaratne KP, Halpe NL. 1968. Sensitivity to primaquine in Ceylonese children due to deficiency of erythrocytic glucose-6-phosphate dehydrogenase. Ceylon Med J 13:134–138. [PubMed] [Google Scholar]
- 276.Aung-Than-Batu, Hla-Pe U, Thein-Than. 1970. Primaquine induced hemolysis in G6PD deficient Burmese. Trans R Soc Trop Med Hyg 64:785–786. doi: 10.1016/0035-9203(70)90023-4. [DOI] [PubMed] [Google Scholar]
- 277.Charoenlarp P, Areekul S, Harinasuta T, Sirivorasarn P. 1972. The hemolytic effect of single dose of 45 mg of primaquine in G-6-PD deficient Thais. J Med Assoc Thai 55:631–638. [PubMed] [Google Scholar]
- 278.Chan TK, Todd D, Tso SC. 1976. Drug-induced haemolysis in glucose-6-phosphate dehydrogenase deficiency. Br Med J 2:1227–1229. doi: 10.1136/bmj.2.6046.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Bancone G, Malleret B, Suwanarusk R, Chowwiwat N, Chu CS, McGready R, Rénia L, Nosten F, Russell B. 2017. Asian G6PD-Mahidol reticulocytes sustain normal Plasmodium vivax development. J Infect Dis 216:263–266. doi: 10.1093/infdis/jix278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Kheng S, Muth S, Tylor WR, Tops N, Kosal K, Sothea K, Souy P, Kim S, Char CM, Vanna C, Ly P, Ringwald P, Kheiu V, Kerleguer A, Tor P, Baird JK, Bjorge S, Menard D, Christophel E. 2015. Tolerability and safety of weekly primaquine against relapse of Plasmodium vivax in Cambodians with glucose-6-phosphate dehydrogenase deficiency. BMC Med 13:203. doi: 10.1186/s12916-015-0441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Bruce-Chwatt LJ, Black RH, Canfield CJ, Clyde DF, Peters W, Wernsdorfer WH (ed). 1981. Chemotherapy of malaria, 2nd ed, p 261 World Health Organization monograph series no. 27 World Health Organization, Geneva, Switzerland. [Google Scholar]
- 282.World Health Organization. 2010. Guidelines for the treatment of malaria, 2nd ed, p 194 Global Malaria Programme, World Health Organization, Geneva, Switzerland. [Google Scholar]
- 283.World Health Organization. 2018. Point-of-care G6PD testing to support safe use of primaquine for the treatment of vivax malaria, p 25 WHO Evidence Review Group meeting report. World Health Organization, Geneva, Switzerland: Accessed 27 December 2018 https://www.who.int/malaria/mpac/mpac-march2015-erg-g6pd.pdf?ua=1. [Google Scholar]
- 284.Recht J, Ashley EA, White NJ. 2018. Use of primaquine and glucose-6-phosphate dehydrogenase deficiency testing: divergent policies and practices in malaria endemic countries. PLoS Negl Trop Dis 12:e0006230. doi: 10.1371/journal.pntd.0006230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.World Health Organization. 2015. Guidelines for the treatment of malaria, 3rd ed, p 317 Global Malaria Programme, World Health Organization, Geneva, Switzerland. [Google Scholar]
- 286.World Health Organization. 2018. Guide to G6PD deficiency rapid diagnostic testing to support P. vivax radical cure, p 26 Global Malaria Programme, World Health Organization, Geneva, Switzerland. [Google Scholar]
- 287.Ley B, Thriemer K, Jaswal J, Poirot E, Alam MS, Phru CS, Khan WA, Dysoley L, Qi G, Kheong CC, Shamsudin UK, Chen I, Hwang J, Gosling R, Price RN. 2017. Barriers to routine G6PD testing prior to treatment with primaquine. Malar J 16:329. doi: 10.1186/s12936-017-1981-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Beutler E, Mitchell M. 1968. Special modifications of the fluorescent spot screening method for glucose-6-phosphate dehydrogenase deficiency. Blood 32:816–818. [PubMed] [Google Scholar]
- 289.Bancone G, Chu CS, Chowwiwat N, Somsakchaicharoen R, Wilaisrisak P, Charunwatthana P, Bansil P, McGray S, Domingo GJ, Nosten FH. 2015. Suitability of capillary blood for quantitative assessment of G6PD activity and performances of G6PD point-of-care tests. Am J Trop Med Hyg 92:818–824. doi: 10.4269/ajtmh.14-0696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Baird JK, Dewi M, Subekti D, Elyazar I, Satyagraha AW. 2015. Noninferiority of glucose-6-phosphate dehydrogenase deficiency diagnosis by a point of care rapid test vs the laboratory fluorescent spot test demonstrated by copper inhibition in normal red blood cells. Transl Res 165:677–688. doi: 10.1016/j.trsl.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Wolf BH, Weening RS, Schutgens RB, van Noorden CJ, Vogels IM, Nagelkerke NJ. 1987. Detection of glucose-6-phosphate dehydrogenase deficiency in erythrocytes: a spectrophotometric assay and fluorescent spot test compared with a cytochemical method. Clin Chim Acta 168:129–136. doi: 10.1016/0009-8981(87)90281-6. [DOI] [PubMed] [Google Scholar]
- 292.Henriques G, Phommasone K, Tripura R, Peto TJ, Raut S, Snethlage C, Sambo I, Sanaan N, Nguon C, Adhikari B, Pongvongsa T, Imwong M, von Seidlein L, Day NP, White NJ, Dondorp AM, Newton P, Ley B, Mayxay M. 2018. Comparison of glucose-6-phosophate dehydrogenase status by fluorescent spot test and rapid diagnostic test in Lao PDR and Cambodia. Malar J 17:243. doi: 10.1186/s12936-018-2390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Espino FE, Bibit JA, Sornillo JB, Tan A, von Seidlein L, Ley B. 2016. Comparison of three screening kits for G6PD enzyme deficiency: implications for its use in radical cure of vivax malaria in remote and resource-poor areas. PLoS One 11:e30148172. doi: 10.1371/journal.pone.0148172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Satyagraha AW, Sadhewa A, Elvira R, Elyazar I, Feriandika D, Antonjaya U, Oyong D, Subekti D, Rozi IE, Domingo GJ, Harahap AR, Baird JK. 2016. Assessment of point-of-care diagnostics for G6PD deficiency in malaria endemic rural eastern Indonesia. PLoS Negl Trop Dis 10:e0004457. doi: 10.1371/journal.pntd.0004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Ley B, Bancone G, von Seidlein L, Thriemer K, Richards JS, Domingo GJ, Price RN. 2017. Methods for the field evaluation of quantitative G6PD diagnostics: a review. Malar J 16:361. doi: 10.1186/s12936-017-2017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Williams DP, Bhattacharyya R. 1935. Notes on an experiment of the prophylactic and curative value of atebrin and plasmochin therapy in a tea garden in Assam. Ind Med Gaz 70:8–14. [PMC free article] [PubMed] [Google Scholar]
- 297.Feldman HA, Packer H. 1946. Pamaquine naphthoate as a prophylactic for malarial infections. Fed Proc 5:244. [PubMed] [Google Scholar]
- 298.Arnold J, Alving AS, Hockwald RS, Clayman CB, Dern RJ, Beutler E, Jeffery GM. 1954. The effect of continuous and intermittent primaquine therapy on the relapse rate of Chesson strain of vivax malaria. J Lab Clin Med 44:429–438. [PubMed] [Google Scholar]
- 299.White NJ, Qiao LG, Qi G, Luzzatto L. 2012. Rationale for recommending a lower dose of primaquine as a Plasmodium falciparum gametocytocide in populations where G6PD deficiency is common. Malar J 11:418. doi: 10.1186/1475-2875-11-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Desai M, Hill J, Fernandes S, Walker P, Pell C, Gutman J, Kayentao K, Gonzalez R, Webster J, Greenwood B, Cot M, Ter Kuile FO. 2018. Prevention of malaria in pregnancy. Lancet Infect Dis 18:e119–e132. doi: 10.1016/S1473-3099(18)30064-1. [DOI] [PubMed] [Google Scholar]
- 301.Gonzalez R, Pons-Duran C, Piqueras M, Aponte JJ, Ter Kuile FO, Menendez C. 2018. Mefloquine for preventing malaria in pregnant women. Cochrane Database Syst Rev 11:CD011444. doi: 10.1002/14651858.CD011444.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Meremikwu MM, Donegan S, Sinclair D, Esu E, Oringanje C. 2012. Intermittent preventive treatment for malaria in children living in areas of seasonal transmission. Cochrane Database Syst Rev 2012:CD003756. doi: 10.1002/14651858.CD003756.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Gutman J, Kovacs S, Dorsey G, Stergachis A, Ter Kuile FO. 2017. Safety, tolerability, and efficacy of repeated doses of dihydroartemisinin-piperaquine for prevention and treatment of malaria: a systematic review and meta-analysis. Lancet Infect Dis 17:184–193. doi: 10.1016/S1473-3099(16)30378-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Wells TNC, Burrows JN, Baird JK. 2010. Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol 26:145–151. doi: 10.1016/j.pt.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 305.Hemingway J, Shretta R, Wells TNC, Bell D, Djimde AA, Achee N, Qi G. 2016. Tools and strategies for malaria control and elimination: what do we need to achieve a grand convergence in malaria? PLoS Biol 14:e1002380. doi: 10.1371/journal.pbio.1002380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Deye GA, Gettayacamin M, Hansukjariya P, Im-Erbsin R, Sattabongkot J, Rothstein Y, Macareo L, Fracisco S, Bennett K, Magill AJ, Ohrt C. 2012. Use of a rhesus Plasmodium cynomolgi model to screen for anti-hypnozoite activity of pharmaceutical substances. Am J Trop Med Hyg 86:931–935. doi: 10.4269/ajtmh.2012.11-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Zeeman AM, der Wel AV, Kocken CH. 2013. Ex vivo culture of Plasmodium vivax and Plasmodium cynomolgi and in vitro of Plasmodium knowlesi blood stages. Methods Mol Biol 923:35–49. doi: 10.1007/978-1-62703-026-7_4. [DOI] [PubMed] [Google Scholar]
- 308.Dembele L, Gego A, Zeeman AM, Franetich JF, Silvie O, Rametti A, Le Grand R, Dereuddre-Bosquet N, Sauerwein R, van Gemert GJ, Vaillant JC, Thomas AW, Snounou G, Kocken CH, Mazier DH. 2011. Towards an in vitro model of Plasmodium hypnozoites suitable for drug discovery. PLoS One 6:e18162. doi: 10.1371/journal.pone.0018162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Roth A, Maher SP, Conway AJ, Ubalee R, Chaumeau V, Andolina C, Kaba SA, Vantaux A, Makowski MA, Thomson-Luque R, Adapa SR, Singh N, Barnes SJ, Cooper CA, Rouillier M, McNamara CW, Mikolajczak SA, Sather N, Witkowski B, Campo B, Kappe SHI, Lanar DE, Nosten F, Davidson S, Jiang RHY, Kyle DE, Adams JH. 2018. A comprehensive model for assessment of liver stage therapies targeting Plasmodium vivax and Plasmodium falciparum. Nat Commun 9:1837. doi: 10.1038/s41467-018-04221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Dembélé L, Franetich J-F, Lorthiois A, Gego A, Zeeman A-M, Kocken CHM, Le Grand R, Dereuddre-Bosquet N, van Gemert G-J, Sauerwein R, Vaillant J-C, Hannoun L, Fuchter MJ, Diagana TT, Malmquist NA, Scherf A, Snounou G, Mazier D. 2014. Persistence and activation of malaria hypnozoites in long-term primary hepatocyte cultures. Nat Med 20:307–312. doi: 10.1038/nm.3461. [DOI] [PubMed] [Google Scholar]
- 311.Cubi R, Vembar SS, Biton A, Franetich J-F, Bordessoulles M, Sossau D, Zanghi G, Bosson-Vanga H, Benard M, Moreno A, Dereuddre-Bosquet N, Le Grand R, Scherf A, Mazier D. 2017. Laser capture microdissection enables transcriptomic analysis of dividing and quiescent liver stages of Plasmodium relapsing species. Cell Microbiol 19:e12735. doi: 10.1111/cmi.12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Chattopadhyay R, Velmurugan S, Chakiath C, Andrews Donkor L, Milhous W, Barnwell JW, Collins WE, Hoffman SL. 2010. Establishment of an in vitro assay for assessing the effects of drugs on the liver stages of Plasmodium vivax malaria. PLoS One 5:e14275. doi: 10.1371/journal.pone.0014275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Pewkliang Y, Rungin S, Lerdpanyangam K, Duangmanee A, Kanjanasirirat P, Suthivanich P, Sa-Ngiamsuntorn K, Borornpinyo S, Sattabongkot J, Patrapuvich R, Hongeng S. 2018. A novel immortalized hepatocyte-like cell line (imHC) support in vitro liver stage development of the human malaria parasite Plasmodium vivax. Malar J 17:50. doi: 10.1186/s12936-018-2198-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Schafer C, Dambrauskas N, Steel RW, Carbonetti S, Cheunchob V, Flannery EL, Vigdorovich V, Oliver BG, Roobsoon W, Maher SP, Sattabongkaot J, Kappe SHI, Mikolajczak SA, Sather DN. 2018. A recombinant antibody against Plasmodium vivax UIS4 for distinguishing replicating from dormant liver stages. Malar J 17:370. doi: 10.1186/s12936-018-2519-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Moreno-Sabater A, Perignon JL, Mazier D, Lavazec C, Soulard V. 2018. Humanized mouse models infected with human Plasmodium species for antimalarial drug discovery. Expert Opin Drug Discov 13:131–140. doi: 10.1080/17460441.2018.1410136. [DOI] [PubMed] [Google Scholar]
- 316.Saito T, Gutierrez Rico EM, Kikuchi A, Kaneko A, Kumondai M, Akai F, Saigusa D, Oda A, Hirasawa N, Hiratsuka M. 2018. Functional characterization of 50 CYP2D6 allelic variants by assessing primaquine 5-hydroxylation. Drug Metab Pharmacokinet 33:250–257. doi: 10.1016/j.dmpk.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 317.Jin X, Potter B, Luong TL, Nelson J, Vuong C, Potter C, Xie L, Zhang J, Zhang P, Sousa J, Li Q, Pybus BS, Kreishman-Deitrick M, Hickman M, Smith PL, Paris R, Reichard G, Marcsisin SR. 2016. Pre-clinical evaluation of CYP 2D6 dependent drug-drug interactions between primaquine and SSRI/SNRI antidepressants. Malar J 15:280. doi: 10.1186/s12936-016-1329-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Ganesan S, Tekwani BL, Sahu R, Tripathi LM, Walker LA. 2009. Cytochrome P-450-dependent toxic effects of primaquine on human erythrocytes. Toxicol Appl Pharmacol 24:14–22. doi: 10.1016/j.taap.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 319.Jin X, Pybus BS, Marcsisin R, Logan T, Luong TL, Sousa J, Matlock N, Collazo V, Asher C, Carroll D, Olmeda R, Walker LA, Kozar MP, Melendez V. 2014. An LC-MS based study of the metabolic profile of primaquine, an 8-aminoquinoline antiparasitic drug with an in vitro primary human hepatocyte culture model. Eur J Drug Metab Pharmacokinet 39:139–146. doi: 10.1007/s13318-013-0139-8. [DOI] [PubMed] [Google Scholar]
- 320.Harvey JW, Keitt AS. 1983. Studies of the efficacy and potential hazards of methylene blue therapy in analine-induced methemoglobinemia. Br J Haematol 54:29–41. doi: 10.1111/j.1365-2141.1983.tb02064.x. [DOI] [PubMed] [Google Scholar]
- 321.Karadsheh KS, Shaker Q, Ratroat B. 2001. Metoclopramide-induced methemoglobinemia in a patient with co-existing deficiency of glucose-6-phosphate dehydrogenase and NADH-cytochrome b5 reductase: failure of methylene blue treatment. Haematologica 86:659–660. [PubMed] [Google Scholar]
- 322.Foltz LM, Dalal BI, Wadsworth LD, Broady R, Chi K, Eisenhauer E, Kobayashi K, Kollmannsburger C. 2006. Recognition and management of methemoglobinemia and hemolysis in a G6PD-deficient patient on experimental anticancer drug triapine. Am J Hematol 81:210–211. doi: 10.1002/ajh.20547. [DOI] [PubMed] [Google Scholar]
- 323.Mullick P, Kumar A, Dayal JM, Babbar S, Kumar A. 2007. Aniline-induced methaemoglobinaemia in a glucose-6-phosphate dehydrogenase enzyme deficient patient. Anaesth Intensive Care 35:286–288. doi: 10.1177/0310057X0703500222. [DOI] [PubMed] [Google Scholar]
- 324.McDonagh EM, Bautista JM, Youngster I, Altman RB, Klein TE. 2013. PharmGKB summary: methylene blue pathway. Pharmacogenet Genomics 23:498–508. doi: 10.1097/FPC.0b013e32836498f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Müller O, Mockenhaupt FP, Marks B, Meissner P, Coulibaly B, Kuhnert R, Buchner H, Schirmer RH, Walter-Sack I, Sié A, Mansmann U. 2013. Haemolysis risk in methylene blue treatment of G6PD-sufficient and G6PD-deficient West African children with uncomplicated falciparum malaria: a synopsis of four RCTs. Pharmacoepidemiol Drug Saf 22:376–385. doi: 10.1002/pds.3370. [DOI] [PubMed] [Google Scholar]
- 326.Mandi G, Witte S, Meissner P, Coulibaly B, Mansmann U, Rengelshausen J, Schiek W, Jahn A, Sanon M, Wüst K, Walter-Sack I, Mikus G, Burhenne J, Riedel K-D, Schirmer H, Kouyaté B, Müller O. 2005. Safety of the combination of chloroquine and methylene blue in healthy adult men with G6PD deficiency from Burkina Faso. Trop Med Int Health 10:32–38. doi: 10.1111/j.1365-3156.2004.01356.x. [DOI] [PubMed] [Google Scholar]
- 327.Lu G, Nagbanshi M, Goldau N, Jorge MM, Meissner P, Jahn A, Mockenhaupt FP, Muller O. 2018. Efficacy and safety of methylene blue in the treatment of malaria: a systematic review. BMC Med 16:59. doi: 10.1186/s12916-018-1045-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Pluta K, Jeleń M, Morak-Młodawska B, Zimecki M, Artym J, Kocięba M, Zaczyńska E. 2017. Azaphenothiazines—promising phenothiazine derivatives. An insight into nomenclature, synthesis, structure elucidation and biological properties. Eur J Med Chem 138:774–806. doi: 10.1016/j.ejmech.2017.07.009. [DOI] [PubMed] [Google Scholar]
- 329.Wen S, Harvard KE, Gueye CS, Canavati SE, Chancellor A, Ahmed BN, Leburi J, Lek D, Namgay R, Surya A, Thakur GD, Whittaker MA, Gosling RD. 2016. Targeting populations at higher risk of malaria: a survey of national malaria elimination programmes in the Asia Pacific. Malar J 15:271. doi: 10.1186/s12936-016-1319-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Mercado CE, Ekapirat N, Dondorp AM, Maude RJ. 2017. An assessment of national surveillance systems for malaria elimination in the Asia Pacific. Malar J 16:127. doi: 10.1186/s12936-017-1774-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Baird JK. 2017. Asia-Pacific malaria is singular, pervasive, diverse, and invisible. Int J Parasitol 47:371–377. doi: 10.1016/j.ijpara.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Dellicour S, Tatem AJ, Guerra CA, Snow RW, ter Kuile FO. 2010. Quantifying the number of pregnancies at risk of malaria in 2007: a demographic study. PLoS Med 7:e1000221. doi: 10.1371/journal.pmed.1000221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Baird JK, Battle KE, Howes RE. 2018. Primaquine ineligibility in anti-relapse therapy of Plasmodium vivax malaria: the problem of G6PD deficiency and cytochrome P-450 2D6 polymorphisms. Malar J 17:42. doi: 10.1186/s12936-018-2190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Watson J, Taylor WRJ, Bancone G, Chu CS, Jittamala P, White NJ. 2018. Implications of current therapeutic restrictions for primaquine and tafenoquine in the radical cure of vivax malaria. PLoS Negl Trop Dis 12:e0006440. doi: 10.1371/journal.pntd.0006440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Rijken MJ, McGready R, Boel ME, Poespoprodjo R, Singh N, Syafruddin D, Rogerson S, Nosten F. 2012. Malaria in pregnancy in the Asia-Pacific region. Lancet Infect Dis 12:75–88. doi: 10.1016/S1473-3099(11)70315-2. [DOI] [PubMed] [Google Scholar]
- 336.Poespoprodjo JR, Fobia W, Kenangalem E, Lampah DA, Warikar N, Seal A, McGready R, Sugiarto P, Tjitra E, Anstey NM, Price RN. 2008. Adverse pregnancy outcomes in an area where multidrug-resistant Plasmodium vivax and Plasmodium falciparum infections are endemic. Clin Infect Dis 46:1374–1381. doi: 10.1086/586743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Kenangalem E, Karyana M, Burdarm L, Yeung S, Simpson JA, Tjitra E, Anstey NM, Poespoprodjo JR, Price RN, Douglas NM. 2016. Plasmodium vivax infection: a major determinant of severe anaemia in infancy. Malar J 15:321. doi: 10.1186/s12936-016-1373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Moore KA, Simpson JA, Scoullar MJL, McGready R, Fowkes F. 2017. Quantification of the association between malaria in pregnancy and stillbirth: a systematic review and meta-analysis. Lancet Glob Health 5:e1101–e1112. doi: 10.1016/S2214-109X(17)30340-6. [DOI] [PubMed] [Google Scholar]
- 339.Dombrowski JG, de Souza RM, Silva NRM, Barateiro A, Epiphanio S, Gonçalves LA, Marinho CRF. 2018. Malaria during pregnancy and newborn outcome in an unstable transmission area in Brazil: a population-based record linkage study. PLoS One 13:e0199415. doi: 10.1371/journal.pone.0199415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.McGready R, Lee SJ, Wiladphaingern J, Ashley EA, Rijken MJ, Boel M, Simpson JA, Paw MK, Pimpanpanarak M, Mu O, Singhasivanon P, White NJ, Nosten FH. 2012. Adverse effects of falciparum and vivax malaria and the safety of antimalarial treatment in early pregnancy: a population-based study. Lancet Infect Dis 12:388–396. doi: 10.1016/S1473-3099(11)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Weathersby AB. 1973. Clay G. Huff: with great esteem. Exp Parasitol 34:iii–xiv. [PubMed] [Google Scholar]
- 342.Taylor WRJ, Thriemer K, von Seidlein L, Yuentrakul P, Assawariyathipat T, Assefa A, Auburn S, Chand K, Chau NH, Cheah PY, Dong LT, Dhorda M, Degaga TS, Devine A, Ekawati LL, Fahmi F, Hailu A, Hasanzai MA, Hien TT, Khu H, Ley B, Lubell Y, Marfurt J, Mohammad H, Moore KA, Naddim MN, Pasaribu AP, Pasaribu S, Promnarate C, Rahim AG, Sirithiranont P, Solomon H, Sudoyo H, Sutanto I, Thanh NV, Tuyet-Trinh NT, Waithira N, Woyessa A, Yamin FY, Dondorp A, Simpson JA, Baird JK, White NJ, Day NP, Price RN. 18 July 2019. Short-course primaquine for the radical cure of Plasmodium vivax malaria: a multicentre, randomised, placebo-controlled non-inferiority trial. Lancet doi: 10.1016/S0140-6736(19)31285-1. [DOI] [PMC free article] [PubMed] [Google Scholar]