

Abstract
The prevalence of obesity has steadily risen over the past decades, even doubling in more than 70 countries. High levels of body fat (adiposity) and obesity are associated with endocrine and hormonal dysregulation, cardiovascular compromise, hepatic dysfunction, pancreatitis, changes in drug metabolism and clearance, inflammation, and metabolic stress. It is thus unsurprising that obesity can affect the development of and survival from a wide variety of malignancies. This review focuses on acute lymphoblastic leukemia, the most common malignancy in children, to explore the multiple mechanisms connecting acute lymphoblastic leukemia, obesity, and adipocytes, and the implications for leukemia therapy.
The prevalence of obesity has steadily risen over the past decades (1). Obesity is associated with cancer mortality (2) and the development of all major leukemia subtypes in adults (3). Although childhood obesity has not been linked to acute lymphoblastic leukemia (ALL) incidence, pediatric obesity has been shown in multiple cohorts to have an adverse impact on ALL relapse and survival (4). A major question derived from these cohort studies was whether adipocytes (fat cells), independent of confounding environmental or socioeconomic factors, cause poorer survival. To test this, we developed mouse models of ALL and obesity and showed that obesity directly influences the development and cure of ALL (5–7). Given that up to one in three pediatric patients with newly diagnosed ALL are overweight or obese (8), understanding and breaking the obesity–ALL connection could yield significant improvements in survival.
Obesity-Associated Hormones and Leukemia
Systemic Signals
Obesity is associated with a number of complex physiological changes, many of which remain poorly understood. Among them, high insulin and insulin-like growth factor-1 (IGF-1) levels likely contribute to ALL incidence and outcome. Although increased insulin and free IGF-1 have not been directly associated with ALL survival, a large body of in vitro and ex vivo data strongly suggest they may be key mediators for ALL outcomes.
Insulin . Hyperinsulinemia associated with obesity-induced insulin resistance is likely a major mechanistic link between obesity and cancer. Insulin increases glucose uptake, protein synthesis, cell proliferation, and tissue growth. Insulin resistance impairs the effects of insulin on glucose metabolism, and the growth-promoting effects of insulin are even enhanced in the compensatory hyperinsulinemic state.
Insulin signaling through its receptor initiates a complex set of signaling cascades, including activation of phosphoinositide 3-kinase and protein kinase B (PI3K/AKT), mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription (STAT), and extracellular signal-regulated kinase (ERK). These pathways are often dysregulated in ALL, with PI3K/AKT and STAT3 overexpression associated with poorer survival and chemotherapy resistance in pediatric B-precursor ALL (B-ALL) (9–11). Even more concerning, insulin promotes in vitro ALL resistance to daunorubicin, a common ALL chemotherapy agent, but not to several other chemotherapies (12). These implicated pathways and resistance patterns indicate a potential agent- and/or regimen-specific impact of hyperinsulinemia on outcome from ALL.
IGF-1. IGF-1 may be a critical link between obesity and cancer in general and has been hypothesized to link fetal size and developing childhood ALL (13). Obese patients often have low or normal total concentrations but high free (active) IGF-1 because of lower levels of IGF binding proteins. The IGF-1 receptor (IGF1R) shares homology with insulin receptor, stimulating overlapping pathways including PI3K/AKT. IGF1R stimulation results in ALL proliferation and chemotherapy resistance (14,15), whereas its inhibition slows proliferation and enhances apoptosis (16). The micro-RNAs miR-99a and miR-100, which act in part by inhibiting IGF1R expression, are lower in ALL cells, and their restoration impairs ALL proliferation (15). Finally, IGF1R overexpression relieves stromal dependence of ALL cells (17).
Adipose Signals
One of the most obvious characteristics of obesity is excess body fat. Adipocytes secrete a large number of cytokines and hormones (collectively termed adipokines), which affect a myriad of paracrine and endocrine targets.
CXCL12 . We observed in a mouse model of obesity and ALL that, after chemotherapy, ALL cells had migrated into adipose tissues (Figure 1) (5). We showed that this was mediated via adipocytes secretion of CXCL12 (also known as SDF1α), an important factor in lymphocyte migration (18). Importantly, this migration was prevented by blocking the CXCL12 receptor, CXCR4. As described below, this intercalation of ALL cells and adipocytes in bone marrow and adipose tissue microenvironments could result in a distinct, chemoprotective niche from the metabolic protection described below.
Figure 1.
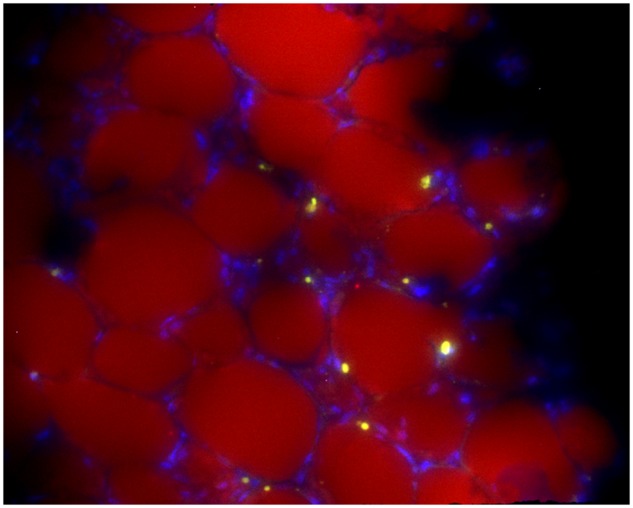
GFP+ 8093 acute lymphoblastic leukemia cells in the perirenal fat pad of a transplanted obese mouse that developed progressive leukemia during vincristine treatment. Lipid is stained with Nile Red, and the image is counterstained with 4', 6-diamidino-2-phenylindole. Image was taken on a Leica DM RXA2 with a x20 objective with x1.6 Optovar magnification (x32 final). Image is representative of fat pads obtained from the other four mice after vincristine treatment. Reprinted with permission from (5).
Leptin . Leptin is an adipokine secreted in proportion to obesity and adiposity (19,20). Whereas leptin in the bone marrow enhances ALL cell engraftment (21), higher leptin receptor expression on ALL blasts is associated with improved prognosis in patients (22,23). Further, fasting induces leptin receptor expression, which stimulates ALL cell differentiation and inhibits initiation and progression (24). Therefore, although elevated leptin levels in obesity may promote ALL initiation, they are unlikely to directly worsen ALL survival.
Adiponectin . Adiponectin is one of the most abundant systemic hormones (25). Unlike most adipokines, adiponectin circulates in inverse proportion to obesity and generally has beneficial metabolic effects. Adiponectin simulates the AMP-activated protein kinase (AMPK) pathway, which in ALL cells would be expected to promote apoptosis (26). In vitro, adiponectin does not influence proliferation of lymphoid precursors in vitro (23), and thus does not appear to play a major role in ALL chemoresistance.
Visfatin . Visfatin, also known as nicotinamide phosphoribosyltransferase and pre‐B cell colony‐enhancing factor, is secreted by adipocytes and elevated in obesity. However, its intracellular form, which has enzymatic activity, is likely more relevant to cancer cells. So, although visfatin expression is upregulated in many cancer cells, including ALL, and its inhibition is cytotoxic (27), it is unlikely to mediate obesity and ALL survival.
The adipocyte secretome therefore has multiple points of interaction with ALL, yet no clear direct influence on ALL cell survival. However, adipocytes also secrete a myriad of other hormones and cytokines (eg, adipsin, RBP4, FGF21, resistin, omentin), most of which have yet to be explored in ALL. Leptin and other adipokines also contribute to obesity-associated insulin resistance and inflammation, which themselves can influence ALL outcomes. The secretome may therefore play an indirect role either as a mediator or indicator of the systemic impact of obesity.
Providing Metabolic “Fuel” for Leukemia Cells
Obesity is characterized by excess fuel storage. The obese, insulin-resistant state, particularly when it is not completely compensated by hyperinsulinemia, is associated with increased levels of circulating glucose, free fatty acids, and certain amino acids (28,29). In the adipose tissue microenvironment, interstitial levels of adipocyte-released fuels are likely extremely high.
Glucose
Many cancers, including ALL, exhibit increased aerobic glycolysis, termed the Warburg effect (30). This serves to support both energy production and macromolecule synthesis in rapidly dividing cells. Targeting glucose uptake or glycolysis in cancer cells, as has been done in BCR/ABL+ ALL cells, can reduce proliferation, impair nucleotide synthesis, and increase sensitivity to chemotherapy (31). A recent study showed that acute myeloid leukemia cells reprogram cells in their microenvironment, impairing their glucose uptake and resulting in increased available glucose for the cancer cells (32). Additionally, clinical studies revealed that hyperglycemia during induction was associated with decreased duration of complete remission in adult patients with ALL (33), although this association was not detected in children (34). Thus, it seems likely that higher glucose levels seen in obesity could contribute to ALL cell proliferation and chemotherapy resistance and thus help explain the links between obesity and poorer ALL outcome.
Free Fatty Acids (FFA)
Arguably, the primary role of adipocytes is the storage and provision of lipids. In the obese state, adipocytes are insulin resistant, resulting in higher circulating triglycerides and FFA. Many cancers rely heavily on FFA as both a source of energy (through beta-oxidation) and a precursor for synthesis of macromolecules, such as phospholipids. Cancers often overexpress enzymes involved in lipogenesis, such as fatty acid synthase and acetyl-coenzyme A carboxylase (35,36), and lipid uptake, including lipoprotein lipase (37), fatty acid binding protein 4, and CD36 (38). Inhibition of CD36 and fatty acid binding protein 4 can impair cancer proliferation and induce apoptosis (35,39). Further, some cancers induce nearby adipocyte release of FFA (40,41), an effect we have observed in ALL cells (42). In fact, we found that ALL cells take up adipocyte-derived FFA and incorporate them into phospholipid membranes and lipid droplets. The direct connection between adipocyte provision of FFA to ALL cells as a source of energy and chemotherapy resistance and proliferation is currently being investigated.
Amino Acids
Glutamine (GLN) is an important fuel and macromolecule precursor for most cancers, including ALL. Generally lacking asparagine (ASN) synthetase, ALL cells depend on uptake of extracellular ASN. Asparaginase treatment exploits these needs by hydrolyzing ASN and to a lesser extent, GLN, reducing their availability in the ALL microenvironment. Adipocytes release both ASN and GLN into the local microenvironment (6,43), thereby potentially fueling ALL cells and circumventing asparaginase activity.
Branched chain amino acids (BCAA), including leucine, isoleucine, and valine, are important for protein synthesis and energy and as signaling molecules in cancer cells. Circulating BCAA levels are increased in obesity, insulin resistance, and type 2 diabetes (44), although the reasons for this are not clear. Importantly, exogenous BCAA partially reduces reliance on endogenous BCAA synthesis, although BCAA metabolism has not yet been studied in ALL. Thus, BCAA, ASN, and GLN all may contribute to enhanced ALL cell metabolic health and chemotherapy resistance in obesity.
Inflammation, Obesity, and Cancer
Obesity is associated with chronic inflammation, a well-known contributor to solid tumor oncogenesis (45). Adipocytes secrete pro-inflammatory cytokines such as IGF-1, interleukin (IL)-1β, IL-6, IL-7, and tumor necrosis factor α (TNFα) (46), as well as FFA. Obese adipose tissue accumulates immune cells and induces their polarization toward a pro-inflammatory phenotype, further promoting systemic inflammation. Inflammatory cytokines activate traditional leukemia signal transduction cascades, primarily PI3K/AKT, MAPK, and STAT3 pathways (47). IL-7, which is increased in morbidly obese subjects, is associated with T-cell ALL (T-ALL) oncogenesis (48) and glucocorticoid chemotherapy resistance (49). Obesity-associated inflammation and its downstream signaling overlaps with obesity-induced systemic triggers, all of which may contribute to poorer survival for obesity-associated ALL.
Pharmacokinetics and Pharmacodynamics (PK/PD) in the Obese
Several physical and physiological changes in obesity may affect the PK of drugs. Increased adiposity can alter a drug’s volume of distribution in either direction, depending on its lipophilicity. Alterations in volume of distribution can affect a drug’s peak concentration and area-under-the-curve (AUC), both of which can impair efficacy or increase toxicity, depending on the direction of change. Obesity-associated alterations in gastrointestinal, hepatic, and renal function can further affect chemotherapy PK/PD. Chemotherapy dosing is routinely based on body surface area, which increases disproportionately slower with increasing weight and body fat mass. Extensive PK modeling has not been done for most chemotherapies in obese subjects, and there is almost no data in extreme obesity. In 2012, the American Society of Clinical Oncology recommended that “full weight-based cytotoxic chemotherapy doses be used to treat obese patients with cancer” (50). Limited PK data from the St Jude Total trials showed no influence of body mass index (BMI) on the PK of many common ALL chemotherapies (methotrexate, cytarabine, mercaptopurine) (51). However, two core chemotherapy classes integral to ALL treatment were not assessed and are discussed below. For a more extensive review of the literature on obesity and chemotherapy PK/PD, we refer the reader to (52).
Vincristine (VCR)
Vincristine is part of the backbone of ALL therapy. For yet unknown reasons, a high degree of variability is observed in the PK of VCR among individuals (53). Across ALL regimens, VCR dosing is capped at 2 mg, such that patients larger than an average 8-year-old (∼1 m2) receive a capped dose. We found that diet-induced obesity in mice altered the PK of VCR, leading to a lower overall AUC of the drug in blood over time, a faster clearance from spleen and bone marrow, and accumulation in adipose tissue (54). This is particularly worrisome, as decreased VCR AUC has been linked with higher relapse risk in children with ALL (55). In adolescents, one study reported no impact of increased size on VCR PK (56). Better understanding of body fat’s contribution to VCR PK is necessary to optimize dosing across ages and in the obese.
Anthracyclines
Daunorubicin and doxorubicin are commonly used to treat ALL. PK studies showed no effect of BMI or body fat on the plasma clearance of either drug in children (57,58), whereas obese adults were found to have lower systemic clearance and higher AUC of doxorubicin (59). In contrast, we recently reported that adipocytes absorb and metabolize daunorubicin to its largely inactive form, daunorubicinol (Figure 2) (60). We found human adipocytes express high levels of the aldo-keto reductase and carbonyl reductase enzymes responsible for this inactivation, leading to a reduced concentration of active daunorubicin within the nearby environment. Although this may not alter the plasma PK of daunorubicin (and potentially other anthracyclines), it may result in subtherapeutic concentrations in the leukemia-adipose marrow microenvironment. Further work is ongoing to evaluate this effect in patients.
Figure 2.
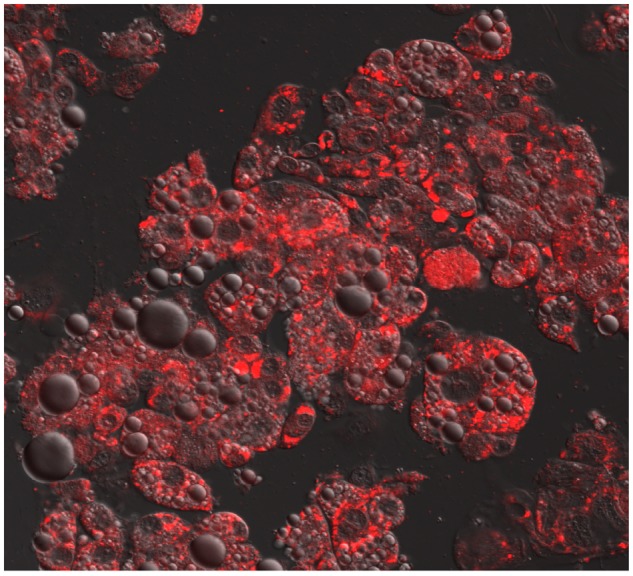
Confocal microscopy of adipocytes treated with 60 nM daunorubicin (red) for 4 hours. Photo taken on a LSM 700 confocal system mounted on an AxioObserver.Z1 microscope equipped with a 63/1.4 Plan-APOCHROMAT objective lens and controlled with ZEN 2009 software (Carl Zeiss Microscopy). Reprinted with permission from (60).
Breaking the Link Between Obesity and Leukemia Outcome
Although the evidence for obesity to worsen ALL outcomes is strong, clinical and preclinical data provide a glimmer of hope that this association may be reversible. An improved understanding of the obesity-ALL biology could provide new opportunities to develop nonchemotherapy integrative approaches to augment therapy and reduce risk for relapse.
Targeted Therapy for Obesity-Associated ALL
Multiple commercially available biologics are available to target mTOR and the other obesity-associated leukemia signaling pathways described above. Metformin is one of the best characterized and stimulates AMPK, a potent mTOR inhibitor. Metformin has pro-apoptotic and antiproliferative activity in leukemia cell lines (61,62). When tested with traditional chemotherapy, in vitro synergy was observed for some T-ALL cell lines, representing a variety of leukemia phenotypes (63). Metformin had promising results in a phase 1 study of relapsed pediatric ALL patients (64) and is currently being tested by the Children’s Oncology Group to target constitutive Janus kinase (JAK)/STAT activation due to cytokine receptor-like factor 2 (CRLF2) fusions (NCT02723994). If successful, a similar approach could be envisioned for ALL with obesity-mediated pathway activation. Alternatively, future targeted therapy may instead focus on obesity’s adverse influence on metabolism of traditional chemotherapy, such as incorporating drugs targeting enzyme-mediated sequestration and deactivation of anthracyclines. Although our understanding of the biology of obesity-associated leukemia remains incomplete, data offer promising glimpses for future augmentation of chemotherapy via pathway inhibition.
Metabolic Manipulation of ALL
Adipocytes directly and obesity indirectly enhance fuel availability to rapidly proliferating leukemia cells. Restricting fuels, such as with a low-glycemic index or calorie-restricting diets, could prove efficacious, particularly in obese patients, although data remain limited. Glucose restriction was as effective as the drug rapamycin in vitro for inhibiting mTOR and inducing apoptosis in multiple cell lines (65). Serum restriction, which simulates the hormone and growth factor changes of calorie restriction, can also sensitize ALL cells to chemotherapy (66). In mouse models, intermittent fasting completely halted the engraftment and progression of B-ALL and markedly reduced both for T-ALL (24). We have shown that switching obese mice from a high-fat to a low-fat diet when starting chemotherapy substantially improved ALL outcome with vincristine, although not with L-asparaginase or dexamethasone (66). Identifying the most advantageous interventions, and how best to incorporate these into standard chemotherapy regimens remain important goals.
Based on the above, we launched a prospective obesity-intervention trial during ALL induction (NCT02708108). In this trial, we are investigating a metabolic intervention consisting of a 10% dietary calorie restriction and a 10% increase in energy expenditure through exercise. The intervention takes place during the first month of induction chemotherapy, with the goals of reducing body fat accretion and decreasing the incidence of minimal residual disease. To our knowledge, this is the first trial in pediatric ALL to investigate the use of metabolic modification as an integrated treatment modality to increase chemotherapy efficacy.
The evidence that obesity impairs ALL outcome is strong, and our understanding of the mechanisms responsible for this association is growing. In the era of immune therapy and biologics for treatment of childhood leukemia, metabolic therapy offers a potential avenue with few side effects for augmenting chemotherapy to reduce relapse and improve survival.
Notes
Affiliations of authors: Children’s Center for Cancer and Blood Diseases, Children’s Hospital Los Angeles, Los Angeles, CA Department of Pediatrics, Keck School of Medicine, University of Southern California (EO); Division of Pediatric Endocrinology, UCLA Children’s Discovery and Innovation Institute, David Geffen School of Medicine UCLA, Los Angeles, CA (JLS, SDM).
The authors declare no conflicts of interest. This work was supported by Gabrielle’s Angel Foundation No. 95 (EO), and the National Cancer Institute CA201444 and CA213129 (SDM).
References
- 1. G. B. D. Obesity Collaborators. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;3771:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Calle EE, Rodriguez C, Walker-Thurmond K, et al. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;34817:1625–1638. [DOI] [PubMed] [Google Scholar]
- 3. Larsson SC, Wolk A.. Overweight and obesity and incidence of leukemia: a meta-analysis of cohort studies. Int J Cancer. 2008;1226:1418–1421. [DOI] [PubMed] [Google Scholar]
- 4. Orgel E, Genkinger JM, Aggarwal D, et al. Association of body mass index and survival in pediatric leukemia: a meta-analysis. Am J Clin Nutr. 2016;1033:808–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Behan JW, Yun JP, Proektor MP, et al. Adipocytes impair leukemia treatment in mice. Cancer Res. 2009;6919:7867–7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehsanipour EA, Sheng X, Behan JW, et al. Adipocytes cause leukemia cell resistance to L-Asparaginase via release of glutamine. Cancer Res. 2013;7310:2998–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yun JP, Behan JW, Heisterkamp N, et al. Diet-induced obesity accelerates acute lymphoblastic leukemia progression in two murine models. Cancer Prev Res (Phila PA). 2010;310:1259–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Withycombe JS, Smith LM, Meza JL, et al. Weight change during childhood acute lymphoblastic leukemia induction therapy predicts obesity: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2015;623:434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morishita N, Tsukahara H, Chayama K, et al. Activation of AKT is associated with poor prognosis and chemotherapeutic resistance in pediatric B-precursor acute lymphoblastic leukemia. Pediatr Blood Cancer. 2012;591:83–89. [DOI] [PubMed] [Google Scholar]
- 10. Steelman LS, Abrams SL, Whelan J, et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/AKT/mTOR and JAK/STAT pathways to leukemia. Leukemia. 2008;224:686–707. [DOI] [PubMed] [Google Scholar]
- 11. Chen IM, Harvey RC, Mullighan CG, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. 2012;11915:3512–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pan J, Chen C, Jin Y, et al. Differential impact of structurally different anti-diabetic drugs on proliferation and chemosensitivity of acute lymphoblastic leukemia cells. Cell Cycle. 2012;1112:2314–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Callan AC, Milne E.. Involvement of the IGF system in fetal growth and childhood cancer: an overview of potential mechanisms. Cancer Causes Control. 2009;2010:1783–1798. [DOI] [PubMed] [Google Scholar]
- 14. Gusscott S, Jenkins CE, Lam SH, et al. IGF1R derived PI3K/AKT signaling maintains growth in a subset of human T-cell acute lymphoblastic leukemias. PLoS One. 2016;118:e0161158.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li XJ, Luo XQ, Han BW, et al. MicroRNA-100/99a, deregulated in acute lymphoblastic leukaemia, suppress proliferation and promote apoptosis by regulating the FKBP51 and IGF1R/mTOR signalling pathways. Br J Cancer. 2013;1098:2189–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Medyouf H, Gusscott S, Wang H, et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J Exp Med. 2011;2089:1809–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McCubrey JA, Steelman LS, Mayo MW, et al. Growth-promoting effects of insulin-like growth factor-1 (IGF-1) on hematopoietic cells: overexpression of introduced IGF-1 receptor abrogates interleukin-3 dependency of murine factor-dependent cells by a ligand-dependent mechanism. Blood. 1991;784:921–929. [PubMed] [Google Scholar]
- 18. Pramanik R, Sheng X, Ichihara B, et al. Adipose tissue attracts and protects acute lymphoblastic leukemia cells from chemotherapy. Leuk Res. 2013;375:503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;3345:292–295. [DOI] [PubMed] [Google Scholar]
- 20. Frederich RC, Hamann A, Anderson S, et al. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;112:1311–1314. [DOI] [PubMed] [Google Scholar]
- 21. Battula VL, Chen Y, Cabreira Mda G, et al. Connective tissue growth factor regulates adipocyte differentiation of mesenchymal stromal cells and facilitates leukemia bone marrow engraftment. Blood. 2013;1223:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hino M, Nakao T, Yamane T, et al. Leptin receptor and leukemia. Leuk Lymphoma. 2000;36(5-6):457–461. [DOI] [PubMed] [Google Scholar]
- 23. Yokota T, Oritani K, Takahashi I, et al. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;965:1723–1732. [PubMed] [Google Scholar]
- 24. Lu Z, Xie J, Wu G, et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat Med. 2017;231:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang ZV, Scherer PE.. Adiponectin, the past two decades. J Mol Cell Biol. 2016;82:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leclerc GM, Leclerc GJ, Kuznetsov JN, et al. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS One. 2013;88:e74420.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takao S, Chien W, Madan V, et al. Targeting the vulnerability to NAD(+) depletion in B-cell acute lymphoblastic leukemia. Leukemia. 2018;323:616–625. [DOI] [PubMed] [Google Scholar]
- 28. Giesbertz P, Daniel H.. Branched-chain amino acids as biomarkers in diabetes. Curr Opin Clin Nutr Metab Care. 2016;191:48–54. [DOI] [PubMed] [Google Scholar]
- 29. Frohnert BI, Jacobs DR Jr, Steinberger J, et al. Relation between serum free fatty acids and adiposity, insulin resistance, and cardiovascular risk factors from adolescence to adulthood. Diabetes. 2013;629:3163–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boag JM, Beesley AH, Firth MJ, et al. Altered glucose metabolism in childhood pre-B acute lymphoblastic leukaemia. Leukemia. 2006;2010:1731–1737. [DOI] [PubMed] [Google Scholar]
- 31. Liu T, Kishton RJ, Macintyre AN, et al. Glucose transporter 1-mediated glucose uptake is limiting for B-cell acute lymphoblastic leukemia anabolic metabolism and resistance to apoptosis. Cell Death Dis. 2014;510:e1470.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ye H, Adane B, Khan N, et al. Subversion of systemic glucose metabolism as a mechanism to support the growth of leukemia cells. Cancer Cell. 2018;344:659–673 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weiser MA, Cabanillas ME, Konopleva M, et al. Relation between the duration of remission and hyperglycemia during induction chemotherapy for acute lymphocytic leukemia with a hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone/methotrexate-cytarabine regimen. Cancer. 2004;1006:1179–1185. [DOI] [PubMed] [Google Scholar]
- 34. Roberson JR, Spraker HL, Shelso J, et al. Clinical consequences of hyperglycemia during remission induction therapy for pediatric acute lymphoblastic leukemia. Leukemia. 2009;232:245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tabe Y, Yamamoto S, Saitoh K, et al. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. 2017;776:1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang C, Ma J, Zhang N, et al. The acetyl-CoA carboxylase enzyme: a target for cancer therapy? Expert Rev Anticancer Ther. 2015;156:667–676. [DOI] [PubMed] [Google Scholar]
- 37. van’t Veer MB, Brooijmans AM, Langerak AW, et al. The predictive value of lipoprotein lipase for survival in chronic lymphocytic leukemia. Haematologica. 2006;911:56–63. [PubMed] [Google Scholar]
- 38. Shafat MS, Oellerich T, Mohr S, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood. 2017;12910:1320–1332. [DOI] [PubMed] [Google Scholar]
- 39. Rozovski U, Harris DM, Li P, et al. STAT3-activated CD36 facilitates fatty acid uptake in chronic lymphocytic leukemia cells. Oncotarget. 2018;930:21268–21280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nieman KM, Kenny HA, Penicka CV, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;1711:1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Balaban S, Shearer RF, Lee LS, et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017;51:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tucci J, Sheng X, Mittelman SD.. Acute lymphoblastic leukemia cells stimulate adipocyte lipolysis and utilize adipocyte-derived free-fatty acids for proliferation. Cancer Research. 2014;74(19 Suppl):Abstract number 4339. [Google Scholar]
- 43. Frayn KN, Khan K, Coppack SW, et al. Amino acid metabolism in human subcutaneous adipose tissue in vivo. Clin Sci. 1991;805:471–474. [DOI] [PubMed] [Google Scholar]
- 44. Herman MA, She P, Peroni OD, et al. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem. 2010;28515:11348–11356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Coussens LM, Werb Z.. Inflammation and cancer. Nature. 2002;4206917:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Makki K, Froguel P, Wolowczuk I.. Adipose tissue in obesity-related inflammation and insulin resistance: Cells, cytokines, and chemokines. ISRN Inflamm. 2013;2013:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen J. Multiple signal pathways in obesity-associated cancer. Obes Rev. 2011;1212:1063–1070. [DOI] [PubMed] [Google Scholar]
- 48. Zenatti PP, Ribeiro D, Li W, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;4310:932–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Delgado-Martin C, Meyer LK, Huang BJ, et al. JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia. 2017;3112:2568–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Griggs JJ, Mangu PB, Anderson H, et al. Appropriate chemotherapy dosing for obese adult patients with cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2012;3013:1553–1561. [DOI] [PubMed] [Google Scholar]
- 51. Hijiya N, Panetta JC, Zhou Y, et al. Body mass index does not influence pharmacokinetics or outcome of treatment in children with acute lymphoblastic leukemia. Blood. 2006;10813:3997–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hall RG 2nd, Jean GW, Sigler M, et al. Dosing considerations for obese patients receiving cancer chemotherapeutic agents. Ann Pharmacother. 2013;4712:1666–1674. [DOI] [PubMed] [Google Scholar]
- 53. Groninger E, Meeuwsen-de BT, Koopmans P, et al. Pharmacokinetics of vincristine monotherapy in childhood acute lymphoblastic leukemia. Pediatr Res. 2002;521:113–118. [DOI] [PubMed] [Google Scholar]
- 54. Behan JW, Avramis VI, Yun JP, et al. Diet-induced obesity alters vincristine pharmacokinetics in blood and tissues of mice. Pharmacol Res. 2010;615:385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lönnerholm G, Frost B-M, Abrahamsson J, et al. Vincristine pharmacokinetics is related to clinical outcome in children with standard risk acute lymphoblastic leukemia. Br J Haematol. 2008;1424:616–621. [DOI] [PubMed] [Google Scholar]
- 56. Isenalumhe LL, Margol AS, Louie S, et al. Comparison of vincristine pharmacokinetics (PK) in adolescent/young adult (AYA) versus younger patients defined by tanner stage during treatment for acute lymphoblastic leukemia (ALL). Blood. 2015;12623:3725–3725. [Google Scholar]
- 57. Thompson P, Wheeler HE, Delaney SM, et al. Pharmacokinetics and pharmacogenomics of daunorubicin in children: a report from the Children’s Oncology Group. Cancer Chemother Pharmacol. 2014;744:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thompson PA, Rosner GL, Matthay KK, et al. Impact of body composition on pharmacokinetics of doxorubicin in children: a Glaser Pediatric Research Network study. Cancer Chemother Pharmacol. 2009;642:243–251. [DOI] [PubMed] [Google Scholar]
- 59. Rodvold KA, Rushing DA, Tewksbury DA.. Doxorubicin clearance in the obese. J Clin Oncol. 1988;68:1321–1327. [DOI] [PubMed] [Google Scholar]
- 60. Sheng X, Parmentier JH, Tucci J, et al. Adipocytes sequester and metabolize the chemotherapeutic daunorubicin. Mol Cancer Res. 2017;1512:1704–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Green AS, Chapuis N, Trovati Maciel T, et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood. 2010;11620:4262–4273. [DOI] [PubMed] [Google Scholar]
- 62. Grimaldi C, Chiarini F, Tabellini G, et al. AMP-dependent kinase/mammalian target of rapamycin complex 1 signaling in T-cell acute lymphoblastic leukemia: therapeutic implications. Leukemia. 2012;261:91–100. [DOI] [PubMed] [Google Scholar]
- 63. Rosilio C, Lounnas N, Nebout M, et al. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. 2013;3361:114–126. [DOI] [PubMed] [Google Scholar]
- 64. Trucco M, Barredo JC, Goldberg J, et al. A phase I window, dose escalating and safety trial of metformin in combination with induction chemotherapy in relapsed refractory acute lymphoblastic leukemia: metformin with induction chemotherapy of vincristine, dexamethasone, PEG-asparaginase, and doxorubicin. Pediatr Blood Cancer. 2018;659:e27224. [DOI] [PubMed] [Google Scholar]
- 65. Pradelli LA, Beneteau M, Chauvin C, et al. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene. 2010;2911:1641–1652. [DOI] [PubMed] [Google Scholar]
- 66. Tucci J, Alhushki W, Chen T, et al. Switch to low-fat diet improves outcome of acute lymphoblastic leukemia in obese mice. Cancer Metab. 2018;61:15.. [DOI] [PMC free article] [PubMed] [Google Scholar]