

SUMMARY
Intestinal epithelial cells are the first line of defense against enteric pathogens, yet bacterial pathogens, such as Listeria monocytogenes, can breach this barrier. We show that Listeria adhesion protein (LAP) induces intestinal epithelial barrier dysfunction to promote bacterial translocation. These disruptions are attributed to the production of pro-inflammatory cytokines TNF-α and IL-6, which is observed in mice challenged with WT and isogenic strains lacking the surface invasion protein Internalin A (ΔinlA), but not a lap− mutant. Additionally, upon engagement of its surface receptor Hsp60, LAP activates canonical NF-κB signaling, facilitating myosin light-chain kinase (MLCK)-mediated opening of the epithelial barrier via cellular redistribution of the epithelial junctional proteins claudin-1, occludin, and E-cadherin. Pharmacological inhibition of MLCK or NF-κB in cells or genetic ablation of MLCK in mice prevents mislocalization of junctional proteins and L. monocytogenes translocation. Thus, L. monocytogenes uses LAP to exploit epithelial defenses and cross the intestinal epithelial barrier.
In Brief
The pathogen Listeria monocytogenes crosses the intestinal barrier during infection. Drolia et al. show that L. monocytogenes employs Listeria adhesion protein (LAP) to exploit epithelial innate defenses and induce intestinal barrier dysfunction. LAP induces activation of NF-κB and MLCK, resulting in cellular redistribution of epithelial junction proteins and bacterial translocation.
Graphical Abstract
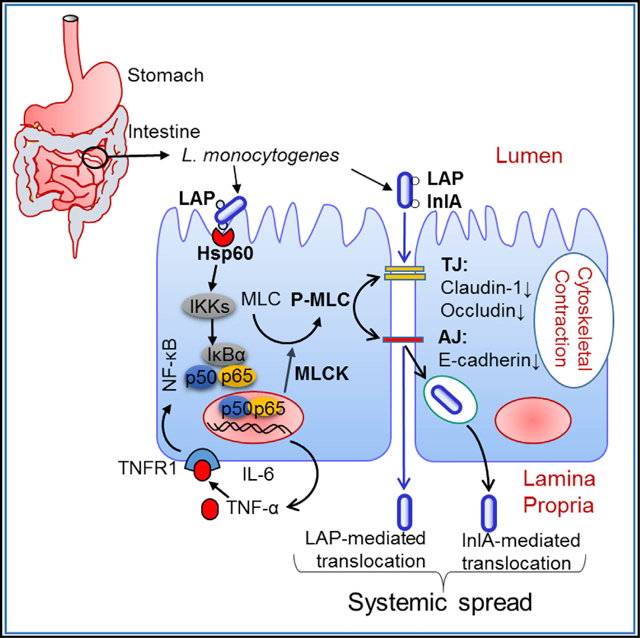
INTRODUCTION
The breaching of host barriers (intestinal, blood-brain, and placental) is a key mechanism of the intracellular bacterium Listeria monocytogenes during infection (Radoshevich and Cossart, 2018). The epithelial cell invasion of L. monocytogenes is mediated by two proteins, Internalin A (InlA) and Internalin B (InlB) (Dramsi et al., 1995; Lecuit et al., 2001). Following entry, the bacterium resides in a phagosome, which is lysed by listeriolysin O (LLO, encoded by the hly gene) to access the cytosol (Portnoy et al., 1988). The bacterium utilizes ActA protein to stimulate cellularactin polymerization via the recruitment of the Arp2/3 complex for cell-to-cell spread (Kocks et al., 1992; Tilney and Portnoy, 1989), which is also aided by Internalin C (InlC) (Rajabian et al., 2009).
The gastrointestinal tract is the primary route of infection for L. monocytogenes, and crossing the intestinal epithelial barrier is the first step. The importance of the InlA-mediated L. monocytogenes invasion and crossing of the intestinal epithelial barrier has been reviewed (Radoshevich and Cossart, 2018). The host cell receptor for InlA is E-cadherin (E-cad) (Mengaud et al., 1996). The InlA/E-cad interaction exhibits a species specificity that is attributed to a variation at amino acid sequence position 16, at which proline is substituted by glutamic acid in the host species’ E-cad (Lecuit et al., 1999). Therefore, InlA does not interact with the E-cad of the non-permissive hosts—mouse or rat—but it interacts with the E-cad of permissive hosts—humans, guinea pigs, rabbits, and gerbils (Disson et al., 2008; Lecuit et al., 2001). E-cad located at the adherens junction (AJ) is inaccessible to bacteria in the intestinal lumen. E-cad access is proposed to occur during villous epithelial ‘‘cell extrusion’’ (Pentecost et al., 2006), during which the apical junctional complex proteins are redistributed to the lateral membranes (Marchiando et al., 2011) and around mucus-expelling goblet cells (Nikitas et al., 2011). However, a ΔinlA mutant, after intra-gastric (ig) inoculation, showed high bacterial burdens in the liver and spleen of wild-type (WT) mice (Bierne et al., 2002) and in the small intestine, cecum, colon, and MLN of transgenic mice expressing “humanized” E-cad (Disson et al., 2008). This suggests that the humanized E-cad allele is only relevant to InlA-mediated bacterial invasion and that L. monocytogenes use alternate routes to translocate across the gut mucosa. Furthermore, ig inoculation of L. monocytogenes expressing murinized InlA (InlAm) with high affinity for E-cad did not show significantly higher bacterial burdens in the liver, spleen, MLN, and small intestine of mice compared to mice that were ig inoculated with the WT for up to 48 hr post-infection (pi) (Wollert et al., 2007). This suggests that the InlA-E-cad interaction may not be essential for the intestinal barrier crossing, at least during the early phase of infection, which is confirmed in a co-infection study using InlAm, WT, or ΔinlA in mice (Bou Ghanem et al., 2012).
As stated above, the interaction between InlA and E-cad in mice and rats is not fully functional (Lecuit et al., 2001), yet L. monocytogenes can cross the intestinal barrier to disseminate to the MLN, liver, and spleen in orally infected mice (Bierne et al., 2002; Burkholder et al., 2009; Czuprynski et al., 2003; Bou Ghanem et al., 2012). Though the murine M cells in Peyer’s patches (PPs) are considered the main route for translocation, L. monocytogenes was able to spread through a rat ligated ileal loop with or without PPs or in a PP null mouse (Chiba et al., 2011; Pron et al., 1998), utilizing InlB (Chiba et al., 2011), but not InlA or LLO (Corr et al., 2006). These findings reinforce L. monocytogenes’ ability to translocate across the intestinal epithelium independent of InlA and M cells.
Our group has identified Listeria adhesion protein (LAP), a 104-kDa alcohol acetaldehyde dehydrogenase (lmo1634) that promotes adhesion of pathogenic Listeria species to intestinal cells (Jagadeesan et al., 2010; Jaradat et al., 2003). Heat shock protein 60 (Hsp60) is the epithelial receptor for LAP (Jagadeesan et al., 2011; Wampler et al., 2004). LAP also promoted translocation across the human enterocyte-like Caco-2 cell monolayer in a Transwell culture device, as the lap− strain exhibited a significantly reduced translocation competency compared to that of the WT strain, while the ΔinlA translocated similarly to the WT and WT translocation was also severely impaired in an Hsp60-suppressed Caco-2 cell line (Burkholder and Bhunia, 2010).
The present study investigates whether LAP contributes to L. monocytogenes translocation across the intestinal barrier in vivo and elucidates the underlying molecular mechanism. We used a mouse model in which InlA-E-cad interaction is not fully functional and the Caco-2 cell model in which the InlA-E-cad interaction is active. We demonstrate that LAP contributes to L. monocytogenes translocation into the lamina propria (LP) and systemic dissemination in mice and across the Caco-2 cell barrier by increasing epithelial permeability. Further, we show that the increased epithelial permeability directly correlates with the increased expression TNF-α and IL-6 in mice or cells infected with the WT and the ΔinlA strains, but not with the lap− strain. By using genetic models and pharmacological inhibitors, we establish that LAP directly binds to Hsp60 to activate canonical NF-κB signaling, thereby facilitating the myosin light chain kinase (MLCK)-mediated opening of the intestinal cell-cell barrier via the cellular redistribution of the major junctional proteins, claudin-1, occluding, and E-cad, and bacterial translocation in both model systems.
RESULTS
LAP Contributes to Bacterial Translocation across the Intestinal Barrier and Systemic Dissemination
We orally challenged A/J mice with a WT strain F4244 (serotype 4b), an isogenic lap-insertion mutant strain (lap−), and a ΔinlA deletion strain and enumerated Listeria in the extra-intestinal tissues at 24 and 48 hr pi. The dissemination of lap− was significantly impaired in the liver, spleen, and MLN, and undetectable in the kidneys and blood (Figures 1A–1C, S1A, and S1B). Histopathology indicated mild-to-moderate multifocal hepatic and splenic necrosis, and neutrophilia in the white and red pulp in spleens of mice challenged with the WT or ΔinlA at 48 hr pi (Figures S1C and S1D), while these signs were absent in the lap−-challenged mice. Thus, the lap− strain exhibits a dissemination defect in trafficking to extra-intestinal sites.
Figure 1. LAP Contributes L. monocytogenes Translocation across the Intestinal Barrier and Systemic Dissemination.
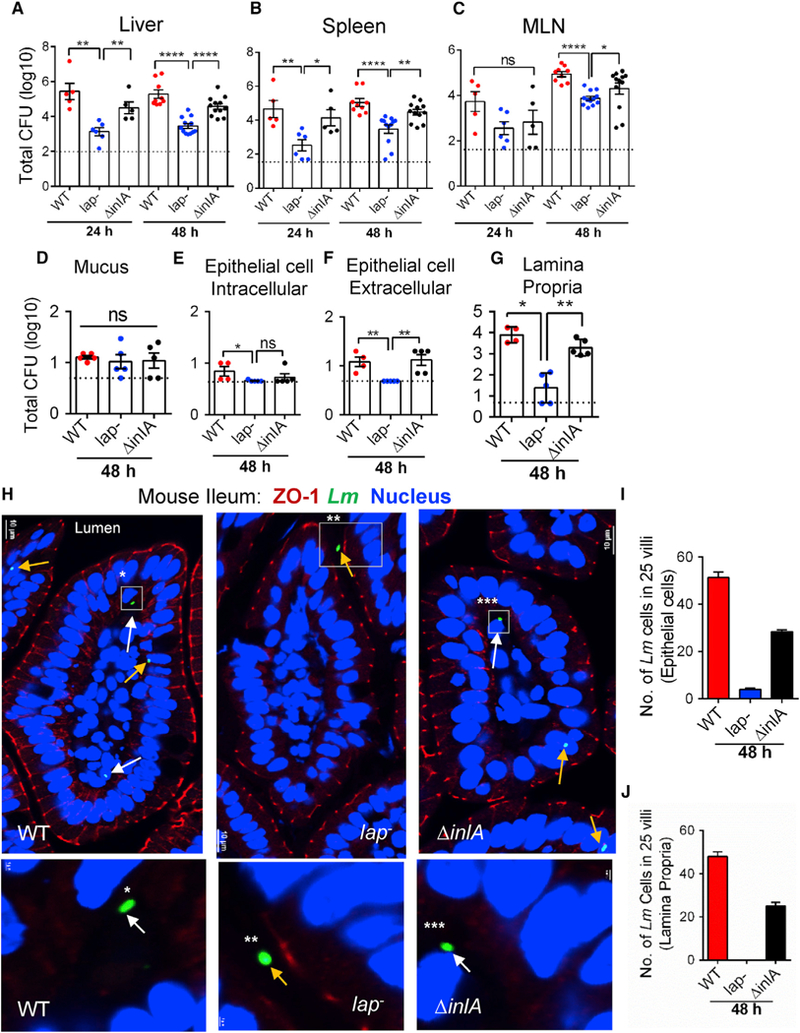
(A–G) Listeria counts (Total CFU) in liver (A), spleen (B), MLN (C), ileal mucus (D), epithelial cell intracellular (E), epithelial cell extracellular (F), and lamina propria (G) of A/J mice (n = 4–12) at 24 and 48 hr pi from three independent experiments. Dashed horizontal lines indicate the detection limit.
(H) Images of ileal villi (48 hr pi) stained for ZO-1 (red), L. monocytogenes (Lm) (green, arrows), and nucleus (blue). Scale bar, 10 μm. The panels below are enlargements of the boxed areas. Scale bar, 1 μm. White arrows indicate bacteria in the lamina propria and yellow arrows on the epithelial cells or in the lumen.
(I and J) Lm counts from epithelial cells (I) and lamina propria (J) of villi images (n = 75) from three mice for each treatment.
See also Figure S1. All error bars represent SEM. ****p < 0.0001; **p < 0.01; *p < 0.05; ns, no significance.
Next, we enumerated L. monocytogenes in the mucus, epithelial cells, and LP fractions of the ileal mucosa at 48 hr pi (Bou Ghanem et al., 2012). Relative to the WT strain, the total number of lap− bacteria present in the mucus did not change; however, significantly lower lap− counts were observed in the epithelial fractions and in the LP (Figures 1D–1G). Immunostaining of ileal tissue sections confirmed increased bacterial counts in epithelial cells and in the LP of mice challenged with the WT or ΔinlA strains (Figures 1H–1J and S1E). In contrast, very few lap− bacteria were found on the epithelial cells and were absent in the LP, but were predominantly confined to the lumen. As expected, the lap− strain was found in the ileal PPs (Figures S1F and S1G). These data suggest that LAP contributes to L. monocytogenes translocation across the intestinal barrier into the underlying LP in a mouse model.
LAP Contributes to Intestinal Barrier Dysfunction
Next, we analyzed intestinal permeability to the paracellular marker 4-kDa FITC-dextran (FD4), gavaged orally 4–5 hr prior to sacrifice, in Listeria-infected mice. Relative to the uninfected controls, the FD4 concentrations were significantly increased in the serum, and the urine in WT and ΔinlA-challenged mice 48 hr pi, while the levels in lap−-challenged mice were comparable to uninfected controls (Figures 2A and 2B).
Figure 2. LAP Contributes to Intestinal Barrier Dysfunction.
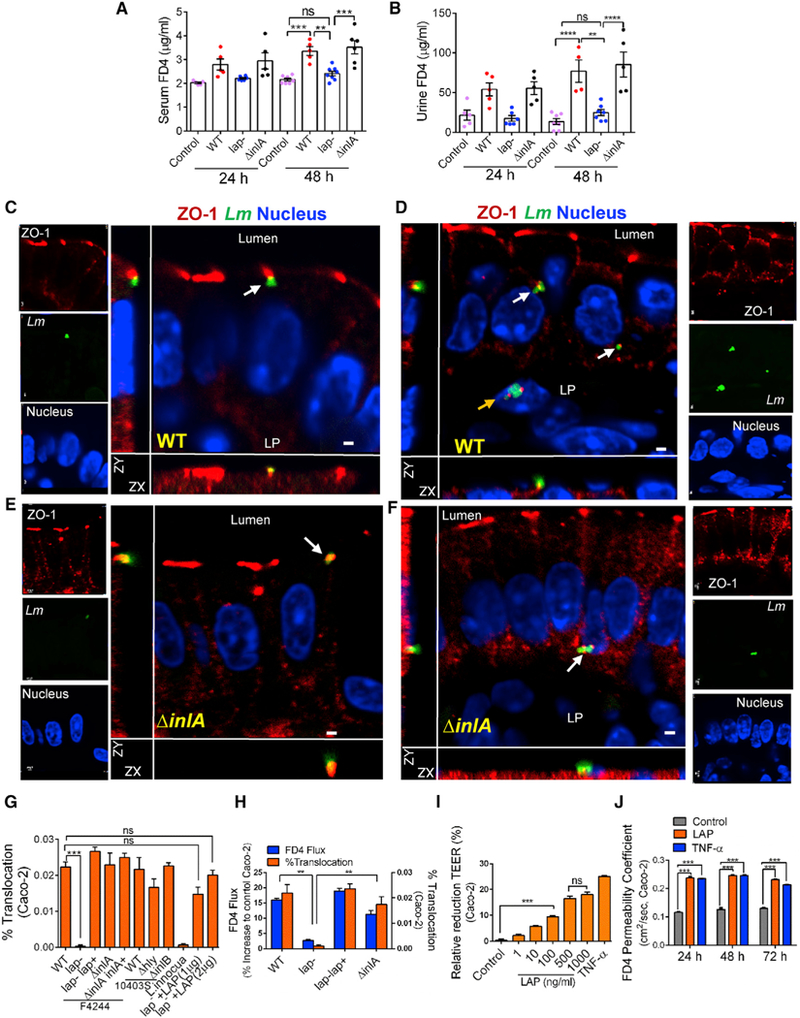
(A and B) 4-kDa FITC-dextran (FD4) permeability through the intestinal epithelium of uninfected (control) and L. monocytogenes-infected A/J mice in serum (A) and urine (B).
(C–F) Lm localization in the mouse ileal epithelial cell junction (48 hr pi). ZO-1 (red), Lm (green, arrows), and nucleus (blue) infected with WT (C and D) or ΔinlA (E and F). Merged images show co-localization of Lm and ZO-1 (arrow), and Lm exiting the epithelial cell (D and F, arrows) and in the lamina propria (LP) (D, yellow arrow). Scale bar, 1 μm.
(G and H) L. monocytogenes WT (F4244 and 10403s) and their isogenic mutant strain translocation across Caco-2 cell barrier or lap− with exogenously added recombinant LAP (lap− +LAP, 1 and 2 μg/mL) and L. innocua (G). (H) FD4 flux (Y1 axis) and bacterial translocation (Y2 axis) after Listeria infection (MOI 50).
(I and J) Purified LAP pre-treatment effect on the Caco-2 TEER 48 hr pi (I) and FD4 permeability (J). Human TNF-α (10 ng/mL) was used as a control.
See also Figure S2 and Videos S1, S2, S3, S4, and S5. All error bars represent SEM (n = 5–7). ****p < 0.0001; ***p < 0.001; **p < 0.01; ns, no significance.
Tight junction (TJ) plays a crucial role in the maintenance of intestinal paracellular permeability. Thus, we examined bacterial localization near TJ. Immunostaining revealed WT and ΔinlA co-localization with the TJ protein ZO-1 at the epithelial cell-cell junction in the ileal tissue (Figures 2C–2F; Videos S1, S2, S3, and S4). In contrast, the lap− strain was predominantly confined in the lumen of the ileal mucosa and was not found to interact with the TJ (Figures 1H, S1E, and S2A; Video S5).
Analysis of translocation phenotypes of mutant strains for other important virulence factors, the ΔinlB and Δhly strains, across the Caco-2 monolayers revealed similar translocation rates to their respective WT strains (Figure 2G). By contrast, the lap− exhibited a severely attenuated translocation phenotype while the lap−lap+ complemented strain restored translocation. Furthermore, the attenuated translocation of the lap− also correlated with decreased FD4 flux and increased transepithelial electrical resistance (TEER) across Caco-2 cells (Figures 2H and S2B). Conversely, the lap−lap+ strain showed increased FD4 flux and decreased Caco-2 TEER, equal to the WT. Importantly, the translocation defect observed by the lap− strain was not due to decreased cytotoxic effects (Figure S2C).
To delineate the intracellular role of LAP from its role at the bacterial cell surface, we determined the relevant concentration of LAP available on the bacterial surface during the infection of Caco-2 cells by using purified recombinant LAP as the standard. Approximately 1 mg LAP was associated with the cell wall of WT bacteria at 1 × 107 CFU (colony-forming units), and an equivalent CFU was used in our translocation assays to achieve a multiplicity of infection (MOI) of 50 (Figure S2D). Pre-incubation of the lap− strain with purified LAP (lap− +LAP), at a concentration that is available on the bacterial surface (1 mg), resulted in a strong association of the protein with the bacterial cell wall (Figure S2E) and rescued the translocation defect of the lap− strain across the Caco-2 barrier (Figure 2G).
Additionally, treatment of Caco-2 monolayers with LAP significantly reduced TEER when LAP was added to the apical, basolateral, or both compartments (Figure S2F) and TEER value reduced progressively with increasing concentration of LAP (Figure 2I). Consequently, FD4 permeability increased, equivalent to TNF-α as a positive control, over a 72-hr period (Figure 2J). Collectively, these data confirm that the LAP increases epithelial permeability and shows a positive correlation between epithelial permeability and Listeria translocation in mouse and human Caco-2 cell models.
LAP Upregulates TNF-α and IL-6 Expression in Intestinal Cells
Cytokines play a crucial role in the modulation of inflammatory responses in the gastrointestinal tract, and pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-6, IL-8, and IFN-γ, induce disturbances in the intestinal cell-cell junction barrier promoting permeation of luminal antigens (Ma et al., 2004). L. monocytogenes upregulates IL-8, MCP1, GM-CSF, and TNF-α in epithelial cells (Jung et al., 1995), and the intestinal host response is InlA independent (Lecuit et al., 2007). Therefore, we compared the expression of 40 inflammatory mediators in culture supernatants of Caco-2 cells infected with WT or lap− using a cytokine dot-blot array (Figure S3A). Densitometry of the arrays revealed that TNF-α, IL-6, IL-8, IFN-γ, and the chemokine MCP-2 were downregulated, in particular TNF-α and IL-6 by 26% ± 1% and 47% ± 2%, respectively, when infected with the lap− compared to the WT (Figures S3A and S3B; Table S1). The lap− strain produced significantly lower TNF-α and IL-6 than the WT in Caco-2 cells (Figures 3A and 3B). Treatment of Caco-2 cells with purified LAP produced high levels of TNF-α and IL-6 without causing any cytotoxic effects (Figures 3A, 3B, and S3C). In the ileal mucosa, levels of TNF-α and IL-6 transcripts (mRNA) and proteins were also significantly higher in mice infected with WT and ΔinlA than in those infected with the lap− (Figures 3C–3H).
Figure 3. LAP Upregulates TNF-α and IL-6 Expression in Intestinal Cells.
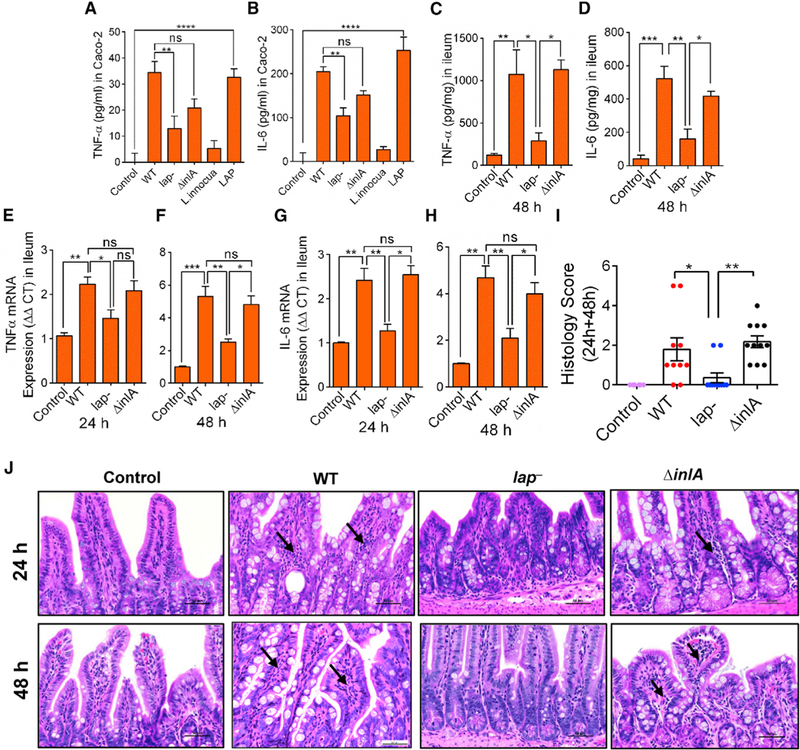
(A–D) ELISA analysis of human TNF-α (A) and IL-6 (B) in Caco-2 cell supernatants (n = 4–6), and mouse TNF-α (C) and IL-6 (D) in mouse (n = 3–4) ileal tissues, at 48 hr pi. L. innocua (nonpathogen) and the purified LAP (1 μg/mL) were used as controls (A and B).
(E–H) mRNA levels for TNF-α at 24 (E) and 48 hr pi (F) and IL-6 at 24 (G) and 48 hr pi (H) in the ileal mucosa of mice (n = 3–4). Values were normalized to gapdh with the average for untreated samples set at 1.
(I and J) Histology score of ileal sections at combined 24 and 48 hr pi (I) and representative hematoxylin and eosin (H&E)-stained images (J) (scale bar, 50 mm) from control uninfected mice (n = 6) or Listeria-infected mice (n = 10–11). Polymorphonuclear and mononuclear cells (arrows) infiltrating the base of the villous lamina propria are evident (J).
See also Figure S3 and Table S1. All error bars represent SEM. ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; ns, no significance.
Ileal tissue histopathology revealed significantly increased numbers of polymorphonuclear and mononuclear cells infiltrating the base of the villous LP, and increased involvement of the submucosa, in mice challenged with the WT and ΔinlA compared to the lap− strain at 24 and 48 hr pi (Figures 3I and 3J). These data suggest that LAP contributes to TNF-α and IL-6 production in Caco-2 cells and mouse ileal mucosa.
LAP Contributes to Listeria-Induced NF-κB Activation for Increased Epithelial Permeability
NF-κB is a central regulator of pro-inflammatory cytokines, including TNF-a and IL-6 (Oeckinghaus et al., 2011), and it is strongly activated in Caco-2 cells upon L. monocytogenes infection independent of invasion (Baldwin et al., 2003); thus, we examined whether LAP contributes to NF-κB(p65) activation. We observed significantly lower levels of nuclear p65 and P-p65 in the lap−-infected cells relative to the WT or ΔinlA-infected cells at 30 min pi (Figures 4A and 4B). The total cellular p65 was not changed in the WT, lap−-infected, or ΔinlA-infected cells (Figure S4A). Purified LAP treatment also increased IKKα level in the cytosol, and p65 and P-p65 in the nucleus, in a dose-dependent manner (Figures 4C and 4D), indicating LAP’s involvement in Listeria-induced NF-κB activation in Caco-2 cells.
Figure 4. LAP Contributes to Listeria-Induced NF-κB Activation for Increased Epithelial Permeability.
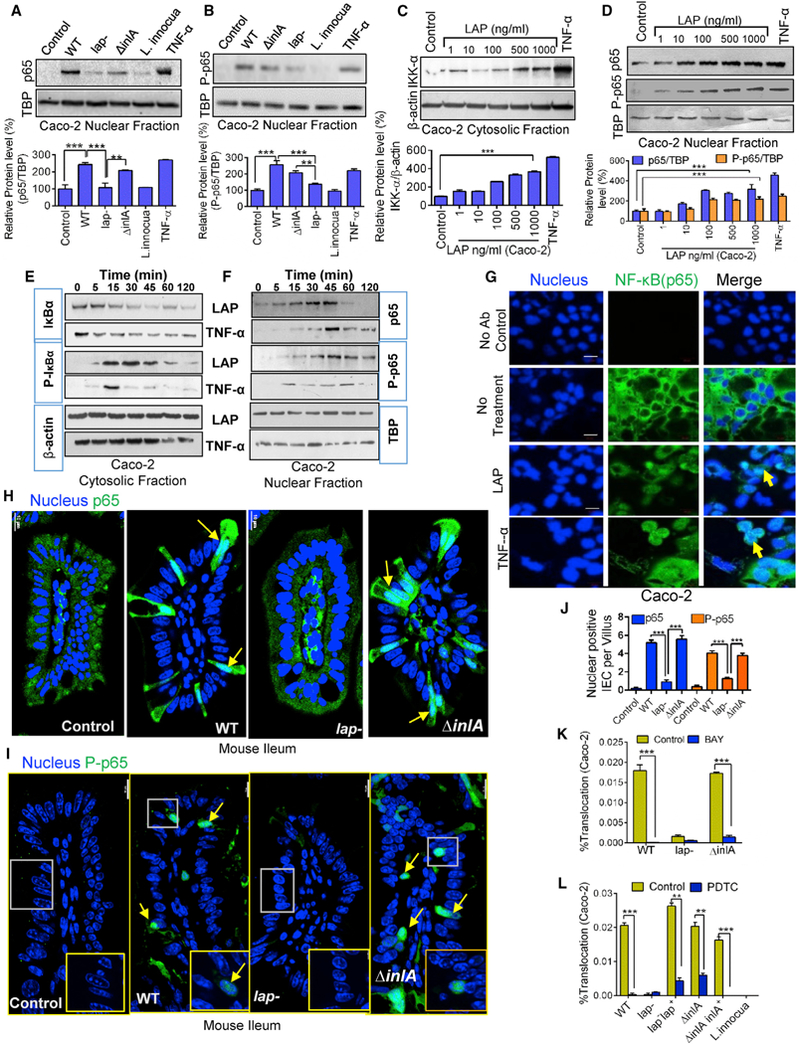
(A and B) Immunoblot and densitometry plots (n = 3) of Listeria-induced p65 (A) and P-p65 (B) levels in the nuclear extracts of uninfected (control) Caco-2 cells or those infected with L. monocytogenes and L. innocua (MOI 50, 30 min). TBP (TATA-binding protein) is a nuclear loading control.
(C and D) Immunoblot and densitometry plots (n = 3) showing IKK-α levels in the cytosolic extracts (C) and those of p65 and P-p65 in the nuclear extracts (D) of LAP-treated (30 min) Caco-2 cells.
(E and F) Immunoblots showing time-dependent expression of IκBα and P-IκBα in the cytosolic extracts (E) and p65 and P-p65 in the nuclear extracts (F) of Caco-2 cells treated with LAP (1 μg/mL) or TNF-α (10 ng/mL).
(G) Images of Caco-2 cells treated with LAP (1 μg/mL) or human TNF-α (10 ng/mL) for 30 min stained for p65 (green) and the nuclei (blue). Arrows show the nuclear localization of p65. Scale bar, 20 μm.
(H and I) Images of ileal villi stained forp65 (H, green) and P-p65 (I, green) and the nuclei (blue) from uninfected mice (control) or mice infected with Listeria 48 hr pi (see Figure 1). Arrows indicate the nuclear localization of p65 and P-p65. Scale bar, 10 mm.
(J) Enumeration of IECs per villus (n = 10–15 villi from 2–3 mice/treatment) positive for p65 and P-p65 in the nucleus of (H) and (I).
(K and L) Listeria (MOI 50) translocation through Caco-2 barrier following pretreatment with BAY (10 μM, 30 min) (K) or PDTC (100 μM, 30 min) (L) (n = 6).
See also Figure S4. All error bars represent SEM. ***p < 0.001; **p < 0.01.
NF-κB is inactive in unstimulated cells and remains associated with the inhibitors of κB (IκB, IκBα, and IκBβ) in the cytoplasm. Treatment of Caco-2 cells with LAP rapidly degraded IκBα within 15 min and most was degraded within 45 min, while phospho-IκBα (P-IκBα) concomitantly appeared at 15 min (Figures 4E and S4B). The IκBα degradation paralleled with a significant increase in nuclear p65 and P-p65 at 30 min pi (Figures 4F and S4C). Human TNF-α treatment also exhibited similar IκBα degradation kinetics and a concomitant increase in cytosolic P-IκBα and nuclear p65 and P-p65 levels (Figures 4E, 4F, S4B, and S4C). Immunostaining confirmed p65 sequestration in the nucleus after 30 min of LAP or TNF-α treatment (Figure 4G). Thus, LAP in Caco-2 cells stimulates IκBα degradation and facilitates rapid translocation of p65 to the nucleus, a hallmark of NF-κB activation.
Analysis of intestinal epithelial cells (IECs) of mice infected with WT or ΔinlA revealed increased NF-κB activity, as immunostaining confirmed nuclear abundance of p65 and P-p65 in IECs (Figures 4H–4J). In contrast, the lap−-infected mice showed the presence of very low nuclear positive p65 and P-p65 cells, and most of the p65 were sequestered in the cytoplasm of IECs.
L. monocytogenes-induced NF-κB pathway is the most dominant host response in macrophage, which is the major resident cell in LP; therefore, we verified the ability of LAP to activate NF-κB in the murine macrophage RAW 264.7-NF-κB-luciferase reporter cell line. LAP activated NF-κB in a dose-dependent manner (Figure S4D). Likewise, purified recombinant InlB protein activated NF-κB in RAW 264.7 cells, but InlA was neutral (Figures S4E and S4F). Heat (Figure S4F) and Proteinase K (Figures S4G and S4H) abolished, while polymyxin-B (Figures S4I and S4J) treatment retained, the NF-κB activation ability of LAP and InlB, indicating susceptibility of these proteins to thermal denaturation and proteolytic enzymes, and absence of LPS contamination, respectively. Pretreatment of LAP with an anti-LAP mAb also significantly reduced LAP-mediated NF-κB response (Figure S4K). Besides, the lap− strain showed lower NF-κB activity than the WT strain (Figure S4L). Collectively, these results suggest that LAP activates NF-κB in Caco-2 cells, IECs of mice, and murine macrophage cell line (RAW 264.7), while InlA is unresponsive (this study). InlB activates NF-κB in macrophage (Mansell et al., 2000), but not in epithelial cell lines (Caco-2, HeLa, Hep G2, and LoVo). Among the other virulence proteins, LLO activates NF-κB in HEK293 cell line (Tsuchiya et al., 2005) and in endothelial cells of transgenic mice (Kayal et al., 2002), while InlC interferes with NF-κB activation in murine macrophages in vivo (Gouin et al., 2010).
We next examined the effect of the NF-κB inhibitors BAY and PDTC on LAP-induced epithelial permeability. Both inhibitors significantly inhibited LAP-mediated NF-κB activation (Figure S4M), restored Caco-2 TEER (Figure S4N), and reduced WT and ΔinlA translocation by 80%–90% across Caco-2 cell barrier (Figures 4K and 4L). Both inhibitors independently did not affect Caco-2 TEER (Figures S4O and S4P) and, more importantly, did not affect WT invasion of Caco-2 cells (Figures S4Q and S4R). As a positive control, we pretreated Caco-2 cells with cytochalasin D, which inhibits actin polymerization, bacterial invasion, and cell-to-cell spread (Tilney and Portnoy, 1989) but increases TJ permeability (Stevenson and Begg, 1994). Cytochalasin D treatment induced a very low TEER (Figure S4S) and markedly increased the translocation competencies of the WT and the isogenic lap−, lap− lap+, and ΔinlA across the Caco-2 cell barrier (Figure S4T), despite a low observed invasion (Figure S4U). Neutralization of TNF-α and IL-6 with antibodies also restored purified LAP-mediated drop in Caco-2 TEER (Figure S4V). Taken together, these data demonstrate that LAP-mediated NF-κB activation is critical during L. monocytogenes- induced epithelial permeability.
LAP-Mediated NF-κB Activation is Hsp60 Receptor Dependent
Hsp60 acts as an IKK-interacting protein and mediates NF-κB-dependent signaling via interaction with IKKα/β in the cytoplasm (Chun et al., 2010). Hsp60 is the receptor for LAP, and to determine the contribution of Hsp60 in Listeria-mediated NF-κB activation, we used Hsp60 knockdown (hsp60::shRNA, ~70% knockdown) Caco-2 cells (Burkholder and Bhunia, 2010) (Figures S5A and S5B) and determined the levels of IKK-β in the cytoplasmic, and p65 and P-p65 in the nuclear, fraction in Listeria-infected or purified LAP-treated cells. Knockdown of Hsp60 resulted in significant decreases of the WT (Figure 5A) or purified LAP-mediated (Figure 5B) IKK-β, p65, and P-p65 expression and p65 nuclear translocation (Figure 5C). Additionally, blocking of surface Hsp60 with an anti-Hsp60-specific mAb prior to purified LAP treatment showed a significant reduction in LAP-mediated IKK-β and p65 expression in Caco-2 cells (Figure 5D), indicating LAP (ligand) and Hsp60 (receptor) interaction contributes to Listeria-induced NF-κB activation in Caco-2 cells.
Figure 5. LAP-Induced NF-kB Activation Is Hsp60 Receptor Dependent.
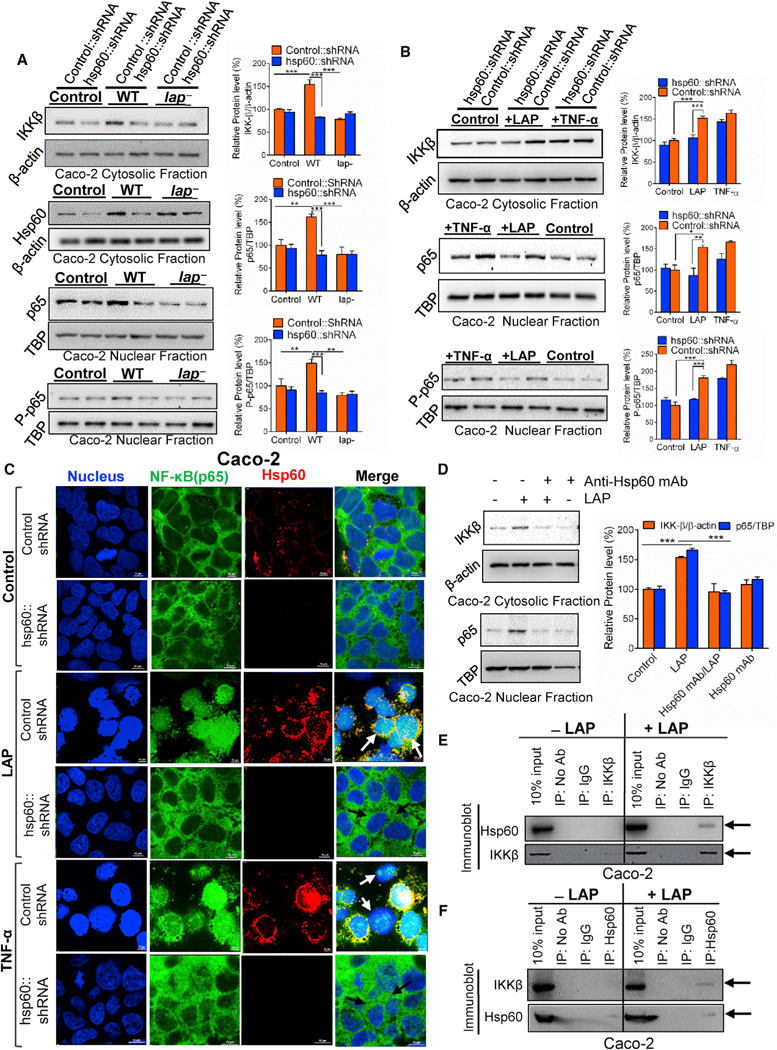
(A and B) Immunoblot and densitometry plots (n = 3) of cytosolic IKK-β and Hsp60 and nuclear p65 and P-p65 In vector control short hairpin RNA (shRNA) (Control::shRNA) or hsp60 knockdown (hsp60::shRNA) Caco-2 cells after infection with Listeria (MOI 50,30 min) or no infection (control) (A). (B) is the same as (A) except the Caco-2 cells were treated with purified LAP (1 μg/mL) or human TNF-α (10 ng/mL) for 30 min (B).
(C) Localization of p65 (green), Hsp60 (red), and nucleus (blue) in vector-control shRNA (Control shRNA) or hsp60 knockdown (hsp60::shRNA) Caco-2 cells treated with purified LAP (1 μg/mL) or TNF-α (10 ng/mL) or untreated (control) for 30 min. In LAP and TNF-α-treated control cells, p65 is in the nucleus (white arrows), and it is in the cytoplasm of hsp60 knockdown cells (dark arrows). Scale bar, 10 μm.
(D) Immunoblot and densitometry plots (n = 3) of cytosolic IKK-β and nuclear p65 in Caco-2 cells incubated with anti-Hsp60 mAb (1 μg/mL, 1 hr) to block surface Hsp60 prior to LAP (1 μg/mL, 30 min) treatment.
(E and F) Immunoblots showing the interaction of Hsp60 with IKK-β in LAP (1 μg/mL, 30 min)-treated Caco-2 cells. IKK-β (E) or Hsp60 (F) was immunoprecipitated from Caco-2 cell lysates and immunoprobed with anti-Hsp60 (E) or anti-IKKβ (F) mAb. Arrows in (E) and (F) indicate co-precipitated IKK-β and Hsp60, respectively, in the LAP-treated cells. The 10% input lane (E and F) represents Caco-2 lysate without immunoprecipitation (IP). The IP: No Ab lane (E and F) represents IP reactions without addition of antibody. Rabbit IgG (IP: IgG lane) (E) or normal mouse IgG (IP: IgG lane) (F) was used asan isotype control IP reaction.
See also Figure S5. All error bars represent SEM. ***p < 0.001; **p < 0.01; *p < 0.05.
To examine the possible interactions between Hsp60 and IKK in Caco-2 cells, we next immunoprecipitated IKKβ from purified LAP-treated or -untreated Caco-2 cell lysates. Immunoblotting the lysates revealed co-precipitation of Hsp60 and IKK-β in LAP-treated cell lysates (Figure 5E). Reverse immunoprecipitation using an anti-Hsp60 antibody also co-precipitated IKK-β and Hsp60 in the LAP-treated cell lysates (Figure 5F). Thus, Hsp60 ultimately resides in the IKK complex in LAP-treated Caco-2 cells. Furthermore, LAP treatment did not change the total cellular Hsp60 levels but decreased the membrane Hsp60 and increased the cytosolic Hsp60 levels (Figure S5C), and increased total cellular and cytosolic IKK-β levels significantly (Figure S5C). Thus, LAP induces internalization of surface-expressed Hsp60 in Caco-2 cells, which may facilitate interaction of Hsp60 with the IKK complex.
The primary function of Hsp60 is to act as a molecular chaperone within the cell cytoplasm and mitochondrial matrix. We have previously (Burkholder and Bhunia, 2010), and also in this study (Figure S5C), demonstrated membrane localization of Hsp60 in Caco-2 cells. In the mouse intestine, Hsp60 has been localized at high levels in the crypts and villi (Berger et al., 2016). Here, we confirmed apical and the plasma membrane localization of Hsp60 in the mouse ileal villi by immunostaining and western blotting (Figures S5D and S5E). These results suggest that apically expressed surface Hsp60 is accessible to bacteria located in the intestinal lumen.
LAP Induces Junctional Protein Dysregulation for Increased Epithelial Permeability and L. monocytogenes Translocation
MLCK phosphorylates myosin II regulatory light chain (MLC), which regulates paracellular permeability via cytoskeleton rearrangement and modulates TJ protein expression (Clayburgh et al., 2005; Hecht et al., 1996). Moreover, pro-inflammatory cytokines can induce TJ dysfunction via activation of MLCK (Ma et al., 2005). We observed a time-dependent increase in MLCK expression with purified LAP treatment and MLCK and P-MLC expression during L. monocytogenes infection in Caco-2 cells (Figures 6A and 6B).
Figure 6. LAP Induces Junctional Protein Dysregulation through MLCK Activation.
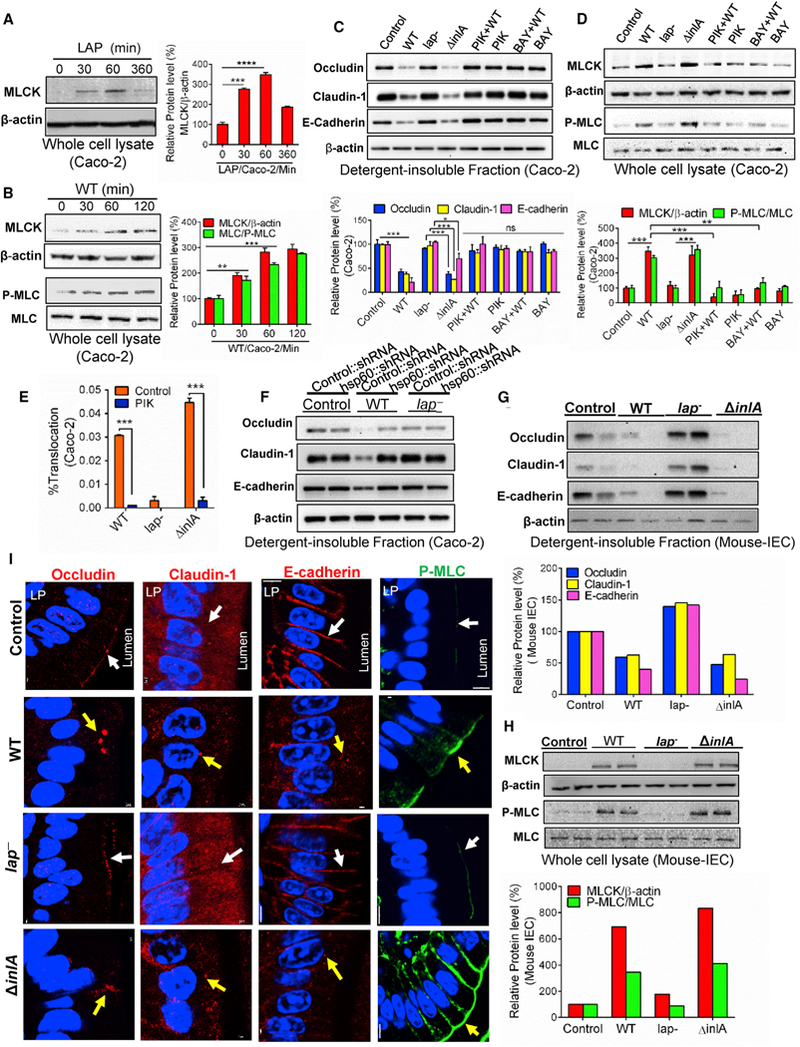
(A and B) Immunoblot and densitometry plots (n = 3) of MLCK (A and B), P-MLC (B), or MLC (B) in the whole-cell lysate of Caco-2 cells treated with purified LAP (1 μg/mL) (A) or WT (MOI 50) (B).
(C and D) Immunoblot and densitometry plots (n = 3) of proteins from the detergent-insoluble fraction (C) and whole-cell lysate of Caco-2 cells infected with Listeria, or pre-treated with the MLCK inhibitor, PIK (150 μM), or the NF-κB inhibitor, BAY (10 μM), prior to infection with WT.
(E) Translocation (n = 6) of Listeria (MOI 50, 2 hr) through Caco-2 monolayers pretreated with the MLCK inhibitor, PIK (150 μM).
(F) Immunoblots of cell junction proteins in the detergent-insoluble fraction of control or hsp60 knockdown Caco-2 cells infected with Listeria or uninfected (control).
(G and H) Immunoblot and densitometry plots of cell junction proteins in the detergent-insoluble fraction (G), and MLCK, P-MLC, and MLC in whole-cell lysates (H) of purified ileal IECs from two mice (A/J) (see Figure 1) at 48 hr pi.
(I) Images of the mouse (A/J) ileal villi sections (48 hr pi) showing membrane localization of occludin, claudin-1, E-cadherin (red), and P-MLC (green). White arrows represent normal presentation while yellow arrows represent alterations or mislocalization. Nuclei, blue. Scale bar, 50 μm. LP, lamina propria.
See also Figure S6. Caco-2 cell infection (MOI 50, 45 min) (C, D, and F). All error bars represent SEM. ***p < 0.001; **p < 0.01, *p < 0.05; ns, no significance.
Next, we analyzed the time-dependent distribution of TJ (claudin-1, occludin, and ZO-1) and AJ (E-cad and β-catenin) proteins in the detergent-insoluble and-soluble fractions from Caco-2 cells infected with WT. In the detergent-insoluble fractions, the WT strain significantly reduced occludin, claudin-1, and E-cad expression at 45 min pi compared to the uninfected control without significant alterations in ZO-1 and β-catenin (Figure S6A), while in the detergent-soluble fractions, a concomitant increased expression of occludin at 60 min pi and claudin-1 and E-cad at 45 min pi was observed (Figure S6B). The total protein levels of claudin-1, occludin, ZO-1, E-cad, and β-catenin in WT-infected Caco-2 cells did not change, at all time points during infection (5–120 min pi) (Figure S6C). These data indicate an infection of Caco-2 cells with the WT strain caused a sub-cellular redistribution of occludin, claudin-1, and E-cad without affecting the total cellular junctional protein levels.
Analysis of cell junction proteins of Caco-2 cells with WT and ΔinlA infection at 45 min pi revealed significantly decreased expression of occludin and claudin-1 in the detergent-insoluble fractions (Figure 6C) and increased total cellular MLCK and P-MLC compared to the uninfected cells (Figure 6D). Notably, E-cad expression was also significantly reduced in WT (~88%) and ΔinlA (~20%)-infected cells (Figure 6C). In contrast, the expression of these proteins in the lap−-infected cells was similar to uninfected Caco-2 cells. Thus, LAP contributes to the subcellular redistribution of occludin, claudin-1, and E-cad and increased expression of MLCK and P-MLC in Caco-2 cells, and the direct interaction between InlA and E-cad also contributes to E-cad internalization, not only in Caco-2 cells (this study), but also in Jeg-3 cells (Bonazzi et al., 2008).
We next examined the effect of the NF-κB inhibitor BAY or the MLCK inhibitor PIK on LAP-mediated junctional protein mislocalization. Both inhibitors prevented L. monocytogenes-induced redistribution of occludin, claudin-1, and E-cad (Figure 6C) and decreased the total MLCK and P-MLC expression to baseline levels (Figure 6D). These inhibitors independently had no effect on the expression of these junctional proteins (Figure 6C) or Caco-2 TEER (Figures S3O, S6D, and S6E). Additionally, consistent with our observations with the NF-κB inhibitors (Figures 3K and 3L), the MLCK inhibitors PIK and ML-9 significantly reduced the translocation competencies of WT and ΔinlA across the Caco-2 cell barrier (Figures 6E and S6F) and restored purified LAP-mediated drop in Caco-2 TEER (Figure S6G) without affecting WT invasion of Caco-2 cells (Figures S6H and S6I). Additionally, knockdown of Hsp60 also prevented L. monocytogenes LAP-induced redistribution of occludin, claudin-1, and E-cad (Figures 6F and S6J–S6L) and restored the drop in Caco-2 TEER significantly (Figure S6M).
Analysis of ileal IECs of mice infected with WT and ΔinlA revealed reduced occludin, claudin-1, and E-cad expression in the detergent-insoluble fractions (Figure 6G) and increased total cellular MLCK and P-MLC (Figure 6H), but not in mice challenged with the lap− strain. Immunostaining of the ileal tissue sections confirmed membrane mislocalization of occludin, claudin-1, and E-cad and increased expression of P-MLC in IECs of mice challenged with WT and ΔinlA, but not the lap− strain (Figure 6I). Collectively, these results indicate that LAP induces MLCK expression resulting in phosphorylation of MLC, which in turn promotes the MLCK-triggered opening of the epithelial barrier via cellular redistribution (mislocalization) of occludin, claudin-1, and E-cad.
L. monocytogenes Translocation and Epithelial Permeability Did Not Increase in MLCK Knockout Mice
We orally challenged MLCK knockout mice lacking the 210-kDa long chain (MLCK−/−) and its parental strain (C57BL/6 mice, MLCK+/+) with WT, lap− or ΔinlA and enumerated bacterial burdens in the intestinal and extra-intestinal tissues at 48 hr pi. The bacterial burden in the liver, spleen, and MLN of the MLCK+/+ mice challenged with the lap− or the MLCK−/− mice challenged with either WT or lap− was significantly reduced compared to that in the same tissues of the MLCK+/+ mice that were challenged with WT or ΔinlA (Figures 7A–7C). In the mucus, WT, lap− or ΔinlA counts were similar for both MLCK+/+ and MLCK−/− mice (Figure 7D). In the epithelial cell of both the MLCK+/+ and MLCK−/− mice, WT counts were similar, but the lap− count was significantly reduced (Figures 7E and 7F), suggesting that the epithelial intracellular invasion of L. monocytogenes was not affected in MLCK−/− mice. As expected, the lap− counts in LP were significantly lower than the WT or ΔinlA in MLCK+/+ mice; however, both lap− and WT strain showed similar but significantly reduced translocation to the LP in MLCK−/− mice relative to MLCK+/+ mice (Figure 7G). Thus, L. monocytogenes in the MLCK−/− mice exhibits a defect in translocating from the ileal mucosa to the underlying LP and systemic dissemination possibly due to decreased translocation in this mouse strain.
Figure 7. L. monocytogenes Translocation and Epithelial Permeability Did Not Increase in MLCK Knockout Mice.
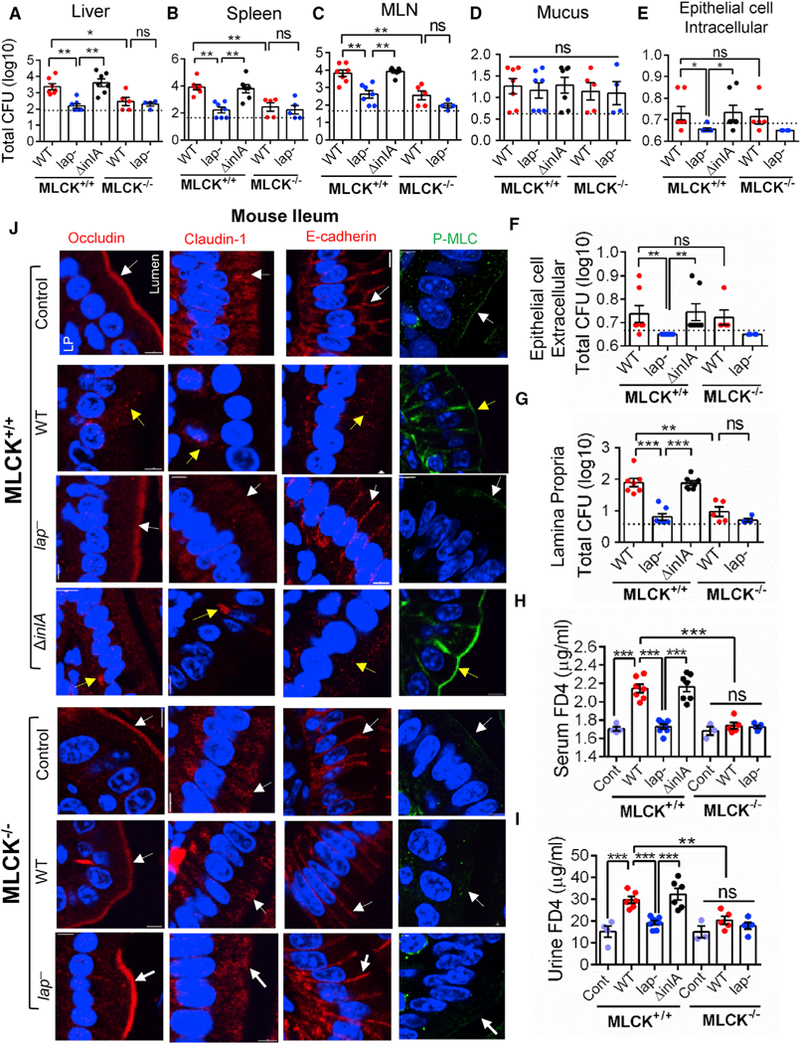
(A–G) Listeria counts (Total CFU) in liver (A), spleen (B), MLN (C), ileal mucus (D), epithelial cell intracellular (E), epithelial cell extracellular (F), and lamina propria (G) of WT C57BL/6 (MLCK+/+) or MLCK knockout (MLCK−/−) mice (n = 4–7 male and female) at 48 hr pi. Dashed lines indicate the detection limit.
(H and I) Analysis of FD4 permeability through the intestinal epithelium of uninfected (Cont) and WT, lap “-infected, and ΔinlA-infected MLCK+/+ or MLCK−/− mice (n = 5–7) in serum (H) and urine (I) at 48 hr pi.
(J) Images of the mouse (C57BL/6, MLCK+/+, and MLCK−/−) ileal villi sections (48 hr pi) showing membrane localization of occludin, claudin-1, E-cadherin (red), and P-MLC (green). White arrows represent normal presentation, while yellow arrows represent alterations or mislocalization. Nuclei, blue. Scale bar, 50 μm. LP, lamina propria.
All error bars represent SEM. ***p < 0.001; **p < 0.01; *p < 0.05; ns, no significance.
We measured FD4 permeability to assess the relationship between bacterial translocation and epithelial permeability in MLCK−/− mice. Listeria-infected MLCK+/+ mice that received FD4 orally 4–5 hr before sacrifice displayed significantly increased FD4 in the sera and urine in WT and ΔinlA-infected mice compared to the lap− -infected or uninfected mice (Figures 7H and 7I). In the MLCK−/− mice, there were no significant differences in the FD4 levels in the sera and urine of mice challenged with the WT or lap− strain or in the uninfected controls.
Immunostaining of ileal tissues confirmed membrane mislocalization of occludin, claudin-1, and E-cad, and increased expression of P-MLC in MLCK+/+ in IECs of mice challenged with WT and ΔinlA, but not with the lap− strain or the MLCK−/− mice challenged with either the WT or lap− (Figure 7J). In summary, besides A/J mice, the translocation defect of the lap− strain is also seen in the C57BL/6 mouse and MLCK contributes to LAP-induced junctional protein dysregulation, epithelial barrier dysfunction, and L. monocytogenes translocation.
DISCUSSION
The precise mechanism by which L. monocytogenes crosses the epithelial barrier during the gastrointestinal phase of infection is not fully understood. Previous work demonstrated that L. monocytogenes used two pathways: InlA-mediated uptake (Radoshevich and Cossart, 2018) and the M cell trans-cytosis (Corr et al., 2006; Pron et al., 1998). However, recent studies have demonstrated that L. monocytogenes expressing non-functional InlA (encoding a premature stop codon) infected fetuses of pregnant guinea pigs and mice after oral administration (Holch et al., 2013) and human newborns with neonatal listeriosis (Gelbicova et al., 2015). Additionally, in a PP null mouse, L. monocytogenes colonized the ileum and disseminated to the MLN, liver, and spleen in a ligated loop assay (Chiba et al., 2011). These findings provide strong evidence that L. monocytogenes also uses alternate routes to translocate across the gut barrier.
Here, we used a mouse model and the human Caco-2 cell model and established that LAP contributes to translocation of L. monocytogenes from the intestinal lumen, across the gut epithelium into the underlying LAP and subsequent systemic dissemination in a mouse model. By using both model systems, we show that LAP binding to Hsp60 activates canonical NF-κB signaling, which facilitates MLCK-mediated opening of the intestinal epithelial barrier via the cellular redistribution of the major TJ proteins (claudin-1 and occludin) and AJ protein (E-cad), and increases intestinal permeability. Furthermore, LAP-mediated epithelial cytokine production (TNF-α and IL-6) in the early phase (24–48 hr) of infection promotes epithelial barrier dysfunction and L. monocytogenes translocation, avoiding the innate immune defenses without showing an overt inflammatory response.
We were intrigued that the LAP-Hsp60 interaction contributed to NF-κB activation despite the primary role of mitochondrial Hsp60 to act as a molecular chaperone and assist in the folding of imported proteins to their native conformation. Increasing evidence also supports a role for eukaryotic Hsp60 in “moonlighting,” which is the property of proteins possessing two or more distinct biological activities. Extracellular eukaryotic Hsp60 is recognized by TLR2 and TLR4 (Vabulas et al., 2001), which leads to NF-κB activation, and plays a role in innate immunity. We and others have demonstrated that Hsp60 acts as a cell surface receptor for Staphylococcus aureus (Dziewanowska et al., 2000), HIV (Speth et al., 1999), and L. monocytogenes (Wampler et al., 2004). Cytosolic Hsp60 also regulates apoptosis activity in cancer cells (Xanthoudakis et al., 1999) and cell survival functions by interacting with IKK in the cytoplasm and activating NF-κB (Chun et al., 2010). Our results suggest that membrane or surface-expressed Hsp60 at the intestinal interface exhibits an immune sensor function that leads to NF-κB activation. Intestinal epithelial TLR2 and TLR10 (Regan et al., 2013) and NOD2 (Kobayashi et al., 2005) also activate NF-κB at the intestinal interface following L. monocytogenes infection. Therefore, one question remains: why must a LAP-Hsp60 interaction that activates NF-κB happen to increase epithelial permeability with all of these redundant pathways? One possible explanation is that the relative distribution of these receptors in the intestine contributes to the differences in the levels of NF-κB activation according to their respective ligands. Notably, we observed earlier a dramatic increase in apical Hsp60 expression following L. monocytogenes infection in Caco-2 cell plasma membranes (Burkholder and Bhunia, 2010). Here, we observed high Hsp60 expression in the membrane of mouse IECs. NOD2 is not expressed in Caco-2 (Hisamatsu et al., 2003), and Caco-2 cells are not responsive to TLR2 or TLR4 agonists because of the low expression of TLR2, the absence of TLR4, and the high expression of the Toll inhibitory protein Tollip (Abreu et al., 2003). An alternate explanation is that the interaction of LAP with Hsp60, in addition to NF-κB activation, may also elicit other signal transduction pathways that contribute to increased paracellular permeability. The present study reveals a moonlighting function of LAP-Hsp60 interaction and links to a wider phenomenon of bacterial recognition during innate immunity.
Our data suggest that in the intestinal phase of infection, the LAP activates NF-κB and exploits the epithelial innate defenses to cross the intestinal epithelial barrier. Strikingly, L. monocytogenes has also evolved mechanisms to counter innate response to overcome host defense. The InlC interferes with innate immune responses in murine macrophages in vivo (Gouin et al., 2010); thus, after translocation to LP, L. monocytogenes may use InlC to dampen immune responses for the systemic spread. This well-orchestrated regulation of NF-κB-mediated response at different stages of infection may allow an efficient spread of L. monocytogenes to peripheral tissues.
Here, we demonstrate that during L. monocytogenes infection, LAP activates MLCK to redistribute junctional proteins, claudin-1, occludin, and E-cad to facilitate bacterial translocation across the gut epithelium in the early stage of infection (24–48 hr). InlA uses E-cad as the receptor, but it is intriguing to learn that L. monocytogenes infection redistributes E-cad to facilitate bacterial translocation. In the absence of InlA-E-cad-mediated uptake (in a mouse model), the bacterium can use the LAP-Hsp60-mediated translocation pathway to gain access for successful infection. This flexibility may make the bacterium far less susceptible to clearance by the innate immune defense by providing easier access to the LP. Our results from Caco-2 cells (functional InlA-E-cad) further suggest that LAP-Hsp60-mediated translocation may be an important precursor event for InlA-dependent epithelial translocation that provides access to E-cad in the AJ of the permissive model. Alternatively, LAP-Hsp60-mediated translocation may serve as an active mechanism for InlA-independent translocation in InlA-permissive hosts such as humans, rabbits, guinea pigs, and gerbils in addition to epithelial invasion via “villous cell extrusion” (Pentecost et al., 2006) and at the empty goblet cell junction (Nikitas et al., 2011).
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-NF-κB p65 | Cell Signaling | Cat #8242, RRID: AB_10859369 |
| Rabbit monoclonal anti-Phospho-NF-κB p65 (Ser536) | Cell Signaling | Cat # 3033, RRID: AB_331284 |
| Mouse monoclonal anti-IκBα | Cell Signaling | Cat # 4814, RRID: AB_390781 |
| Mouse monoclonal anti-IKKα | Cell Signaling | Cat # 11930, RRID: AB_2687618 |
| Rabbit monoclonal anti-IKKβ | Cell Signaling | Cat # 8943, RRID: AB_11024092 |
| Rabbit monoclonal anti-phospho-IκB-α (Ser32) | Cell Signaling | Cat # 2859, RRID: AB_561111 |
| Goat anti-rabbit IgG (HRP-linked) | Cell Signaling | Cat # 7074, RRID: AB_2099233 |
| Horse anti-mouse IgG (HRP-linked) | Cell Signaling | Cat # 7076, RRID: AB_330924 |
| Mouse monoclonal anti-Hsp60 | Enzo life Science | Cat # ADI-SPA-806F, RRID: AB_11177888 |
| Rabbit monoclonal anti-TATA banding protein | Novus Biologicals | Cat # NBP1-96038. RRID: AB_11015773 |
| Mouse monoclonal anti-β-actin | Santa Cruz Biotechnology | Cat # sc-47778, RRID: AB_626632 |
| Normal rabbit IgG | Santa Cruz Biotechnology | Cat # sc-2027, RRID: AB_737197 |
| Normal mouse IgG | Santa Cruz Biotechnology | Cat # sc-2025, RRID: AB_737182 |
| Mouse monoclonal anti-LAP | Our lab (mAb-H7) | N/A |
| Mouse monoclonal anti-InlA | Marcelo Mendonca, University of Pelotas, Brazil | N/A |
| Rabbit polyclonal anti-InlB | Our lab (PAb404) | N/A |
| Mouse monoclonal anti-ZO-1 | Thermo Fisher Scientific | Cat # 3391-00, RRID: AB_2533147 |
| Rabbit polyclonal anti-Listeria | Our Lab | N/A |
| Rabbit polyclonal anti-Hsp60 | Our Lab (Burkholder and Bhunia, 2010) | N/A |
| Goat anti-mouse IgG (H+L), F(ab’)2 Fragment (Alexa Fluor 555 Conjugate) antibody | Cell Signaling | Cat # 4409, RRID: AB_1904022 |
| Goat anti-rabbit IgG (H+L), F(ab’)2 Fragment (Alexa Fluor 555 Conjugate) antibody | Cell Signaling | Cat #4413, RRID: AB_10694110 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed-FITC Conjugate | Thermo Fisher Scientific | Cat # F-2765, RRID: AB_2536525 |
| Rabbit polyclonal anti-claudin-1 | Thermo Fisher Scientific | Cat # 51-9000, RRID: AB_2533916 |
| Mouse monoclonal anti-claudin-1 | Santa Cruz Biotechnology | Cat # sc-166338, RRID: AB_2244863 |
| Rabbit polyclonal anti-occludin | Thermo Fisher Scientific | Cat # 71-1500, RRID: AB_2533977 |
| Mouse monoclonal anti-Beta-catenin | Thermo Fisher Scientific | Cat # MA1-301, RRID: AB_1070649 |
| Rat monoclonal anti-ZO-1 | Supernatant from the Anti-ZO-1 hybridoma R40.76 | RRID: AB_628459 |
| Rabbit monoclonal anti-pMLC(Ser19) | Santa Cruz Biotechnology | Cat # sc-293109, RRID: AB_10847539 |
| Rabbit polyclonal anti-MLCK | Thermo Fisher Scientific | Cat # PA5-15177, RRID: AB_2298066 |
| Rabbit monoclonal anti-E-cadherin(Human) | Cell Signaling | Cat # 3195, RRID: AB_2291471 |
| Mouse monoclonal anti-E-cadherin(Mouse) | Thermo Fisher Scientific | Cat # 33-4000, RRID: AB_2533118 |
| Rabbit polyclonal anti-MLC-2 | Cell Signaling | Cat # 3672, RRID: AB_10692513 |
| Rabbit monoclonal anti-MEK ½ | Cell Signaling | Cat # 8727, RRID:AB_10829473 |
| Rabbit polyclonal anti-Pan Cadherin | Cell Signaling | Cat # 4068, RRID: AB_10693605 |
| Goat anti-rat IgG (H+L), (Alexa Fluor 555 Conjugate) antibody | Cell Signaling | Cat # 4417, RRID: AB_10696896 |
| Rabbit monoclonal human TNF-α neutralizing antibody | Cell Signaling | Cat # 7321, RRID: AB_10925386 |
| Rabbit polyclonal human IL-6 neutralizing antibody | Thermo Fisher Scientific | Cat # P620, RRID: AB_223481 |
| Bacterial and Virus Strains | ||
| L. monocytogenes F4244 (WT) | CDC, Atlanta, GA | N/A |
| L. monocytogenes KB208 (lap−) | (Jagadeesan et al., 2010) | N/A |
| L. monocytogenes CKB208 (lap−lap+) | (Jagadeesan et al., 2010) | N/A |
| L. monocytogenes AKB301 (ΔinlA) | (Burkholder and Bhunia 2010) | N/A |
| L. monocytogenes AKB302 (ΔinlA inlA+) | (Burkholder and Bhunia 2010) | N/A |
| L. monocytogenes 10403S | Dr. D.A. Portnoy, UC-Berkeley | N/A |
| L. monocytogenes DPL2161(ALLO) | Dr. D.A. Portnoy, UC-Berkeley | N/A |
| L. monocytogenes DPL4406(AinlB) | Dr. D.A. Portnoy, UC-Berkeley | N/A |
| L. innocua F4248 | CDC, Atlanta, GA | N/A |
| Clear coli BL21(DE3) Electrocompetent Cells | Lucigen Corporation | Cat# 60810 |
| E. coli BL21(DE3) +pET32a-ELAP-2 | (Jagadeesan et al., 2010) | N/A |
| E. coli (Clear coli BL21-DE3)+pET32a-ELAP-2 | This Study | N/A |
| E. coli pET28b-1 | Dr. Pascale Cossart (Institut Pasteur, France) | N/A |
| Biological Samples | ||
| Epithelial cells isolated from ileum of 8–10 week old A/J mice | N/A | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human TNF-α | R&D Systems | Cat # 210-TA |
| Recombinant mouse TNF-α | R&D Systems | Cat # 410-MT |
| LAP from E. coli BL21(DE3 )+pET32a-ELAP-2 | (Jagadeesan et al., 2010) | N/A |
| LAP from Clear coli BL21(DE3 )+pET32a-ELAP-2 | This study | N/A |
| InlA | Dr. Marcelo Mendonca, University of Pelotas, Brazil | N/A |
| InlB from E. coli pET28b-1 | Dr. Pascale Cossart (Institut Pasteur, France) | N/A |
| Modified Oxford agar Base | Neogen Corporation | Cat # 7428 |
| Modified Oxford agar Base supplement | Neogen Corporation | Cat # 7991 |
| Buffered Listeria enrichment broth | Neogen Corporation | Cat # 7675 |
| Buffered Listeria enrichment broth supplement | Neogen Corporation | Cat # 7980 |
| Collagenase | Worthington BioChem Corp | Cat # LS004188 |
| DNase 1 | Worthington BioChem Corp | Cat # LS002007 |
| RNAlater | Thermo Fisher Scientific | Cat # AM7021 |
| DAPI | Cell Signaling | Cat # 4083 |
| FITC-labeled 4 kDa dextran | Sigma-Aldrich | Cat # 46944 |
| PDTC | Sigma-Aldrich | Cat # P8765 |
| ML-9 | Sigma-Aldrich | Cat # C1172 |
| Dreverse PIK | Dr. J.R. Turner, Harvard Medical School | N/A |
| Bay-11–7085 | Thermo Fisher Scientific | Cat #50–101-2177 |
| Cytochalasin-D | Sigma-Aldrich | Cat # C2618 |
| Polymyxin B | Sigma-Aldrich | Cat # P4932 |
| LPS (E. coli Serotype R515, Re, TLR grade) | Enzo Life Sciences | Cat # ALX-581 −007 |
| Proteinase K | Sigma-Aldrich | Cat # P2308 |
| Protein G agarose beads | Micro-Protein Technologies | Cat # G100R2 |
| Critical Commercial Assays | ||
| Human Inflammation Antibody Array kit | Ray Biotech | Cat # AAH-INF-3–2 |
| Human TNF-α ELISA kit | Ray Biotech | Cat # ELH-TNFa-1 |
| Human IL-6 ELISA kit | Ray Biotech | Cat # ELH-IL6–1 |
| Mouse TNF-α ELISA kit | Ray Biotech | Cat # ELM-TNFa-1 |
| Mouse IL-6 ELISA kit | Ray Biotech | Cat # ELM-IL6–1 |
| Platinum SYBR Green One-Step qRT-PCR kit | Thermo Fisher Scientific | Cat # 11–736-051 |
| Turbo DNA-free Kit | Thermo Fisher Scientific | Cat # AM1907 |
| BCA assay kit | Thermo Fisher Scientific | Cat # PI23235 |
| M-PER extraction kit | Thermo Fisher Scientific | Cat # PI78501 |
| NE-PER extraction reagent | Thermo Fisher Scientific | Cat # PI78835 |
| Mem-Per Plus Membrane Protein Extraction Kit | Thermo Fisher Scientific | Cat # PI89842 |
| Renilla-firefly Luciferase assay kit | Thermo Fisher Scientific | Cat # PI16185 |
| Chromogenic LAL Endotoxin Assay Kit | Genscript | Cat # L00350 |
| LDH Cytotoxicity Assay Kit | Thermo Fisher Scientific | Cat # PI88954 |
| Toxin-Eraser Endotoxin Removal Kit | Genscript | Cat # L00338 |
| Alexa Fluor 647 Phalloidin | Cell Signaling | Cat # 8940S |
| Experimental Models: Cell Lines | ||
| Cell line: Caco-2 | ATTC | Cat # HTB37 |
| Cell line: Caco-2 presenting stable suppression of hsp60 mRNA via short hairpin RNA (shRNA) mediated targeting | (Burkholder and Bhunia 2010) | N/A |
| Cell line: Caco-2 presenting a non-targeting control shRNA vector | (Burkholder and Bhunia 2010) | N/A |
| Cell line: RAW 264.7 NF-κB luciferase reporter cell line | Novus Biologicals | Cat # NBP2–26253; |
| Experimental Models: Organisms/Strains | ||
| Mouse: A/J | The Jackson Laboratory | Stock # 000646 |
| Mouse: C57BL/6 | Bred at Purdue transgenic mouse facility | N/A |
| Mouse: MLCK knockout | (Clayburgh et al., 2005) | N/A |
| Oligonucleotides | ||
| qPCR Primers for murine TNF-α, IL-6 and Gapdh see Table S2 | (Gilbert et al., 2012) | N/A |
| Software and Algorithms | ||
| Bio-Rad quantity one | Bio-Rad | http://www.bio-rad.com/en-ch/product/quantity-one-1-d-analysis-software |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism 6.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Microsoft Excel | Microsoft | https://products.office.com/en-us/excel |
| Nikon Elements | Nikon | https://www.nikoninstruments.com/Products/Software/NIS-Elements-Basic-Research |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Arun K. Bhunia (bhunia@ purdue.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
A/J mice (female, 8–10 week-old; Jackson Laboratory) that are highly sensitive to oral L. monocytogenes challenge (Burkholder et al., 2009; Czuprynski et al., 2003) were used. The use of A/J mice allowed us to use a 108 CFU (10-fold lesser inoculum) to cause a systemic infection. For experiments with C57BL/6 mice, 6–8 week-old, male or female, wild-type C57BL/6 (MLCK+/+), or the 210-kDa MLCK−/− mice (Clayburgh et al., 2005), bred in our facility were used. Mice were housed in individual cages, provided ad libitum feed and water, and acclimatized for 5 days (A/J) before the experiments. On the day of the challenge, food and water were removed from the cages 5 hr prior to oral gavage to prevent mechanical blockage of the Listeria inoculum by food in the stomach, which may cause the inoculum to aspirate into the lungs. The 6-hr grown L. monocytogenes WT, lap−, and ΔinlA strains, each resuspended in 200 μL of phosphate-buffered saline (PBS, pH 7.4) containing approximately 1 × 108 CFU for A/J mice and 1 × 109 CFUforC57BL/6 MLCK+/+or the MLCK−/− mice were administered orally to randomly selected mice using a stainless steel ball-end feeding needle (Popper). The control mice received only PBS. The food was returned 1 hr pi, and the mice were sacrificed 24 hr and 48 hr pi using CO2 asphyxiation. All animal procedure (IACUC Protocol no. 1201000595) was approved by the Purdue University Animal Care and Use Committee, who adheres to the recommendations of the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Bacterial Strains and Growth Conditions
The bacterial strains are listed in the Key Resources Table L. monocytogenes F4244 (WT) serovar4b, the isogenic lap-deficient insertion mutant KB208 (lap−), the lap-complemented CKB208 (lap− lap+) (Jagadeesan et al., 2010), the ΔinlA in-frame deletion mutant (AKB301) and its complement (ΔinlA inlA+; AKB302) (Burkholder and Bhunia, 2010), the 10403s WT strain serovar ½a, and the in-frame deletion mutant strains, Δhly (DP-L2161) and the ΔinlB (DP-L4406) were used. The 10403s and its derivative strains were kindly provided by Dr. Portnoy at UC-Berkeley. All of the L. monocytogenes strains were grown in Tryptic soy broth containing 0.6% yeast extract (TSBYE; BD Bioscience) at 37°C with shaking for 12–16 hr unless otherwise indicated. The lap− strain was grown in TSBYE containing erythromycin (Em; 5 μg/ml) at 42°C, the lap−lap+ strain in TSBYE containing Em (5 μg/ml) and chloramphenicol (Cm; 5 mg/ml) at 37°C, and the ΔinlA inlA+ strain in TSBYE containing Cm (5 μg/ml) at 37°C. L. innocua F4248 was grown in TSBYE at 37°C for 12–16 hr.
Cell Lines
The human colon carcinoma Caco-2 cell line (ATTC # HTB37) from 25–35 passages were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) (Thermo Fisher Scientific) supplemented with 4 mM L-glutamine, 1 mM sodium pyruvate and 10% fetal bovine serum (FBS; Atlanta Biologicals). Caco-2 cells presenting stable suppression of hsp60 mRNA and Caco-2 presenting a non-targeting control shRNA vector were previously developed using shRNA (Burkholder and Bhunia, 2010) and cultured in DMEM supplemented with 4 mM L-glutamine, 1 mM sodium pyruvate, 10% FBS and 800 μg/ml Geneticin; G418. The NF-κB luciferase reporter cell line (Novus Biologicals) was cultured in DMEM supplemented with 4 mM L-glutamine, 1 mM sodium pyruvate, 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 3 μg/ml puromycin. All cell lines were maintained at 37°C with 5% CO2.
METHOD DETAILS
Enumeration of L. monocytogenes in Mouse Organs
The organs were harvested aseptically and homogenized using a tissue homogenizerin 4.5 mL (spleen, MLN, kidney) or 9 mL (liver) of buffered-Listeria enrichment broth (BLEB) containing 0.1% Tween 20 and selective antimicrobial agents (Neogen). To enumerate Listeria, the samples were serially diluted in PBS and plated onto modified Oxford (MOX; Neogen) agar plates. In specific experiments, small sections of liver, spleen and ileal tissue samples (1 cm) were cut into two parts, with one part fixed overnight in 10% formalin for histopathology or immunostaining and the other stored in RNA later (Thermo Fisher Scientific) for gene expression analysis. Urine excreted voluntarily during CO2 asphyxiation was collected in a plastic bag. Blood was collected using a 1 mL syringe with a 21G needle by cardiac puncture. To enumerate the Listeria in the blood, 50 μl of blood was diluted with 450 μl of BLEB immediately following collection and samples were serially diluted and plated as above.
To enumerate bacteria in the mucus, epithelial cell and the lamina propria fractions from the ileal tissue, a previously described protocol (Bou Ghanem et al., 2012) with minor modifications was used. Briefly, for separation of the mucus fraction, the ileum section (10 cm) was first flushed with sterile PBS, visible Peyer’s patches were removed and cut longitudinally. The tissue sections were then washed three times by incubating for 2 min in a tube containing 3 mL of 6 mM N-acetylcysteine (Sigma-Aldrich) and then shaken vigorously before transferring to a fresh tube. The washes (9 ml) were pooled and centrifuged for 20 min at 12,000 ×g. The pellets were resuspended in 0.5 mL of PBS, vortexed and serial dilutions were plated on MOX agar plates. To enumerate bacteria in the epithelial fraction, the ileal tissue from above were cut into small pieces (1 cm each) with a scalpel and incubated at 37°C with shaking in a tube containing 5 mL of RPMI (Invitrogen) supplemented with 5 mM EDTA, and 1 mM DTT for a total of 3 times. Each time, the tissues were transferred to fresh tubes containing 5 mL of RPMI supplemented with 5 mM EDTA, and 1 mM DTT. The combined three washes (15 ml) was centrifuged at low speed (1,200× g) to pellet the cells. The cell pellets and the supernatant fluids were processed separately to enumerate the intracellular and extracellular bacteria in the epithelial fraction. To enumerate intracellular bacteria, the pellet from either the epithelial fraction or the lamina propria fraction (extraction protocol mentioned below) was suspended in 5 mL of RPMI-5 containing 25 mg/ml gentamicin and the single cell suspension was incubated at 37°C with 5% CO2 for 30 min to kill any extracellular L. monocytogenes. The single cell suspension was then centrifuged at low speed (1,200× g) to pellet the cells and the pellets were washed twice in PBS. The pellets were then suspended in 0.5 mL PBS, vortexed to lyse the cells, serially diluted and plated on MOX agar plates. To quantify extracellular bacteria the supernatant from the washes of the epithelial cell fraction or the lamina propria fraction (extraction protocol mentioned below) was pooled and centrifuged at 12,000 x g for 20 min. The pellets were then resuspended in 0.5 mL PBS, vortexed and plated on MOX agar plates. To enumerate bacteria in the lamina propria fraction, the DTT and EDTA from the intestinal pieces were removed by two successive washes in 25 mL sterile PBS. The tissue pieces were then incubated in a sterile tube of digestion solution containing 4 mL of RPMI supplemented with 5% FBS and 1 mg/ml type IV collagenase and 40 μg/ml DNase I (both from Worthington) at 37°C for 40 min with shaking. This step was repeated in a fresh tube containing the digestion solution until the tissue pieces were completely dissolved. The combined digestion solution was centrifuged at low speed (1,200× g) to pellet the cells. The pellet and the supernatant were processed as described above to enumerate the intra-cellular and extracellular bacteria in the lamina propria fraction.
Immunofluorescence Staining and Confocal Microscopy
The mouse ileal-tissue sections were fixed with 10% formalin and embedded in paraffin. The tissues were sectioned (5 μm- thick), deparaffinized, and rehydrated for antigen retrieval by immersing the slides in boiling sodium citrate buffer (10 mM, pH 6.0) for 10 min. The tissue sections were permeabilized and blocked with PBS containing 0.3% Triton X-100 (Sigma-Aldrich) and 3% normal goat serum (Cell signaling) and immunostained with specific antibodies (Key Resources Table) by incubating overnight at 4°C. Following antibody incubation, slides were rinsed with PBS (3 cycles, 5 min), and were incubated with FITC or Alexa Fluor 555-conjugated secondary antibody for 2 hr at room temperature followed by washing three times with PBS (3 cycles, 5 min). The nuclei were stained with DAPI (500 ng/ml; Cell signaling) and slides were mounted in ProLong antifade reagent (Cell Signaling). The p65 and P-p65 nuclear positive cells were counted and expressed as average nuclear positive cells per villus. Images are representative of five different fields from 2–3 mice per treatment.
For antibody labeling in cells, Caco-2 cells were grown to 40%–50% confluence in four-chambered slides (Millipore). At the end of the treatment, the cells were fixed with 3.7% formaldehyde in PBS for 20 min and permeabilized and blocked with PBS containing 0.3% Triton X-100 and 3% BSA (Sigma-Aldrich) for 1 hr at room temperature and then incubated with respective antibodies (Key Resources Table) overnight at 4°C. Following antibody incubation, the cells were washed with PBS (3 cycles, 5 min) and incubated with FITC or Alexa Fluor 555-conjugated secondary antibody for 2 h at room temperature. The nuclei were stained with DAPI (500 ng/ ml; Cell signaling) and slides were mounted in ProLong antifade reagent (Cell Signaling).
All images were acquired using a Nikon A1R confocal microscope (equipped with 405nm/Argon/561nm lasers) using a Plan APO VC 60X/1.40 NA oil immersion objective with the Nikon Elements software (Nikon Instruments) at the Purdue Bindley Bioscience Imaging Facility. The X-Z and Y-Z cross-sections were produced by orthogonal reconstructions from z stack scanning at 0.15mm intervals taken with 60X objective in 5 mm thick paraffin embedded tissue section. Three-dimensional reconstructions were performed using Nikon elements software (Nikon Instruments).
Analysis of In Vivo Intestinal Permeability
The mice were orally gavaged with non-metabolizable 4 kDa FITC-labeled dextran (FD4; 15 mg/100 μl, Sigma-Aldrich) 4–5 hr prior to sacrifice. Serum and urine (50 μl each), collected above, were mixed with an equal volume of PBS, and fluorescence was measured (Em: 485 nm; Ex: 520 nm; Spectramax, Molecular Devices) and the FD4 concentration was calculated using a standard curve generated by serially diluting FD4 in PBS. The serum and urine from the mice that were uninfected and not administered FD4 were used to determine the background levels.
Epithelial Permeability, Bacterial Translocation, Invasion and Pharmacological Inhibitors
Caco-2 cells were grown as monolayers on Transwell inserts with 3.0 mm pores (Corning-Costar) for up to 14–21 days. TEER was measured to monitor the monolayer integrity (Millicells Voltmeter, Millipore). A TEER value of at least 200 Ω/cm2 (±10) was used as the basal value to monitor the monolayer integrity (Burkholder and Bhunia, 2010). Bacterial cells were washed three times in PBS and resuspended in DMEM-FBS (10%) at an MOI of ~50 and were added to the apical side of the Transwell system, and after 2 hr incubation period at 37°C in 5% CO2, the liquid was collected from the basal well, and then translocated bacteria were enumerated by plating (Burkholder and Bhunia, 2010). For analysis of FD4 flux, non-metabolizable 4 kDa FITC-labeled dextran (FD4; 5 mg/ml, Sigma-Aldrich) was added with bacteria (MOI, ~50) resuspended in DMEM-FBS (10%) and added to the apical side. After 2 hr incubation at 37°C in 5% CO2, the liquid was collected from the basal well and fluorescence was measured (Em: 485 nm; Ex: 520 nm; Spectramax, Molecular Devices). For pharmacological inhibition of NF-κB, Caco-2 cells were pretreated with inhibitors (PDTC, 100 mM, R&D Systems; BAY-11–7085, 10 μM, Sigma) for 30 min. For inhibition of MLCK, Caco-2 cells were pretreated with inhibitors (ML-9, 20 mM, Sigma; Dreverse PIK [Owens et al., 2005], 150 μM) for 30 min. In specified experiments, ML-9 and Dreverse PIK (permeant peptide inhibitor of kinase) were maintained for the duration of the experiment. For inhibition of actin polymerization, Caco-2 cells were pretreated with Cytochalasin D (1 μg/ml; Sigma-Aldrich) for 1 h. For neutralization of cytokines (TNF-α and IL-6), Caco-2 cells were incubated with anti-TNF-α (Cell signaling), anti-IL-6 (Thermo Fisher) antibodies (1 μg/ml each) or together for 24 h.
For re-association of externally added LAP to the lap− mutant, bacteria were harvested from 1 mL of overnight grown culture, and the pellet was washed three times in PBS before the addition of 1 or 2 mg/ml of purified LAP. The mixture was incubated for 30 min at 30°C with continuous shaking and then pelleted, washed five times in the PBS, resuspended in DMEM, and used in the aforementioned translocation assay.
To determine the effect of LAP on Caco-2 permeability at specified time points (0, 24, 48 and 72 hr) following incubation with recombinant purified LAP (1 μg/ml), 500 μL of FD4 (1 mg/ml in DMEM) was added to the apical side and the fluorescence readings (Em: 485 nm; Ex: 520 nm; Spectramax, Molecular Devices) for the basal medium (100 μl) were measured. Human TNF-α (10 ng/ml, R&D Systems) was added to both apical and basal sides and used as a positive control.
For bacterial invasion analysis, monolayers were washed with PBS after 1 hr of infection (MOI, ~50) and incubated with DMEM-FBS (10%) containing gentamicin (50 μg/ml) for 1 hr. Caco-2 cells were lysed with 0.1% Triton X, and the internalized bacteria were enumerated by plating.
Caco-2 Cell Viability Assay
To determine Caco-2 viability, cell culture supernatants from Caco-2 cells infected with WT, lap−, lap−lap+, DinlA and DinlA inlA+ strains (MOI ~50,2 hr) grown on Transwell inserts or treated with the purified recombinant LAP for 24 or 48 hr were assayed for lactate dehydrogenase release (Thermo Fisher Scientific). Two controls were included for calculation of percent cytotoxicity (LDH release). The low control consisted of supernatant from untreated Caco-2 cells with no exposure to bacteria. High control was from cells treated with 0.1% Triton X-100 for one minute.
Recombinant Protein Purification
Recombinant proteins (LAP and InlB) containing endogenous His, S and Trx tags derived from the pET-32a/pET28 cloning vector (Novagen) from E. coli BL21 or ClearColi (Lucigen) were purified using a Ni-affinity column. In ClearColi, LPS lacks secondary acyl chain thus eliminating endotoxicity. Briefly, for LAP purification, E. coli BL21 or ClearColi were each grown in 1 L LB broth (BD) with ampicillin (50 μg/ml) for 3 hr at 37°C. For InlB purification, E. coli pET28b-1 was grown in 1 L LB containing 30 mg/ml of kanamycin at 37°Cfor3 hr, and IPTG (1 mM) induced at 20°Cfor 12 hr. After sonication (total 7 min, with cycles of 30 s sonication and 15 s pulse; Branson Sonifier), the supernatants were purified by Ni-column. The Toxin-Eraser Endotoxin Removal Kit (Genscript) was used to remove LPS and the Toxin-Sensor Chromogenic LAL Endotoxin Assay Kit (Genscript) was used to detect any residual LPS in the samples. Protein concentrations were measured by Bradford assay, and the purity was monitored by SDS-PAGE (12.5%-acrylamide). Purified recombinant InlA was provided by Marcelo Mendonca (University of Pelotas, Brazil).
Cytokine Array and ELISA
A semiquantitative membrane-based RayBio Human Inflammation Antibody Array kit (Ray Biotech) was used to analyze a panel of 40 inflammatory mediators in Caco-2 cell supernatant infected either with the WT or lap− strain (MOI, ~50) at 37°C for 1 hr. After killing the extracellular bacteria by gentamicin (50 μg/ml), the Caco-2 cells were incubated for an additional 7 hr at 37°C (Jung et al., 1995). Recombinant LAP (1 μg/ml) from ClearColi was incubated for 8 h with Caco-2 cells. After immunoblotting, the reaction intensity was quantified using NIH ImageJ software. The data were normalized and expressed as the mean fold changes as a ratio of lap−/WT ± SEM. For the ELISA, Caco-2 cell supernatants were centrifuged (2,000 rpm at 4°C for 10 min) following treatment as above, and the quantification of TNF-α and IL-6 in was performed using human TNF-α and IL-6 ELISA kits (Ray Biotech) as per manufacturer’s instruction. The quantification of TNF-α and IL-6 protein levels was performed in the ileal tissue lysates from mice using mouse TNF-α and IL-6 ELISA kits (Ray Biotech) as per manufacturer’s instruction.
RNA Preparation and qRT-PCR
Total RNA was isolated from the mouse ileal tissues using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions and treated with the TURBO DNA-free Kit (Thermo Fisher Scientific) to remove residual genomic DNA. The transcript levels were determined using Superscript III Platinum SYBR Green One-Step qRT-PCR kit (Thermo Fisher Scientific). Primers were obtained from IDT (Table S2) and their recommended thermal cycling conditions were used. GAPDH was used as a house-keeping gene control for the ileal tissues. The 2-ΔΔCt method was used to calculate the relative changes in gene expression. The relative expression in each figure refers to the induction levels of the gene of interest relative to GAPDH, and these levels were then compared with that of an untreated control calibrator sample.
Histopathology
Thin tissue sections from above were stained with hematoxylin and eosin, and a board-certified veterinary pathologist microscopically examined the slides and the interpretations were based on standard histopathological morphologies. The pathologist, who was blinded to the bacterial strain, compared the ileal sections to the controls. To determine the extent of the inflammation, the mouse ileal tissues were scored on a scale of 0–3 for three parameters yielding a maximum score of 9. The scoring parameters were the amount of polymorphonuclear leukocyte infiltrate, mononuclear infiltrate and involvement of the submucosa. To grade the amount of polymorphonuclear leukocyte infiltrate and mononuclear infiltrate the following histomorphological scale was used: 3 = markedly increased, 2 = moderately increased, 1 = slightly increased and 0 = normal. To grade the involvement of the submucosa the following histomorphological scale was used: 3 = 50% or greater of the submucosal diameter, 2 = 10%–50%, 1 = < 10% and 0 = normal. The necrosis score for liver and spleen, 1 = <3 microscopic foci, 2 = >3 microscopic foci and 3 = massive necrosis. The inflammation scores of the livers and spleens were as follows: 1 = mild inflammation, 2 = moderate to marked inflammation associated with the foci of necrosis.
Immunoblotting
To extract the proteins from Caco-2 cells, cells were seeded in 6-well plates for 14–21 days. Following treatment, the cells were washed, scraped from the bottom of 6-well plates, suspended in PBS, and pelleted by centrifugation. Total protein from Caco-2 cells was extracted using the M-PER Extraction Kit (Thermo Fisher Scientific). Detergent-insoluble (membrane) and detergent-soluble (cytosolic) proteins were isolated using a Mem-Per Eukaryotic Protein Extraction Kit (Thermo Fisher Scientific) while the cytosolic and nuclear proteins were extracted using NE-PER Extraction Reagent (Thermo Fisher Scientific). To extract proteins from ileal epithelial cells, the epithelial cell fraction from ileal tissues were isolated as described above (Bou Ghanem et al., 2012) and the detergent-insoluble and the detergent-soluble proteins were isolated using a Mem-Per Eukaryotic Protein Extraction Kit.
To isolate the cell wall-associated proteins bacterial pellets were resuspended in 0.5 mL protein extraction buffer (0.5% SDS, 10 mM Tris at pH 6.9), and incubated at 37°C for 30 min with agitation. The samples were centrifuged (14,000 × g, 10 min, 4°C), and the supernatants (containing cell wall-associated proteins) were retained. Halt proteases and phosphatase inhibitors (Thermo Fisher Scientific) were used during all of the protein extraction procedures. The protein concentrations were determined by BCA assay (Thermo Fisher Scientific), and separated on SDS-PAGE gels (10%–12.5% polyacrylamide) and electro-transferred to polyvinylidene difluoride (PVDF) membrane (Millipore). The membranes were then blocked in 5% nonfat dry milk (NFDM) in 0.1 M Tris-buffered saline, pH 7.5 (TBS) containing 0.1% Tween 20 (TBST) for at least 1 hr. All of the primary antibodies were diluted in 5% bovine serum albumin (BSA) or 5% NFDM in TBST and incubated overnight. Secondary antibodies (1:2000 in 5% NFDM in TBST) incubated for 1 hr at 37°C, and a chemiluminescence method was performed using LumiGLO reagent (Cell Signaling). The membranes were exposed to X-ray films or visualized using the Chemi-Doc XRS system (Bio-Rad). To immunoprobe the same membrane with another antibody, the originally bound antibodies from the membranes were removed by incubating the membranes in Restore Western Blot Stripping Buffer (Thermo Fisher Scientific) according to the manufacturer’s protocol. To compare the reaction intensities, the average band densities were determined using Quantity One software (Bio-Rad). Densitometry reports represent the mean ± SEM after normalization to the loading control and are presented as %change of protein with the average for untreated cells (control) set at 100%. Immunoblots and densitometric reports are representative of 3 independent experiments. The antibodies used in this study are listed in Key Resources Table.
Luciferase Assay
The NF-kB Luciferase reporter RAW 264.7 cell line (Novus Biologicals), which expresses an optimized Renilla luciferase reporter gene (RenSP) under the transcriptional control of an NF-κB response element, was used. The cells were seeded (1 × 105 cells/well) into 96-well luminometer-compatible plates for 16 hr and then treated with analytes for 6 hr or infected with the WT, lap−, lap− lap+ or the ΔinlA strains (MOI ~10, 6 hr). Media from each well were aspirated, and then 100 μL of ice-cold PBS was added to each well. The plates were then frozen solid at −80°C overnight to completely lyse the cells, thawed back to room temperature, and luciferase assays were performed using the LightSwitch Luciferase assay kit (Novus Biologicals). Luminescence was measured as the relative luminescence units (RLU) using Spectramax (Spectramax, Molecular Devices) and reported as the relative fold change compared with that of the control cells that were treated with media alone. Recombinant human or mouse TNF-a (R&D Systems) and polymyxin B, LPS (E. coli Serotype R515, Re, TLR grade), and proteinase K (each from Sigma-Aldrich) were used.
Immunoprecipitation
Caco-2 cells were treated with or without purified LAP (1 μg/ml) for 30 min, rinsed with cold PBS, and lysed in Nondiet P-40 (NP-40) lysis buffer (20 mM Tris HCl, pH 8,137 mM NaCl, 1% NP-40, 2 mM EDTA). The cell lysates were pre-cleared with 10 μl protein G agarose beads (MicroProtein Technologies) for 1 h and the lysates were incubated with 2 μg of anti-IKKβ, 2 μg of anti-Hsp60, normal rabbit serum or normal mouse serum overnight at 4°C. The lysates were further mixed with 20 ml protein G agarose beads for 3 hr at 4°C. The beads were washed five times with 1 mL NP-40 lysis buffer. The protein precipitates were analyzed by immunoblotting, and the complexes were visualized by chemiluminescence assay.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experimental data were analyzed using GraphPad Prism (La Jolla, CA) software. For mouse microbial counts, statistical significance was assessed by Mann-Whitney test. In other experiments, comparisons between two datasets were performed using the unpaired Student t test. When comparisons between more than two datasets were performed, a one-way or a two-way analysis of variance with Tukey’s multiple-comparison test were performed. All data are representative of at least 3 independent experiments and specific numbers of mice per group are noted in corresponding figure legends. Unless otherwise indicated, data for all experiments are presented as the mean ± standard error of the mean (SEM).
Supplementary Material
Highlights.
LAP induces intestinal barrier dysfunction for L. monocytogenes translocation
LAP activates NF-κB and upregulates TNF-α and IL-6 for increased epithelial permeability
LAP interaction with its receptor, Hsp60, is crucial for NF-κB activation
LAP activates MLCK for cellular redistribution of claudin-1, occludin, and E-cadherin
ACKNOWLEDGMENTS
The research was supported by funds from the USDA (grant # 1935-42000072-02G), the AgSEED, and PRF at Purdue University. The authors acknowledge the use of the Bindley Bioscience Center and Histology Research Laboratory facilities at Purdue University. We thank A.K. Singh and G. Moreira for cloning of lap in E. coli ClearColi; G. Singh and all members of our lab in particular; S. Yu, X. Bai, C. To, D. Liu, W. Lv, M. Mathipa, L. Xu, T. Qi, J. Ren, and M.M. Chavez for help with the animal study; H. Kim for helpful discussions; J.C. Kershaw for critical reading of the manuscript; A.B. Taylor and J.A. Schaber for assistance with confocal microscopy; D. Brooks, G. Shafer, and V. Bernal-Crespo for histopathology; and Dr. Daniel A. Portnoy, UC Berkeley, for providing 10403s and its derivative strains.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures, two tables, and five videos and can be found with this article online at https://doi.org/10.1016/j.chom.2018.03.004.
DECLARATION OF INTERESTS
A provisional patent (62/569,020) has been issued.
REFERENCES
- Abreu MT, Thomas LS, Arnold ET, Lukasek K, Michelsen KS, and Arditi M (2003). TLR signaling at the intestinal epithelial interface. J. Endotoxin Res. 9, 322–330. [DOI] [PubMed] [Google Scholar]
- Baldwin DN, Vanchinathan V, Brown PO, and Theriot JA (2003). Agene-expression program reflecting the innate immune response of cultured intestinal epithelial cells to infection by Listeria monocytogenes. Genome Biol. 4, R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger E, Rath E, Yuan D, Waldschmitt N, Khaloian S, Allgauer M, Staszewski O, Lobner EM, Schottl T, Giesbertz P, et al. (2016). Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 7, 13171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierne H, Mazmanian SK, Trost M, Pucciarelli MG, Liu G, Dehoux P, Jaansch L, Garcia-del Portillo F, Schneewind O, and Cossart P; European Listeria Genome Consortium (2002). Inactivation of the srtA gene in Listeria monocytogenes inhibits anchoring of surface proteins and affects virulence. Mol. Microbiol. 43, 869–881. [DOI] [PubMed] [Google Scholar]
- Bonazzi M, Veiga E, Pizarro-Cerda J, and Cossart P (2008). Successive post-translational modifications of E-cadherin are required for InlA-mediated internalization of Listeria monocytogenes. Cell. Microbiol. 70, 2208–2222. [DOI] [PubMed] [Google Scholar]
- Bou Ghanem EN, Jones GS, Myers-Morales T, Patil PD, Hidayatullah AN, and D’Orazio SE (2012). InlA promotes dissemination of Listeria monocytogenes to the mesenteric lymph nodes during food borne infection of mice. PLoS Pathog. 8, e1003015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkholder KM, and Bhunia AK (2010). Listeria monocytogenes uses Listeria adhesion protein (LAP) to promote bacterial transepithelial translocation and induces expression of LAP receptor Hsp60. Infect. Immun. 78, 5062–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkholder KM, Kim K-P, Mishra KK, Medina S, Hahm B-K, Kim H, and Bhunia AK (2009). Expression of LAP, a SecA2-dependent secretory protein, is induced under anaerobic environment. Microbes Infect. 11, 859–867. [DOI] [PubMed] [Google Scholar]
- Chiba S, Nagai T, Hayashi T, Baba Y, Nagai S, and Koyasu S (2011). Listerial invasion protein internalin B promotes entry into ileal Peyer’s patches in vivo. Microbiol. Immunol. 55, 123–129. [DOI] [PubMed] [Google Scholar]
- Chun JN, Choi B, Lee KW, Lee DJ, Kang DH, Lee JY, Song IS, Kim HI, Lee S-H, Kim HS, et al. (2010). Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS One 5, e9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, and Turner JR (2005). Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J. Clin. Invest. 115, 2702–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corr S, Hill C, and Gahan CGM (2006). An in vitro cell-culture model demonstrates internalin- and hemolysin-independent translocation of Listeria monocytogenes across M cells. Microb. Pathog. 41, 241–250. [DOI] [PubMed] [Google Scholar]
- Czuprynski CJ, Faith NG, and Steinberg H (2003). A/J mice are susceptible and C57BL/6 mice are resistant to Listeria monocytogenes infection by intragastric inoculation. Infect. Immun. 71, 682–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disson O, Grayo S, Huillet E, Nikitas G, Langa-Vives F, Dussurget O, Ragon M, Le Monnier A, Babinet C, Cossart P, and Lecuit M (2008). Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455, 1114–1118. [DOI] [PubMed] [Google Scholar]
- Dramsi S, Biswas I, Maguin E, Braun L, Mastroeni P, and Cossart P (1995). Entry of Listeria monocytogenes into hepatocytes requires expression of InIB, a surface protein of the internalin multigene family. Mol. Microbiol. 16, 251–261. [DOI] [PubMed] [Google Scholar]
- Dziewanowska K, Carson AR, Patti JM, Deobald CF, Bayles KW, and Bohach GA (2000). Staphylococcal fibronectin binding protein interacts with heat shock protein 60 and integrins: role in internalization by epithelial cells. Infect. Immun. 68, 6321–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbíčová T, Koláčková I, Pantůček R, and Karpíšková R (2015). A novel mutation leading to a premature stop codon in inlA of Listeria monocytogenes isolated from neonatal listeriosis. New Microbiol. 38, 293–296. [PubMed] [Google Scholar]
- Gilbert S, Zhang R, Denson L, Moriggl R, Steinbrecher K, Shroyer N, Lin J, and Han X (2012). Enterocyte STAT5 promotes mucosal wound healing via suppression of myosin light chain kinase-mediated loss of barrier function and inflammation. EMBO Mol. Med. 4, 109–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouin E, Adib-Conquy M, Balestrino D, Nahori M-A, Villiers V, Colland F, Dramsi S, Dussurget O, and Cossart P (2010). The Listeria monocytogenes InlC protein interferes with innate immune responses by targeting the IkappaB kinase subunit IKKalpha. Proc. Natl. Acad. Sci. USA 107, 17333–17338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht G, Pestic L, Nikcevic G, Koutsouris A, Tripuraneni J, Lorimer DD, Nowak G, Guerriero V Jr., Elson EL, and Lanerolle PD (1996). Expression of the catalytic domain of myosin light chain kinase increases paracellular permeability. Am. J. Physiol. 271, C1678–C1684. [DOI] [PubMed] [Google Scholar]
- Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, and Podolsky DK (2003). CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology 124, 993–1000. [DOI] [PubMed] [Google Scholar]
- Holch A, Ingmer H, Licht TR, and Gram L (2013). Listeria monocytogenes strains encoding premature stop codons in inlA invade mice and guinea pig fetuses in orally dosed dams. J. Med. Microbiol. 62, 1799–1806. [DOI] [PubMed] [Google Scholar]
- Jagadeesan B, Koo OK, Kim KP, Burkholder KM, Mishra KK, Aroonnual A, and Bhunia AK (2010). LAP, an alcohol acetaldehyde dehydrogenase enzyme in Listeria, promotes bacterial adhesion to enterocyte-like Caco-2 cells only in pathogenic species. Microbiology 156, 2782–2795. [DOI] [PubMed] [Google Scholar]
- Jagadeesan B, Fleishman Littlejohn AE, Amalaradjou MAR, Singh AK, Mishra KK, La D, Kihara D, and Bhunia AK (2011). N-terminal Gly(224)-Gly(4n) domain in Listeria adhesion protein interacts with host receptor Hsp60. PLoS ONE 6, e20694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaradat ZW, Wampler JW, and Bhunia AW (2003). A Listeria adhesion protein-deficient Listeria monocytogenes strain shows reduced adhesion primarily to intestinal cell lines. Med. Microbiol. Immunol. (Berl.) 192, 85–91. [DOI] [PubMed] [Google Scholar]
- Jung HC, Eckmann L, Yang S-K, Panja A, Fierer J, Morzycka-Wroblewska E, and Kagnoff MF (1995). A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J. Clin. Invest. 95, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayal S, Lilienbaum A, Join-Lambert O, Li X, Israël A, and Berche P (2002). Listeriolysin O secreted by Listeria monocytogenes induces NF-kappaB signalling by activating the IkappaB kinase complex. Mol. Microbiol. 44, 1407–1419. [DOI] [PubMed] [Google Scholar]
- Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, and Flavell RA (2005). Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734. [DOI] [PubMed] [Google Scholar]
- Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, and Cossart P (1992). L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68, 521–531. [DOI] [PubMed] [Google Scholar]
- Lecuit M, Dramsi S, Gottardi C, Fedor-Chaiken M, Gumbiner B, and Cossart P (1999). A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J. 18, 3956–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit M, Vandormael-Pournin S, Lefort J, Huerre M, Gounon P, Dupuy C, Babinet C, and Cossart P (2001). A transgenic model for listeriosis: role of internalin in crossing the intestinal barrier. Science 292, 1722–1725. [DOI] [PubMed] [Google Scholar]
- Lecuit M, Sonnenburg JL, Cossart P, and Gordon JI (2007). Functional genomic studies of the intestinal response to a foodborne enteropathogen in a humanized gnotobiotic mouse model. J. Biol. Chem. 282, 15065–15072. [DOI] [PubMed] [Google Scholar]
- Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, and Said HM (2004). TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G367–G376. [DOI] [PubMed] [Google Scholar]
- Ma TY, Boivin MA, Ye D, Pedram A, and Said HM (2005). Mechanism of TNF-alpha modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G422–G430. [DOI] [PubMed] [Google Scholar]
- Mansell A, Braun L, Cossart P, and O’Neill LAJ (2000). A novel function of InIB from Listeria monocytogenes: activation of NF-kappaB in J774 macrophages. Cell. Microbiol. 2, 127–136. [DOI] [PubMed] [Google Scholar]
- Marchiando AM, Shen L, Graham WV, Edelblum KL, Duckworth CA, Guan Y, Montrose MH, Turner JR, and Watson AJ (2011). The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology 140, 1208–1218.e1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J, Ohayon H, Gounon P, Mege R-M, and Cossart P (1996). E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 84, 923–932. [DOI] [PubMed] [Google Scholar]
- Nikitas G, Deschamps C, Disson O, Niault T, Cossart P, and Lecuit M (2011). Transcytosis of Listeria monocytogenes across the intestinal barrier upon specific targeting of goblet cell accessible E-cadherin. J. Exp. Med. 208, 2263–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus A, Hayden MS, and Ghosh S (2011). Crosstalk in NF-κB signaling pathways. Nat. Immunol. 12, 695–708. [DOI] [PubMed] [Google Scholar]
- Owens S-E, Graham WV, Siccardi D, Turner JR, and Mrsny RJ (2005). A strategy to identify stable membrane-permeant peptide inhibitors of myosin light chain kinase. Pharm. Res. 22, 703–709. [DOI] [PubMed] [Google Scholar]
- Pentecost M, Otto G, Theriot JA, and Amieva MR (2006). Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog. 2, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Jacks PS, and Hinrichs DJ (1988). Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167,1459–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pron B, Boumaila C,Jaubert F, Sarnacki S,Monnet JP,Berche P,and Gaillard JL (1998). Comprehensive study of the intestinal stage of listeriosis in a rat ligated ileal loop system. Infect. Immun. 66, 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radoshevich L, and Cossart P (2018). Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 16, 32–46. [DOI] [PubMed] [Google Scholar]
- Rajabian T, Gavicherla B, Heisig M, Muller-Altrock S, Goebel W, Gray-Owen SD, and Ireton K (2009). The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat. Cell Biol. 11, 1212–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan T, Nally K, Carmody R, Houston A, Shanahan F, Macsharry J, and Brint E (2013). Identification of TLR10 as a key mediator of the inflammatory response to Listeria monocytogenes in intestinal epithelial cells and macrophages. J. Immunol. 191, 6084–6092. [DOI] [PubMed] [Google Scholar]
- Speth C, Prohaszka Z, Mair M, Stockl G, Zhu X, Jobstl B, Fust G, and Dierich MP (1999). A 60 kD heat-shock protein-like molecule interacts with the HIV transmembrane glycoprotein gp41. Mol. Immunol. 36, 619–628. [DOI] [PubMed] [Google Scholar]
- Stevenson BR, and Begg DA (1994). Concentration-dependent effects of cytochalasin D on tight junctions and actin filaments in MDCK epithelial cells. J. Cell Sci. 107, 367–375. [DOI] [PubMed] [Google Scholar]
- Tilney LG, and Portnoy DA (1989). Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J. Cell Biol. 109, 1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya K, Kawamura I, Takahashi A, Nomura T, Kohda C, and Mitsuyama M (2005). Listeriolysin O-induced membrane permeation mediates persistent interleukin-6 production in Caco-2 cells during Listeria monocytogenes infection in vitro. Infect. Immun. 73, 3869–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Häcker H, and Wagner H (2001). Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J. Biol. Chem. 276, 31332–31339. [DOI] [PubMed] [Google Scholar]
- Wampler JL, Kim KP, Jaradat Z, and Bhunia AK (2004). Heat shock protein 60 acts as a receptor for the Listeria adhesion protein in Caco-2 cells. Infect. Immun. 72, 931–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert T, Pasche B, Rochon M, Deppenmeier S, van den Heuvel J, Gruber AD, Heinz DW, Lengeling A, and Schubert WD (2007). Extending the host range of Listeria monocytogenes by rational protein design. Cell 129, 891–902. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S, Roy S, Rasper D, Hennessey T, Aubin Y, Cassady R, Tawa P, Ruel R, Rosen A, and Nicholson DW (1999). Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J. 18, 2049–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.