

Abstract
Therapeutic options for patients with lower-risk myelodysplastic syndrome (MDS) who have failed prior therapies are limited particularly after hypomethylating agent. Several studies have indicated that deregulation of innate immunity signaling is critical in the pathogenesis of MDS. This process involves Toll-like receptor stimulation, cytokine overexpression, and nuclear factor-kB (NF-kB) activation. Since ruxolitinib, a JAK1/JAK2 inhibitor, suppresses NF-kB expression, we conducted a phase 1 dose-escalation study to determine the safety and efficacy of ruxolitinib in previously treated lower-risk MDS patients with evidence of NF-kB activation. Nineteen patients, 8 with chronic myelomonocytic leukemia and 11 with MDS, were enrolled. No dose limiting toxicity was observed and the maximum tolerated dose was 20 mg twice daily. Responses were restricted to MDS patients with an overall response rate of 22% [hematological improvement in platelets (HI-P) =2, hematological improvement in erythrocytes (HI-E) =1, partial cytogenetic response (PCyR) =1]. Of these patients, 2 relapsed (HI-P and PCyR) and 2 continue to be in HI-P and HI-E, respectively, with ongoing therapy. Meaningful improvement in bone marrow dysplasia was only seen in a patient who achieved HI-E. Phosphorylated p65 (pp65) decreased in 6 of 15 patients (40%) including the 2 patients with continued response to treatment and increased in a patient who relapsed after a short-lived HI-P. This suggests potential correlation between reduction in pp65 expression and response duration. In conclusion, ruxolitinib was well-tolerated in previously treated lower-risk MDS patients with evidence of NF-kB activation and resulted in low but significant frequency of responses. (NCT01895842)
Keywords: myelodysplastic syndromes, NF-kB, toll-like receptors, TNF-α, IL-8
INTRODUCTION
Myelodysplastic syndromes (MDS) are a diverse group of clonal myeloid disorders characterized by ineffective hematopoiesis, cytopenias, and a tendency towards transformation to acute myeloid leukemia (AML) [1]. Based on the International Prognostic Scoring System (IPSS), patients with MDS are prognostically stratified into either lower-risk (low and intermediate-1) or higher-risk (intermediate-2 and high) disease [2]. By the revised IPSS, lower-risk includes very low, low, and a fraction of intermediate-risk patients [3, 4].
Three drugs are currently used in MDS. Lenalidomide has activity in patients with lower-risk MDS who have transfusion-dependent anemia and deletion of chromosome 5q [5–7]. Hypomethylating agents (HMAs) are considered the standard of care in the treatment of higher-risk MDS and symptomatic lower-risk patients who have failed or were not candidates for growth factors or lenalidomide [8–15]. Prognosis of lower-risk MDS patients who failed one line of therapy is heterogeneous with a median OS among those with HMA-failure of about 14 to 17 months [16–19]. Given the limited therapeutic options available for lower-risk MDS patients at the time of HMA-failure who are not candidates for allogeneic stem cell transplantation (SCT), there is a clear unmet need for novel therapeutic strategies for this poor-risk population.
Several studies have indicated that deregulation of innate immune and inflammatory signaling play a key role in the pathogenesis of MDS characterized by NF-kB activation [20, 21]. Ruxolitinib is a selective JAK1/JAK2 inhibitor FDA approved for the treatment of both intermediate and high-risk myelofibrosis and polycythemia vera patients resistant or intolerant to hydroxyurea [22–24]. Based on the interconnection between the NF-kB and JAK-STAT pathways and the ability of ruxolitinib to suppress NF-kB expression, as demonstrated in preclinical studies,[25] we conducted a phase 1 study to determine the safety and efficacy of ruxolitinib in patients with lower-risk MDS who failed at least one prior therapy.
MATERIALS AND METHODS
The clinical trial registration number is NCT01895842. The study was conducted according to the standards of Good Clinical Practice, per institutional research policies and procedures, and in accordance with the Declaration of Helsinki. Approval for this study was obtained from The University of Texas M.D. Anderson Cancer Center’s institutional review board.
Patient Population
Patients with previously treated low or intermediate-1 risk MDS, classified using IPSS,[2] with evidence of NF-kB and/or JAK-STAT activation determined by either presence of a JAK2 mutation or phosphorylated p65 (pp65), a clinical surrogate of NF-kB activation, in ≥5% of bone marrow (BM) cells were enrolled [26, 27]. Patients with elevated levels of β2-microglobulin (β2M) [2 × upper limit of normal (ULN)] were also candidates. Patients had to have adequate hepatic and renal function [serum creatinine ≤ 2 × ULN or 24-hour creatinine clearance ≥ 60 mL/min, total bilirubin ≤ 2 × ULN, aspartate aminotransferase and alanine aminotransferase ≤2.5 × ULN or ≤ 5 × ULN if there if hepatic involvement]. Prior therapy with growth factors, lenalidomide, HMAs, or other investigational agents were allowed with a 4-week wash out period required before the start of ruxolitinib. Major exclusion criteria were treatment-naïve low or intermediate-risk MDS patients, uncontrolled medical comorbidities including uncontrolled active systemic infections, concomitant use of potent CYP3A4 inhibitors, white blood cell count > 30 × 103 K/μL, and prior SCT. All patients provided a written informed consent prior to study enrollment.
Study Design and Treatment
Because of potential hematological toxicity, the primary objective was to determine the maximum tolerated dose (MTD) and dose-limiting toxicity (DLT) of ruxolitinib in patients with low or intermediate-1 risk MDS. The secondary objective was to assess the clinical and molecular activity of ruxolitinib.
Patients were assigned to 4 dose levels of ruxolitinib [5, 10, 15, and 20 mg twice a day (BID)] using a 3+3 dose-escalation design to determine the MTD. The starting dose of ruxolitinib was 5 mg orally BID on a 28 days cycle. After completion of the first dose level, patients at subsequent dose levels received initially the prior dose level for 1 month with dose escalation on the second month (Table 1). Once the MTD was determined, 7 additional patients were treated at the MTD to further evaluate the safety and efficacy of ruxolitinib.
Table 1.
Dose Escalation Schema
| Dose Level | Ruxolitinib (mg orally twice daily on a 28 day cycle) |
|---|---|
| 1 | 5 |
| 2 | 5 first cycle then 10 in subsequent cycles |
| 3 | 10 first cycle then 15 in subsequent cycles |
| 4 | 15 first cycle then 20 in subsequent cycles |
Assessment of Safety and Efficacy
Responses were based on the 2006 International Working Group criteria [28]. OS was measured from start of therapy to date of death or last follow-up. All treatment-emergent adverse events (TEAEs) were recorded and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0. Hematological toxicity was defined using the following criteria: >50% increase in transfusion needs in patients who were transfusion dependent prior to ruxolitinib therapy, lasting for >12 weeks; need for any transfusion for >12 weeks in patients who were transfusion independent prior to therapy; unexpected neutropenic fever or grade 4 bleeding related to study drug; peripheral cytopenias (ANC <500 k/μL or platelets <10 k/μL) in the context of a hypocellular BM (<10% cellularity). Toxicities were assessed during the first 2 cycles of therapy to determine the MTD and DLT. DLTs were defined as any grade ≥3 non-hematologic treatment-related adverse event (TRAE) or grade ≥3 hematologic TRAE as defined above. MTD was defined as the highest dose at which <33% of patients experience a DLT.
Patients were eligible for a one dose level reduction if they experienced either a grade ≥ 3 non-hematologic toxicity or hematologic toxicity. If hematological toxicity was observed, subsequent cycles were administered after count recovery (ANC >500 k/μL and platelet count > 30 k/μL unless lower at baseline). Patients were taken off study if they required >2 dose reductions or drug discontinuation for >6 weeks. Doses below 5 mg were not allowed.
Correlative and Cytomorphological Studies
Upon enrollment, BM aspirates were evaluated for age-adjusted cellularity, blast count, dysplasia (defined by MDS dysplasia score described by Della Porta, et al),[29] and NF-κB activation using pp65 immunofluorescence as previously described [30]. These characteristics were compared to the last BM samples obtained during treatment. Details regarding method of pp65 detection and analysis, cytogenetic studies, and molecular analysis were previously described [30].
Statistical Analysis
The efficacy population included all patients who completed 1 cycle of therapy and were assessed at least once for disease response. The safety population included all patients who received at least one dose of ruxolitinib. The sample size was calculated using the Simon’s two-stage design. Two sided, exact 95% confidence intervals (CIs) were calculated for response rates and pp65 expression. OS was estimated using Kaplan-Meier method. Safety was assessed by analyzing the reported incidence of TEAEs.
RESULTS
Patient Characteristics
From February 2014 to December 2016, 20 patients were enrolled. One patient withdrew consent prior to receiving the study drug. Of the remaining 19 patients, 8 (42%) were classified as CMML, 6 (32%) had MDS with multilineage dysplasia (MDS-MLD), 2 (11%) had MDS with isolated del (5q), 1 (5%) had myelodysplastic syndrome/myeloproliferative neoplasm overlap with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T), 1(5%) had MDS-MLD in the context of systemic mastocytosis [systemic mastocytosis with an associated hematological neoplasm (SM-AHN)], and 1 (5%) patient had therapy-related MDS (T-MDS). Patient characteristics are summarized in Table 2.
Table 2.
Patient Characteristics
| Variable | Total Population (N=19) | ||
|---|---|---|---|
| N (%) | Median [range] | Mean [95% CI] | |
| Age (years) | 71 [53-87] | ||
| Male | 16 (84) | ||
| Diagnosis | |||
| CMML | 8 (42) | ||
| WHO classification 2016 | |||
| CMML-0 | 3 (16) | ||
| CMML-1 | 5 (26) | ||
| CMML- dysplastic | 5 (26) | ||
| CMML- proliferative | 3 (16) | ||
| MDS | 11 (58) | ||
| MDS-MLD | 6 (32) | ||
| MDS-isolated 5q | 2 (10) | ||
| MDS/MPN-RS-T | 1 (5) | ||
| MDS-MLD with SM-AHN | 1 (5) | ||
| T-MDS | 1 (5) | ||
| IPSS classification | |||
| Low | 6 (32) | ||
| Intermediate-1 | 13 (68) | ||
| IPSS-Revised classification | |||
| Very low | 1 (5) | ||
| Low | 11 (58) | ||
| Intermediate | 4 (21) | ||
| High | 3 (16) | ||
| Transfusion dependence | 16 (84) | ||
| PRBCs | 12 (63) | ||
| PRBCs + platelets | 3 (16) | ||
| Platelets | 1 (5) | ||
| Karyotype | |||
| Diploid | 13 (68) | ||
| Intermediate | 6 (32) | ||
| Hgb (g/dl) | 9.1 [8.4-9.8] | ||
| Platelets (× 109/L) | 127 [74-178] | ||
| ANC (×109/L) | 4.7 [2.5-6.9] | ||
| Bone marrow blasts (%) | 2 [0-6] | ||
| Blasts ≥ 5% | 3 (16) | ||
| B2M level (mg/L) | 4.4 [3.4-5.5] | ||
| No. of Prior therapy | 2 [1-4] | ||
| Hypomethylating agents | 14 (74) | ||
| Growth Factors | 7 (37) | ||
| Lenalidomide | 4 (21) | ||
| Investigational agents | 3 (16) | ||
| Bortezomib | 3 (16) | ||
| JAK2 positive at baseline | 8 (42) | ||
Abbreviations: CMML: chronic Myelomonocytic leukemia; MDS: myelodysplastic syndromes; MLD: multilineage dysplasia; MPN-RS-T: myeloproliferative neoplasm with ring sideroblasts and thrombocytosis; SM-ANH: systemic mastocytosis with an associated hematological neoplasm; T-MDS: therapy-related myelodysplastic syndrome; IPSS: International Prognostic Scoring System; PRBCs: packed red blood cells; Hgb: Hemoglobin; ANC: Absolute Neutrophil Count; B2M: β2-microglobulin; N: Number; CI: Confidence Interval.
Using IPSS, 6 patients (32%) had low risk and 13 (68%) had intermediate-1 risk disease. Median age was 71 years (range, 53-87) with male predominance. Sixteen patients (84%) were transfusion dependent at enrollment: 12 (63%) required red blood cells (PRBCs), 3 (16%) required PRBCs and platelets, and 1 (5%) required platelets only. Median number of prior therapies was 2 (range, 1-4) of which 14 patients (74%) received prior HMA therapy [azacitidine (N=9), decitabine (N=4), and both (N=1)]. Thirteen patients (68%) had diploid cytogenetics and 6 (32%) had intermediate karyotype, with del5q (10%) and trisomy 8 (10%) being the most commonly reported chromosomal abnormalities. Inclusion criteria included elevated B2M in 9 (47%) patients, pp65 in 16 (84%), and presence of JAK2 mutation in 8 (42%) patients; 13 patients (68%) fulfilled ≥2 of these criteria. Sequencing data was available for 16 patients (84%); details of the mutations detected are present in Table 3. Patients received a median of 4 cycles (range, 1-24) of ruxolitinib.
Table 3.
Bone Marrow Characteristics
| IWG | Response | WHO Classification 2016 | Cellularity (%) | Blast (%) | Della Porta MDS Displasia Score | Pp65(%) | Cytogenetics | Somatic mutations | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pre-Rx | Post-Rx | Pre-Rx | Post-Rx | Pre-Rx | Post-Rx | Pre-Rx | Post-Rx | Pre-Rx | Post-Rx | Pre-Rx | Post-Rx | |||
| Responders | PCyR | MDS/MPN-RS-T | 80 | 80 | 0 | 0 | 15 | 12 | 8 | NA | 46,XX,inv(3)(q21q25)[18]46,XX[2] | 46,XX,inv(3)(q21q25)[20] | JAK2 | NA |
| *HI-P+ | MDS-MLD-RS | 45 | 30 | 0 | 0 | 4 | 4 | 63 | 33 | 46,XX[20] | 46,XX[20] | None | NA | |
| HI-P | T-MDS | 100 | 40 | 0 | 0 | 13 | 11 | 7 | 15 | 46,XY[20] | 46,XY[20] | ASXL1, EZH2, KRAS, TET2 | NA | |
| *HI-E+ | MDS-MLD | 100 | 50 | 0 | 0 | 11 | 0 | 17 | 10 | 46,XY[20] | 46,XY[20] | MDM2 | NA | |
| No Responders | NR | CMML0 | 30 | 65 | 2 | 2 | 11 | 6 | 21 | 30 | 46,XY[20] | 46,XY[20] | None | NA |
| NR | Del5q | 30 | 50 | 2 | 3 | 6 | 6 | 17 | 36 | 47,XY,del(5)(q22q35),+21[20] | 47,XY,del(5)(q22q35),+21[20] | None | NA | |
| NR | MDS-MLD (SM-AHN) | 70 | 70 | 4 | 8 | 10 | 12 | 16 | 63 | 46,XY,der(13)t(13;13)(p12;q14)[18]46,idem,der(22)t(13;22)(q14;p11.2)[2] | 46,XY,der(13)t(13;13)(p12;q14)[18]46,idem,der(22)t(13;22)(q14;p11.2)[2] | TET2, DNMT3A, KRAS | NA | |
| NR | CMML0 | 100 | 95 | 5 | 11 | 12 | 9 | ND | ND | 46,XY[20] | 46,XY[20] | NRAS | NRAS, KRAS | |
| NR | MDS-MLD | 100 | NA | 2 | 3 | 5 | 6 | 22 | 50 | 46,XY[20] | 46,XY[20] | JAK2 | JAK2 | |
| NR | MDS-MLD | NA | 75 | 5 | 3 | 12 | 9 | 55 | 90 | 46,XY[20] | 46,XY[20] | ASXL1, JAK2 | ASXL1, JAK2, KRAS, NPM1, NRAS | |
| NR | CMML1 | 95 | 95 | 4 | 9 | 10 | 12 | 17 | 32 | 46,XY[20] | 46,XY[20] | FLT3 | FLT3 | |
| NR | CMML0 | 65 | 40 | 0 | 1 | 7 | 8 | 45 | 40 | 46,XY[20] | 46,XY,del(3)(q13.2q24)[3]/46,XY[17] | ASXL1 | ASXL1, IDH2 | |
| NR | CMML1 | 95 | 90 | 2 | 2 | 6 | 6 | 0 | 0 | 46,XY[20] | 46,XY[20] | MYD88, NRAS, TET2 | NA | |
| NR | MDS-MLD-RS | 95 | 100 | 3 | 3 | 6 | 11 | 40 | 20 | 46,XY[20] | 46,XY[20] | JAK2, TET2 | NA | |
| NR | CMML1 | 70 | 90 | 2 | 6 | 11 | 17 | 20 | 55 | 47,XX,+8[19]/46,XX[1] | 47,XX,+8[20] | JAK2 TET2 | NA | |
| NR | CMML1 | NA | NA | 1 | 0 | 3 | 3 | 9 | 4 | 46,XY[20] | 46,XY[20] | None | NA | |
| NR | MDS-MLD | NA | NA | 1 | 1 | 8 | 8 | 46 | NA | 46,XY[20] | 46,XY[20] | ASXL1, DNMT3A, JAK2 | NA | |
| NR | Del5q | 65 | 50 | 6 | 14 | 13 | 12 | 26 | 1 | 46,XY,del(5)(q13q33)[12]46,idem,del(12)(p12p13)[4]46,idem,der(12)t(12;22)(p11.2;q11.2),der(22)t(12;22)del(12)(p11.2)[4] | 46,XY,del(5)(q13q33)[5]/46,idem,del(12)(p12p13)[9]/46,idem,der(12)t(12;22)(p11.2;q11.2),der(22)t(12;22)del(12)(p11.2)[6] | JAK2, TP53 | NA | |
| Not | NE | CMML1 | NA | NA | 1 | 0 | NA | NA | NA | NA | 47,XY,+8,del(13)(q12q22)[8]/46,XY[4] | 47,XY,+8,del(13)(q12q22)[20] | JAK2, TET2 | NA |
+: ongoing response to therapy on study.
Abbreviations: IWG: International working group; PCyR: partial cytogenetic response; HI-P: Hematological improvement of platelets; HI-E: hematological improvement of erythrocytes; NR: no response; NE: not evaluable; Pre-Rx: before therapy; Post-Rx: after therapy; pp65: phosphorylated p65; MDS: myelodysplastic syndromes; CMML: chronic myelomonocytic leukemia; MDS-MLD: MDS with multilineage dysplasia; MDS/MPN-RS-T: myelodysplastic syndrome/myeloproliferative neoplasm with overlap with ring sideroblasts and thrombocytosis; SM-ANH: systemic mastocytosis with an associated hematological neoplasm; T-MDS: therapy-related myelodysplastic syndrome; NA: not available; ND: not done.
Safety and Toxicity
All patients were evaluable for toxicity. TEAEs are summarized in Table 4. The most common non-hematological TEAEs were hyperglycemia (79%), fatigue (74%), and elevated liver function tests (58%). Thirteen patients (68%) experienced a grade ≥3 non-hematologic TEAE, with hyperglycemia (16%) and infections (16%) reported as the most common toxicities. Thrombocytopenia (74%) and anemia (100%) were the most common hematologic TEAEs and were experienced at grade ≥3 in 47% and 74% of the patients, respectively.
Table 4.
Treatment Emergent Adverse Events
| Adverse Events | Number of patients (%) | |
|---|---|---|
| All Grades | Grade ≥3 | |
| Anemia | 19 (100) | 14 (74) |
| Hyperglycemia | 15 (79) | 3 (16) |
| Thrombocytopenia | 14 (74) | 9 (47) |
| Fatigue | 14 (74) | 0 |
| Elevated LFTs | 11 (58) | 1 (5) |
| Elevated creatinine | 9 (47) | 1 (5) |
| Hyperuricemia | 7 (37) | 2 (10) |
| Hyperkalemia | 7 (37) | 2 (10) |
| Hemorrhage | 7 (37) | 0 |
| Dyspnea | 6 (31) | 2 (10) |
| Hypocalcemia | 6 (31) | 1 (5) |
| Fluid retention | 6 (31) | 0 |
| Nausea/Vomiting | 6 (31) | 0 |
| Infection | 5 (26) | 3 (16) |
| Diarrhea | 5 (26) | 0 |
| Rash | 5 (26) | 0 |
| Dizziness | 5 (26) | 0 |
| Neutropenia | 4 (21) | 2 (10) |
| Hyperbilirubinemia | 4 (21) | 0 |
| Peripheral neuropathy | 4 (21) | 0 |
| Hypoalbuminemia | 4 (21) | 0 |
| Musculoskeletal pain | 4 (21) | 0 |
| Hypomagnesemia | 4 (21) | 0 |
| Hypotension | 3 (16) | 2 (10) |
| Hypertension | 3 (16) | 2 (10) |
| Mouth sores | 3 (16) | 1 (5) |
| Hyponatremia | 3 (16) | 0 |
| Anorexia | 3 (16) | 0 |
| Cough | 3 (16) | 0 |
| Iron overload | 2 (10) | 2 (10) |
| Hypophosphatemia | 2 (10) | 1 (5) |
| Elevated INR | 2 (10) | 1 (5) |
| Hyperphosphatemia | 2 (10) | 0 |
| Constipation | 2 (10) | 0 |
| Headache | 2 (10) | 0 |
| Hypokalemia | 1 (5) | 1 (5) |
| Stroke | 1 (5) | 1 (5) |
Abbreviations: LFTs: Liver function tests; INR: International normalized ratio.
TEAEs leading to treatment interruption of ruxolitinib occurred in 5 patients (26%) [10 mg (N=1) and 20 mg (N=4)]. Of these patients, 3 experienced prolonged significant thrombocytopenia (>6 weeks), despite dose reduction in 2 patients from 20 mg to 15 mg, resulting in treatment discontinuation. No DLTs were observed and therefore the MTD was declared to be 20 mg BID. Toxicities were reversible. No withdrawal syndrome was observed.
Response and Survival
Eighteen patients were evaluable for response with the remaining patient excluded due to insufficient BM specimen after cycle 1. The overall response rate (ORR) was 22% of which 2 patients (11%) achieved hematological improvement of platelets (HI-P), 1 (5.5%) achieved hematological improvement in erythrocytes (HI-E), and 1 (5.5%) patient achieved partial cytogenetic response (PCyR); time to best response was 4, 8, 16, and 8 weeks respectively. All responses occurred among patients with MDS with no responses noted in patients diagnosed with CMML. Among responders, 2 patients relapsed (achieving HI-P and PCyR) after a response duration of 1 and 4 months, respectively; including 1 patient who transformed to AML after achieving HI-P. The remaining 2 patients continue to be in HI-P and HI-E after 20.5+ and 5.1+ months of follow-up, respectively. Prior to therapy, 3 of the 4 responders were transfusion dependent on platelets (HI-P, N=1) and PRBCs (HI-P, N=2 and HI-E, N=1). With therapy, the patient who achieved HI-E and 1 of the 2 patients who achieved HI-P became transfusion independent to PRBCs and platelets, respectively. However, both patients who achieved HI-P still remained transfusion dependent on PRBCs while on therapy.
Median OS for the whole cohort was 14.5 months (95% CI: 8.5-20.45) with a 1-year OS rate of 63% (Figure 1A). When stratified by diagnoses, median OS was 14.8 (95% CI: 13.2-16.35) and 8.1 (95% CI: 7.6-8.6) in the MDS and CMML subgroup of patients, respectively (p=0.45) (Figure 1A). Among the MDS subgroup, there was a non-significant statistical improvement in the median OS among responders when compared to non-responders [Not reached vs 13.6 months (95%CI: 10-17), p=0.2] (Figure 1B).
Figure 1. Outcomes of patients treated with ruxolitinib.
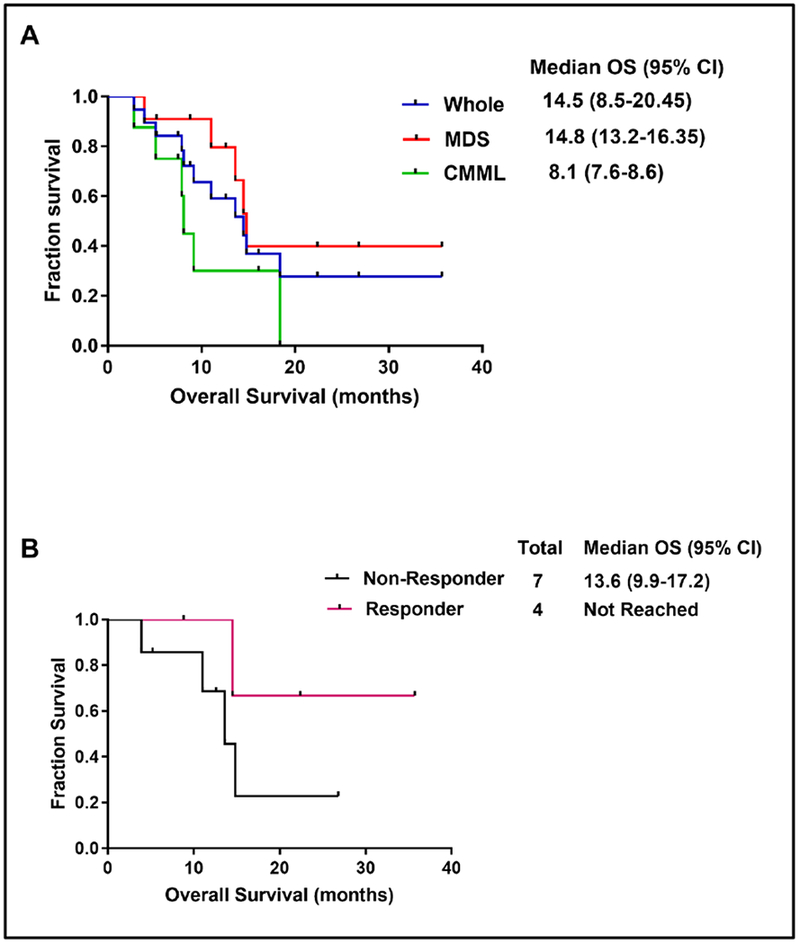
(A) Overall survival for the whole cohort. (B) Overall survival for myelodysplastic syndrome patients stratified by response to therapy.
With a median follow-up of 12 months (range, 4-36), 3 patients (16%) remain on study including the 2 patients with ongoing response to therapy (achieving HI-E and HI-P) and 1 patient who has stable disease with no response to therapy (PRBC-transfusion dependent) after receiving 7 cycles of ruxolitinib. The remaining 16 patients (84%) were taken off study due to lack of response (N=8), toxicity (N=3), disease progression (N=2), SCT (N=1), death (N=1), and development of a second malignancy (N=1).
Two patients received SCT; 1 patient had no response to therapy after 2 cycles and proceeded with haploidentical SCT and 1 received matched-related donor SCT after progression to AML. Both transplant patients died; the former died 6 months post-SCT due to transplant complications and the latter died 10 months post-SCT from AML. A total of 11 patients (58%) died including only 1 death occurring on study due to acute renal failure and tumor lysis syndrome deemed unrelated to ruxolitinib. The remaining causes of death included AML transformation (N=3), sepsis (N=2), disease progression (N=1), SCT complications (N=1), hemorrhage (N=1), unrelated stroke complications (N=1), and unknown etiology (N=1).
Correlative Studies
Seventeen patients (89%) had detectable BM pp65 at the time of initial evaluation with a mean pp65 positivity of 25% (95%CI: 16-34.5). Sequential measurement of the pp65 level was available in 15 patients [responders (N=3) and non-responders (N=12)] (Table 3). Among responders, the pp65 level decreased in the 2 patients who have ongoing response to therapy achieving HI-P (63% to 33%) and HI-E (17% to 10%), respectively. In the third patient who achieved a brief HI-P and then progressed to AML, the pp65 level increased from 7% to 15%. In the non-responder group, the pp65 level increased in 7 patients, decreased in 4 patients, and remained unchanged in 1 patient (Figure 2).
Figure 2. Changes in the phosphorylated p65 level with ruxolitinib therapy.

Bone marrow samples were evaluated for phosphorylated p65 (pp65) levels at baseline and after 2 to 8 cycles of ruxolitinib therapy. Each line represents one patient and red asterisks indicate patients who responded to therapy.
Cytomorphological Analysis
BM characteristics are summarized in Table 3. Among the four responders, the patient who achieved HI-E was the only responder to show resolution of dysplasia after 6 cycles of treatment (Della Porta dysplasia score from 11 to 0) (Figure 3). This improvement in dysplasia was associated with a reduction in BM cellularity (100% to 50%) and pp65 level (17% to 10%). The remaining 3 patients (HI-P=2, PCyR=1) failed to show significant morphologic changes with therapy.
Figure 3. Resolution of bone marrow dysplasia after achieving hematological improvement of erythrocytes with single-agent ruxolitinib.

(A) Assessment of megakaryocytic dysplasia on bone marrow (H&E ×500 magnification); from hypolobated dysplastic megakaryocytes at baseline (left panel) to megakaryocytes with multilobated nuclei in the last follow-up (right panel). (B) Assessment of granulocytic dysplasia on bone marrow (Wright-Giemsa stain ×500 magnification); from hypogranular granulocytes at baseline (left panel) to granulocytes with proper granulation in the last follow-up (right panel).
In the non-responder group (N=14), nine patients showed no improvement in dysplasia, 3 patients had mild improvement in dysplasia (Della Porta dysplasia score decreased from 11 to 6 in 1 patient and from 12 to 9 in 2 patients), and 2 patients showed increased dysplasia (Della Porta dysplasia score increased from 6 to 11 and from 11 to 17, respectively). Molecular studies were available for comparison in 5 patients, all of them non-responders, as demonstrated in Table 3.
DISCUSSION
Single-agent ruxolitinib was found to be generally well-tolerated in the treatment of patients with lower-risk MDS who failed at least one prior therapy. Although the majority of patients experienced at least one high-grade toxicity, no DLTs were observed and therefore ruxolitinib given at a dose of 20 mg orally BID was declared as the MTD. Ruxolitinib demonstrated modest clinical activity with an ORR of 22% noted only among MDS patients who had evidence of NF-kB activation with no clinical activity found in patients with CMML.
Anemia and thrombocytopenia were the most frequently reported hematological toxicities, consistent with the previous studies of ruxolitinib in myelofibrosis and in line with its known mechanism of action [22, 24, 31]. Although thrombocytopenia was generally managed with dose reductions or brief interruptions of therapy, 3 patients required prolonged treatment interruption (>6 weeks) due to severe thrombocytopenia leading to treatment discontinuation. This incidence of prolonged thrombocytopenia is higher than that reported in the COMFORT trials which may be attributed to the significant differences in disease biology.
Increase in the rates of intramedullary apoptosis of hematopoietic stem and progenitor cells (HSPCs) is the main mechanism of ineffective hematopoiesis leading to the cytopenias observed in lower-risk MDS [20, 27, 32]. Aberrant activation of the innate immune signals through overexpression of several TLRs results in increased cytokine secretion, including TNF-α, leading to the constitutive activation of NF-kB and overexpression of IL-8 in the BM of MDS patients [20, 21, 33–38]. The NF-kB family of transcription factors activate multiple target genes which mostly suppress apoptosis and promote proliferation of MDS HSPCs thereby playing a critical role in the pathogenesis of higher-risk MDS and progression to AML [20, 39–41]. Despite its anti-apoptotic properties, NF-kB has also been found to promote intramedullary apoptosis in lower-risk MDS through inducing overexpression of TNF-α which in turn stimulates HSPCs to express the death receptor ligand, FasL, increasing their apoptotic rates [20, 32, 42, 43].
Given these preclinical findings, inhibition of the NF-kB pathway and therefore preventing the expression of apoptogenic cytokines has been an appealing strategy in the treatment of lower-risk MDS patients, particularly those who failed prior lines of therapy. We hypothesized that ruxolitinib could induce NF-kB inhibition in MDS cells. It should be noted that we did not perform preclinical testing to support this. In a recently published phase 1 trial, bortezomib induced HI-E in 20% of the lower-risk MDS patients who had evidence of NF-kB activation [30]. This response was associated with significant reduction in pp65 expression and improvement of BM dysplasia indicating the potential clinical role of NF-kB inhibition in MDS therapy.
In our study, meaningful improvement of dysplasia occurred only in the patient who achieved HI-E, similar to the phase 1 bortezomib results.[30] This finding is consistent with the preclinical studies which showed that blockade of MYD88 mediated innate immunity signaling, including IL-8, in HSPCs isolated from patients with lower-risk MDS prompted erythroid differentiation [21, 37]. Whether the clinical activity of NF-kB inhibitors is restricted to the improvement in erythropoiesis in lower-risk MDS patients is yet to be determined.
Ruxolitinib exerted an inhibitory effect on the NF-kB pathway demonstrated by the reduction in the pp65 level in 2 out of the 3 tested responders. The level of pp65 decreased in the two patients who continue to be in HI-P and HI-E with ongoing therapy, but increased in the third patient who had a short-lived response (HI-P) and progressed to AML. This observation may suggest potential correlation between reduction in pp65 levels and response durability among responders, however the small number of patients precludes any definitive conclusions.
We failed to find similar results among our CMML patients. Padron et al conducted a multiinstitution phase 1 study to determine the safety and efficacy of ruxolitinib in patients with CMML [44]. In contrast to our study, they reported activity of ruxolitinib in CMML with hematological responses achieved in 4 patients (3 MPN-CMML and 1 MDS-CMML) [44]. This difference in response rates may potentially be related to the discrepancy in the types of CMML patients enrolled in both studies. In our study, 16% of our CMML patients were of the proliferative subtype and 75% received prior HMA therapy as opposed to 70% and 40%, respectively, in the former study [44]. This confirms the extreme heterogeneity noted among CMML patients and the distinct clinical behavior of the dysplastic and proliferative subtypes of CMML which may not only affect the disease course but also response to therapy.
In conclusion, ruxolitinib was well-tolerated with modest activity in lower-risk MDS patients who failed at least one prior therapy, including HMAs, and had evidence of NF-kB activation. Prolonged thrombocytopenia mandating drug discontinuation was not an infrequent finding with ruxolitinib therapy. Among responders, reduction in pp65 expression may potentially correlate with response duration. Future studies are needed to confirm the efficacy of ruxolitinib in the salvage treatment of lower-risk MDS patients and determine the subgroup of patients who would benefit the most from therapy. Studies of JAK2 inhibitors in combination with hypomethylating agents in MDS/MPN are ongoing.
HIGHLIGHTS.
Deregulation of innate immune signaling is important in the pathogenesis of MDS.
Ruxolitinib is active in previously treated lower-risk MDS with NF-kB activation.
Reduction in pp65 expression may correlate with response duration.
ACKNOWLEDGEMENTS:
This study was funded by Incyte Corporation, the University of Texas MD Anderson Cancer Center Support Grant CA016672, and the University of Texas MD Anderson MDS/AML Moon Shot.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE OF CONFLICTS OF INTEREST: Authors disclose no relevant conflicts related to this article.
REFERENCES
- [1].Tefferi A, Vardiman JW, Myelodysplastic syndromes, N Engl J Med 361(19) (2009) 1872–85. [DOI] [PubMed] [Google Scholar]
- [2].Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, Sanz M, Vallespi T, Hamblin T, Oscier D, Ohyashiki K, Toyama K, Aul C, Mufti G, Bennett J, International scoring system for evaluating prognosis in myelodysplastic syndromes, Blood 89(6) (1997) 2079–88. [PubMed] [Google Scholar]
- [3].Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, Bennett JM, Bowen D, Fenaux P, Dreyfus F, Kantarjian H, Kuendgen A, Levis A, Malcovati L, Cazzola M, Cermak J, Fonatsch C, Le Beau MM, Slovak ML, Krieger O, Luebbert M, Maciejewski J, Magalhaes SM, Miyazaki Y, Pfeilstocker M, Sekeres M, Sperr WR, Stauder R, Tauro S, Valent P, Vallespi T, van de Loosdrecht AA, Germing U, Haase D, Revised international prognostic scoring system for myelodysplastic syndromes, Blood 120(12) (2012) 2454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Benton CB, Khan M, Nogueras González G, Nazha A, Piao J, Ning J, Aung FM, Jabbour EJ, Kadia T, Borthakur G, Ravandi F, Komrokji RS, Steensma DP, DeZern AE, Roboz GJ, Sekeres MA, Andreeff M, Kantarjian HM, Garcia-Manero G, Sequential Stratification Prognostic Score for Patients with Intermediate-Risk Myelodysplastic Syndrome, Blood 130(Suppl 1) (2017) 4265–4265. [Google Scholar]
- [5].List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, Powell B, Greenberg P, Thomas D, Stone R, Reeder C, Wride K, Patin J, Schmidt M, Zeldis J, Knight R, I. Myelodysplastic Syndrome-003 Study, Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion, N Engl J Med 355(14) (2006) 1456–65. [DOI] [PubMed] [Google Scholar]
- [6].List A, Kurtin S, Roe DJ, Buresh A, Mahadevan D, Fuchs D, Rimsza L, Heaton R, Knight R, Zeldis JB, Efficacy of lenalidomide in myelodysplastic syndromes, N Engl J Med 352(6) (2005) 549–57. [DOI] [PubMed] [Google Scholar]
- [7].Wei S, Chen X, Rocha K, Epling-Burnette PK, Djeu JY, Liu Q, Byrd J, Sokol L, Lawrence N, Pireddu R, Dewald G, Williams A, Maciejewski J, List A, A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide, Proc Natl Acad Sci U S A 106(31) (2009) 12974–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Garcia-Manero G, Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management, Am J Hematol 90(9) (2015) 831–41. [DOI] [PubMed] [Google Scholar]
- [9].Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, Helmer R 3rd, Shen L, Nimer SD, Leavitt R, Raza A, Saba H, Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study, Cancer 106(8) (2006) 1794–803. [DOI] [PubMed] [Google Scholar]
- [10].Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O’Brien S, Cortes J, Faderl S, Bueso-Ramos C, Ravandi F, Estrov Z, Ferrajoli A, Wierda W, Shan J, Davis J, Giles F, Saba HI, Issa JP, Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia, Blood 109(1) (2007) 52–7. [DOI] [PubMed] [Google Scholar]
- [11].Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, Stone RM, Nelson D, Powell BL, DeCastro CM, Ellerton J, Larson RA, Schiffer CA, Holland JF, Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B, J Clin Oncol 20(10) (2002) 2429–40. [DOI] [PubMed] [Google Scholar]
- [12].Jabbour E, Short NJ, Montalban-Bravo G, Huang X, Bueso-Ramos C, Qiao W, Yang H, Zhao C, Kadia T, Borthakur G, Pemmaraju N, Sasaki K, Estrov Z, Cortes J, Ravandi F, Alvarado Y, Komrokji R, Sekeres MA, Steensma DP, DeZern A, Roboz G, Kantarjian H, Garcia-Manero G, Randomized phase 2 study of low-dose decitabine vs low-dose azacitidine in lower-risk MDS and MDS/MPN, Blood 130(13) (2017) 1514–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, Byrd J, Backstrom J, Zimmerman L, McKenzie D, Beach C, Silverman LR, M.D.S.S.S.G. International Vidaza High-Risk, Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study, Lancet Oncol 10(3) (2009) 223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fili C, Malagola M, Follo MY, Finelli C, Iacobucci I, Martinelli G, Cattina F, Clissa C, Candoni A, Fanin R, Gobbi M, Bocchia M, Defina M, Spedini P, Skert C, Manzoli L, Cocco L, Russo D, Prospective phase II Study on 5-days azacitidine for treatment of symptomatic and/or erythropoietin unresponsive patients with low/INT-1-risk myelodysplastic syndromes, Clin Cancer Res 19(12) (2013) 3297–308. [DOI] [PubMed] [Google Scholar]
- [15].Garcia-Manero G, Jabbour E, Borthakur G, Faderl S, Estrov Z, Yang H, Maddipoti S, Godley LA, Gabrail N, Berdeja JG, Nadeem A, Kassalow L, Kantarjian H, Randomized open-label phase II study of decitabine in patients with low- or intermediate-risk myelodysplastic syndromes, J Clin Oncol 31(20) (2013) 2548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jabbour EJ, Garcia-Manero G, Strati P, Mishra A, Al Ali NH, Padron E, Lancet J, Kadia T, Daver N, O’Brien S, Steensma DP, Sekeres MA, Gore SD, Dezern A, Roboz GJ, List AF, Kantarjian HM, Komrokji RS, Outcome of patients with low-risk and intermediate-1-risk myelodysplastic syndrome after hypomethylating agent failure: a report on behalf of the MDS Clinical Research Consortium, Cancer 121(6) (2015) 876–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prebet T, Thepot S, Gore SD, Dreyfus F, Fenaux P, Vey N, Outcome of patients with low-risk myelodysplasia after azacitidine treatment failure, Haematologica 98(2) (2013) e18–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jabbour E, Garcia-Manero G, Batty N, Shan J, O’Brien S, Cortes J, Ravandi F, Issa JP, Kantarjian H, Outcome of patients with myelodysplastic syndrome after failure of decitabine therapy, Cancer 116(16) (2010) 3830–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Montalban-Bravo G, Garcia-Manero G, Jabbour E, Therapeutic choices after hypomethylating agent resistance for myelodysplastic syndromes, Curr Opin Hematol 25(2) (2018) 146–153. [DOI] [PubMed] [Google Scholar]
- [20].Ganan-Gomez I, Wei Y, Starczynowski DT, Colla S, Yang H, Cabrero-Calvo M, Bohannan ZS, Verma A, Steidl U, Garcia-Manero G, Deregulation of innate immune and inflammatory signaling in myelodysplastic syndromes, Leukemia 29(7) (2015) 1458–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dimicoli S, Wei Y, Bueso-Ramos C, Yang H, Dinardo C, Jia Y, Zheng H, Fang Z, Nguyen M, Pierce S, Chen R, Wang H, Wu C, Garcia-Manero G, Overexpression of the toll-like receptor (TLR) signaling adaptor MYD88, but lack of genetic mutation, in myelodysplastic syndromes, PLoS One 8(8) (2013) e71120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, McQuitty M, Hunter DS, Levy R, Knoops L, Cervantes F, Vannucchi AM, Barbui T, Barosi G, JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis, N Engl J Med 366(9) (2012) 787–98. [DOI] [PubMed] [Google Scholar]
- [23].Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, Harrison CN, Pane F, Zachee P, Mesa R, He S, Jones MM, Garrett W, Li J, Pirron U, Habr D, Verstovsek S, Ruxolitinib versus standard therapy for the treatment of polycythemia vera, N Engl J Med 372(5) (2015) 426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, Talpaz M, Winton EF, Harvey JH Jr., Arcasoy MO, Hexner E, Lyons RM, Paquette R, Raza A, Vaddi K, Erickson-Viitanen S, Koumenis IL, Sun W, Sandor V, Kantarjian HM, A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis, N Engl J Med 366(9) (2012) 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Heine A, Wolf D, Brossart P, Ruxolitinib Inhibits Dendritic Cell Function By Interfering With The MAP-Kinase/NF-Kb Signalling Pathways, Blood 122(21) (2013) 3463–3463. [Google Scholar]
- [26].Zheng C, Yin Q, Wu H, Structural studies of NF-kappaB signaling, Cell Res 21(1) (2011) 183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Huang B, Yang XD, Lamb A, Chen LF, Posttranslational modifications of NF-kappaB: another layer of regulation for NF-kappaB signaling pathway, Cell Signal 22(9) (2010) 1282–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD, Pinto A, Beran M, de Witte TM, Stone RM, Mittelman M, Sanz GF, Gore SD, Schiffer CA, Kantarjian H, Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia, Blood 108(2) (2006) 419–25. [DOI] [PubMed] [Google Scholar]
- [29].Della Porta MG, Travaglino E, Boveri E, Ponzoni M, Malcovati L, Papaemmanuil E, Rigolin GM, Pascutto C, Croci G, Gianelli U, Milani R, Ambaglio I, Elena C, Ubezio M, Da Via MC, Bono E, Pietra D, Quaglia F, Bastia R, Ferretti V, Cuneo A, Morra E, Campbell PJ, Orazi A, Invernizzi R, Cazzola M, Rete N Ematologica Lombarda Clinical, Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes, Leukemia 29(1) (2015) 66–75. [DOI] [PubMed] [Google Scholar]
- [30].Daher M, Hidalgo Lopez JE, Randhawa JK, Jabbar KJ, Wei Y, Pemmaraju N, Borthakur G, Kadia T, Konopleva M, Kantarjian HM, Hearn K, Estrov Z, Reyes S, Bueso-Ramos CE, Garcia-Manero G, An exploratory clinical trial of bortezomib in patients with lower risk myelodysplastic syndromes, Am J Hematol 92(7) (2017) 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, Vaddi K, Levy R, Tefferi A, Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis, N Engl J Med 363(12) (2010) 1117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sanz C, Richard C, Prosper F, Fernandez-Luna JL, Nuclear factor k B is activated in myelodysplastic bone marrow cells, Haematologica 87(9) (2002) 1005–6. [PubMed] [Google Scholar]
- [33].Wei Y, Dimicoli S, Bueso-Ramos C, Chen R, Yang H, Neuberg D, Pierce S, Jia Y, Zheng H, Wang H, Wang X, Nguyen M, Wang SA, Ebert B, Bejar R, Levine R, Abdel-Wahab O, Kleppe M, Ganan-Gomez I, Kantarjian H, Garcia-Manero G, Toll-like receptor alterations in myelodysplastic syndrome, Leukemia 27(9) (2013) 1832–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kuninaka N, Kurata M, Yamamoto K, Suzuki S, Umeda S, Kirimura S, Arai A, Nakagawa Y, Suzuki K, Kitagawa M, Expression of Toll-like receptor 9 in bone marrow cells of myelodysplastic syndromes is down-regulated during transformation to overt leukemia, Exp Mol Pathol 88(2) (2010) 293–8. [DOI] [PubMed] [Google Scholar]
- [35].Maratheftis CI, Andreakos E, Moutsopoulos HM, Voulgarelis M, Toll-like receptor-4 is up-regulated in hematopoietic progenitor cells and contributes to increased apoptosis in myelodysplastic syndromes, Clin Cancer Res 13(4) (2007) 1154–60. [DOI] [PubMed] [Google Scholar]
- [36].Velegraki M, Papakonstanti E, Mavroudi I, Psyllaki M, Tsatsanis C, Oulas A, Iliopoulos I, Katonis P, Papadaki HA, Impaired clearance of apoptotic cells leads to HMGB1 release in the bone marrow of patients with myelodysplastic syndromes and induces TLR4-mediated cytokine production, Haematologica 98(8) (2013) 1206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wei Y, Chen R, Dimicoli S, Bueso-Ramos C, Neuberg D, Pierce S, Wang H, Yang H, Jia Y, Zheng H, Fang Z, Nguyen M, Ganan-Gomez I, Ebert B, Levine R, Kantarjian H, Garcia-Manero G,Global H3K4me3 genome mapping reveals alterations of innate immunity signaling and overexpression of JMJD3 in human myelodysplastic syndrome CD34+ cells, Leukemia 27(11) (2013) 2177–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Akira S, Uematsu S, Takeuchi O, Pathogen recognition and innate immunity, Cell 124(4) (2006) 783–801. [DOI] [PubMed] [Google Scholar]
- [39].Kerbauy DM, Lesnikov V, Abbasi N, Seal S, Scott B, Deeg HJ, NF-kappaB and FLIP in arsenic trioxide (ATO)-induced apoptosis in myelodysplastic syndromes (MDSs), Blood 106(12) (2005) 3917–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Braun T, Carvalho G, Coquelle A, Vozenin MC, Lepelley P, Hirsch F, Kiladjian JJ, Ribrag V, Fenaux P, Kroemer G, NF-kappaB constitutes a potential therapeutic target in high-risk myelodysplastic syndrome, Blood 107(3) (2006) 1156–65. [DOI] [PubMed] [Google Scholar]
- [41].Fabre C, Carvalho G, Tasdemir E, Braun T, Ades L, Grosjean J, Boehrer S, Metivier D, Souquere S, Pierron G, Fenaux P, Kroemer G, NF-kappaB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia, Oncogene 26(28) (2007) 4071–83. [DOI] [PubMed] [Google Scholar]
- [42].Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, Hirst M, Hogge D, Marra M, Wells RA, Buckstein R, Lam W, Humphries RK, Karsan A, Identification of miR-145 and miR-146a as mediators of the 5q-syndrome phenotype, Nat Med 16(1) (2010) 49–58. [DOI] [PubMed] [Google Scholar]
- [43].Kumar MS, Narla A, Nonami A, Mullally A, Dimitrova N, Ball B, McAuley JR, Poveromo L, Kutok JL, Galili N, Raza A, Attar E, Gilliland DG, Jacks T, Ebert BL, Coordinate loss of a microRNA and protein-coding gene cooperate in the pathogenesis of 5q-syndrome, Blood 118(17) (2011) 4666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Padron E, Dezern A, Andrade-Campos M, Vaddi K, Scherle P, Zhang Q, Ma Y, Balasis ME, Tinsley S, Ramadan H, Zimmerman C, Steensma DP, Roboz GJ, Lancet JE, List AF, Sekeres MA,Komrokji RS, Myelodysplastic C Syndrome Clinical Research, A Multi-Institution Phase I Trial of Ruxolitinib in Patients with Chronic Myelomonocytic Leukemia (CMML), Clin Cancer Res 22(15) (2016) 3746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]