

Abstract
Drug-tolerance is an acute defense response prior to a fully drug-resistant state and tumor relapse, however there are few therapeutic agents targeting drug-tolerance in the clinic. Here we show that miR-147b initiates a reversible tolerant-state to the EGFR inhibitor osimertinib in non-small cell lung cancer. With miRNA-seq analysis we find that miR-147b is the most upregulated microRNA in osimertinib-tolerant and EGFR mutated lung cancer cells. Whole transcriptome analysis of single-cell derived clones reveals a link between osimertinib-tolerance and pseudohypoxia responses irrespective of oxygen levels. Further metabolomics and genetic studies demonstrate that osimertinib-tolerance is driven by miR-147b repression of VHL and succinate dehydrogenase linked to the tricarboxylic acid cycle and pseudohypoxia pathways. Finally, pretreatment with a miR-147b inhibitor delays osimertinib-associated drug tolerance in patient-derived three-dimensional (3D) structures. This link between miR-147b and tricarboxylic acid cycle may provide promising targets for preventing tumor relapse.
Introduction
Relapsed disease following conventional treatments remains one of the central problems in cancer management, including epidermal growth factor receptor (EGFR)-based targeted therapy1,2. Tumor cells overcome anti-EGFR treatment by acquisition of drug binding-deficient mutations of EGFR and bypass through other protein tyrosine kinase signaling pathways3. For example, a majority of tumours from EGFR-mutant non-small cell lung cancer (NSCLC) patients acquired resistance mutations such as EGFRT790M or EGFRC797S when the patients were treated with EGFR tyrosine kinase inhibitors (TKIs), gefitinib or erlotinib and osimertinib, respectively4,5. Recently, it has been found that EGFRT790M-positive drug-resistant cells can emerge from EGFRT790M-negative drug-tolerant cells that survive initial drug treatment6,7. Thus, targeting drug-tolerant cells might be a new strategy to block drug resistance8,9. With success in applying osimertinib in the first-line treatment of EGFRT790M-positive NSCLC10, it is therefore crucial to identify the changes driving drug-tolerance. However, the molecules driving drug-tolerance towards EGFR TKIs are not well studied.
Aberrantly regulated metabolic pathways lead to tumorigenesis and advantageous survival of tumor cells11–15. The tricarboxylic acid (TCA) cycle is a central pathway in the metabolism of sugars, lipids and amino acids16. A dysfunctional TCA cycle induces oncogenesis by activating pseudohypoxia responses which express hypoxia-associated proteins regardless of the oxygen status17–19. For example, succinate accumulation caused by functional loss of the TCA cycle enzyme succinate dehydrogenase (SDH) stabilizes hypoxia-inducible factor 1alpha (HIF1alpha) via prolyl-hydroxylase (PHD) inhibition20,21. In addition, loss of function of Von Hippel-Lindau (VHL) also induces the pseudohypoxia response through decreased ubiquitination and proteasomal degradation of HIF1alpha22. Compared to other cancers, NSCLC is well vascularized and tumor cells depend on high levels of the iron-sulfur cluster biosynthetic enzymes to reduce oxidative damage due to exposure to high oxygen23. Most recently, it was shown that drug-tolerant persister cancer cells were vulnerable to lipid hydroperoxidase GPX4 inhibition due to a disabled antioxidant program24. However, our understanding of changes conferring drug-tolerance remain limited. To address this knowledge gap, we explored which signaling pathways initiate anticancer drug-tolerance and how this shapes cancer metabolism and tumor relapse. In this study, we have discovered that a subpopulation of tumor cells adopts a tolerance strategy to defend against EGFR-based anticancer treatments by altering microRNA-147b (miR-147b)-dependent dysregulation of the TCA cycle and pseudohypoxia responses. We have revealed that miR-147b, by targeting VHL and SDH, is critical to tolerance-mediated tumor relapse.
Results
Lung cancer cells adopt a tolerance strategy to EGFR inhibitors
Due to an advantage for visualizing in vivo-like structures in 3D structures, we established lung 3D structures in immortalized tracheobronchial epithelial AALE cells and EGFR mutated lung cancer HCC827 cells (Fig. 1a–c and Supplementary Fig. 1a–c). Compared with adult lung tissues, AALE-derived lung 3D structures express higher levels of lung progenitor cell gene inhibitor of DNA binding 2 (ID2) on day 15 followed by decreased expression on day 24 by qRT-PCR analysis (Supplementary Fig. 1d and Supplementary Table 1). In contrast, the 3D structures from AALE express lower levels of type I and II pneumocyte markers including surfactant protein C (SFTPC), HOP homeobox (HOPX) and NK2 homeobox 1 (NKX2.1) (transcription termination factor 1, TTF-1) on day 15 followed by increasing expressions on day 24 (Supplementary Fig. 1d). The gene expression levels of ID2, SFTPC, HOPX and NKX2.1 in lung 3D structures are comparable to those in adult lung tissues, which is consistent to previous finding of lung 3D structures differentiated from pluripotent stem cells 25. Similarly, 3D structures from lung adenocarcinoma patient-derived xenograft tumor (PDX_LU_10) (Supplementary Table 2) on day 25 express tumor and lung-relevant genes including carcinoembryonic antigen related cell adhesion molecule 5 (CEACAM5), Lin-28 homolog B (LIN28B), SFTPC, and HOPX, which are comparable to those in the parental tumor (Supplementary Fig. 1e). Collectively, our data suggest that lung 3D structures cultured over time are relevant to clinical tissues.
Figure 1. NSCLC cells adopt a tolerance strategy against EGFR-TKIs.
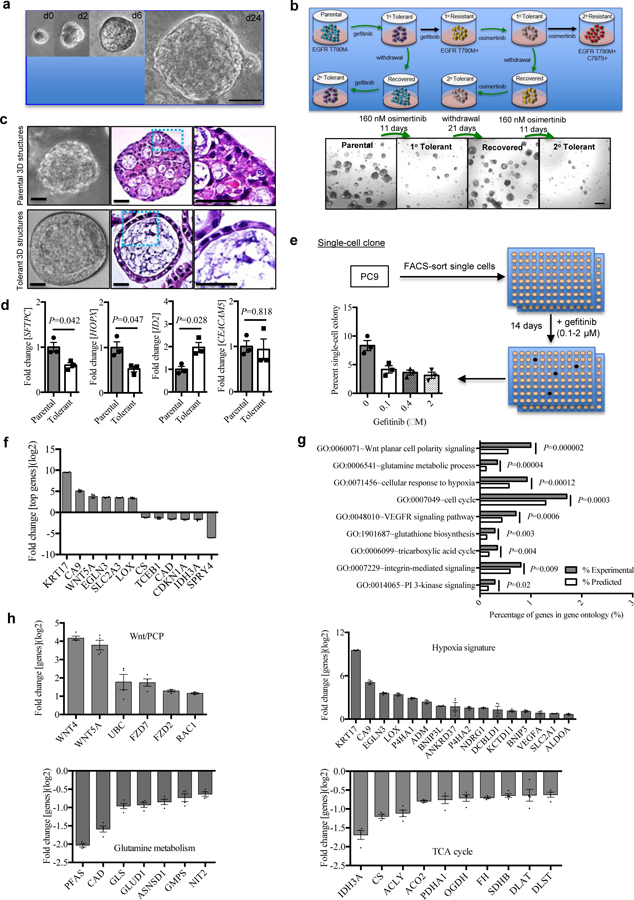
a, Representative phase contrast images of 3D structures from AALE cells cultured according to the protocol at top of the panel. Scale bar, 50 μm. Repeated six times with similar results.
b, Top, the scenario of anti-EGFR tolerance and resistance in lung cancer. The tumor cells treated with the EGFR-TKI gefitinib or osimertinib enter a reversible drug-tolerant cycle (in green arrows, 1° Tolerant) with a brief therapy withdrawal (up to 21 days) followed by reinstatement of the 160 nM dose (2° Tolerant). Alternatively, the tumor cells treated continuously with gefitinib or osimertinib without therapy interruption undergo drug-tolerance briefly and go into a drug-resistance state in which cells do not respond to gefitinib (1° Resistant)/osimertinib (2° Resistant). Bottom, osimertinib treatment response on HCC827 3D structures. Representative images of Parental cells, 1° Tolerant cells (derived from the Parental cells treated with 160 nM osimertinib for 11 days), Recovered cells (derived from the 1° Tolerant cells with a therapy withdrawal up to 21 days) and 2° Tolerant cells (derived from the Recovered cells by reinstatement of the 160 nM dose for 11 days). Scale bar, 200 μm. Repeated six times with similar results.
c, Representative phase contrast microscopy (left panel) and H&E staining of HCC827 3D structures derived from parental (top) and osimertinib-tolerant (bottom) cells. Images in blue dotted squares (middle panel) were amplified (right panel) and shown. Scale bar, 50 μm. Repeated six times with similar results.
d, qRT-PCR analysis of SFTPC, HOPX, ID2 and CEACAM5 expression in single cell clone HCC827-derived 3D structures in the presence of osimertinib. Single cell clone derived cells were plated with geltrex and treated with 100 nM osimertinib (tolerant) or vehicle (parental) for 24 days. Gene expression for surviving 3D structures were analyzed. n=3 independent biological replicates.
e, Single-cell clonogenicity of PC9 cells treated with gefitinib. A single cell was sorted by FACS into a 96-well plate and treated with 0.1, 0.4, and 2 μM gefitinib or the vehicle for 14 days. The frequency of colony formation was calculated as a ratio of the total number of colonies to the total number of wells plated with a single cell in a 96-well plate. n=3 independent biological replicates.
f, qRT-PCR analysis of top upregulated and downregulated genes in gefitinib-tolerant clones (n=2) compared with vehicle-treated parental single cell clone (n=1) in PC9. The gene expression in parental sensitive clone was calibrated as 1. ACTB was used as endogenous control. n=4 independent biological replicates.
g, Whole transcriptome and gene ontology analysis of gefitinib-tolerant clones compared with the parental single cell clone in PC9. n=4 independent biological replicates.
h, qRT-PCR analysis of genes in top regulated signaling pathways including Wnt planar cell polarity signaling, glutamine metabolic process, cellular response to hypoxia, and tricarboxylic acid cycle in gefitinib-tolerant clones compared with parental the single cell clone in PC9. The gene expression in parental sensitive clone was calibrated as 1. ACTB was used as endogenous control. n=4 independent biological replicates.
Data are mean ± s.e.m. and were analysed with unpaired two-tailed t-test with Welch’s correction (d); modified one-tailed Fisher’s Exact test (g).
Using both 3D structures and monolayer cultures, we treated HCC827 cells with serially-diluted osimertinib for three days to observe their acute treatment responses. We found that a subpopulation of tumor cells survived cytotoxic doses (0.01–2 µM) of osimertinib treatment initially (Supplementary Fig. 2a). Surprisingly, a small percentage of cells could survive longer than 2–3 weeks in both monolayer and 3D culture models when they were treated with 160 nM of osimertinib continuously (Supplementary Fig. 2b). However, different from drug-resistant cells, most of those surviving 3D structures disappeared when they were treated with three-fold higher concentration of osimertinib for additional 9 days (Supplementary Fig. 2c). This indicates that some tumor cells adopt a new strategy different from that applied for drug-resistance to protect themselves during the early-stage of treatment response to anti-EGFR therapy. To further understand the protective strategy applied by some tumor cells, after 11-days treatment, we withdrew osimertinib on HCC827 3D structures and found that the initially surviving 3D structures recovered with increasing size within the following 21 days. Those recovered 3D structures remained similarly sensitive to osimertinib when they were exposed to the same dose of osimertinib again (Fig. 1b). Another two lung cancer cell lines with EGFR mutations, PC9 and H1975 cells entered a similar “tolerance cycle” when gefitinib / osimertinib treatments alternated with treatment withdrawal (Supplementary Fig. 2d–e). This suggests that a subpopulation of tumor cells enter a reversible tolerant state to defend against EGFR-TKIs at the early stages of anti-EGFR treatment.
To understand whether the above tolerance is conferred by acquisition of EGFR T790M mutation, we performed pyrosequencing for quantitative analysis of EGFR exon 19 and 20 sequence variations (Supplementary Fig. 3). We found that the drug-tolerant cells demonstrated comparable EGFR exon 19 and 20 sequence to the parental cells in PC9 rather than EGFRT790M-positive gefitinib-resistant PC9 cells. This indicates the tolerant strategy adopted by tumor cells against EGFR-TKI might be mediated by mechanisms different from EGFRT790M mutation. Furthermore, single HCC827 cells mixed with geltrex were plated in 96-well plate and divided into two groups. After 24-hours, one half of cells were treated with 100 nM osimertinib for 21 days (tolerant 3D structures) and the other half of cells were treated with DMSO as control (parental 3D structures) (Fig. 1c). Then we looked at the microscopic features of 3D structures from parental cells and osimertinib-tolerant cells in HCC827 by H&E staining in histology. The parental cell-derived 3D structures showed an adenocarcinoma-like structure. Unexpectedly, a “ring-like” structure was found in the osimertinib-tolerant 3D structures (Fig. 1c). To understand the gene expression in those structures, we performed qRT-PCR analysis on the parental 3D structures and tolerant ones derived from single HCC827 cells. Both parental and osimertinib-tolerant 3D structures express comparable levels of CEACAM5 (Fig. 1d). However, osimertinib-tolerant 3D structures expressed two-fold lower levels of SFTPC and HOPX but up to two-fold higher levels of ID2 (Fig. 1d), suggesting that osimertinib-tolerant 3D structures are enriched for stem-associated genes.
To better understand the transcriptomic changes and tumor heterogeneity conferring osimertinib or gefitinib tolerance in lung cancer, we developed single cell-derived clones in PC9 (Fig. 1e). A single cell was sorted into a 96-well plate at one cell per well by fluorescence-activated cell sorting (FACS). On the following day, the cells were treated with 0.1, 0.4 and 2 μM gefitinib or the vehicle for 14 days (n=192 wells per group). The frequency of colony formation was 8.3%±0.7% and 3.6%±0.3% in the vehicle-treated and all three gefitinib-treated groups, respectively (Fig. 1e). One parental single cell-derived clone treated with vehicle that was sensitive to gefitinib and two drug-tolerant single cell-derived clones treated with 0.4 μM gefitinib were randomly selected and applied for the following whole transcriptome analysis by microarray. We found the top changed genes included upregulated expression of KRT17 (keratin 17), CA9 (carbonic anhydrase 9), WNT5A (Wnt family member 5A), EGLN3 (Egl-9 family hypoxia inducible factor 3), SLC2A3 (solute carrier family 2 member 3), and LOX (lysyl oxidase), as well as downregulated expression of SPRY4 (sprouty RTK signaling antagonist 4) and IDH3A (isocitrate dehydrogenase 3 (NAD(+)) alpha) (Fig. 1f and Supplementary Data 1). Gene ontology analysis demonstrated the top differentially-expressed signaling pathways in the gefitinib-tolerant single-cell clones, including Wnt planar cell polarity (Wnt/PCP) signaling, glutamine metabolic process, cellular response to hypoxia, cell cycle, VEGFR signaling pathway, glutathione derivative biosynthesis, TCA cycle, integrin-mediated signaling and PI 3-kinase signaling (Fig. 1g and Supplementary Table 3). The gene signatures for activated Wnt/PCP signaling and the hypoxia response as well as inactivated glutamine metabolic process and the TCA cycle were validated by qRT-PCR (Fig. 1h). An activated Wnt/PCP signaling pathway has been linked to drug resistance in many studies 26. It was unexpected that activated hypoxia responses as well as inactivated metabolic processes, such as glutamine process and the TCA cycle are among the top signaling pathways relevant to drug-tolerance. Our data suggests that these pathways might cooperatively maintain a “tolerance signature” in EGFR mutant lung cancer cells when they were exposed to EGFR-TKIs.
To exclude the possibility that pre-existing cellular heterogeneity could be responsible for this tolerance, we made single cell clones first followed by exposure to 2 µM gefitinib. In parallel, as in the previous experiment, PC9 cells were cloned in the same concentration of gefitinib (parental clones) as control. All tested single cell-derived clones generate gefitinib-tolerant clones at a frequency of 1.9 ~ 2.1% (n=4 clones), which is comparable to that in parental PC9 clones (2.2 ± 0.1%) (Supplementary Fig. 4a). A similar frequency of osimertinib-tolerance was found between single-cell clones and parental clones in PC9 cells (Supplementary Fig. 4a). Consistently, both single-cell clones from HCC827 and parental clones demonstrated a comparable frequency of osimertinib-tolerance (Supplementary Fig. 4b). All our data strongly suggest that drug-tolerance is spontaneously acquired rather than a reflection of pre-existing cellular heterogeneity, which is consistent with previous findings 8. In addition, compared with PC9 cells tolerant to gefitinib (Fig. 1h), the cells tolerant to osimertinib express similar genes in hypoxia pathway and the TCA cycle (Supplementary Fig. 4c–d). This suggests that lung cancer cells utilize similar strategies to protect themselves from drug-induced cytotoxicity when the cells are treated with either gefitinib or osimertinib. Collectively, our data has demonstrated that drug-tolerance is acquired spontaneously by a small population of lung cancer cells.
MicroRNA-147b initiates anticancer drug tolerance
To test which microRNAs (miRNAs) are linked to osimertinib-tolerance, we performed miRNA-seq analysis in two paired osimertinib-tolerant cells and osimertinib-sensitive parental cells from HCC827 and PC9. A list of differentially expressed miRNAs (n=45) relevant to osimertinib-tolerance was derived from this analysis (Supplementary Table 4). The top upregulated miRNAs included miR-181a-2–3p, miR-147b, miR-574–5p and the top downregulated miRNAs included miR-7641–1, miR-4454 and miR-125b-1–3p (Fig. 2a). It has been reported that overexpression of miR-181a and miR-574 confers chemoresistance in lung and other cancers27–29. However, miR-147b is a miRNA that has not been well studied in drug tolerance. Thus, we focused on miR-147b in our following drug-tolerance study.
Figure 2. miR-147b initiates drug-tolerance.
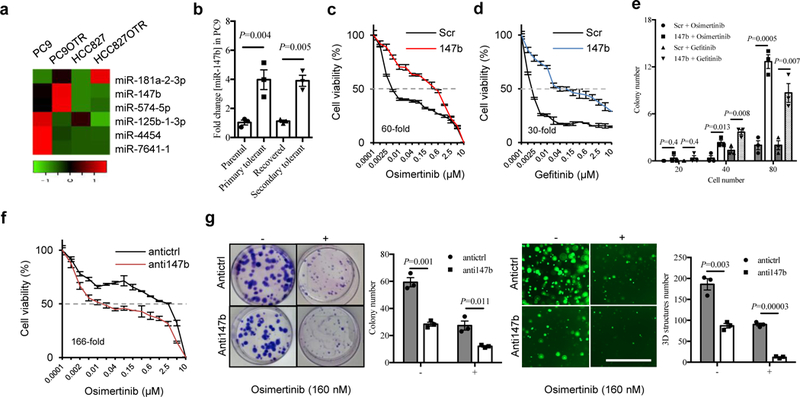
a, A heat map showing top upregulated and downregulated miRNAs in two paired osimertinib-tolerant (OTR) and parental cells in PC9 and HCC827 by miRNA-seq analysis.
b, qRT-PCR analysis of miR-147b expressions in parental, recovered, primary and secondary osimertinib-tolerant cells in PC9. The parental tumor cells treated with 160 nM EGFR-TKI osimertinib for 6 days enter a drug-tolerant state (primary tolerant cells) with a brief therapy withdrawal up to 18 days (recovered cells) followed by reinstatement of the 160 nM dose for 11 days (secondary tolerant cells). The relative miR-147b expression level in the parental cells were calibrated as 1. miR-423 was used as endogenous control. n=3 independent biological replicates.
c-d, Osimertinib (c) and gefitinib (d) treatment response on scrambled control (Scr) and miR-147b-overexpressing cells (147b) in HCC827 for 3 days. n=3 independent biological replicates.
e, Osimertinib (40 nM) and gefitinib (40 nM) treatment response on scrambled control and miR-147b-overexpressing cells in HCC827 by colony formation assay. 20, 40 and 80 cells were plated in 10-cm dish and the colonies were stained with Giemsa on day 10 and the total number of colonies were quantified. n=3 independent biological replicates.
f, Osimertinib treatment response on H1975 cells with miR-147b knockdown (anti147b) and scrambled control (antictrl). The cell viability was measured on day 4. n=3 independent biological replicates.
g, Osimertinib (160 nM) treatment response on H1975 cells with miR-147b knockdown. Left, the monolayer colonies were treated for 10 days and stained with Giemsa. Right, the 3D structures were treated for 14 days. -, vehicle; +, osimertinib. Scale bar, 1000 μm. n=3 independent biological replicates.
Data are mean ± s.e.m. and were analysed with one-way ANOVA (b); unpaired two-tailed t-test with Holm-Sidak’s correction (e,g).
As expected, our qRT-PCR analysis validated the up to five-fold upregulation of miR-147b expression in gefitinib- and osimertinib-tolerant cells compared with parental cells in both PC9 and HCC827 (Fig. 2b and Supplementary Fig. 5a). Furthermore, the expression levels of miR-147b decreased in the recovered primary drug-tolerant cells in PC9 cells upon osimertinib withdrawal for 18 days. miR-147b expression levels rose in the recovered cells when osimertinib was administered again after 11 days (Fig. 2b). Next, we established parental 3D structures from PDX lung tumors harboring EGFR mutations (Supplementary Table 2) and created osimertinib-tolerant 3D structures (Supplementary Fig. 5b) by continuous treatment with 100 nM osimertinib for 21 days. Consistently, miR-147b expression levels in osimertinib-tolerant PDX 3D structures showed up to five-fold increase compared with parental PDX 3D structures (n=5) (Supplementary Fig. 5c). In addition, hypoxia genes including ANGPTL4 (angiopoietin like 4), LOX, ENO1, LDHA (lactate dehydrogenase A), VEGFA (vascular endothelial growth factor A), and SLC2A1 (solute carrier family 2 member 1) were also upregulated in osimertinib-tolerant PDX 3D structures (Supplementary Fig. 5d). To understand effects of 3D structures culture stages on outcome of drug-tolerance, we made established 3D structures (grown for 24 days) first followed by osimertinib treatment for additional 21 days. Our data showed that drug-tolerant cells derived from 3D structures on day 24 form comparable structures and express similar levels of miR-147b and pseudohypoxia genes compared to those derived from 3D structures on day 1 (Supplementary Fig. 5e–g). Thus, our data indicate that the 3D structures culture stage does not affect the outcome of drug-tolerance. Then we asked whether heterogeneity is existed in the initial 3D structures with respect to expressions for miR-147b and pseudohypoxia genes. To answer the question, using initial 3D structures established from single cell-derived HCC827 3D structures on day 2, 4 and 6, we performed qRT-PCR analysis on miR-147b and pseudohypoxia gene expression. Our data demonstrated that there is no significant difference regarding to miR-147b and pseudohypoxia genes expressions in the initial 3D structures (Supplementary Fig. 5h). Collectively, our data suggests that miR-147b expression levels are relevant to the reversible drug-tolerance.
EGFR and KRAS mutations are widely known as mutually exclusive in lung cancer patients, and mutations in KRAS are associated with a lack of sensitivity to gefitinib 30. EGFR-TKI tolerant cells still respond to EGFR inhibitors at higher concentrations (Supplementary Fig. 2c) because they harbor the same EGFR activating mutation as their parental cells (Supplementary Fig. 3), thus we hypothesized that miR-147b expression might be distinguishable in patients with mutated EGFR rather than mutated RAS. To validate this hypothesis, we performed whole transcriptome RNA-seq analysis on a cohort of lung adenocarcinoma cell lines for miRNA profiles relevant to EGFR mutations using a public dataset (Supplementary Table 5) 31. We found the top upregulated miRNAs include miR-147b, miR-936, miR-141, miR-559, and miR-200c in EGFR mutant cell lines (n=8) compared with RAS mutant cell lines (n=17) (Supplementary Fig. 6a–b). Consistently, qRT-PCR analysis demonstrated that miR-147b expression levels in lung cancer cell lines with TKI sensitizing or resistant EGFR mutations (n=7) were higher than those in EGFR wild-type lung cancer cell lines (n=5) (Supplementary Fig. 6c and Supplementary Table 6). Interestingly, the miR-147b expression levels in cancer cells (HCC827GR, PC9ER and H1975) with EGFRT790M were even higher than those (HCC827, H3255, PC9 and H1650) with EGFR sensitizing mutations (Supplementary Fig. 6c). Next, analysis of lung adenocarcinoma patient-derived xenografts showed that miR-147b expression levels in EGFR mutant PDX tumors (176 ± 38) were up to four-fold higher than those in the EGFR wild-type lung cancers (54 ± 16) (P<0.05) (Supplementary Fig. 6d). This is consistent with our data for human lung cancer cell lines (Supplementary Fig. 6a). Further analysis of lung adenocarcinoma tissues in The Cancer Genome Atlas (TCGA) dataset 32,33 showed that the median read counts of miR-147b in EGFR mutant tumors (median=1.16, n=31) are 1.7-fold higher than those in KRAS mutant tumors (median=0.68, n=75) (P=0.2) (Supplementary Fig. 6e–f and Supplementary Table 7). The above data suggests that miR-147b might be a potent marker in EGFR mutant lung cancers.
Furthermore, to study the functional roles of miR-147b in regulating drug-tolerance, we overexpressed lentiviral miR-147b in HCC827 cells. We found that the enforced overexpression of miR-147b enhanced drug-tolerance by 60-fold and 30-fold at the half-maximum inhibitory concentration (IC50) of osimertinib and gefitinib, respectively (Fig. 2c–d). As expected, miR-147b overexpression on HCC827 cells rescued decreased colony-formation induced by treatments with osimertinib or gefitinib (Fig. 2e). Conversely, knocking down miR-147b by lentiviral infection on H1975 cells increased their sensitivity towards osimertinib by 166-fold at the IC50 (Fig. 2f). As expected, miR-147b knockdown almost abolished all the drug-tolerant colonies and 3D structures in the presence of osimertinib within 12 – 21 days (Fig. 2g). This suggests that miR-147b is critical for regulating drug-tolerance. Furthermore, a spheroid-formation assay and limiting-dilution analysis showed that knocking down miR-147b decreased the frequency of tumor-initiating cell (TIC) by seven-fold from 1/11.8 (8.5%) to 1/83.1 (1.2%) (Supplementary Fig. 7a–c). Consistently, miR-147b knockdown decreased expression levels of stem-associated genes in Wnt/PCP signaling pathway by qRT-PCR analysis, including WNT5A, FZD2 and FZD7 34 (Supplementary Fig. 7d). In addition, miR-147b knockdown also downregulated expression levels for SLC2A3 and LOX as well as upregulated expression levels for SPRY4 and IDH3A (Supplementary Fig. 7d). The dysregulated gene profile is consistent to those dysregulated in drug-tolerant cells (Fig. 1f). Furthermore, using a CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 approach, we knocked out miR-147b in H1975 cells (Supplementary Fig. 8a) and demonstrated that miR-147b knockout could consistently reduce cell viability in 3D structures and decrease osimertinib-tolerance in H1975 cells (Supplementary Fig. 8b–d). Thus, EGFR-TKIs tolerance is conferred by miR-147b.
miR-147b-VHL axis confers drug-tolerance
To study which genes are repressed by miR-147b directly, we performed sequence-based target prediction using the TargetScan tool. The predicted targets were then analyzed to match the signaling pathways for drug-tolerance (Fig. 3a). Our data had shown that VHL and SDHD are the top two most upregulated targets upon miR-147b knockdown in H1975 cells in the list of predicted targets for miR-147b (Fig. 3a). They are matched to the signaling pathways, cellular response to hypoxia and the TCA cycle, respectively (Fig. 3a). However, expression levels for other predicted targets relevant to a “tolerance gene signature” including ISCU (iron-sulfur cluster assembly enzyme) and TCEA3 (transcription elongation factor A3) (involved in cellular response to hypoxia) as well as NDUFA4 (NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4, 9kDa) (involved in the TCA cycle) were not upregulated significantly in cells with miR-147b knockdown (Fig. 3a). This indicated that VHL and SDH are potential targets of miR-147b in the context of drug-tolerance.
Figure 3. miR-147b-VHL axis mediates drug-tolerance through impaired VHL activity.
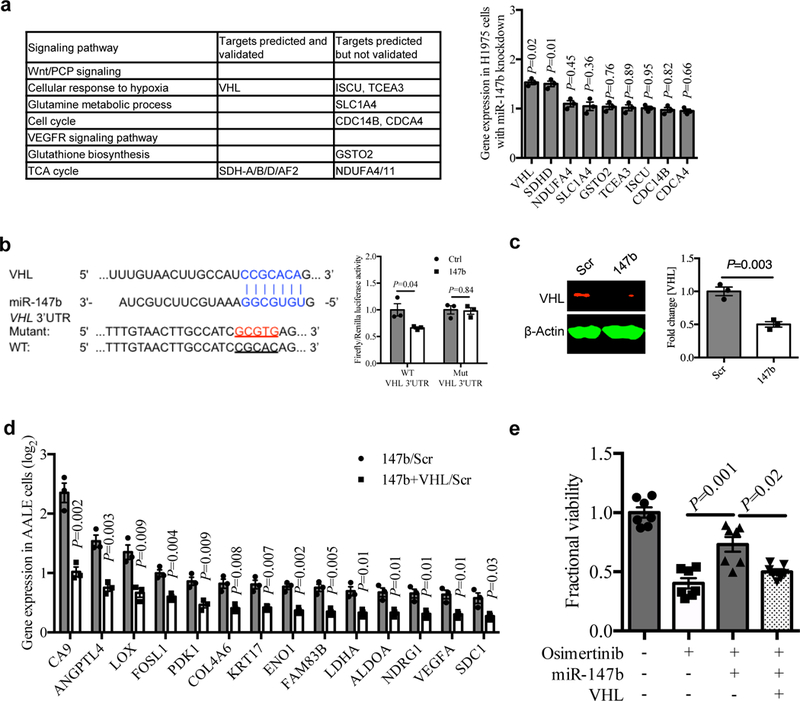
a, Left, gene candidates predicted for miR-147b by the TargetScan tool were shown in signaling pathways enriched for gefitinib-tolerance in PC9 single-cell clones in fig. 1f. Right, qRT-PCR analysis for the predicted gene candidates for miR-147b in H1975 cells with miR-147b knockdown compared with scrambled control. n=3 independent biological replicates.
b, Left, computational prediction of RNA duplex formation between miR-147b and the 3’UTR (untranslated region) of VHL mRNA. Mutations generated within the 3’UTR for the luciferase assay are shown in red. Right, dual-luciferase reporter assay in miR-147b-overexpressing AALE cells. The Firefly luciferase and Renilla luciferase activities were measured 48 hours post co-transfection with miR-147b or control vector and wild-type (WT) or mutant (Mut) VHL 3’UTR. n=3 independent biological replicates.
c, Western blot analysis and quantification of VHL in miR-147b-overexpressing AALE cells. β-Actin was used as loading control. n=3 independent biological replicates.
d, qRT-PCR analysis for fold change of hypoxia gene expression in AALE cells with miR-147b overexpression relative to scrambled control (147b/Scr) and cells with co-overexpression of miR-147b and VHL relative to scrambled control (147b+VHL/Scr). ACTB was used as endogenous control. n=3 independent biological replicates.
e, Fractional viability of HCC827 cells treated with vehicle, osimertinib (20 nM), miR-147b vector, VHL vector or combinations. The cell viability was measured on day 3. The relative cell viability treated with vehicle on day 3 was calibrated as 1. n=7 independent biological replicates.
Data are mean ± s.e.m. and were analysed with unpaired two-tailed t-test (a,b,c,d); Kruskal-Wallis test (e).
Next, we designed a dual-luciferase assay based on the VHL 3’UTR, wild-type and mutant in those predicted 3’UTR miR-147b binding sites (Fig. 3b). We found that the 3’UTR luciferase activity of VHL was downregulated when miR-147b was overexpressed in AALE cells. However, the luciferase activity for 3’UTR mutant VHL did not change upon overexpression of miR-147b (Fig. 3b). Then we asked whether miR-147b is more likely to be experimentally validated among the top candidate VHL-regulating miRNAs emerging from the TargetScan tool (Supplementary Fig. 9a). We performed a correlation analysis for VHL and non-coding gene expression in 60 human lung adenocarcinoma cell lines using RNA-seq data 31. Our results demonstrated that miR-147b is the most negatively correlating miRNA, supporting our findings that miR-147b can regulate VHL negatively (r = −0.34, P = 0.002) (Supplementary Fig. 9b–c and Supplementary Table 8).
Furthermore, we checked the VHL protein level in miR-147b overexpressing cells in AALE. The VHL protein levels decreased only two-fold when miR-147b was overexpressed in AALE cells (Fig. 3c). In the cytoplasm, an E3 ubiquitin ligase containing the VHL tumor suppressor protein targets HIF1alpha for destruction in the presence of oxygen35. Loss of VHL function leads to the alteration of numerous direct HIF1alpha-mediated transcriptional programs that alter cellular metabolism and induces angiogenesis independent of oxygen levels 36. Thus, we hypothesize that the changes required for miR-147b induced pseudohypoxia depend on the activity of VHL. To test this hypothesis, we overexpressed VHL in miR-147b overexpressing cells on AALE. As expected, gain-of-function of VHL decreased expression levels of pseudohypoxia genes induced by miR-147b. Those perturbed genes included CA9, ANGPTL4, LOX, FOSL1 (FOS like 1, AP-1 transcription factor subunit), PDK1 (pyruvate dehydrogenase kinase 1), COL4A6 (collagen type IV alpha 6 chain), ENO1 (enolase 1), FAM83B (family with sequence similarity 83 member B), LDHA, ALDOA (aldolase, fructose-bisphosphate A), NDRG1 (n-Myc downstream regulated 1), VEGFA and SDC1 (syndecan 1) (Fig. 3d). Further, functional assay showed that the enhanced osimertinib-tolerance induced by miR-147b-overexpression was reduced upon VHL overexpression on HCC827 cells (Fig. 3e). Taken together, these data indicate that the activity of VHL for repressing “pseudohypoxia gene signature” mediates drug-tolerance initiated by miR-147b.
Tricarboxylic acid pathways mediate drug tolerance and depend on miR-147b
In addition to the functional roles of VHL-mediated “pseudohypoxia gene signature” in drug-tolerance, we hypothesized that another predicted target of miR-147b, SDH might also mediate drug-tolerance induced by miR-147b through its impact on the TCA cycle. To test this hypothesis, we first designed a dual-luciferase assay based on the SDHD 3’UTR, wild-type and mutant in the predicted miR-147b 3’UTR binding sites (Fig. 4a). We found that luciferase activity of 3’UTR SDHD wild type rather than mutant SDHD was downregulated upon overexpression of miR-147b on AALE cells (Fig. 4a). This strongly suggests that SDHD is a direct target repressed by miR-147b.
Figure 4. miR-147b-SDH axis mediates drug tolerance through SDH enzyme activity in the TCA cycle.
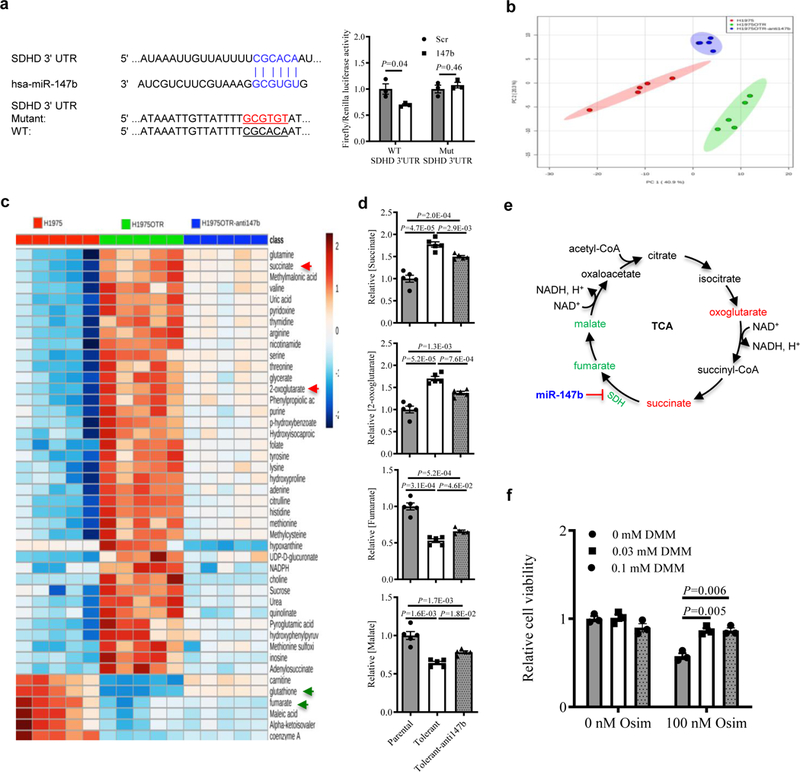
a, Left, computational prediction of RNA duplex formation between miR-147b and the 3’UTR of SDHD mRNA. Mutations generated within the 3’UTR for the luciferase assay are shown in red. Right, dual-luciferase reporter assay in miR-147b-overexpressing AALE cells. The Firefly luciferase and Renilla luciferase activities were measured 48 hours post co-transfection with miR-147b or control vector and wild-type (WT) or mutant (Mut) SDHD 3’UTR. n=3 independent biological replicates.
b, Principal component analysis (PCA) of parental cells, osimertinib-tolerant cells (H1975OTR) and tolerant cells with miR-147b knockdown (H1975OTR-anti147b) in H1975 cell monolayers. The tolerant cells were derived from the parental cells treated with 100 nM osimertinib continuously for 21 days. n=5 independent biological replicates.
c, A heat map showing top metabolites levels across cells of H1975, H1975OTR and H1975OTR-anti147b. n=5 independent biological replicates.
d, Levels of succinate, 2-oxoglutarate, fumarate, and malate in cells of H1975, H1975OTR and H1975OTR-anti147b. The relative levels in the parental H1975 cells were calibrated as 1. n=5 independent biological replicates.
e, Schematic of the interaction among miR-147b and SDH enzyme leading to dysregulated TCA cycle metabolites for drug-tolerance to EGFR tyrosine kinase inhibitors. Upregulated levels of oxoglutarate and succinate (in red) as well as downregulated levels of fumarate and malate (in green) in drug-tolerant cells are highlighted.
f, SDH inhibitor promotes drug-tolerance to osimertinib in H1975 cells. Vehicle or 100 nM osimertinib (osim)-treated cells were co-incubated with 0, 0.03 and 0.1 mM membrane-permeable dimethyl malonate (DMM) for 3 days. The cell viability was measured on day 4. n=3 independent biological replicates.
Data are mean ± s.e.m. and were analysed with unpaired two-tailed t-test (a,f); one-way ANOVA (d).
SDHD, one of the subunits of SDH complex, catalyzes the conversion of succinate to fumarate and regulates both the TCA cycle and the ETC. We asked whether miR-147b-SDHD axis mediated drug-tolerance could impact on the metabolite changes in metabolic pathways. To answer this question, the human lung adenocarcinoma cell line H1975 harboring with EGFR T790M; L858R mutations was used for a metabolomics study. Cells with either EGFR L858R or EGFR T790M are sensitive to osimertinib. The osimertinib-tolerant cells (H1975OTR) were derived from parental H1975 treated with 100 nM osimertinib for 21 days in monolayer cultures. H1975OTR cells are stable and continue to proliferate even in the presence of 100 nM osimertinib. As a control, H1975 cells were treated with vehicle for 21 days. Then we performed a LC/MS metabolomics study using the paired H1975 and H1975OTR cells (Fig. 4b). Analysis of metabolite levels demonstrated that the metabolites in both the TCA cycle and the electron transport chain (ETC) related redox reactions were perturbed in drug-tolerant cells. We observed up to a two-fold rise of succinate and 2-oxoglutarate levels but up to a two-fold decrease of fumarate and malate levels in drug-tolerant cells (Fig. 4c–d, Supplementary Fig. 10a and Supplementary Table 9). In addition, the levels of oxidized nicotinamide adenine dinucleotide (NAD+) decreased 26% in drug-tolerant cells compared to the parental cells (Supplementary Fig. 10a). This is consistent with a previous finding showing that decreased NAD+ levels induced a pseudohypoxia state in aging37. Furthermore, reduced glutathione (GSH), the master antioxidant, decreased 86% in drug-tolerant cells compared with the parental cells (Fig. 4c). Our data suggest that the metabolic changes in the TCA cycle might be important in regulating drug-tolerance. Then we asked whether the perturbed metabolite changes could be rescued by blocking miR-147b in drug-tolerant cells. To address this question, we knocked down miR-147b in drug-tolerant cells in H1975 and analyzed the metabolic changes with LC/MS tool. As expected, the increased levels of succinate and 2-oxoglutarate as well as the decreased levels of metabolites such as fumarate, malate, NAD+ and GSH were partially rescued by knocking down miR-147b on osimertinib-tolerant cells (Fig. 4c–e, Supplementary Fig. 10a, and Supplementary Table 9). These data confirmed our hypothesis that one role of miR-147b is through repressing the enzyme activity of SDH.
Then we asked whether the metabolomic changes in the monolayer cultures are reproducible in the 3D structure models. To address this question, we established drug-tolerant and parental 3D structures by continuous treatments with 100 nM osimertinib or vehicle for 21 days on H1975 cells and performed a LC/MS metabolomics study. Consistently, the levels of fumarate, malate, and NAD+ were reduced in osimertinib-tolerant 3D structures. Knockdown of miR-147b rescued the decreased levels of the above metabolites in those tolerant 3D structures (Supplementary Fig. 10b–c). Our data suggest that the metabolic changes due to the depression of SDH by miR-147b might regulate drug-tolerance (Fig. 4e). To further confirm the functional roles of SDH activity in mediating drug-tolerance, we treated H1975 cells with membrane-permeable dimethyl malonate (DMM), one of the inhibitors of SDH in the presence of 100 nM osimertinib. Our results demonstrated that DMM effectively rescued the decreased drug-tolerance to osimertinib (Fig. 4f). Collectively, our data have demonstrated that repressed SDH activity by miR-147b mediates osimertinib-tolerance in lung cancer.
Blocking miR-147b overcomes drug tolerance
We asked whether miR-147b is a druggable target in lung cancer. First, we knocked down miR-147b with a lentiviral miRNA inhibitor in H1975 cells and transplanted those cells into nude mice. The tumor growth in the cohort with miR-147b knockdown was up to two-fold slower compared with that in the control group (Supplementary Fig. 11a–b). This indicates that blocking miR-147b inhibits tumor growth in vivo.
Furthermore, to understand functional roles of miR-147b in regulating drug-tolerance via regulation of a pseudohypoxia signaling pathway, we blocked miR-147b by administration of locked nucleic acid (LNA) miRNA inhibitors as well as perturbing pseudohypoxia signaling with small molecule activators and inhibitors. First, LNA-miR-147b inhibitor treatment increased the sensitivity of drug-tolerant 3D structures to osimertinib by 30-fold compared with the control group in H1975 (Supplementary Fig. 11c–d). The small molecule dimethyloxaloylglycine (DMOG) was reported to activate a pseudohypoxia response through repressing its negative regulator PHD2. As expected, treatment with a single dose of 10 µM DMOG induced upregulated expression of pseudohypoxia genes in H1975 (Supplementary Fig. 11e). Further functional assays demonstrated that co-treatment with DMOG rescued reduced osimertinib-tolerance caused by LNA-miR-147b inhibitor (Fig. 5a). Consistent with the functional rescue experiment, the reduced levels of “pseudohypoxia genes” induced by miR-147b knockdown were rescued significantly by co-treatments with DMOG in H1975 3D structures (Fig. 5b). Then we hypothesized that blocking the pseudohypoxia signaling pathway with small molecules might further enhance drug sensitivity induced by miR-147b inhibitor. To address this question, we applied another small molecule R59949 to this study due to its role in inhibiting pseudohypoxia response through activation of PHD2. We confirmed that treatment with a single dose of 30 µM R59949 induced downregulation of pseudohypoxia genes compared with vehicle treated H1975 cells (Supplementary Fig. 11f). And co-administration of R59949 and LNA-miR-147b inhibitor showed stronger inhibition on drug-tolerance to EGFR-TKI compared with a single agent of LNA-miR-147b inhibitor (Fig. 5c). Consistently, the reduced expression levels of “pseudohypoxia genes” induced by LNA-miR-147b inhibitor were further inhibited by co-treatment with R59949 in H1975 cells (Supplementary Fig. 11g). This strongly supports our idea that miR-147b and miR-147b-induced pseudohypoxia signaling pathway are druggable targets to overcome osimertinib-tolerance in lung cancer.
Figure 5. Blocking miR-147b overcomes drug-tolerance.
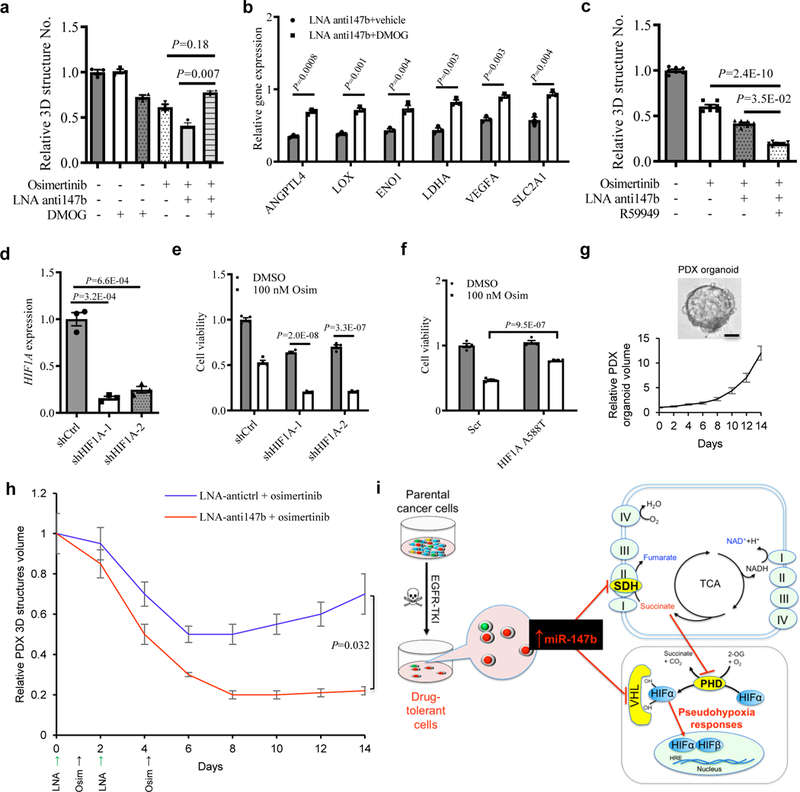
a, Fractional viability of H1975 3D structures treated with osimertinib (25 nM), LNA miR-147b inhibitor (LNA-anti147b, 90 nM), DMOG (10 μM) or combinations for 14 days. n=3 independent biological replicates.
b, qRT-PCR analysis for hypoxia gene expression in H1975 cells treated with 90 nM LNA miR-147b inhibitor (LNA-anti147b) and 10 µM DMOG or vehicle for three days. The relative gene expression in scrambled control cells treated with vehicle was calibrated as 1. n=3 independent biological replicates.
c, Fractional viability of H1975 3D structures treated with 25 nM osimertinib, 90 nM LNA-anti147b, 30 µM R59949 or combinations for 14 days. n=7 independent biological replicates.
d, qRT-PCR analysis of HIF1A in H1975 cells with shRNAs against HIF1A. H1975 cells were transfected with shRNAs against HIF1A (shHIF1A-1 and -2) or scrambled control (shCtrl) and selected with 0.5 μg/ml puromycin. GAPDH was used as endogenous control. n=3 independent biological replicates.
e, Cell viability of H1975 cells with HIF1A knockdown treated with osimertinib. The cells with shRNAs against HIF1A (shHIF1A-1 and shHIF1A-2) and scrambled control cells (shCtrl) were treated with 100 nM osimertinib or vehicle for 3 days. The cell viability was analyzed on day 4. n=4 independent biological replicates.
f, Cell viability of H1975 cells with constitutive active HIF1A mutant treated with osimertinib. The cells were transfected with HIF1A A588T and scrambled control cells (Scr) followed by 600 μg/ml neomycin selection. Then the cells were treated with 100 nM osimertinib or vehicle for 3 days. The cell viability was analyzed on day 4. n=4 independent biological replicates.
g, Derivation and growth of 3D structures from lung PDX tumors. (top) Representative phase contrast microscopy for parental EGFR mutant lung PDX-derived 3D structures in PDX_LU_10 3D structures. Repeated six times with similar results. (Bottom) growth curve of PDX 3D structures. The 3D structures size was measured every two days. The media were replenished every three days till day 14. n=3 independent biological replicates. Scale bar, 50 µm.
h, Pretreatment response on lung PDX_LU_10 3D structures with LNA miR-147b inhibitor (anti147b) and osimertinib. The 3D structures were established at medium size seven days after seeding 2000 single-cells into 3D cultures in 96-well plate. This timepoint was recorded as day 0. Then the 3D structures were administrated with LNA anti147b or antictrl (90 nM) on day 0 and day 2 or osimertinib (25 nM) on day 1 and day 4. The vehicle treated group did not receive treatments with LNA or osimertinib. The 3D structures’ size was measured every two days. The media were replenished every three days till day 14. n=3 independent biological replicates.
i, Schematic for miR-147b-driven drug-tolerance model. miR-147b is enriched in a subpopulation of parental lung cancer cells entering drug-tolerant status when they are treated with EGFR-TKIs. miR-147b mediates drug-tolerance through repressing activities of VHL and SDH leading to activated pseudohypoxia response. TKI, tyrosine kinase inhibitor; SDH, succinate dehydrogenase; TCA, tricarboxylic acid; PHD, prolyl-hydroxylase.
Data are mean ± s.e.m and were analysed with Kruskal-Wallis test (a,c); unpaired two-tailed t-test (b,d,e,f,h).
To understand roles of HIF-1 or HIF-2 in the osimertinib tolerant state, we knocked down HIF1A and HIF2A/EPAS1 (endothelial PAS domain protein 1) using lentiviral shRNAs in H1975 cells and investigated their effect on osimertinib response. Our results showed that HIF1A knockdown increased cell sensitivity up to 2.6-fold towards osimertinib (Fig. 5d–e). However, HIF2A knockdown did not change drug sensitivity towards osimertinib significantly (Supplementary Fig. 12a–b). Furthermore, to better understand whether gain of HIF-1 is sufficient to induce a tolerant state, we overexpressed constitutive active HIF1A using mutant HIF1A A588T in H1975 cells. As expected, overexpression of HIF1A A588T increased drug-tolerance towards osimertinib by up to two-fold (Fig. 5f). Thus, our results have now demonstrated that HIF1A rather than HIF2A is sufficient to induce an osimertinib tolerant state.
Last, we asked whether we could delay drug-tolerance to EGFR-TKIs by targeting miR-147b. To address this question, 3D structures obtained from PDX lung tumors were tested. Among these PDX lung tumors, one EGFR T790M mutated PDX tumor-derived 3D structures (PDX_LU_10) at passage two was tested in the following functional study (Fig. 5g). We established PDX 3D structures at medium size one week after seeding single-cells into 3D cultures. We recorded this timepoint as day 0 before the administration of LNAs or osimertinib. As expected, the PDX 3D structures increased their volumes up to ten-fold within 14 days in the vehicle-treated group (Fig. 5g). With the administration of 25 nM osimertinib to PDX 3D structures on day 1 and 4, the size of the tumor 3D structures decreased 50% on day 6 and then started to recover gradually with a 40% increase on day 14. To test whether early perturbation of miR-147b could delay drug-tolerance to osimertinib, we pretreated the 3D structures with 90 nM LNA miR-147b inhibitor on day 0 and repeated the treatment on day 2. These pretreatments with LNA miR-147b inhibitor further decreased osimertinib-tolerance on day 8 by 80% compared with control cells. Furthermore, the PDX 3D structures volume increased no more than 10% of that in control cells with the single agent of osimertinib from day 8 to day 14 (Fig. 5h). Our data suggests that early treatment on EGFR mutant lung cancer with miR-147b inhibitor might delay drug-tolerance to EGFR-TKIs compared with single EGFR-TKI treatment.
Discussion
Dysregulated cancer metabolism has recently gained attention for its potential role in promoting therapeutic resistance by a drug-tolerance strategy in a novel manner24. Here we have demonstrated that blocking miR-147b and reactivation of the TCA cycle pathway provides a promising strategy to prevent drug-tolerance-mediated tumor relapse (Fig. 5i).
NSCLC cells demonstrate higher levels of both glycolysis and glucose oxidation relative to adjacent benign lung38,39. Increasing evidence suggests that the metabolic enzymes and the catalyzed metabolites in the TCA cycle are not only involved in tumorigenesis but also drug resistance, such as isocitrate dehydrogenase, SDH and succinate20,40. In our study, the reciprocal changes of metabolites in the TCA cycle such as increased levels of succinate and 2-oxoglutarate as well as decreased levels of malate and fumarate in osimertinib-tolerant cells indicate that silenced activity for SDH is linked to drug-tolerance. In addition, small molecule inhibitor R59949 silencing SDH activity could enhance drug-tolerance, which is comparable to the function of miR-147b overexpression in drug-tolerant cells. It is not surprising that accumulated succinate due to loss of function of SDH could activate pseudohypoxia signaling pathway through repressing PHD2 as this was reported previously 20. This is consistent with our findings that the miR-147b-SDH axis could increase the gene expression for pseudohypoxia signaling pathways. It has also been reported that miR-147 overexpression could protect cells from hypoxia-induced injuries 41. A hypoxia response is linked to tumor cell survival and drug-resistance in many cancers 42,43. Other factors in our data set might also activate pseudohypoxia responses and drug-tolerance, including VHL inactivation, reduced NAD+ and decreased GSH, which are consistent with previous findings24,37,44,45. In addition, these pseudohypoxia responses may further perturb the TCA cycle and cooperatively regulate drug-tolerance.
Certain miRNAs regulate tumor cells growth as oncomiRs 46–50. We have demonstrated that higher miR-147b expression levels link to activated EGFR mutations in both cancer cell lines and lung cancer PDX tumors. This suggests that miR-147b may promote drug-tolerance to EGFR-TKIs either through reactivation of the EGFR downstream signaling pathway or through bypass by another receptor tyrosine kinase that sustains downstream signaling despite inhibition of EGFR3,51. Another study supports our hypothesis showing that miR-147 reverses EGFR inhibitor resistance by inducing a mesenchymal-to-epithelial transition in KRAS mutant cancers 52.
In addition, VHL binds directly to hydroxylated Akt and inhibits Akt activity53 suggesting that miR-147b-VHL axis might confer drug-tolerance through activating Akt activity. In addition to the non-coding RNAs, other upstream transcription factors could also control VHL levels, such as ID2, which binds to the VHL ubiquitin ligase complex, displaces VHL-associated Cullin 2, and impairs HIF2alpha ubiquitylation and degradation 54. The crosstalk between miR-147b and other transcription factors regulating VHL needs to be studied further in the future.
Our findings have revealed that tumor cells protect themselves with a drug-tolerance mechanism when they are treated with EGFR-TKIs. These findings align with previous experimental data showing that tumor cells enter into a tolerant state when they are treated with TKIs in lung and other cancers8,9,24. These tolerant cells precede and evolve into resistant cells over time by acquiring EGFR resistant mutations6,7. Whether those cells tolerant to each generation of EGFR-TKIs are derived from the same subpopulation of cells needs to be investigated in the future. In addition, we found that these tolerant cells are slow cycling and are enriched in expression of stem-associated genes in the Wnt/PCP signaling pathway, such as WNT5A, FZD2 and FZD7. These findings are conceptually similar to a recent report that post-drug transition to stable resistance consists of dedifferentiation55. It is not clear whether drug-tolerant cells and TICs are derived from the same cellular origin. TICs display heterogeneous phenotypes due to different genotypes in tumors56. Thus, the genetic backgrounds, such as mutations of EGFR and RAS need to be taken into consideration to better understand the association between drug-tolerant cells and TICs in the future.
In summary, we have determined that cancer cells adopt a tolerance strategy to defend against EGFR inhibition by expressing miR-147b, disrupting the TCA cycle and activating the pseudohypoxia response. Targeting miR-147b and its relevant TCA/hypoxia pathways could provide a new strategy to prevent anticancer drug resistance.
METHODS
Cell Culture.
Human lung EGFR-wild type cell lines H358, H460, A549, H1299, and H69 (ATCC) as well as EGFR-mutant cell lines H1650, H1975, HCC827, HCC827GR, PC9, PC9ER, and H3255 (provided by Dr. Susumu Kobayashi from Beth Israel Deaconess Medical Center (BIDMC) of Harvard Medical School) were cultured in DMEM (high glucose) (Gibco, Cat #11995–065) with 10% FBS (Sigma-Aldrich, Cat #F2442), 2 mM L-glutamine and 1% penicillin-streptomycin. Immortalized tracheobronchial epithelial AALE cells (provided by Dr. William C. Hahn from Dana-Farber Cancer Institute of Harvard Medical School) were derived as previously described57 and maintained in SAGM media (Lonza, Cat #CC-3118). Primary 3D cultures of lung cancer patient-derived xenograft tumor (PDX_LU_10) were established at BIDMC of Harvard Medical School and maintained in advanced DMEM/F12 (Gibco, Cat #12634–010) with glutamax (1x) (Gibco, Cat #35050–061), HEPES (1x) (Gibco, Cat #15630080), 1.25 mM N-Acetylcysteine (Sigma-Aldrich, Cat #A9165), 10 mM Nicotinamide (Sigma-Aldrich, Cat #N0636), 10 µM Forskolin (Sigma-Aldrich, Cat #F6886), B27 (1x) (Gibco, Cat #17504–044), 5 ng/ml Noggin (Sigma-Aldrich, Cat #PHC1506), 100 ng/ml FGF10 (Gibco, Cat #PHG0204), 20 ng/ml FGF2 (Gibco, Cat #PHG0026), 50 ng/ml EGF (Gibco, Cat #10605-HNAE-250), 10 ng/ml PDGFA (Gibco, Cat #PHG0035), 10 ng/ml FGF7 (Gibco, Cat #PHG0094), 1% penicillin-streptomycin and 10 µM Y-27632 (Selleck Chemicals, Cat #S1049). Each cell line was maintained in a 5% CO2 atmosphere at 37°C. Cell line identities were confirmed by STR fingerprinting and all were found negative for mycoplasma using the MycoAler Kit (Lonza).
Ethical approval.
All tissues were obtained under written informed patient consent and were de-identified. Only de-identified human tumor samples implanted in immunodeficient mice were used to generate PDXs for The Jackson Laboratory (JAX) PDX Resource and Yale University. The procedures for patient samples and data collection were conducted with approval by The Jackson Laboratory Institutional Review Board (IRB) (reference #121200011) and Yale University Human Investigation Committee. All relevant ethical regulations were followed.
Mice.
All research involving animals complied with protocols approved by the Institutional Animal Care and Use Committees (IACUC) from BIDMC, The Jackson Laboratory and Yale University. For establishing subcutaneous tumors from PDXs, 4–6 weeks old female NSG immunodeficient mice (The Jackson Laboratory, strain # NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, stock # 005557, homozygous for both the Prkdcscid and Il2rgtm1Wjl alleles) were used as part of commercial production of the PDX at The Jackson Laboratory or implanted as part of this study at Yale Cancer Center. No littermate controls were used. For generating xenograft tumors by transplanting H1975 cells with miR-147b knockdown, 4–6 weeks old female nude immunodeficient mice (The Jackson Laboratory, strain # NU/J, stock # 002019, homozygous for Foxn1nu) were used for subcutaneous injections at the BIDMC. For subcutaneous xenograft tumor assay, 100,000 cells in serum-free medium and growth factor reduced Matrigel (BD, Cat #354230) (1:1) were inoculated into the flank of nude mice. The xenograft tumor formation was blindly monitored by calipers twice a week. The recipient mice were monitored and euthanized when the tumors reached 1 cm in diameter or the mice have lost 15% weight. See reporting summary.
Patient-derived Xenograft Tumor Specimens.
Patient tumor material was obtained through a network of collaborating cancer research centers to develop the JAX PDX Resource. The collaborating centers were responsible for any necessary IRB approvals and patient consents to allow their tumor tissue to be used in research. These responsibilities are acknowledged in the JAX PDX Consortium Agreement, which was signed by the participating member prior to submitting their first patient specimen for model development. The JAX PDX Resource received coded patient tumor samples, with all personal identifiers removed. No JAX investigator has access to patient consent forms. Successful PDX models were assigned a unique number with all reference to the donating center and date of specimen collection removed to further protect patient privacy. Four of the six patients from the JAX PDX Resources did not receive any chemotherapy or radiotherapy before tissue collection. The other two patients received chemotherapy with or without radiotherapy prior to sample collection. Besides, patients with advanced NSCLC who developed progression after initial response to EGFR TKI were consented and enrolled to a Yale University IRB approved protocol, in accordance with ethical guidelines, allowing the collection and analysis of clinical data, fresh tissue, and the generation of patient-derived xenografts. All Yale patients received prior radiotherapy. Three of the four received prior chemotherapy before sample collection. The detailed prior treatment information is listed in Supplementary Table 2.
PDXs from JAX PDX Resource were generated under protocol 12027 approved by The Jackson Laboratory IACUC before study initiation. The models were generated using subcutaneous tumor implantation in 4–6 weeks old female NSG host mice. Detailed methods for model generation are described elsewhere 58. Information and data for PDX models from the JAX PDX Resource are publicly available from the PDX Portal hosted by Mouse Tumor Biology Database (MTB; http://www.tumor.informatics.jax.org/mtbwi/pdxSearch.do) 59. Tumor samples from PDXs were generated at The Jackson Laboratory and the Yale Cancer Center by subcutaneous implantation of previously passaged tumors in up to 5 female NSG mice. When tumor samples reached 1000 mm3 they were shipped to the laboratory in frozen media of DMEM with 90% FBS and 10% DMSO in dry ice. Samples were washed with cold phosphate buffer saline (PBS) with antibiotics (Sigma-Aldrich, Cat #A5955, St. Louis, MO) three times, chopped with a sterile blade, and incubated in 0.001% deoxyribonuclease (DNase) (Sigma-Aldrich, Cat #D4513), 1 mg/ml collagenase/dispase (Sigma-Aldrich, Cat #11097113001), 200 U/ml penicillin, 200 µg/ml streptomycin, 0.5 µg/ml amphotericin B (2% antibiotics, Sigma-Aldrich, Cat #A5955) in DMEM/F12 medium (Gibco, Cat #11320–033, GrandIsland, NY) at 37°C water bath for 3 hr with intermittent shaking. After incubation, the suspensions were repeatedly triturated, passed through 70 µm and 40 µm cell-strainers (BD Falcon, San Jose, CA), and centrifuged at 122 g for 5 min at 4°C. Cells were resuspended in red blood cell lysis buffer (eBioscience, Cat #00-4333-57, San Diego, CA) for 4 min at room temperature with intermittent shaking, before resuspension in serum-free medium. After lysis, cell viability was evaluated by trypan blue dye exclusion. Live single cells accounted for 90% of the whole population and dead cells accounted for less than 10%. Each tumor sample yielded ~1×105 to 1×106 cells, depending on the sample size.
Antibodies.
For immunofluorescence staining, primary mouse anti-human ZO-1 (1:100, clone #ZO1–1A12, lot #Q1215680, Cat #33–9100) was from Thermo Fisher Scientific. Secondary donkey anti-mouse IgG conjugated with Alexa Fluor 488 (1:500, lot #1741782, Cat #A-21202) was from Life Technologies. For western blot, primary polyclonal rabbit anti-VHL antibody (1:100, lot #RD2197039, Cat #PA5–27322) was from Thermo Fisher Scientific. Mouse anti-β-actin (1:5,000, clone #C4, lot #I1018, Santa Cruz, Cat #sc-47778) was used as loading control. IRDye 680RD goat anti-rabbit (1: 20,000, lot #C70406–04, Cat #LI-COR926–68171, LI-COR Biosciences) and IRDye 800CW goat-anti-mouse (1: 20,000, lot #C70310–02, Cat #LI-COR827–08364, LI-COR Biosciences) were used as secondary antibodies.
Spheroids and 3D Structures.
For spheroid formation, single-cell suspensions (10,000 cells/well) were plated in 6-well ultra-low attachment (Corning) or non-treated cell culture plates (Nunc) in DMEM/F12 medium containing 2 mM L-glutamine, 15 mM HEPES, 1 mg/ml NaHCO3, 0.6% Glucose, 1% NEAA, 4 mg/ml BSA (Sigma-Aldrich, Cat #A7979), ITS (0.05 mg/ml insulin/transferrin/selenium, Gibco Cat #41400045), 1% antibiotics (Sigma-Aldrich), 50 ng/ml EGF and 20 ng/ml FGF2 (Invitrogen). Fresh medium was replenished every 3 days. Spheroids were cultured for 10–14 days and then quantified. For passaging, spheroids were digested by accutase (Chemicon, Cat # SCR005) into single cells and re-plated into the above plates. For limiting dilution assays, 200, 600 and 1800 cells were plated to assess spheroid formation.
For 3D structures formation, single-cell suspensions (2000 cells/well/20 µl) were co-plated with geltrex (25 µl) in 96-well non-treated clear plates (Corning, Cat #08-772-53). The plate was incubated for 20 minutes at 37°c followed by adding 100 µl complete growth media. The complete growth media was advanced DMEM/F12 with glutamax (1x), HEPES (1x), 1.25 mM N-Acetylcysteine, 10 mM Nicotinamide, 10 µM Forskolin, B27 (1x), 5 ng/ml Noggin, 100 ng/ml FGF10, 20 ng/ml FGF2, 50 ng/ml EGF, 10 ng/ml PDGFA, 10 ng/ml FGF7, 1% penicillin-streptomycin and 10 µM Y-27632. Y-27632 was used only for the initial three days because Y27632 is a rock inhibitor preventing apoptosis of single cells 60. PDGFA and FGF7 were not used until day 7 in 3D structure cultures because they are important for alveolarization during late lung development 61,62. FGF10 is essential for maintenance of lung progenitor cells and branching morphogenesis as well as tissue homeostasis in the adult lung 63. EGF and FGF2 are mitogens for growth of epithelial cells and used for maintaining lung tumor-initiating cells previously by us 13. Noggin binds and inactivates bone morphogenetic protein-4 and is involved in the development of the lungs 64. The media was changed every three days in 24 days. The 3D structures were photographed with a microscope (Evos FL, Life Technology) and the size was measured by ImageJ 1.51s software.
Colony Formation Assay in Plate.
Single cells were plated in 10 cm dish in triplicates with 20, 40, 80 cells or 300 cells per dish. Fresh medium was replenished every 3 days. The cells were incubated for 10–12 days followed by Giemsa (Sigma-Aldrich) staining. The plates were air-dried, taken photos, and the total number of colonies was analyzed by openCFU (http://opencfu.sourceforge.net).
Single Cell-Derived Clones of PC9 and HCC827 Cells.
In PC9 and HCC827 cells, a single cell was sorted into a 96-well plate at one cell per well by fluorescence-activated cell sorting (FACS) using a FACSAria (BD). The single cell in each well was confirmed under a microscope 12 hour after sorting. Gefitinib or osimertinib were administrated on both parental clones and single-cell clones. In parental clones, the single cells were treated immediately with 0.1, 0.4 and 2 µM gefitinib, osimertinib or the vehicle for 14 days on the second day (n=192 wells per group). In single-cell clones, clones were made first, and then exposed to 0.1–2 µM gefitinib, osimertinib or vehicle for 14 days. Drug responses of the surviving clones were determined by measuring an IC50. The frequency of colony formation was calculated as a ratio of the total number of colonies (consisting of more than 50 cells) to the total number of wells plated with a single cell. Medium and small molecule inhibitors were replenished every three days. One parental single-cell derived clone treated with vehicle that was sensitive to gefitinib and two gefitinib-tolerant single-cell derived clones were randomly selected and applied for the following whole transcriptome analysis by microarray. Four single-cell clones from PC9 and HCC827 were established from the above were used for drug-tolerance assay.
Compounds.
Osimertinib (S7297) and gefitinib (S1025) were purchased from Selleck Chemicals. DMOG 65 (Cat #400091) was from Calbiochem. R59949 66 (Cat #D5794) and dimethyl malonate 67,68 (DMM, Cat #136441) were purchased from Sigma-Aldrich.
Compound Treatment.
Cell viability experiments were performed in 96-well format using opaque white plates (Corning). For 2D monolayer cell cultures and 3D structures, cells were plated into 96-well plates with 100–2000 cells per well in three-four replicates on day 0. Twenty-four hours after seeding, cells or 3D structures were exposed to compounds at indicated concentrations for 72 h. Cellular ATP levels (as a surrogate for viability) were measured using CellTiter-Glo (Cat #G7570, Promega) or CellTiter-Glo 3D (Cat #G9681, Promega). For co-treatment experiments, spent medium was removed 24 h after cell seeding and replaced with medium containing a single concentration of the modulator of interest (for example, osimertinib).
To establish gefitinib and osimertinib tolerant cells, PC9 single-cells were treated with 20 nM osimertinib and 40 nM gefitinib for 12–14 days, HCC827 cell monolayers and 3D structures were treated with 20–160 nM osimertinib for 12–21 days and H1975 cell monolayers and 3D structures were treated with 25 nM-1 μM osimertinib for 12–21 days. To study effects of 3D structure culture stages on outcome of drug-tolerance, both single-cells (grown for 1 day) and established 3D structures from HCC827 cells (grown for 24 days) were made first followed by 100 nM osimertinib treatment for additional 21 days. Medium was replenished every three days.
RNA Extraction and Real-Time PCR.
Total RNA was extracted from solid tissues and cultured cells using mirVana™ miRNA Isolation Kit (Ambion #AM1561) according to the manufacturer’s instructions. A total of 10 ng RNA each sample was input for consecutive reactions including Poly(A) Tail reaction, Ligation reaction, Reverse Transcription reaction and miR-Amp reaction using the Taqman Advanced miRNA cDNA synthesis kit (Applied Biosystems #A28007). Then miRNA expression was assessed by Taqman Advanced microRNA Assay and the Taqman Fast Advanced miRNA master mix (Applied Biosystems #4444557). The PCR reaction plate was run in a real-time PCR instrument (Roche Lightcycler 480 system) according to the manufacturer’s instructions. Three biological replicates were applied for each sample. MiRNA expression was assessed by Taqman Fast Advanced MicroRNA Assay, and the gene expression of mRNAs was evaluated by Taqman Probes (Applied Biosystems). Taqman miRNA and gene-expression probes were shown in Supplementary Table 1. hsa-miR-423–5p and ACTB or GAPDH were used as endogenous controls for analyses of miRNA and gene-expression, respectively.
Pyrosequencing for quantitative analysis of sequence variations.
The parental cells, gefitinib-tolerant cells and gefitinib-resistant cells in PC9 were extracted for DNA (QIAamp DNA blood mini kit, Cat #51104, Qiagen) and analyzed for pyrosequencing according to the manufacturer’s protocol 69,70. 10 ng input DNA was amplified using PyroMark PCR kit (Qiagen) and biotinylated primers (EGFR exon 19 forward 5′-GCATGTGGCACCATCTCA-3′ and reverse 5′-AAAAGGTGGGCCTGAGGTT-3′; or EGFR exon 20 forward 5′-ATGGCCAGCGTGGACAAC-3′ and reverse 5′-TTTGTGTTCCCGGACATAGTC-3′) on the following PCR program: 95°C for 15 min; 45 cycles of 94°C for 30 s, 56°C for 30 s, 72°C for 30 s; and 72°C for 5 min. The PCR product was incubated with Streptavidin Sepharose beads (GE Healthcare) and PyroMark Binding Buffer (Qiagen) at room temperature for 30 min followed by hybridization to the sequencing primers (exon 19 deletion, 5′-ATTCCCGTCGCTATC-3′; exon 20 T790M, 5′-GATGCCCAGCAGGCG-3′). The pyrosequencing reaction was performed on the PyroMark Q24 (Qiagen) platform using PyroMark Gold Q24 reagents (Cat #970802, Qiagen). The pyrosequencing result was analyzed using PyroMark Q24 software. Samples with more than 5% mutated alleles were scored as positive.
High-Throughput Sequencing.
The total RNA samples (1 µg) were processed by LC Sciences for microRNA sequencing (miRNA-seq). All RNA samples were analyzed for quality on an Agilent 2100 Bioanalyzer. For miRNA-seq analysis 71, paired osimertinib-tolerant cells and parental cells (treated with 20 nM osimertinib or vehicle for 14 days) from HCC827 and PC9 cells were applied. The RNA samples were blindly processed utilizing Illumina’s TruSeq small RNA sample preparation protocol for small RNA library generation (Part# 15004197 Rev. F, Cat #RS-200–9002DOC). The subsequent sequencing was performed on the HiSeq 2500 platform for 1 × 50-nt single-end sequencing and the sequencing adaptor was trimmed from the raw reads. To identify the known miRNAs, the remaining sequences were aligned to the miRBase (release 21.0) (http://www.mirbase.org/) using Bowtie (http://bowtie-bio.sourceforge.net/manual.shtml) (version: 1.2.1, release: 06/12/2017) 72. Matched sequences with < 1 mismatch were known miRNAs. In addition, the unmatched sequences were used to predict the candidate novel miRNAs using miRDeep2 (Version 2.0.0.8) 73,74. The hairpin RNA structures containing the unmatched sequences were predicated, complying with the criteria of pre-miRNAs in order to identify the potentially novel miRNAs. The expression level of miRNAs was determined by normalizing the reads to tags per million (TPM) counts. The TPM was calculated as follows: normalized expression, TPM = (actual miRNA count/number of total clean read) × 106. The miRNA differential expression, based on the normalized TPM expression value, was analyzed using Student’s two-tailed t-test. The significance threshold was set to 0.05 for each test.
Whole Transcriptome Analysis by microarray.
The Illumina Whole Human Genome Microarray Kit (HumanHT-12 v4 Expression BeadChip, Cat #BD-103–0204) was used to identify differentially-expressed genes in single-cell clones from PC9. Amplification of RNA, hybridization, image processing, and raw data extraction: The Illumina TotalPrep RNA Amplification kit (Ambion, UK) was used for all samples using 200 ng of total RNA as starting material. Briefly, the procedure consisted of a reverse transcription step using an oligo (Dt) primer bearing a T7 promoter and the high yield ArrayScriptTM reverse transcriptase. The cDNA then underwent second strand synthesis and clean-up to become a template for in vitro transcription with T7 RNA Polymerase and biotin-NTP mix. Labelled cRNA was then cleaned up and 1.5 µg were hybridized to humanHT12_V4 beadarrays (Illumina, CA, USA) for 16 hr at 55°C. Following hybridization, beadarrays were washed and stained with streptavidin-Cy3 (GE Healthcare, UK). Fluorescent images were obtained with a Beadarray reader and processed with the BeadScan software (version: 3.5, Illumina, CA, USA). The whole transcriptome raw data were obtained from the GenomeStudio software (version: 2011.1) with the subtraction of the background. All mRNA raw data were normalized based on the Cross-Correlation method 75. Significantly changed mRNAs were blindly identified based on average fold change cutoff of 1.5 and the cutoff of the p value cross all replicates at 0.05.
Two-Color Western Blot and Chemical Reagents.
Cells were harvested and lysed with RIPA buffer (Radio Immuno Precipitation Assay buffer) supplemented with protease and phosphatase inhibitor cocktail (Roche). Protein concentrations of the extracts were measured using BCA assay (Pierce) and equalized with the extraction reagent. Equal amount of the extracts was loaded and subjected to SDS-PAGE, transferred onto Immobilon-FL PVDF membranes. The PVDF membranes were air-dried for 1hr at room temperature followed by rehydration. The membranes were blocked with Odyssey Blocking Buffer for 1 hr and then incubated with primary antibodies in cold room overnight. Then IRDye 680RD goat anti-rabbit (1:20,000, LI-COR926–68171) and IRDye 800CW goat-anti-mouse (1: 20,000, LI-COR827–08364) were used as secondary antibodies. Then the images were scanned with Odyssey Family Imaging System (LI-COR Biosciences). Western blot quantification was performed by Image Studio Lite (V5.0, release/March 3, 2015, LI-COR Biosciences).
Transfection by LNAs in vitro.
Tumor cells were plated at 2,000 cells in complete growth medium in a 96 well plate to reach 50–60% confluence. 0~120 nM of fluorescein-conjugated LNA anti-miR-147b (Sequence: AGCAGAAGCATTTCCGCACA) (Cat #4100977–011) or negative control (Sequence: TAACACGTCTATACGCCCA) (Cat #199006–011, Exiqon) with PureFection (System Biosciences, Cat #LV750A-1) were applied for transfection. The transfected cells were harvested after culturing for 48 and 72 hr.
HIF1A and EPAS1 shRNAs and cDNA transfection.
H1975 cells were seeded in a 6-well plate at 100,000 cells per well one day prior to transfection. A mixture of 2.5 μg pGFP-C-shLenti vector targeting HIF1A (OriGene, Cat #320380), EPAS1 (OriGene, Cat #TL315484), scrambled negative control (Cat #TR30021), lentiviral vector targeting HIF1A mutant A588T (OriGene, Cat #RC402571), control vector and 7.5 µL of PureFection were used for transfection. The transfected cells were selected and maintained in 0.5 µg/ml puromycin (for shRNAs) or 600 µg/ml neomycin (for HIF1A A588T) in DMEM containing with 10% FBS for 9 days. Then the stable cells were passaged into 96-well plate at 3,000 cells per well followed by treatment with 100 nM osimertinib for 3 days. hsa-HIF1A targeting sequences: shRNA 1: AGCTTGCTCATCAGTTGCCACTTCCACAT, shRNA 2: AGGCCACATTCACGTATATGATACCAACA, shRNA 3: TACGTTGTGAGTGGTATTATTCAGCACGA, shRNA 4: ACAAGAACCTACTGCTAATGCCACCACTA. hsa-EPAS1 targeting sequences: shRNA 1: GTATGAAGAGCAAGCCTTCCAGGACCTGA, shRNA 2: AGCACTGCTTCAGTGCCATGACAAACATC, shRNA 3: CCTGGTGGCAGCACCTCACATTTGATGTG, shRNA 4: GGCTGTGTCTGAGAAGAGTAACTTCCTAT.
Transient Transfection and Dual-Luciferase Assay.
PureFection was used for transient transfection. 100 ng of wild-type or mutant 3’UTR reporter constructs of VHL or SDHD constructs (GeneCopoeia) were co-transfected into H1975 cells with 120 nM of LNA antimiR-147b or negative control (Exiqon). Firefly and Renilla luciferase activities were measured 48 hr post-transfection using Dual-Luciferase Reporter System (Promega). The firefly luminescence was normalized to Renilla luminescence as an internal control for transfection efficiency. MiR-147b binding site CGCAC was substituted with GCGTG in mutated VHL and binding site CGCACA was substituted with GCGTGT in mutated SDHD.
Lentiviral-mediated miRNA and VHL Overexpression or Knockdown Infection.
For lentiviral overexpression or knockdown of miR-147b, cells (AALE, HCC827, H1975 and PC9ER) were infected with the lentiviral particles (Applied Biological Material Inc, ABM) for 48 h in the presence of 1:100 Viralplus transduction enhancer (ABM) and 8 μg ml−1 polybrene (Sigma-Aldrich). Two days after infection, puromycin was added to the media at 0.5 μg ml−1, and cell populations were selected for 1–2 weeks. For lentiviral overexpression of VHL, cells (HCC827) at 70% confluence were transduced with VHL lentiviral particles (1.6 × 108 TU ml−1, ABM) or blank control lentiviral particles (2 × 108 TU ml−1, ABM) together with polybrene. Then the infected cells were passaged and selected by puromycin (Invitrogen) at 0.5 μg ml−1 for 1–2 weeks.
crRNA:tracrRNA transfection.
H1975-Cas9 cells were generated with pLenti-EF1a-Cas9 lentiviral particles (ABM, Cat #K003) and maintained in 0.5 μg/ml puromycin in DMEM containing with 10% FBS. H1975-Cas9-intergrated cells were seeded in a 96-well plate at 3,000 cells per well one day prior to transfection. Edit-R-synthetic crRNA (CRISPR RNA) targeting MIR147B (GE Healthcare Dharmacon, Cat #crRNA-413428, 413429, 413430 and 413431), non-targeting control (Cat #U-007501–01-20) and tracrRNA (trans-activating CRISPR RNA) (Cat #U-002005–20) were individually resuspended in 10 mM Tris-HCl pH7.5 to a concentration of 100 µM. crRNA and tracrRNA were obtained at equimolar ratio and diluted to 2.5 μM using 10 mM Tris-HCl pH7.5. A final concentration of 50 nM crRNA:tracrRNA complex was used for transfection. Cells were transfected using 0.4 µL/well of DharmaFECT Duo transfection reagent (GE Healthcare Dharmacon, Cat #T-2010–02). hsa-miR-147b targeting sequences: crRNA 1: 5’ AGAGTACTCTATAAATCTAG 3’, crRNA 2: 5’ TTTCTGCACAAACTAGATTC 3’, crRNA 3: 5’ AGATTCTGGACACCAGTGTG 3’, and crRNA 4: 5’ GCAGAAGCATTTCCGCACAC 3’.
H&E Staining and Immunofluorescence.
Samples were formalin-fixed, paraffin-embedded, sectioned, and stained with hematoxylin-eosin (H&E) according to standard histopathological techniques. For immunofluorescence, 3D structures were fixed and then incubated with mouse anti-ZO-1 (Thermo Fisher Scientific), washed, then incubated with anti-mouse IgG-Alexa Fluor 488 (Invitrogen). The 3D structures were counterstained with Hoechst 33342. Z-stack images were acquired with 2 μm slice interval and 3-D projection was created with a confocal microscope (Zeiss LSM 880).
Metabolite extraction.
For collecting adherent cells from 10-cm dishes, the metabolomics samples were prepared according to a previous method 76. Briefly, the growing cells at 80% confluence were incubated with 80% methanol at −80°C for 15 min. The cell lysate/methanol mixture were transferred to 15 mL conical tubes and centrifuged at 4500 g at 4 °C for 15 min in cold room to pellet cell debris and proteins. The centrifugation was repeated twice, and all three extractions were pooled together. The supernatants were completely dried by speedVac and were further processed for LC-MS analysis. Five biological replicates were used in each group and the analysis was normalized with the same number of cells of each group.
For collecting 3D structures, the above method was modified. Briefly, single cells mixed with geltrex were plated into six-well low attachment plates (Nunc) and incubated with complete media for 21 days. Next, the 3D structures/geltrex mixtures were incubated with TrypLE Express (Gibco) at 37°C for 5 min to separate geltrex from 3D structures. The supernatants were aspirated after centrifuge at 188 g for 5 min. Then the 3D structures pellets were incubated with 80% methanol at −80°C for 30 min. The cell lysate/methanol mixture were transferred to 15 mL conical tubes and centrifuged at 4500 g at 4 °C for 15 min to pellet cell debris and proteins. The centrifugation was repeated twice, and all three extractions were pooled. The supernatants were completely dried by speedVac and were further processed for LC-MS analysis. Five biological replicates were used in each group and the analysis was normalized with the same number of cells of each group.
Targeted Mass Spectrometry.
Samples were blindly re-suspended using 20 mL HPLC grade water for mass spectrometry. 5–7 μL were injected and analyzed using a hybrid 5500 QTRAP triple quadrupole mass spectrometer (AB/SCIEX) coupled to a Prominence UFLC HPLC system (Shimadzu) via selected reaction monitoring (SRM) of a total of 274 unique endogenous water-soluble metabolites for steady-state analyses of samples. Some metabolites were targeted in both positive and negative ion mode for a total of 306 SRM transitions using positive/negative ion polarity switching. ESI voltage was +4900V in positive ion mode and –4500V in negative ion mode. The dwell time was 3 ms per SRM transition and the total cycle time was 1.65 seconds. Approximately 9–13 data points were acquired per detected metabolite. Samples were delivered to the mass spectrometer via hydrophilic interaction chromatography (HILIC) using a 4.6 mm i.d × 10 cm Amide XBridge column (Waters) at 400 μL/min. Gradients were run starting from 85% buffer B (HPLC grade acetonitrile) to 42% B from 0–5 minutes; 42% B to 0% B from 5–16 minutes; 0% B was held from 16–24 minutes; 0% B to 85% B from 24–25 minutes; 85% B was held for 7 minutes to re-equilibrate the column. Buffer A was comprised of 20 mM ammonium hydroxide/20 mM ammonium acetate (pH=9.0) in 95:5 water: acetonitrile. Peak areas from the total ion current for each metabolite SRM transition were integrated using MultiQuant v2.1 software (AB/SCIEX). Further informatics analysis was blindly performed with online MetaboAnalyst 3.0 software (https://www.metaboanalyst.ca/)77.
Statistical Analysis.
No statistical methods were used to predetermine sample size. For mouse experiments, the mice were not randomized. The investigators performing tumor volume measurements were blinded. All experiments were performed in three to seven biological replicates, and independently reproduced as indicated in figure legends. Data are presented as the means ± S.E.M. Unless otherwise stated, statistical significance was determined by a Student’s two-tailed t-test by GraphPad Prism v. 6.0.2. P<0.05 was considered statistically significant. For analysis of transcriptome between gefitinib-tolerant PC9 cells and parental gefitinib-sensitive cells, two-tailed t-test with Welch’s correction was applied. For two samples that are not normally distributed, Mann-Whitney test was applied for a comparison. Fisher’s exact test was applied for an association analysis between miR-147b expression levels and EGFR/KRAS mutations in lung adenocarcinoma tissues from the TCGA dataset. Spearman correlation test was used for a correlation analysis between VHL and its upstream candidate VHL-regulating miRNAs emerging from the TargetScan (version: release 7.2, March 2018) analysis. The TIC frequencies were estimated using ELDA software (version 4.11.0.3973 vom 02.01.2018) 78. The enrichment of Gene Ontology (version: releases/2016-09-30) functional annotations using DAVID Bioinformatics tool (v6.8, Oct. 2016) was performed by modified Fisher’s exact test on the microarray data from PC9 single-cell clones. The enrichments were based on all evidence codes.
Supplementary Material
Acknowledgements
We thank Dr. Susumu Kobayashi for discussion and providing EGFR mutated lung cancer cell lines. We thank Slack Laboratory members for comments and sharing materials, Yale SPORE in Lung Cancer team for feedback, Dr. Alan Jiao and Daniel Foster and Soo Mi Lee for reading and feedback on the manuscript and Drs. Henry Yang, Min Gong, Danielle Cook and Emily Poulin for technical support. F.J.S. and K.P. acknowledge NIH-YALE SPORE in Lung Cancer P50CA196530. Additional support to F.J.S. was provided by the P50CA196530-03S1 and the BIDMC-JAX collaboration project, and support from the Ludwig Center at Harvard. C.J.B. acknowledges NIH P30CA034196. J.M.A. acknowledges NIH 5P01CA120964 and 5P30CA006516. W.C.Z. acknowledges NIH-YALE SPORE in Lung Cancer Career Development Program Award and NRSA 5T32HL007893-20.
Footnotes
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data Availability.
Information and data for PDX models from the JAX PDX Resource are publicly available from the PDX Portal hosted by Mouse Tumor Biology Database (MTB; http://www.tumor.informatics.jax.org/mtbwi/pdxSearch.do) 59. Data from this study have been deposited in the Gene Expression Omnibus (GEO) databases under the following accession: GSE103155 (microarray: single cell clones) and GSE103352 (miRNAseq). The results shown in this manuscript were part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/32. Analyses for an association between miRNA profiles and EGFR mutations as well as an association between VHL and MIR147b on a cohort of human lung adenocarcinoma cell lines were derived from a public RNA-seq dataset in the ArrayExpress database under accession number E-MTAB-2706 31.The heatmap for miRNA expression was generated according to Heatmapper website server (http://www2.heatmapper.ca/expression/)79. The genetic mutation status was confirmed by cansar portal (v3.0 beta) (https://cansar.icr.ac.uk/) and cancer Catalogue Of Somatic Mutations In Cancer (COSMIC) (http://cancer.sanger.ac.uk/cosmic/sample/overview?id=722040). The data that support the findings of this study are available from the corresponding author upon request.
Supplementary information is available in the online version of the paper.
Competing interests
The authors declare no competing financial interests.
REFERENCES
- 1.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. The New England journal of medicine 352, 786–792 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Niederst MJ & Engelman JA Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal 6, re6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thress KS, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 21, 560–562 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2, e73 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hata AN, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 22, 262–269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramirez M, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun 7, 10690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma SV, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith MP, et al. Inhibiting Drivers of Non-mutational Drug Tolerance Is a Salvage Strategy for Targeted Melanoma Therapy. Cancer cell 29, 270–284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soria JC, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. The New England journal of medicine (2017). [DOI] [PubMed]
- 11.Go MK, Zhang WC, Lim B & Yew WS Glycine decarboxylase is an unusual amino acid decarboxylase involved in tumorigenesis. Biochemistry 53, 947–956 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Ward PS & Thompson CB Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer cell 21, 297–308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang WC, et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 148, 259–272 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Jain M, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vander Heiden MG, Cantley LC & Thompson CB Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raimundo N, Baysal BE & Shadel GS Revisiting the TCA cycle: signaling to tumor formation. Trends in molecular medicine 17, 641–649 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vyas S, Zaganjor E & Haigis MC Mitochondria and Cancer. Cell 166, 555–566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabharwal SS & Schumacker PT Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat Rev Cancer 14, 709–721 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacKenzie ED, et al. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol 27, 3282–3289 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selak MA, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer cell 7, 77–85 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Nowicki S & Gottlieb E Oncometabolites: tailoring our genes. FEBS J 282, 2796–2805 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaelin WG Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2, 673–682 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Alvarez SW, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hangauer MJ, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dye BR, et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 4(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhan T, Rindtorff N & Boutros M Wnt signaling in cancer. Oncogene 36, 1461–1473 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, et al. MicroRNA-181a regulates epithelial-mesenchymal transition by targeting PTEN in drug-resistant lung adenocarcinoma cells. Int J Oncol 47, 1379–1392 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Sun FD, Wang PC, Luan RL, Zou SH & Du X MicroRNA-574 enhances doxorubicin resistance through down-regulating SMAD4 in breast cancer cells. Eur Rev Med Pharmacol Sci 22, 1342–1350 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Galluzzi L, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res 70, 1793–1803 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Pao W, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2, e17 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klijn C, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 33, 306–312 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anaya J OncoRank: A pan-cancer method of combining survival correlations and its application to mRNAs, miRNAs, and lncRNAs. PeerJ Preprints 4, e2574v2571 (2016). [Google Scholar]
- 34.Asad M, et al. FZD7 drives in vitro aggressiveness in Stem-A subtype of ovarian cancer via regulation of non-canonical Wnt/PCP pathway. Cell Death Dis 5, e1346 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ivan M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Frew IJ & Krek W pVHL: a multipurpose adaptor protein. Sci Signal 1, pe30 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Gomes AP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeBerardinis RJ & Chandel NS Fundamentals of cancer metabolism. Sci Adv 2, e1600200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hensley CT, et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Calvert AE, et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep 19, 1858–1873 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Han L, Dong Z, Liu N, Xie F & Wang N Maternally Expressed Gene 3 (MEG3) Enhances PC12 Cell Hypoxia Injury by Targeting MiR-147. Cell Physiol Biochem 43, 2457–2469 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Keith B, Johnson RS & Simon MC HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12, 9–22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samanta D, Gilkes DM, Chaturvedi P, Xiang L & Semenza GL Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci U S A 111, E5429–5438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Lim JH, et al. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 38, 864–878 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer 8, 865–873 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Rupaimoole R & Slack FJ MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 16, 203–222 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Esquela-Kerscher A & Slack FJ Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer 6, 259–269 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Zhang WC, et al. Tumour-initiating cell-specific miR-1246 and miR-1290 expression converge to promote non-small cell lung cancer progression. Nat Commun 7, 11702 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams BD, Parsons C, Walker L, Zhang WC & Slack FJ Targeting noncoding RNAs in disease. J Clin Invest 127, 761–771 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang WC & Slack FJ ADARs Edit MicroRNAs to Promote Leukemic Stem Cell Activity. Cell Stem Cell 19, 141–142 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Wilson TR, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 487, 505–509 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee CG, McCarthy S, Gruidl M, Timme C & Yeatman TJ MicroRNA-147 induces a mesenchymal-to-epithelial transition (MET) and reverses EGFR inhibitor resistance. PLoS One 9, e84597 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo J, et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 353, 929–932 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee SB, et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature 529, 172–177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaffer SM, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Curtis SJ, et al. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell 7, 127–133 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lundberg AS, et al. Immortalization and transformation of primary human airway epithelial cells by gene transfer. Oncogene 21, 4577–4586 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Shultz LD, et al. Human cancer growth and therapy in immunodeficient mouse models. Cold Spring Harbor protocols 2014, 694–708 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krupke DM, et al. The Mouse Tumor Biology Database: A Comprehensive Resource for Mouse Models of Human Cancer. Cancer research 77, e67–e70 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watanabe K, et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25, 681–686 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Padela S, et al. A critical role for fibroblast growth factor-7 during early alveolar formation in the neonatal rat. Pediatr Res 63, 232–238 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Bostrom H, et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85, 863–873 (1996). [DOI] [PubMed] [Google Scholar]
- 63.Sekine K, et al. Fgf10 is essential for limb and lung formation. Nature genetics 21, 138–141 (1999). [DOI] [PubMed] [Google Scholar]
- 64.Krause C, Guzman A & Knaus P Noggin. Int J Biochem Cell Biol 43, 478–481 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Temes E, et al. Activation of HIF-prolyl hydroxylases by R59949, an inhibitor of the diacylglycerol kinase. J Biol Chem 280, 24238–24244 (2005). [DOI] [PubMed] [Google Scholar]
- 67.Mills EL, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470 e413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dervartanian DV & Veeger C Studies on Succinate Dehydrogenase. I. Spectral Properties of the Purified Enzyme and Formation of Enzyme-Competitive Inhibitor Complexes. Biochim Biophys Acta 92, 233–247 (1964). [PubMed] [Google Scholar]
- 69.Gong M, et al. Pyrosequencing enhancement for better detection limit and sequencing homopolymers. Biochem Biophys Res Commun 401, 117–123 (2010). [DOI] [PubMed] [Google Scholar]
- 70.Kim HJ, et al. Clinical investigation of EGFR mutation detection by pyrosequencing in lung cancer patients. Oncol Lett 5, 271–276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shi J, et al. Deep RNA Sequencing Reveals a Repertoire of Human Fibroblast Circular RNAs Associated with Cellular Responses to Herpes Simplex Virus 1 Infection. Cell Physiol Biochem 47, 2031–2045 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robinson MD & Oshlack A A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trapnell C, et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31, 46–53 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chua SW, et al. A novel normalization method for effective removal of systematic variation in microarray data. Nucleic Acids Res 34, e38 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yuan M, Breitkopf SB, Yang X & Asara JM A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7, 872–881 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xia J & Wishart DS Using MetaboAnalyst 3.0 for Comprehensive Metabolomics Data Analysis. Curr Protoc Bioinformatics 55, 14 10 11–14 10 91 (2016). [DOI] [PubMed] [Google Scholar]
- 78.Hu Y & Smyth GK ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 347, 70–78 (2009). [DOI] [PubMed] [Google Scholar]
- 79.Babicki S, et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44, W147–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.