

SUMMARY
Unlike the adult mammalian retina, Müller glia (MG) in the adult zebrafish retina are able to dedifferentiate into a ‘‘stem cell’’-like state and give rise to multipotent progenitor cells upon retinal damage. We show that miR-216a is downregulated in MG after constant intense light lesioning and that miR-216a suppression is necessary and sufficient for MG dedifferentiation and proliferation during retina regeneration. miR-216a targets the H3K79 methyltransferase Dot1l, which is upregulated in proliferating MG after retinal damage. Loss-of-function experiments show that Dot1l is necessary for MG reprogramming and mediates MG proliferation downstream of miR-216a. We further demonstrate that miR-216a and Dot1l regulate MG-mediated retina regeneration through canonical Wnt signaling. This article reports a regulatory mechanism upstream of Wnt signaling during retina regeneration and provides potential targets for enhancing regeneration in the adult mammalian retina.
Graphical Abstract
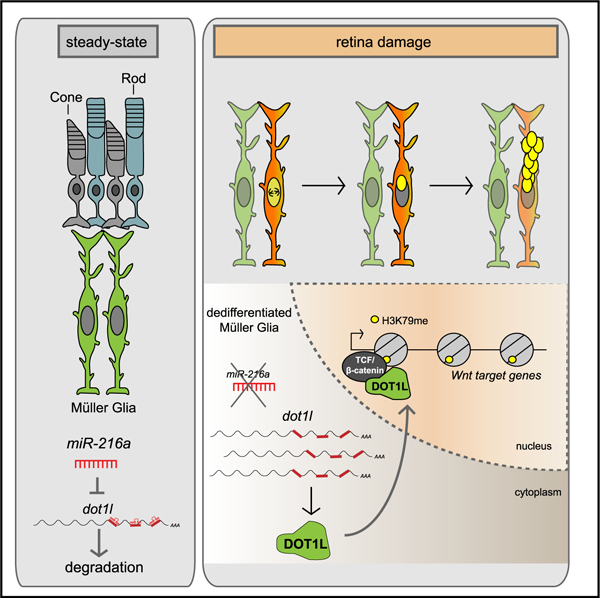
In Brief
Unlike the adult mammalian retina, Müller glia in the adult zebrafish retina are able to reprogram into a stem cell-like state and give rise to multipotent progenitor cells upon retinal damage. Kara et al. show that miR-216a suppression stimulates Müller glia reprogramming through upregulation of the H3K79 methyltransferase Dot1l and activation of Wnt/β-catenin signaling.
INTRODUCTION
A promising strategy to restore impaired vision due to degenerative retinal disorders is to induce endogenous repair mechanisms to regenerate lost cell types. It is unfortunate that mammals are unable to spontaneously regenerate retinal neurons; instead, damage often induces reactive gliosis (Bringmann et al., 2009). However, retinal damage in teleost fish, including zebrafish, initiates a robust spontaneous regenerative response that restores both retinal structure and function (Goldman, 2014). Given that the cells and structure of the retina are highly conserved among vertebrates, understanding the molecular mechanisms that allow zebrafish to spontaneously regenerate damaged retinas is key to develop novel therapeutic strategies for retinal damage and disease in humans.
In zebrafish, Müller glia (MG) are the source of regenerated neurons in the retina (Bernardos et al., 2007; Fausett and Goldman, 2006). After injury, MG dedifferentiate, undergo asymmetric cell division, and generate a population of proliferating neuronal progenitor cells (Nagashima et al., 2013; Ramachandran et al., 2010; Thummel et al., 2008). MG-derived neural progenitors are able to differentiate into any of the lost retinal cell types and fully restore visual function in the zebrafish retina. Understanding the major cellular events and identifying key differentially expressed genes is a current focus of much research, but the precise molecular mechanisms that regulate retina regeneration remain largely unknown (Goldman, 2014; Lenkowski and Raymond, 2014; Rajaram et al., 2014b).
MicroRNAs (miRNAs) are a family of highly conserved small noncoding RNAs that post-transcriptionally regulate gene expression and play important roles in many cellular processes during development and regeneration (Thatcher and Patton, 2010; Wienholds and Plasterk, 2005; Zhao and Srivastava, 2007). We recently showed that the major miRNA processing enzyme, Dicer, is required for retina regeneration in zebrafish and profiled dynamic miRNA expression patterns in the retina during regeneration induced by constant intense light damage (Rajaram et al., 2014a). Here, we report that miR-216a acts as a gatekeeper for MG reprogramming, maintaining MG in a quiescent state in undamaged retina. miR-216a suppression is necessary and sufficient for MG dedifferentiation and proliferation. We identify the disruptor of telomeric silencing-1-like (Dot1l) as a bona fide target of miR-216a and demonstrate that the miR-216a/Dot1l regulatory axis mediates the initiation of retina regeneration through the Wnt/β-catenin pathway. Previous studies in multiple species have revealed that Dot1l is able to regulate the transcription of Wnt-target genes by directly interacting with T cell factor (TCF)/β-catenin complexes (Castaño Betancourt et al., 2012; Mahmoudi et al., 2010; Mohan et al., 2010). Our work uncovers a requirement for Dot1l downstream of miR-216a during MG dedifferentiation, proliferation, and retina regeneration.
RESULTS
miR-216a Is Suppressed in Dedifferentiated MG during Early Retina Regeneration
We previously demonstrated a general requirement for the Dicer-dependent miRNA biogenesis pathway during retina regeneration induced by constant intense light damage in adult zebrafish (Rajaram et al., 2014a). miR-216a belongs to a highly conserved miRNA family with previously characterized functions in gliogenesis during retina development (Olena et al., 2015). We sought to test whether miR-216a may also regulate the reprogramming of MG during retina regeneration. We determined the expression levels of miR-216a in quiescent and proliferating MG and non-MG cells in the retina. We used fluorescence-activated cell sorting (FACS) to isolate GFP+ quiescent MG from undamaged Tg(gfap:gfp) retinas, GFP+ dedifferentiated MG after 45 h of intense light damage using Tg(1016tuba1a:gfp) retinas (Bernardos and Raymond, 2006; Fausett and Goldman, 2006), and GFP− cells from both sorts (non-MG) (Figure 1A). Glial fibrillary acidic protein (GFAP) is expressed in quiescent MG; the Tg(1016tuba1a:gfp) transgenic line specifically marks dedifferentiated MG and MG-derived neural progenitors in actively regenerating retinas (Fausett and Goldman, 2006). We chose 45 h post-light damage to focus on the initial reprogramming of MG when MG undergoes the first round of division between 35 and 52 h of intense light damage (Rajaram et al., 2014c). Quantitative real-time PCR analysis showed that miR-216a is expressed at significantly higher levels in quiescent MG compared to the non-MG cell population in undamaged retinas (Figure 1B). After damage, however, miR-216a was significantly downregulated in dedifferentiated MG in regenerating retinas compared to quiescent MG. When we compared non-MG cell populations in undamaged and intense light-damaged retinas, we did not detect any significant changes in the expression levels of miR-216a, indicating that miR-216a expression is more highly expressed in MG than in other retinal cell types. Of note, we discovered that miR-216b is also expressed in MG and undergoes a significant decrease in expression after damage. miR-216b differs from miR-216a at two positions, one of which is in the seed region, meaning it targets a different set of mRNAs. Based on our prior work and because we identified Dot1l as a target of miR-216a and not miR-216b, we focus here on miR-216a.
Figure 1. Suppression of miR-216a Is Required for MG Dedifferentiation and Proliferation during Retina Regeneration.

(A) Schematic for post-mitotic and dedifferentiated MG sorting. Adult zebrafish were dark adapted for 2 weeks and then exposed to constant intense light lesioning for 45 h. For post-mitotic MG isolation, GFP+ cells were sorted from dark adapted Tg(gfap:gfp) retinas. For dedifferentiated MG isolation, GFP+ cells were isolated from 45 h light lesioned Tg(1016tuba1a:GFP) retinas.
(B) Fold changes in miR-216a levels in FACS-purified MG were determined by qPCR. miR-216a is enriched in post-mitotic MG (GFP+) from undamaged retinas in Tg(gfap:gfp) fish. After 45 h of light damage, miR-216a is downregulated ~5-fold in dedifferentiated MG (GFP+) in Tg(1016tuba1a:gfp) fish. miR-216a expression did not change in non-MG cells (GFP−) during regeneration. Data are from 5 independent experiments with 3 technical replicates of qPCR. MG were purified from 18 and 20 light-damaged fish in each experiment. Error bars represent the SEMs. *p < 0.05 and **p < 0.01 using 2-way ANOVA with Fisher’s least significant difference (LSD) post hoc test.
(C) Control miRNA (siRNA against luciferase [siLuc]) or miR-216a was injected and electroporated into the left eyes of Tg(1016tuba1a:gfp) zebrafish before intense light exposure (0 h). After 45 h, retinas were collected, sectioned, and immunostained using antibodies against GFP and PCNA. Nuclei were counterstained with TOPRO (blue). miR-216a gain of function abolished tuba1a:GFP transgene expression and significantly reduced the number of INL PCNA+ proliferating cells.
(D) Quantification of PCNA+ cells in the INL and ONL. Error bars represent the SEMs (n = 5–6 fish); **p < 0.01 using Student’s t test.
(E) Overexpression of miR-216a reduced the number of GFP+/PCNA+ proliferating progenitor cells after 60 h of intense light damage using Tg(1016tuba1a:gfp) zebrafish.
(F) Quantification of total GFP+ and PCNA+ cells. Error bars represent the SEMs (n = 10 fish); *p < 0.03 and **p < 0.003 using Student’s t test.
GCL, ganglion cell layer; INL, inner nuclear layer; ns, not significant; ONL, outer nuclear layer.
Scale bars, 50 µm.
miR-216a Suppression Is Required for MG Dedifferentiation and Proliferation
To test whether miR-216a suppression is required for MG dedifferentiation and proliferation during retina regeneration, we performed an overexpression analysis of miR-216a. We injected and electroporated miR-216a mimics or a control miRNA intravitreally into the eye of Tg(tuba1a:gfp) transgenic fish before intense light damage (0 h) and assessed the effects on MG dedifferentiation and proliferation at 45 h of light exposure (Figure 1C). While there were numerous GFP+ dedifferentiated MG in control miRNA-injected retinas at 45 h of light damage, there was a striking absence of GFP+ dedifferentiated MG in miR-216a-overexpressing retinas (Figure 1C). We then analyzed the effect of miR-216a overexpression on MG proliferation using proliferating cell nuclear antigen (PCNA) as a marker of DNA replication. Compared to control miRNA overexpressing retinas, which had clusters of PCNA+ cells in the inner nuclear layer (INL), excess miR-216a resulted in significantly decreased numbers of proliferating MG (Figures 1C and 1D). The proliferation of rod progenitor cells in the outer nuclear layer (ONL) was not affected (Figures 1C and 1D).
We then analyzed the effects of miR-216a overexpression on neural progenitor cell proliferation (Figure 1F). At 60 h of intense light exposure, there were significantly fewer dedifferentiated MG marked by GFP, as well as decreased numbers of PCNA+ proliferating progenitors in the INL of miR-216a-overexpressing Tg(tuba1a:GFP) retinas compared to controls (Figure 1G). This suggests that the inhibitory effects of miR-216a overexpression on the proliferation of MG and MG-derived progenitors are observed at later stages of regeneration, consistent with the model that the suppression of miR-216a is a critical step in both the initiation of MG dedifferentiation and the generation of proliferating progenitor cells during retina regeneration. To ensure that the effects we observed were not indirectly a consequence of unexpected apoptosis due to the overexpression of miR-216a, we conducted terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays. We did not observe any changes in apoptotic cells between control and miR-216a overexpressed retinas (Figure S2).
Dot1l Is a Direct Target of miR-216a In Vivo
Suppression of miR-216a in dedifferentiated MG predicts an upregulation of mRNA targets that carry miR-216a binding sites in their 3′ UTRs. To investigate the molecular mechanism through which miR-216a regulates MG reprogramming, we used the target prediction algorithm TargetScanFish and identified 61 potential target genes based on spatiotemporal expression patterns using RNA sequencing (RNA-seq) transcriptome analysis in purified MG before and after intense light lesioning (data not shown). We further narrowed the list down to those with validated upregulation by quantitative real-time PCR and those containing at least two miRNA recognition elements (MREs) for miR-216a. Dot1l emerged as a strong candidate target of miR-216a and a potential regulator of retina regeneration, since it was previously shown that it functions as an activator of canonical Wnt-dependent transcription in zebrafish (Mahmoudi et al., 2010), and canonical Wnt activation is necessary for MG dedifferentiation and proliferation during retina regeneration (Ramachandran et al., 2011).
There are 3 MREs for miR-216a in the 3′ UTR of dot1l (Figure 2A). To test whether miR-216a directly targets dot1l, we performed GFP reporter assays in embryos. We cloned the dot1l 3′ UTR downstream of the GFP coding sequence and in vitro transcribed reporter mRNAs. We then injected the reporter mRNAs, either alone or with co-injected miR-216a, mimics into 1-cell stage zebrafish embryos. At 24 h post-fertilization (hpf), GFP expression levels were significantly lower in the embryos co-injected with miR-216a compared to those injected with only the reporter mRNA (Figures 2B and 2C). This indicates that miR-216a can directly target the 3′ UTR of dot1l mRNAs. To test whether this targeting is via the MREs predicted by the algorithm, we mutated the miR-216a seed sites in all 3 MRE sites. Reporter assays using the mutated reporter construct did not show any changes in the levels of GFP fluorescence upon co-injection with miR-216a (Figures 2D and 2E). This result shows that dot1l mRNAs can be targeted by miR-216a through the indicated MREs.
Figure 2. Dot1l Is a Direct Target of miR-216a.

(A) Schematic of the reporter mRNA consisting of the coding sequence of GFP fused to the dot1l 3′ UTR. Three predicted miRNA recognition elements (MREs) are indicated. Predicted base pairing between MREs (shown in green) and the miR-216a sequence (shown in red).
(B) Embryos injected at the 1-cell stage with 100 pg of GFP-dot1l 3′ UTR, with or without 100 pg miR-216a, were examined for GFP expression at 1 day post fertilization (dpf). GFP expression was apparent in embryos injected with GFP-dot1l 3′ UTR, but it was reduced in embryos co-injected with miR-216a.
(C) Quantification of relative fluorescence in 1 dpf embryos injected with GFP reporter only or co-injected with miR-216a and the GFP reporter. Data represent means ± SEMs from 3 independent experiments. **p < 0.01 (Student’s t test).
(D) One-dpf-old embryos injected at the 1-cell stage with 100 pg of GFP-dot1l 3′ UTR carrying mutations in all miR-216a MREs. Embryos were injected with the mutant reporter, either alone or co-injected with miR-216a.
(E) Quantification of relative fluorescence in 1 dpf embryos injected with mutant GFP reporter alone or with co-injection of miR-216a. Data represent means ± SEMs from 3 independent experiments. **p < 0.01 (Student’s t test).
Scale bars, 500 µm.
To determine whether Dot1l is expressed in a manner that is consistent with regulation by miR-216a, we analyzed mRNA expression levels of dot1l in quiescent MG using Tg(gfap:gfp) retinas and in dedifferentiated MG using Tg(tuba1a:gfp) retinas (Figure 3A). Quantitative real-time PCR analysis of RNA from sorted cell populations showed a ~2-fold upregulation of dot1l transcripts in dedifferentiated MG compared to post-mitotic MG (Figure 3B). No changes in dot1l expression were observed in other retinal cells (GFP− cells). We also investigated whether miR-216a is able to target endogenous dot1l in the retina during regeneration by injection and electroporation of miR-216a mimics into the dorsal retina of fish exposed to 24 h of constant intense light damage (Figure 3C). Increased miR-216a levels (Figure 3D) led to a significant decrease (~95%) in endogenous dot1l mRNA levels, as shown by quantitative real-time PCR analysis (Figure 3E). We also showed co-localization of Dot1l with PCNA by performing immunohistochemistry on Tg(tuba1a:gfp) fish after 51 h of light exposure (Figure S1). Despite weak signals with the available antibodies, we observed punctate nuclear localization of Dot1l protein in tuba1a:GFP+/PCNA+ dedifferentiated and proliferating MG. Our data support the hypothesis that miR-216a regulates dot1l in the adult retina.
Figure 3. miR-216 Targets Dot1l in the Retina during Photoreceptor Regeneration.

(A) For post-mitotic MG isolation, GFP+ cells were sorted from dark adapted undamaged Tg(gfap:gfp) retinas. For dedifferentiated MG isolation, GFP+ cells were isolated from 45-h light-lesioned Tg(1016tuba1a:GFP) retinas.
(B) Fold changes in dot1l levels in FACS-purified MG were determined by qPCR. After 45 h of light damage, dot1l is upregulated in dedifferentiated MG (GFP+) in Tg(1016tuba1a:gfp) fish. Dot1l expression did not change in non-MG cells (GFP−) during regeneration. Data represent the means ± SEMs from 15 undamaged fish, and dedifferentiated MG were purified from 18 light-damaged fish.
(C) Experimental scheme to test the effects of miR-216a overexpression on dot1l levels. Wild-type adult zebrafish were dark adapted and then either control miRNA or miR-216a was injected and electroporated into the left eyes before intense light exposure. After 24 h of light exposure, retinas were dissected for RNA isolation.
(D) Fold changes in miR-216a levels in control miRNA (small interfering RNA [siRNA] against luciferase [siLuc]) or miR-216a mimic electroporated retinas were quantified by qPCR. miR-216a levels were upregulated by 15-fold in miR-216a mimic-injected retinas compared to controls. Data represent the means ± SEMs from 6 retinas.
(E) Fold changes in dot1l levels in siLuc or miR-216 mimic electroporated retinas were quantified by qPCR. After 24 h of light damage, dot1l was downregulated in miR-216a-overexpressing retinas ~20-fold. Data represent means ± SEMs from 3 independent experiments. Six retinas were pooled for RNA isolation in each experiment.
**p < 0.01 (Student’s t test), p = 0.0093.
Scale bars, 50 µm.
See also Figures S1 and S2.
Dot1l Is Necessary for Proliferation during Retina Regeneration
Given that excess miR-216a inhibited the dedifferentiation and proliferation of MG, we hypothesized that Dot1l is required for the early phases of retina regeneration. To analyze the loss-of-function of Dot1l during retina regeneration, we first generated Dot1l null alleles by CRISPR-Cas9. However, homozygous mutants displayed embryonic lethality similar to homozygous Dot1l mice (Jones et al., 2008). Thus, we knocked down Dot1l in adult zebrafish retinas by injecting and electroporating previously characterized morpholinos (MOs) against dot1l into the retina before intense light damage (Mahmoudi et al., 2010) (Figure 4A). We used the Tg(tuba1a:GFP) transgenic line to assess the dedifferentiation of MG, cell-cycle re-entry, and proliferation of MG-derived progenitors. At 45 h of constant intense light damage, we detected significantly fewer dedifferentiated MG, as well as decreased numbers of PCNA+ proliferating progenitors in retinas electroporated with the dot1l MO, compared to injection of a control MO (Figures 4B and 4C). To test for specificity and possible off-target effects of MOs, we used a second dot1l MO (MO-dot1l-2) and again assessed the effect of dot1l knockdown on regenerating retinas after intense light damage (Figure S3A). dot1l-MO-2 injected retinas also displayed significantly reduced numbers of PCNA+ proliferating neural progenitors at 45 h of light damage. Furthermore, we showed that the effects of the Dot1l MO on MG proliferation are dose dependent, arguing in favor of specificity (Figure 4D). Lastly, we showed that the increase in MG proliferation and dedifferentiation upon dotl1 MO injection was not a consequence of unrelated apoptosis since TUNEL staining showed no significant difference in cell death compared to control MOs (Figures S2C and 2D).
Figure 4. Dot1l Is Required for MG Dedifferentiation and Proliferation during Retina Regeneration.

(A) Control MO or dot1l MOs were injected and electroporated into the left eyes of Tg(1016tuba1a:gfp) zebrafish before intense light exposure (0 h). After 45 h, retinas were collected, sectioned, and immunostained using antibodies against GFP and PCNA. Nuclei were counterstained with TOPRO (blue).
(B) Dot1l loss-of-function reduced tuba1a:GFP transgene expression and the number of INL PCNA+ proliferating cells.
(C) Quantification of GFP+ dedifferentiated MG and PCNA+ proliferating progenitors in MO-ctl and MO-dot1l electroporated retinas. Data represent means ± SEMs, n = 5–6 fish; **p < 0.01 by 2-tailed Mann-Whitney U test.
(D) Dose response to MO-dot1l. Increasing amounts of MO-dot1l MOs were injected and analyzed as in (B) and (C). Each data point represents an individual fish; *p < 0.05, **p < 0.01, and ***p < 0.001 by 1-way ANOVA.
(E) Control vehicle (DMSO) or iDot1l (Dot1l inhibitor EPZ004777) were injected intravitreally into the left eyes of Tg(1016tuba1a:gfp) zebrafish before intense light exposure (0 h). After 45 h, retinas were collected, sectioned, and immunostained using antibodies against GFP and PCNA. Nuclei were counterstained with TOPRO (blue).
(F) Quantification of total GFP+ and PCNA+ cells. Data represent means ± SEMs, n = 10 fish; *p = 0.0111; ****p < 0.0001 by 2-tailed Mann-Whitney U test.
(G) Dose response to iDot1l. Increasing amounts of the Dot1l inhibitor were injected and analyzed as in (E) and (F). Each data point represents an individual fish; *p < 0.05, **p < 0.01, and ***p < 0.001 by 1-way ANOVA.
GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer;. Scale bars, 50 µm.
See also Figures S2 and S3.
To test whether the requirement for Dot1l is through its histone methyltransferase activity, we used a small molecule inhibitor of Dot1l catalytic activity that competitively binds to the S-adenosylmethionine-binding pocket of Dot1l (iDot1l; EPZ004777) (Daigle et al., 2011). Compared to control vehicle-injected retinas, the intravitreal injection of iDot1l led to a significant reduction in the number of dedifferentiated MG, as well as the number of proliferating progenitors in the INL after 45 h of intense light damage (Figures 4E and 4F). The effects of the Dot1l inhibitor were dose dependent (Figure 4G). Together, the knockdown experiments and pharmacologic inhibition experiments argue that Dot1l is required for MG reprogramming’s acting as an epigenetic modifier required for dedifferentiation and proliferation of MG-derived neural progenitors during retina regeneration.
Suppression of miR-216a Is Sufficient for Retina Regeneration through Targeting Dot1l
Next, we investigated whether miR-216a downregulation is sufficient to drive MG dedifferentiation and the formation of neural progenitors. To test this, we suppressed the miR-216a levels by injection and electroporation of antisense miR-216a MOs (MO-216a) in undamaged Tg(tuba1a:gfp) retinas (Figure 5A). At 51 h post-injection (hpi), we showed that miR-216a suppression significantly increased the dot1l mRNA and protein levels in the retinas (Figures 5B–5D). Then, we assessed whether the loss of miR-216a function results in the initiation of a regenerative response similar to what happens after retinal damage. While we did not detect any de-differentiated GFP+ MG in control MO-injected retinas, miR-216a suppression resulted in a significant increase in the number of dedifferentiated MG in undamaged Tg(tuba1a:gfp) retinas (Figures 5E and 5F). We also detected significantly higher numbers of proliferating PCNA+ cells in miR-216a-suppressed retinas compared to control MO-injected retinas (Figures 5E and 5G). By showing the colocalization of PCNA+ and tuba1a-GFP+ cells, we confirmed that proliferating cells upon miR-216 suppression are dedifferentiated MG (Figure 5H).
Figure 5. miR-216a Suppression Stimulates MG Dedifferentiation and Proliferation in the Uninjured Retina through Regulating Dot1l.

(A) Undamaged Tg(1016tuba1a:gfp) zebrafish were injected and electroporated with control MOs (n = 5), miR-216a MOs (n = 6), or both (n = 5). Either eyes for immunostaining or retinas for expression analysis were collected at 51 h post-injection (hpi).
(B) Fold changes in dot1l levels in MO-ctl or MO-216a electroporated retinas were quantified by qPCR. At 51 hpi, dot1l was significantly upregulated in miR-216a overexpressing retinas ~2-fold. Data represent means ± SEMs; *p < 0.05 using Student’s t test, p = 0.0365.
(C) Representative western blot for Dot1l and a-tubulin in control and miR-216a MO-injected retinas at 51 hpi.
(D) Quantification for the relative levels of Dot1l protein in miR-216a MO-injected retinas compared to controls. Data represent means ± SEMs from 3 independent experiments, *p < 0.05 using Student’s t test.
(E) miR-216a MOs significantly increased the number of PCNA+ and GFP+ cells in the INL, while there was no significant difference between miR-216a MO+ dot1l MO co-injected eyes and control eyes.
(F and G) Quantification of GFP+ dedifferentiated MG (F) and PCNA+ proliferating progenitors (G) in MO-ctl, MO-216a, and MO-216a + MO-dot1l electroporated retinas. Data represent means ± SEMs (n = 6 fish). **p < 0.01, PCNA p = 0.0098, and GFP p = 0.0039 by 1-way ANOVA with Dunnett’s multiple comparisons test.
(H) Representative retinal sections showing colocalization of tuba1a-GFP+ and PCNA+ cells in miR-216 MO injected undamaged retinas at 51 hpi.
GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer.
Scale bar, 50 µm.
Our data are most consistent with a model whereby miR-216a suppression stimulates MG proliferation through targeting dot1l. If true, then co-suppressing dot1l in the presence of MO-216a should block MG proliferation. To test this, we combined the intravitreal injection and electroporation of both dot1l and miR-216a MOs in uninjured Tg(tuba1a:gfp) retinas. Compared to control retinas, we no longer observed a significant increase in the number of dedifferentiated and proliferating MG at 51 h post injection after double knockdown of both dot1l and miR-216a (Figures 5E–5G). This result demonstrates that miR-216a must be downregulated to activate Dot1l during retina regeneration and that Dot1l is required for the initiation of MG proliferation.
Wnt/β-Catenin Signaling Is Required Downstream of miR-216a/Dot1l during Retina Regeneration
Dot1l is a histone methyltransferase responsible for H3K79me3 modification associated with gene activation (Feng et al., 2002; Jones et al., 2008; Shanower et al., 2005; Steger et al., 2008; van Leeuwen et al., 2002). Dot1l serves as a co-activator of Wnt/β-catenin signaling (Mahmoudi et al., 2010; Mohan et al., 2010), which is known to be activated during retina regeneration and is required for the formation of MG-derived progenitors (Meyers et al., 2012; Ramachandran et al., 2011). Since miR-216a suppression is necessary for MG-dependent retina regeneration, we wanted to test whether miR-216a suppression is also required for the activation of Wnt/β-catenin signaling. We assessed β-catenin accumulation in MG after light damage since β-catenin accumulation as a result of Wnt signaling activation is necessary for MG dedifferentiation and proliferation (Ramachandran et al., 2011). We injected and electroporated miR-216a mimics or control miRNAs in the retinas of adult wild-type fish before intense light damage (Figure 6A). At 51 h of light damage, β-catenin accumulation was clearly observed in MG associated with PCNA+ neural progenitors in control retinas. However, no β-catenin accumulation was observed in miR-216a-overexpressing retinas (Figure 6B). These results support the hypothesis that the miR-216a/Dot1l regulatory pathway regulates retina regeneration through canonical Wnt signaling. To activate Wnt signaling, we pharmacologically stabilized β-catenin using a glycogen synthase kinase-3β (GSK-3β) inhibitor and confirmed that at the concentration tested, the inhibitor is able to induce MG proliferation in undamaged retinas, as expected (Ramachandran et al., 2011) (Figure S4). We then tested whether the proliferation defects after knock down of Dot1l could be rescued by the activation of Wnt signaling. We intravitreally injected either the GSK-3β inhibitor or the control vehicle (DMSO) in the presence of either control MOs or dot1l MOs before intense light damage (Figure 7A). At 51 h of light damage, dot1l depletion resulted in significantly decreased numbers of PCNA+ proliferating progenitors compared to control retinas (Figures 7B and 7C). However, co-injection of dot1l MOs and the GSK-3β inhibitor showed no defects in the number of neural progenitors. Next, we tested whether in undamaged retinas Dot1l is required for the induction of MG proliferation through the activation of Wnt/β-catenin signaling (Figure 7D). While the stabilization of β-catenin by the injection of the GSK-3β inhibitor induced MG proliferation, Dot1l inhibition by the co-injection of the Dot1l inhibitor led to no significant increase in MG proliferation. This indicates that Dot1l regulates MG proliferation through Wnt/β-catenin signaling during intense light damage-induced retina regeneration.
Figure 6. miR-216a Gain of Function Impairs β-Catenin Accumulation in MG after Intense Light Damage.

(A) Control miRNA or miR-216a was injected and electroporated into the left eyes of wild-type zebrafish before intense light exposure (0 h). After 45 h, retinas were collected, sectioned, and immunostained using antibodies against β-catenin (red) and PCNA (green). Nuclei were counterstained with TOPRO (blue).
(B) β-Catenin colocalized with PCNA+ proliferating MG after 45 h of intense light damage. β-Catenin accumulation was not detected in miR-216a-over-expressing retinas. Scale bar, 50 µm.
Figure 7. miR-216a and Dot1l Regulate Retinal Regeneration through the Wnt/β-Catenin Pathway.

(A) Control MOs or Dot1l MOs were injected and electroporated into the left eyes of wild-type zebrafish, followed by either 4% DMSO or GSK-β-inhibitor (1 mM) before intense light exposure (0 h). Eyes were collected after 51 h of intense light lesioning and immunostained using antibodies against PCNA. Nuclei were counterstained with TOPRO (blue).
(B) dot1l MOs significantly decreased the number of PCNA+ cells in the INL, while there was no significant difference between dotl1l MO + GSK-3β-inhibitor co-injected eyes and control eyes.
(C) Quantification of PCNA+ proliferating progenitors in MO-ctl + DMSO, MO-dot1l + DMSO, MO-ctl + GSK-3β-inhibitor, and MO-dot1l + GSK-3β-inhibitor electroporated retinas. Activation of Wnt signaling rescued the decrease in the number of proliferating progenitors upon dot1l knockdown after 51 h of intense light lesioning. Data represent means ± SEMs; n = 9–11 fish. *p < 0.05, p = 0.0167 (MO-ctl + DMSO versus MO-dot1l + DMSO) by 1-way ANOVA with Dunnett’s multiple comparisons test.
(D) GSK-3β-inhibitor alone, Dot1l inhibitor (iDot1l) alone, or the combination was injected into the left eyes of Tg(1016tuba1a:GFP) zebrafish; DMSO alone was used as a control. At 51 h post-injection, eyes were collected for PCNA immunostaining. GSK-3β-inhibitor alone induced MG proliferation in undamaged eyes while co-injection of the Dot1l inhibitor led to no significant changes in number of proliferating MG. Data represent means ± SEMs. Each data point represents an individual fish. ****p < 0.0001, by 1-way ANOVA with Dunnett’s multiple comparisons test.
(E) Control MO or miR-216a MO was injected and electroporated into the left eyes of Tg(1016tuba1a:GFP) zebrafish, followed by either 4% DMSO or XAV939 (10 µM). Eyes were collected at 51 h post-injection and immunostained using antibodies against GFP for dedifferentiated MG and PCNA for proliferating progenitors. Nuclei were counterstained with TOPRO (blue).
(F) Suppression of miR-216a by MO-216a injection stimulates MG proliferation; however, upon co-injection with XAV939, no significant increase in the number of proliferating progenitors was detected.
(G) Quantification of PCNA+ proliferating progenitors in MO-ctl + DMSO, MO-216a + DMSO, MO-ctl + XAV939, and MO-216a + XAV939 electroporated retinas. Inhibition of Wnt signaling reversed the increase in the number of progenitors upon miR-216a knockdown after 51 h of intense light lesioning. Data represent means ± SEMs; n = 18–21 fish. **p < 0.01, p = 0.0089 (MO-ctl + DMSO versus MO-216 + DMSO) by 1-way ANOVA with Dunnett’s multiple comparisons test.
GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer. Scale bars, 50 µm.
See also Figure S4.
Finally, we investigated whether miR-216a depletion in undamaged retinas would induce MG proliferation through modulating Wnt/β-catenin signaling (Figure 7E). We pharmacologically inhibited canonical Wnt signaling by injecting XAV939, a tankyrase inhibitor, which stabilizes Axin and stimulates β-catenin degradation (Huang et al., 2009). We injected either XAV939 (10 µM) or control vehicle (DMSO) along with the co-injection of either control or miR-216a MO into undamaged retinas. Intravitreal injection of XAV939 at a 10-µM concentration was previously shown to be sufficient to prevent injury-dependent β-catenin accumulation in MG (Ramachandran et al., 2011). As above, miR-216a MO injection in undamaged retinas caused the spontaneous proliferation of MG, as detected by the presence of significantly higher numbers of PCNA+ proliferating cells in the INL at 51 hpi (Figures 7F and 7G). However, the co-injection of XAV939 and MO-216a suppressed the increase in the number of PCNA+ proliferating cells compared to control MOs or DMSO-injected retinas. These results demonstrate that Wnt/β-catenin signaling is required for spontaneous MG proliferation initiated by the depletion of miR-216a.
DISCUSSION
MG dedifferentiation and re-entry into the cell cycle are key events during retina regeneration. In zebrafish, MG are capable of eliciting a robust spontaneous regenerative response upon damage, while in mammals, MG lack this ability and typically become reactive and undergo hypertrophy (Bringmann et al., 2006). Understanding the molecular mechanisms of MG activation during regeneration is necessary to develop therapeutic strategies for retinal diseases in humans. Here, we identify a chromatin-mediated mechanism that regulated the initiation of retina regeneration in adult zebrafish. We show that suppression of miR-216a in MG is required for dedifferentiation and proliferation upon constant intense light damage, leading to the derepression of the H3K79 methyltransferase Dot1l, which is required for regeneration. Furthermore, we demonstrate that miR-216a and Dot1l regulate MG activation through Wnt/β-catenin signaling. Our data provide a mechanism through which miR-216a serves as a gatekeeper for MG dedifferentiation and proliferation by suppressing Dot1l during retina regeneration.
Although many individual miRNAs have been identified that regulate cell fate in development and disease, only a few miRNAs have been shown to be functionally involved in modulating retina regeneration (Rajaram et al., 2014a, 2014b; Ramachandran et al., 2010). We show that miR-216a is expressed in quiescent MG in the adult zebrafish retina and must be repressed to allow MG dedifferentiation and re-entry into the cell cycle. The expression of miR-216a does not change in non-MG cells of the retina. miR-216a was first reported to modulate retinal gliogenesis by targeting snx5 (sorting nexin 5) during development (Olena et al., 2015). miR-216a is suppressed in the central retina to allow MG specification through the activation of Notch signaling. In that model, miR-216a targets snx5 to block the association of the Notch ligand Delta, preventing Delta endocytosis and thereby regulating Notch signaling. These experiments predict that miR-216a functions in cells containing the Notch ligand Delta. We did not detect any significant changes in snx5 expression levels in FACS-purified MG populations before and after intense light damage (data not shown). In addition, a previous study showed that while Notch receptors are present in proliferating neural progenitors, Notch ligands were detected in cells adjacent to the proliferating progenitors during retina regeneration (Wan et al., 2012). Therefore, miR-216a plays distinct roles during development versus regeneration regulating distinctly different targets.
Previous reports have shown that it is possible to stimulate MG proliferation in undamaged zebrafish retinas through the manipulation of various factors such as tumor necrosis factor α (TNF-α) (Nelson et al., 2013), GSK-3β (Ramachandran et al., 2011), leptin, interleukin-6 (IL-6) (Zhao et al., 2014), and γ-aminobutyric acid (GABA) (Rao et al., 2017). Although the interdependence of these factors has yet to be determined, they hold great promise as therapeutic agents to induce a regenerative response in mammals. Here, we show that miR-216a is an endogenous inhibitor of retina regeneration, and suppression of miR-216a is sufficient to induce a regenerative response in the absence of damage. This suggests that miR-216a may be upstream of the many signaling pathways that are required for MG proliferation. Stimulation of MG proliferation upon suppression of miR-216a was blocked by the co-suppression of canonical Wnt signaling (Figures 7F and 7G). In addition, excess miR-216a levels resulted in decreased β-catenin accumulation in MG after intense light damage (Figure 6). These findings suggest that miR-216a serves as an inhibitory factor in quiescent MG that needs to be suppressed to turn on canonical Wnt signaling and allow MG to dedifferentiate and proliferate.
It is perhaps not surprising that epigenetic modifications accompany cell fate changes in MG during retina regeneration. Analysis of DNA methylation profiles in quiescent MG and MG-derived progenitors have shown that many pluripotency- and regeneration-associated genes are hypomethylated during zebrafish retina regeneration (Powell et al., 2013). In mouse MG, these genes are also hypomethylated, suggesting that DNA methylation is not an epigenetic barrier for retina regeneration in mammals. Accessible chromatin in mouse mature MG decreases relatively rapidly, coincident with the loss of neurogenic capacity in postnatal development (Jorstad et al., 2017; Ueki et al., 2015). This suggests that changes in histone modifications may underlie MG reprogramming during retina regeneration in zebrafish, but the precise chromatin modifying enzymes have remained unknown. Our data support the hypothesis that the H3K79 methyltransferase Dot1l is required for MG dedifferentiation and proliferation and is regulated by miR-216a. First, 3′ UTR reporter assays showed that miR-216a targets the 3′ UTR of dot1l (Figure 2). Second, we showed that Dot1l is upregulated in dedifferentiated MG compared to post-mitotic MG after 45 h of constant intense light damage (Figures 3A–3C). Third, MO knockdown and inhibition of Dot1l H3K79 methyltransferase activity showed that Dot1l upregulation is required for MG activation during early regeneration (Figures 4B–4E). Lastly, suppression of miR-216a alone is sufficient to stimulate a regenerative response in the undamaged retina, while co-suppressing Dot1l prevented the MG proliferation.
Previous studies in multiple species have revealed that Dot1l is the only methyltransferase that catalyzes the histone H3-lysine 79 (H3K79) mono-, di-, and trimethylation (Feng et al., 2002; Jones et al., 2008; Shanower et al., 2005; Steger et al., 2008; van Leeuwen et al., 2002). Dot1l-mediated H3K79 methylation is associated with the transcription of Wnt-target genes, which is mediated by TCF transcription factors and the co-activator β-catenin (Castaño Betancourt et al., 2012; Clevers, 2006; Mahmoudi et al., 2010; Mohan et al., 2010). A direct interaction between Dotl1l and β-catenin-dependent TCF4 complexes was identified in zebrafish intestinal stem cells and in mouse small intestinal crypts (Mahmoudi et al., 2010). The hypothesis is that recruitment of Dot1l to Wnt-target genes by β-catenin leads to H3K79 methylation and the preferential activation of the transcription of Wnt target genes. The interaction of Dot1l with β-catenin was confirmed in Drosophila embryos by demonstrating the presence of β-catenin within Dot1L-containing protein complexes and a requirement for H3K79me3 in regulating Wnt-target genes (Mohan et al., 2010). In addition, in a genome-wide association study (GWAS), a Dot1l polymorphism was linked to the reduced risk of osteoarthritis. Furthermore, it was shown that Dot1l interacts with Tcf4 in articular chondrocytes and is required for Wnt-dependent chondrogenesis (Castaño Betancourt et al., 2012). In zebrafish, retinal damage including both intense light and mechanical damage leads to the activation of Wnt/β-catenin signaling in MG (Meyers et al., 2012; Ramachandran et al., 2011). In this study, we show that canonical Wnt signaling is required along with Dot1l activity during MG dedifferentiation and proliferation. Our data show that the activation of Wnt signaling by stabilizing β-catenin alleviates defects due to Dot1l knockdown during retina regeneration (Figures 7A–7C). In addition, while β-catenin stabilization is able to induce MG proliferation in the absence of retinal damage, inhibiting Dot1l methyltransferase activity by co-injecting the Dot1l inhibitor significantly reduced the number of proliferating MG (Figure 7D).
Despite the experiments cited above, it remains controversial whether Dot1l solely mediates its effects via association with TCF4 transcription factors and β-catenin. Dot1l has also been reported to associate with the c-Myc-p300 complex to activate epithelial-mesenchymal transition regulators (Cho et al., 2015). Because Wnt activation is required for retina regeneration (Gallina et al., 2016; Osakada et al., 2007; Ramachandran et al., 2011; Sanges et al., 2013; Yao et al., 2016, 2018) and because we have shown that the loss of Dot1l inhibits retina regeneration, we favor the hypothesis that in MG during retina regeneration, the association of Dot1l with TCF4 transcription factors and β-catenin activates Wnt genes. It remains possible that Dot1l-mediated H3K789me3 modifications may track with basal RNA polymerase II transcription and show the enrichment of H3K79me3 modifications on activated Wnt genes. It is also possible that the activation of Wnt genes by Dot1l is not MG specific.
We used MOs to knock down the expression of both miR-216a and Dot1l. There have been concerns raised about the concordance between mutant and MO-induced phenotypes in zebrafish (Stainier et al., 2015). To ensure specificity, we used multiple MOs and MOs that have been previously published. However, most directly, we used suppression/rescue experiments with combinations of MOs. The ability to suppress the effects of MOs against miR-216a by co-injection of MOs against dot1l is the best evidence of specificity, regardless of how many different MOs are used. In addition, we used pharmacologic inhibitors to complement the Dot1l MO experiments and obtained nearly identical results. For Dot1l, we also created CRISPR-Cas9 mutants, but, unfortunately, they are embryonic lethal. We are now attempting to create MG conditional knockouts of Dot1l.
Our experiments focused on miR-216a due to previous work and because we identified dot1l as a target of miR-216a. However, miR-216b is also repressed during retina regeneration. Because miR-216b has a different seed sequence, it targets a different set of mRNAs (it does not target dot1l). Of note, our MO-based experiments deplete both miR-216a and miR-216b, but all of the overexpression and rescue experiments were only done with miR-216a. It will be interesting to identify and test the targets of miR-216b and to determine the mechanisms that underlie the repression of both miRNAs because they are produced from the same polycistronic transcript.
Here, we report the discovery of an MG reprogramming mechanism driven by the H3K79 methyltransferase Dot1l that is under the regulation of miR-216a. miR-216a is repressed in response to photoreceptor loss in adult zebrafish, thereby de-repressing dot1l. Retinitis pigmentosa and age-related macular degeneration involve photoreceptor dysfunction that eventually leads to the loss of vision. Previously, mouse retina regeneration could be induced in young mice (~2 weeks) by the overexpression of the transcription factor Ascl1 and co-administration of the histone deacetylase inhibitor trichostatin-A (Jorstad et al., 2017). It will be interesting to test whether Dot1l plays a role in additional chromatin modifications in mammalian MG and whether suppressing miR-216a can induce mammalian MG proliferation and neural progenitor production.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James G. Patton (james.g.patton@vanderbilt.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Zebrafish
Wild-type (AB)(Walker, 1999), Tg(1016tuba1a:gfp) (Fausett and Goldman, 2006), Tg(gfap:gfp)mi2001 (Bernardos and Raymond, 2006) lines were maintained at 28.5°C on a 14:10 hour light:dark cycle. All experiments with zebrafish were performed with the approval of the Vanderbilt University Institutional Animal Care and Use Committee. Adult zebrafish used for the experiments were between 5–12 months old and sex matched between experimental groups. Constant intense light lesioning to induce cone and rod photoreceptor cell death was performed as previously described (Rajaram et al., 2014c). Briefly, adult fish were dark adapted for 14 days, transferred to clear tanks placed between two fluorescent lights with light intensity at ~20,000 lux for 16h-3days. The tank temperature was maintained at 30–33°C.
METHOD DETAILS
Plasmid construction and embryo injections
The dot1l 3′UTR was amplified from cDNA by PCR with the following primers: dot1l-3′utr-fp: 5′-AGACTTGAATTCCCTTCCAG GAACTGAGTTTAACC-3′ dot1l-3′utr-rp: 5′-AGTCTGCTCGAGCAGCTCCACAGGTAAATGATCC-3′. The 3′UTR was cloned downstream of the GFP coding sequence in the PCS2+ vector. miRNA recognition elements (MREs) within the dot1l 3′UTR were deleted using the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene). mRNAs were in vitro synthesized from linearized constructs using mMESSAGE mMACHINE® SP6 Transcription Kit (Life Technologies). In vitro transcribed RNA was purified by NucAway™ Spin Columns (Life Technologies). For reporter assays, GFP-dot1l-3′UTR mRNA was injected at 100pg/embryo concentration either alone or with a synthetic miR-216a duplex (Dharmacon) at 100pg/embryo.
Morpholino and miRNA mimic injection & electroporation
Lissamine tagged morpholinos (MOs) (Gene Tools) were injected intravitreally and electroporated into adult zebrafish eyes prior to light lesioning as described (Thummel et al., 2006). The following 3′-Lissamine-tagged MOs were used: Gene Tools standard control MO, Dot1 MO, Dot1l MO-2, miR-216a MO and their sequences are listed on Table S1.
Duplex mature miRNAs (Thermo scientific) were injected and electroporated into eyes prior to start of light lesioning as previously described (Rajaram et al., 2014b). Double stranded mature miRNAs were synthesized with 3′-UU overhangs for the following target sequences: miR-216 and control (siLuc) (sequences are listed on Table S1). Electroporation was performed using the Gene Pulser Xcell™ Electroporation Systems (Biorad).
Fluorescence activated cell sorting (FACS)
FACS was used to isolate GFP+ and GFP− cells from the retinas of undamaged Tg(gfap:gfp)mi2001 and Tg(1016tuba1a:gfp) fish using BD FACSAria III (BD Biosciences) at the VUMC Flow Cytometry Shared Resource. Retinas were dissociated as previously described (Rajaram et al., 2014b) with the following changes. After the retinas were dissected, they were collected in Leibovitz L-15 media (ThermoFisher #21083–027) and treated with 1mg/ml hyaluronidase (Sigma #H3884) at room temperature for 15 minutes on a rocker. Cells from the dissociated retinas were stained with propidium iodide to detect dead cells. 12 adult fish were used for each FACS experiment. As a quality control, sorted GFP+ cells were re-analyzed to check the purity of the cell population by re-sorting.
Western blots
Protein was collected from whole retinas by dissociation in 1x RIPA buffer with 1x SDS (Life Technologies) for 3 hours at 4°C followed by centrifugation at 12,000 rpm for 20 minutes. Protein concentrations were determined using BCA assays (BIO-RAD). 10 µg of protein was separated on 12% MINI-PROTEAN TGX® pre-cast gels (BIO-RAD) and transferred to PVDF membranes using the Trans-Blot® Turbo Transfer System (BIO-RAD). Membranes were blocked in 5% milk in 1x TBS-T at room temperature for 1 hour. Primary antibodies against Dot1l (1:300; ab228766, Abcam) and a-tubulin (1:10,000; ab15246, Abcam) were incubated at 4°C overnight. Secondary anti-rabbit antibodies (1:50,000; 7074S, Cell Signaling Technologies) were incubated in 5% milk in 1x TBS-T at room temperature for 1 hour. For imaging, SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) was incubated with the blots for 60 s before exposure.
RT-PCR
Total RNA was isolated from FAC-sorted cells using TRIzol-LS (ThermoFisher # 10296028). Taqman small RNA assays (Life Technologies) were used to perform qRT-PCR of the indicated miRNAs. 5ng of total RNA was used per RT reaction and 1.33 mL of 1:2 diluted resultant cDNA was used in 10 µL qPCR reaction in technical triplicates. qPCR reactions were conducted in either 96-well plates using Bio-Rad CFX96 Real-time system or in 384-well plates using Bio-Rad CFX384 Real-time System. All quantifications were normalized to an endogenous U6 snRNA control. Fold changes were calculated using the ΔC(t) method, where Δ = C(t) miRNA − C(t)U6 snRNA, and ΔΔC(t) = ΔC(t)condition1 − ΔC(t) condition2, and FC = 2—ΔΔC(t). Taqman probe #: U6 snRNA: 001973; hsa-miR-216a:002220. For RT-PCR of mRNAs, RNA was DNase treated (TURBO DNAfree kit ThermoFisher #AM1907), converted to cDNA using Maxima first strand cDNA synthesis kit (Thermo Scientific) and qPCR was performed using SYBR Green (Biorad). All qPCR primers spanned exon-exon junctions (IDT). miRNA realtime PCR was perfomed using Taqman probes as per the manufacturer’s instructions (Life Technologies). Relative RNA expression during regeneration were determined using the ΔΔCt method and normalized to 18 s rRNA levels and U6 snRNA levels for mRNAs and miRNAs respectively. Real-time PCR was performed on a Biorad CFX 96 Real time system. The primer sequences are listed in Table S1.
Pharmacological treatment
To stimulate Wnt signaling, GSK-3β Inhibitor I (Calbiochem; CAS 327036–89-5) was intravitreally injected at 1mM in 4%DMSO. Wnt signaling was blocked by the tankyrase inhibitor/axin stabilizing agent XAV939 (Cayman chemical; 10mM stock in DMSO) intravitreally injected at 10 µM in 4%DMSO. For catalytic inhibition of Dot1l, EPZ004777 (Calbiochem; CAS 1338466–77-5) was intravitreally injected.
Immunohistochemistry
Adult zebrafish eyes were collected and fixed in either 4% paraformaldehyde at 4°C overnight or a fixant containing 9 parts 95% ethanol:1 part 37% formaldehyde (for dot1l IHC), cryoprotected in 30% sucrose/1X PBS before embedding. 10–12 micron sections were obtained using a cryostat (Leica), collected on charged Histobond slides (VWR), dried and stored at −80°C. For IHC, slides were warmed to room temperature, rehydrated in 1X PBS and blocked (3% Donkey serum, 0.1% Triton X-100 in 1X PBS) for 1–2h at room temperature before incubating with primary antibodies overnight at 4°C. The following primary antibodies were used: mouse anti-PCNA monoclonal antibody (1:500, Sigma), anti-PCNA polyclonal antibody (1:500, Abcam), rabbit anti-GFP polyclonal antiserum (1:1000, Torrey Pines Biolabs), mouse anti-β-catenin antibody (1:500, BD Bioscience) and rabbit anti-dot1l polyclonal antibody (1:200, Bethyl labs). After primary antibody incubation, sections were washed and incubated with secondary antibody and nuclear stain TOPRO 3 (1:1000, Invitrogen) at room temperature. Secondary antibodies were donkey anti-mouse AF488 (1:200), donkey anti-mouse AF647 (1:200), donkey anti-mouse Cy-3 (1:100), donkey anti-rabbit Cy3 (1:100) and donkey anti-rabbit AF488 (1:200)(Jackson Immuno). Slides were washed, dried and coverslipped with Vectashield (Vector labs). Antigen retrieval was performed for β-catenin and PCNA IHC as previously described (Rajaram et al., 2014b).
Imaging and image processing
For imaging of immunofluorescent staining, a META Zeiss LSM 510 Meta confocal microscope was used. Images were processed using ImageJ software 4.13. Fluorescence intensity measurements for the GFP reporter assays were done using ImageJ software (Gavet and Pines, 2010).
QUANTIFICATION AND STATISTICAL ANALYSIS
For fluorescence intensity quantifications, ‘‘integrated density,’’ ‘‘area’’ and ‘‘mean gray value’’ GFP+ region, as well as background, were measured for each image. Corrected fluorescence intensity of the selected region was calculated according to the formula: ‘‘Corrected fluorescence intensity= Integrated Density - (Area of selected region * mean fluorescence of background).’’ Data were represented as a mean of corrected fluorescence intensity for each experimental condition and statistical analyses were performed using a two-tailed Student’s t test. For immunostaining and cell quantification, only retina sections that comprised optic nerves were used. All cell counts were done in the central-dorsal retina, at a linear distance of ~300 microns from the optic nerve. Cells from one to four sections were quantified and averaged from each eye. In all figures, data are represented as mean ± standard error of the mean (s.e.m). Significance was calculated either by the non-parametric Mann–Whitney U test or one-way ANOVA with Dunnett’s multiple comparisons test for cell quantifications using the Graphpad Prism. Statistical tests and number of animals used for each experiment are descirbed in the figure legends.
DATA AND CODE AVAILABILITY
This study did not generate/analyze datasets.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-PCNA monoclonal antibody | Sigma | P8825; RRID: AB_477413 |
| Rabbit anti-PCNA polyclonal antibody | Abcam | 18197; RRID: AB_444313 |
| Rabbit anti-GFP polyclonal antiserum | Torrey Pines Biolabs | TP401; RRID: AB_10013661 |
| Mouse anti-β-catenin antibody | BD Bioscience | 610153; RRID: AB_397554 |
| Rabbit anti-dot1l polyclonal antibody | Bethyl labs | A300-954A; RRID: AB_2261948 |
| Donkey anti-mouse AF488 | Jackson Immuno | 715-545-150 |
| Donkey anti-mouse AF647 | Jackson Immuno | 715-605-150 |
| Donkey anti-mouse Cy-3 | Jackson Immuno | 715-165-150 |
| Donkey anti-rabbit Cy3 | Jackson Immuno | 711-165-152 |
| Donkey anti-rabbit AF488 | Jackson Immuno | 711-545-152 |
| Rabbit anti-KMT4/Dot1l polyclonal antibody | Abcam | Ab228766 |
| Rabbit anti-alpha tubulin antibody | Abcam | Ab15246; RRID: AB_301787 |
| Anti-rabbit HRP-conjugated antibody | Cell Signaling Technologies | 7074S;RRID: AB_2099233 |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| Leibovitz L-15 media | ThermoFisher | 21083-027 |
| Hyaluronidase | Sigma-Aldrich | H3884 |
| Papain dissociation kit | Worthington Biochemical Corp | LK003178 |
| DNase I | NEB | M0303S |
| Leupeptin trifluoroacetate salt | Sigma-Aldrich | L2023-10MG |
| GSK-3β Inhibitor I | Calbiochem | 361540 |
| Tankyrase inhibitor/axin stabilizing agen XAV939 | Cayman chemical | 13596 |
| Dot1 inhibitor EPZ004777 | Calbiochem | 532282 |
| TO-PRO-3 Iodide | Invitrogen | T3605 |
| Vectashield | Vector Labs | H-1000 |
|
Critical Commercial Assays | ||
| TRIzol-LS | ThermoFisher | 10296028 |
| Taqman probe: U6 snRNA | Life Technologies | 001973 |
| Taqman probe: hsa-miR-216a | Life Technologies | 002220 |
| TURBO DNAfree kit | ThermoFisher | AM1907 |
| Maxima first strand cDNA synthesis kit | Thermo Scientific | K1641 |
| SsoAdvanced Universal SYBR® Green Supermix | Bio-Rad | 1725271 |
| QuikChange Lightning Site-Directed Mutagenesis Kit | Stratagene | 210518 |
| mMESSAGE mMACHINE® SP6 Transcription Kit | Life Technologies | AM1340 |
| NucAway™ Spin Columns | Life Technologies | AM10070 |
|
Experimental Models: Organisms/Strains | ||
| Zebrafish: Wild-type (AB) | Walker, 1999 | N/A |
| Zebrafish: Tg(1016tuba1a:gfp) | Fausett and Goldman, 2006 | N/A |
| Zebrafish: Tg(gfap:gfp)mi2001 | Bernardos and Raymond, 2006 | N/A |
|
Oligonucleotides | ||
| See Table S1 for oligonucleotides used. | N/A | N/A |
|
Recombinant DNA | ||
| PCS2+ vector | Turner and Weintraub, 1994 | N/A |
|
Software and Algorithms | ||
| ImageJ software 4.13 | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
miR-216a is suppressed in Müller glia during retina regeneration
miR-216a suppression stimulates Müller glia reprogramming and proliferation
Dot1l is a target of miR-216a and required for Müller glia proliferation
miR-216a and Dot1l regulate Müller glia reprogramming through canonical Wnt signaling
ACKNOWLEDGMENTS
We would like to thank members of the Patton laboratory for helpful discussions. Transgenic zebrafish lines were shared by Pamela Raymond (Tg(gfap:GFP)mi2001) and Daniel Goldman (Tg(1016tuba1a:GFP)). This work was supported by grants from the NIH (RO1 EY024354, R21 EY019759, and UO1 EY027265 to J.G.P.), a Vanderbilt Vision Research Center NEI Core Grant (P30-EY008126), and additional support from the Stevenson family endowment and the Gisela Mosig Endowment for Biological Sciences to Vanderbilt University.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.061.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bernardos RL, and Raymond PA (2006). GFAP transgenic zebrafish. Gene Expr. Patterns 6, 1007–1013. [DOI] [PubMed] [Google Scholar]
- Bernardos RL, Barthel LK, Meyers JR, and Raymond PA (2007). Latestage neuronal progenitors in the retina are radial Müller glia that function as retinal stem cells. J. Neurosci 27, 7028–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, and Reichenbach A (2006). Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res 25, 397–424. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Iandiev I, Pannicke T, Wurm A, Hollborn M, Wiedemann P, Osborne NN, and Reichenbach A (2009). Cellular signaling and factors involved in Müller cell gliosis: neuroprotective and detrimental effects. Prog. Retin. Eye Res 28, 423–451. [DOI] [PubMed] [Google Scholar]
- Castaño Betancourt MC, Cailotto F, Kerkhof HJ, Cornelis FMF, Doherty SA, Hart DJ, Hofman A, Luyten FP, Maciewicz RA, Mangino M, et al. (2012). Genome-wide association and functional studies identify the DOT1L gene to be involved in cartilage thickness and hip osteoarthritis. Proc. Natl. Acad. Sci. USA 109, 8218–8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho MH, Park JH, Choi HJ, Park MK, Won HY, Park YJ, Lee CH, Oh SH, Song YS, Kim HS, et al. (2015). DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat. Commun 6, 7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480. [DOI] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, et al. (2011). Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausett BV, and Goldman D (2006). A role for alpha1 tubulin-expressing Müller glia in regeneration of the injured zebrafish retina. J. Neurosci 26, 6303–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, and Zhang Y (2002). Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol 12, 1052–1058. [DOI] [PubMed] [Google Scholar]
- Gallina D, Palazzo I, Steffenson L, Todd L, and Fischer AJ (2016). Wnt/β-catenin-signaling and the formation of Müller glia-derived progenitors in the chick retina. Dev. Neurobiol 76, 983–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, and Pines J (2010). Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 18, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman D (2014). Müller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci 15, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S-MA, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, et al. (2009). Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620. [DOI] [PubMed] [Google Scholar]
- Jones B, Su H, Bhat A, Lei H, Bajko J, Hevi S, Baltus GA, Kadam S, Zhai H, Valdez R, et al. (2008). The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet 4, e1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorstad NL, Wilken MS, Grimes WN, Wohl SG, VandenBosch LS, Yoshimatsu T, Wong RO, Rieke F, and Reh TA (2017). Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 548, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkowski JR, and Raymond PA (2014). Müller glia: Stem cells for generation and regeneration of retinal neurons in teleost fish. Prog. Retin. Eye Res 40, 94–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi T, Boj SF, Hatzis P, Li VS, Taouatas N, Vries RG, Teunissen H, Begthel H, Korving J, Mohammed S, et al. (2010). The leukemia-associated Mllt10/Af10-Dot1l are Tcf4/β-catenin coactivators essential for intestinal homeostasis. PLoS Biol 8, e1000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers JR, Hu L, Moses A, Kaboli K, Papandrea A, and Raymond PA (2012). β-catenin/Wnt signaling controls progenitor fate in the developing and regenerating zebrafish retina. Neural Dev 7, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan M, Herz HM, Takahashi YH, Lin C, Lai KC, Zhang Y, Washburn MP, Florens L, and Shilatifard A (2010). Linking H3K79 trimethylation to Wnt signaling through a novel Dot1-containing complex (DotCom). Genes Dev 24, 574–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima M, Barthel LK, and Raymond PA (2013). A self-renewing division of zebrafish Müller glial cells generates neuronal progenitors that require N-cadherin to regenerate retinal neurons. Development 140, 4510–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Ackerman KM, O’Hayer P, Bailey TJ, Gorsuch RA, and Hyde DR (2013). Tumor necrosis factor-alpha is produced by dying retinal neurons and is required for Muller glia proliferation during zebrafish retinal regeneration. J. Neurosci 33, 6524–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olena AF, Rao MB, Thatcher EJ, Wu SY, and Patton JG (2015). miR-216a regulates snx5, a novel notch signaling pathway component, during zebrafish retinal development. Dev. Biol 400, 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osakada F, Ooto S, Akagi T, Mandai M, Akaike A, and Takahashi M (2007). Wnt signaling promotes regeneration in the retina of adult mammals. J. Neurosci 27, 4210–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C, Grant AR, Cornblath E, and Goldman D (2013). Analysis of DNA methylation reveals a partial reprogramming of the Müller glia genome during retina regeneration. Proc. Natl. Acad. Sci. USA 110, 19814–19819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaram K, Harding RL, Bailey T, Patton JG, and Hyde DR (2014a). Dynamic miRNA expression patterns during retinal regeneration in zebrafish: reduced dicer or miRNA expression suppresses proliferation of Müller glia-derived neuronal progenitor cells. Dev. Dyn 243, 1591–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaram K, Harding RL, Hyde DR, and Patton JG (2014b). miR-203 regulates progenitor cell proliferation during adult zebrafish retina regeneration. Dev. Biol 392, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaram K, Summerbell ER, and Patton JG (2014c). Technical brief: constant intense light exposure to lesion and initiate regeneration in normally pigmented zebrafish. Mol. Vis 20, 1075–1084. [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Fausett BV, and Goldman D (2010). Ascl1a regulates Müller glia dedifferentiation and retinal regeneration through a Lin-28-dependent, let-7 microRNA signalling pathway. Nat. Cell Biol 12, 1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Zhao XF, and Goldman D (2011). Ascl1a/Dkk/beta-catenin signaling pathway is necessary and glycogen synthase kinase-3beta inhibition is sufficient for zebrafish retina regeneration. Proc. Natl. Acad. Sci. USA 108, 15858–15863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MB, Didiano D, and Patton JG (2017). Neurotransmitter-Regulated Regeneration in the Zebrafish Retina. Stem Cell Reports 8, 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanges D, Romo N, Simonte G, Di Vicino U, Tahoces AD, Fernández E, and Cosma MP (2013). Wnt/β-catenin signaling triggers neuron reprogramming and regeneration in the mouse retina. Cell Rep 4, 271–286. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanower GA, Muller M, Blanton JL, Honti V, Gyurkovics H, and Schedl P (2005). Characterization of the grappa gene, the Drosophila histone H3 lysine 79 methyltransferase. Genetics 169, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stainier DYR, Kontarakis Z, and Rossi A (2015). Making sense of anti-sense data. Dev. Cell 32, 7–8. [DOI] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, et al. (2008). DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol 28, 2825–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher EJ, and Patton JG (2010). Small RNAs have a big impact on regeneration. RNA Biol 7, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Bai S, Sarras MP Jr., Song P, McDermott J, Brewer J, Perry M, Zhang X, Hyde DR, and Godwin AR (2006). Inhibition of zebrafish fin regeneration using in vivo electroporation of morpholinos against fgfr1 and msxb. Dev. Dyn 235, 336–346. [DOI] [PubMed] [Google Scholar]
- Thummel R, Kassen SC, Enright JM, Nelson CM, Montgomery JE, and Hyde DR (2008). Characterization of Müller glia and neuronal progenitors during adult zebrafish retinal regeneration. Exp. Eye Res 87, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DL, and Weintraub H (1994). Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev 8, 1434–1447. [DOI] [PubMed] [Google Scholar]
- Ueki Y, Wilken MS, Cox KE, Chipman L, Jorstad N, Sternhagen K, Simic M, Ullom K, Nakafuku M, and Reh TA (2015). Transgenic expression of the proneural transcription factor Ascl1 in Müller glia stimulates retinal regeneration in young mice. Proc. Natl. Acad. Sci. USA 112, 13717–13722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, and Gottschling DE (2002). Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109, 745–756. [DOI] [PubMed] [Google Scholar]
- Walker C (1999). Haploid screens and gamma-ray mutagenesis. Methods Cell Biol 60, 43–70. [DOI] [PubMed] [Google Scholar]
- Wan J, Ramachandran R, and Goldman D (2012). HB-EGF is necessary and sufficient for Müller glia dedifferentiation and retina regeneration. Dev. Cell 22, 334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienholds E, and Plasterk RHA (2005). MicroRNA function in animal development. FEBS Lett 579, 5911–5922. [DOI] [PubMed] [Google Scholar]
- Yao K, Qiu S, Tian L, Snider WD, Flannery JG, Schaffer DV, and Chen B (2016). Wnt Regulates Proliferation and Neurogenic Potential of Müller Glial Cells via a Lin28/let-7 miRNA-Dependent Pathway in Adult Mammalian Retinas. Cell Rep 17, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao K, Qiu S, Wang YV, Park SJH, Mohns EJ, Mehta B, Liu X, Chang B, Zenisek D, Crair MC, et al. (2018). Restoration of vision after de novo genesis of rod photoreceptors in mammalian retinas. Nature 560, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, and Srivastava D (2007). A developmental view of microRNA function. Trends Biochem. Sci 32, 189–197. [DOI] [PubMed] [Google Scholar]
- Zhao XF, Wan J, Powell C, Ramachandran R, Myers MG Jr., and Goldman D (2014). Leptin and IL-6 family cytokines synergize to stimulate Müller glia reprogramming and retina regeneration. Cell Rep 9, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets.