

Abstract
The natural product pepticinnamin E potently inhibits protein farnesyl transferases and has potential applications in treating cancer and malaria. Pepticinnamin E contains a rare N-terminal cinnamoyl moiety and several nonproteinogenic amino acids including an unusual 2-chloro-4-O-methyl-3,4-dihydroxy-l-phenylalanine. The biosynthesis of pepticinnamin E has remained uncharacterized because its original producing strain is unavailable. Here, we identified a gene cluster (pcm) for this natural product in a new producer, Actinobacteria bacterium OK006, using a targeted rediscovery strategy. We demonstrated that the pcm cluster is responsible for the biosynthesis of pepticinnamin E, a nonribosomal peptide and polyketide hybrid. We also characterized a key O-methyltransferase that modifies 3,4-dihydroxy-l-phenylalanine. Our work identified the gene cluster for pepticinnamins for the first time and sets the stage for elucidating the unique chemistries required for biosynthesis.
Keywords: genome mining, natural product, nonribosomal peptide, O-methyltransferase, nonproteinogenic amino acids
Graphical Abstract
Inaccessibility of the producing strain for the farnesyl-transferase inhibitor, pepticinnamin E, has precluded biosynthetic studies. A new producer of pepticinnamin E was discovered by mining bacterial genomes for a characteristic enzyme, leading to the identification of the biosynthetic gene cluster. Biosynthesis of the compound involves a promiscuous catechol O-methyltransferase.
Living organisms synthesize an astounding number of natural products, many of which have important uses in medicine and biotechnology. While tens of thousands of biosynthetic gene clusters have been identified from the genomes of bacteria, relatively few have been characterized in terms of the natural products they produce. Additionally, while many bioactive natural products have been isolated, their biosynthetic gene clusters remain undetermined. Examples include the neurotoxin tetrodotoxin,[1] the psychoactive drug ibotenic acid,[2] and the anticancer agent acivicin,[3] isolated from marine animals, mushrooms, and bacteria, respectively. The lack of knowledge regarding the biosynthesis of these natural products limits the ability to study and engineer these compounds.
The pepticinnamins were discovered in 1992 as farnesyl-transferase inhibitors.[4] Farnesyl-transferase inhibitors have been explored for cancer therapy as well as potential treatments for malaria and trypanosomiasis.[5] Six pepticinnamin variants A−F have been isolated, among which pepticinnamin E is the most abundant and thoroughly characterized. Pepticinnamin E is composed of a cinnamoyl tail, a highly modified tripeptide core, and an ester-linked diketopiperazine (Figure 1).[6] The tripeptide core contains three nonproteinogenic aromatic amino acids: d-tyrosine, N-methyl-2-chloro-4-O-methyl-3,4-dihydroxy-l-phenylalanine (N-Me-2-Cl-4-O-Me-l-DOPA), and N-methyl-l-phenylalanine (N-Me-l-Phe). The diketopiperazine consists of a d-serine and a glycine. Total synthesis of pepticinnamin E and its epimer revealed that the second amino acid in the natural product possesses (S) stereochemistry and is N-Me-2-Cl-4-O-Me-l-DOPA.[7] Pepticinnamin E acts as a natural bisubstrate inhibitor of protein farnesyl transferase and several analogs induce apoptosis in tumor cells.[8]
Figure 1.
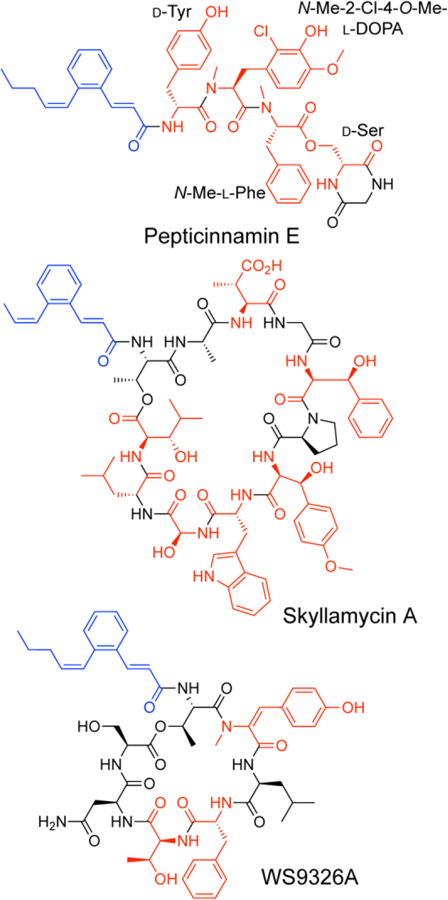
Structures of NRPS/PKS hybrid natural products containing cinnamoyl moieties. Cinnamoyl moieties are highlighted in blue, and nonproteinogenic amino acids in red.
The unique structure of pepticinnamin E suggests that its biosynthesis requires novel enzymatic chemistry. For example, the 2-Cl-4-O-Me-l-DOPA is a rare nonproteinogenic amino acid in nonribosomal peptides. Diketopiperazine formation is an unusual strategy for release of nonribosomal peptides longer than a dipeptide from the assembly line enzymes, nonribosomal peptide synthetases (NRPSs). Identifying the biosynthetic gene cluster for a given natural product generally requires access to the producing microbe or the genome sequence of the producer. However, the original strain that pepticinnamin was isolated from, Streptomyces sp. OH-4652, has not been sequenced and is not readily accessible, posing a significant challenge for studying the biosynthesis of pepticinnamin E.
To overcome this challenge, we set out to identify new pepticinnamin producers by bioinformatics. Many natural products have been identified from more than one bacterial strain due to horizontal gene transfer of their biosynthetic gene clusters. This phenomenon has caused substantial natural product rediscovery problems.[9] We reasoned that we could take advantage of this phenomenon and the growing microbial genome databases to identify the biosynthetic gene clusters of natural products whose original producers are unavailable. Using this targeted rediscovery strategy, we identified a new bacterial producer for pepticinnamin E, Actinobacteria bacterium OK006, and determined the biosynthetic gene cluster responsible for its biosynthesis. Additionally, we reconstituted the activity of the methyltransferase responsible for the formation of 2-Cl-4-O-Me-l-DOPA and characterized the substrate scope of this enzyme.
Analysis of the pepticinnamin E structure revealed that it shares a similar N-terminal cinnamoyl lipid moiety as the natural products skyllamycin A and WS9326A (Figure 1).[10] The biosynthetic gene clusters for skyllamycin A and WS9326A have been identified and are both polyketide synthase (PKS)-NRPS hybrids (Figure 2 A&B).[11] The formation of the benzene ring in the cinnamoyl moiety of skyllamycin A has been proposed to be catalyzed by the putative oxidoreductase Sky4 or phytoene dehydrogenase-like enzyme Sky28.[11a] Therefore, we used Sky28 as a biosynthetic “hook” to identify gene clusters that might encode enzymes for the synthesis of cinnamoyl moiety. Specifically, Sky28 (AEA30271.1) was used as a query for a blastp search against the Joint Genomic Institute’s Integrated Microbial Genome Database. The top 500 hits were inspected for proximity of NRPS genes, and further analyzed by antiSMASH3.0 and MultiGeneBlast against the MiBiG database.[12] This analysis enabled us to identify a putative 45 kilobase pair gene cluster for pepticinnamin biosynthesis (pcm) in A. bacterium OK006, a Streptomyces strain isolated from the root endosphere of a poplar tree.[13] This cluster encodes a gene product Pcm9 that is 58% identical to Sky28. It also harbors a number of NRPS and PKS genes. The core PKS genes in this cluster share high sequence identity with those in the clusters for skyllamycin and WS9326 that are involved in biosynthesis of the cinnamoyl moiety (Figure 2C, Table S1).
Figure 2.
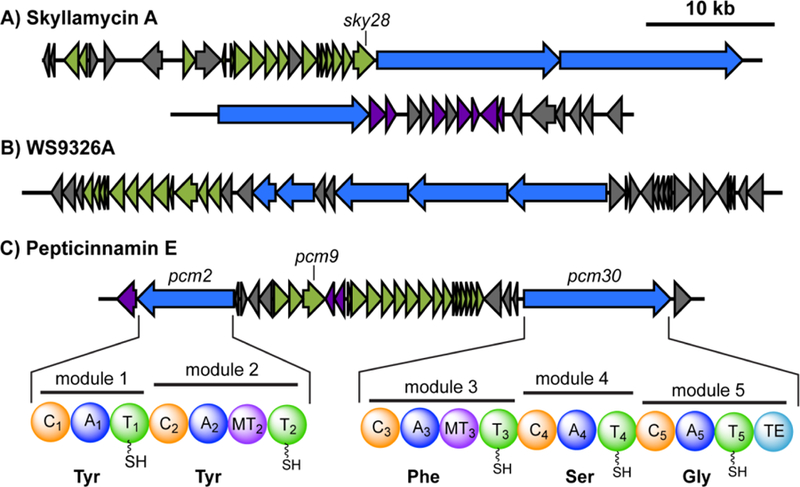
Identification of a candidate biosynthetic gene cluster for pepticinnamin E by bioinformatics. Using signature genes for the cinnamoyl moieties in the gene clusters for skyllamycin (A, sky28) and WS9326A (B), the pepticinnamin gene cluster containing a sky28 homolog pcm9 was identified (C). Genes are colored by function as follows: NRPS, blue; PKS, green; tailoring enzymes, purple; and others, grey. Predicted domain structure of the NRPS genes in the pcm cluster, pcm2 and pcm30, is shown in (C). Abbreviations are as follows: C: condensation; A: adenylation; T: thiolation or peptidyl carrier protein (PCP); MT: N-methyltransferase; TE: thioesterase.
We conducted further analysis of the two NRPSs, Pcm2 and Pcm30, in this cluster using NRPSpredictor2,[14] and identified a total of five modules, two in Pcm2 and three in Pcm30. Substrate prediction based on bioinformatics suggested that the adenylation (A) domains in modules 1−5 prefer tyrosine, tyrosine, phenylalanine, serine, and glycine, respectively (Figure 2C, Table S2). The predicted substrate preference matches the composition of the core peptide in pepticinnamin E. Additionally, module 2 in Pcm2 and module 3 in Pcm30 both contain a methyltransferase domain (Figure 2C), consistent with the N-methyl groups present in the peptide backbone of pepticinnamin E at the second and third amino acids.
We also identified three potential tailoring enzymes for the formation of 2-Cl-4-O-Me-l-DOPA in pepticinnamin E: a flavin-dependent halogenase, Pcm1, a biopterin-dependent hydroxylase, Pcm11, and a SAM-dependent methyltransferase, Pcm10. These enzymes are of particular interest because halogenation of aromatic residues typically occurs at the 3- and 4-postions instead of the 2-position found in 2-Cl-4-O-Me-l-DOPA. Additionally, hydroxylations in bacterial nonribosomal peptide pathways are often catalyzed by cytochrome P450 or non-heme iron/α-ketoglutarate-dependent enzymes;[15] biopterin-dependent hydroxylases, essential for the metabolism of aromatic amino acids and synthesis of neurotransmitters in mammals, are relatively rare in bacteria.[16] Based on these bioinformatic evidences, we hypothesized that this cluster encodes biosynthetic enzymes for pepticinnamin production. We set out to test this hypothesis and characterize the unusual tailoring enzymes.
Having identified a candidate biosynthetic gene cluster for pepticinnamin E from A. bacterium OK006, we first examined the ability of this strain to produce pepticinnamin E. The bacterium was cultured in different media to elicit pepticinnamin production, and the resulting cell pellet was extracted with methanol. The extract was then analyzed by liquid chromatography coupled high-resolution mass spectrometry (LC-HRMS) analysis. We identified peaks with mass to charge ratios (m/z) of 908.356 and 930.345, corresponding to [M+H]+ and [M+Na]+ adducts of pepticinnamin E. In-source mass fragmentation yielded product ions with m/z of 603.223 and 547.191, which correspond to the b and y ions at the 2-Cl-4-O-Me-l-DOPA residue, respectively (Figures 3A and S1). 1H NMR analysis of isolated pepticinnamin E also supports the reported structure (Figure S2 and Table S3). These results demonstrated that A. bacterium OK006 is a new producer of pepticinnamin E. To validate that the pcm cluster is responsible for the biosynthesis of pepticinnamin E, we disrupted pcm2 by generating insertional mutants with a single crossover apramycin resistance cassette. Attempts at double crossover mutants were unsuccessful. Growth and extraction of the mutant strains under the same conditions as the wildtype bacterium yielded no pepticinnamin E detectable by UV absorption or MS (Figure 3B). This result confirms that pcm is the biosynthetic gene cluster for pepticinnamin E.
Figure 3.
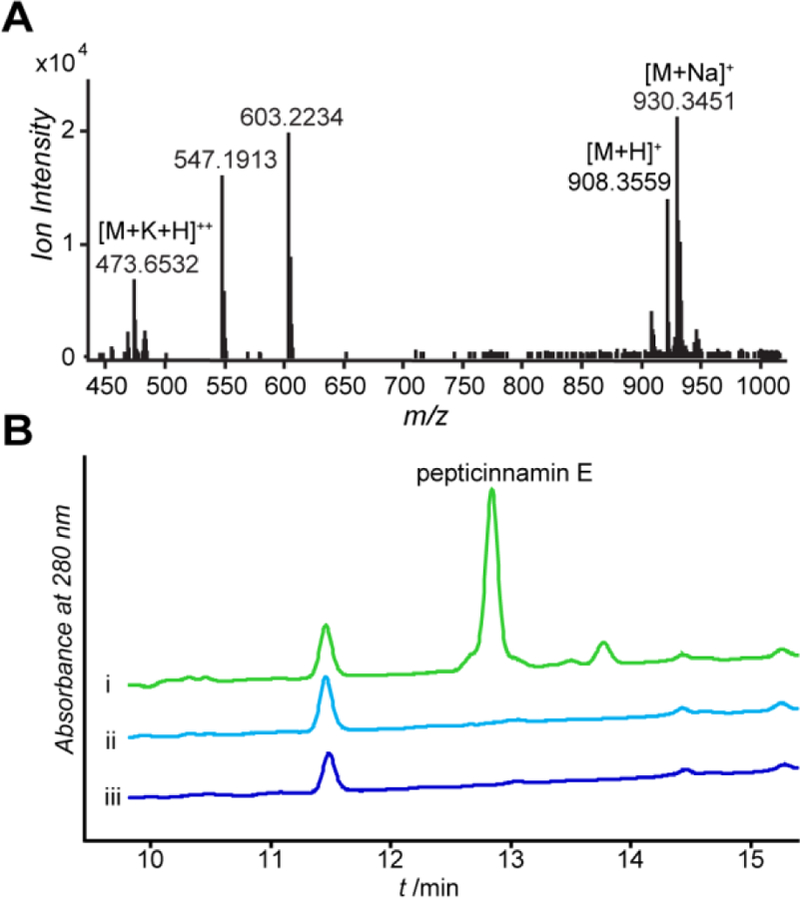
The pcm cluster is responsible for pepticinnamin E biosynthesis. (A) High-resolution mass spectrum of pepticinnamin E in the culture extract of A. bacterium OK006. In-source fragmentation ions were also observed (Figure S1). (B) Pepticinnamin E is produced by wildtype A. bacterium OK006, but not the pcm2 insertional mutants. The UV traces at A280nm of cell extracts are shown for (i) wildtype A. bacterium OK006 and (ii, iii) two independent pcm2::aprR mutant strains.
Pcm10 shares 58% sequence identity with SafC, a characterized catechol 4-O-methyltransferase in the biosynthesis of saframycin MX1.[17] Therefore, we hypothesized that Pcm10 catalyzes O-methylation of l-DOPA and derivatives of l-DOPA. To characterize the function of Pcm10, we cloned, expressed, and purified a His-tagged version in E. coli. The activity of Pcm10 was reconstituted in a buffered solution containing S-adenosyl methionine (SAM) and l-DOPA at 28 °C. A methyl-l-DOPA product was detected by LC-HRMS and its production was abolished in assays containing boiled Pcm10, indicating that Pcm10 converts l-DOPA to methyl-l-DOPA. To identify the position of methylation, the methyl-l-DOPA product was purified from a 10 mL overnight Pcm10 reaction and subjected to 1H NMR analysis (Figure S3). The 1H shifts are consistent with those reported for 4-methyl-l-DOPA,[18] but differ from those of 3-methyl-l-DOPA.[19] This result indicated that Pcm10 methylates l-DOPA at the 4-hydroxy position, in agreement with the structure of pepticinnamin E. No methyltransfer activity was observed when Pcm10 was incubated with l-tyrosine, suggesting that hydroxylation of l-tyrosine occurs prior to 4-methylation.
To probe the substrate scope of Pcm10, we tested catechol and its derivatives, including dopamine, caffeic acid, and chlorogenic acid as substrates for Pcm10. Pcm10 did not modify catechol, but converted ~50% of dopamine and greater than 70% of caffeic acid and chlorogenic acid to their methylated products during the incubation period (Figures 4, S4 and S5). The activity of Pcm10 toward these substrates is comparable to that of l-DOPA as a substrate under the same conditions. The regiochemistry of the methylated products for dopamine, caffeic acid, or chlorogenic acid was not determined. The shape of the product peaks for these non-native substrates suggests that Pcm10 may not be regioselective toward these substrates (Figure S4). These results indicate that Pcm10 is a promiscuous methyltransferase of modified catechols. SafC can also methylate catechol derivatives and is regioselective for l-DOPA, but not the non-native substrates.[17] These observations are explained by a recent structural study that revealed the active site of SafC is plastic; it can accommodate a variety of catechol substrates, and the regioselectivity of SafC is largely substrate-dependent.[20]
Figure 4.
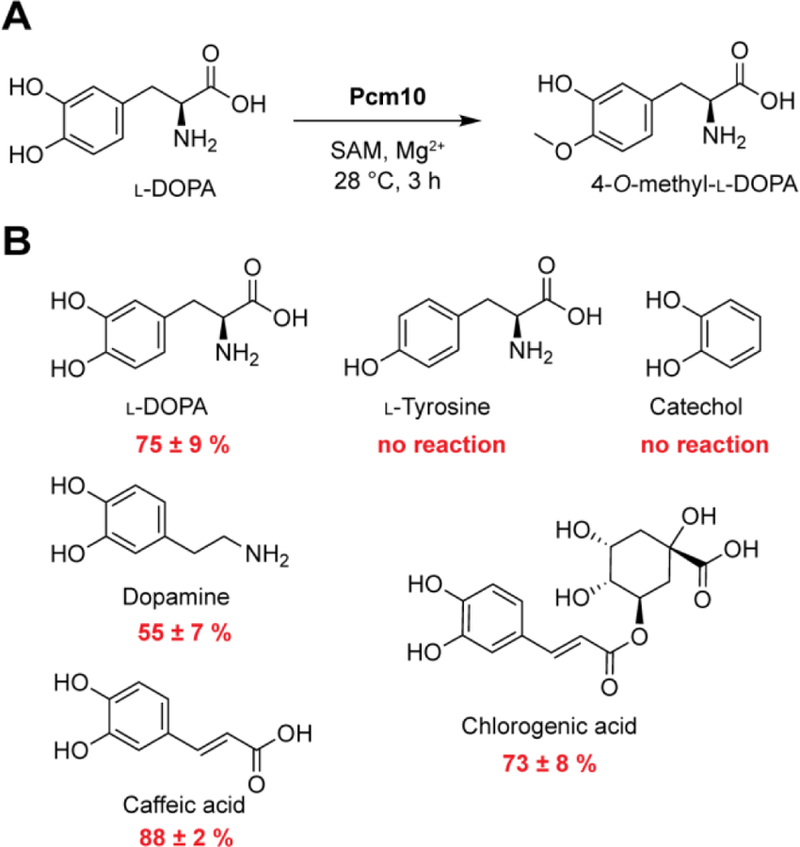
Pcm10 is a promiscuous O-methyltransferase that modifies catechol derivatives. (A) Pcm10 regioselectively methylates the 4-hydroxy position of LDOPA. (B) Pcm10 converts a variety of catechol derivatives to their methylated products.
We conducted additional bioinformatic analyses to characterize the timing of hydroxylation and other tailoring reactions in the generation of 2-Cl-4-O-Me-l-DOPA. The amino acid precursor is likely activated by the second A domain of Pcm2 (Pcm2-A2). Pcm2-A2 shares 56% sequence identity with the previous A domain (Pcm2-A1) that likely activates Tyr1 of pepticinnamin E. It also possesses a nearly identical 10 amino acid specificity code to Pcm2-A1 and another Tyr-activating A domain in the NRPS NosD,[21] suggesting that l-Tyr is the preferred substrate for Pcm2-A2. Further, phylogenetic analysis of Pcm1 revealed that it groups with flavin-dependent halogenases SgcC3 and BhaA[22] that prefer thiolation (T) domain-linked substrates rather than those that prefer free substrates (Figure S6). Lastly, the promiscuity of Pcm10 toward modified catechols suggests that methylation by Pcm10 could occur on substrates (l-DOPA or 2-chloro-l-DOPA) loaded on Pcm2 as T-linked thioesters. Therefore, in the biosynthesis of 2-Cl-4-O-Me-l-DOPA, l-Tyr is likely first loaded to the second T domain of Pcm2 followed by hydroxylation by Pcm11, chlorination by Pcm1, and O-methylation by Pcm10. Modification of T domain-tethered substrates has also been reported in many NRPS biosynthetic pathways.[23] This strategy could control the flux of the amino acid precursor into natural product biosynthesis and reduce the formation of unnecessary intermediates.
Based on our experimental results and bioinformatic analyses, we propose a pathway for pepticinnamin E biosynthesis (Figure 5). Each module is likely responsible for the activation and loading of the corresponding amino acid, condensation with the downstream amino acid, and methylation of the amide NH in the second and third amino acids. Besides the canonical NRPS chemistry, a few domains in the pathway may catalyze unusual chemistry. For example, two out of five amino acids in pepticinnamin E, Tyr1 and Ser4, exhibit d-stereochemistry. Although d-amino acids in nonribosomal peptides often result from the epimerase activity of condensation (C) domains, the C domains in the pcm pathway exhibit no significant similarity to those with dual epimerase/condensation functions (Figure S7). In fact, phylogenetic analysis revealed that C domains in modules 2−5 all group with LCL domains that catalyze amide formation between two l amino acids, while the first C domain groups with starter domains (Figure S7). Additionally, N-Me-Phe3 and Ser4 are connected via an ester linkage. A standalone C domain SgcC5 in the C-1027 gene cluster has been shown to catalyze ester bond formation between the benzoxazolinate and the enediyne core.[24] Other C domains have also been associated with nonribosomal peptide chain extension via ester bonds in the pathways for kutzneride,[25] valinomycin,[26] and cereulide.[27] Thus, the fourth C domain (C4) in Pcm30 may directly catalyze the ester bond or facilitate the rearrangement from an amide to an ester. However, like in SgcC5, no significant changes in the conserved residues are observed in Pcm30-C4. The ester linkage allows the amine of Ser4 to condense with Gly5, forming the C-terminal diketopiperazine. The diketopiperazine formation may be catalyzed by the terminal thioesterase (TE) domain of Pcm30. However, sequence analysis of Pcm30-TE identified the conserved GxSxG motif but did not suggest any unusual features. Phylogenetic analysis revealed no distinct grouping with any subtypes of TE domains (Figure S8). The functions and mechanisms of the C and TE domains in the pepticinnamin pathway remain to be characterized.
Figure 5.
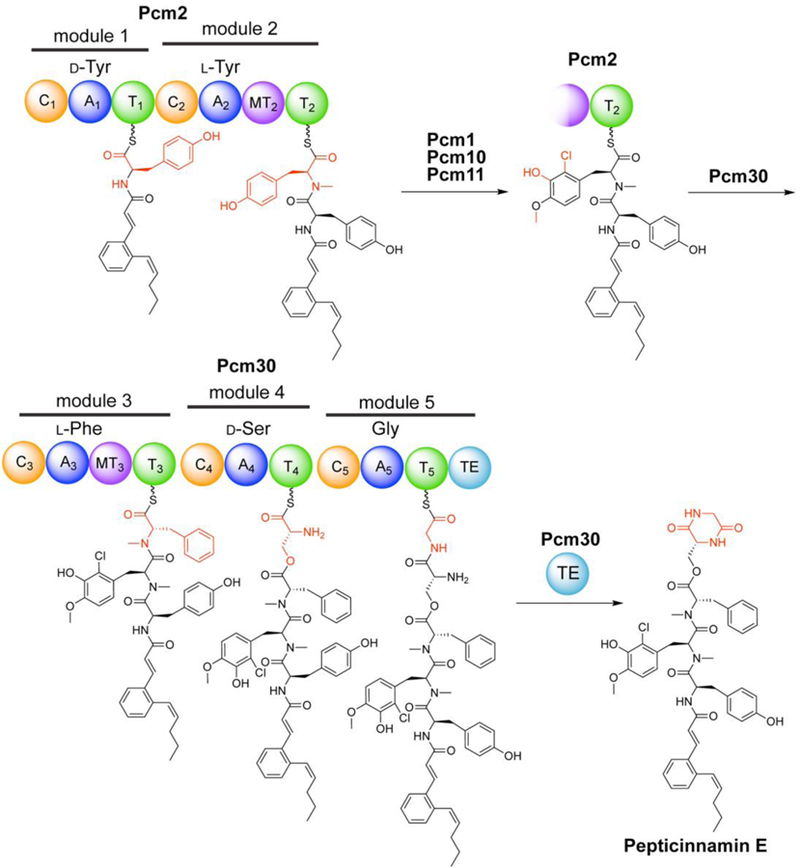
Proposed biosynthetic pathway of pepticinnamin E.
In summary, we have employed a bioinformatic approach to identify the biosynthetic gene cluster of pepticinnamin E and discover a new producing bacterium for this natural product. We also identified and characterized an O-methyltransferase enzyme in the biosynthesis of the rare nonproteinogenic amino acid 2-Cl-4-O-Me-l-DOPA and demonstrated that this enzyme is promiscuous toward a variety of catechol derivatives. Our work sets the stage for elucidating the biosynthetic chemistry of pepticinnamin E, especially the timing and mechanisms of biopterin-dependent hydroxylation, chlorination, and diketopiperazine formation.
Experimental Section
Cultivation of A. bacterium OK006 and extraction of pepticinnamin E:
Actinobacteria bacterium OK006 was cultivated on ISP Medium No. 4 (ISP4) plates (BD Difco) at 28 °C. Liquid cultures were grown in Tryptic Soy Broth (TSB; BD Difco) at 28 °C for 24 hours with aeration. For production of pepticinnamin E, a 1 mL sample of this TSB liquid culture was used to inoculate 200 mL of pepticinnamin production medium (20 g L−1 dextrose, 10 g L−1 asparagine, 0.2 g L−1 magnesium sulfate heptahydrate, 0.5 g L−1 potassium phosphate dibasic (anhydrous), 0.02 g L−1 iron(II) sulfate heptahydrate, and 5.84 g L−1 sodium chloride), and grown for 5 days at 28 °C with shaking at 250 RPM. To extract pepticinnamin E, cultures were centrifuged at 4,000 x g for 1 h to harvest bacterial cells. The cell pellets were resuspended in methanol and gently rocked for 2 h. Following centrifugation at 4,500 x g for 30 min, the methanol extract was filtered through 0.2 μm syringe filters and evaporated to dryness under reduced pressure. The resulting extracts were dissolved in methanol for analysis by liquid chromatography coupled high-resolution mass spectrometry (LC-HRMS). For LC-HRMS analysis, 10 μL of sample was analyzed using an Agilent Technologies 1260 Infinity II LC system coupled to an Agilent Technologies 6520 Accurate-Mass Quadruple-Time of Flight (Q-TOF). Samples were separated on a Gemini NX-C18 column (Phenomenex, 110 Å, 5 µm, 50 mm x 2 mm) at a flow rate of 0.4 mL min−1 over a linear gradient of 2 to 98% acetonitrile containing 0.1% formic acid over 20 minutes. Electrospray ionization mass spectrometry (ESI-MS) was conducted under positive ion mode with the following parameters: gas temperature 350 °C, drying gas 12 L min−1, nebulizer 50 lb in−2, fragmentor 175 V, skimmer 65 V, capillary cap 3500 V, octopole RF 750 V.
Reconstitution of Pcm10 methyltransferase activity and analysis by LC-HRMS:
The activity of Pcm10 was reconstituted as follows. Each 100 μL reaction contained 50 mM Tris, pH 8.3, 200 mM NaCl, 1 mM MgCl2, 1.6 mM SAM, and 1 mM substrate. Reactions were initiated by addition of 4 μL of Pcm10 to a final concentration of 9.2 μM, incubated for 1.5 h at 28 °C, 750 RPM, and quenched by the addition of 1 volume of 10% trichloroacetic acid to precipitate proteins in the assay. Reactions were stored at −20 °C prior to LC-HRMS analysis, at which point the precipitated proteins were removed by centrifugation at 21,700 x g for 5 min. The supernatant was diluted 1:2 in H2O and 10 μL of the diluted sample was analyzed using an Agilent Technologies 6520 Accurate-Mass Q-TOF LC-HRMS. Samples were separated on a Kinetex C18 column (Phenomenex, 100 Å, 5 µm, 150 mm x 4.6 mm) at a flow rate of 0.4 mL min−1 over a linear gradient of 5 to 95% acetonitrile containing 0.1% formic acid over 25 min. ESI-MS was conducted under positive ion mode with the following parameters: gas temperature 350 °C, drying gas 12 L min−1, nebulizer 50 lb in−2, fragmentor 175 V, skimmer 65 V, capillary cap 3500 V, octopole RF 750 V. Assays containing catechol as a substrate were analyzed using ESI-MS under negative ion mode and the following parameters: gas temperature 350 °C, drying gas 5 L min−1, nebulizer 20 lb in−2, fragmentor 175 V, skimmer 65 V, capillary cap 3500 V, octopole RF 750 V.
Analysis of the substrate scope of Pcm10 was performed under the same conditions using 1 mM of different substrates. After 3 h of incubation, the reactions were quenched with 10% trichloroacetic acid and stored at −20 °C. Prior to HPLC analysis, samples were centrifuged at 21,000 x g for 5 minutes to remove the protein precipitants and neutralized with a NaOH solution. A 20 μL sample was injected on a Kinetex C18 column (Phenomenex, 100Å, 5 µm, 150 mm x 4.6 mm) and analyzed by Shimadzu analytical HPLC using a gradient of 2.5% to 20% solvent B over 10 min for l-DOPA and l-Tyr, and a gradient of 2.5% to 50% solvent B over 10 min for catechol, caffeic acid, chlorogenic acid, and dopamine (solvent A: water containing 0.1% trifluoroacetic acid; solvent B: acetonitrile containing 0.1% trifluoroacetic acid). Substrate consumption and product formation in the reaction was calculated based on the integrated peak areas of UV absorbance at 280 nm at specific retention times.
Supplementary Material
Acknowledgements
The authors thank Dale A. Pelletier (Oak Ridge National Laboratory) for sharing the Actinobacteria bacterium OK006 strain. The authors also thank Katie Acken for assistance with culturing this strain. This work is supported by National Science Foundation (CHE1654678), National Institutes of Health (R00GM099904 and DP2HD094657), the Packard Fellowship for Science and Engineering, and the Rita Allen Foundation Scholars Award. K.S.M. acknowledges support from a Burroughs Wellcome Graduate Fellowship.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Suehiro M, Yakushigaku zasshi 1994, 29, 428–434. [PubMed] [Google Scholar]
- [2].Wieland T, Science 1968, 159, 946–952. [DOI] [PubMed] [Google Scholar]
- [3].Haňka L, Dietz A, Antimicrobial agents and chemotherapy 1973, 3, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Omura S, Van Der Pyl D, Inokoshi J, Takahashi Y, Takeshima H, The Journal of antibiotics 1993, 46, 222–228. [DOI] [PubMed] [Google Scholar]
- [5] a).Ohkanda J, Buckner FS, Lockman JW, Yokoyama K, Carrico D, Eastman R, de Luca-Fradley K, Davies W, Croft SL, Van Voorhis WC, Journal of medicinal chemistry 2004, 47, 432–445; [DOI] [PubMed] [Google Scholar]; b) SHN Moorthy N, Sousa SF, Ramos MJ, Fernandes PA, Current medicinal chemistry 2013, 20, 4888–4923; [DOI] [PubMed] [Google Scholar]; c) Wiesner J, Kettler K, Sakowski J, Ortmann R, Katzin AM, Kimura EA, Silber K, Klebe G, Jomaa H, Schlitzer M, Angewandte Chemie International Edition 2004, 43, 251–254. [DOI] [PubMed] [Google Scholar]
- [6].Shiomi K, Yang H, Inokoshi J, Van Der Pyl D, Nakagawa A, Takeshima H, Omura S, The Journal of antibiotics 1993, 46, 229–234. [DOI] [PubMed] [Google Scholar]
- [7].Hinterding K, Hagenbuch P, Rétey J, Waldmann H, Angewandte Chemie International Edition 1998, 37, 1236–1239. [DOI] [PubMed] [Google Scholar]
- [8] a).Thutewohl M, Waldmann H, Bioorganic & medicinal chemistry 2003, 11, 2591–2615; [DOI] [PubMed] [Google Scholar]; b) Thutewohl M, Kissau L, Popkirova B, Karaguni I-M, Nowak T, Bate M, Kuhlmann J, Müller O, Waldmann H, Bioorganic & medicinal chemistry 2003, 11, 2617–2626. [DOI] [PubMed] [Google Scholar]
- [9].Baltz RH, Journal of Industrial Microbiology and Biotechnology 2006, 33, 507–513. [DOI] [PubMed] [Google Scholar]
- [10] a).Toki S, Agatsuma T, Ochiai K, Saitoh Y, Ando K, Nakanishi S, Lokker NA, Giese NA, Matsuda Y, The Journal of antibiotics 2001, 54, 405–414; [DOI] [PubMed] [Google Scholar]; b) Schubert V, Di Meo F, Saaidi PL, Bartoschek S, Fiedler HP, Trouillas P, Süssmuth RD, Chemistry–A European Journal 2014, 20, 4948–4955; [DOI] [PubMed] [Google Scholar]; c) Hayashi K, Hashimoto M, Shigematsu N, Nishikawa M, Ezaki M, Yamashita M, Kiyoto S, Okuhara M, Kohsaka M, Imanaka H, The Journal of antibiotics 1992, 45, 1055–1063. [DOI] [PubMed] [Google Scholar]
- [11] a).Pohle S, Appelt C, Roux M, Fiedler H-P, ssmuth RD, Journal of the American Chemical Society 2011, 133, 6194–6205; [DOI] [PubMed] [Google Scholar]; b) Johnston CW, Skinnider MA, Wyatt MA, Li X, Ranieri MR, Yang L, Zechel DL, Ma B, Magarvey NA, Nature communications 2015, 6, 8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12] a).Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Müller R, Wohlleben W, Nucleic acids research 2015, 43, W237–W243; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, De Bruijn I, Chooi YH, Claesen J, Coates RC, Nature chemical biology 2015, 11, 625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Klingeman DM, Utturkar S, Lu T-YS, Schadt CW, Pelletier DA, Brown SD, Genome announcements 2015, 3, e01344–01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Röttig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O, Nucleic acids research 2011, 39, W362–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15] a).Uhlmann S, Süssmuth RD, Cryle MJ, ACS chemical biology 2013, 8, 2586–2596; [DOI] [PubMed] [Google Scholar]; b) Jiang W, Cacho RA, Chiou G, Garg NK, Tang Y, Walsh CT, Journal of the American Chemical Society 2013, 135, 4457–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16] a).Fitzpatrick PF, Biochemistry 2003, 42, 14083–14091; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang W, Ames BD, Walsh CT, Biochemistry 2011, 50, 5401–5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nelson JT, Lee J, Sims JW, Schmidt EW, Applied and environmental microbiology 2007, 73, 3575–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schmidt EW, Nelson JT, Fillmore JP, Tetrahedron letters 2004, 45, 3921–3924. [Google Scholar]
- [19].Seisser B, Zinkl R, Gruber K, Kaufmann F, Hafner A, Kroutil W, Advanced Synthesis & Catalysis 2010, 352, 731–736. [Google Scholar]
- [20].Siegrist J, Netzer J, Mordhorst S, Karst L, Gerhardt S, Einsle O, Richter M, Andexer JN, FEBS letters 2017, 591, 312–321. [DOI] [PubMed] [Google Scholar]
- [21].Hoffmann D, Hevel JM, Moore RE, Moore BS, Gene 2003, 311, 171–180. [DOI] [PubMed] [Google Scholar]
- [22] a).Lin S, Van Lanen SG, Shen B, Journal of the American Chemical Society 2007, 129, 12432–12438; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Puk O, Huber P, Bischoff D, Recktenwald J, Jung G, Süßmuth RD, van Pée K-H, Wohlleben W, Pelzer S, Chemistry & biology 2002, 9, 225–235; [DOI] [PubMed] [Google Scholar]; c) Schmartz PC, Zerbe K, Abou-Hadeed K, Robinson JA, Organic & biomolecular chemistry 2014, 12, 5574–5577; [DOI] [PubMed] [Google Scholar]; d) Weichold V, Milbredt D, van Pée KH, Angewandte Chemie International Edition 2016, 55, 6374–6389. [DOI] [PubMed] [Google Scholar]
- [23] a).Jiang W, Heemstra JR Jr, Forseth RR, Neumann CS, Manaviazar S, Schroeder FC, Hale KJ, Walsh CT, Biochemistry 2011, 50, 6063–6072; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Haslinger K, Peschke M, Brieke C, Maximowitsch E, Cryle MJ, Nature 2015, 521, 105–109; [DOI] [PubMed] [Google Scholar]; c) Kittilä T, Kittel C, Tailhades J, Butz D, Schoppet M, Büttner A, Goode RJ, Schittenhelm RB, van Pee K-H, Süssmuth RD, Chemical science 2017, 8, 5992–6004; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Patteson JB, Dunn ZD, Li B, Angewandte Chemie 2018, 130, 6896–6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lin S, Van Lanen SG, Shen B, Proceedings of the National Academy of Sciences 2009, 106, 4183–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT, Proceedings of the National Academy of Sciences 2007, 104, 16498–16503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cheng YQ, ChemBioChem 2006, 7, 471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ehling-Schulz M, Vukov N, Schulz A, Shaheen R, Andersson M, Märtlbauer E, Scherer S, Applied and Environmental Microbiology 2005, 71, 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.