

Abstract
Background:
Interleukin-6 (IL-6) can initiate intracellular signaling in skeletal muscle through binding to the IL-6-receptor and interaction with the transmembrane gp130 protein. Circulating IL-6 has established effects on skeletal muscle mass and metabolism in both physiological and pathological conditions. However, the effects of circulating IL-6 on skeletal muscle function are not well understood. The purpose of this study was to determine if chronically elevated systemic IL-6 was sufficient to disrupt skeletal muscle force, fatigue, and mitochondrial function. Additionally, we examined the role of muscle gp130 signaling during IL-6 over-expression.
Methods:
Systemic IL-6 overexpression for 2-weeks was achieved by electroporation of an IL-6 over-expression plasmid or empty vector into the quadriceps of either C57BL/6 (WT) or skeletal muscle gp130 knockout (KO) male mice. Tibialis anterior muscle in situ functional properties and mitochondrial respiration were determined.
Results:
IL-6 accelerated in situ skeletal muscle fatigue in the WT; a 18.5% reduction in force within 90s of repeated submaximal contractions and a 7% reduction in maximal tetanic force following 5 minutes. There was no difference between KO and KO+IL-6 fatigue. IL-6 reduced WT muscle mitochondrial respiratory control ratio (RCR) 36% and COX activity 42%. IL-6 had no effect on either KO RCR or COX activity. IL-6 also had no effect on body weight, muscle mass, or tetanic force in either genotype.
Conclusions:
These results provide evidence that 2 weeks of elevated systemic IL-6 is sufficient to increase skeletal muscle fatigability and decrease muscle mitochondrial content and function and these effects require muscle gp130 signaling.
Keywords: Muscle Fatigability, Mitochondrial Function, Inflammation
INTRODUCTION
Perceived fatigue and disrupted metabolic homeostasis occur with chronic disease and contribute to reduced life quality and poorer prognoses (Deans & Wigmore, 2005; Kuller et al., 2008; Sin & Man, 2003; Vidt, 2006). Fatigue remains the most frequently reported symptom in cancer patients (al-Majid & McCarthy, 2001; Chang, Hwang, Feuerman, & Kasimis, 2000; Kilgour RD, 2010; Laird et al., 2011; Monga et al., 1997; Stewart, Skipworth, & Fearon, 2006). The difficulty in determining if such fatigue has central, peripheral, or musculoskeletal origins has served as a significant barrier to understanding fatigue’s etiology. Skeletal muscle fatigability, or the ability to sustain force over time, relies on several physiological phenomena, most notably, adequate ATP production by mitochondrial respiration and glycolytic pathways and the accumulation of their metabolic biproducts (Fitts, 1994; Fitts & Holloszy, 1977; Holloszy & Booth, 1976; Munkvik, Lunde, & Sejersted, 2009; Rutherford, Manning, & Newton, 2016; Thompson, Balog, Riley, & Fitts, 1992).
While there is significant overlap between the regulation of skeletal muscle force production and fatigability, they can be modulated independent of one another. Skeletal muscle mitochondrial dysfunction and increased fatigability have been demonstrated to occur independent of changes to muscle size and strength in tumor-bearing mice suggesting that cancer-induced muscle weakness and fatigue have distinct etiologies (Brown et al., 2017; Ramage & Skipworth, 2018; VanderVeen, Hardee, Fix, & Carson, 2018). Classically, the contractile properties and fiber-type distribution has been associated with its fatigue properties; however, changes in mitochondrial respiration may regulate muscle function independent of changes in myosin heavy chain expression (Allen, Lamb, & Westerblad, 2008; Fitts, 1994; Westerblad & Allen, 2003). The heterogeneity of skeletal muscle allows for differential recruitment patterns with unique functional properties dependent on the relative abundance of each fiber type (Allen et al., 2008; Altenburg, Degens, van Mechelen, Sargeant, & de Haan, 2007; R. Close, 1964; R. I. Close, 1972; Fitts, 1994; Fitts & Holloszy, 1977; Westerblad & Allen, 2003). Higher percentages of type I or type IIa myosin ATPase isoform within a muscle has a slower, more oxidative, fatigue resistant phenotype, while higher percentage of type IIb/x myosin ATPase isoform results in a fast, more glycolytic, higher force production, fatigable phenotype (Fitts, 1994; Westerblad & Allen, 2003). While type I muscle fibers are susceptible to disuse atrophy (Coyle, Martin, Bloomfield, Lowry, & Holloszy, 1985; Iqbal, Ostojic, Singh, Joseph, & Hood, 2013; Thomason & Booth, 1990), chronic inflammatory diseases such as cancer-induced muscle wasting have been associated with preferential atrophy of type II muscle fibers (Carson, Hardee, & VanderVeen, 2016; Wang & Pessin, 2013). The tibialis anterior (TA), which has primarily type II muscle fibers, has been shown to have increased muscle fatigability and reduced muscle force in tumor-bearing mice (VanderVeen et al., 2018). We have previously shown that the TA is susceptible to inflammation induced muscle wasting (Hardee, Fix, et al., 2018); whether elevated inflammatory cytokines alone can disrupt the TA skeletal muscle function has not been determined.
While tumor necrosis factor α (TNF-α) has been eloquently shown to disrupt murine skeletal muscle contractile function in vivo and ex vivo (Gilliam, Moylan, Ferreira, & Reid, 2011; Hardin et al., 2008; Li et al., 2000; Reid, Lannergren, & Westerblad, 2002; Reid & Moylan, 2011), the capacity for other inflammatory cytokines to disrupt skeletal muscle function has not been well described. The pleiotropic inflammatory cytokine interleukin-6 (IL-6) has been shown to be both pro- and anti-inflammatory and regulates several immune functions as well as whole-body metabolism during physiological and pathological conditions (Bonetto et al., 2011; Carson & Baltgalvis, 2010; Gao et al., 2017; Hardee, Counts, et al., 2018; Puppa, Gao, Narsale, & Carson, 2014). IL-6 promotes intracellular signaling through binding to its receptor (IL-6r) which allows for its β subunit (glycoprotein 130; gp130) to homodimerize and activate downstream intracellular signaling (Garbers, Aparicio-Siegmund, & Rose-John, 2015; Rose-John, 2018; Schwantner, Dingley, Ozbek, Rose-John, & Grotzinger, 2004). The anti-inflammatory nature of IL-6 contributes to the acute phase response and skeletal muscle’s exercise adaptations, though chronically elevated IL-6 accelerates inflammation and contributes to several disease comorbidities including skeletal muscle wasting (Carson & Baltgalvis, 2010; Garbers et al., 2015; Spate & Schulze, 2004). While our understanding of IL-6 in exercise and disease has improved, whether the detrimental effects of chronically elevated circulating IL-6 directly affect skeletal muscle through gp130-dependent mechanisms is currently unknown.
Systemic IL-6 overexpression can accelerate muscle mass loss in tumor-bearing mice, but the effects of IL-6 on skeletal muscle mass and function in tumor-free mice remains inconclusive (Puppa et al., 2012; White et al., 2011; White et al., 2012). Mitochondrial function has emerged as an intriguing regulator of inflammation-induced skeletal muscle metabolic dysfunction in both humans (Marzetti et al., 2017; Merlini, Bonaldo, & Marzetti, 2015) and murine models of cancer-induced wasting (Boland, Chourasia, & Macleod, 2013; Brown et al., 2017; Carson et al., 2016; Hardee et al., 2016). We have previously shown that 2 weeks of elevated circulating IL-6 decreased murine skeletal muscle mitochondrial protein expression and loss of muscle gp130 signaling increased mitochondrial fission and reduced mitochondrial fusion under basal conditions (Fix et al., 2018; Puppa et al., 2014; Puppa et al., 2012). Additionally, skeletal muscle fatigability has been demonstrated to occur synergistically with elevated muscle IL-6 production and increased plasma IL-6 levels immediately following exercise in active adult men (Febbraio, Hiscock, Sacchetti, Fischer, & Pedersen, 2004; Febbraio & Pedersen, 2002); however, a causal role for IL-6 to accelerate skeletal muscle fatigue has not been investigated. Furthermore, we have previously shown that gp130 expression is greater in the TA compared to the soleus and gastrocnemius suggesting greater IL-6 sensitivity in faster muscles (Puppa et al., 2014). Interestingly, skeletal muscle weakness in tumor-bearing mice was associated with increased skeletal muscle STAT3 and SOCS3, two primary downstream targets of IL-6 (VanderVeen et al., 2018). The purpose of the current study was to determine if chronically elevated systemic IL-6 for two weeks was sufficient to disrupt skeletal muscle force, fatigue, and mitochondrial function in the TA. Additionally, we examined if the muscle gp130 receptor mediates the effects of IL-6 on skeletal muscle. We hypothesized that chronically elevated circulating IL-6 can disrupt muscle mitochondrial content and function and induce muscle weakness and fatigue through the activation of muscle gp130 signaling.
METHODS
Ethical Approval
All animal experiments were approved by the University of South Carolina’s Institutional Animal Care and Use Committee (reference no. 101081) and complied with the principles and standards for reporting animal experiments outlined for Experimental Physiology (Grundy, 2015).
Animals
Male C57BL/6 mice were originally purchased from Jackson Laboratories and were bred at the University of South Carolina’s Animal Resources Facility. Male mice on a C57BL/6 background were bred with gp130fl/fl mice as previously described (Fix et al., 2018; Hardee, Fix, et al., 2018; Puppa et al., 2014). Gp130fl/fl (WT) male mice were bred with Cre-expressing mice driven by myosin light chain (MLC), resulting in a skeletal muscle floxed gp130 heterozygote cre knockout mouse with a skeletal muscle specific deletion of gp130 (KO). A total of 52 mice were divided into 4 groups, WT (n=14), WT+IL-6 (n=15), KO (n=12), and KO+IL-6 (n=11) for this study. All animals were group housed (<5 per cage) and kept on a 12:12-h light-dark cycle. All animals were fasted 5 hours prior to tissue collection. Mice were anesthetized with a ketamine-xylazine-acepromazine cocktail, and hindlimb muscles and select organs were carefully dissected and snap frozen in liquid nitrogen and stored at −80°C until further analysis.
IL-6 Overexpression
In vivo intramuscular electroporation of an IL-6 plasmid was performed to increase circulating IL-6 levels in mice as previously described (Hardee, Counts, et al., 2018; Hardee, Fix, et al., 2018; White et al., 2012). Both WT and KO mice were randomized into Vector or IL-6 treatment groups; however, all mice were subjected to in vivo electroporation of the quadriceps muscle. Briefly, mice were electroporated with 50μg of the IL-6 plasmid driven by the CMV promoter, or empty control vector, into the quadriceps muscle. To accomplish this, mice were anaesthetized with a 2% mixture of isoflurane and oxygen (1 l/min), the leg was shaved, and a small incision was made over the quadriceps muscle. Fat was dissected away from the muscle, and the plasmid was injected in a 50-μl volume of phosphate-buffered saline (PBS). A series of eight 50-ms, 100-V pulses were used to promote uptake of the plasmid into myofibers, and then the incision was closed with a wound clip. Both vector control and IL-6 groups received the appropriate plasmid starting at 11 wk of age, and a second electroporation on the opposite leg was performed at 12 wk of age to maintain systemically elevated plasma IL-6 levels. The tibialis anterior (TA) and extensor digitorum longus (EDL) muscles used in the study were not subjected to electroporation. Mice were euthanized 2 weeks after the initial plasmid electroporation. A dose of 50 μg has been shown to increase circulating IL-6 levels similar to what’s been observed with tumor-bearing mice (Baltgalvis et al., 2008; Hardee, Counts, et al., 2018; Puppa et al., 2012; White et al., 2012). Additionally, two weeks of elevated IL-6 was sufficient to accelerate cachexia in tumor-bearing mice and disrupt muscle metabolism in tumor-free mice (Puppa et al., 2012).
Plasma IL-6
Plasma IL-6 was quantified as previously published (Hardee, Fix, et al., 2018). Briefly, blood samples were centrifuged at 10,000 g for 10 min at 4°C. Plasma was collected and stored at −80°C until analysis. A commercially available IL-6 enzyme-linked immunosorbent assay kit was obtained from BD Biosciences (San Diego, CA). Briefly, a Costar clear 96-well plate (Corning, NY) was coated with IL-6 capture antibody and allowed to incubate overnight. The plate was then blocked with assay diluent buffer and IL-6 standards and plasma samples were added to the plate. The wells were then incubated with streptavidinhorseradish peroxidase reagent. After several washes, 3,3,5,5-tetramethylbenzidine substrate was added, and the reaction was developed for 20 min. The reaction was stopped with sulfuric acid, and absorbance was measured. Samples that fell below the curve not reported; however, mice were given a plasma IL-6 value of 7.8 pg/mL (low detection limit) for statistical analysis. All of the mice given the IL-6 plasmid fell within the detection limit (7.8–1000 pg/mL).
Analysis of Muscle Function
At ~13 weeks of age, mice were anesthetized with 2% isoflurane inhalation and anesthesia was maintained at 1.5 % isoflurane throughout the duration of the procedure (~1 hour). The mouse was place on the apparatus maintained at 37°C throughout the entirety of the procedure (Bonetto, Andersson, & Waning, 2015). Functional analysis of the TA muscle in situ, which maintains the host nerve and blood supply, has been previously described (VanderVeen et al., 2018). The sciatic nerve was exposed proximal to the knee and maintained using warmed mineral oil. The sciatic nerve was then subjected to a single stimulus to determine the optimal length (Lo). Once Lo was obtained, a force-frequency curve was generated, and maximal tetanic force was determined. After a 5-minute rest following the force-frequency response protocol, the TA was subjected to an intermittent fatigue protocol consisting of 0.5 second submaximal stimulation (50Hz) every second for 5 minutes for a total of 300 submaximal contractions. Immediately after the submaximal contractions, the TA was subjected to a maximal stimulation (200Hz) to elicit a maximal contraction. Fatigue was measure by the % reduction in maximal force following the 5-minute contraction protocol (Allen et al., 2008; Fitts, 1994). Disrupted fatigue properties were assessed as change in force throughout the force-time tracing (Crilly, Tryon, Erlich, & Hood, 2016).
Immunohistochemistry for myosin heavy chain IIA, IIX, and IIB
Immunohistochemistry for myosin heavy chain (MHC) type IIa, IIx, and IIb was performed as previously described (Goodman, Kotecki, Jacobs, & Hornberger, 2012). Transverse muscle sections (8μm) of the TA were blocked in 10% IgG Fab (Jackson Immunology) in PBS (5% BSA + 5% Triton X100) for 1 h at room temperature and then incubated overnight at 4°C with primary antibodies [mouse IgG1 monoclonal anti-type IIa MHC (clone SC-71; 1:100) and mouse IgM monoclonal anti-type IIb MHC (clone BF-F3, 1:10). All MHC antibodies were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Secondary antibodies (biotinylated anti-mouse IgG FITC, IgM AMCA; Thermo Fischer) were incubated with the sections for 1 hour at RT. Slides were air dried and covered in glycerol mounting medium containing DABCO and coverslipped. At least 8 random, non-overlapping digital images at 20X magnification were taken, and fibers stained positive or absent for MHC type IIa, IIx, and IIb were tabulated using imaging software (ImageJ; NIH). The analyses were performed by an investigator blinded to the treatment groups.
Cytochrome C Oxidase Activity
Cytochrome C Oxidase Activity was measured to determine changes in mitochondrial content across all groups. Due to tissue availability, the Extensor Digitorum Longus (EDL) muscle, a highly glycolytic muscle, was used. The whole EDL was homogenized in extraction buffer (0.1 M KH2P04/Na2HP04, 2 mM EDTA, pH 7.2) and cytochrome-c oxidase (COX) activity was determined by measuring the rate of oxidation of fully reduced cytochrome c at 550nm using Sigma Aldrich Kit (#CYTOCOX1) and spectrophotometer (Eppendorf) as previously described (VanderVeen et al., 2018).
Respiratory Control Ratio
A randomly selected cohort of 4 mice from each treatment group were used for analysis of mitochondrial function. Mitochondrial respiration was measured polarographically in a respiration chamber (Hansatech Instruments) maintained at 37°C as previously described (Hughey, Hittel, Johnsen, & Shearer, 2011; Kwon et al., 2015; Perry, Kane, Lanza, & Neufer, 2013). A 7–10 mg piece of TA muscle was mechanically tweezed with forceps under a dissecting microscope in ice-cold buffer X (60 mM K-MES, 35 mM KCl, 7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 0.5 mM DTT, 20 mM taurine, 5.7 mM ATP, 15 mM phosphocreatine, and 6.56 mM MgCl2, pH 7.1). The fiber bundle was then incubated in 50uM saponin for 30 minutes and washed 3 times for 5 minutes in respiration buffer (105mM K-MES, 3mM KCl, 1mM EGTA, 10mM K2HPO4, 5mM MgCl2, 0.005mM Glutamate, 0.002mM Malate, 0.05% BSA, 20mM Creatine, pH 7.1). Fiber bundles were then placed into the oxygraph machine in 20mM creatine respiration buffer at 37 degrees and provided with 5mM of pyruvate and 2mM of malate to measure complex I mediated mitochondrial respiration (Saks et al., 1998; Walsh et al., 2001). Two minutes following pyruvate and malate, 0.25mM of ADP was injected into the chamber to induce STATE 3 respiration for a duration of 5 minutes. 10ug/mL of Oligomycin was then injected to induce steady state 4 respiration for a duration of 10 minutes. Respiratory Control Ratio (RCR) was calculated by dividing state 3 by state 4 respirations.
Western blot analysis
Western blot analysis was performed as previously described (Fix et al., 2018). Briefly, the proximal portion of the TA muscle was homogenized, and protein concentration was determined using the Bradford standard curve method. Homogenates were fractionated on SDS-polyacrylamide gels and transferred to PVDF membrane. After the membranes were blocked, antibodies Total OXPHOS Cocktail (Abcam, Cambridge, United Kingdom), were incubated at dilutions of 1:5000 overnight at 4°C in 1% TBST milk. Anti-rabbit IgG-conjugated secondary antibodies (Cell Signaling Technology) were incubated with the membranes at 1:5000 dilutions for 1 h in 1% TBST milk.
Enhanced chemiluminescence developed by autoradiography was used to visualize the antibody–antigen interactions. Blots were analyzed by measuring the integrated optical density (IOD) of each band with ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
Values are presented as means plus / minus standard deviation (SD). A Bartlett’s test was used to determine significantly different standard deviations (p<0.05). First, to understand the effects of IL-6, a pre-planned t-test was performed between WT and WT+IL-6. Second, to understand if IL-6 directly affects skeletal muscle, a Two-Way ANOVA was performed. If an interaction was observed a Tukey’s multiple comparisons test was performed. For analysis of circulating IL-6 levels and skeletal muscle fatigue properties, a repeated measures Two-Way ANOVA was performed. A power analysis was performed to determine the sample size needed to observe statistical significance using a two-way ANOVA (G*Power, Dusseldorf, Germany). Power (1-β) was set to 0.8 and error of probability (α) was set at 0.05. Based on previously published results and our preliminary data, to achieve significance for an estimated 15 ± 5 % difference in skeletal muscle fatigability between groups with an effect size of 2, a sample size of 13 is needed for each group. Based on our preliminary results, to achieve a significant interaction in skeletal muscle respiration between groups with an effect size of 3.6, a sample size of 4 is needed for each group. A Pearson’s correlation was run to determine a relationship between several properties of skeletal muscle function and key markers of muscle metabolic health. Significance was set at p<0.05.
RESULTS
The effect of IL-6 on body weight and muscle mass
The electroporation of the IL-6 plasmid into the quadriceps muscle was sufficient to increase circulating IL-6 to the level previously reported in tumor-bearing mice (Carson & Baltgalvis, 2010), but remained below the levels observed with infection (Copeland et al., 2005). Following electroporation treatment, IL-6 was increased in WT+IL-6 mice at one-week (range 7.8–154.5 pg/mL) and two-weeks (range 15.6–371.0 pg/mL) compared to WT vector controls (below the detection limit, 7.8 pg/mL; Table 1). Skeletal muscle specific gp130 knockout (KO) mice had increased circulating IL-6 levels at one-week (range 63.1–218.0 pg/mL) and two-weeks (KO+IL-6 range 99.1–204.5 pg/mL) when compared to KO vector controls. Circulating IL-6 was affected by skeletal muscle gp130 loss. There was an interaction for KO-IL-6 to have increased circulating IL-6 by 55.5 % and 88.0% compared to WT+IL-6 at one and two-weeks, respectively (Table 1).
Table 1.
Animal Characteristics
| WT |
KO |
|||
|---|---|---|---|---|
| Control | IL-6 | Control | IL-6 | |
| n | 14 | 15 | 12 | 11 |
| Body Weights (g) | ||||
| Pre | 23.3 ± 1.5 | 23.4 ± 1.9 | 24.7 ± 2.0 | 24.0 ± 1.7 |
| Mid | 24.0 ± 1.5 | 23.4 ± 1.4 | 24.9 ± 2.0 | 24.1 ± 1.3 |
| Post | 24.9 ± 2.0 | 24.0 ± 2.1 | 25.1 ± 1.3 | 24.3 ± 1.7 |
| Plasma IL-6 (pg/mL) | ||||
| Pre | - | - | - | - |
| Mid | - | 63 ± 52* | - | 98 ± 55& |
| Post | - | 75 ± 43* | - | 141 ± 31& |
| Tissues at Sacrifice | ||||
| Hindlimb (mg) | 207 ± 14 | 200 ± 14 | 214 ± 11@ | 206 ± 14@ |
| Hindlimb/Tibia (mg/mm) |
12.2 ± 0.7 | 12.0 ± 0.7 | 12.7 ± 0.6 | 12.3 ± 0.8 |
| Tibia Length (mm) | 17.0 ± 0.3 | 16.8 ± 0.2# | 16.8 ± 0.2 | 16.7 ± 0.2# |
| Epididymal Fat (mg) | 362 ± 139 | 313 ± 96 | 308 ± 52 | 290 ± 85 |
| Spleen (mg) | 71 ± 12 | 122 ± 26# | 93 ± 68 | 199 ± 92# |
Values are means ± SD. Male C57BL/6 (WT) mice. Male skeletal muscle specific gp130 knockout (KO) mice. Age in weeks (wks). Body weight in grams (g). Plasma interleukin-6 (IL-6) in picograms/milliliters (pg/mL). All vector controls and pre-IL-6 levels fell below the detection limit and therefore weren’t reported. Combined weight of the soleus, plantaris, gastrocnemius, and tibialis anterior (hindlimb) in milligrams (mg). Tibia length in millimeters (mm). Epididymal fat pad weight in milligrams. Spleen weight in milligrams. Significance set at p<0.05 (Two-Way ANOVA).
Significant main effect of IL-6.
Significant main effect of KO. & Significantly different from all groups.
WT-IL-6 significant interaction from all groups.
KO-IL-6 significant interaction from all groups.
Body weight and muscle weight were not affected by elevated IL-6; however, muscle weight was increased in the KO (Table 1). There was a main effect of KO to increase hindlimb mass 3% regardless of IL-6 treatment; however, this difference was lost when hindlimb mass (mg) was normalized to body weight (g) (WT – 8.3 ± 0.1 mg/g, KO – 8.5 ± 0.1 mg/g; p=0.98). High circulating IL-6 has been previously shown to impair bone growth and development in mice (De Benedetti et al., 2006). Tibia length was affected by IL-6; there was a main effect of IL-6 to reduce tibia length 1% regardless of genotype. Hindlimb mass corrected for tibia length was not different across all groups. Spleen weight was affected by IL-6. There was a main effect of IL-6 in increase spleen weight by 91% regardless of genotype.
The effect of IL-6 on muscle mitochondrial content and function
Mitochondrial function was assessed in a subset of mice from each treatment group with a range of plasma IL-6 between 12.0 – 289.9 pg/mL (n=4 per group). Elevated IL-6 levels disrupted complex 1 mediated mitochondrial respiration in the TA muscle (Figure 1). State 3 oxygen consumption was reduced by IL-6; however, muscle gp130 loss rescued the IL-6 suppression of state 3 respiration (Figure 1A). There was an interaction for WT+IL-6 to have State 3 respiration reduced by 21.5%, 17.1%, and 16.1% compared to WT, KO, and KO+IL-6, respectively. State 4 respiration was not affected by IL-6 treatment or genotype (Figure 1B). The IL-6 suppression of State 3 respiration resulted in a reduction in muscle respiratory control ratio (State 3/State 4, RCR; Figure 1C). There was an interaction for WT+IL-6 to have RCR respiration reduced by 36.0%, 30.5%, and 36.7% compared to WT, KO, and KO+IL-6, respectively.
Figure 1.
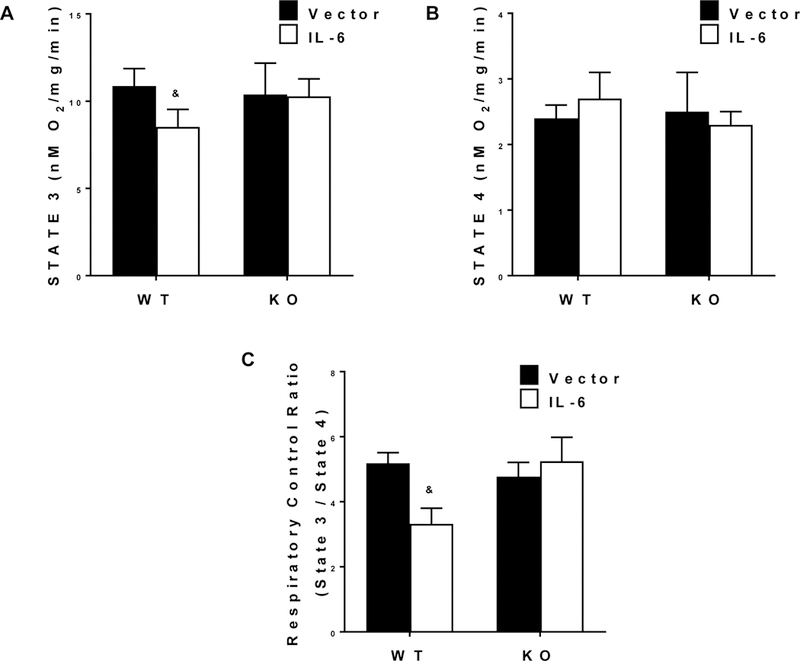
The effect of IL-6 on skeletal muscle mitochondrial function. Tibialis anterior (TA) respiratory control ratio (n=4 per group). Values are means plus / minus standard deviation (SD). A) 0.25mM of ADP was injected into the chamber to induce STATE 3 respiration for a duration of 5 minutes. B) 10ug/mL of Oligomycin was injected to induce steady STATE 4 respiration for a duration of 10 minutes. C) Respiratory Control Ratio (RCR) was calculated by dividing STATE 3 by STATE 4 respirations. & Significantly different from all groups. Significance was set at p<0.05. Two-Way ANOVA.
Mitochondrial content, measured by COX activity was reduced by IL-6 in the EDL muscle, and the IL-6 suppression of COX activity was rescued by muscle gp130 loss (Figure 2A). There was an interaction for WT+IL-6 to have reduced COX activity (n=8 for each group) by 42.2%, 43.4%, and 38.7% compared to WT, KO, and KO+IL-6, respectively. The expression of TA muscle mitochondrial complex proteins was reduced by IL-6 (Figure 2C). There were significant interactions found between the IL-6 treatment and mouse genotype for the expression of mitochondrial complexes I, II, III and IV. Complex I expression was reduced by 27.2% and 29.7% in WT+IL-6 compared to WT and KO+IL-6, respectively. Complex II expression was reduced 49.6%, 44%, and 49.8% in WT+IL-6 compared to WT, KO, and KO+IL-6, respectively. Complex III expression was reduced by 26.8% in WT+IL-6 compared to KO+IL-6. Complex IV was reduced by 39.4% in WT+IL-6 compared to WT. Complex V was not affected by either the IL-6 treatment or genotype.
Figure 2.
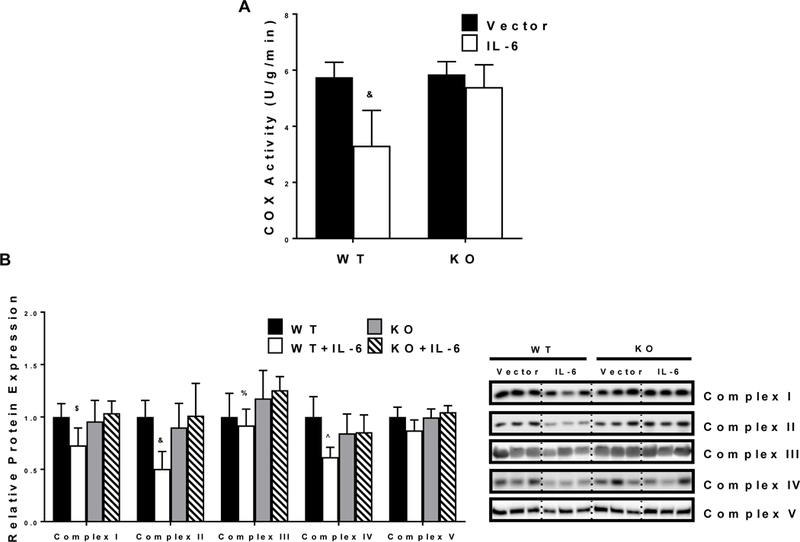
The effect of IL-6 on muscle mitochondrial content. A) Extensor digitorum longus (EDL) COX activity (WT n=14, WT+IL-6 n=15, KO n=12, KO+IL-6 n=11). B) Relative mitochondrial protein expression of Complexes 1–5 in the TA (n=6 per group). Representative western blot images are shown to the right of the graph. Values are means plus / minus standard deviation (SD). C57BL/6 (WT). Skeletal muscle gp130 knockout (KO). Interleukin-6 (IL-6). $Significantly different from WT and KO + IL-6. &Significantly different from all groups. %Significantly different from KO + IL-6. ^Significantly different from WT. Significance was set at p<0.05. Two-Way ANOVA.
The effect of IL-6 on skeletal muscle force production
Absolute muscle force was not affected by either the IL-6 treatment or muscle gp130 loss. There was no effect of IL-6 on muscle force production across the range of frequencies (10–200 Hz) tested in WT controls (Figure 3A). IL-6 reduced absolute force at 80hz by 13.1% in the KO (Figure 3B). Maximal tetanic force (Po; Table 2) was not altered by either the IL-6 treatment or muscle gp130 loss. Analysis of specific tension (sPo) showed no effect of IL-6 on force production across a range of frequencies (10–200 Hz) in either the WT (Figure 3C) or KO (Figure 3D). Specific tetanic force (sPo) was reduced by muscle gp130 loss (Table 2). There was a main effect of KO to reduce sPo by 8.7% regardless of IL-6 treatment. Muscle twitch characteristics were not affected by IL-6 (Table 2); however, there was a main effect of KO to have 1/2 RT reduced by 13.4% and TPT reduced by 5.6%. Additionally, there was a main effect of KO to have +dP/dt increased by 10.9% (Table 2). Fiber-type frequency (i.e. type IIa, type IIb or type IIx) was not affected by IL-6 or muscle gp130 loss (Table 3).
Figure 3.
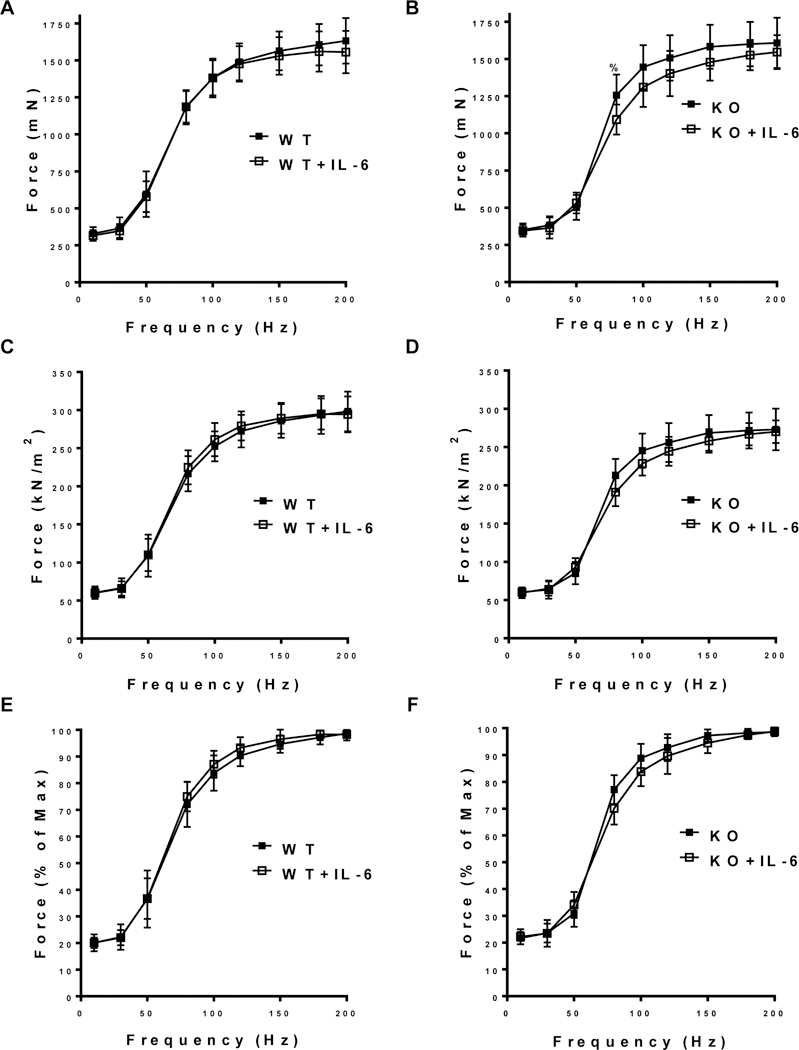
The effect of IL-6 on skeletal muscle force production. A) Absolute force-frequency curve of the TA in WT and WT+IL-6 in millinewtons (mN). B) Absolute force-frequency curve of the TA in KO and KO+IL-6 in mN. C) Specific force-frequency curve of the TA in WT and WT+IL-6 in kilonewtons/meters2 (kN/m2). D) Specific force-frequency curve of the TA in KO and KO+IL-6 in kN/m2. E) Relative force-frequency curve of the TA in WT and WT+IL-6 (% of maximal force). B) Relative force-frequency curve of the TA in KO and KO+IL-6 (% of maximal force). WT n=14, WT+IL-6 n=15, KO n=12, KO+IL-6 n=11. Values are means plus / minus standard deviation (SD). C57BL/6 (WT). Skeletal muscle gp130 knockout (KO). Interleukin-6 (IL-6). %Significantly different from KO + IL-6. Significance was set at p<0.05. Pre-planned t-test.
Table 2.
Contractile Properties
| WT |
KO |
|||
|---|---|---|---|---|
| Control | IL-6 | Control | IL-6 | |
| TA Weight (mg) | 48.4 ± 3.1 | 46.0 ± 3.4 | 49.8 ± 3.2@ | 48.8 ± 3.6@ |
| Twitch Properties | ||||
| 1/2 RT | 8.7 ± 1.6 | 9.0 ± 1.5 | 7.7 ± 1.2@ | 7.8 ± 1.7@ |
| TPT | 15.7 ± 1.4 | 16.2 ± 1.4 | 14.8 ± 0.8@ | 15.1 ± 1.1@ |
| Force Production | ||||
| Po | 1,655 ± 155 | 1,585 ± 136 | 1,628 ± 152 | 1,563 ± 100 |
| sPo | 302 ± 25 | 300 ± 22 | 276 ± 24@ | 273 ± 14@ |
| Contraction Rates | ||||
| + dP/dt | 23,233 ± 1675 | 22,222 ± 3421 | 25,652 ± 2824@ | 24,709 ± 2020@ |
| − dP/dt | 20,221 ± 3222 | 18,664 ± 3253 | 19,069 ± 3437 | 20,045 ± 1740 |
Values are means ± SD. Male C57BL/6 (WT n=14, WT+IL-6 n=15) mice. Male skeletal muscle specific gp130 knockout (KO n=12, KO+IL-6 n=11) mice. Tibialis anterior (TA) weight in milligrams (mg). 1/2 Relaxation time (RT) in milliseconds. Time to peak twitch (TPT) in milliseconds. Maximal tetanic force (Po) in millinewtons. Specific tetanic force (sPo) in kilonewtons/meter2. Rate of contraction at 200 Hz (+dP/dt). Rate of Relaxation at 200 Hz (-dP/dt). Significance set at p<0.05 (Two-Way ANOVA).
Significant main effect of KO.
Table 3.
Fiber-type Distribution
| WT |
KO |
|||
|---|---|---|---|---|
| Control | IL-6 | Control | IL-6 | |
| Type IIa | 10.9 ± 4.7 | 11.2 ± 6.8 | 9.5 ± 6.5 | 6.8 ± 3.1 |
| Type IIb | 38.4 ± 7.1 | 40.6 ± 4.3 | 38.9 ± 7.7 | 35.4 ± 9.2 |
| Type IIx | 50.7 ± 8.5 | 48.2 ± 5.4 | 51.8 ± 13.4 | 58.0 ± 11.3 |
Values are % means ± SD. C57BL/6 (WT). Skeletal muscle specific gp130 knockout (KO). n=5 for each group
The effect of IL-6 on skeletal muscle fatigability
Skeletal muscle fatigue was increased by the IL-6 treatment. Following 5 minutes of submaximal contraction maximal tetanic force was reduced 7% in WT+IL-6 compared to WT (pre-planned t-test; Figure 4C). Skeletal muscle fatigability was increased by the IL-6 treatment, and muscle gp130 loss rescued the IL-6 induction of fatigue (Figure 4D). There was an interaction for WT+IL-6 to have reduced relative force throughout 55–90 seconds of the contraction period compared to WT and KO+IL-6 (Figure 4 A, B). There was also an interaction for WT+IL-6 to have relative force reduced by 18.5% and 18.0% at 90 seconds of contraction compared to WT and KO+IL-6, respectively (Figure 4D).
Figure 4.
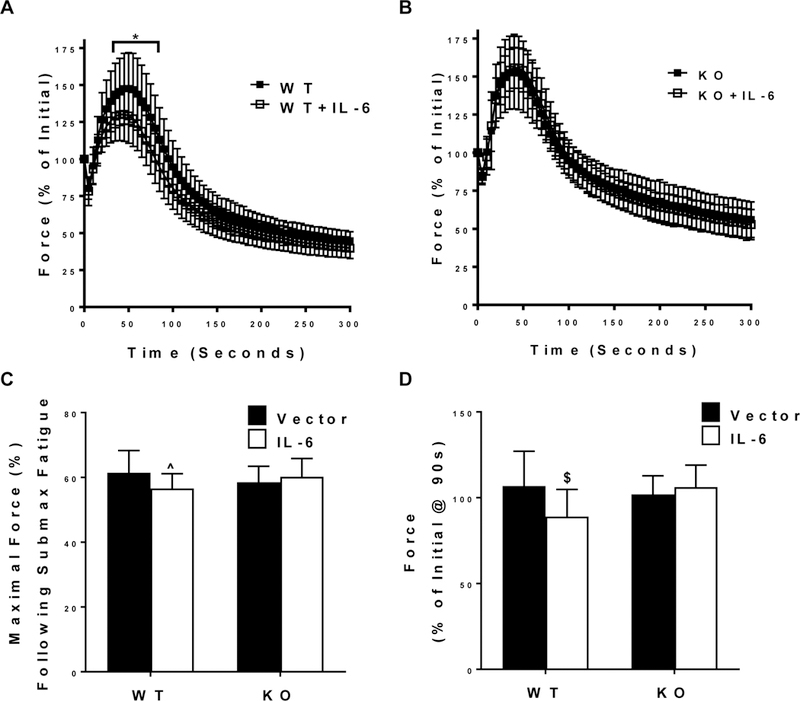
The effect of IL-6 on submaximal contraction-induced skeletal muscle fatigability. TA muscle’s fatigability during a 5-minute submaximal (50 Hz) fatigue test. A) Relative (% of Initial) force-time tracing of the TA muscle’s fatigability during a 5-minute submaximal (50 Hz) fatigue test from WT and WT + IL-6 mice. B) Relative (% of Initial) force-time tracing of the TA muscle’s fatigability during a 5-minute submaximal (50 Hz) fatigue test from KO and KO + IL-6 mice. C) Relative (% of maximal tetanic force) force after the 5-minute submaximal contraction-induced fatigue test in all mice. D) Relative (% of Initial) force after 90 seconds of a submaximal (50 Hz) fatigue test in all mice. WT n=14, WT+IL-6 n=15, KO n=12, KO+IL-6 n=11. Values are means plus / minus standard deviation (SD). C57BL/6 (WT). Skeletal muscle specific gp130 knockout (KO). $Significantly different from WT and KO+IL-6. Significance was set at p<0.05.
DISCUSSION
IL-6 has an established role in exercise- and disease-induced changes to systemic and skeletal muscle metabolism (Febbraio et al., 2004; Febbraio & Pedersen, 2002; Lightfoot & Cooper, 2016; Wolf, Rose-John, & Garbers, 2014). Although elevating circulating IL-6 for two weeks through overexpression of an IL-6 plasmid has been shown to accelerate cachexia in tumor-bearing mice and disrupt muscle metabolism in tumor-free mice (Carson & Baltgalvis, 2010; Puppa et al., 2012; White et al., 2012), whether IL-6 disrupts skeletal muscle mitochondrial function and muscle fatigability has not been measured. The current study investigated if chronically elevated systemic IL-6 was sufficient to disrupt skeletal muscle force, fatigue, and mitochondrial content and function in a muscle with primarily type II fibers directly through muscle skeletal muscle gp130 signaling. Our results demonstrate that 2 weeks of elevated circulating IL-6 accelerated submaximal contraction-induced skeletal muscle fatigue of the TA muscle. Conversely, IL-6 was unable to induced submaximal fatigue in skeletal muscle specific gp130 KO mice suggesting that IL-6-induced fatigue occurred through direct skeletal muscle gp130 activation. Additionally, our results demonstrate that 2 weeks of elevated IL-6 was sufficient to disrupt skeletal muscle mitochondrial function and decrease mitochondrial content through gp130-dependent signaling, mirroring the submaximal fatigue findings.
While chronic inflammation is a nonspecific immune response, cytokines are inflammatory mediators that have been implicated in the progression of several diseases and associated comorbidities (Deans & Wigmore, 2005; Fearon, Glass, & Guttridge, 2012; Lightfoot & Cooper, 2016; Zhou, Liu, Liang, Li, & Song, 2016). In addition to tumor growth and development, elevated circulating IL-6 has been linked to disrupted protein turnover associated with cancer-induced muscle wasting (Carson & Baltgalvis, 2010; Cron, Allen, & Febbraio, 2016; Narsale & Carson, 2014). IL-6 has been shown to regulate skeletal muscle metabolism through the activation of GLUT4 translocation and accelerated lipolysis (Cron et al., 2016). We have previously reported that IL-6 can reduce mitochondrial content in both tumor-free and tumor-bearing mice (Puppa et al., 2012). The current study extends these findings demonstrating that elevated circulating IL-6 accelerates submaximal contraction-induced skeletal muscle fatigue in a muscle with predominately type II fibers. A potential mechanism for IL-6 regulation of fatigue is through the disruption of skeletal muscle metabolic homeostasis. Chronic activation of muscle AMPK and suppressed mitochondrial biogenesis can occur during cancer-induced muscle wasting (White et al., 2013; White et al., 2012). Here we report that elevated circulating IL-6 is sufficient to reduce TA muscle RCR, which is a strong indicator of mitochondrial function (Hughey et al., 2011; Pettersen et al., 2017; Saks et al., 1998). Additionally, we found that IL-6 reduced COX activity in the EDL suggesting a reduction in mitochondrial content in a muscle with predominately type II fibers. A recent study reported that weight stable and cachectic (>5% body weight loss) cancer patients with elevated IL-6, had reduced nuclear factor erythroid 2-related factor 2 (Nrf2) expression associated with reduced antioxidant production (Brzeszczynska et al., 2016). Furthermore, loss of Nrf2 alone was sufficient to reduce gastrocnemius force production during a submaximal contraction-induced fatigue protocol (Crilly et al., 2016).
Signaling through the skeletal muscle gp130 receptor has been an active area of investigation in disease-induced muscle wasting (Mihara M, 2012; Miller et al., 2016; Puppa et al., 2014). In this regard, constitutively activated STAT3, a direct downstream gp130 target, can inhibit myotube growth in vitro and induce muscle mass loss in vivo (Bonetto A, 2012). In addition, increased STAT3 signaling can regulate mitochondrial respiration by disrupting the electron transport chain and increasing reactive oxygen species production (Carson et al., 2016; Wegrzyn et al., 2009). We recently found that skeletal muscle gp130 signaling can regulate protein expression related to basal mitochondria dynamics without affecting skeletal muscle fatigability (Fix et al., 2018). We extend these findings to demonstrate that the IL-6 disruption of submaximal contraction-induced skeletal muscle fatigue and mitochondrial content and function are regulated through muscle gp130 signaling. Therefore, determining whether chronically elevated IL-6 directly effects skeletal muscle mitochondrial function via modulating STAT3 signaling is an intriguing area of future investigation. Additionally, since both mitochondrial content and function were reduced by IL-6, whether the observed reduction in respiration was simply due to a loss of mitochondrial content could not be determined. In order to determine if IL-6 directly reduced an individual mitochondrion’s oxygen consumption, future studies measuring respiration in isolated mitochondria is needed (Perry et al., 2013).
IL-6 can also regulate the muscle macro- and microenvironment. IL-6 directly regulates fibroblast proliferation, liver glucose metabolism, lipolysis, and immune cell function (Choi, Kang, Yang, & Pyun, 1994; Cron et al., 2016; Febbraio et al., 2004; Lightfoot & Cooper, 2016; Narsale & Carson, 2014; Pedersen et al., 2003; Sundararaj et al., 2009). While the IL-6r is predominately expressed in hepatocytes, megakaryocytes, and leukocytes, the signal transducing gp130 β-subunit is detectable in every cell type (Wolf et al., 2014). There is an established liver-skeletal muscle metabolic cross-talk involving lactate recycling and glucose availability which can alter muscle fatigue properties indirectly (Rui, 2014). Similar studies examining systemic IL-6 overexpression showed IL-6 induced hepatic insulin resistance while having no effect on skeletal muscle insulin sensitivity (Glund & Krook, 2008). IL-6 has an established role in several pathophysiological and physiological phenomena; however, it’s role in cancer and skeletal muscle wasting is quite contentious. While several clinical studies have identified a direct relationship between circulating IL-6 and weight loss in cancer patients, others show either no change in circulating IL-6 with malignancy or no relationship to weight loss (Barber, Fearon, & Ross, 1999; Iwase, Murakami, Saito, & Nakagawa, 2004; Martin et al., 1999). These discrepancies have been recapitulated in preclinical investigations suggesting that the role of IL-6 in cachexia progression is dependent on tumor type. The results of the current study found that 2 weeks of systemic IL-6 overexpression had no effect on muscle mass or body weight, though regardless of treatment gp130 loss resulted in increased muscle mass. The range of IL-6 levels in the experimental groups in the current study (range 15.6–371.0 pg/mL) are similar to previously published values in tumor-bearing mice (Baltgalvis et al., 2008; Strassmann, Fong, Kenney, & Jacob, 1992; VanderVeen et al., 2018) as well as comparable to cancer patients (Iwase et al., 2004; Martin et al., 1999), but remain below the levels observed with severe infection (Copeland et al., 2005). IL-6 can regulate basal muscle protein synthesis and suppress the acute induction of protein synthesis by eccentric contraction (Gao et al., 2017; Hardee, Counts, et al., 2018); however, we observed no changes in muscle strength or contraction rates with IL-6 overexpression. Interestingly skeletal muscle lacking gp130 showed reduced specific tension, 1/2 RT, and TPT with no apparent effect on muscle fatigability which is similar to TNF-α overexpression (Reid et al., 2002). IL-6 also increased submaximal contraction-induced fatigue, which was not dependent on fiber type distribution or mass changes. Our results found that muscle fatigability, force, and mass can be regulated independently, which points to muscle weakness and fatigue with chronic disease having distinct etiologies.
Aging and several chronic inflammatory diseases have been associated with preferential atrophy of type II muscle fibers in both humans and animal models (Carson et al., 2016; Lexell, 1995; Wang & Pessin, 2013). Our lab previously showed that the tibialis anterior (TA), which has primarily type IIB muscle fibers (Bloemberg & Quadrilatero, 2012), had reduced force and increased muscle fatigability in tumor-bearing mice associated with increased STAT3 and SOCS3 activation (Murphy, Chee, Trieu, Naim, & Lynch, 2012; VanderVeen et al., 2018). While more work is needed to understand the fiber-type specificity of IL-6, COXIV and Cytochrome C protein expression in the murine quadriceps muscle was susceptible to IL-6 (Puppa et al., 2012), but was unchanged in the gastrocnemius (White et al., 2011). Additionally, gp130 protein expression was greater in the TA compared to the soleus and gastrocnemius (Puppa et al., 2014). Muscle dysfunction with cachexia has been identified in both type I/IIA and type IIX/B predominate muscles; however, type I/IIA fibers have been suggested to be protected against the early stages of inflammation induced wasting (Roberts, Ahn, et al., 2013; Roberts, Frye, Ahn, Ferreira, & Judge, 2013; Wang & Pessin, 2013). Additional work is needed to understand the IL-6 sensitivity of oxidative muscles since the results of the current study were limited to analysis of muscles that are predominately type IIB (TA and EDL).
We report that chronically elevated IL-6 increased skeletal muscle fatigability and disrupted aspects of oxidative metabolism independent of fiber-type and mass changes. IL-6 accelerated submaximal fatigue and reduced mitochondrial content and function through muscle gp130 signaling. While the effect of TNF-α on skeletal muscle function has been extensively characterized, to the best of our knowledge this is the first study investigating if IL-6 is sufficient to regulate skeletal muscle fatigue and muscle mitochondrial content and function. Future studies are warranted to establish the mechanisms regulating IL-6 induced maximal contraction-induced fatigability and disrupted mitochondrial quality control which contribute to submaximal contraction-induced fatigue.
NEW FINDINGS.
What is the central question of this study?
Interleukin-6 has been associated with muscle mass and metabolism with both physiological and pathological conditions. A causal role for IL-6 to induce fatigue and disrupt mitochondrial function has not been determined.
What is the main finding and its importance?
We demonstrate that chronically elevated IL-6 increased skeletal muscle fatigability and disrupted mitochondrial content and function independent of fiber-type and mass changes.
Acknowledgements:
The authors have no acknowledgements.
Funding: The research described in this report was supported by R01CA-121249A501 (National Cancer Institute) to JAC.
Footnotes
Competing interests: The authors have no conflict of interest to disclose.
REFERENCES
- al-Majid S, & McCarthy DO (2001). Cancer-induced fatigue and skeletal muscle wasting: the role of exercise. Biol Res Nurs, 2(3), 186–197. doi: 10.1177/109980040100200304 [DOI] [PubMed] [Google Scholar]
- Allen DG, Lamb GD, & Westerblad H (2008). Skeletal muscle fatigue: cellular mechanisms. Physiol Rev, 88(1), 287–332. doi: 10.1152/physrev.00015.2007 [DOI] [PubMed] [Google Scholar]
- Altenburg TM, Degens H, van Mechelen W, Sargeant AJ, & de Haan A (2007). Recruitment of single muscle fibers during submaximal cycling exercise. J Appl Physiol (1985), 103(5), 1752–1756. doi: 10.1152/japplphysiol.00496.2007 [DOI] [PubMed] [Google Scholar]
- Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ, & Carson JA (2008). Interleukin-6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol, 294(2), R393–401. doi: 10.1152/ajpregu.00716.2007 [DOI] [PubMed] [Google Scholar]
- Barber MD, Fearon KC, & Ross JA (1999). Relationship of serum levels of interleukin-6, soluble interleukin-6 receptor and tumour necrosis factor receptors to the acute-phase protein response in advanced pancreatic cancer. Clin Sci (Lond), 96(1), 83–87. [PubMed] [Google Scholar]
- Bloemberg D, & Quadrilatero J (2012). Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS One, 7(4), e35273. doi: 10.1371/journal.pone.0035273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland ML, Chourasia AH, & Macleod KF (2013). Mitochondrial dysfunction in cancer. Front Oncol, 3, 292. doi: 10.3389/fonc.2013.00292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Andersson DC, & Waning DL (2015). Assessment of muscle mass and strength in mice. Bonekey Rep, 4, 732. doi: 10.1038/bonekey.2015.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A AT, Jin X, Zhang Z, Zhan R, Puzis L, Koniaris LG, Zimmers TA (2012). JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. American Journal of Physiology Endocrinology and Metabolism, 303(3), E410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, & Zimmers TA (2011). STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One, 6(7), e22538. doi: 10.1371/journal.pone.0022538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, … Greene NP (2017). Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. Journal of cachexia, sarcopenia and muscle doi: 10.1002/jcsm.12232 [DOI] [PMC free article] [PubMed]
- Brzeszczynska J, Johns N, Schilb A, Degen S, Degen M, Langen R, … Ross JA (2016). Loss of oxidative defense and potential blockade of satellite cell maturation in the skeletal muscle of patients with cancer but not in the healthy elderly. Aging (Albany NY), 8(8), 1690–1702. doi: 10.18632/aging.101006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson JA, & Baltgalvis KA (2010). Interleukin 6 as a key regulator of muscle mass during cachexia. Exerc Sport Sci Rev, 38(4), 168–176. doi: 10.1097/JES.0b013e3181f44f11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson JA, Hardee JP, & VanderVeen BN (2016). The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer-induced muscle wasting. Semin Cell Dev Biol, 54, 53–67. doi: 10.1016/j.semcdb.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang VT, Hwang SS, Feuerman M, & Kasimis BS (2000). Symptom and quality of life survey of medical oncology patients at a veterans affairs medical center: a role for symptom assessment. Cancer, 88(5), 1175–1183. [DOI] [PubMed] [Google Scholar]
- Choi I, Kang HS, Yang Y, & Pyun KH (1994). IL-6 induces hepatic inflammation and collagen synthesis in vivo. Clin Exp Immunol, 95(3), 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close R (1964). Dynamic Properties of Fast and Slow Skeletal Muscles of the Rat during Development. The Journal of physiology, 173, 74–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close RI (1972). Dynamic properties of mammalian skeletal muscles. Physiol Rev, 52(1), 129–197. [DOI] [PubMed] [Google Scholar]
- Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D, Inflammation, & the Host Response to Injury, I. (2005). Acute inflammatory response to endotoxin in mice and humans. Clin Diagn Lab Immunol, 12(1), 60–67. doi: 10.1128/CDLI.12.1.60-67.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle EF, Martin WH 3rd, Bloomfield SA, Lowry OH, & Holloszy JO (1985). Effects of detraining on responses to submaximal exercise. J Appl Physiol (1985), 59(3), 853–859. [DOI] [PubMed] [Google Scholar]
- Crilly MJ, Tryon LD, Erlich AT, & Hood DA (2016). The role of Nrf2 in skeletal muscle contractile and mitochondrial function. J Appl Physiol (1985), 121(3), 730–740. doi: 10.1152/japplphysiol.00042.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cron L, Allen T, & Febbraio MA (2016). The role of gp130 receptor cytokines in the regulation of metabolic homeostasis. J Exp Biol, 219(Pt 2), 259–265. doi: 10.1242/jeb.129213 [DOI] [PubMed] [Google Scholar]
- De Benedetti F, Rucci N, Del Fattore A, Peruzzi B, Paro R, Longo M, … Teti A (2006). Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum, 54(11), 3551–3563. doi: 10.1002/art.22175 [DOI] [PubMed] [Google Scholar]
- Deans C, & Wigmore SJ (2005). Systemic inflammation, cachexia and prognosis in patients with cancer. Curr Opin Clin Nutr Metab Care, 8(3), 265–269. [DOI] [PubMed] [Google Scholar]
- Fearon KC, Glass DJ, & Guttridge DC (2012). Cancer cachexia: mediators, signaling, and metabolic pathways. Cell metabolism, 16(2), 153–166. doi: 10.1016/j.cmet.2012.06.011 [DOI] [PubMed] [Google Scholar]
- Febbraio MA, Hiscock N, Sacchetti M, Fischer CP, & Pedersen BK (2004). Interleukin-6 is a novel factor mediating glucose homeostasis during skeletal muscle contraction. Diabetes, 53(7), 1643–1648. [DOI] [PubMed] [Google Scholar]
- Febbraio MA, & Pedersen BK (2002). Muscle-derived interleukin-6: mechanisms for activation and possible biological roles. FASEB J, 16(11), 1335–1347. doi: 10.1096/fj.01-0876rev [DOI] [PubMed] [Google Scholar]
- Fitts RH (1994). Cellular mechanisms of muscle fatigue. Physiol Rev, 74(1), 49–94. doi: 10.1152/physrev.1994.74.1.49 [DOI] [PubMed] [Google Scholar]
- Fitts RH, & Holloszy JO (1977). Contractile properties of rat soleus muscle: effects of training and fatique. Am J Physiol, 233(3), C86–91. [DOI] [PubMed] [Google Scholar]
- Fix DK, Hardee JP, Gao S, VanderVeen BN, Velazquez KT, & Carson JA (2018). The Role of gp130 in Basal and Exercise Trained Skeletal Muscle Mitochondrial Quality Control. J Appl Physiol (1985) doi: 10.1152/japplphysiol.01063.2017 [DOI] [PMC free article] [PubMed]
- Gao S, Durstine JL, Koh HJ, Carver WE, Frizzell N, & Carson JA (2017). Acute myotube protein synthesis regulation by IL-6-related cytokines. Am J Physiol Cell Physiol, 313(5), C487–C500. doi: 10.1152/ajpcell.00112.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbers C, Aparicio-Siegmund S, & Rose-John S (2015). The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol, 34, 75–82. doi: 10.1016/j.coi.2015.02.008 [DOI] [PubMed] [Google Scholar]
- Gilliam LA, Moylan JS, Ferreira LF, & Reid MB (2011). TNF/TNFR1 signaling mediates doxorubicin-induced diaphragm weakness. Am J Physiol Lung Cell Mol Physiol, 300(2), L225–231. doi: 10.1152/ajplung.00264.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glund S, & Krook A (2008). Role of interleukin-6 signalling in glucose and lipid metabolism. Acta Physiol (Oxf), 192(1), 37–48. doi: 10.1111/j.1748-1716.2007.01779.x [DOI] [PubMed] [Google Scholar]
- Goodman CA, Kotecki JA, Jacobs BL, & Hornberger TA (2012). Muscle fiber type-dependent differences in the regulation of protein synthesis. PLoS One, 7(5), e37890. doi: 10.1371/journal.pone.0037890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. Experimental physiology, 100(7), 755–758. doi: 10.1113/EP085299 [DOI] [PubMed] [Google Scholar]
- Hardee JP, Counts BR, Gao S, VanderVeen BN, Fix DK, Koh HJ, & Carson JA (2018). Inflammatory signalling regulates eccentric contraction-induced protein synthesis in cachectic skeletal muscle. Journal of cachexia, sarcopenia and muscle, 9(2), 369–383. doi: 10.1002/jcsm.12271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardee JP, Fix DK, Wang X, Goldsmith EC, Koh HJ, & Carson JA (2018). Systemic IL-6 regulation of eccentric contraction-induced muscle protein synthesis. Am J Physiol Cell Physiol doi: 10.1152/ajpcell.00063.2018 [DOI] [PMC free article] [PubMed]
- Hardee JP, Mangum JE, Gao S, Sato S, Hetzler KL, Puppa MJ, … Carson JA (2016). Eccentric contraction-induced myofiber growth in tumor-bearing mice. J Appl Physiol (1985), 120(1), 29–37. doi: 10.1152/japplphysiol.00416.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin BJ, Campbell KS, Smith JD, Arbogast S, Smith J, Moylan JS, & Reid MB (2008). TNF-alpha acts via TNFR1 and muscle-derived oxidants to depress myofibrillar force in murine skeletal muscle. J Appl Physiol (1985), 104(3), 694–699. doi: 10.1152/japplphysiol.00898.2007 [DOI] [PubMed] [Google Scholar]
- Holloszy JO, & Booth FW (1976). Biochemical adaptations to endurance exercise in muscle. Annu Rev Physiol, 38, 273–291. doi: 10.1146/annurev.ph.38.030176.001421 [DOI] [PubMed] [Google Scholar]
- Hughey CC, Hittel DS, Johnsen VL, & Shearer J (2011). Respirometric oxidative phosphorylation assessment in saponin-permeabilized cardiac fibers. Journal of visualized experiments : JoVE(48). doi: 10.3791/2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal S, Ostojic O, Singh K, Joseph AM, & Hood DA (2013). Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve, 48(6), 963–970. doi: 10.1002/mus.23838 [DOI] [PubMed] [Google Scholar]
- Iwase S, Murakami T, Saito Y, & Nakagawa K (2004). Steep elevation of blood interleukin-6 (IL-6) associated only with late stages of cachexia in cancer patients. Eur Cytokine Netw, 15(4), 312–316. [PubMed] [Google Scholar]
- Kilgour RD VA, Trutschnigg B, Hornby L, Lucar E, Bacon SL, Morais JA (2010). Cancer-related fatigue: the impact of skeleatl muscle mass and strength in patients with advanced cancer. J Cachexia Saropenia Muscle, 1, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuller LH, Tracy R, Belloso W, De Wit S, Drummond F, Lane HC, … Group, I. S. S. (2008). Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med, 5(10), e203. doi: 10.1371/journal.pmed.0050203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon OS, Smuder AJ, Wiggs MP, Hall SE, Sollanek KJ, Morton AB, … Powers SK (2015). AT1 receptor blocker losartan protects against mechanical ventilation-induced diaphragmatic dysfunction. J Appl Physiol (1985), 119(10), 1033–1041. doi: 10.1152/japplphysiol.00237.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird BJ, Scott AC, Colvin LA, McKeon AL, Murray GD, Fearon KC, & Fallon MT (2011). Pain, depression, and fatigue as a symptom cluster in advanced cancer. J Pain Symptom Manage, 42(1), 1–11. doi: 10.1016/j.jpainsymman.2010.10.261 [DOI] [PubMed] [Google Scholar]
- Lexell J (1995). Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci, 50 Spec No, 11–16. [DOI] [PubMed] [Google Scholar]
- Li X, Moody MR, Engel D, Walker S, Clubb FJ Jr., Sivasubramanian N, … Reid MB (2000). Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragm. Circulation, 102(14), 1690–1696. [DOI] [PubMed] [Google Scholar]
- Lightfoot AP, & Cooper RG (2016). The role of myokines in muscle health and disease. Curr Opin Rheumatol, 28(6), 661–666. doi: 10.1097/BOR.0000000000000337 [DOI] [PubMed] [Google Scholar]
- Martin F, Santolaria F, Batista N, Milena A, Gonzalez-Reimers E, Brito MJ, & Oramas J (1999). Cytokine levels (IL-6 and IFN-gamma), acute phase response and nutritional status as prognostic factors in lung cancer. Cytokine, 11(1), 80–86. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Lorenzi M, Landi F, Picca A, Rosa F, Tanganelli F, … Bossola M (2017). Altered mitochondrial quality control signaling in muscle of old gastric cancer patients with cachexia. Exp Gerontol, 87(Pt A), 92–99. doi: 10.1016/j.exger.2016.10.003 [DOI] [PubMed] [Google Scholar]
- Merlini L, Bonaldo P, & Marzetti E (2015). Editorial: Pathophysiological Mechanisms of Sarcopenia in Aging and in Muscular Dystrophy: A Translational Approach. Front Aging Neurosci, 7, 153. doi: 10.3389/fnagi.2015.00153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M HM, Yoshida H, Suzuki M, Shiina M (2012). IL-6/IL-6 receptor system and its role in physiological and pathlogical conditions. Clinical Science, 122, 143–159. [DOI] [PubMed] [Google Scholar]
- Miller A, McLeod L, Alhayyani S, Szczepny A, Watkins DN, Chen W, … Jenkins BJ (2016). Blockade of the IL-6 trans-signalling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma. Oncogene doi: 10.1038/onc.2016.437 [DOI] [PubMed]
- Monga U, Jaweed M, Kerrigan AJ, Lawhon L, Johnson J, Vallbona C, & Monga TN (1997). Neuromuscular fatigue in prostate cancer patients undergoing radiation therapy. Arch Phys Med Rehabil, 78(9), 961–966. [DOI] [PubMed] [Google Scholar]
- Munkvik M, Lunde PK, & Sejersted OM (2009). Causes of fatigue in slow-twitch rat skeletal muscle during dynamic activity. Am J Physiol Regul Integr Comp Physiol, 297(3), R900–910. doi: 10.1152/ajpregu.91043.2008 [DOI] [PubMed] [Google Scholar]
- Murphy KT, Chee A, Trieu J, Naim T, & Lynch GS (2012). Importance of functional and metabolic impairments in the characterization of the C-26 murine model of cancer cachexia. Dis Model Mech, 5(4), 533–545. doi: 10.1242/dmm.008839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narsale AA, & Carson JA (2014). Role of interleukin-6 in cachexia: therapeutic implications. Curr Opin Support Palliat Care, 8(4), 321–327. doi: 10.1097/SPC.0000000000000091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK, Steensberg A, Keller P, Keller C, Fischer C, Hiscock N, … Febbraio MA (2003). Muscle-derived interleukin-6: lipolytic, anti-inflammatory and immune regulatory effects. Pflugers Arch, 446(1), 9–16. doi: 10.1007/s00424-002-0981-z [DOI] [PubMed] [Google Scholar]
- Perry CG, Kane DA, Lanza IR, & Neufer PD (2013). Methods for assessing mitochondrial function in diabetes. Diabetes, 62(4), 1041–1053. doi: 10.2337/db12-1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen K, Andersen S, Degen S, Tadini V, Grosjean J, Hatakeyama S, … Bjorkoy G (2017). Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci Rep, 7(1), 2046. doi: 10.1038/s41598-017-02088-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppa MJ, Gao S, Narsale AA, & Carson JA (2014). Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J, 28(2), 998–1009. doi: 10.1096/fj.13-240580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppa MJ, White JP, Velazquez KT, Baltgalvis KA, Sato S, Baynes JW, & Carson JA (2012). The effect of exercise on IL-6-induced cachexia in the Apc ( Min/+) mouse. Journal of cachexia, sarcopenia and muscle, 3(2), 117–137. doi: 10.1007/s13539-011-0047-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramage MI, & Skipworth RJE (2018). The relationship between muscle mass and function in cancer cachexia: smoke and mirrors? Curr Opin Support Palliat Care doi: 10.1097/SPC.0000000000000381 [DOI] [PubMed]
- Reid MB, Lannergren J, & Westerblad H (2002). Respiratory and limb muscle weakness induced by tumor necrosis factor-alpha: involvement of muscle myofilaments. Am J Respir Crit Care Med, 166(4), 479–484. doi: 10.1164/rccm.2202005 [DOI] [PubMed] [Google Scholar]
- Reid MB, & Moylan JS (2011). Beyond atrophy: redox mechanisms of muscle dysfunction in chronic inflammatory disease. The Journal of physiology, 589(Pt 9), 2171–2179. doi: 10.1113/jphysiol.2010.203356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BM, Ahn B, Smuder AJ, Al-Rajhi M, Gill LC, Beharry AW, … Judge AR (2013). Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J, 27(7), 2600–2610. doi: 10.1096/fj.12-222844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BM, Frye GS, Ahn B, Ferreira LF, & Judge AR (2013). Cancer cachexia decreases specific force and accelerates fatigue in limb muscle. Biochemical and biophysical research communications, 435(3), 488–492. doi: 10.1016/j.bbrc.2013.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose-John S (2018). Interleukin-6 Family Cytokines. Cold Spring Harb Perspect Biol, 10(2). doi: 10.1101/cshperspect.a028415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L (2014). Energy metabolism in the liver. Compr Physiol, 4(1), 177–197. doi: 10.1002/cphy.c130024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford G, Manning P, & Newton JL (2016). Understanding Muscle Dysfunction in Chronic Fatigue Syndrome. J Aging Res, 2016, 2497348. doi: 10.1155/2016/2497348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saks VA, Veksler VI, Kuznetsov AV, Kay L, Sikk P, Tiivel T, … Kunz WS (1998). Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol Cell Biochem, 184(1–2), 81–100. [PubMed] [Google Scholar]
- Schwantner A, Dingley AJ, Ozbek S, Rose-John S, & Grotzinger J (2004). Direct determination of the interleukin-6 binding epitope of the interleukin-6 receptor by NMR spectroscopy. The Journal of biological chemistry, 279(1), 571–576. doi: 10.1074/jbc.M311019200 [DOI] [PubMed] [Google Scholar]
- Sin DD, & Man SF (2003). Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation, 107(11), 1514–1519. [DOI] [PubMed] [Google Scholar]
- Spate U, & Schulze PC (2004). Proinflammatory cytokines and skeletal muscle. Curr Opin Clin Nutr Metab Care, 7(3), 265–269. [DOI] [PubMed] [Google Scholar]
- Stewart GD, Skipworth RJ, & Fearon KC (2006). Cancer cachexia and fatigue. Clin Med (Lond), 6(2), 140–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassmann G, Fong M, Kenney JS, & Jacob CO (1992). Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest, 89(5), 1681–1684. doi: 10.1172/JCI115767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundararaj KP, Samuvel DJ, Li Y, Sanders JJ, Lopes-Virella MF, & Huang Y (2009). Interleukin-6 released from fibroblasts is essential for up-regulation of matrix metalloproteinase-1 expression by U937 macrophages in coculture: cross-talking between fibroblasts and U937 macrophages exposed to high glucose. The Journal of biological chemistry, 284(20), 13714–13724. doi: 10.1074/jbc.M806573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason DB, & Booth FW (1990). Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol (1985), 68(1), 1–12. doi: 10.1152/jappl.1990.68.1.1 [DOI] [PubMed] [Google Scholar]
- Thompson LV, Balog EM, Riley DA, & Fitts RH (1992). Muscle fatigue in frog semitendinosus: alterations in contractile function. Am J Physiol, 262(6 Pt 1), C1500–1506. doi: 10.1152/ajpcell.1992.262.6.C1500 [DOI] [PubMed] [Google Scholar]
- VanderVeen BN, Hardee JP, Fix DK, & Carson JA (2018). Skeletal muscle function during the progression of cancer cachexia in the male Apc(Min/+) mouse. J Appl Physiol (1985), 124(3), 684–695. doi: 10.1152/japplphysiol.00897.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidt DG (2006). Inflammation in renal disease. Am J Cardiol, 97(2A), 20A–27A. doi: 10.1016/j.amjcard.2005.11.012 [DOI] [PubMed] [Google Scholar]
- Walsh B, Tonkonogi M, Soderlund K, Hultman E, Saks V, & Sahlin K (2001). The role of phosphorylcreatine and creatine in the regulation of mitochondrial respiration in human skeletal muscle. The Journal of physiology, 537(Pt 3), 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, & Pessin JE (2013). Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care, 16(3), 243–250. doi: 10.1097/MCO.0b013e328360272d [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, … Larner AC (2009). Function of mitochondrial Stat3 in cellular respiration. Science, 323(5915), 793–797. doi: 10.1126/science.1164551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerblad H, & Allen DG (2003). Cellular mechanisms of skeletal muscle fatigue. Adv Exp Med Biol, 538, 563–570; discussion 571. [DOI] [PubMed] [Google Scholar]
- White JP, Baltgalvis KA, Puppa MJ, Sato S, Baynes JW, & Carson JA (2011). Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol, 300(2), R201–211. doi: 10.1152/ajpregu.00300.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Puppa MJ, Gao S, Sato S, Welle SL, & Carson JA (2013). Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab, 304(10), E1042–1052. doi: 10.1152/ajpendo.00410.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, … Carson JA (2012). IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle, 2, 14. doi: 10.1186/2044-5040-2-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf J, Rose-John S, & Garbers C (2014). Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine, 70(1), 11–20. doi: 10.1016/j.cyto.2014.05.024 [DOI] [PubMed] [Google Scholar]
- Zhou J, Liu B, Liang C, Li Y, & Song YH (2016). Cytokine Signaling in Skeletal Muscle Wasting. Trends Endocrinol Metab, 27(5), 335–347. doi: 10.1016/j.tem.2016.03.002 [DOI] [PubMed] [Google Scholar]