

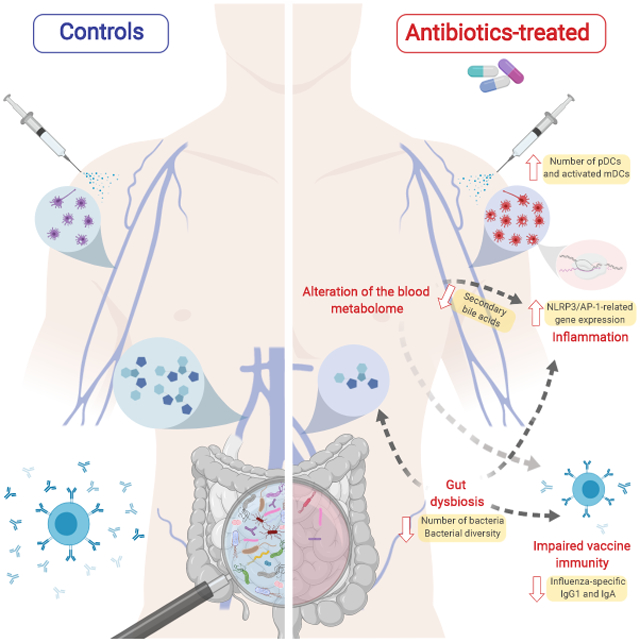
Summary
Emerging evidence indicates a central role for the microbiome in immunity. However, causal evidence in humans is sparse. Here we administered broad spectrum antibiotics to healthy adults prior and subsequent to seasonal influenza vaccination. Despite a 10,000-fold reduction in gut bacterial load and long-lasting diminution in bacterial diversity, antibody responses were not significantly affected. However, in a second trial of subjects with low pre-existing antibody titers, there was significant impairment in H1N1-specific neutralization and binding IgG1 and IgA responses. In addition, in both studies antibiotics treatment resulted in: (i) Enhanced inflammatory signatures (including AP-1/NR4A expression), observed previously in the elderly, and increased dendritic cell activation; (ii) Divergent metabolic trajectories, with a 1000-fold reduction in serum secondary bile acids which was highly correlated with AP-1/NR4A signaling and inflammasome activation. Multi-omics integration revealed significant associations between bacterial species and metabolic phenotypes, highlighting a key role for the microbiome in modulating human immunity.
Graphical Abstract
Introduction
Trillions of bacteria reside in our guts, outnumbering the eukaryotic cells in our bodies by a factor of 10 to 1 (Savage, 1977). Emerging evidence suggests a potent role for this microbial community, our so called ‘second genome’, in shaping physiology through its diverse effects on host metabolism (Nicholson et al., 2012; Sonnenburg and Backhed, 2016), enteric immunity (Belkaid and Hand, 2014; Steinhoff, 2005), autoimmune and allergic inflammation (Kostic et al., 2014; Mitre et al., 2018; Scher et al., 2013), and even communication with the central nervous system (Carabotti et al., 2015). In addition to promotion of autoimmunity and allergy, variations in microbiome composition have been linked to reduced efficacy in various immune interventions, including prevention of HIV infection (Klatt et al., 2017) and anti-PD1 cancer immunotherapy (Gopalakrishnan et al., 2018; Routy et al., 2018; Zitvogel et al., 2018).
However, much of the evidence in support of the microbiome’s impact on the immune system comes from work in mouse models or correlative studies in humans (Belkaid and Hand, 2014; Keeney et al., 2014), and there remains little causal evidence for the impact of deliberate perturbations in the microbiome on the physiological states of humans during disease and health. Developing a more comprehensive understanding of the mechanisms by which the microbiome can shape human physiology is critical in order to harness the microbiome-host axis in the therapeutic modulation of immune disorders. This is particularly relevant in the context of vaccination, since the efficacy of many vaccines is known to vary significantly by geographical region (Collins and Belkaid, 2018; Levine, 2010; Littman, 2018), and microbiome composition is also strongly geographically dependent (Yatsunenko et al., 2012). Understanding how the gut microbiota can impact responsiveness to vaccination therefore has significant implications for increasing vaccine efficacy and improving global public health.
In order to improve our understanding of the biological mechanisms responsible for protective immune responses, immunologists have begun to use high-throughput cellular and molecular technologies to comprehensively profile responses to vaccination in the nascent field of systems vaccinology (Hagan and Pulendran, 2018; Pulendran, 2014; Pulendran et al., 2010; Rappuoli et al., 2018). One biologically significant observation to arise from such studies was the identification in our analysis of immune responses to seasonal influenza vaccine of an association between expression of TLR5, a toll-like receptor capable of recognizing bacterial flagellin, on day 3 post-vaccination and the subsequent magnitude of the day 28 antibody response (Nakaya et al., 2011). This finding prompted follow-up experimentation in antibiotics-treated and TLR5 knockout mice which demonstrated that abrogation of TLR5-mediated sensing of flagellin from the intestinal microbiota resulted in impaired plasma cell and antibody responses to inactivated influenza vaccination (Oh et al., 2014). This established a potential role of the gut flora in influencing humoral responses to vaccines.
Thus, in order to understand if antibiotics-driven depletion of the gut flora can influence responses to vaccination in humans, here we performed extensive profiling of innate and adaptive immune responses in antibiotics-treated and control subjects vaccinated with the trivalent inactivated influenza vaccine (TIV) over two separate seasons. We show that the profound antibiotics-driven perturbation of gut microbiome resulted in a significant impact on the H1N1-specific IgG1 response in subjects with low pre-existing immunity to influenza. Furthermore, antibiotics treatment had a striking effect on the plasma metabolome, with microbiome-associated disturbances in bile acid metabolism highly correlated with elevated cellular and transcriptional signatures of inflammation. Taken together these findings reveal unappreciated roles for the microbiome in modulating human immunity.
Results
Antibiotics use results in a profound reduction in gut bacterial load and persistent changes in bacterial diversity
To investigate the role of the human gut microbiota in shaping immune responses to seasonal influenza vaccine, during the 2014-2015 season we enrolled a total of 22 healthy individuals aged 18-45. We randomized 11 subjects to a five-day broad-spectrum antibiotic regimen consisting of an oral cocktail of neomycin, vancomycin, and metronidazole, with the aim of depleting gram-negative (neomycin), gram-positive (neomycin and vancomycin), and anaerobic (metronidazole) bacteria in the gastrointestinal tract. Antibiotic treatment extended from three days prior to vaccination until one day after. We collected biological samples at regular intervals up to one year after immunization and measured key aspects of the immune system as indicated (Figure 1A).
Figure 1. Study overview and evaluation of the effects of antibiotic use on the gut microbiome of healthy adults.

(A) Study overview. A total of 22 study participants aged 18-45 received a trivalent influenza vaccine (TIV) on day 0. Eleven subjects were randomized to a five-day oral broad-spectrum antibiotic regimen between day −3 and day 1. Samples were collected and analyses performed at regular intervals (black circles) as illustrated in the diagram.
(B) Normalized copy number of bacterial 16s ribosomal RNA per gram of stool. Each line corresponds to an individual subject. Controls are shown in blue, antibiotics-treated subjects in red. Median values and distributions for each time point are illustrated in the form of box plots.
(C-D) Flagellin (C) and LPS (D) concentrations per gram of stool. Each thin line represents a single subject, thick lines represent geometric means.
(E) Relative abundance of bacterial families in the antibiotics-treated group at different time points. Each vertical bar corresponds to a study participant. On day 1 and day 3, data is available for 9 and 10 out of 11 individuals only, respectively.
(F) Dimensional reduction of the Bray-Curtis distance between microbiome samples, using the principal coordinates analysis (PCoA) ordination method. Each circle corresponds to a single individual.
(G) Alpha-diversity estimates. “Observed” diversity represents the number of OTUs (richness) present in each sample (left panel). “Shannon” diversity takes into account both richness and evenness of OTUs within a sample (right panel). Each circle corresponds to a single individual in the antibiotics-treated group. Median values and interquartile ranges shown in box plots.
Where calculated, comparisons between control and antibiotics-treated groups at specific time points were performed by Mann-Whitney tests. Wilcoxon matched-pairs signed rank tests were used to compare time points within the same group (panel G). *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
See also Figure S1.
We began our analysis by quantifying the effect of antibiotic use on the gut microbiota. On the day of vaccination, antibiotics-treated subjects showed a three to four-log reduction in the total number of 16S rRNA copies per gram of stool, a surrogate measure of the relative abundance of bacteria in the gut (Figure 1B). On day 1 (the last day of antibiotics) the total bacterial content in the antibiotic administered cohort reached its nadir, before gradually recovering to baseline levels between days 7 and 30. In accord, we found that fecal concentrations of both flagellin and LPS were also significantly reduced at early time points after antibiotic administration with similar kinetics (Figure 1C, D). As expected, the control group did not show significant changes in 16s copy numbers, flagellin, or LPS concentrations over time (Figure 1B-D).
Given the profound contraction in total bacterial load, we asked which bacterial species were most affected. Family-level analysis revealed striking changes in microbial community composition, with relative abundances significantly altered up to one month post antibiotics use. Enterobacteriaceae dominated the gut microbiota of treated subjects at days 0, 1, and 3, although two individuals in this cohort appeared to resist disturbance and did not show evident signs of alteration in numbers or composition of their microbiota overall (Figure 1E), likely related to questionable compliance with antibiotics. By day 7, Enterobacteriaceae remained elevated in several subjects, albeit in lower proportions. Streptococcaceae were also more abundant at early time points, while Lachnospiraceae, Ruminococcaceae, Bacteroidaceae, and Veillonellaceae diminished between days 0 and 3. By day 30, relative levels of Lachnospiraceae, Enterobacteriaceae and Ruminococcaceae remained perturbed but eventually returned to baseline by day 90 (Figure 1E and Table S1).
We also quantified differences in overall microbial community structure through principal cooordinates analysis (PCoA) of the beta (between-sample) diversity (Figure 1F). On day 0, samples from the antibiotic cohort had diverged from controls, with the largest distance between the two groups measured on day 3. Interestingly, at day 180 the microbial composition of the two groups still showed substantial dissimilarity, suggesting only partial recovery of the original microbial composition in the antibiotic group. We also examined alpha (within-sample) diversity to compare microbial richness and evenness in the antibiotic group samples pre- and postantibiotics. Both measures were heavily compromised after the use of antibiotics, with the smallest number of different OTUs recorded on day 3 (Figure 1G). Notably, species richness and biodiversity were not fully recovered at six months, indicating long-lasting loss of unique bacterial species, consistent with previous studies (Becattini et al., 2016; Dethlefsen and Relman, 2011; Modi et al., 2014).
Effect of antibiotics on antibody responses to influenza vaccination
We next examined how the profound and durable effect on the gut microbiota impacted adaptive immune responses to TIV immunization. First, we evaluated the serum antibody responses of vaccinated individuals by measuring microneutralization (MN) titers. We found that both cohorts had similar MN titers for each of the three influenza strains included in the vaccine at all time points, suggesting that antibiotics-induced perturbation of the gut ecosystem did not significantly alter the ability of TIV-specific antibodies to neutralize homologous vaccine strains in vitro (Figure 2A). Analysis of seroconversion rates on day 30, which describe the proportion of subjects who developed a ≥ 4-fold increase in titer between pre- and post-vaccination sera, also showed comparable results between the two groups (Figure S2A). However, we noticed a generally low percentage of seroconversion overall, possibly due to high baseline titers observed for some subjects.
Figure 2. Impact of antibiotics treatment on humoral responses to TIV.

(A-B) Microneutralization (MN) titers against the 3 influenza strains contained in the 2014-2015 (A) and 2015-2016 (B) TIV formulations. Geometric means are presented in thick lines, while shades are for geometric standard deviations (SD).
(C) IgG1-binding to A/California H1 for phase 1 (left panel) and phase 2 (right panel) measured by ELISA. Violin plots show sample distributions. Each circle represents an individual subject, while medians are presented in thick lines.
(D) Relative concentration of A/California H1 HA-specific IgG1 for phase 2 determined using a high-throughput Luminex-based assay (Brown et al., 2012). Violin plots show sample distributions. Each circle represents an individual subject, while medians are presented in thick lines.
(E) Scatterplot of A/California H1 HA-specific IgG1 measured by ELISA vs A/California H1 HA-specific IgG1 measured by Luminex for phase 2 subjects on days 0, 7, 30. Each dot represents one subject.
(F) Off-rate measurements in seconds (sec) by surface plasmon resonance (SPR) to assess antibody affinity to A/California H1. The data is presented as reciprocally transformed and as fold change over the baseline. Each line represents one subject.
(G) A/California H1 HA-specific IgA isotype binding capacity measured by SPR and presented as maximum resonance units (max RU) for phase 2 subjects. Violin plots show sample distributions. Each circle represents an individual subject, while medians are presented in thick lines.
(H) Relative concentration of A/California H1 HA-specific IgA1 for phase 2 determined using a high-throughput Luminex-based assay, as for panel D. Violin plots show sample distributions. Each circle represents an individual subject, while medians are presented in thick lines.
(I) Scatterplot of A/California H1 HA-specific IgA isotype binding capacity measured by SPR vs A/California H1 HA-specific IgA1 measured by Luminex for phase 2 subjects on days 0 and 30. Each dot represents one subject. See STAR Methods for further details.
Where calculated, comparisons between control and antibiotics-treated groups at specific time points were performed by Mann-Whitney tests (panels A-D, F-H). Wilcoxon matched-pairs signed rank tests were used to compare time points within the same group (panel F). Pearson correlation was used for panels E and I. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
See also Figures S2 and S3.
To determine if the lack of impact on antibody responses was due to pre-existing immunity to influenza, one year later we enrolled 11 additional subjects whose MN titers before vaccination were ≤ 320 for each of the three vaccine strains contained in the 2015-2016 TIV formulation, and who did not receive TIV in the prior three influenza seasons. Five of these individuals were randomized to receive the same antibiotics regimen as in phase 1. A comparison of MN titers at baseline for the participants in the two phases of our study is presented in Table S2. Importantly, the gut microbiome of the five subjects in the phase 2 antibiotic group suffered decreased microbial richness and diversity in a similar fashion to phase 1 individuals (Figure S1A-C). However, in this second experiment we also found an impairment in the H1N1 A/California-specific neutralization on day 90 and day 180 post vaccination for the subjects in the antibiotic group, while no differences were observed for the other two influenza strains (Figure 2B). Predictably, given the lower baseline titers, seroconversion rates were much higher in the second cohort and reached 100% in both control and antibiotics groups for two out of three vaccine strains (Figure S2B).
Remarkably, we also noticed that the antibiotics-treated subjects in phase 2 exhibited significantly reduced concentrations of vaccine-induced IgG1 antibodies specific to the H1N1 A/California strain (Figure 2C, right panel) on day 7, as measured by ELISA. This was in contrast with phase 1, where the magnitude of IgG1 response was not strongly affected by antibiotic treatment (Figure 2C, left panel). In an effort to confirm these results, we asked an independent laboratory to validate our initial findings using a high-throughput Luminex-based assay (Brown et al., 2012). There was a striking correlation between the two independent measurements, with antibiotic-treated subjects in phase 2 showing a significant impairment in the production of H1N1-specific IgG1 titers on days 7 and 30 post vaccination by Luminex assay (Figure 2D-E), thus confirming our previous results. In agreement with the ELISA data, no significant effect of antibiotic treatment on IgG1 production was detected for phase 1 participants by Luminex (data not shown). While vaccine-induced IgG1 production accounts for the vast majority of total IgG responses specific to influenza (Frasca et al., 2013), we asked whether the concentration of other IgG subclasses would be affected by antibiotic treatment, but found no significant effect on IgG2 (Figure S2C) or IgG3 antibodies (data not shown). Importantly, antibody affinity to the H1N1 A/California strain was also altered as a consequence of antibiotic use (Figure 2F).
The marked effects of antibiotic treatment on the magnitude of vaccine-induced antibody responses were not confined to IgG1 only. Using surface plasmon resonance (SPR), we detected lower H1N1-specific IgA antibody responses in the phase 2 antibiotic group before and after vaccination (Figure 2G). These differences were again confirmed by Luminex (Figure 2H-I) and not observed in the phase 1 antibiotic group (Figure S2D).
We next asked whether other important aspects of adaptive immunity were also affected. Seasonal influenza vaccination results in the differentiation of antigen-specific B cells into plasmablasts (PBs) (Wrammert et al., 2008) and “activated B cells” (ABCs) which are committed to the memory B cell pathway (Ellebedy et al., 2016). We assessed the frequencies of vaccine-specific PBs and ABCs in the blood and observed no difference between control and antibiotics-treated groups (Figure S3A-C). In addition, we assessed the frequencies of blood follicular T cell (Tfh)-like cells, which are crucial for providing B cell help (Crotty, 2014), and observed no differences (Figure S3D). Furthermore, we examined the possible effects of antibiotic use on B cell somatic hypermutation (SHM) by sorting single vaccine-induced PBs 7 days after vaccination and generating TIV-reactive human monoclonal antibodies (hmAbs) from antibiotics-treated subjects in both phases of our study. We then compared the number of mutations in light and heavy chains to those in flu-positive hmAbs isolated from control subjects, or from healthy controls who received a quadrivalent influenza vaccine (QIV) for the 2014-2015 flu season, but found no significant differences (Figure S3E). Taken together these results demonstrate that disturbance of the gut microbial community can impair antibody responses to influenza vaccination in absence of significant pre-existing humoral immunity.
Impact of antibiotics use on vaccine induced blood transcriptional signatures
In order to evaluate the potential impact of antibiotic use on the magnitude and kinetics of vaccine-induced transcriptional responses, we identified differentially expressed genes in each cohort on days 1-7 post-vaccination, relative to day 0 (day of vaccine administration) (Figure 3A). The response kinetics were the same in both groups: vaccine-induced changes in gene expression peaked at day 1, then decreased by day 3, and had a second increase at day 7. The control group had a modest increase in differentially expressed genes at all time points relative to the antibiotics group, potentially due to a slightly higher number of subjects (17 versus 15).
Figure 3. Transcriptional Responses to TIV in Control and Antibiotics-Treated Subjects.

(A) Number of genes differentially expressed (log2 fold-change > 0.2 and t-test p value < 0.01) relative to day 0 in control and antibiotics-treated subjects on days 1, 3, and 7 post-vaccination.
(B) BTMs significantly enriched (FDR < 0.05, NES ≥ 2) in control and antibiotics-treated subjects post-vaccination. GSEA (Subramanian et al., 2005) was used to identify positive (red) or negative (green) enrichment of BTMs using ranked gene lists, where genes were ordered by t-statistic based on post-vaccination fold change relative to day 0.
(C-D) Temporal expression patterns of genes within modules M127 and M156.1 among antibiotics-treated (red) and control subjects (blue). Black line represents the mean fold change of all genes.
See also Figure S4 for comparison of responses in phases 1 and 2.
We then ran Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) to identify the transcriptional pathways regulated in response to TIV in both groups (Figure 3B). For this, we utilized a set of blood transcriptional modules (BTMs) identified previously by our group through large-scale network integration of publicly available human blood transcriptome datasets (Li et al., 2014). Both control and antibiotics-treated subjects exhibited very similar enrichment patterns. These patterns were also maintained between phases 1 and 2, suggesting that antibiotics treatment does not strongly alter the transcriptional pathways induced by vaccination even in subjects with low pre-existing titers (Figure S4A). On day 1, we observed enrichment of modules associated with the innate immune response, including dendritic cell activation, antigen presentation, and interferon signaling. By day 7 a signature of the adaptive response emerged, with enrichment of cell cycle and plasma cell-related modules reflecting the expansion of plasmablasts. These observations were consistent with the activated pathways identified in our analysis of healthy adult vaccinees in previous influenza seasons (Nakaya et al., 2015). We further examined the temporal profiles of the individual genes involved in these canonical influenza vaccine response pathways, and saw no effect of antibiotics use on the kinetics or magnitude of gene expression related to interferon signaling (Figure 3C) or plasma cell and immunoglobulin expression (Figure 3D). Collectively these findings suggest that antibiotics-treatment has little effect on transcriptional responses to influenza vaccination.
Antibiotics use induces pro-inflammatory transcriptional and cellular responses in blood
We next explored whether administration of antibiotics alone could induce changes in the blood transcriptome. We performed GSEA based on changes in gene expression in antibiotics-treated subjects between the screening and day 0 time point (Figure 4A). We identified expression changes in a substantial number of modules following antibiotics use, including increases in inflammatory signaling and activation of dendritic cells, and decreases in B cell and cell cycle-related expression, which were largely consistent in both phases of the study (data not shown). We also measured frequencies of several innate immune cell populations by flow cytometry, and observed significant increases in two dendritic cell subsets, CD80+ CD16+ mDCs and CD123++ pDCs, post-antibiotics (Figure 4B). These changes were consistent with the enrichment in dendritic cell activation detected by gene expression (Figure 4A). Both populations were elevated in antibiotics-treated subjects throughout the course of antibiotics (days 0 and 1) and began to return to control levels following the cessation of antibiotics, further suggesting that these changes were driven by the antibiotics-induced perturbations in the gut microbiome.
Figure 4. Transcriptional and Cellular Responses to Antibiotics Administration.

(A) BTMs significantly enriched (FDR < 0.05) following antibiotics use. GSEA (Subramanian et al., 2005) was used to compute the normalized enrichment score (NES) of BTMs using ranked gene lists, where genes were ordered by t-statistic based on day 0 versus screening (day −21) fold change in antibiotics-treated subjects. Enriched modules are colored according to their high-level functional annotation.
(B) Kinetics of dendritic cell subsets following antibiotics administration and vaccination. Solid lines represent mean fold change and shaded areas represent 95% confidence interval.
(C) Kinetics of AP-1/NR4A related BTMs following antibiotics administration and vaccination. Solid lines represent average module expression among antibiotics-treated (red) and control subjects (blue), and bar plots represent average module expression among young (< 65 years, light blue) and elderly (≥ 65 years, maroon) subjects vaccinated with TIV during the 2010-2011 influenza season (Nakaya et al., 2015). Error bars represent standard error of the mean (SEM).
(D) Genes in AP-1/NR4A related BTMs. Each “edge” (gray line) represents a coexpression relationship, as described in Li et al. (Li et al., 2014); colors represent the day 0 versus screening (day −21) log2 fold change (positive – red, negative – green).
(E) Spearman correlation of AP-1/NR4A target genes (from TRANSFAC (AP-1) or Pei et al. (Pei et al., 2006) (NR4A)) with their corresponding transcription factors on day 1. For AP-1 the average expression of FOS/JUN was used, and for the NR4A family the average of NR4A1/2/3 was used to compute the correlation.
Where calculated, comparisons between control and antibiotics-treated groups at specific time points were performed by Student’s t-tests. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Of particular interest among the pathways activated in response to antibiotics were several modules associated with AP-1 (FOS/JUN) and Nur (NR4A) transcription factors (Figure 4C-D). These modules remained elevated in antibiotics-treated subjects on day 1 post-vaccination, whereas they were downregulated in control subjects following vaccination (Figure 4C). Surprisingly, when we compared the kinetics of these modules to those from young and elderly adults vaccinated during the 2010-2011 influenza season (light blue and maroon bars, respectively) (Nakaya et al., 2015), we observed that while young subjects showed decreased expression of these modules on day 1 as in the control subjects, antibiotics-treated subjects behaved similarly to elderly vaccinees, who exhibited upregulation of these pathways postvaccination. These modules also included several pro-inflammatory cytokines, including IL6, IL8, and CXCL2, which were induced following antibiotics (Figure 4D). To further explore the potential role of AP-1 and NR4A in regulating these transcriptional responses, we identified putative target genes of these transcription factors either in the TRANSFAC database (Matys et al., 2003) or through literature search. We observed strong correlations between these transcription factors and several of their respective target genes on day 1 (Figure 4E). These results indicate that antibiotics-driven depletion of the gut microbiome may drive inflammatory signaling in innate immune cells in a manner consistent with age-associated changes in immune responses.
Antibiotics use perturbs the blood metabolome
The gut microbiota is well known to play an important role in the metabolism of various nutrients in the diet, including aiding in the digestion of complex carbohydrates and synthesis of vitamins (Levy et al., 2017; Nicholson et al., 2012; Tremaroli and Backhed, 2012). Therefore, we sought to examine whether we could detect changes in the blood metabolome of subjects following antibiotics use through untargeted high-resolution metabolomics. Antibiotics-treated subjects did display a large perturbation in their metabolome relative to control subjects as measured by Euclidean distance between the screening and day 0 time points over the most variable metabolite features (Figure 5A). Enrichment analysis of differential features using mummichog software (Li et al., 2013) revealed antibiotics-induced changes in multiple pathways, including bile acid metabolism and metabolism of tryptophan (Figure 5B). This is consistent with established functions of the gut microbiota in regulating the bile acid pool (Ridlon et al., 2014) as well as in the control of tryptophan metabolism towards indole, kynurenine, and serotonin pathways (Agus et al., 2018). Because our untargeted approach is unable to reliably identify specific metabolites, we performed an independent liquid chromatography-mass spectrometry (LC-MS) analysis using validated chemical standards to further characterize changes in individual bile acids following antibiotics-treatment. Because the microbiota is responsible for converting primary bile acids synthesized in the liver to secondary bile acids in the gut (Ridlon et al., 2006), we expected to see an increase in the ratio of primary to secondary bile acids following antibiotics treatment. Indeed, there was an elevation of primary and dramatic reduction in secondary bile acid levels in the serum of antibiotics-treated subjects (Figure 5C).
Figure 5. Impact of antibiotics administration and influenza vaccination on the blood metabolome.

(A) Euclidean distance across the most variable metabolite features (coefficient of variation >8% across all time points) between the day 0 and screening (day −21) time point in control (blue) and antibiotics-treated (red) subjects. Error bars represent standard error of the mean (SEM).
(B) Metabolic pathways significantly enriched following antibiotics administration. Mummichog software (Li et al., 2013) was used to identify enriched pathways (p<0.05 by permutation test) based on differential metabolite features (p<0.05 by Student’s t-test) between the day 0 and screening (day −21) time point in antibiotics-treated subjects.
(C) Fold change of primary and secondary bile acids in antibiotics-treated subjects on days 0-7 relative to the screening time point (day −21).
(D) Euclidean distance across the most variable metabolite features (coefficient of variation >8% across all time points) on days 1-7 post-vaccination relative to day 0 in control and antibiotics-treated subjects. Error bars represent standard error of the mean (SEM).
(E) Metabolic pathways significantly enriched (p<0.05) following influenza vaccination in control and antibiotics-treated subjects.
(F) Metabolic trajectories along the first two principal components for control and antibiotics-treated subjects for days 0-7 relative to the screening time point. Here metabolic trajectories refer to the trajectory of each subject according to the changes in abundance across all differential metabolite features (p<0.01) throughout the time course of the study (days 0-7) when projected in the principal component space.
Where calculated, comparisons between control and antibiotics-treated groups at specific time points were performed by Student’s t-tests. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
See also Figure S4 for comparison of responses in phases 1 and 2.
Next, we investigated the impact of gut microbiome depletion on the metabolic response to influenza vaccination. In control subjects, metabolic changes in response to vaccination peaked at day 1 (Figure 5D). Antibiotics-treated subjects had similar magnitude of response at day 1, however they maintained a large shift in their metabolome beyond control subjects, and still exhibited considerable differences compared to day 0 at day 7 post-vaccination. When we examined the enriched metabolic pathways following vaccination, we observed very little overlap in enriched pathways between the two groups (Figure 5E). Thus, antibiotics significantly altered the metabolic response to influenza vaccination in the blood.
To further explore the differences in metabolic responses, we performed a principal component analysis (PCA) on fold change values of differentially abundant metabolite peaks (p<0.01) in either group across all time points. Indeed, antibiotics-treated subjects showed profoundly altered metabolic trajectories relative to control subjects, both before and after vaccination (Figure 5F). These differences were observed in both phases of the study (Figure S4C). Importantly, we note that the two antibiotics-treated subjects whose trajectories grouped with control subjects were the same subjects who displayed little change in their microbiome (Figure 1), suggesting poor compliance with the antibiotic regimen.
Perturbation of secondary bile acids is associated with elevated NLRP3 inflammasome signaling in antibiotics-treated subjects
Given that the loss of intestinal microbiota strongly altered the plasma metabolome, we wanted to explore potential connections between this metabolic disturbance and the cellular and transcriptional changes we observed following antibiotics treatment (Figure 4). In particular, dysbiosis of bile acid metabolism, which was significantly altered in antibiotics-treated subjects, has been associated with inflammatory bowel diseases (IBD) (Pavlidis et al., 2015) and could potentially play an immunomodulatory role in this setting. Indeed, integrative analysis of the transcriptomic and metabolomic data showed significant inverse correlations between the levels of individual secondary bile acids and many of the inflammatory BTMs identified in Figure 4 which were induced following antibiotics-treatment (Figure 6A). Lithocholic acid (LCA), which showed a 1,000-fold reduction in the plasma following antibiotics treatment (Figure 6E), had the strongest association with inflammatory responses, including modules involved in AP-1 signaling such as M35.0, which was upregulated post-vaccination in antibiotics-treated subjects (Figure 6C) but downregulated in control subjects (Figure 6D). LCA is the most potent agonist of the bile acid receptor TGR5 (Kawamata et al., 2003), and TGR5 signaling has been shown to inhibit activation of the NLRP3 inflammasome in mice (Guo et al., 2016). Furthermore, AP-1 is known to play a critical role in the IL-1B-mediated induction of pro-inflammatory cytokines such as IL-6 (Cahill and Rogers, 2008). As such, we also investigated expression changes in genes related to inflammasome and IL-1B signaling and found robust associations with LCA and other secondary bile acids (Figure 6A-B). These findings highlight a potential mechanism by which the microbiome may regulate inflammatory responses in humans.
Figure 6. Perturbation of secondary bile acids is associated with elevated NLRP3 inflammasome signaling in antibiotics-treated subjects.

(A) Circos plot showing the log2 fold change in secondary bile acids, antibiotics-induced BTMs (Figure 4A), and inflammasome signaling genes on days 0-7 relative to screening (day −21) in control and antibiotics-treated subjects. Lines indicate significant correlations (p<0.01, Pearson correlation across all time points).
(B) Scatterplot of FOSB expression versus fold change of LCA in the plasma (all time points). Each dot represents one subject.
(C-D) Fold change of genes in M35.0 in (C) antibiotics-treated and (D) control subjects. Each “edge” (gray line) represents a coexpression relationship; colors represent the day 1 versus screening (day −21) log2 fold change (positive – yellow, negative – blue).
(E) Fold change of LCA in the plasma among antibiotics-treated (red) and control (blue) subjects. Each thin line represents a single subject, thick lines represent geometric means.
See also Figure S4 for comparison of LCA fold changes in phases 1 and 2.
MMRN analysis suggests the gut microbiome regulates inflammatory signaling and antibody responses to vaccination through distinct pathways
The gut microbiome can impact immune responses directly through translocation of microbial stimuli to systemic compartments (Brenchley et al., 2006), or indirectly via secondary messengers such as metabolites. We thus measured the concentrations of flagellin and anti-LPS antibodies in the serum (Figure S1D-F), but could find no differences between control and antibiotics-treated subjects, arguing against translocation of microbial products to the periphery. To explore the putative indirect effects, we constructed a multiscale, multifactorial response network (MMRN) as described previously (Li et al., 2017) (see also STAR Methods and Figure S5) which integrated transcriptional, metabolic, microbiomic, and cellular measurements to identify associations between these data types during antibiotics use and influenza vaccination. Bacteria-metabolite associations dominated the most significant associations in the network (Figure 7A), demonstrating the substantial capability of the gut microbiome to shape host metabolism.
Figure 7. MMRN analysis suggests distinct functions of the gut microbiome in regulating inflammatory signaling and H1N1-specific IgG1 responses.

(A) Distribution of p values between the different data types included in the MMRN.
(B) Sub-network visualization of the day 0 vs screening connections in the MMRN, containing nodes associated with either LCA or H1N1-specific IgG1. Each node is a cluster of features from one data type. The links between nodes (gray) were established by significant association (FDR < 0.05) using partial least square regression and permutation test. The network was queried through an enrichment based approach to identify positive (red) or negative (green) associations (FDR < 0.05, NES > 2.6) between antibiotics-induced (day 0 vs screening) changes in the nodes with either the day 0 vs screening change in LCA or the day 30 abundance of H1N1-specific IgG1. Individual cluster features are provided in Table S3. See Figure S5 and STAR Methods for details of MMRN construction.
(C) Pie charts showing the family membership of OTUs within bacterial clusters B0, B1, and B4.
(D) Scatterplots of plasma levels of LCA versus changes in bacterial load (measured by 16s rRNA qPCR), LPS, and flagellin (day 0 versus screening fold change). Each dot represents one subject.
(E) Scatterplots of the day 7 and day 30 abundance of H1N1-specific IgG1 versus flagellin measured in the stool (day 0 versus screening fold change). Each dot represents one subject.
Next, to identify the network nodes most associated with the observed differences in bile acid metabolism and H1N1-specific IgG1 responses, we queried the network through an enrichment-based approach using features ranked by correlation of their day 0 versus screening fold change with either the antibiotics-induced change in LCA or the day 30 abundance of H1N1-specific IgG1 (Figure 7B). In agreement with our previous analysis, there was a strong negative association between decreases in LCA and increases in transcriptional cluster G3, containing many BTMs involved in inflammatory responses, dendritic cell activation, and AP-1 signaling. Furthermore, LCA was also closely associated with metabolite cluster M79 which was enriched in bile acid biosynthesis, suggesting that our untargeted metabolomics analysis was accurately capturing the metabolic pathways altered by perturbation of the microbiome.
Despite disturbances in both bile acid metabolism and IgG1 responses in antibiotics-treated subjects, there was little overlap in metabolic and transcriptional clusters associated with both LCA and H1N1-specific IgG1 abundance. There was a sole metabolite cluster, M84, enriched in androgen and estrogen metabolism, which was negatively associated with both parameters. Consistent with this low overlap, IgG1 titers were weakly correlated with changes in abundance of bile acids (data not shown). Instead, commonly associated features were dominated by bacteria, with three bacterial clusters positively associated with both parameters. Composition analysis of these clusters showed a high degree of membership in families Lachnospiraceae and Ruminococcaceae (Figure 7C), both of which were drastically reduced following antibiotics treatment (Figure 1E). Interestingly, among the clusters associated uniquely with IgG1 response were two clusters highly enriched in fatty acid metabolism (M26 and M58), which is emerging as a key regulator of immune function (Ganeshan and Chawla, 2014; Sinclair et al., 2017).
The central role of bacterial clusters in the network prompted us to further examine the importance that gut bacterial content has on these two pathways. Indeed, changes in LCA were strongly associated with bacterial load, flagellin, and LPS content in the stool (Figure 7D) and the abundance of H1N1-specific IgG1 also significantly correlated with decreases in flagellin post-antibiotics treatment (Figure 7E). Interestingly, this positive correlation between flagellin and IgG1 titers was only significant in phase 2 subjects with low baseline titers, highlighting the resilience of immune memory to severe dysbiosis. Together, these results suggest that secondary bile acid-associated inflammatory signaling and impaired IgG1 responses to influenza vaccination arise as independent effects mediated by antibiotics-driven loss of key bacterial communities in the gut.
Discussion
By performing this trial in two separate cohorts with differing baseline levels of neutralizing antibodies, we were able to gain insight into the impact of pre-existing humoral immunity on microbiome-mediated changes in adaptive response to vaccination. In subjects with high baseline titers there was minimal impact on the antibody response, but in the second cohort of subjects with low baseline titers we observed a striking impact of antibiotics treatment on the amount of H1N1-specific IgG1 following vaccination. Noticeably this effect was only observed with the response to H1N1 and not against the other strains. The reason for this is unclear at present, but it is possible that H3N2 and B antibodies responses following vaccination in adults are primarily recall memory responses. Recent work has shown adults have high H3N2 and B subtype memory due to prior exposure by vaccination or infection (Khurana et al., 2019).
These results provide important context to our previous findings in mice (Oh et al., 2014), where loss of TLR5-mediated sensing of flagellin through TLR5 knockout or antibiotics administration resulted in significant reduction in antibody titers following influenza vaccination. The mice used in these studies were naïve to influenza, whereas virtually all humans have been previously exposed, and it is possible that the mechanisms involved in recall responses are more resilient to changes in the gut microbiota as compared to a primary response. These results highlight the true adaptive nature of the human immune system, which appears able to largely maintain appropriate response to stimulus or challenge despite significant physiological disturbances consequent to perturbations in the microbiome. This also highlights the importance of immune memory and the environment in shaping the immune system. Previous work has shown that immune variation within the human population is driven primarily by environmental factors such as prior immune exposure, rather than genetic factors (Brodin et al., 2015). The results of this study are consistent with the concept of immune responses in adults being largely determined by immune history and resilient to transient changes in the microbiome. However, it will be extremely important in the future to extend such types of analyses to infant populations, where the immature state of the immune system may be more susceptible to such perturbation, and not subject to imprinting. This is already evinced by reports of correlation between autoimmune disorders such as asthma and type 1 diabetes and delivery method-associated gut microbial differences in infants (Dominguez-Bello et al., 2010; Giongo et al., 2011; Thavagnanam et al., 2008)
With regards to innate immune responses, we observed that antibiotics-administration alone induced significant cellular and transcriptional changes within the immune system, promoting a pro-inflammatory state. Integrative analysis revealed a strong association of these inflammatory responses with decreases in bacterial-mediated production of secondary bile acids. In addition to TGR5-mediated inhibition of the NLRP3 inflammasome (Guo et al., 2016), secondary bile acids such as LCA have also been shown to suppress pro-inflammatory cytokine production and NFkB target expression through binding to both the vitamin D receptor (Bakke and Sun, 2018; Makishima et al., 2002) and pregnane X receptor (Shah et al., 2007; Staudinger et al., 2001). Together, these results suggest that the inflammatory responses detected in peripheral blood following antibiotics treatment are driven by increased inflammasome signaling as a consequence of impairments in bile acid metabolism by the gut flora.
Interestingly, the pro-inflammatory state induced by antibiotics-administration showed similarity to early transcriptional responses to influenza vaccination in elderly adults. The composition of the microbiome is known to change with age, with decreases in the phylum Firmicutes and overall diversity (Salazar et al., 2017). The impact of age-related changes in the microbiome on bile acid metabolism is not well established (Frommherz et al., 2016), but there is evidence that the gut microbiota of elderly subjects are less capable of producing other immunomodulatory metabolites, such as short-chain fatty acids (Rampelli et al., 2013). These upregulated pathways (AP-1 and NR4A signaling) are also known to be triggered by bacterial LPS, and gut permeability has been shown to increase with age in mice (Thevaranjan et al., 2017). Further investigations into the direct and indirect mechanisms by which gut microbiota can influence the immune system in the elderly would be of value in understanding how age-associated changes in the microbiome can contribute to chronic inflammation and immunosenescence.
Finally, we constructed a MMRN which integrated responses to antibiotics treatment and influenza vaccination across multiple data types. The findings suggest that the microbiome modulates bile acid metabolism and its consequent effects on inflammation through a different mechanism from its impact on antibody responses. Interestingly, the IgG1 response was significantly associated with two metabolic clusters highly enriched in fatty acid metabolism, which is emerging as an important orchestrator of immune responses (Ganeshan and Chawla, 2014). These results highlight the capability of the microbiome to exert diverse effects on immune function, not only through direct interaction with immune cells in the gut, but also through indirect mechanisms such as regulating the systemic availability of critical metabolites.
Here we have demonstrated the potential for antibiotic-driven perturbation of the microbiome to influence immune responses to vaccination in healthy adults. As antibiotics and vaccines represent two of the most widely used medical interventions, this has important implications for clinical practice and public health. These findings should inform further research seeking to better understand mechanisms that control the interplay between the gut microbiota and our immune system.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bali Pulendran (bpulend@stanford.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
During the 2014-2015 and 2015-2016 seasons, we enrolled a total of 33 healthy adults who were randomized into antibiotics-treated (n=16) and control (n=17) groups. Subjects were males and non-pregnant females between the ages of 18-40 who met the eligibility criteria as listed on clinicaltrials.gov (). Subject demographics are listed in Table S4. The antibiotics treatment consisted of a cocktail of neomycin, vancomycin, and metronidazole, all given orally, for five days. Antibiotic treatment started 3 days before the day of vaccination and continued until one day after for the antibiotics-treated group. All the study participants were vaccinated with Fluzone for the 2014-2015 or 2015-2016 season. Written informed consent was obtained from each subject and protocols were approved by Institutional Review Boards of Emory University.
METHOD DETAILS
Treatment protocol and specimen collection
This study was conducted in Atlanta, GA and consisted of two separate phases. In phase 1, a total of 22 subjects were enrolled in spring of 2015 and randomized into antibiotics-treated (n=11) and control (n=11) groups. In phase 2 conducted in the spring of 2016, subjects who did not receive IIV in the past 3 influenza seasons, were pre-screened for MN titers < 320 for at least two out of the three influenza strains contained in the vaccine. A total of 11 subjects in phase 2 were enrolled and split into antibiotics-treated (n=5) and control (n=6) groups. All subjects but one (in whom we were unable to obtain pre-vaccination blood sample) were vaccinated with a trivalent influenza vaccine (TIV). The influenza strains contained in the vaccines are listed in Table S5. The antibiotic regimen consisted of a cocktail of three different antibiotics, neomycin sulfate (500 mg three times a day), vancomycin (125 mg four times a day), and metronidazole (500 mg three times a day) administered orally for 5 days. Pill count was performed at days 0, 1 and 3. Safety labs were performed prior to vaccination and 7 days later. Written informed consent was obtained from each subject prior to any study procedure and the study was approved by the Emory University Institutional Review Board. Blood and stool samples were collected at regular intervals as indicated in Figure 1.
Cells, plasma and RNA isolation
Peripheral blood mononuclear cells (PBMCs) and plasma were isolated from fresh blood (CPTs; Vacutainer® with Sodium Citrate; BD), following the manufacturer’s protocol. PBMCs were frozen in DMSO with 10% FBS and stored at −80°C and then transferred on the next day to liquid nitrogen freezers (−196°C). Plasma samples from CPTs were stored at −80°C. Trizol (Invitrogen) was used to lyse fresh PBMCs (1 ml of Trizol to ~1.5 × 106 cells) and to protect RNA from degradation. Trizol samples were stored at −80°C.
Microbiome analysis
Sample isolation
Second Genome (http://www.secondgenome.com) performed nucleic acid isolation from fecal samples with the MoBio PowerMag® Microbiome kit (Carlsbad, CA) according to manufacturer’s guidelines and optimized for high-throughput processing. All samples were quantified via the Qubit® Quant-iT dsDNA High Sensitivity Kit (Invitrogen, Life Technologies, Grand Island, NY) to ensure that they met minimum concentration and mass of DNA.
Library preparation
To enrich the sample for bacterial 16S V4 rDNA region, DNA was amplified utilizing fusion primers designed against the surrounding conserved regions which are tailed with sequences to incorporate Illumina (San Diego, CA) adapters and indexing barcodes. Each sample was PCR amplified with two differently barcoded V4 fusion primers. Samples that met the post-PCR quantification minimum and were advanced for pooling and sequencing. For each sample, amplified products were concentrated using a solid-phase reversible immobilization method for the purification of PCR products and quantified by qPCR.
Profiling methods
A pool containing 16S V4 enriched, amplified, barcoded samples were loaded into a MiSeq® reagent cartridge, and then onto the instrument along with the flow cell. After cluster formation on the MiSeq instrument, the amplicons were sequenced for 250 cycles with custom primers designed for paired-end sequencing.
qPCR assay
Strain quantification was performed by qPCR with SYBRGreen on the ABI 5900ht. Universal 16S primers were used:
• 16SU-F 5’-ACTCCTACGGGAGGCAGCAGT-3’
• 16SU-R1 5’-TATTACCGCGGCTGCTGGC-3’
Reactions were composed of 10 μL SYBRGreen MasterMix (2x, VeriQuest), 1 μL each primer (10uM), 6 μL PCR-grade water, and 2 μL of DNA template (<100 ng). Reaction conditions began with 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 sec at 95°C, 30 sec at 54°C and 30 sec at 72°C. Each sample was run in triplicate and the mean value was used. Standard curves were generated with each qPCR assay and used to determine sample quantification. The average quantities of 16s copies per sample were normalized to target gene copies per gram of stool.
Flagellin and LPS quantification
The quantifications of bioactive flagellin and lipopolysaccharide (LPS) in feces were conducted as previously described using human embryonic kidney (HEK)-Blue-mTLR5 and HEK-Blue-mTLR4 cells respectively (Invivogen, San Diego, CA, USA) (Chassaing et al., 2015). Fecal samples were prepared at a concentration of 100 mg/mL and homogenized before centrifuging at 8000g for 10 min, the resulting supernatant was serially diluted and measured by the cell lines. Purified native flagellin (Salmonella Typhimurium) and LPS (Sigma-Aldrich) were used for standard curve determination using HEK-Blue-mTLR5 and HEK-Blue-mTLR4 cells, respectively. For measuring flagellin in plasma, 20 μl of diluted plasma (1:5) was added to 180 μl culture medium. After 24 h of stimulation, the cell culture supernatant was applied to QUANTI-Blue medium (Invivogen) and the alkaline phosphatase activity was measured at 640 nm.
Measurement of LPS-specific IgA and IgG antibodies
LPS-specific IgA and IgG levels were measured by ELISA. Microtiter plates were coated with purified LPS (2 μg/well). Plasma samples from this study subjects were diluted 1:200 before applying to the coated plate. After incubation and washing, the wells were incubated with antihuman IgA (KPL) or IgG (GE Healthcare) coupled to horseradish peroxidase. The colorimetric peroxidase substrate tetramethylbenzidine was used to quantify the total IgA and IgG in plasma, and optical density (OD) of each well was read at 450 nm with an ELISA plate reader.
Influenza microneutralization assay
Viral-neutralizing activity was analyzed in a microneutralization (MN) assay in MDCK cells based on the methods of the pandemic influenza reference laboratories of the Centers for Disease Control and Prevention (CDC) with minor modifications provided in the updated SOP issued by the CDC. Antibody-neutralization titers by MN were measured against seasonal influenza vaccine strains of H1N1-A/California/7/2009, H3N2-A/Texas/50/2012, H3N2- A/Switzerland/9715293/2013, B/Massachusetts/2/2012 and B/Phuket/3073/2013. Sera were tested at an initial dilution of 1:20, and those that were negative (<1:20) were assigned a titer of 10. All sera were tested in triplicate, and the geometric mean value was used for analysis.
Measurement of antigen-specific IgG1 and IgG2 antibodies by ELISA
A Hemagglutinin-specific ELISA was used to measure IgG subclasses in human sera. Thermo 2 HB Immulon Microtiter Plates Flat-Bottom (supplier no. 3355) were coated with A/California/07/09 HA0 protein (BEI Resources) overnight. Plates were washed and blocked using Thermo diluent from the Novex Human IgG Subclass Profile Novex (Life Technologies) kit. Sera dilutions (1:500, 1:1000, 1:2000 and 1:4000) were added to coated plates for 1 hr at RT. After washing, either anti- human IgG1 or IgG2 [Southern Biotech mouse anti-human IgG1 (9054-01) and IgG2 (9070-01)] was added and kept for 1 hr at RT. After washing, Goat antimouse IgG Fc-specific Affinipure (Jackson 115-035-071) was added and reactivity revealed by Thermo TMB solution. Plates were read by Molecular Devices plate reader.
Measurement of antigen-specific IgG1 and IgA1 antibodies by Luminex
Antigen-specific antibody subclass/isotypes were determined using a high-throughput Luminex-based assay (Brown et al., 2012). The H1 A/California/07/2009 antigen was provided from Immune Technology Corp. (New York, NY). The antigen was coupled to carboxylate-modified microspheres (Luminex Corp., Austin, TX) by covalent NHS-ester linkages via EDC (ThermoFisher) and Sulfo-NHS (ThermoFisher) per manufacturer’s instructions. These antigen-coated microspheres were added to non-binding 384-well plates (Grenier Bio-One, Kremsmunster, Austria) at 1000 beads per well (45μl). Serum samples were diluted 1:100 in PBS for IgG1; samples were diluted 1:10 for other subclasses prior to incubation with beads. 5μl of diluted serum samples were added to plates with microspheres and incubated on a shaker overnight at 4°C. Microspheres were washed in PBS buffer containing 0.1% BSA and 0.05% Tween-20, and incubated for 1 hour at room temperature with secondary detectors, namely PE-conjugated anti-IgG1 or -IgA1 detection antibodies (Southern Biotech, Birmingham, AL). The microspheres were washed as before and read on an iQue Screener Plus (Intellicyt Corp., Albuquerque, NM).
Binding kinetics and antibody affinity of polyclonal serum antibodies to rHA protein by surface plasmon resonance
Steady-state equilibrium binding of post-vaccination individual sera was monitored at 25°C using a ProteOn surface plasmon resonance biosensor (BioRad) as previously described (Khurana et al., 2011). The rHA protein from the H1N1-A/California/7/2009 prepared in house (Khurana et al., 2010) was coupled to a GLC sensor chip with amine coupling with 500 resonance units (RU) in the test flow cells. Samples of 60 μL freshly prepared sera at 10- and 100-fold dilutions were injected at a flow rate of 50 μL/min (120-sec contact time) for association, and dissociation was performed over a 1200 second interval (at a flow rate of 50 μL/min). Responses from the protein surface were corrected for the response from a mock surface and for responses from a separate, buffer only injection. MAb 2D7 (anti-CCR5) was used as a negative control in these experiments. Total antibody binding was determined directly from the serum sample interaction with rHA protein of the influenza virus by SPR using the BioRad ProteOn manager software as described before. Antibody off-rate constants, which describe the fraction of antigen-antibody complexes that decay per second, were determined directly from the serum/plasma sample interaction with rHA using SPR in the dissociation phase and calculated using the BioRad ProteOn manager software for the heterogeneous sample model as described before. Off-rate constants were determined from two independent SPR runs.
Flow cytometry analysis of immune populations
Flow cytometry analysis was done on either heparinized whole blood (plasmablasts and blood follicular T helper like cells) or cryopreserved PBMCs (activated B cells and innate immune populations).
Whole blood
For whole blood assays, cells were first surface-stained with an appropriate antibody cocktail for 20 min at room temperature followed by erythrocyte lysis using BD FACS lysing solution (BD Biosciences). Cells were then fixed and permeabilized with BD Cytofix/Cytoperm for 30 min on ice and anti-Ki67 PE-CY7 (BD Biosciences) added for 30 min on ice in BD permeabilization buffer. All samples were run on an LSR II (BD Biosciences) and analyzed via the FlowJo software (FlowJo, LLC).
For the identification of plasmablasts, cells were stained with a cocktail of anti-CD19 FITC, anti-CD27 PE, anti-CD3 PerCP, anti-CD38 APC, anti-CD20 Pacific Blue, anti-HLA-DR V500, anti-CD195 BV605, and anti-CD138 PE-CF594 antibodies. Plasmablasts were identified as CD3−, CD19+, CD27++, CD38++ cells and quantified as percentage of total CD19+ B cells.
For the identification of blood Tfh-like cells, a cocktail of anti-CD3 Alexa Fluor 700, anti-CD4 PerCP-Cy5.5, anti-CD45RA BV605, anti-CD183 Alexa Fluor 488, anti-CD196 PE-CF594, anti-CD279 BV421, anti-CD14 APC-H7, anti-CD20 APC-H7, anti-CD185 PE, and anti-CD278 Alexa Fluor 647 antibodies was used. Activated blood Tfh type 1 cells were described previously by Schmitt and Ueno (Schmitt and Ueno, 2013). This subset of circulating Tfh-like cells is defined in our study as CD3+, CD4+, CXCR5+, CXCR3+, CCR6-, ICOS+, PD-1+ and their frequency levels on day 7 after vaccination were presented as log2 fold change over day 0 frequencies.
Cryopreserved PBMCs
2-3 × 106 cells PBMCs were thawed and stained for 20 min at room temperature with an appropriate antibody cocktail. Cells were washed in PBS with 5% fetal bovine serum, fixed with the Cytofix buffer (BD), and then analyzed on the LSR II flow cytometer (BD Biosciences). All flow cytometry analysis was done using the FlowJo software (FlowJo, LLC).
For the identification of activated/non-activated myeloid (mDCs) and plasmacytoid (pDCs) dendritic cells, a cocktail of anti-CD3 FITC, anti-CD19 FITC, anti-CD20 FITC, anti-CD56 FITC, anti-CD80 PE, anti-BDCA-1 PerCP-Cy5.5, anti-CD123 PE-Cy7, anti-CD33 BV421, anti-CD14-BV605, anti-CD11b BV650, anti-CCR7 BV711, anti-CD16 APC, anti-CD11c Alexa700, and HLA-DR APC-Cy7 antibodies was used. Activated (CD80+) CD16+ mDCs were defined within the singlets gate as CD3−CD19−CD20−CD56− cells and then uniquely identified as CD14−HLA−DR+ CD123− CD11c+ CD16+ CD1c− CD80+. pDCs were defined within the singlets gate as CD3−CD19−CD20−CD56− cells and then uniquely identified as CD14−HLA−DR+ CD123+ CD11c−. Activated pDCs are also CD80+.
For the identification of activated B cells (ABCs), a cocktail of anti-CD21 FITC, anti-CD38 PE, anti CD19 PE-CF594, anti-HLA-DR PerCP-Cy5.5, anti-CD27 PE-Cy7, anti-IgD BV421, anti-CD69 BV605, anti-CD20 BV650, anti-CD56 BV711, anti-CD71 APC, anti-CD16 Alexa700, and anti-CD3 APC-Cy7 antibodies was used. ABCs were described previously by (Ellebedy et al., 2016). ABCs were uniquely identified as CD3-CD19+CD20+IgD-CD71+.
ELISPOT measurement of vaccine-specific antibody-secreting cells (ASCs)
ELISPOT plates (Millipore, cat. #MAIPS4510) were coated overnight at 4°C with the administered trivalent influenza vaccine (Fluzone) at 9μg/ml in PBS, subsequently washed, and complete medium added for 2 hours at 37°C to avoid aspecific binding. The following day, in order to enumerate the number of IgG-secreting vaccine-specific plasmablasts, dilutions of washed PBMCs (ranging from 6×105 to 2×104 cells) were added to the ELISPOT plates and incubated overnight at 37°C with 5% CO2. After incubation, plates were washed and alkaline phosphatase-conjugated goat anti-human IgG antibody (Millipore) added for 4 hours at room temperature (RT). Alkaline phosphatase (AP) substrates (Bio-Rad, cat. #170-6432) were then added to each well for spot reveal. Plates were scanned and spots counted using the CTL Immunospot reader software.
Somatic hypermutation (SHM) analysis of influenza-reactive hmAbs
Plasmablasts (CD3-CD19+CD27hiCD38hi) were single cell-sorted into 96-well PCR plates (Bio-Rad) 7 days after vaccination. Monoclonal antibodies sequences were amplified by singlecell PCR amplification of the variable heavy and light chain genes and inserted into a human IgG expression vector, as previously described (Chen et al., 2018; Henry Dunand et al., 2016; Wrammert et al., 2008). mAbs were generated by transfecting a mAb’s respective individual heavy and light chain expression vectors into HEK293T cells and mAbs purified using Protein A agarose beads 6 days after transfection. Purified mAbs were tested for binding to the seasonal vaccine and for binding individual viral components of the seasonal vaccine. SHM data was obtained by comparing mAb sequences to germline sequences deposited in IgBlast.
Microarray experiments
Total RNA from fresh PBMCs (~1.5 × 106 cells) was purified using Trizol® (Invitrogen, Life Technologies Corporation) according to the manufacturer's instructions. All RNA samples were checked for purity using a ND-1000 spectrophotometer (NanoDrop Technologies) and for integrity by electrophoresis on a 2100 BioAnalyzer (Agilent Technologies). Two-round in vitro transcription amplification and labeling was performed starting with 50 ng intact, total RNA per sample, following the Affymetrix protocol. After hybridization on Human U133 Plus 2.0 Arrays (using GeneTitan platform, Affymetrix, or individual cartridges) for 16 h at 45 °C and 60 r.p.m. in a Hybridization Oven 640 (Affymetrix), slides were washed and stained with a Fluidics Station 450 (Affymetrix). Scanning was performed on a seventh-generation GeneChip Scanner 3000 (Affymetrix), and Affymetrix GCOS software was used to perform image analysis and generate raw intensity data.
Plasma metabolomics analysis
Metabolomics analysis was performed similarly as previously described (Li et al., 2013). Briefly, metabolites were extracted by the addition of 130 mL of acetonitrile to 65 mL of plasma, followed by mixing, and 30 min of incubation on ice and centrifugation (13,400 x rpm at 4C) for 10 min. The supernatant was transferred into autosampler vials for LC-MS analysis. Mass spectral data were collected with a 10 min reversed phase gradient on a Thermo Q Exactive mass spectrometer (Thermo Fisher, San Diego, CA) set to record from mass-to-charge ratio (m/z) 85 to 2000. Three technical replicates were run for each sample using reversed phase chromatography (Higgins Analytical, Targa C18 5 mM, 100 3 2.1 mm) using both positive and negative electrospray ionization.
Measurement of bile acids by liquid chromatography-mass spectrometry (LC-MS)
The measurement of bile acids was performed similarly as previously described (Voelker et al., 2013) with some modifications. Briefly, concentrations of primary and secondary bile acids in human plasma were quantified using a Shimadzu CBM-20A HPLC system coupled with a Thermo Q Exactive Focus mass spectrometer. For chromatography, a Phenomenex C18 column (4.6×100mm 2.6 μm) was used. The mobile phase A was 10mM ammonium acetate in water, while mobile phase B was 50:50 methanol:acetonitrile (v/v). The column temperature was held at 60°C, and the gradient was kept at 35% mobile phase B for two minutes before being raised to 55% mobile phase B in 4.5 minutes. Then the gradient was kept at 55% mobile phase B for two minutes. Finally, the column was eluted with 95% mobile phase B for 1 minute. The flow rate was 1.2 mL/min. For mass spectrometry, a Thermo Scientific Q Exactive Focus mass spectrometer with a HESI and operating in the negative ion mode was used. The ion source temperature was set to 400 °C. Quantitation was performed using target SIM with a resolution of 70,000. Standards used included CDCA (chenodeoxycholic acid), DCA (deoxycholic acid), gCDCA (glycochenodeoxycholic acid), gDCA (glycodeoxycholic acid), tCDCA (taurochendeoxycholic acid), tDCA (taurodeoxycholic acid), gLCA (glycolithocholic acid), CA (cholic acid), tCA (taurocholic acid), tLCA (taurolithocholic acid), LCA (lithocholic acid), and gCA (glycocholic acid). For gCDCA, standard was dissolved in acetonitrile to generate ten concentration gradients including 2.5 ng/mL, 5 ng/mL, 10 ng/mL, 50 ng/mL, 200 ng/mL, 500 ng/mL, 1500 ng/mL, 2250 ng/mL, 2500 ng/mL and 5000 ng/mL. For the other 11 bile acids, standards were dissolved in acetonitrile to generate ten concentration gradients including 0.5 ng/mL, 1 ng/mL, 2 ng/mL, 10 ng/mL, 40 ng/mL, 100 ng/mL, 300 ng/mL, 450 ng/mL, 500 ng/mL and 1000 ng/mL.
QUANTIFICATION AND STATISTICAL ANALYSIS
Microbiome data analysis
OTU selection
Sequenced paired-end reads were merged using USEARCH (Edgar, 2010) and the resulting sequences were compared to an in-house strains database using USEARCH (usearch_global). All sequences hitting a unique strain with an identity >=99% were assigned a strain Operation Taxonomic Unit (OTU). To ensure specificity of the strain hits, a difference of >=0.25% between the identity of the best hit and the second best hit was required (e.g. 99.75 versus 99.5). For each strain OTU one of the matching reads was selected as representative and all sequences were mapped by USEARCH (usearch_global) against the strain OTU representatives to calculate strain abundances. The remaining non-strain sequences were quality filtered and dereplicated with USEARCH. Resulting unique sequences were then clustered at 97% by UPARSE (de novo OTU clustering) and a representative consensus sequence per de novo OTU was determined. The UPARSE clustering algorithm comprises a chimera filtering and discards likely chimeric OTUs. All non-strain sequences that passed the quality filtering were mapped to the representative consensus sequences to generate an abundance table for de novo OTUs. Representative OTU sequences were assigned taxonomic classification via bayesian classifier in mothur (Schloss et al., 2009), trained against the Greengenes reference database of 16S rRNA gene sequences clustered at 99%.
Alpha-diversity (within sample diversity) metrics
‘Observed’ diversity is simply the sum of unique OTUs found in each sample, also known as sample richness. Shannon diversity (H) utilizes the richness of a sample along with the relative abundance of the present OTUs to calculate a diversity index according to the following formula:
where pi is the relative abundance of the ith OTU and S is the total number of OTUs.
Beta-diversity (sample-to-sample dissimilarity) metrics
All profiles are inter-compared in a pair-wise fashion to determine a dissimilarity score and store it in a distance dissimilarity matrix. Distance functions produce low dissimilarity scores when comparing similar samples. Abundance-weighted sample pair-wise differences were calculated using the Bray-Curtis dissimilarity. Bray-Curtis dissimilarity is calculated by the ratio of the summed absolute differences in counts to the sum of abundances in the two samples (Bray and Curtis, 1957). The binary dissimilarity values were calculated with the Jaccard index. This metric compares the number of mismatches (OTUs present in one but absent in the other) in two samples relative to the number of OTUs present in at least one of the samples (Jaccard, 1912).
Ordination
Two-dimensional ordinations were created using principal coordinate analysis (PCoA) (Gower, 1966) to graphically summarize the inter-sample relationships. PCoA is a method of twodimensional ordination plotting that is used to help visualize complex relationships between samples. PCoA uses the sample-to-sample dissimilarity values to position the points relative to each other by maximizing the linear correlation between the dissimilarity values and the plot distances.
Transcriptomics data processing and pathway/module analysis
Initial data quality was assessed by background level, 3’ labeling bias, and pairwise correlation among samples. CEL files from outlier samples were excluded and the remaining CEL files of all the samples belonging to the same trial were grouped and normalized in Bioconductor by RMA (Irizarry et al., 2003), which includes global background adjustment and quantile normalization. Probes mapping to multiple genes were discarded, and the remaining probes were collapsed to gene level by selecting the probe for each gene with the highest mean expression across all subjects in both trials. Statistical tests and correlation analyses were performed using MATLAB. Test details and significance cutoffs are reported in figure legends. Enrichment tests of BTMs were performed using GSEA (Subramanian et al., 2005). BTMs were visualized using Cytoscape (Shannon et al., 2003).
Metabolomics and pathway analysis
Metabolite peaks were extracted using the metabolomics software package apLCMS (Yu et al., 2009). Only peaks that were present in > 50% samples were used for further analysis. Pearson correlation coefficient R between the three technical replicates was used as an indicator of analytical quality. Samples were removed if pairwise correlation between 3 technical replicates was R<0.7. Otherwise, average value of technical replicates was used. If a single technical replicate had R<0.7 with the other two, it was removed and other two were averaged. For each sample, data were log2 transformed. A total of 5572 peaks and 5745 peaks were detected for positive and negative ion mode, respectively. Statistical tests, correlation analyses, and PCA were performed using MATLAB. Test details and significance cutoffs are reported in figure legends. Mummichog software (Li et al., 2013) was used to identify enrichment in metabolic pathways, using significant peaks selected as p<0.05 by pairwise t-test between each time point and the baseline (day −21 or day 0).
Construction and query of MMRN
The MMRN integration of transcriptomics, metabolomics, microbiomics, and cell frequencies was performed similarly as previously described (Li et al., 2017), using updated software (hiconet). Briefly, feature clusters were first identified in each data type (excluding metabolomics) through similarity of expression across all time points via the Leiden algorithm (Traag et al., 2019), a common modularity algorithm for community detection. An additional step was performed prior to the clustering for the transcriptomics data, which were collapsed to BTMs, and module activity scores were taken as the mean value of member genes. The metabolomics data were clustered using a hierarchical clustering method customized for LC-MS data by taking into account chromatographic retention time, as described previously (Gardinassi et al., 2018). Associations between the feature clusters of each data type were then estimated by partial least square (PLS) regression. The significance of associations was computed by comparing with permutation data, resampling both features and sample labels. The resulting networks were visualized using Cytoscape (Shannon et al., 2003). To query the network for association with LCA or H1N1-specific IgG1 data, ranked lists of all network features (BTMs, m/z peaks, OTUs, and cell frequencies) were generated using Spearman correlation with either LCA or IgG1 as the ranking metric. Each cluster in the MMRN was then treated as a “geneset”, and enrichment testing was performed via GSEA (Subramanian et al., 2005) to identify clusters whose member features were enriched at either end of the ranked lists (highly correlated with LCA or IgG1).
DATA AND CODE AVAILABILITY
The data generated in this study is available at ImmPort (https://www.immport.org) under accession SDY1086. The microarray data is additionally available at GEO: accession GSE120719.
ADDITIONAL RESOURCES
Clinical trial registration (): https://clinicaltrials.gov/ct2/show/NCT02154061
Supplementary Material
Table S1. Wilcoxon-signed rank tests on the most abundant bacterial families before and after antibiotics administration, related to Figure 1.
Table S2. Comparison of MN antibody titers before vaccination between phase 1 and phase 2, related to Figure 2.
Table S3. Member features and pathway enrichments of nodes in the MMRN, related to Figure 7.
Table S4. Demographic information of study participants, related to STAR Methods.
Table S5. Influenza virus strains contained in the vaccines, related to STAR Methods.
Figure S1. Effects of antibiotic use on the gut microbiome of healthy adults in phase 2, related to Figure 1.
(A) Normalized copy number of bacterial 16s ribosomal RNA per gram of stool for 11 additional subjects recruited during the 2015-2016 influenza season and pre-screened for microneutralization (MN) antibody titers. Each line corresponds to an individual subject. Controls are shown in blue, antibiotics-treated subjects in red. Median values and distributions for each time point are illustrated in the form of box plots.
(B) Relative abundance of bacterial families in 5 antibiotics-treated subjects recruited during the 2015-2016 influenza season at different time points. Each vertical bar corresponds to a study participant. On day 1 and day 180, data is available for 4 and 3 individuals only, respectively.
(C) Alpha-diversity estimates for 5 antibiotics-treated subjects enrolled during the 2015-2016 season. See Figure 1 for further details.
(D-F) Flagellin (D), anti-LPS IgG (E), and anti-LPS IgA (F) relative concentrations assessed in the plasma of the 32 subjects (both 2014-2015 and 2015-2016 influenza seasons) enrolled in the study. Relative measurements are reported as optical density (OD) values.
Figure S2. Additional measurements of humoral responses to TIV, related to Figure 2.
(A-B) MN titers for each 2014-2015 (A) or 2015-2016 (B) TIV strain at day 30 presented as fold changes over baseline values (day 0). Each bar corresponds to an individual subject. Seroconversion rates describe the proportion of subjects who developed a ≥ 4-fold increase in titer between pre- and day 30 post-vaccination sera.
(C) IgG2-binding to A/California H1 for phase 1 (left panel) and phase 2 (right panel) measured by ELISA. Violin plots show sample distributions. Each circle represents an individual subject, while medians are presented in thick lines.
(D) A/California H1 HA-specific IgA isotype binding capacity measured by SPR and presented as maximum resonance units (max RU). Violin plots show sample distributions. Each circle represents an individual subject, while medians are presented in thick lines.
Figure S3. Adaptive cellular responses to TIV, related to Figure 2
(A) Flow cytometry analysis of plasmablasts (PBs) on day 0 and day 7 after vaccination for phase 1 (left panel) and phase 2 (right panel) subjects. Plasmablasts are identified as CD27hi CD38hi cells within total CD19+ B cells.
(B) Ex vivo ELISPOT measurements of TIV-specific IgG antibody-secreting cells (ASC) per million PBMC at days 0 and 7.
(C) Flow cytometry analysis of activated B cells (ABC) as described in Ellebedy et al. (Ellebedy et al., 2016). ABC are defined as the CD71+ CD38int-lo CD20hi subpopulation within total CD19+ B cells. Fold change frequencies (d7/d0) are presented.
(D) Log2 fold change frequency levels (d7/d0) of activated blood T follicular helper 1 (Tfh1)-like cells measured by flow cytometry. Activated blood Tfh1-like cells are defined as CXCR5+ CXCR3+ PD1+ ICOS+ CD4+ T cells, as described previously (Schmitt and Ueno, 2013). (E) Somatic hypermutation (SHM) analysis of vaccine-reactive human monoclonal antibodies (hmAbs) generated from single cell-sorted plasmablasts 7 days after vaccination. The number of mutations in the light (left panel) and heavy (right panel) antibody chains was calculated for a total of 35 hmAbs from 8 antibiotics-treated subjects (red circles) which were found to bind at least one of the TIV strains included in the 2014-2015 formulation and compared versus 223 hmAbs from healthy controls, 212 of which belonged to individuals who received a quadrivalent influenza vaccine (QIV) that same season (light blue squares). HmAbs from controls in this study are highlighted in dark blue.
Where calculated, comparisons between control and antibiotics-treated groups at specific time points were performed by Mann-Whitney tests. NS – not significant.
Figure S4. Comparison of transcriptional and metabolic responses in phases 1 and 2, related to Figures 3, 5, and 6.
(A) Comparison of enrichment scores for BTMs significantly enriched (FDR < 0.05, NES > 2.2) post-vaccination in antibiotics-treated subjects in either phase 1 or 2. See Figure 3B for results of all subjects combined.
(B) Metabolic trajectories along the first two principal components for control (blue) and antibiotics-treated (red) subjects in phase 1 (dark) and phase 2 (light) for days 0-7 relative to the screening time point. See Figure 5F for results of all subjects combined.
(C) Fold change of LCA in the plasma among antibiotics-treated (red) and control (blue) subjects in phases 1 and 2. Each thin line represents a single subject, thick lines represent geometric means. See Figure 6E for results of all subjects combined.
Figure S5. Workflow of MMRN construction, related to Figure 7.
Feature clusters were identified in each data type through clustering algorithms. Associations between the feature clusters of each data type were then estimated by partial least square (PLS) regression. The network was then queried for associations with LCA or H1N1-specific IgG1 data through an enrichment-based approach. See STAR Methods for details.
Microbiome loss impairs antibody response in subjects with low pre-existing immunity
Antibiotics treatment leads to enhanced inflammatory signatures in the blood
Loss of secondary bile acids is linked to AP-1/NR4A and inflammasome activation
Integrative analysis reveals divergent mechanisms of microbiome influence on immunity
Antibiotic use-induced alterations to the gut microbiome can adversely affect immunogenicity and responses to influenza vaccination in humans
Acknowledgements
We are grateful to the Hope Clinic staff who assisted with the clinical work, particularly Mary Bower, Eileen Osinski, and Briyana Domjahn. We would also like to thank Luisa Chan, Kathryn Iverson, and Second Genome for their work in generation and analysis of the microbiome data, Lily Chen and Creative Proteomics for measurement of bile acids by LC-MS, Dmitri Kazmin for data management and curation, Chris Ibegbu and the Emory Immunology/Flow Cytometry Core for technical assistance, Amber N. Wohlabaugh for library preparation and sequencing, Mark Mulligan and Aneesh Mehta for advice about the clinical study design, and Mark Davis, Rafi Ahmed, David Relman, Justin Sonneburg and Taia Wang for helpful discussion. This work was supported by Emory-UGA CEIRS Contract HHSN272201400004C (to B.P., P.I. Walt Orenstein), NIH grants HIPC U19AI090023 (to B.P.), U19AI057266 (to B.P., P.I. Rafi Ahmed), UH2AI132345 (to S.L.), U24AI120134 (S.E.B.), T32AI07290 (to T.H.), the Sean Parker Cancer Institute, the Soffer endowment (B.P), and the Violetta Horton endowment (B.P).
Footnotes
Declaration of Interests: Nothing to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Agus A, Planchais J, and Sokol H (2018). Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 23, 716–724. [DOI] [PubMed] [Google Scholar]
- Bakke D, and Sun J (2018). Ancient Nuclear Receptor VDR With New Functions: Microbiome and Inflammation. Inflamm Bowel Dis 24, 1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becattini S, Taur Y, and Pamer EG (2016). Antibiotic-Induced Changes in the Intestinal Microbiota and Disease. Trends Mol Med 22, 458–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y, and Hand TW (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray JR, and Curtis JT (1957). An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol Monogr 27, 326–349. [Google Scholar]
- Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, et al. (2006). Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12, 1365–1371. [DOI] [PubMed] [Google Scholar]
- Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, et al. (2015). Variation in the human immune system is largely driven by non-heritable influences. Cell 160, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EP, Licht AF, Dugast AS, Choi I, Bailey-Kellogg C, Alter G, and Ackerman ME (2012). High-throughput, multiplexed IgG subclassing of antigen-specific antibodies from clinical samples. J Immunol Methods 386, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill CM, and Rogers JT (2008). Interleukin (IL) 1beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IkappaB kinase alpha pathway targeting activator protein-1. J Biol Chem 283, 25900–25912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabotti M, Scirocco A, Maselli MA, and Severi C (2015). The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol 28, 203–209. [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, and Gewirtz AT (2015). Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YQ, Wohlbold TJ, Zheng NY, Huang M, Huang Y, Neu KE, Lee J, Wan H, Rojas KT, Kirkpatrick E, et al. (2018). Influenza Infection in Humans Induces Broadly Cross-Reactive and Protective Neuraminidase-Reactive Antibodies. Cell 173, 417–429 e410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins N, and Belkaid Y (2018). Do the Microbiota Influence Vaccines and Protective Immunity to Pathogens? Engaging Our Endogenous Adjuvants. Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S (2014). T follicular helper cell differentiation, function, and roles in disease. Immunity 41, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, and Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 108 Suppl 1, 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, and Knight R (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 107, 11971–11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Ellebedy AH, Jackson KJ, Kissick HT, Nakaya HI, Davis CW, Roskin KM, McElroy AK, Oshansky CM, Elbein R, Thomas S, et al. (2016). Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat Immunol 17, 1226–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasca D, Diaz A, Romero M, Mendez NV, Landin AM, and Blomberg BB (2013). Effects of age on H1N1-specific serum IgG1 and IgG3 levels evaluated during the 2011–2012 influenza vaccine season. Immun Ageing 10, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommherz L, Bub A, Hummel E, Rist MJ, Roth A, Watzl B, and Kulling SE (2016). Age-Related Changes of Plasma Bile Acid Concentrations in Healthy Adults--Results from the Cross-Sectional KarMeN Study. PLoS One 11, e0153959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganeshan K, and Chawla A (2014). Metabolic regulation of immune responses. Annu Rev Immunol 32, 609–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardinassi LG, Arevalo-Herrera M, Herrera S, Cordy RJ, Tran V, Smith MR, Johnson MS, Chacko B, Liu KH, Darley-Usmar VM, et al. (2018). Integrative metabolomics and transcriptomics signatures of clinical tolerance to Plasmodium vivax reveal activation of innate cell immunity and T cell signaling. Redox Biol 17, 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, Drew JC, Ilonen J, Knip M, Hyoty H, et al. (2011). Toward defining the autoimmune microbiome for type 1 diabetes. ISME J 5, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gower JC (1966). Some Distance Properties of Latent Root and Vector Methods Used in Multivariate Analysis. Biometrika 53, 325-&. [Google Scholar]
- Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, Zheng M, Zhang X, Xia D, Ke Y, et al. (2016). Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 45, 802–816. [DOI] [PubMed] [Google Scholar]
- Hagan T, and Pulendran B (2018). Will Systems Biology Deliver Its Promise and Contribute to the Development of New or Improved Vaccines? From Data to Understanding through Systems Biology. Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry Dunand CJ, Leon PE, Huang M, Choi A, Chromikova V, Ho IY, Tan GS, Cruz J, Hirsh A, Zheng NY, et al. (2016). Both Neutralizing and Non-Neutralizing Human H7N9 Influenza Vaccine-Induced Monoclonal Antibodies Confer Protection. Cell Host Microbe 19, 800–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, and Speed TP (2003). Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264. [DOI] [PubMed] [Google Scholar]
- Jaccard P (1912). The distribution of the flora in the alpine zone. New Phytol 11, 37–50. [Google Scholar]
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. (2003). A G protein-coupled receptor responsive to bile acids. J Biol Chem 278, 9435–9440. [DOI] [PubMed] [Google Scholar]
- Keeney KM, Yurist-Doutsch S, Arrieta MC, and Finlay BB (2014). Effects of antibiotics on human microbiota and subsequent disease. Annu Rev Microbiol 68, 217–235. [DOI] [PubMed] [Google Scholar]
- Khurana S, Hahn M, Coyle EM, King LR, Lin TL, Treanor J, Sant A, and Golding H (2019). Repeat vaccination reduces antibody affinity maturation across different influenza vaccine platforms in humans. Nat Commun 10, 3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Verma N, Yewdell JW, Hilbert AK, Castellino F, Lattanzi M, Del Giudice G, Rappuoli R, and Golding H (2011). MF59 adjuvant enhances diversity and affinity of antibody-mediated immune response to pandemic influenza vaccines. Sci Transl Med 3, 85ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S, Verma S, Verma N, Crevar CJ, Carter DM, Manischewitz J, King LR, Ross TM, and Golding H (2010). Properly folded bacterially expressed H1N1 hemagglutinin globular head and ectodomain vaccines protect ferrets against H1N1 pandemic influenza virus. PLoS One 5, e11548. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Klatt NR, Cheu R, Birse K, Zevin AS, Perner M, Noel-Romas L, Grobler A, Westmacott G, Xie IY, Butler J, et al. (2017). Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science 356, 938–945. [DOI] [PubMed] [Google Scholar]
- Kostic AD, Xavier RJ, and Gevers D (2014). The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146, 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MM (2010). Immunogenicity and efficacy of oral vaccines in developing countries: lessons from a live cholera vaccine. BMC Biol 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy M, Blacher E, and Elinav E (2017). Microbiome, metabolites and host immunity. Curr Opin Microbiol 35, 8–15. [DOI] [PubMed] [Google Scholar]
- Li S, Park Y, Duraisingham S, Strobel FH, Khan N, Soltow QA, Jones DP, and Pulendran B (2013). Predicting network activity from high throughput metabolomics. PLoS Comput Biol 9, e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Rouphael N, Duraisingham S, Romero-Steiner S, Presnell S, Davis C, Schmidt DS, Johnson SE, Milton A, Rajam G, et al. (2014). Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat Immunol 15, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Sullivan NL, Rouphael N, Yu T, Banton S, Maddur MS, McCausland M, Chiu C, Canniff J, Dubey S, et al. (2017). Metabolic Phenotypes of Response to Vaccination in Humans. Cell 169, 862–877 e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littman DR (2018). Do the Microbiota Influence Vaccines and Protective Immunity to Pathogens? If So, Is There Potential for Efficacious Microbiota-Based Vaccines? Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, and Mangelsdorf DJ (2002). Vitamin D receptor as an intestinal bile acid sensor. Science 296, 1313–1316. [DOI] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, et al. (2003). TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Res 31, 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitre E, Susi A, Kropp LE, Schwartz DJ, Gorman GH, and Nylund CM (2018). Association Between Use of Acid-Suppressive Medications and Antibiotics During Infancy and Allergic Diseases in Early Childhood. JAMA Pediatr, e180315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi SR, Collins JJ, and Relman DA (2014). Antibiotics and the gut microbiota. J Clin Invest 124, 4212–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya HI, Hagan T, Duraisingham SS, Lee EK, Kwissa M, Rouphael N, Frasca D, Gersten M, Mehta AK, Gaujoux R, et al. (2015). Systems Analysis of Immunity to Influenza Vaccination across Multiple Years and in Diverse Populations Reveals Shared Molecular Signatures. Immunity 43, 1186–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakaya HI, Wrammert J, Lee EK, Racioppi L, Marie-Kunze S, Haining WN, Means AR, Kasturi SP, Khan N, Li GM, et al. (2011). Systems biology of vaccination for seasonal influenza in humans. Nat Immunol 12, 786–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, and Pettersson S (2012). Host-gut microbiota metabolic interactions. Science 336, 1262–1267. [DOI] [PubMed] [Google Scholar]
- Oh JZ, Ravindran R, Chassaing B, Carvalho FA, Maddur MS, Bower M, Hakimpour P, Gill KP, Nakaya HI, Yarovinsky F, et al. (2014). TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity 41, 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Powell N, Vincent RP, Ehrlich D, Bjarnason I, and Hayee B (2015). Systematic review: bile acids and intestinal inflammation-luminal aggressors or regulators of mucosal defence? Aliment Pharmacol Ther 42, 802–817. [DOI] [PubMed] [Google Scholar]
- Pei L, Castrillo A, and Tontonoz P (2006). Regulation of macrophage inflammatory gene expression by the orphan nuclear receptor Nur77. Mol Endocrinol 20, 786–794. [DOI] [PubMed] [Google Scholar]
- Pulendran B (2014). Systems vaccinology: probing humanity's diverse immune systems with vaccines. Proc Natl Acad Sci U S A 111, 12300–12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulendran B, Li S, and Nakaya HI (2010). Systems vaccinology. Immunity 33, 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampelli S, Candela M, Turroni S, Biagi E, Collino S, Franceschi C, O'Toole PW, and Brigidi P (2013). Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging (Albany NY) 5, 902–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappuoli R, Siena E, and Finco O (2018). Will Systems Biology Deliver Its Promise and Contribute to the Development of New or Improved Vaccines? Systems Biology Views of Vaccine Innate and Adaptive Immunity. Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, and Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47, 241–259. [DOI] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, Hylemon PB, and Bajaj JS (2014). Bile acids and the gut microbiome. Curr Opin Gastroenterol 30, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. (2018). Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97. [DOI] [PubMed] [Google Scholar]
- Salazar N, Valdes-Varela L, Gonzalez S, Gueimonde M, and de Los Reyes-Gavilan CG (2017). Nutrition and the gut microbiome in the elderly. Gut Microbes 8, 82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DC (1977). Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol 31, 107–133. [DOI] [PubMed] [Google Scholar]
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. (2013). Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2, e01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt N, and Ueno H (2013). Blood Tfh cells come with colors. Immunity 39, 629–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah YM, Ma X, Morimura K, Kim I, and Gonzalez FJ (2007). Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol 292, G1114–1122. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair C, Bommakanti G, Gardinassi L, Loebbermann J, Johnson MJ, Hakimpour P, Hagan T, Benitez L, Todor A, Machiah D, et al. (2017). mTOR regulates metabolic adaptation of APCs in the lung and controls the outcome of allergic inflammation. Science 357, 1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg JL, and Backhed F (2016). Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, et al. (2001). The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A 98, 3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhoff U (2005). Who controls the crowd? New findings and old questions about the intestinal microflora. Immunol Lett 99, 12–16. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thavagnanam S, Fleming J, Bromley A, Shields MD, and Cardwell CR (2008). A meta-analysis of the association between Caesarean section and childhood asthma. Clin Exp Allergy 38, 629–633. [DOI] [PubMed] [Google Scholar]
- Thevaranjan N, Puchta A, Schulz C, Naidoo A, Szamosi JC, Verschoor CP, Loukov D, Schenck LP, Jury J, Foley KP, et al. (2017). Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 21, 455–466 e454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 9, 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V, and Backhed F (2012). Functional interactions between the gut microbiota and host metabolism. Nature 489, 242–249. [DOI] [PubMed] [Google Scholar]
- Voelker T, Wang H, Irish M, Wang J, Harrison S, Reuschel S, Bennett P, and Meng M (2013). Method development and validation of six bile acids for regulated bioanalysis: improving selectivity and sensitivity. Bioanalysis 5, 1229–1248. [DOI] [PubMed] [Google Scholar]
- Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, Zheng NY, Mays I, Garman L, Helms C, et al. (2008). Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 453, 667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Park Y, Johnson JM, and Jones DP (2009). apLCMS--adaptive processing of high-resolution LC/MS data. Bioinformatics 25, 1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Ma Y, Raoult D, Kroemer G, and Gajewski TF (2018). The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 359, 1366–1370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Wilcoxon-signed rank tests on the most abundant bacterial families before and after antibiotics administration, related to Figure 1.
Table S2. Comparison of MN antibody titers before vaccination between phase 1 and phase 2, related to Figure 2.
Table S3. Member features and pathway enrichments of nodes in the MMRN, related to Figure 7.
Table S4. Demographic information of study participants, related to STAR Methods.
Table S5. Influenza virus strains contained in the vaccines, related to STAR Methods.
Figure S1. Effects of antibiotic use on the gut microbiome of healthy adults in phase 2, related to Figure 1.
(A) Normalized copy number of bacterial 16s ribosomal RNA per gram of stool for 11 additional subjects recruited during the 2015-2016 influenza season and pre-screened for microneutralization (MN) antibody titers. Each line corresponds to an individual subject. Controls are shown in blue, antibiotics-treated subjects in red. Median values and distributions for each time point are illustrated in the form of box plots.
(B) Relative abundance of bacterial families in 5 antibiotics-treated subjects recruited during the 2015-2016 influenza season at different time points. Each vertical bar corresponds to a study participant. On day 1 and day 180, data is available for 4 and 3 individuals only, respectively.
(C) Alpha-diversity estimates for 5 antibiotics-treated subjects enrolled during the 2015-2016 season. See Figure 1 for further details.
(D-F) Flagellin (D), anti-LPS IgG (E), and anti-LPS IgA (F) relative concentrations assessed in the plasma of the 32 subjects (both 2014-2015 and 2015-2016 influenza seasons) enrolled in the study. Relative measurements are reported as optical density (OD) values.
Figure S2. Additional measurements of humoral responses to TIV, related to Figure 2.
(A-B) MN titers for each 2014-2015 (A) or 2015-2016 (B) TIV strain at day 30 presented as fold changes over baseline values (day 0). Each bar corresponds to an individual subject. Seroconversion rates describe the proportion of subjects who developed a ≥ 4-fold increase in titer between pre- and day 30 post-vaccination sera.
(C) IgG2-binding to A/California H1 for phase 1 (left panel) and phase 2 (right panel) measured by ELISA. Violin plots show sample distributions. Each circle represents an individual subject, while medians are presented in thick lines.
(D) A/California H1 HA-specific IgA isotype binding capacity measured by SPR and presented as maximum resonance units (max RU). Violin plots show sample distributions. Each circle represents an individual subject, while medians are presented in thick lines.
Figure S3. Adaptive cellular responses to TIV, related to Figure 2
(A) Flow cytometry analysis of plasmablasts (PBs) on day 0 and day 7 after vaccination for phase 1 (left panel) and phase 2 (right panel) subjects. Plasmablasts are identified as CD27hi CD38hi cells within total CD19+ B cells.
(B) Ex vivo ELISPOT measurements of TIV-specific IgG antibody-secreting cells (ASC) per million PBMC at days 0 and 7.
(C) Flow cytometry analysis of activated B cells (ABC) as described in Ellebedy et al. (Ellebedy et al., 2016). ABC are defined as the CD71+ CD38int-lo CD20hi subpopulation within total CD19+ B cells. Fold change frequencies (d7/d0) are presented.
(D) Log2 fold change frequency levels (d7/d0) of activated blood T follicular helper 1 (Tfh1)-like cells measured by flow cytometry. Activated blood Tfh1-like cells are defined as CXCR5+ CXCR3+ PD1+ ICOS+ CD4+ T cells, as described previously (Schmitt and Ueno, 2013). (E) Somatic hypermutation (SHM) analysis of vaccine-reactive human monoclonal antibodies (hmAbs) generated from single cell-sorted plasmablasts 7 days after vaccination. The number of mutations in the light (left panel) and heavy (right panel) antibody chains was calculated for a total of 35 hmAbs from 8 antibiotics-treated subjects (red circles) which were found to bind at least one of the TIV strains included in the 2014-2015 formulation and compared versus 223 hmAbs from healthy controls, 212 of which belonged to individuals who received a quadrivalent influenza vaccine (QIV) that same season (light blue squares). HmAbs from controls in this study are highlighted in dark blue.
Where calculated, comparisons between control and antibiotics-treated groups at specific time points were performed by Mann-Whitney tests. NS – not significant.
Figure S4. Comparison of transcriptional and metabolic responses in phases 1 and 2, related to Figures 3, 5, and 6.
(A) Comparison of enrichment scores for BTMs significantly enriched (FDR < 0.05, NES > 2.2) post-vaccination in antibiotics-treated subjects in either phase 1 or 2. See Figure 3B for results of all subjects combined.
(B) Metabolic trajectories along the first two principal components for control (blue) and antibiotics-treated (red) subjects in phase 1 (dark) and phase 2 (light) for days 0-7 relative to the screening time point. See Figure 5F for results of all subjects combined.
(C) Fold change of LCA in the plasma among antibiotics-treated (red) and control (blue) subjects in phases 1 and 2. Each thin line represents a single subject, thick lines represent geometric means. See Figure 6E for results of all subjects combined.
Figure S5. Workflow of MMRN construction, related to Figure 7.
Feature clusters were identified in each data type through clustering algorithms. Associations between the feature clusters of each data type were then estimated by partial least square (PLS) regression. The network was then queried for associations with LCA or H1N1-specific IgG1 data through an enrichment-based approach. See STAR Methods for details.
Data Availability Statement
The data generated in this study is available at ImmPort (https://www.immport.org) under accession SDY1086. The microarray data is additionally available at GEO: accession GSE120719.