

Abstract
We introduce a genetic correlation by environment interaction model [(rG)xE] which allows for social environmental moderation of the genetic relationship between two traits. To empirically demonstrate the significance of the (rG)xE perspective, we use genome wide information from respondents of the Health and Retirement Study (HRS; n = 8,181; birth years 1920–1959) and the National Longitudinal Study of Adolescent to Adult Health (Add Health; n = 4,347; birth years 1974–1983) to examine whether the genetic correlation (rG) between education and smoking has increased over historical time. Genetic correlation estimates (rGHRS = −0.357; rGAdd Health = −0.729) support this hypothesis. Using polygenic scores for educational attainment, we show that this is not due to latent indicators of intellectual capacity, and we highlight the importance of education itself as an explanation of the increasing genetic correlation. Analyses based on contextual variation the milieus of the Add Health respondents corroborate key elements of the birth cohort analyses. We argue that the increasing overlap with respect to genes associated with educational attainment and smoking is a fundamentally social process involving complex process of selection based on observable behaviors that may be linked to genotype.
Keywords: genetic correlation, pleiotropy, smoking, education, historical time
INTRODUCTION
Sociologists have long been interested in demonstrating that phenomena formerly considered innate, essential, or immutable in fact have social causes (Berger and Luckman 1966; Winant and Omi 1994; Lorber 1994; Martin and Beittel 1998; Ridgeway and Correll 2004; Saperstein and Penner 2012). In this vein, social scientists have argued that the effects of genes on complex human behaviors are contingent upon the environments in which people live, work, and play (Jencks 1980; Domingue et al. 2014; Boardman et al. 2014; Domingue et al. 2015; Branigan, McCallum, and Freese 2013). In this paper, we extend the sociological approach to another aspect of genetic inquiry by illustrating the capacity of the social environment to transform the relationship between two complex human behaviors at the genetic level.
Our arguments focus on “genetic correlation”. Genetic correlation (rG) is a measure of the extent to which the genetic origins of two traits overlap. We innovate by illustrating that social forces can be crucial to clear interpretation of genetic correlation. In particular, we propose that changes in social forces may induce changes in rG. A genetic correlation between two traits may differ as a function of social norms and structural constraints that serve to either enhance or mute the influence of genes on either or both traits. We refer to this process as genetic correlation-by-environment interaction [(rG)xE].
We empirically demonstrate the sociological significance of the (rG)xE perspective by documenting changes in the genetic correlation between educational attainment and smoking as a function of birth cohort, which we use as a measure of one’s environment. We find that the rG between educational attainment and smoking has increased across birth cohorts. We extend these findings using polygenic scores for educational attainment, showing that the genes that predict educational attainment are increasingly associated with smoking. The mediating role of educational attainment has grown across cohorts, while cognition shows comparatively little explanatory power. While our primary analysis focuses on temporal change in rG, we also utilize variation in geospatial social contexts to demonstrate one possible mechanism for the changes we observe across time. We conclude that genetic factors associated with successful navigation of educational systems select people into environments that are increasingly discouraging of smoking.
After describing observed trends in the relationship between educational attainment and smoking among U.S. adults, we introduce the concepts of gene-by-environment interaction and genetic correlation, as these are the foundation upon which (rG)xE rests. We then describe conditions under which (rG)xE will occur, outlining why the genetic correlation between education and smoking is expected to have changed across birth cohorts. Finally, we present our methods and results and conclude with a discussion of the broad sociological relevance of (rG)xE.
Educational attainment and smoking
The environment in which decisions about education and tobacco use are made has changed dramatically across the past century. Changes in the labor market (e.g., industrialization and the decline of farming; the decline of highly paid jobs in manufacturing for low-skill workers; technological changes; etc.) have resulted in increased economic and social returns to education as well as diminished economic security among poorly educated workers (Hout 1988, 2012; Goldin and Katz 2010). Meanwhile, the public’s knowledge and acceptance of the harms that smoking entails have become almost universal in recent decades. Prior to the 1960s, such knowledge was less common and concentrated among highly educated persons (Link 2008). Perhaps owing to public health interventions and anti-smoking policies, smoking is now stigmatized in some social circles (Stuber, Galea, and Link 2008).
The relationship between education and smoking has important consequences for socioeconomic disparities in health, as smoking remains the leading cause of preventable death and disease worldwide (CDC 2016). Further, while smoking prevalence declined sharply across the second half of the twentieth century from 42% of American adults in 1965 to just 17% in 2014 (CDC 2016), a socioeconomic gradient in smoking has emerged. Prior to the Surgeon General’s Report of 1964, which announced the dangers associated with smoking to a previously unaware public, smoking prevalence was roughly equivalent across the socioeconomic spectrum. Since that time socioeconomic disparities in rates of smoking have grown dramatically (Link 2008; Pampel 2009; Pampel et al. 2015). Today, smoking prevalence among high school dropouts is about four times higher (24%) than that among people with a college degree (6%).1 Jha et al. (2006) estimate that disparities in smoking prevalence explain nearly half of the educational disparity in mortality among middle-aged U.S. men. In this study we use tools from the field of genomics to shed light on the growing educational disparities in smoking prevalence. Our novel methods suggest that even the genetic relationship between the two traits is fundamentally social in nature.
Contemporary U.S. society continues to witness substantial geographic variation in smoking prevalence (CDC 2011–2015). Smoking prevalence ranges from 9% in Utah to 26% in West Virginia and Kentucky. Educational attainment also differs across states (U.S. Census Bureau, Decennial Census of Census of Population, 1940 to 2000). Census data from 2000 suggests that the percentage of adults with a high school degree ranges from about 73% in Mississippi to 88% in Alaska and Minnesota. Thus, we can also use geographic variation in cultural contexts to study the degree to which the genetic relationship between smoking and educational attainment has varied as a function of social context.
Genetic effects on complex human behaviors
Gene-by-environment interaction
While voluminous evidence exists to support the idea that nearly all traits (often called phenotypes) are heritable (Polderman et al. 2015; Turkheimer 2000), there is also broad consensus that genetic effects are conditioned by one’s environment (Landecker and Panofsky 2013; Jencks 1980). This environmental moderation of genetic effects is referred to as gene-by-environment interaction (GxE). Empirical analyses of GxE have often conceptualized the environment in terms of proximal, micro-level conditions (e.g., household characteristics: Guo, Roettger, and Cai 2008) or in terms of behaviors (e.g., engagement in exercise: Li et al. 2010). Yet scholars in the field have noted that limited and/or endogenous measures of the environment may be insufficient for a complete accounting of the ways in which genetic influences are shaped by the environment; consequently, there has been a call for a broader definition of the environment as multilevel, multidimensional, and longitudinal (Freese and Shostak 2009; Boardman, Daw, and Freese, 2013; Manuck and McCaffery, 2014). Sociologists are uniquely well-positioned to answer this call given the discipline’s interest in macro-level social structures such as nation-states (Durkheim 1893) and historical eras (Elder 1999; Mills 1959). Indeed, in GxE work, sociologists have often operationalized “E” with aspects of geography (Branigan et al. 2013; Boardman 2009; Boardman et al. 2008), historical time (Tropf and Mandemakers 2017; Boardman, Blalock, and Pampel 2010; Boardman et al. 2011; Conley et al. 2016; Branigan et al. 2013), and demographic characteristics (Belsky et al. 2016; Wedow et al. 2016; Mitchell et al. 2014; Short, Yang, and Jenkins 2013; Briley and Tucker-Drob 2013; Bergen, Gardner, and Kendler 2007; Eley, Lichtenstein, and Stevenson 1999).
GxE scholars have also stressed the need for measures of the environment to be exogenous with respect to genes (Conley and Rauscher 2013; Cook and Fletcher 2013). This is due to the typical problem of selection: genes are not necessarily distributed at random across environments. Whether this selection is active (e.g., individuals with specific genes select certain environments because of those genes), passive (e.g., people inherit their genes and their environments from their parents), or evocative (e.g., genes evoke particular environmental responses), the detection of environmental moderation of genetic effects is greatly complicated in the presence of such selection. The more distal aspects of the environment that sociologists have gravitated towards are less likely than more proximal environmental conditions or behaviors to present such problems of endogeneity.
Genetic correlation
Genetic correlation (rG) summarizes the extent to which genetic factors are implicated in an observed correlation between two traits. At one extreme, two heritable traits may be strongly correlated in the population, yet the genes associated with each trait may be independent (i.e., rG=0). In such a case, the observed correlation between the traits cannot be traced back to genes. On the other hand, genetic correlation may be non-zero when some gene or set of genes influences both traits of interest. This effect is known as pleiotropy (Solovieff et al. 2013).
Two distinct types of pleiotropy are depicted in Figure 1, which provides examples plausible for the relationship between education (ED) and smoking (SM). Biological pleiotropy (Figure 1A) involves one or more genes exerting independent influence on each of the two phenotypes of interest. For example, research has found that one particular genetic variant increases risk of both prostate and colorectal cancers, likely by enhancing tumor growth in both colon and prostate tissue (Wasserman et al. 2008; Pomerantz et al. 2009). Mediated pleiotropy (Figure 1B), on the other hand, exists when a set of genes influence a phenotype that has its own causal effect on the second phenotype. Genes are causally associated with one trait and affect the second trait only through this mediated path. Scholars have identified overlapping genetic effects on nicotine dependence and lung cancer risk (Hung et al. 2010; Thorgiersson et al. 2008). These effects may be due to mediated pleiotropy whereby genes influence nicotine dependence, which then affects lung cancer risk via smoking (Chanock and Hunter 2008). Notice that these forms of pleiotropy are not mutually exclusive—for any pair of phenotypes, one or both could simultaneously play a role in producing genetic correlation.
Figure 1.
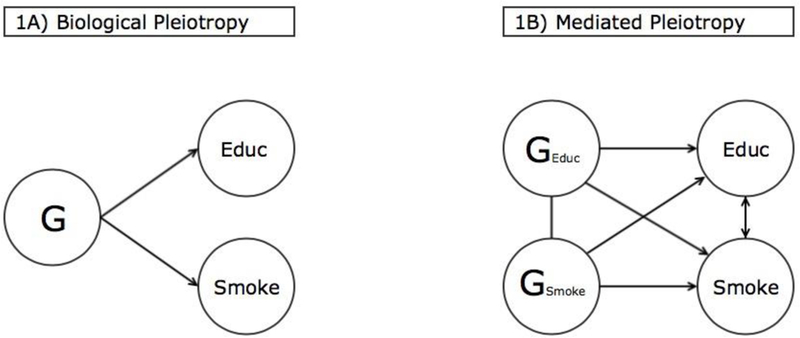
Education, smoking, and types of pleiotropy
Neither are the models depicted in Figure 1 exhaustive. For instance, pleiotropy might occur through parental effects due to the intergenerational transmission of genes and, through environmental conditions, behaviors. Importantly, this type of pleiotropy is generated not through the effects of a child’s genes on his or her own phenotype(s). It is instead produced through a correlation between a child’s genes, which are passed on by parents, and environmental conditions set up by parents that either support or discourage certain behaviors and outcomes. Furthermore, assortative mating can produce pleiotropy if individuals with genes that predict one phenotype tend to select partners with genes predicting another trait. Their offspring will then be more likely to have genes predicting both phenotypes, and in the aggregate, genetic correlation will result. These complementary pleiotropic models are important to consider and they offer alternative explanations as to why genes may be increasingly correlated with both education and smoking over time.
To date, pleiotropic effects have been described almost exclusively as the result of natural selection (Lawson et al. 2011; Marchini et al. 2014). For example, the same genes affect one’s bone size and body mass presumably due to evolutionary forces selecting against a skeleton that cannot support the body’s weight (Marchini et al. 2014). As scholars begin to investigate genetic correlation between traits that are highly contextual, such as that between educational attainment (SSGAC 2016) and smoking (Tobacco and Genetics Consortium 2010), additional considerations may become relevant. In particular, we focus on how differences in structural or cultural climate, across both time and space, are associated with differences in pleiotropic patterns and genetic correlation.
The genetics of educational attainment and smoking
There is substantial evidence pertaining to the moderate heritability of both traits (Branigan et al. 2013; Li et al. 2003; Polderman et al. 2015). In addition, the traits appear to be genetically correlated. For example, McCaffery et al. (2008) estimate a genetic correlation of −0.30 between educational attainment and smoking initiation in a sample of male twins from the U.S. Using molecular data from unrelated individuals, Bulik-Sullivan et al. (2015) estimate a very similar genetic correlation of −0.36 between education and having ever smoked.
There is also evidence that genetic effects on both phenotypes have, in general, shifted over time. Specifically, the heritability of education appears to have increased modestly (Branigan et al. 2013; Heath et al. 1985), as has the heritability of smoking in the U.S. (Boardman et al. 2010; Boardman et al. 2011; Kendler, Thornton, and Pedersen 2013). In addition, the relative effects of genes on educational attainment have shifted across birth cohorts, as have the predictive power of polygenic scores for educational attainment and for smoking (Conley et al. 2016; Domingue et al. 2016).
Given these observed temporal trends, we focus on the study of (rG) in distinct U.S. birth cohorts. Along the lines of earlier work (Elder 1999; Yang 2008; Boardman et al. 2010; Boardman et al. 2011; Liu and Guo 2015; Walter et al. 2016; Conley et al. 2016), we consider historical periods to be discrete social environments into which people have no ability to select themselves. In this way, these analyses are akin to exploiting a natural experiment. Any change in rG across birth cohorts can be interpreted as reflecting a causal effect of the social, institutional, and physical environments that are unique to each birth cohort on rG.
Social processes that could produce (rG)xE
Why might one observe (rG)xE? The mechanisms that drive (rG)xE will depend on the phenotypes and the aspect of the environment under consideration. In the context of smoking and educational attainment, we suspect that institutional selective forces and culture play large roles. Sudden changes to the formal institutions that condition behaviors may lead to changes in the salience of genotype. Alternately, mechanisms driving (rG)xE could involve subtler and more gradual shifts in the cultural regulations to which the phenotype(s) are subject.
In the case of educational attainment and smoking, selection mechanisms driving rG (and therefore (rG)xE) are likely found relatively early in the life course before people make final decisions regarding their education or smoking behaviors. They may therefore be located in latent genetic traits. These latent traits presumably exert influence on a person’s orientation toward the world from day one. Such effects on education and smoking may have increased across birth cohorts if, for example, genetic propensity for risk-taking has become increasingly predictive of both low education and smoking. This is a real possibility, as the risk inherent in a low level of educational attainment has grown (Hout 1988, 2012; Goldin and Katz 2009), and as the health-related dangers of smoking have been widely publicized since the mid-1960s (Link 2008). Similarly, general intelligence could also have become more predictive of both educational attainment and smoking over time via biological pleiotropy. This would be the case if people with greater cognition increasingly attend school at higher rates and/or if their desire to avoid smoking has strengthened relative to others.
Mechanisms driving the rG between education and smoking, and changes across birth cohorts therein, might also be located within the education system itself due to mediated pleiotropy. Mediated pleiotropic effects (Figure 1B) might have grown across cohorts if education has become an increasingly important causal mechanism for smoking. For example, the skills cultivated through schooling might be more important for fostering the efficacy and agency to avoid smoking among later generations who are more likely to have been aware of its dangers at an early age. As anti-smoking norms have proliferated, especially among the highly educated (Pampel, Krueger, and Denney 2010), education and the status associated with it might be more likely to encourage non-smoking.
Finally, while environmental contexts and the formal and cultural institutions governing selection into particular outcomes and behaviors differ across historical eras, inside a particular historical era, these mechanisms may also differ across discrete social contexts such as countries, states, neighborhoods, or schools (Myers et al. 2013). Examining variation across contemporaneous environmental conditions can provide leverage for exploring (rG)xE, since the mechanisms - institutional differences and norms - might be easier to isolate and measure across more proximal conditions.
In sum, we expect that social transformations over the past century have driven the magnitude of pleiotropic effects for education and smoking up such that what these outcomes reflect at the genetic level has become increasingly similar over time. We posit that whether or not the genetic correlation between education and smoking behaviors has increased in tandem with the observed correlation sheds light on the mechanisms underlying the observed trend, and we attempt to explore some of these mechanisms in our analyses below. This study thus provides direction for future research on the sources of educational disparities in smoking and its consequences for health.
DATA, MEASURES, AND METHODS
Data
At present, genetic information is available for only a limited range of social surveys. However, in combination they contain a representative sample of U.S. births from across the 20th century. We combine data from the Health and Retirement Study (birth years 1920–1959) and the National Longitudinal Study of Adolescent to Adult Health (birth years 1974–1983). Both of these surveys sample from the U.S. population, ask respondents similar questions regarding educational attainment and smoking history, and have recently genotyped a relatively large number of respondents. We describe these surveys and the measures constructed for analysis below.
Health and Retirement Study
To estimate genetic effects among earlier birth cohorts, we use data from the Health and Retirement Study (HRS), a panel study of U.S. households that began in 1992 with the intention of monitoring physical, emotional, and economic wellbeing during the transition into retirement and older age (RAND 2016). The HRS is now representative of the U.S. population over age 50, and its participants are surveyed approximately every two years.
As of 2008, 12,507 HRS respondents had been genotyped2. We limit the sample to non-related individuals of European genetic ancestry, as the methods that currently exist for calculating genetic correlation are optimized for this population (SSGAC 2016)3. We also restrict the sample to respondents born between 1920 and 1959 in order to neatly delineate the range of birth years assessed. After making these and other quality control restrictions, our sample from the HRS includes 8,181 people.
National Longitudinal Study of Adolescent to Adult Health
We use the National Longitudinal Study of Adolescent to Adult Health (Add Health) to study genetic relationships within a later birth cohort (Harris 2009). Add Health originated as an in-school survey of a nationally representative sample of U.S. adolescents enrolled in grades 7 through 12 during the 1994–1995 school year. Respondents were born between 1974 and 1983. A subset of the original Add Health respondents has been followed up with in-home interviews, which allows researchers to assess correlates of outcomes in the transition to early adulthood.
In 2008, respondents were invited to provide a saliva sample for future genotyping4. The genetic data currently available to researchers represents a subset of these respondents5. As with the HRS, we limit the sample to non-related individuals of European descent. Our final sample from Add Health includes 4,347 respondents. For more details concerning the quality control of the genetic data in this study, see our Appendix.
Measures
We coded years of education similarly in the HRS and Add Health (Table 1). The resulting quasi-continuous variable ranges from 8 (attended school for 8 years or less) to 20 (earned a PhD or completed professional school). Average educational attainment among HRS respondents is 13.49 years; among Add Health respondents, it is 14.66.
Table 1.
Coding of educational attainment in the HRS and Add Health
| HRS | |||
|---|---|---|---|
| Label | Code | N | % |
| 8 years or fewer, w/o HS diploma, GED, or college experience | 8 | 306 | 3.74 |
| 9+ years, w/o HS diploma, GED, or college experience | 10 | 717 | 8.76 |
| HS diploma or GED, w/o college experience | 12 | 3142 | 38.41 |
| 13 years (with a HS diploma/GED and college experience) | 13 | 660 | 8.07 |
| 14 years (with a HS diploma/GED and college experience) | 14 | 886 | 10.83 |
| 15+ years, w/o BA (with a HS diploma/GED and college experience) | 15 | 433 | 5.29 |
| BA (<=16 years) | 16 | 938 | 11.47 |
| BA (17+ years) | 17 | 276 | 3.37 |
| MA/MBA | 19 | 632 | 7.73 |
| PhD/Law/MD | 20 | 191 | 2.33 |
| Total | 8,181 | 100 | |
| Add Health | |||
| Label | Code | N | % |
| 8th grade or less | 8 | 15 | 0.35 |
| Some high school | 10 | 292 | 6.72 |
| High school graduate | 12 | 689 | 15.85 |
| Some vocational/technical training (post-HS) | 13 | 147 | 3.38 |
| Completed vocational/technical school (post-HS) | 14 | 280 | 6.44 |
| Some college | 15 | 1476 | 33.95 |
| BA | 16 | 919 | 21.14 |
| Some graduate or professional school | 17 | 194 | 4.46 |
| MA | 19 | 257 | 5.91 |
| PhD or professional school | 20 | 78 | 1.79 |
| Total | 4,347 | 100 | |
Notes: HS: High school; GED: General Education Diploma; BA: Bachelor’s degree; MA: Master’s degree; MBA: Master of Business Administration; PhD: Doctoral degree; MD: Medical degree. HRS: Health and Retirement Study (RAND 2016); Add Health: National Longitudinal Study of Adolescent to Adult Health (Harris et al. 2009).
Our second phenotype is a binary indicator of having ever smoked regularly. In their first interview, HRS respondents were asked, “Have you ever smoked cigarettes? By smoking we mean more than 100 cigarettes in your lifetime.” Those who reported that they had are considered to have smoked regularly. In the 2008 Add Health interview, respondents were asked, “Have you ever smoked cigarettes regularly, that is, at least 1 cigarette every day for 30 days?” Both questions are commonly used to assess whether a person has ever smoked regularly (IARC 2008).
One complication is that HRS respondents were older at the time that their smoking history was assessed than were Add Health respondents, and thus had much more time to evolve into regular smokers. Nonetheless, we note that very few HRS respondents (just 8% of ever regular smokers) reported having started to smoke after the age of 24. Other evidence also suggests that the vast majority of smokers take up the habit in their teens (Elders et al. 1999). Thus, despite the different ages at which respondents were asked to recount their smoking history, these measures should largely capture a comparable set of behaviors.
Methods
To assess the hypothesized changes in the genetic correlation between education and smoking across cohorts, we begin by estimating genetic effects on education and smoking through regression-based genome-wide association studies (GWAS). We then estimate genetic correlations using Linkage Disequilibrium Score Regression (LDSC). Finally, we construct polygenic scores for educational attainment to investigate mechanisms driving the change in pleiotropic effects on the two phenotypes.
Genome-wide association study (GWAS)
GWAS is the predominant method for the identification of genetic effects across the genome. We start by noting that the base unit of most molecular genetic analyses is the single nucleotide polymorphism, or SNP. Most genetic diversity across people exists because of differences in the number and type of nucleotides found at each SNP, which determines a person’s genotype at that SNP. GWAS considers the effect of genotype at several million SNPs across the genome on a phenotype of interest. Specifically, GWAS quantifies the extent to which an additional nucleotide of a particular variety (called a risk allele) at a particular SNP is associated with a higher or lower value of the phenotype. In effect, GWAS results consist of a long vector of estimated effects, one per SNP. GWAS results serve a variety of purposes. For gene discovery, extremely large sample sizes are required—much more than those available in the HRS and Add Health. However, when using LDSC to summarize the results of GWAS in estimates of heritability or genetic correlation, much smaller sample sizes can be used (Bulik-Sullivan et al. 2015).
We conduct GWAS for three phenotypes (years of education, our binary indicator of having ever smoked regularly, and height) separately for the HRS and Add Health cohorts. To conduct GWAS we use Rvtests (Zhan et al. 2016), which estimates linear and probit regression equations for each SNP in the data sets:
| [Equation 1A] |
| [Equation 1B] |
| [Equation 1C] |
Genotype is the number of risk alleles present at the genetic marker being analyzed. PC is a matrix of the first ten principal components of the variance-covariance matrix of the genetic data. Including principal components in the regression equations controls for differences in genotype across ancestry groups that could confound the effects of genes on phenotypes. Finally, Controls is a matrix consisting of genetic sex, year of birth, squared year of birth, and interactions between sex and birth year as well as sex and squared birth year.
Linkage disequilibrium score regression (LDSC)
Genotypes at SNPs that are in close proximity to each other tend to be highly correlated. This phenomenon is referred to as linkage disequilibrium, or LD. Its implication is that the effect of one SNP on a phenotype may not be independent of the effects of nearby SNPs. LDSC is a method of adjusting GWAS estimates for the presence of LD (Bulik-Sullivan et al. 2015; Bulik-Sullivan et al. 2016)6. In addition, it provides unbiased estimates of genetic correlation. Briefly, in LDSC, genetic correlation is the correlation between the SNP-length vector of LD-adjusted genetic effects estimated for the first phenotype and those estimated for the second. For more details about LDSC, see the Appendix.
Polygenic scoring
Polygenic scores (PGS) summarize the extent to which a person possesses alleles (i.e., genotypes) associated with a trait of interest. These scores utilize GWAS results to link genetic variation to phenotypic variation. A PGS is essentially a weighted average of SNP effects estimated through GWAS:
| [Equation 2] |
where indicates the number of risk alleles individual i possesses at SNP j, and indicates the weight or effect size of that SNP. To avoid “double counting” the causal effects of SNPs that are strongly correlated with each other, we transform the estimated effects using LDpred (Vilhjálmsson et al. 2015). Quality control of the genetic data relevant to the construction of our PGSs is described in our Appendix.
We construct a polygenic score for years of education, utilizing results from a GWAS in progress of over 700,000 individuals, one of the most highly powered polygenic scores constructed to date. Those who conducted the original GWAS report that the score explains about 10% of the variance in years of education in the HRS (incremental R2 = 0.089) and Add Health (incremental R2 = 0.103) samples. The effect of a standard deviation increase in the PGS for education is reported to predict a 0.77-year increase in educational attainment in the HRS (SE = 0.026), and a similar 0.71-year increase in Add Health (SE = 0.031).
After assigning each respondent in the HRS and Add Health with a polygenic score for years of education, we use that score to predict whether or not the respondent had ever been a regular smoker. We consider
| [Equation 3] |
where PGS is the respondent’s PGS for educational attainment and Controls is a matrix consisting of biological sex, year of birth, and an interaction between sex and birth year. We estimate Equation 3 by cohort. In general, if the coefficient on the PGS estimated among Add Health respondents is higher than that estimated among HRS respondents, we can conclude that the genetic variants that predict educational attainment have become increasingly predictive of smoking behaviors across birth cohorts.
We then consider mediation analyses. In particular, we first assess whether cognition7 mediates the relationship between the score for education and smoking. We are only able to do this for the Add Health cohort, as no suitable measures of cognition are available in the HRS:
| [Equation 4] |
We compare estimated in Equation 4 to that estimated in Equation 3. If it has declined in magnitude to a substantial extent, we can infer that the association between the score and smoking is due to the correlation of cognition with both variables.
Second, we estimate the following model for all three cohorts (early HRS, late HRS, and Add Health):
| [Equation 5] |
If the relative decline in between Equations 3 and 5 remains roughly constant across cohorts, it will indicate that the importance of achieved education for understanding the relationship between the PGS for education and smoking has neither grown nor shrunk. If the extent to which is reduced in Equation 5 compared to Equation 3 grows across cohorts, this will indicate that the original association between the PGS for education and smoking is increasingly explained by the association of these two variables with actual education attained. This result is consistent with growth in mediated pleiotropy: that the effect of education on smoking has increased across cohorts.
We also extend our analysis of temporal variation in the rG between education and smoking to contextual variation in rG during a particular historical era. We limit this analysis to Add Health respondents. Specifically, using data from all students involved in the initial in-school survey, we calculated the average educational attainment of mothers in each school. We then divided the genotyped sample into three groups based on the average education of mothers in the school they attended and we estimate Equations 3 and 5 for each of these three groups.
Finally, we include and examine association between height and years of education as a robustness check. We evaluate change in the genetic correlation between height and years of education. Height is highly heritable (Yang et al. 2010) and somewhat correlated with education8, but we do not expect it to evince any strong rG with educational attainment, or changes therein across birth cohorts.
RESULTS
Temporal trends in phenotypes
Descriptive statistics for the HRS and Add Health samples are provided in Table 2. Birth years range from 1920 through 1959 among HRS respondents (mean = 1938). Add Health respondents were born between 1974 and 1983 (mean = 1979). The HRS sample has a slightly larger female population (57%) compared to the Add Health sample (54%). Note that HRS respondents supplied saliva samples (and phenotypic information) at much older ages (mean age = 68) than did Add Health respondents (mean age = 28).
Table 2.
Descriptive statistics from the HRS and Add Health
| HRS | ||||
|---|---|---|---|---|
| Mean | SD | Min | Max | |
| Years of education | 13.49 | 2.77 | 8 | 20 |
| Ever smoked regularly | 0.57 | 0.49 | 0 | 1 |
| Height in centimeters (n = 8,180) | 169.93 | 9.84 | 127 | 211 |
| Female | 0.57 | 0.49 | 0 | 1 |
| Year of birth | 1938.38 | 9.32 | 1920 | 1959 |
| Age when DNA sample taken | 68.07 | 9.36 | 46 | 88 |
| N = 8,181 | ||||
| Add Health | ||||
| Mean | SD | Min | Max | |
| Years of education | 14.66 | 2.27 | 8 | 20 |
| Ever smoked regularly | 0.53 | 0.5 | 0 | 1 |
| Height in centimeters (n = 4,336) | 171.33 | 9.92 | 123 | 204 |
| Female | 0.54 | 0.50 | 0 | 1 |
| Year of birth | 1978.98 | 1.74 | 1974 | 1983 |
| Age when DNA sample taken | 28.42 | 1.77 | 24 | 34 |
| N = 4,347 | ||||
Notes: HRS: Health and Retirement Study (RAND 2016); Add Health: National Longitudinal Study of Adolescent to Adult Health (Harris et al. 2009).
Average educational attainment is higher among Add Health participants (14.66 years; see Table 2) than those in the HRS (13.49 years). This is consistent with historical trends (US Census Bureau, 1947–2015; Ryan and Bauman 2016, Figure 2). Incidence of having ever smoked regularly is slightly lower among Add Health respondents (53%) than HRS respondents (57%). And, the correlation between years of education and having ever smoked regularly increased substantially between HRS (−0.108, SE = 0.014) and Add Health (−0.368, SE = 0.017). Note also that all phenotypes are estimated to be heritable in these samples.9
Figure 2.
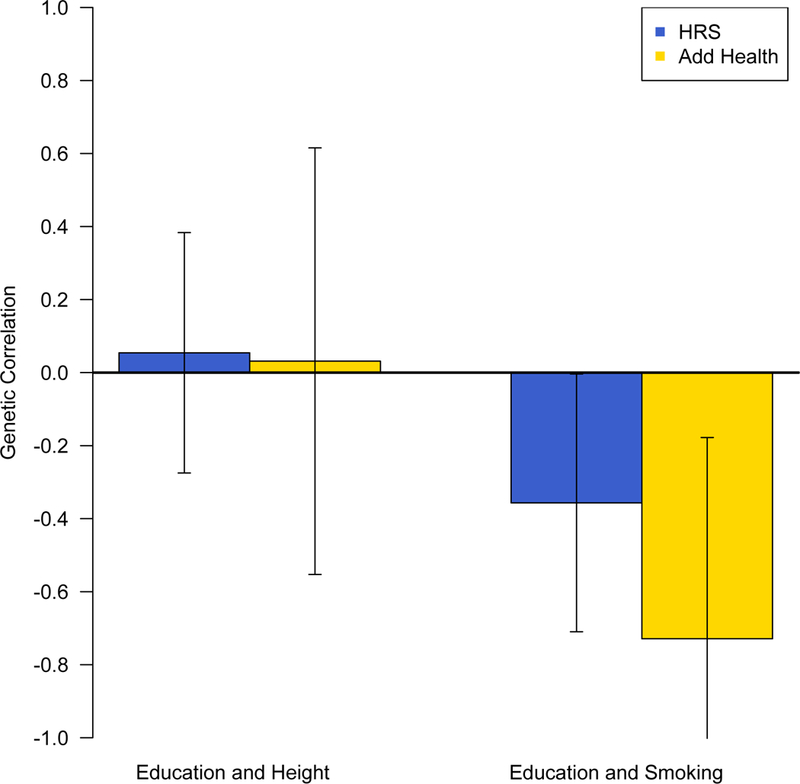
LDSC genetic correlations between education and height and between education and smoking in the HRS and Add Health, with 95% confidence intervals
LDSC: Change in genetic correlation over time
The estimated genetic correlation between education and smoking is negative in both data sources. The sign of the rG estimate indicates that genotypes that are positively associated with years of education attained tend to be negatively associated with smoking. As hypothesized, the rG estimates are greater in magnitude among Add Health respondents than among those in the HRS (Table 3, Figure 2). Specifically, the estimated rG is −0.357 in the HRS (SE = 0.180, p =0.047) and −0.729 in Add Health (SE = 0.281, p = 0.010).
Table 3.
LDSC estimates of pairwise genetic correlations with heritability estimates for each trait
| EDUCATION AND HEIGHT | |||||||
|---|---|---|---|---|---|---|---|
| COHORT | rG | SE | p-value | h2 Education | SE | h2 Height | SE |
| HRS | 0.054 | 0.168 | 0.763 | 0.116 | 0.044 | 0.215 | 0.047 |
| Add Health | 0.031 | 0.298 | 0.924 | 0.197 | 0.088 | 0.207 | 0.085 |
| EDUCATION AND SMOKING | |||||||
| COHORT | rG | SE | p-value | h2 Education | SE | h2 Smoking | SE |
| HRS | −0.357 | 0.180 | 0.047 | 0.116 | 0.044 | 0.129 | 0.047 |
| Add Health | −0.729 | 0.281 | 0.010 | 0.197 | 0.088 | 0.242 | 0.084 |
rG: Genetic correlation; SE: Standard error; h2: heritability
While both estimates are significantly different from 0, the estimates are not statistically significantly different from each other10 (p = 0.265). We interpret this in light of two additional findings. First, the point estimates are comparable to those reported in a supplemental analysis of twins (see Online Supplement). Second, we consider the genetic correlation between education and height as a negative control test (Table 3, Figure 2). Consistent with prior work (Bulik-Sullivan et al. 2015), the genetic correlations between education and height are very near zero in both data sets (HRS: 0.054, SE = 0.168; Add Health: 0.031, SE = 0.298). This lends credence to our main finding of increased pleiotropy between education and smoking over time since we neither expect nor find changes in pleiotropy for our negative control phenotypes.
Polygenic score analyses: Substantive implications of changes in rG
We report the marginal effects on smoking of the educational attainment polygenic score (PGS) for three different cohorts: an early HRS cohort (birth years 1920–1938), a late HRS cohort (birth years 1939–1959), and the Add Health cohort (birth years 1974–1983) (Table 4, Panel A). A one-standard deviation (SD) increase in the educational attainment PGS predicts a much larger reduction in the probability of smoking amongst the later-born. Among the earliest HRS birth cohort, a one-SD increase in the PGS reduces the probability of having ever smoked regularly by 3.3 percentage points; by 4.3 percentage points among the later HRS birth cohort; and by 7.6 percentage points among Add Health respondents. These results are consistent with the findings we report from our LDSC analysis above.
Table 4.
Linear and probit regression models of ever regular smoking, by cohort
| Panel A. Marginal effects estimated through probit regressions of ever regular smoking on a PGS for education | |||
|---|---|---|---|
| Early HRS (1920–1938) | Late HRS (1939–1959) | Add Health (1974–1983) | |
| PGS for education | −0.033*** | −0.043*** | −0.076*** |
| (0.007) | (0.008) | (0.007) | |
| N | 4,190 | 3,990 | 4,350 |
| Panel B. Marginal effects estimated through probit regressions of ever regular smoking on a PGS for smoking | |||
| Early HRS (1920–1938) | Late HRS (1939–1959) | Add Health (1974–1983) | |
| PGS for smoking | 0.053*** | 0.065*** | 0.069*** |
| (0.008) | (0.008) | (0.008) | |
| N | 4,190 | 3,990 | 4,350 |
p<0.001,
p<0.01,
p<0.05; Robust standard errors in parentheses; PGS: Polygenic score
Models control for genetic sex, year of birth, and their interaction. All variables are held at their means to estimate marginal effects.
As a point of comparison we also constructed PGSs for smoking using GWAS results made available through the Tobacco and Genetics Consortium (2010). We then used these scores to predict respondents’ likelihood of having ever smoked regularly. Results are presented in Table 4, Panel B. Note that the marginal effects of a SD increase in the smoking PGS on the probability of having smoked are similar to the marginal effects estimated for a SD increase in the PGS for education. In other words, genes that predict educational attainment are nearly as useful for predicting whether someone has ever smoked as are the genes that have been found to predict smoking itself. Also, the degree to which the education PGS predicts smoking has grown more across cohorts than that of the smoking PGS.
Analysis of mechanisms
Our above analyses using LDSC and polygenic scores suggest that there has been an increase in pleiotropy between education and smoking over time. However, as we outlined above, we are also interested in possible mechanisms that might explain these increases across time. We explore these below.
Verbal cognition
It might be the case that over time, natural intelligence is increasingly associated with both the genes for education and the genes for smoking. Accordingly, we investigate the possibility that verbal cognition mediates the relationship between the PGS for education and smoking among Add Health respondents. (We were not able to assess this for HRS respondents as no suitable measure of cognition was available.) As shown in Table 5, we find very weak evidence that verbal cognition partially mediates the relationship between the PGS for education and regular smoking. In fact, the marginal effect of the PGS declines by only 10.71% between models. These results suggest that cognition does not explain the genetic correlation between education and smoking even in the cohort with the highest observed genetic correlation between these two traits. We take this as evidence that changes in the social environment over time, rather than shifting effects of individual characteristics and selective processes outside of individuals’ control, are far more plausible explanations for the pleiotropic increases we observe.
Table 5.
Mediation model: Marginal effects from probit models of ever regular smoking, controlling for respondent’s verbal cognition score
| Add Health (1974–1983) | ||
|---|---|---|
| Cognition control | ||
| No | Yes | |
| PGS for education | −0.076*** | −0.068*** |
| (0.007) | (0.001) | |
| Verbal cognition score | — | −0.003*** |
| — | (0.001) | |
| % Mediated | 10.708% | |
| N | 4,163 | |
p<0.001,
p<0.01,
p<0.05; Robust standard errors in parentheses; PGS: Polygenic score
Models control for genetic sex, year of birth, and their interaction. All variables are held at their means to estimate marginal effects.
Environmental selection
Next, we consider the degree to which the observed pleiotropy is mediational in nature. Specifically, in Equation 5 we control for educational attainment. The degree to which the observed effect of the educational attainment PGS is mediated through observed attainment increases from 16.46% in the early HRS cohort to 48.28% in the late HRS cohort to 85.20% in the Add Health cohort (Table 6; Figure 3). At the same time, the coefficient estimated for education increases across cohorts, while that estimated for the education PGS remains about the same or even declines. The increase observed across cohorts in the effect of the PGS for education on smoking appears to be largely explained by an increasing relationship between smoking and actual education attained. These results are consistent with the idea that in later birth cohorts, the genes associated with educational attainment are associated with selection into environments that are increasingly not conducive to smoking.
Table 6.
Mediation models: Marginal effects from probit models of ever regular smoking by birth cohort, controlling for respondent’s educational attainment
| Early HRS (1920–1938) | Late HRS (1939–1959) | Add Health (1974–1983) | ||||
|---|---|---|---|---|---|---|
| Educational Control | Educational Control | Educational Control | ||||
| No | Yes | No | Yes | No | Yes | |
| PGS for education | −0.033*** | −0.028*** | −0.043*** | −0.022*** | −0.076*** | −0.011*** |
| (0.007) | (0.008) | (0.008) | (0.008) | (0.007) | (0.008) | |
| Education | — | −0.007** | — | −0.026*** | — | −0.062*** |
| — | (0.003) | — | (0.003) | — | (0.003) | |
| % Mediated | 16.456% | 48.281% | 85.200% | |||
| N | 4,190 | 3,990 | 4,350 | |||
p<0.001,
p<0.01,
p<0.05; Robust standard errors in parentheses; PGS: Polygenic score
Models control for genetic sex, year of birth, and their interaction. All variables are held at their means to estimate marginal effects.
Figure 3.
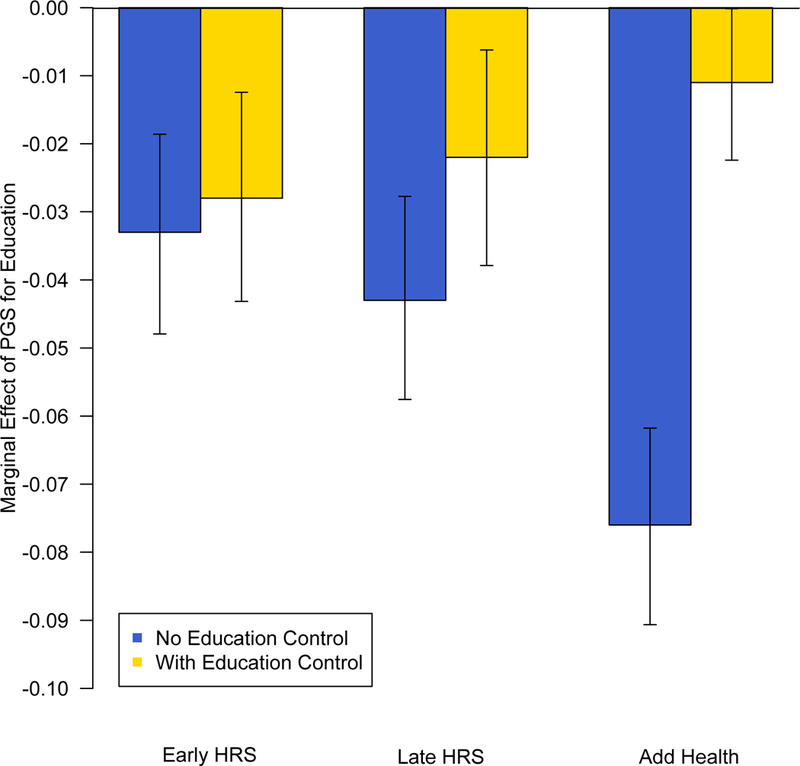
Estimated marginal effects of the polygenic score for educational attainment on probability of having ever smoked regularly, with and without educational attainment as a control, for the early HRS cohort (birth years 1920–1938), the late HRS cohort (1939–1959), and the Add Health cohort (1974–1983)
Exploring spatial variation in genetic correlation
While we find large cohort effects in increasing rG over time, such analyses are not conducive to identification of specific mechanisms that might drive these changes. To further investigate these moderating mechanisms at work in our (rG)xE models, we extend our analysis of temporal variation to another source of environmental variation that, like birth cohort, also reflects smoking-related knowledge and norms: contextual embeddedness. In Add Health (unlike in HRS), we have good measures of school SES context. Thus we divide the Add Health sample into tertiles (low, medium, and high) based on the average educational attainment of mothers in a respondent’s school. We consider average maternal education in a school, which reflects the school’s socioeconomic composition, to be a very rough proxy for collective health lifestyles (Frohlich et al. 2001). We then repeat our mediation analyses for each tertile, first regressing smoking on the PGS for education alone, and then controlling for educational attainment.
In Table 7, we present the results from this analysis. Educational attainment mediates the relationship between the PGS for education and regular smoking more for students from schools with high levels of maternal education, those in which smoking-related norms are most restrictive (akin to later birth cohorts). The percent mediated moves from 55.71% for respondents in schools with low maternal education to 60.94% for those schools with medium maternal education to 67.69% in schools with high maternal education. This 1.22-fold increase in mediation across schools is substantially lower than the 5.18-fold increase found across birth cohorts (Table 6 and Figure 3)11. Though the explanatory power of actual education attained therefore seems to vary more across temporal, socio-historical environments than across social contexts within the same time period, these results add an additional piece to the puzzle. Inside of historical era, which captures the large-scale cultural norms governing selection into smoking, social contexts defined across space, which are easier to measure, act as more proximal mechanisms through which variation in rG occur and operate.
Table 7.
Mediation models: Marginal effects from probit models of ever regular smoking in Add Health by mean parental education in a school, controlling for respondent’s educational attainment
| Low maternal education | Average maternal education | High maternal education | ||||
|---|---|---|---|---|---|---|
| Education control | Education control | Education control | ||||
| No | Yes | No | Yes | No | Yes | |
| PGS for education | −0.070*** | −0.031* | −0.064*** | −0.025* | −0.065*** | −0.021 |
| (0.013) | (0.014) | (0.013) | (0.011) | (0.014) | (0.014) | |
| Years of education | — | −0.054*** | — | −0.066*** | — | −0.071*** |
| — | (0.006) | — | (0.006) | — | (0.007) | |
| % Mediated | 55.7% | 60.9% | 67.7% | |||
| N | 1,392 | 1,447 | 1,332 | |||
p<0.001,
p<0.01,
p<0.05; Robust standard errors in parentheses; PGS: Polygenic score
Models control for genetic sex, year of birth, and their interaction. All variables are held at their means to estimate marginal effects.
School type is determined by average maternal education among the school’s students.
Robustness analyses
Alternative methods for measuring genetic correlation
Besides LDSC, there are two additional methods for measuring genetic correlation. The standard method before the availability of molecular data was the bivariate twin model. In our Online Supplement, we present a full analysis using this method with the Add Health and with a twin sample from the Midlife in the United States (MIDUS) study. Importantly, the results are very similar to those using LDSC: the genetic correlation between education and smoking appears to have increased over time. Second, Genome-wide Complex Trait Analysis (GCTA) can be used with individual-level genetic data. Though we are underpowered to perform this method with our small samples (relative to LDSC), we perform these analyses with the HRS and Add Health data and find similar trends with increasing pleiotropy over time. These results are available from the authors upon request. Taken together, the results provide consistent evidence across methods for the conclusion that rG with respect to education and smoking has increased in magnitude across U.S. birth cohorts in the United States.
DISCUSSION
In this paper, we have shown that genes implicated in educational attainment have increasingly overlapped with genes associated with smoking behaviors across U.S. birth cohorts: the genetic correlation between the two traits has grown in magnitude. Using polygenic scores, we also demonstrated that disparities in smoking due to genes that are associated with educational attainment have grown across birth cohorts.
Why might this be? What mechanisms might drive the changes we observe over historical time? Results from our mediation analyses suggest that the growth of a particular type of pleiotropic effect is responsible for much of this change. We turned first to a common but problematic explanation regarding socioeconomically-stratified health behaviors: intelligence (Herrnstein and Murray 1994). One of the most important findings of this paper is that our results do not support the idea that verbal cognition plays a role in explaining the increased genetic correlation between education and smoking.
Instead, it appears that education itself and/or the status associated with it is now, more so than in the past, likely to encourage non-smoking. Specifically, we find that years of attained education has explained a larger portion of the relationship between the genes that predict education and smoking behavior over time. Thus, the social selection we explore appears to operate in such a way that genes related to education are associated with selection into environments that are increasingly not conducive to smoking. We believe this to be driven by historic macro changes regarding educationally-conditioned smoking norms over time. While we cannot make strong general causal claims based on these analyses, these exercises lend support for the role of genes implicated in educational attainment which is then an increasingly important causal mechanism for smoking.
While historical time is a powerful context, it is also incredibly broad with multiple sources of potential influence. To narrow the range of potential influences and to sharpen the scope of our inquiry, we focused on a more proximate social context (school socioeconomic status) to isolate one potential mechanism. Here the mediating effect of education on smoking was strongest in the environment characterized by the highest school-level SES (as proxied by maternal education), or in the environment with more restrictive anti-smoking norms and requisite resources to enforce anti-smoking behaviors. Thus we show evidence of salient mechanisms in exactly the social contexts in which we expect developmental norms around smoking behavior and the effects of education on smoking to differ substantially: the school environment. We note that if we are pinpointing a culprit for the increases we observe in rG over time, the adolescent processes in school environments are likely important places to look.
Accordingly, this concept of (rG)xE, the environmental moderation of genetic correlation, has broad and innovative relevance to sociology. The sociological perspective, with its comprehensive vision of the environment and its attention to overarching social structures, fits squarely within mainstream gene-by-environment interaction (GxE) research, which has already demonstrated that genetic effects on single phenotypes are environmentally contingent (Boardman et al. 2013). We push the sociological perspective further by illustrating that the social environment also has the power to transform the relationship between complex human behaviors at the genetic level. The (rG)xE model is therefore a logical step forward in conceptualizing gene-environment interplay for social scientists, since it has long been understood that there are multiple competing and interacting influences that shape outcomes. Our results suggest that genes are becoming increasingly important for understanding the link between education and smoking but that this process has been a fundamentally social one. This, we believe, is among the most important contributions of our paper.
The (rG)xE perspective for the first time embeds the genetic antecedents of more than one phenotype within a single conceptual model. Consequently, it highlights that the extent to which outcomes in distinct domains develop as a bundled set based on genes may differ across environmental conditions. Thus, this perspective can be used to identify whether there are genetic bases for other observed correlations and to explore how this genetic overlap differs across environments. In particular, examining variations in the genetic correlation between health conditions and either education or other measures of socioeconomic status, sheds light on the mechanisms producing disparities in health (e.g., Boardman, Domingue, and Daw 2015). Indeed, prior work shows that genes partially explain the overlap between low education and high blood pressure, low cholesterol, or high waist circumference (Vermeiren et al. 2013). Future research could examine how this overlap varies across time or across specific institutional and policy regimes. It may also assess whether declines in the genetic correlation between two traits correspond to similar shifts in the observed correlation, which indicate variation in socioeconomic disparities in health. By extension, assessing (rG)xE reveals something novel about the nature of inequality and how societal conditions either ameliorate or intensify it.
Stratification is not the only research area in which the (rG)xE perspective may be illuminating. In the same way that an intersectional perspective permeates much of the sociological literature (Collins 2015), one might imagine moderating effects of not just time, but of gender or social class, for example, on the genetic correlation between any number of traits (e.g., fertility and education, risk taking behavior and peer association networks, subjective wellbeing and occupational prestige). The novelty of the (rG)xE perspective is that we can better gauge whether outcomes in distinct domains of life become more strongly bundled in some environmental conditions than others.
Despite the significance of our results, it is important to consider some weaknesses of the current study. First, while our results are consistent with the idea that mediated pleiotropy has grown across birth cohorts (such that the causal effect of educational attainment on smoking has increased), we are unable to fully disentangle this from different forms of pleiotropy. It is still possible that other forms of pleiotropy play a role in generating the genetic correlation between education and smoking, especially given the changing patterns in assortative mating and child-rearing strategies that have been observed over birth cohorts (Domingue et al. 2014). Next, we cannot empirically discern whether the causal effect of education on smoking has grown across birth cohorts or whether the reverse is true (i.e., that the effect of smoking on education has grown). While we can make no certain claims about the absence of reverse causation, prior work suggests that our causal ordering is likely to play a larger role (Pampel et al.2010). There is also new pleiotropic evidence suggesting that often the biological mechanisms uncovered for education are the same as those already uncovered in similar studies for smoking (Karlsson and Linner 2017). The social and behavioral pathways that led to this form of pleiotropy remains unknown; we believe that the sociological perspective provides critical insights into this form of correlation.
CONCLUSION
We write this article at a time of controversy in the subfield of social science genetics. Though researchers have increasingly called for the incorporation of biological insights into social science inquiry (Freese, Li, and Wade 2003; Freese and Shostak 2009; Boardman, Daw, and Freese 2013; Shostak and Beckfield 2015), there is also concern that the field has begun to head away from understanding the social origins upon which our subfield was founded. In particular, some worry that in speculating beyond their empirical findings, sociogenomicists may reach for controversial conclusions that hint at biological determinism, claims that are dangerous for broader sociology and the social world.
In this paper, we wish to return the social science of genetics to its roots — roots which begin at the heart of sociological inquiry – while also extending it to the study of relationships between complex human outcomes. We assert that many genetic associations are fundamentally sociological in nature. Given this, genes are far from causal or deterministic. Rather, a deeper understanding of how genes operate in the population tells us far more about the social environment and about social structure than about the effects of any one individual’s genotype or genetic propensity.
We highlight that the study of human genetics that is divorced from an understanding of social forces is likely to produce misleading conclusions. Using this particular study as an example, consider a researcher carrying out a genome wide study of smoking in 1950. That researcher may have uncovered genetic variants relevant to the biology of smoking — such as those involved in the metabolism of nicotine — variants that could in fact have been good candidates for pharmaceutical interventions (Boardman et al. 2010). However, given the social forces at work and the evidence found in this study, if a similar study of smoking was carried out in a younger set of individuals today, a researcher would be more likely to identify variants that are in fact predicting educational attainment. The associations with smoking would be spurious. Individuals who get more schooling are selected into environments where smoking is no longer acceptable. And thus we arrive at the larger sociological contribution of this study: without a deeper understanding of the environment and broad social forces, genetic research may produce unsound conclusions.
As sociologists, we have long been fascinated with understanding and explaining the causes of non-random variation within a population. Our field is well-equipped with a vibrant and rich history that highlights how this non-random variation is in fact driven by the fundamental social forces which define human interactions and behavior. Here we use the tools of population genetics as a way to gain insight into how the social environment might structure genetic correlation over time. In doing so we show not only that there is valuable insight to be gained by incorporating genetic concepts into sociological research, but also how much sociology has to contribute to the field of population genetics.
Supplementary Material
APPENDIX
Quality control of genetic data
Quality control of the genetic data in this study involved three separate quality control processes.
Imputation quality control
We imputed the HRS and Add Health data to the Haplotype Reference Consortium (HRC) reference panel (McCarthy et al. 2016) using the Michigan Imputation Server (Das et al. 2016). Before imputation, we applied quality control filters to both data sets using PLINK, Version 1.9 (Chang et al. 2015). We determined genetic ancestry, and removed all non-European individualsxii. We also removed individuals who were ancestral outliers, who had excessive levels of heterozygosity, and who were missing more than 5% of their genetic data (per chromosome). Next, we dropped all non-autosomal SNPs, all SNPs with missing call rates exceeding 0.02, all SNPs with Hardy-Weinberg equilibrium (HWE) exact test p-value below 10−4, and all SNPs with a minor allele frequency (MAF) below 0.01.
GWAS quality control
Before performing genome-wide association studies, we applied another set of standard QC filters on the imputed genetic data in both the HRS and Add Health. In the HRS, we began with 12,454 individuals and 20,994,118 imputed variants. We removed 3,802 non-European individuals; 3 individuals who were missing more than 5% of their genetic data; and 283 randomly selected individuals who were in pair with genetic relatedness greater than 0.025. We further removed 2,286,958 SNPs with missing call rates exceeding 0.02; 3,632 SNPs with Hardy-Weinberg equilibrium exact test p-values below 10−4; and 12,336,437 SNPs with a minor allele frequency below 0.01. In total then, 8,366 individuals and 6,367,091 SNPs were eligible to be included in our HRS association studies.
In Add Health, we began with 5,191 individuals and 38,909,200 imputed variants. We removed 785 individuals who were missing more than 5% of genetic data; and 0 individuals who, for a given pair, had an observed genetic relatedness greater than 0.025. We further remove 0 SNPs with missing call rates exceeding 0.02; 8,802 SNPs with Hardy-Weinberg equilibrium exact test p-values below 10−4; and 31,333,710 SNPs with a minor allele frequency below 0.01. In total for Add Health, 4,406 individuals and 7,566,688 SNPs were eligible to be included in our association studies.
LDpred quality control
Our LDpred polygenic scores in the HRS and Add Health were composed only of unrelated European-ancestry individuals. For the HRS, there were 12,094,964 SNPs in the meta-analysis of educational attainment. Of these, 1,535,195 SNPs were genotyped in HRS with a call rate above 98% and have a HWE test p-value greater than 10−4. 48,878 strand-ambiguous SNPs, 128,822 SNPs with minor allele frequencies less than 1%, and 47 SNPs with a high minor allele frequency discrepancy between the meta-analysis and the HRS prediction cohort were excluded, leaving 1,357,448 SNPs that were used to construct the scores. For Add Health, of the 12,094,964 SNPs in the meta-analysis, 704,105 were genotyped in Add Health with a call rate above 98% and had a HWE test p-value greater than 10−4. Zero strand-ambiguous SNPs, 300 SNPs with minor allele frequency less than 1%, and 10 SNPs with a substantial discrepancy in minor allele frequency between the meta-analysis and Add Health prediction cohort were excluded, leaving 703,795 SNPs that were used to construct the Add Health scores.
LDSC additional methods
In addition to the results from GWAS, LDSC requires as an input a reference data set detailing the structure of LD in a population of similar ancestry. The reference data set specifies an LD score for each SNP, which measures the extent to which the SNP is in LD with other genetic markers. We use a reference panel constructed by Finucane et al. (2015) using data from Europeans involved in the 1,000 Genomes Project. After merging the SNPs from our data sets with this reference panel, we were able to use about 1,105,000 SNPs from the HRS and roughly 1,163,000 SNPs from Add Health with LDSC.
LDSC uses the following moment formula to generate estimates of heritability and genetic covariance:
| [Equation S1] |
where zkj is the effect of genotype at SNP j on phenotype k (k = 1, 2) estimated via GWAS and expressed as a z-score, Intercept is the regression intercept, Nk is the number of respondents in the GWAS of phenotype k, M is the number of SNPs included in the GWAS, is the genetic covariance between phenotypes 1 and 2, and is the LD score of SNP j, which is a measure of the extent to which SNP j is in LD with other SNPs. By simple algebraic rules, one can estimate the genetic covariance between phenotypes 1 and 2 with the coefficient generated through from a regression of on , multiplied by M. Heritability can be thought of as a special case of genetic covariance: it is the genetic covariance of a phenotype with itself. Thus, estimating the heritability of phenotypes 1 () and 2 () with LDSC involves regressing on and on , respectively.
LDSC estimates the genetic correlation, , between two phenotypes as the estimated genetic covariance between the phenotypes of interest scaled by their estimated heritabilities:
| [Equation S2] |
Standard errors for both heritabilities and genetic correlation are estimated by using a block jackknife over all SNPs.
Footnotes
Author’s own calculations using data from the 2015 National Health Interview Survey (NHIS), which was compiled by the Minnesota Population Center and the State Health Access Data Assistance Center (2016).
Saliva samples were first collected from HRS respondents during in-home interviews in 2006, at which time a random subsample of households was asked to participate; of those invited, 83% consented to genotyping. In 2008, the genetic sample was expanded to all remaining households; 84% agreed to participate. Genotyping was performed using Illumina’s Human Omni2.5-Quad BeadChip technology by the NIH Center for Inherited Disease Research (Health and Retirement Study 2017).
We recognize the limitations and inherent problems with this approach, as does the broader human genetics field. Efforts to collect such samples are already underway.
A total of n=12,234 (78%) consented. Genotyping was performed at the Institute for Behavioral Genetics in Boulder, CO using Illumina’s Human Omni1-Quad-BeadChip (McQueen et al. 2015).
At the time of writing, we had permission to use the latest release of the Add Health data, which was in a “Freeze 2” hold. In Freeze 2, 7,598 individuals were genotyped using the Illumina Human Omni 1 chip, and 2,098 individuals were genotyped on the Illumina Human Omni 2.5 chip. Because nearly 85% of subjects genotyped using the Omni 2.5 chip were of non-European ancestry, we excluded individuals on this chip both to reduce bias introduced by batch effects and also to avoid an unnecessary loss of SNPs when taking the intersection of SNPs common to both chips.
LDSC also adjusts results for confounding of genetic effects by ancestry (population stratification).
Our measure of cognition is a modified version of the Peabody Picture Vocabulary Test (Dunn and Dunn 2007). The test was administered when respondents were ages 12–20 during the first in-home follow-up survey. In this test, an interviewer reads a word aloud, and a respondent selects the illustration that best fits the word’s meaning. Eighty-seven items were included on this computer-adapted test, and scores were age-standardized.
Among men in the HRS, the correlation between years of education and height is 0.134 (p<0.001). For women, the correlation is 0.133 (p<0.001). For Add Health men, r = 0.108 (p<0.001) and for women, r = 0.112 (p<0.001).
LDSC estimates narrow-sense heritability, which is the proportion of variance in a phenotype that can be explained by additive genetic effects. Such estimates are likely biased downward in comparison to broad-sense heritability estimates from twin studies, which include additive as well as non-linear genetic effects (see Online Supplement for twin-based estimates in comparable data sets). Despite this, LDSC estimates of genetic correlation remain unbiased (Bulik-Sullivan 2015; see also “Heritability and Genetic Correlation wiki here: https://github.com/bulik/ldsc/wiki/Heritability-and-Genetic-Correlation). The heritability of height, computed via LDSC, is roughly identical among HRS and Add Health respondents (Table 3), while the heritability of both education and smoking is higher among those born in later years. This is in line with previous research finding increases in the heritability of education (Heath et al. 1985; Branigan et al. 2013) and smoking (Domingue et al. 2016) over time.
We note that, while we are well powered to distinguish genetic correlation from 0 in each data set, we are not well powered to distinguish correlations from each other. Future research should revisit the question of statistical significance when larger samples are available.
This is unsurprising in light of outside data showing more drastic changes in these phenotypes over time than across places. For instance, in 1940 just 25% of Americans ages 25 and above had a high school degree. By 2000, this percentage had increased to 80%. Comparatively, in 2000, the percentage of Americans who had attained a high school degree ranged only modestly across states, from 73% to 89% (U.S. Census Bureau, Decennial Census of Census of Population, 1940 to 2000). Thus, our findings regarding temporal mediation vs. spatial mediation seem to be in line with larger demographic trends.
Non-Europeans were removed before imputation in Add Health, but after imputation in the HRS.
REFERENCES
- Belsky Daniel W., Moffitt Terrie E., Corcoran David L., Domingue Benjamin, Harrington Hona Lee, Hogan Sean, Houts Renate, Ramrakha Sandhya, Sugden Karen, Williams Benjamin S., Poulton Richie, and Caspi Avshalom. 2016. “The Genetics of Success: How Single-Nucleotide Polymorphisms Associated with Educational Attainment Relate to Life-Course Development.” Psychological Science 27(7):957–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergen Sarah E., Gardner Charles O., and Kendler Kenneth S.. 2007. ” Age-Related Changes in Heritability of Behavioral Phenotypes over Adolescence and Young Adulthood: A Meta-Analysis.” Twin Research and Human Genetics 10(3):423–433. [DOI] [PubMed] [Google Scholar]
- Berger Peter L. and Luckmann Thomas. 1966. “The Social Construction of Reality: A Treatise in the Sociology of Knowledge.” Garden City, NY: First Anchor. [Google Scholar]
- Boardman Jason D., Saint Onge Jarron M., Haberstick Brett C., Timberlake David S., and Hewitt John K.. 2008. “Do Schools Moderate the Genetic Determinants of Smoking?” Behavior Genetics 38:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman Jason D. 2009. “State-Level Moderation of Genetic Tendencies to Smoke.” American Journal of Public Health 99(3):480–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman Jason D., Blalock Casey L., and Pampel Fred C. 2010. “Trends in the Genetic Influences on Smoking.” Journal of Health and Social Behavior 51(1):108–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman Jason D., Blalock Casey L., Pampel Fred C., Hatemi Peter K., Heath Andrew C., and Eaves Lindon J.. 2011. “Population Composition, Public Policy, and the Genetics of Smoking.” Demography 48(4):1517–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman Jason D., Daw Jonathan, and Freese Jeremy. 2013. “Defining the Environment in Gene–Environment Research: Lessons From Social Epidemiology. “American Journal of Public Health 103(1):S64–S72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman Jason D., Domingue Benjamin W., Blalock Casey L., Haberstick Brett C., Harris Kathleen Mullan, and McQueen Matthew B.. 2014. “Is the Gene-Environment Interaction Paradigm Relevant to Genome-Wide Studies? The Case of Education and Body Mass Index.” Demography 51(1):119–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boardman Jason D., Domingue Benjamin W., and Daw Jonathan. 2015. “What Can Genes Tell Us About the Relationship Between Education and Health?” Social Science & Medicine 127:171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boker Steven, Neale Michael, Maes Hermine, Wilde Michael, Spiegel Michael, Brick Timothy, Spies Jeffrey, Estabrook Ryne, Kenny Sarah, Bates Timothy, Mehta Paras, and Fox John. 2011. “OpenMx: An Open Source Extended Structural Equation Modeling Framework.” Psychometrika 76(2):307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branigan Amelia R., McCallum Kenneth J., and Freese Jeremy. 2013. “Variation in the Heritability of Educational Attainment: An International Meta-Analysis.” Social Forces 92(1):109–140. [Google Scholar]
- Briley Daniel A. and Tucker-Drob Elliot M.. 2013. “Genetic and Environmental Continuity in Personality Development: A Meta-Analysis.” Psychological Bulletin 140(5):1303–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan Brendan K., Loh Po-Ru, Finucane Hilary K., Ripke Stephan, Yang Jian, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson Nick, Daly Mark J., Prices Alkes L., and Neale Benjamin M.. 2015. “LD Score Regression Distinguishes Confounding from Polygenicity in Genome-Wide Association Studies.” Nature Genetics 47:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan Brendan K., Finacane Hilary K., Anttila Verneri, Gusav Alexander, Day Felix R., Loh Po-Ru, Consortium ReproGen, Psychiatric Genomics Consortium, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3, Duncan Laramie, Perry John R. B., Patterson Nick, Robinson Elise B., Daly Mark J., Price Alkes L., and Neale Benjamin M.. 2016. “An Atlas of Genetic Correlations across Human Diseases and Traits.” Nature Genetics 47:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control (CDC). 2016. “Trends in Current Cigarette Smoking Among High School Student and Adults, United States, 1965–2014.” Accessed March 27, 2017: https://www.cdc.gov/tobacco/data_statistics/tables/trends/cig_smoking/
- Centers for Disease Control (CDC). 2011–2015. “Map of Current Cigarette Use Among Adults.” Accessed March 27, 2017: https://www.cdc.gov/statesystem/cigaretteuseadult.html
- Chang Christopher C., Chow Carson C., Tellier Laurent CAM, Vattikuti Shashaank, Purcell Shaun M., and Lee James J.. 2015. “Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets.” GigaScience 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins Patricia H. 2015. “Intersectionality’s Definitional Dilemmas.” Annual Review of Sociology 41:1–20. [Google Scholar]
- Conley Dalton and Rauscher Emily. 2013. “The Effect of Daughters on Partisanship and Social Attitudes toward Women.” Sociological Forum 28(4):700–718. [Google Scholar]
- Conley Dalton, Laidley Thomas M., Boardman Jason D., and Domingue Benjamin W.. 2016. “Changing Polygenic Penetrance on Phenotypes in the 20th Century Among Adults in the US Population.” Scientific Reports 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin Cook, C. and Fletcher Jason M.. 2013. “Interactive Effects of In Utero Nutrition and Genetic Inheritance on Cognition: New Evidence using Sibling Comparisons.” Economics & Human Biology 13:144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Sayantan, Forer Lukas, Sebastian Schönherr Carlo Sidore, Locke Adam E., Kwong Alan, Vrieze Scott I., Chew Emily Y., Levy Shawn, Matt McGue David Schlessinger, Stambolian Dwight, Loh Po-Ru, Iacono William G., Swaroop Anand, Scott Laura J., Cucca Francesco, Kronenberg Florian, Boehnke Michael, Abecasis Gonçalo R., and Fuchsberger Christian. 2016. “Next Generation Genotype Imputation Service and Methods.” Nature Genetics 48(10):1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingue Benjamin W., Fletcher Jason, Conley Dalton, and Boardman Jason D.. 2014. “Genetic and Educational Assortative Mating Among US Adults.” Proceedings of the National Academy of Sciences 111(22):7996–8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingue Benjamin W., Conley Dalton, Fletcher Jason, and Boardman Jason D.. 2016. “Cohort Effects in the Genetic Influence on Smoking.” Behavior Genetics 46(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingue Benjamin W., Belsky Daniel W., Conley Dalton, Harris Kathleen Mullan, and Boardman Jason D.. 2015. “Polygenic Influence on Educational Attainment: New Evidence from the National Longitudinal Study of Adolescent to Adult Health.” AERA Open 1(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkheim Emile. 1997[1893]. The Division of Labor in Society. New York, NY: The Free Press. [Google Scholar]
- Jr Elder Glen H., 1999. Children of the Great Depression: Social Change in Life Experience. Boulder, CO: Westview Press. [Google Scholar]
- Eley Thalia C., Lichtenstein Paul, and Stevenson Jim. 1999. “Sex Differences in the Etiology of Aggressive and Nonaggressive Antisocial Behavior: Results from Two Twin Studies.” Child Development 70(1):155–168. [DOI] [PubMed] [Google Scholar]
- Freese Jeremy, Jui-Chung Allen Li, and Wade Lisa D.. 2003. “The Potential Relevances of Biology to Social Inquiry.” Annual Review of Sociology 29:233–256. [Google Scholar]
- Freese Jeremy and Shostak Sara. 2009. “Genetics and Social Inquiry.” Annual Review of Sociology 35:107–128. [Google Scholar]
- Frohlich Katherine L., Corin Ellen, and Potvin Louise. 2001. “A Theoretical Proposal for the Relationship Between Context and Disease.” Sociology of Health & Illness 23(6):776–797. [Google Scholar]
- Goldin Claudia and Katz Lawrence F.. 2009. The Race Between Education and Technology. Cambridge, MA: Harvard University Press. [Google Scholar]
- Guo Guang, Roettger Michael E., and Cai Tianji. 2008. “The Integration of Genetic Propensities into Social Control Models of Delinquency and Violence among Male Youths.” American Sociological Review 73(4):543–568. [Google Scholar]
- Harris Kathleen Mullan, Halpern Carolyn T., Whitsel Eric A., Hussey Jon, Tabor Joyce, Entzel Pamela, and Udry J. Richard. 2009. The National Longitudinal Study of Adolescent to Adult Health: Research Design. Accessed March 27, 2017: http://www.cpc.unc.edu/projects/addhealth/design.
- Heath Andrew C., Kendler Kenneth S., Eaves Lindon J., and Markell David. 1985. “The Resolution of Cultural and Biological Inheritance: Informativeness of Different Relationships.” Behavior Genetics 15(5):439–465. [DOI] [PubMed] [Google Scholar]
- Herrnstein Richard J. and Murray Charles. 1994. The Bell Curve: Intelligence and Class Structure in American Life. New York, NY: The Free Press. [Google Scholar]
- Hout Michael. 1988. “More Universalism, Less Structural Mobility: The American Occupational Structure in the 1980s.” American Journal of Sociology 93(6):1358–1400. [Google Scholar]
- Hout Michael. 2012. “Social and Economic Returns to College Education in the United States.” Annual Review of Sociology 38:379–400. [Google Scholar]
- Hung Rayjean J., McKay James D., et al. 2008. “A Susceptibility Locus for Lung Cancer Maps to Nicotinic Acetylcholine Receptor Subunit Genes on 15q25.” Nature 452:633–637. [DOI] [PubMed] [Google Scholar]
- Jencks Christopher. 1980. “Structural Versus Individual Explanations of Inequality: Where Do We Go from Here?” Contemporary Sociology 9(6):762–767. [Google Scholar]
- Jha Prabhat, Peto Richard, Zatonski Witold, Boreham Jillian, Jarvis Martin J., and Lopez Alan D.. 2006. “Social Inequalities in Male Mortality, and in Male Mortality from Smoking: Indirect Estimation from National Death Rates in England and Wales, Poland, and North America.” Lancet 368(9533):367–370. [DOI] [PubMed] [Google Scholar]
- Kendler Kenneth S., Thornton Laura M., and Pedersen Nancy L.. 2000. “Tobacco Consumption in Swedish Twins Reared Apart and Reared Together.” Arch Gen Psychiatry 57(9):886–892. [DOI] [PubMed] [Google Scholar]
- Landecker Hannah and Panofsky Aaron. 2013. “From Social Structure to Gene Regulation, and Back: A Critical Introduction to Environmental Epigenetics for Sociology.” Annual Review of Sociology 39:333–357. [Google Scholar]
- Lawson Elizabeth A., Eddy Kamryn T., Donoho Daniel, Misra Madhusmita, Miller Karen K., Meenaghan Erinne, Lydecker Janet, Herzog David, and Klibanski Anne. 2011. “Appetite-Regulating Hormones Cortisol and Peptide YY are Associated with Disordered Eating Psychopathology, Independent of Body Mass Index.” European Journal of Endocrinology 164(2):253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Ming D., Cheng Rong, Ma Jennie Z., and Swan Gary E.. 2003. “A Meta-Analysis of Estimated Genetic and Environmental Effects on Smoking Behavior in Male and Female Adult Twins.” Addiction 98(1):23–31. [DOI] [PubMed] [Google Scholar]
- Li Shengxu, Zhao Jing Hua, Luan Jian’an, Ekelund Ulf, Luben Robert N., Khaw Kay-Tee, Wareham Nicholas J., and Loos Ruth J. F.. 2010. “Physical Activity Attenuates the Genetic Predisposition to Obesity in 20,000 Men and Women from EPIC-Norfolk Prospective Population Study.” PLoS Med 7(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link Bruce G. 2008. “Epidemiological Sociology and the Social Shaping of Population Health.” Journal of Health and Social Behavior 49(4):367–384. [DOI] [PubMed] [Google Scholar]
- Liu Hexuan and Guo Guang. 2015. “Lifetime Socioeconomic Status, Historical Context, and Genetic Inheritance in Shaping Body Mass in Middle and Late Adulthood.” American Sociological Review 80(4):705–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber Judith. Paradoxes of Gender. 1994. New Haven, CT: Yale University Press. [Google Scholar]
- Manuck Stephen B. and McCaffery Jeanne M.. 2014. “Gene-Environment Interaction.” Annual Review of Psychology 65:41–70. [DOI] [PubMed] [Google Scholar]
- Marchini Marta, Sparrow Leah M., Cosman Miranda N., Dowhanik Alexandra, Krueger Carston B., Hallgrimsson Benedikt, and Rolian Campbell. 2014. “Impacts of Genetic Correlation on the Independent Evolution of Body Mass and Skeletal Size in Mammals.” BMC Evolutionary Biology 14:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin William G. and Beittel Mark. 1998. “Toward a Global Sociology? Evaluating Current Conceptions, Methods, and Practices.” The Sociological Quarterly 39(1):139–161. [Google Scholar]
- McCaffery Jeanne M., Papandonatos George D., Lyons Michael J., Koenen Karestan C., Tsuang Ming T., and Niaura Raymond. 2008. “Educational Attainment, Smoking Initiation and Lifetime Nicotine Dependence among Male Vietnam-era Twins.” Psychological Medicine 38(9):1287–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy Shane, Das Sayantan, Kretzschmar Warren, et al. , Durbin Richard, Abecasis Gonçalo, and Marchini Jonathan. 2016. “A Reference Panel of 64,976 Haplotypes for Genotype Imputation.” Nature Genetics 48:1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGue Matt and Bouchard Thomas J. Jr. 1984. “Adjustment of Twin Data for the Effects of Age and Sex.” Behavior Genetics 14(4):325–343. [DOI] [PubMed] [Google Scholar]
- Wright Mills, C.. 1959. The Sociological Imagination. New York, NY: Oxford University Press. [Google Scholar]
- Mitchell Colter, Hobcraft John, McLanahan Sara S., Siegel Susan Rutherford, Berg Arthur, Brooks-Gunn Jeanne, Garfinkel Irwin, and Notterman Daniel. 2014. “Social Disadvantage, Genetic Sensitivity, and Children’s Telomere Length.” Proceedings of the National Academy of Sciences 111(16):5944–5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers JL, Cerda M, Galea S, Keyes KM, Aiello AE, Uddin M, Wildman DE, Koenen KC. 2013. “Interaction Between Polygenic Risk for Cigarette Use and Environmental Exposures in the Detroit Neighborhood Health Study.” Translational Psychiatry 3:e290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omi Michael and Winant Howard. 1994. “Racial Formation in the United States: From the 1960s to the 1990s.” New York, NY: Routledge. [Google Scholar]
- Pampel Fred C. 2009. “The Persistence of Educational Disparities in Smoking.” Social Problems 56(3):526–542. [Google Scholar]
- Pampel Fred C., Krueger Patrick M., and Denney Justin T.. 2010. “Socioeconomic Disparities in Health Behaviors.” Annual Review of Sociology 36:349–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pampel Fred C., Legleye Stephanie, Goffette Céline, Piontek Daniela, Kraus Ludwig, and Khlat Myriam. 2015. “Cohort Changes in Educational Disparities in Smoking: France, Germany, and the United States.” Social Science & Medicine 127:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomin R, Campos J, Corley R, Emde RN, Fulker DW, Kagan J, Reznick JS, Robinson J, Zahn-Waxler C, DeFries JC.1990. “Individual Differences During the Second Year of Life: The MacArthur Longitudinal Twin Study” Pp.431–455 in Colombo John and Fagan Jeffrey (Eds.), Individual Differences in Infancy: Reliability, Stability, and Predictability. Hillsdale, NJ: Earlbaum. [Google Scholar]
- Polderman Tinca J. C., Benyamin Beben, de Leeuw Christiaan A., Sullivan Patrick F., Bochovan Arjen van, Visscher Peter M., and Posthuma Danielle. 2015. “Meta-Analysis of the Heritability of Human Traits Based on Fifty Years of Twin Studies.” Nature Genetics 47(7):702–709. [DOI] [PubMed] [Google Scholar]
- Pomerantz Mark M., Ahmadiyeh Nasim, Jia Li, Herman Paula, Verzi Michael P., Doddapaneni Harshavardhan, Beckwith Christine A., Chan Jennifer A., Hills Adam, David Matt, Yao Keluo, Kehoe Sarah M., Lenz Heinz-Josef, Haiman Christopher A., Yan Chunli, Henderson Brian E., Frenkel Baruch, Barretina Jordi, Bass Adam, Tabernero Josep, Baselga José, Regan Meredith M., Manak J. Robert, Shivdasani Ramesh, Coetzee Gerhard A., and Freedman Matthew L.. 2009. “The 8q24 Cancer Risk Variant rs6983267 Shows Long-Range Interaction with MYC in Colorectal Cancer.” Nature Genetics. 41:882–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAND. 2016. RAND HRS Data, Version P. 2016. Produced by the RAND Center for the Study of Aging, with funding from the National Institute on Aging and the Social Security Administration. Santa Monica, CA: RAND. [Google Scholar]
- Ridgeway Cecilia L. and Correll Shelley J.. 2004. “Unpacking the Gender System a Theoretical Perspective on Gender Beliefs and Social Relations.” Gender & Society 18(4):510–531. [Google Scholar]
- Ryan Camille L. and Bauman Kurt. 2016. “Educational Attainment in the United States: 2015.” Current Population Reports 20. [Google Scholar]
- Saperstein Aliya and Penner Andrew M.. 2012. “Racial Fluidity and Inequality in the United States.” American Journal of Sociology 118(3): 676–727. [Google Scholar]
- Short Susan E., Yang Yang Claire, and Jenkins Tania M.. 2013. “Sex, Gender, Genetics, and Health.” American Journal of Public Health 103(S1):S93–S101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shostak Sara and Beckfield Jason. 2015. “Making a Case for Genetics: Interdisciplinary Visions and Practices in the Contemporary Social Sciences” Pp.95–125 in Perry Brea L. (Ed.), Genetics, Health, and Society (Advances in Medical Sociology, Volume 16) Emerald Group Publishing Limited. [Google Scholar]
- Social Science Genetic Association Consortium. 2016. Genome-Wide Association Study Identifies 74 Loci Associated with Educational Attainment.” Nature 533:539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovieff Nadia, Cotsapas Chris, Lee Phil H., Purcell Shaun M., and Smoller Jordan W., 2013. “Pleiotropy in Complex Traits: Challenges and Strategies.” Nature Reviews Genetics 14(7):483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber Jennifer, Galea Sandro, and Link Bruce G.. 2009. “Stigma and Smoking: The Consequences of Our Good Intentions.” Social Service Review 83(4):585–609. [Google Scholar]
- Thorgeirsson Thorgeir E., Geller Frank, Sulem Patrick, Rafnar Thorunn, Wiste Anna, Magnusson Kristinn P., Manolescu Andrei, Thorleifsson Gudmar, Stefansson Hreinn, et al. , Thorsteinsdottir Unnur, Kong Augustine, and Stefansson Kari, et al. 2008. “A Variant Associated with Nicotine Dependence, Lung Cancer and Peripheral Arterial Disease.” Nature 452:638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobacco and Genetics Consortium. 2010. “Genome-Wide Meta-Analyses Identify Multiple Loci Associated with Smoking Behavior.” Nature Genetics 42(5):441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropf Felix C. and Mandemakers Jornt J.. 2017. “Is the Association between Education and Fertility Postponement Causal? The Role of Family Background Factors.” Demography 54(1):71–91. [DOI] [PubMed] [Google Scholar]
- Turkheimer Eric. 2000. “Three Laws of Behavior Genetics and What They Mean.” Current Directions in Psychological Science 9:160–164. [Google Scholar]
- U.S. Bureau of the Census. 1947–2015. Current Population Survey and 1940 Decennial Census. Washington, D.C: U.S. Government Printing Office. [Google Scholar]
- Vermeiren Angelique P. A., et al. 2013. “The Association Between APOE 4 and Alzheimer-type Dementia Among Memory Clinic Patients is Confined to those with a Higher Education. The DESCRIPA Study.” Journal of Alzheimer’s Disease 35:1–2. [DOI] [PubMed] [Google Scholar]
- Vilhjálmsson Bjarni J., Yang Jian, Finucane Hilary K., Gusev Alexander, Sara Lindström Stephan Ripke, Genovese Giulio, et al. , Holmans Peter A., Lee Phil, Bulik-Sullivan Brendan. 2015. “Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores.” American Journal of Human Genetics 97(4): 576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter Stephan, Ivan Mejía-Guevara Karol I. Estrada, Liu Sze Y., and Glymour M. Maria. 2016. “Association of a Genetic Risk Score with Body Mass Index Across Different Birth Cohorts.” JAMA 316(1):63–69. [DOI] [PubMed] [Google Scholar]
- Wasserman Nora F., Aneas Ivy, and Nobrega Marcelo A.. 2010. “An 8q24 Gene Desert Variant Associated with Prostate Cancer Risk Confers Differential In Vivo Activity to a MYC Enhancer.” Genome Research 20(9):1191–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedow Robert, Briley Daniel A., Short Susan E., Boardman Jason D.. 2016. “Gender and Genetic Contributions to Weight Identity Among Adolescents and Young adults in the US.” Social Science & Medicine 165:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Yang. 2008. “Social Inequalities in Happiness in the United States, 1972 to 2004: An Age-Period-Cohort Analysis.” American Sociological Review 73(2):204–226. [Google Scholar]
- Yang Jian, Benyamin Beben, McEvoy Brian P., Gordon Scott, Henders Anjali K., Nyholt Dale R., Madden Pamela A., Heath Andrew C., Martin Nicholas G., Montgomery Grant W., Goddard Michael E., and Visscher Peter M.. 2010. “Common SNPs Explain a Large Proportion of the Heritability for Human Height.” Nature Genetics 42(7):565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.