

Abstract
Background:
Modeling of human arrhythmias using induced pluripotent stem cell-derived cardiomyocytes has focused on single cell phenotypes. However, arrhythmias are the emergent properties of cells assembled into tissues, and the impact of inherited arrhythmia mutations on tissue-level properties of human heart tissue has not been reported.
Methods:
Here, we report an optogenetically-based, human engineered tissue model of catecholaminergic polymorphic ventricular tachycardia (CPVT), an inherited arrhythmia caused by mutation of the cardiac ryanodine channel (RYR2) and triggered by exercise. We developed a hiPSC-CM-based platform to study the tissue-level properties of engineered human myocardium. We investigated pathogenic mechanisms in CPVT, by combining this novel platform with genome editing.
Results:
In our model, CPVT tissues were vulnerable to develop reentrant rhythms when stimulated by rapid pacing and catecholamine, recapitulating hallmark features of the disease. These conditions elevated diastolic Ca2+ levels and increased temporal and spatial dispersion of Ca2+ wave speed, creating a vulnerable arrhythmia substrate. Using Cas9 genome editing, we pinpointed a single catecholamine-driven phosphorylation event, RYR2-S2814 phosphorylation by Ca2+-calmodulin-dependent protein kinase II (CaMKII), that is required to unmask the arrhythmic potential of CPVT tissues.
Conclusions:
Our study illuminates the molecular and cellular pathogenesis of CPVT and reveals a critical role of CaMKII-dependent reentry in the tissue-scale mechanism of this disease. We anticipate that this approach will be useful to model other inherited and acquired cardiac arrhythmias.
Keywords: Catecholaminergic polymorphic ventricular tachycardia (CPVT), Stem cell, Disease modeling
INTRODUCTION
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited arrhythmia predominantly caused by autosomal dominant mutation of the gene encoding the cardiac ryanodine receptor (RYR2), the main intracellular calcium release channel of cardiomyocytes.1 Typically, CPVT patients are asymptomatic at rest but develop potentially lethal ventricular tachycardia during exercise or emotional distress (Figure 1A and Supplemental Figure 1A). In wild type cells, when the cardiac action potential opens the voltage sensitive L-type Ca2+ channel located in the plasma membrane, the resulting local influx of Ca2+ triggers release of Ca2+ from the sarcoplasmic reticulum via RYR2 (Figure 1B). The resulting increase in cytoplasmic Ca2+ leads to sarcomere contraction. As the cell enters diastole, RYR2 closes and cytosolic Ca2+ is pumped back into the sarcoplasmic reticulum. In cells carrying CPVT mutations, RYR2 releases more Ca2+ into the cytoplasm, resulting in elevated diastolic Ca2+ that drives exchange of sodium and calcium through the plasma membrane via the sodium calcium exchanger (NCX1),3 leading to after-depolarizations that may trigger additional action potentials. The molecular mechanism by which catecholamine stimulation unmasks the arrhythmic nature of CPVT mutations is not known, although catecholamine-induced activation of Ca2+-calmodulin-dependent protein kinase II (CaMKII) has been implicated.4,5 The mechanisms by which RYR2 mutation and consequent cellular abnormalities such as after-depolarizations cause the organ-level phenotype of ventricular tachycardia is also uncertain. One theory is that triggered activity in the His-Purkinje system initiates ventricular tachycardia.6,7 However, in a mouse CPVT model, ventricular arrhythmias required the CPVT-causing mutation in working myocardium, in addition to the His-Purkinje system.8 suggesting that CPVT-causing mutations produce a substrate for re-entrant and/or focal arrhythmias in the working myocardium. Understanding how single cell Ca2+ handling abnormalities lead to tissue, organ, and clinical manifestations of CPVT have been hampered by limitations of currently available disease models.
Figure 1. Characterization of Ca2+ oscillations in iPSC-CM clusters.
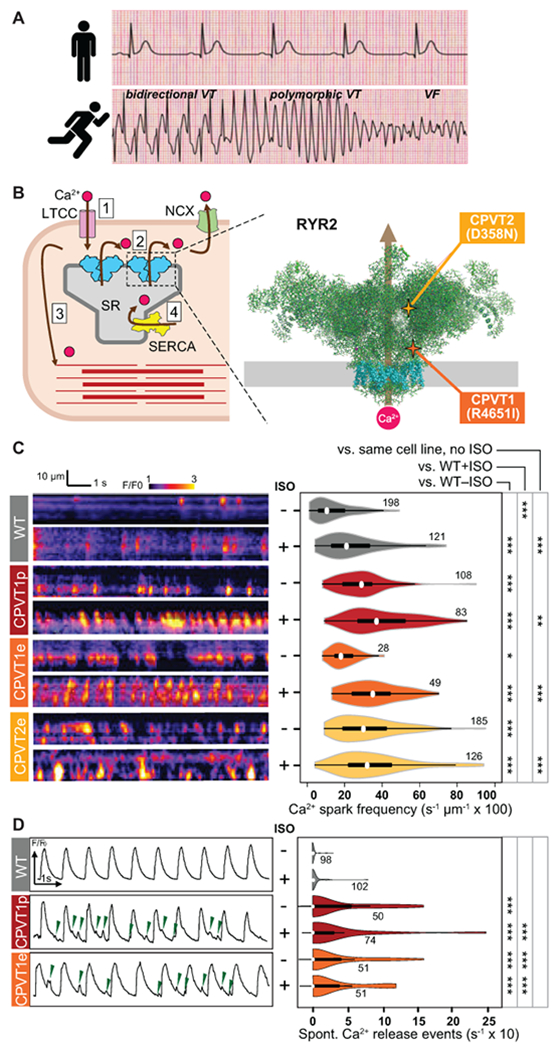
A. CPVT patients have normal resting electrocardiograms but severe, potentially life-threatening arrhythmias with exercise. VT, ventricular tachycardia. VF, ventricular fibrillation. Traces are idealized sketches shown for illustration purposes. B. CPVT pathophysiology. Left, cartoon of cardiomyocyte Ca2+-induced Ca2+ release. 1. Action potential opens L-type Ca2+ channel (LTCC); 2. Ca2+ induces opening of RYR2 and release of Ca2+ from the sarcoplasmic reticulum (SR); 3. Elevated intracellular Ca2+ induces myofilament contraction; 4. Ca2+ is cleared from the cytosol by SERCA and NCX. Right, CPVT mutations in RYR2 increase diastolic Ca2+ leak. Cartoon shows RYR2 structure based on CryoEM data2. Grey rectangle indicates the sarcoplasmic reticulum membrane. Transmembrane domains are shown in cyan. CPVT-causing RYR2 mutations in this study are highlighted. C. Incidence of Ca2+ sparks in quiescent iPSC-CMs. Left, representative confocal line scans of Fluo-4 signal within individual iPSC-CMs in cell clusters. Right, quantitative analysis. Number by each shape denotes number of cells examined. D. Incidence of spontaneous Ca2+ release events in spontaneously beating iPSC-CMs. Left, representative Ca2+ signal traces, spatially averaged over confocal line scans within individual iPSC-CMs in cell clusters. Green arrowheads indicate spontaneous Ca2+ release events. Right, quantitative analysis. Number by each shape denotes number of cells examined. Steel Dwass non-parametric test with multiple testing correction: *, P<0.05; **, P<0.01, ***, P<0.001
The advent of induced pluripotent stem cell (iPSC) technology and efficient methods to differentiate iPSCs to cardiomyocytes (iPSC-CMs) have created exciting opportunities to study inherited arrhythmias.9 iPSC-CMs have been generated from patients with CPVT5,10–13 as well as other inherited arrhythmias and have been shown to capture key features of these diseases, including abnormal action potential duration and drug responses.9 However, current iPSC-CM models of arrhythmia have been limited to isolated cells or cell clusters, leaving a large gap to modeling clinical arrhythmias, which are the emergent properties of cells assembled into myocardial tissue.14
We15–17 and others18 have developed engineered cardiac microphysiological systems that induce stem cell-derived and primary cardiomyocytes to adopt native-like laminar tissue architecture, permitting tissue level measurement of contractility. Here, we integrated an engineered cardiac tissue with optogenetics, optical mapping, and iPSC-CMs to create a human tissue model of CPVT. We used this model and Cas9-mediated genome editing to investigate the molecular and cellular mechanisms underlying exercise-induced ventricular tachycardia in this disease.
METHODS
Please refer to the Supplemental Methods for a full description of experimental procedures. The authors declare that all supporting data are available within the article and its online supplementary files. Materials will be shared upon reasonable request.
Human induced pluripotent stem cells and differentiation of iPSC-CMs
Patients provided informed consent to participate in this study under protocols approved by the Boston Children’s Hospital Institutional Review Board. Fibroblasts were cultured from skin biopsies and reprogrammed by episomal transfection with reprogramming factors. iPSC lines were maintained in mTeSR1 medium (STEMCELL Technologies) and passaged in versene solution (15040066, Thermo Fisher Scientific) every five days on culture dishes pre-coated with Matrigel (hESC-Qualified Matrix, LDEV-Free, Corning) diluted 1:100.
The procedures for CRISPR/Cas9 genome editing using a wild-type PGP1 human iPSC line containing doxycycline-inducible Cas9 was described in detail in a recent publication.19 Guide RNA and homology-directed repair constructs are provided in the Supplemental Methods. We predicted Cas9-gRNA off-target sites using http://crispr.mit.edu/.20 Amplicons containing the top 10 predicted sites were amplified from iPSCs and Sanger sequenced.
iPSCs differentiated to cardiomyocytes by modulating WNT signaling with CHIR99021 and IWR-1. iPSC-CMs were enriched by culture in lactate-containing media. Commercial hiPSC derived cardiomyocytes (hiPSC-CMs, Cor4U; Axiogenesis, Cologne, Germany) were cultured according to manufacturer’s instructions.
Opto-Muscular Thin Film (Opto-MTF) Construction
Micro-molded gelatin was fabricated on glass coverslips such that the gelatin in the base region of MTFs would firmly attach to the glass coverslips but the gelatin in the cantilever region could be easily peeled. Cantilevers (1 mm wide × 2 mm long) were laser engraved into the dehydrated micro-molded gelatin. Gelatin chips were UVO-treated for 90 seconds and re-hydrated in a 2 mM MES solution of pH 4.5 with 1 mg/ml collagen and 0.1 mg/mg fibronectin. After 2 hours at room temperature, the collagen and fibronectin solution was replaced with PBS. The gelatin chips were stored at 4ºC until cell seeding.
Dissociated iPSC-CMs were plated on gelatin chips and then transduced with ChR2 lentiviral vector, in which the cardiac troponin T promoter drives ChR2-eYFPP was constructed based on the FCK(1.3)GW plasmid with the cardiac troponin T (cTnT) promoter, ChR2, and enhanced yellow fluorescent tag.21,22
Calcium Imaging of Cell Clusters
iPSC-CMs were seeded on Matrigel-coated glass bottom dishes for 5 days. After loading with Fluo-4 (F14201, Thermo Fisher Scientific) or Fluovolt (Thermo Fisher Scientific), cells were imaged with an Olympus FV1000 using line scan mode (10 msec/line, 1000 lines per recording). Where indicated, cells were treated with PKA Inhibitor (myristoylated 14-22 amide, 1 µM) or CaMKII inhibitor (myristolated Autocamtide-2-related Inhibitory Peptide; AIP). Isoproterenol and dantrolene were each used at 1 µM.
Optical Recording of Opto-MTFs
Opto-MTFs were imaged using a modified tandem-lens macroscope (Scimedia) for simultaneous Ca2+ imaging and contractility measurement with optogenetic stimulation (Supplemental Figure 2). Optogenetic stimulation was delivered using optical fibers mounted 500 µm above the gelatin chips.
Three days after transduction with ChR2 lentivirus, opto-MTFs were loaded with X-Rhod-1. Culture media was replaced with Tyrode’s solution and maintained at 37°C. Where indicated, opto-MTFs were treated with 6 μM dantrolene or 2 μM AIP.
Opto-MTFs were stimulated 10 ms optical pulses over a range of frequencies from 0.7 to 3 Hz using a custom LabVIEW program (National Instruments). For each recording, Ca2+ and dark field images were simultaneously acquired with 2000 frames and 400 frames at a frame rate of 200 Hz and 100 Hz over 10 s and 4 s, respectively.
Post-processing of the raw calcium and darkfield imaging data were conducted with custom software written in MATLAB (MathWorks).
Statistical Analysis
Statistical analysis was performed using JMP Pro 14 (SAS Institute, Inc.).
The significance between groups from cell cluster measurements was evaluated using the Steel Dwass non-parametric test with multiple testing correction. Statistical analysis of western blots was performed using the Tukey-Kramer honestly significant difference test for multiple groups, or Student’s t-test for two groups.
For tissue-level differences, between-group differences in occurrence of re-entry were evaluated using Pearson’s chi-squared test and the Bonferroni correction for multiple comparisons with corrected p-values, by dividing p-values by number of possible pairwise comparison tests (i.e., 0.05/6, 0.01/6, 0.001/6 for four different tissues and six possible pairwise comparison tests, 0.05/3, 0.01/3, 0.001/3 for three different tissues and three possible pairwise comparison tests). Heterogeneities of calcium handling in the iPSC-CMs used in this study and neonatal rat ventricular myocytes or commercial iPSC-CMs were compared using one-way ANOVA followed by Tukey-Kramer honestly significant difference test. Tissue-level functional differences were tested with Student’s t-test corrected with Benjamini-Hochberg multiple testing correction (false discovery rate, FDR of 20%)23.
RESULTS
Ca2+ Handling in Individual iPSC-CM within a Cell Cluster
We obtained skin fibroblasts from a CPVT patient. This patient had a normal resting electrocardiogram but exercise-induced bidirectional and polymorphic ventricular tachycardia (Supplemental Figure 1A). Genotyping revealed that the patient had a heterozygous point mutation in RYR2 that caused substitution of isoleucine for arginine at position 4651 (R4651I; Figure. 1B, Supplemental Figure 1B & 1C). Clinical genotyping did not implicate other candidate inherited arrhythmia genes. The fibroblasts were reprogrammed into iPSCs (line CPVT1p, where p indicates patient-derived; Supplemental Figure 1C). We used Cas9 genome editing19 to introduce the patient mutation into a wild-type (WT) iPSC line, PGP1, yielding a CPVT line (PGP1-RYR2R4651I, abbreviated CPVT1e, where e denotes engineered) otherwise isogenic to WT (Supplemental Figure 1B & 1D). In addition, we similarly used Cas9 genome editing to introduce a second CPVT-causing mutation, substitution of asparagine for aspartate at position 358 (D358N) into PGP1, yielding a second engineered CPVT line (PGP1-RYR2D358N, abbreviated CPVT2e; Figure. 1B, Supplemental Figure 1B & 3). Sequencing of the top 10 predicted off-target genome editing sites for CPVT1e and CPVT2e did not identify unintended genome modification (Supplemental Figure 4). These iPSCs had normal karyotype and robustly differentiated into iPSC-CMs with comparable efficiency (Supplemental Figure 5).
We analyzed Ca2+ handling of WT, CPVT1p, CPVT1e, and CPVT2e iPSC-CMs by loading spontaneously beating iPSC-CM clusters (3-10 cells/cluster) with the Ca2+-sensitive dye Fluo-4 and performing confocal line scan imaging within individual cells of a cluster. These clusters exhibited periods of spontaneous rhythmic beating interspersed with periods of quiescence. When recorded during periods of quiescence, both patient-derived and genome-edited isogenic CPVT1 iPSC-CMs had more frequent Ca2+ sparks than WT,24 and this was further exacerbated by isoproterenol (ISO), a beta-sympathomimetic (Figure 1C). The genome-edited isogenic CPVT2 iPSC-CM line also had more frequent Ca2+ sparks than WT, albeit this was not further exacerbated by ISO (Figure 1C). During spontaneous beating, we observed spontaneous Ca2+ release events interspersed between Ca2+ transients (Figure 1D, left). Quantitative analysis showed greater frequency of spontaneous Ca2+ release events in beating CPVT iPSC-CMs, with and without ISO stimulation (Figure 1D, right). These spontaneous Ca2+ release events correlated with membrane depolarization (Supplemental Figure 5, H–J), consistent with after-depolarizations previously reported in CPVT iPSC-CMs and CMs4,5,12. These results confirm that both CPVT mutations tested are sufficient to increase Ca2+ release from RYR2, consistent with Ca2+ handling abnormalities reported in iPSC-CMs with other CPVT-causing RYR2 mutations.5,10–13
Opto-MTF Engineered Heart Tissue Model of Inherited Arrhythmia
Since clinical arrhythmias emerge from the collective behavior of cardiomyocytes assembled into tissues, to better model inherited arrhythmias we integrated muscular thin films (MTF),25 optogenetics, and optical mapping to yield “opto-MTFs”, a platform that permits simultaneous assessment of myocardial Ca2+ transient propagation and contraction (Figure 2A and Supplemental Figure 2 & 6). iPSC-CMs were seeded on 10 × 10 mm micro-molded gelatin MTFs16 (Supplemental Figure 6), so that they assembled into tissue with parallel cell alignment characteristic of native myocardium (Figure 2B and Supplemental Video 1). Lentivirus was used to program iPSC-CMs to express channelrhodopsin (ChR2), a light-gated channel that allows cardiomyocytes to be paced with 488 nm light, as described previously.25,26 ChR2 expression in cardiomyocytes was shown previously to alter minimally the electrophysiological properties of the tissue.21 Using 10 msec pulses of 488 nm light, we optically stimulated a ~0.79 mm2 region, containing ~500 cells, at one end of the opto-MTFs. This local activation stimulated Ca2+ transient propagation across the opto-MTFs in the direction of the long axis of muscle fibers. Propagation of Ca2+ transients was recorded using the Ca2+ sensitive fluorescent dye X-Rhod-1 at a resolution of 100 µm x 100 µm (Figure. 2C, 2D, and Supplemental Figure 7; Supplemental Video 2 & 3). Ca2+ transient propagation into two film cantilevers at the far end of the opto-MTF induced iPSC-CM contraction, curving the cantilever and permitting measurement of mechanical systole (Figure 2C, 2D, and Supplemental Figure 7; Supplemental Video 2 & 3).15–17 Each chip was composed of three individual MTFs (3 × 10 mm) that were isolated from one another, permitting independent assays within the same chip (Supplemental Figure 8; Supplemental Video 4 & 5). Spatiotemporal characteristics of the opto-MTFs, including propagating Ca2+ transient activation time and propagation speed (CaS) and direction, were measured from the Ca2+ imaging data (Figure 2E). At a pacing rate of 1.5 Hz, the heterogeneity in CaS across space and time observed for the iPSC-CMs used in this study was comparable to or less than that observed using neonatal rat ventricular myocytes or commercial human stem cell-derived cardiomyocytes (Figure 2E and Supplemental Figure 9).
Figure 2. Opto-MTF engineered heart tissue for arrhythmia modeling.
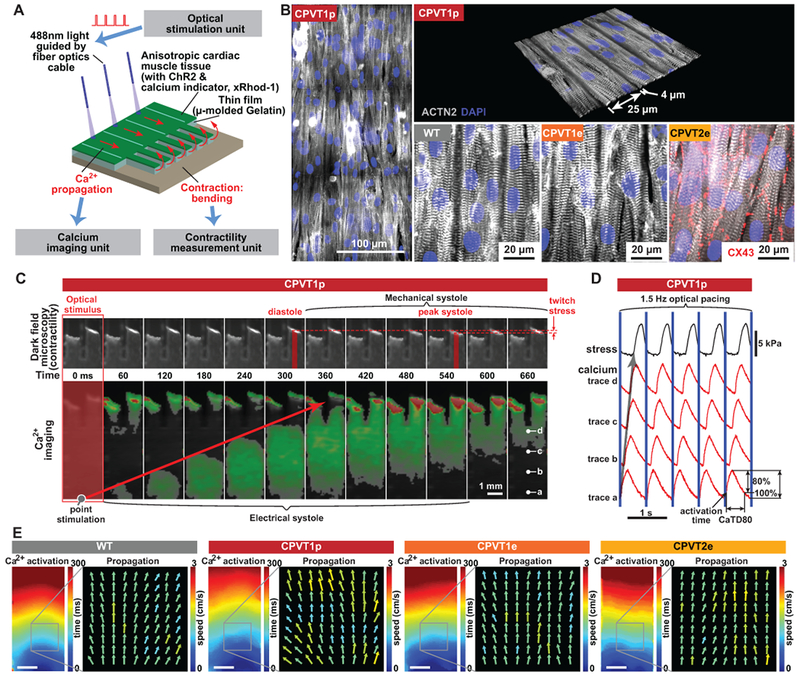
A. Schematic of opto-MTF system to optically pace and optically measure tissue-level Ca2+ wave propagation and contraction. Cardiomyocytes programmed to express ChR2 are seeded on micro-molded gelatin with flexible cantilevers on one end. Focal illumination using optical fibers excites cells, resulting in Ca2+ wave propagation along the MTF and into the cantilevers. Ca2+ wave propagation is measured by fluorescent imaging of the Ca2+-sensitive dye X-Rhod-1, and mechanical contraction by darkfield imaging of the cantilevers. B. Confocal images of ACTN2-stained opto-MTFs. Micro-molded gelatin induces iPSC-CMs to grow with their long axis aligned with the long axis of the MTF. C. Excitation-contraction coupling in CPVT1p opto-MTFs. Representative time lapse images show Ca2+ wave propagation and mechanical systole recorded induced by optogenetic point stimulation. D. Ca2+ traces recorded at the points labeled a-d in the right-most image of panel C. Blue lines indicate optical pacing at the stimulation point. Activation time is the time to the maximal Ca2+ signal upstroke velocity. CaTD80 is the duration of the Ca2+ transient at 80% decay. E. Spatial maps of activation time and Ca2+ wave speed and direction for WT, CPVT1p, CPVT1e, CPVT2e opto-MTFs at 1.5 Hz pacing, demonstrating well-ordered tissue behavior under these conditions. Bar, 1 mm.
We used the opto-MTF platform to characterize the properties of CPVT iPSC-CMs assembled into engineered heart tissues. The spontaneous beating frequency of CPVT tissues was not different from that of WT tissues (Supplemental Figure 10A). Spontaneous beats in both WT and CPVT originated from tissue edges, as expected from reduced source-to-load mismatch present at these sites. Whereas spontaneously beating, unstimulated CPVT iPSC-CMs in cell clusters composed of a few cells often exhibited Ca2+ transients with aberrant waveforms (Figure 1D), we did not observe any abnormal Ca2+ traces in spontaneously beating CPVT or control tissues recorded at lower resolution (Supplemental Figure 10B–E). These data indicate that Ca2+ transient abnormalities observed in single cells do not coordinate in spontaneously beating tissues to yield detectable abnormalities in tissue level propagating Ca2+ transients (referred to hereafter in this study as “calcium waves”). This quiescent baseline behavior of spontaneously beating CPVT tissues parallels the normal baseline phenotype of CPVT patients, who have few arrhythmias in the absence of exercise or emotional stress.
High Pacing Rate and β-adrenergic Stimulation Induce Reentry in CPVT Opto-MTFs
CPVT patients are at risk for developing ventricular tachycardia during exercise or emotional stress. To simulate key features of these provocative conditions in vitro, we treated opto-MTFs with increasing optical pacing frequency (1-3 Hz) or β-adrenergic stimulation (0-10 µM ISO). Remarkably, the three CPVT iPSC-CM lines but not WT opto-MTFs were vulnerable to spiral wave reentry, a cause of tachycardia at the tissue level (Figure 3A–B; Supplemental Video 6–9). The combination of both high pacing rate and ISO most potently evoked reentry (Figure 3C). During reentry, the opto-MTF cantilevers generated less stress and induced asynchronous contraction (Supplemental Figure 11 and Supplemental Video 6, 7, 10), mimicking the loss of forceful coordinated contraction that impairs cardiac output in clinical ventricular tachycardia. Dantrolene, a small molecule that inhibits release of Ca2+ through ryanodine receptors, inhibits CPVT arrhythmias.27,28 Consistent with prior studies,10,27 dantrolene reduced abnormal Ca2+ handling in CPVT iPSC-CMs (Supplemental Figure 12). Dantrolene likewise suppressed re-entry in CPVT1e opto-MTFs (Supplemental Figure 13). These data show that assembly of CPVT iPSC-CMs into opto-MTFs models key features of the disease at a tissue level. Furthermore, the data implicate reentry as an arrhythmia mechanism in CPVT.
Figure 3. Characterization of CPVT opto-MTFs.
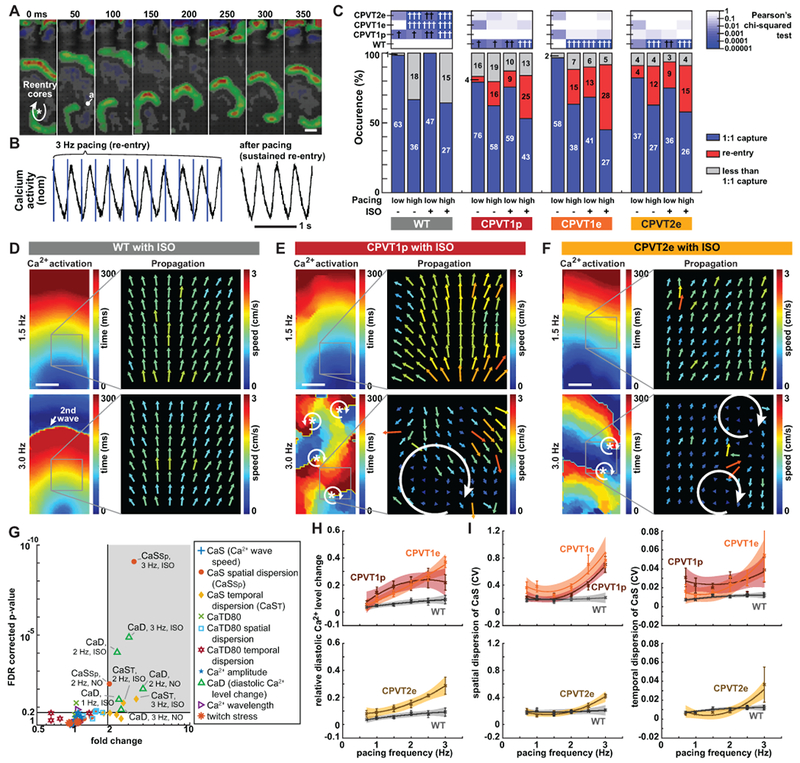
A. Time lapse images of CPVT1e opto-MTF Ca2+ wavefront propagation. Ca2+ wavefronts, calculated from the temporal derivative of Ca2+ signals, show spiral wave reentry. B. Representative Ca2+ signal traces during re-entry. Re-entrant Ca2+ signals were sustained even after pacing was discontinued. C. Occurrence of re-entry in CPVT and WT opto-MTFs. hi, ≥ 2 Hz pacing; lo, < 2 Hz pacing. High pacing rate and ISO increased re-entry occurrence. Pearson’s chi-squared test with same pacing and ISO conditions. The Bonferroni correction for multiple comparisons was applied for six possible pairwise tests performed with a corrected p threshold: †, P<0.05/6. ††, P<0.01/6. †††, P<0.001/6. Bars are labeled with sample numbers. D–F, Spatial maps of Ca2+ wave activation time and velocity in WT (D), CPVT1p (E), or CPVT2e (F) opto-MTFs. The same tissue is shown with 1.5 Hz or 3 Hz pacing. 3 Hz pacing and ISO increased spatiotemporal heterogeneity of CPVT tissues. G. Volcano plot shows 60 tissue-level parameters of Ca2+ propagation in WT vs. CPVT1p and CPVT1e opto-MTFs. Each of the ten markers represents the indicated property measured at three different pacing rates (1, 2, and 3 Hz) with and without ISO. Shaded region indicates parameters with FDR-adjusted P<0.20 and more than 2-fold change. H–I. Relative diastolic Ca2+ level change from basal condition (H) and spatial and temporal dispersion of Ca2+ wave propagation speed (I) as a function of pacing frequency, under ISO stimulation. Smooth lines are quadratic functions fit to the data. Shaded areas and error bars represent the 95% confidence interval for the fit and SEM, respectively. Data from tissues with 1:1 capture were included in G to I (n=12 WT, 33 CPVT1p, 13 CPVT1e, 15 CPVT2e from > 3 harvests). Bar in a, d, e, and f = 1 mm.
To better understand the development of reentry in CPVT tissues, we analyzed optical mapping data of Ca2+ wave propagation. When challenged with 3 Hz pacing and ISO, wild-type opto-MTFs retained organized Ca2+ waves, as shown by coherent Ca2+ activation maps and relatively homogenous Ca2+ wave propagation speed and direction (Figure 3D). At 1.5 Hz pacing with or without ISO, CPVT1p, CPVT1e, and CPVT2e opto-MTFs also were relatively coordinated (Figure 3E & 3F, top). However, rapid pacing of the same CPVT tissues induced reentry in which Ca2+ waves propagated in a circular pattern, associated with increased spatial dispersion of CaS (Figure 3E & 3F, bottom, Supplemental Figure 14, Supplemental Video 11).
To characterize the tissue-level mechanisms that make CPVT tissues vulnerable to reentry, we compared WT to CPVT1 tissues for 10 different parameters (CaS, spatial and temporal dispersion of CaS, Ca2+ transient duration at 80% recovery (CaTD80), spatial and temporal dispersion of CaTD80, relative diastolic Ca2+ level change from basal condition, Ca2+ wave amplitude, Ca2+ wavelength, and twitch stress) at 3 different pacing rates (1, 2, and 3 Hz) with and without ISO (Figure 3G and Supplemental Figure 15). For these analyses into the conditions that made CPVT1 tissues vulnerable reentry substrates, we focused on recordings with 1:1 capture and without active reentry. Under higher pacing rates and ISO stimulation, CPVT1 tissues differed from WT (greater than two-fold change, nominal P<0.05, and FDR<0.2) in three parameters: relative diastolic Ca2+ level, spatial dispersion of CaS, and temporal dispersion of CaS. Specifically, significant differences were observed in relative diastolic Ca2+ level at 3 Hz ± ISO, 2 Hz ± ISO, and 1 Hz + ISO; temporal dispersion of CaS at 3 Hz + ISO and 2 Hz + ISO, and spatial dispersion of CaS at 3 Hz + ISO and 2 Hz - ISO. We further analyzed the effect of pacing rate on these three parameters in ISO-stimulated CPVT and WT tissue (Figure 3H & 3I, and Supplemental Figure 15). In ISO-stimulated WT tissues, diastolic Ca2+ levels and spatial and temporal dispersion of CaS were relatively insensitive to changes in pacing rate from 1 to 3 Hz. In contrast, as CPVT1p, CPVT1e, and CPVT2e tissues were paced at higher rates, they developed greater relative diastolic Ca2+ level and greater spatial and temporal dispersion of CaS (Figure 3H & 3I, and Supplemental Figure 15). Dispersion of CaS was also manifest by irregular Ca2+ activation maps observed in CPVT tissue paced at 3 Hz (Supplemental Video 11 and Supplemental Figure 14); importantly, these irregular Ca2+ activation maps were not observed when the same tissues were paced at 2 Hz (Supplemental Video 12 and Supplemental Figure 14). Since heterogeneity increases tissue vulnerability to reentry,14 this analysis indicates that rapid pacing and ISO stimulation create a vulnerable arrhythmia substrate by increasing spatial and temporal heterogeneity in CaS. This increased tissue heterogeneity might be linked to pacing-dependent elevation of diastolic Ca2+, which drives after-depolarizations and impairs gap junction function29 thereby potentially reducing intercellular connectivity.
We next analyzed events that initiated reentry in the vulnerable CPVT substrate. We identified eight recordings that captured the initiation of reentry (Supplemental Table I). The rare reentry events that we observed at low pacing rates (Figure 3C) were initiated by spontaneous Ca2+ waves originating from edges of the tissue that collided with pacing-driven Ca2+ waves (Supplemental Video 10 and Supplemental Figure 16). High pacing rates suppressed these spontaneous Ca2+ waves, and reentry events were initiated by regional conduction block (Figure 4 and Supplemental Videos 13–14). ISO-treated CPVT1p tissue paced at 2 Hz showed well-ordered Ca2+ wave propagation (Figure 4A & 4B). When the same tissue was paced at 3 Hz, there was greater dispersion of propagation velocity, including regions with functional conduction block (Figure 4C, grey areas in velocity vector field). Ca2+ waves propagating around the focal block reentered the original path, resulting in rotor formation. A similar example of reentry initiation with functional conduction block in CPVT1e and CPVT2e tissues are shown in Supplemental Video 8, 15, 16 and Supplemental Figure 17. Although the mechanism underlying functional, regional conduction block in CPVT tissue requires further study, they were associated with elevated relative diastolic Ca2+ (Figure 3G & 3H), which could reduce iPSC-CM excitability30 as well as reduce cell-cell coupling.29
Figure 4. Initiation of re-entry in CPVT opto-MTFs.
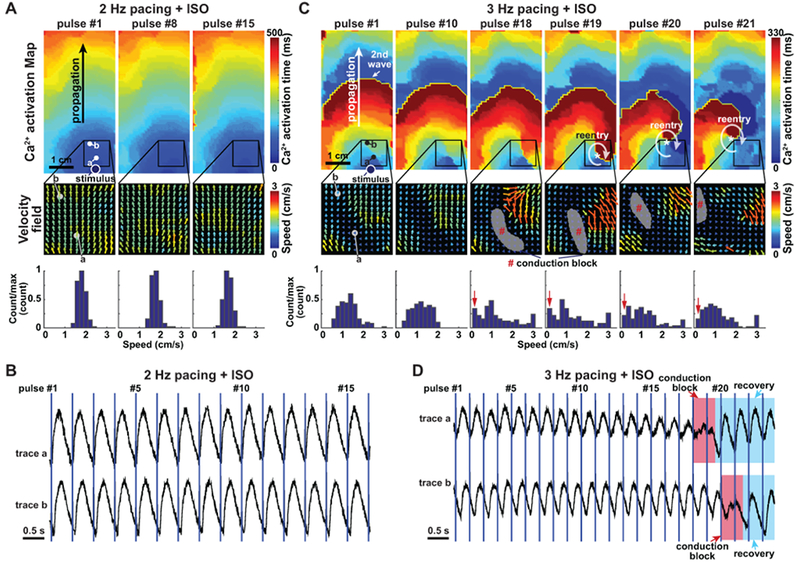
A. CPVT1p opto-MTF at 2 Hz pacing with ISO. The Ca2+ activation map and velocity fields were well-ordered. Speed histogram reflects narrow range of values. B. Ca2+ tracings from points a and b in panel A. C. The same opto-MTF as in panel A, paced at 3 Hz with ISO. There is greater heterogeneity in the velocity field and disorganization of the Ca2+ activation map. Localized conduction block that permitted reentrant Ca2+ wave propagation become evident at pulse #18 and #19. Histograms indicate greater spatial dispersion of speed. D. Ca2+ tracings at points a and b in panel C. Red shading indicates conduction block overlying recording points, and blue shading indicates recovery from block.
Together, our engineered tissue model demonstrates that CPVT tissue is vulnerable to circus movement with reentry. Rapid pacing and isoproterenol stimulation increased relative diastolic Ca2+ and spatiotemporal dispersion of CaS. In this context, localized conduction block initiated rotor formation.
CaMKII Phosphorylation of RYR2-S2814 is Required to Unmask CPVT Arrhythmic Potential
Although catecholamine stimulation is well known to provoke arrhythmia in CPVT, the molecular targets through which β-adrenergic stimulation unmasks the latent arrhythmic potential of RYR2 mutations were incompletely defined. β-adrenergic stimulation activates numerous signaling pathways, including Ca2+-calmodulin-dependent kinase II (CaMKII) and protein kinase A (PKA; Figure 5A). When we inhibited PKA using a potent, cell-permeable peptide,31 the incidence of Ca2+ sparks was not significantly reduced in CPVT1p iPSC-CMs (Figure 5B). In contrast, CaMKII inhibition with cell-permeable autocamtide-2 related inhibitory peptide (AIP; Supplemental Figure 18A), a highly selective and potent CaMKII inhibitor,32 strongly reduced the incidence of Ca2+ sparks and after-depolarizations in CPVT1p, CPVT1e, and CPVT2e iPSC-CMs (Figure 5B and Supplemental Figure 18B & 18C). This effect is consistent with prior reports using a small molecule inhibitor4,5 and with a recently reported CaMKII-dependent, PKA-independent pathway that mediates arrhythmogenesis provoked by adrenergic stimulation.33 To model the effect of CaMKII inhibition at the tissue level, we treated CPVT1 and CPVT2 opto-MTFs with the AIP. AIP inhibited development of re-entry provoked by rapid pacing and ISO (Figure 5C and Supplemental Video 17). Analysis of diastolic Ca2+ level (Figure 5D) and spatial and temporal dispersion of Ca2+ wave speed (Figure 5E) showed that these key parameters of CPVT tissue vulnerability were normalized by AIP. These data suggest that CaMKII is a key signaling molecule in the pathogenesis of CPVT.
Figure 5. CaMKII inhibition suppresses the CPVT arrhythmic phenotype.
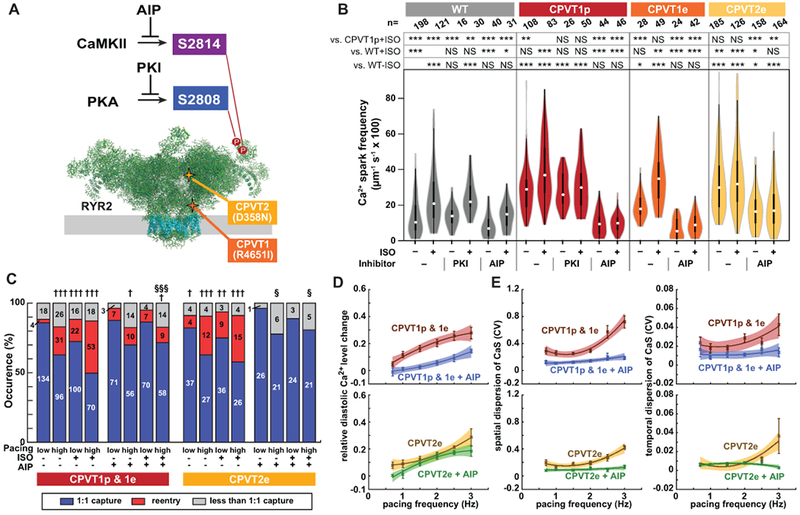
A. Schematic of RYR2 (two subunits of tetramer shown). Three key residues are highlighted: S2808, the target of PKA phosphorylation; S2814, the target of CaMKII phosphorylation; and R4651, mutated in CPVTp. B. iPSC-CMs in isolated cell clusters were treated with ISO and selective CaMKII (AIP) or PKA (PKI) inhibitors. Ca2+ sparks were imaged by confocal line scanning within individual iPSC-CMs. Data without inhibitors are the same as in Fig. 1c. Steel-Dwass non-parametric test with multiple testing correction: *, P<0.05; **, P<0.01; ***, P<0.001. NS, not significant. C. Occurrence of re-entry in CPVT1 and CPVT2 engineered tissues treated with a CaMKII inhibitor (AIP). Pearson’s chi-squared test: † vs WT with same pacing and ISO conditions; § vs. CPVT with same pacing and ISO conditions. The Bonferroni correction for multiple comparisons was applied for three possible pairwise tests performed with a corrected p threshold: †, §, P<0.05/3. ††, P<0.01/3. †††, §§§, P<0.001/3. Bars are labeled with samples sizes. D–E. Relative diastolic Ca2+ level change from basal condition (D), and spatial and temporal dispersion of Ca2+ wave propagation speed (E) as a function of pacing frequency under ISO stimulation in CPVT1 and CPVT2 before and after AIP loading. Only tissues responding 1:1 to every stimulus were included in d and e (n=33 CPVT1p, 15 CPVT1p + AIP, 13 CPVT1e, 15 CPVT2e, and 9 CPVT2e + AIP from > 3 harvests).
CaMKII targets multiple proteins that directly or indirectly impact Ca2+-handling.34 One important CaMKII target is serine 2814 (S2814) on RYR2 itself35 (Figure 5A). RYR2-S2814 phosphorylation by CaMKII enhances diastolic RYR2 Ca2+ leak and is pro-arrhythmic.36 To test the hypothesis that CaMKII-mediated phosphorylation of RYR2-S2814 is essential for expression of CPVT mutations, we used Cas9 genome editing to replace S2814 with alanine (S2814A) in both RYR2 alleles, in RYR2 wild-type and RYR2R4651I/+ backgrounds and showed that these mutations block this phosphorylation event (Supplemental Figure 4 & 19). We refer to these mutant alleles as WT-S2814A and CPVT1e-S2814A, respectively. RYR2 is also phosphorylated on S2808 by PKA,37 and this phosphorylation event has also been implicated in increasing RYR2 diastolic Ca2+ leak.38 Therefore, we used genome editing to generate analogous RYR2-S2808A homozygous mutant lines, named WT-S2808A and CPVT1e-S2808A (Supplemental Figure 4 & 19). In keeping with the effect of CaMKII inhibitory peptide, in quiescent iPSC-CMs in cell clusters the incidence of Ca2+ sparks in CPVT1e-S2814A was less than CPVT1e and comparable to WT, either at baseline or with ISO stimulation (Figure 6A & 6B). In contrast, the incidence of these events was similar in CPVT1e-S2808A and CPVT1e iPSC-CMs (Figure 6A & 6B), consistent with the ineffectiveness of pharmacological PKA inhibition (Figure 5B). Similar results were obtained from spontaneously beating cell clusters (Figure 6C & 6D). These data indicate that CaMKII phosphorylation of RYR2-S2814 is required to unmask the latent arrhythmic potential of the CPVT R4651I mutation.
Figure 6. CaMKII phosphorylation of RYR2-S2814 is required to unmask the CPVT arrhythmic phenotype.
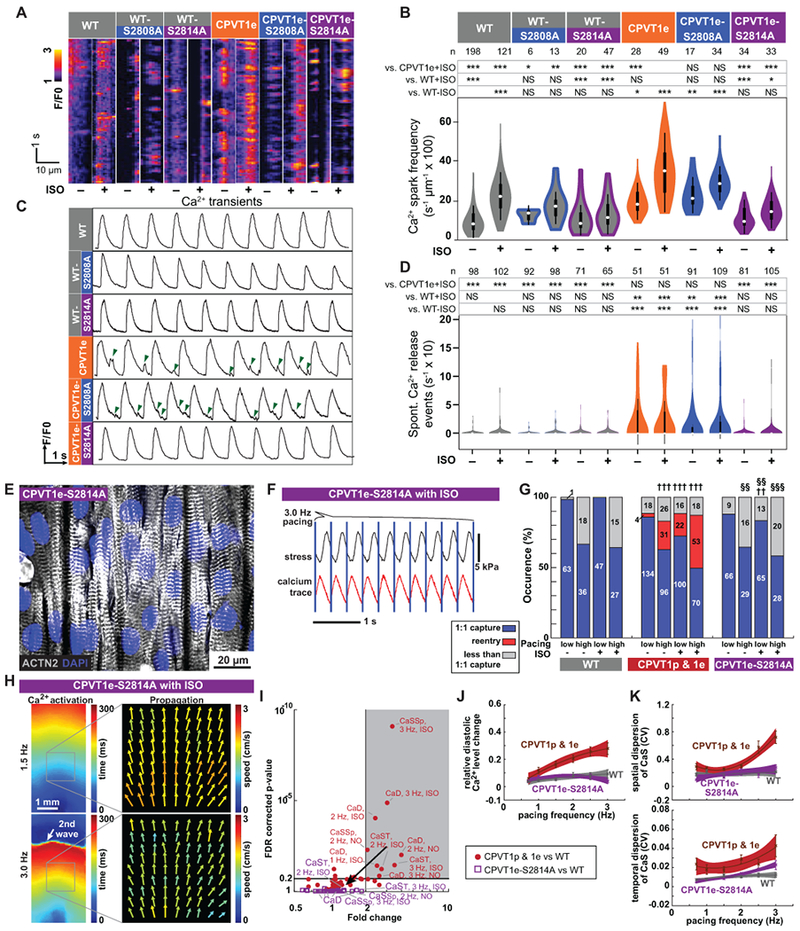
A–B. Incidence of Ca2+ sparks in quiescent iPSC-CMs in cell clusters. Representative traces (A). Quantitative analysis (B). WT and CPVT1e data are the same as in Figure 5B. Steel-Dwass non-parametric test with multiple testing correction: *, P<0.05; **, P<0.01; ***, P<0.001. NS, not significant. C–D. Incidence of spontaneous Ca2+ release events in spontaneously beating iPSC-CMs in cell clusters. Representative traces, (C). Green arrowheads indicate spontaneous Ca2+ release events. Quantitative analysis, (D). WT and CPVT1e data are the same as in Figure 1D. Statistics and symbols as in panel B. E. Confocal image of opto-MTF constructed using CPVT1e-S2814A iPSC-CMs. Myocytes are aligned by micro-molded gelatin substrate. F. Representative CPVT1e-S2814A opto-MTF. Ca2+ transients and systolic contraction were coupled 1:1 with 3 Hz optical stimuli (blue lines). G. Occurrence of re-entry in CPVT1e-S2814A compared to WT (††, †††) and CPVT1e (§§, §§§) opto-MTFs under matching conditions using Pearson’s chi-squared test. The Bonferroni correction for multiple comparisons was applied for three possible pairwise tests performed with a corrected p threshold: ††, §§, P<0.01/3. †††, §§§, P<0.001/3. Bars are labeled with sample numbers. H. Spatial maps of the same CPVT1e-S2814A opto-MTF paced at 1.5 Hz or 3.0 Hz, in the presence of ISO. Ca2+ activation time and Ca2+ wave propagation speed were well-organized and relatively homogeneous compared to CPVT1 (see Figure 3). I. Volcano plot of 60 tissue-level parameters of Ca2+ wave propagation (please see Figure 3). Unlike CPVT1 tissue parameters, CPVT1e-S2814A tissue parameters were not statistically different from those of WT. J–K. Relative diastolic Ca2+ level change from basal condition (J), and spatial and temporal dispersion of Ca2+ wave propagation speed (K) as a function of pacing frequency under ISO stimulation in CPVT1e-S2814A compared to CPVT and WT. Smooth lines are quadratic functions fit to the data; shaded areas and error bars show the 95% confidence interval for the fit and SEM, respectively. Only tissues responding 1:1 to every stimulus were included in i to k (n=12 WT, 33 CPVT1p, 13 CPVT1e, 15 CPVT2e, and 18 CPVT1e-S2814A from > 3 harvests).
We modeled the effect of ablation of the RYR2-S2814 CaMKII phosphorylation site on the development of reentry in CPVT engineered heart tissue. We fabricated opto-MTFs from CPVT1e-S2814A iPSC-CMs (Figure 6E). Unlike CPVT1p and CPVT1e tissues, rapid pacing and ISO did not induce reentry in CPVT1e-S2814A tissues (Figure 6F & 6G; Supplemental Video 18). These tissues retained well-ordered Ca2+ waves without regions of conduction block (Figure 6H), which we observed in CPVT1p, CPVT1e, and CPVT2e opto-MTFs under the same conditions (Figure 3 & 4). We analyzed Ca2+ wave propagation and other tissue properties of opto-MTFs between CPVT1e-S2814A and WT (purple, Figure 6I), compared results to the prior analysis of the differences between CPVT and WT (Figure 3G, shown in red in Figure 6I). The RYR2-S2814A substitution did not alter global CaS (Supplemental Figure 15), but notably it normalized the three parameters (relative diastolic Ca2+ level change from basal condition, spatial dispersion of CaS, and temporal dispersion of CaS) that were significantly different between CPVT and WT under higher pacing rates and ISO. These three parameters became progressively higher in ISO-treated CPVT than WT tissues with increasing pacing rate (Figure 3H & 3I and Supplemental Figure 15), but these abnormalities were abolished by the RYR2-S2814A substitution (Figure 6J & 6K and Supplemental Figure 15). These data show that preventing RYR2-S2814 phosphorylation normalizes pacing- and ISO-induced CaS heterogeneity and relative diastolic Ca2+ level, resulting in a substrate that is less vulnerable to tissue-level reentry.
DISCUSSION
By integrating optogenetics, tissue engineering, lab-on-a-chip technology, and induced pluripotent stem cell technology, we have created an engineered human tissue model to study arrhythmia mechanisms and to test anti-arrhythmic therapy. We combined this platform with patient-derived and genome-edited iPSC-CMs to model the inherited arrhythmia CPVT at the tissue level. Similar strategies could be used to model other forms of inherited arrhythmia, or to develop high fidelity tissue level models for cardiovascular drug toxicity studies.
Our engineered tissue model recapitulated key features of the clinical phenotype. First, under baseline conditions the CPVT engineered tissues did not exhibit ectopic Ca2+ wave initiation, consistent with the normal baseline electrocardiograms typically observed in patients at rest. This contrasts with the abnormal phenotype of unstimulated CPVT iPSC-CMs or dissociated murine CMs39. Second, adrenergic stimulation and rapid pacing induced re-entrant rhythms in the CPVT engineered tissues, mimicking exercise-induced ventricular tachycardia that is a hallmark of the clinical phenotype.
Our CPVT models provide insights into disease pathogenesis. Although it was previously known that adrenergic stimulation of CaMKII was important for arrhythmogenesis in CPVT,4,5 our study identified a single CaMKII target site that must be phosphorylated to unmask the latent arrhythmic potential of the RYR2-R4651I CPVT mutation. Interestingly, this CaMKII target site is on RYR2 itself, at S2814. Phosphorylation of this site is known to increase RYR2 Ca2+ release and susceptibility to arrhythmia.35,36 Our results provide a molecular pathway that explains how exercise provokes arrhythmia in CPVT patients and suggest that inhibiting this pathway will protect patients from developing lethal arrhythmia. We demonstrated that CaMKII activity is required to unmask the abnormal phenotype of several independent CPVT-causing RYR2 mutations (please also see the accompanying study by Bezzerides et al.), although further work will be required to demonstrate the precise requirement for RYR2-S2814 phosphorylation across multiple genotypes. The accompanying study by Bezzerides et al. further demonstrated that CaMKII inhibition by cardiac targeted gene therapy effectively suppresses ventricular arrhythmia in a CPVT mouse model. Together, these studies suggest that CaMKII inhibition may be a translatable therapy for CPVT.
The CPVT engineered heart tissue model provided insights into mechanisms by which RYR2 mutation may cause arrhythmia at the tissue level. In isolated cell clusters, CPVT mutations are well established to cause aberrant Ca2+ release from the sarcoplasmic reticulum.5,10–13 The resulting elevation of cytosolic Ca2+ drives depolarizing Na+-Ca2+ exchange through the sodium-calcium exchanger (NCX1), resulting in after-depolarizations that can trigger subsequent action potentials.1 In an ex vivo study of a mouse model of CPVT, triggered activity originating from His-Purkinje cells was reported to be the source of ventricular tachycardia, setting up reentry and mediating the transition from polymorphic ventricular tachycardia to ventricular fibrillation.6 However, triggered activity in His-Purkinje cells alone is not sufficient for ventricular arrhythmia, since a recent study demonstrated that the CPVT-inducing mutation must be present in working myocardium in addition to His-Purkinje to yield ventricular arrhythmia.8 In the working myocardium, the two conditions necessary for focal activity to occur are unlikely to be fulfilled. First, the intracellular Ca2+ waves associated with increased diastolic Ca2+ and Ca2+ release events rarely cross cell borders24, and therefore do not form delayed afterdepolarizations synchronized among the quantity of cells necessary to elicit a triggered action potential. Second, electrotonic interaction between potential foci and neighboring tissue (so-called “source-to-load mismatch”) is expected – in contrast to observations made in single cells or clusters of few cells - to damp the amount of local depolarization and to counteract or prevent ectopic action potential formation.40 Likely for these same reasons, simulated exercise did not stimulate ectopic impulse initiation in our engineered CPVT tissues. Rather, high pacing rate and ISO stimulation markedly increased heterogeneity of Ca2+ wave propagation and elevated diastolic Ca2+ in CPVT tissue, resulting in local depolarization, propagation block and subsequent reentry. This is consistent with computational modeling studies, in which subthreshold afterdepolarizations increased repolarization heterogeneity and locally impaired cardiomyocyte excitability.30 Thus our data based on Ca2+ measurements, in combination with computational electrical modeling and mouse models, suggest that CPVT mutations cause heterogeneity of action potential repolarization and excitability, creating a vulnerable substrate within working myocardium that sustains re-entrant arrhythmias. While re-entrant spiral waves were initiated by collision of optically stimulated propagating Ca2+ transients with regions of focal conduction block in our experiments, triggered activity arising within the His-Purkinje system is likely to play a role as initiating mechanism in whole hearts. Further studies in animal models are required to test this hypothesis.
In conclusion, we developed a human engineered tissue platform to elucidate the pathogenesis of inherited arrhythmias, and used this platform to gain molecular and pathophysiological insights into CPVT. It is important to point out important limitations of the current engineered tissue model. One limitation is that we were unable to measure membrane voltage because currently available optical voltage sensors have greater toxicity, inadequate signal-to-noise properties, or spectral incompatibility with our optical actuator. Improved optical voltage sensors, or refinements to the patterned substrate that permit multi-electrode array recordings, will allow measurement of membrane voltage and/or extracellular electrograms and provide additional insights into the mechanisms responsible for re-entry. A second limitation is that current stem cell-derived cardiomyocytes are immature compared to adult cardiomyocytes41, and consequently findings made in iPSC-CM systems, such as dynamic conduction block precipitating re-entry, require further validation in animal models and in patients. Ongoing efforts to improve iPSC-CM maturity, combined with continued development of physiological, tissue-level arrhythmia assays, will further advance our ability to model of human arrhythmias in in vitro systems. Finally, the molecular and cellular mechanisms that lead to heterogeneous calcium transient propagation and dynamic conduction block in CPVT opto-MTFs stimulated with ISO and rapid pacing are unclear. Addressing these questions will be fertile ground for future studies.
Supplementary Material
CLINICAL PERSPECTIVE.
What is new?
Engineered heart tissue fabricated from human pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) effectively modeled Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) caused by dominant mutations in the cardiac ryanodine receptor (RYR2), including induction of arrhythmias by conditions that simulate exercise.
Using selective pharmacology and genome editing, we identified activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) and CaMKII-mediated phosphorylation of RYR2 at serine 2814 as critical events that are required to unmask the latent arrhythmic potential of CPVT-causing RYR2 mutations, highlighting a molecular pathway that links β-adrenergic stimulation to arrhythmogenesis in this disease.
What are the clinical implications?
This study identifies CaMKII activation as a central event in the triggering of arrhythmias in CPVT patients, suggesting that CaMKII inhibition will be an effective and translatable strategy to treat CPVT caused by RYR2 mutations.
Under exercise conditions, CPVT mutations create heterogeneities in cardiac impulse propagation that make the working myocardium vulnerable to re-entrant arrhythmias initiated by triggered beats emanating from the His-Purkinje system.
The human engineered heart tissue platform will be useful to dissect mechanisms and treatment responses in diverse forms of arrhythmia.
Acknowledgments
D.Z. and S-J.P. contributed equally to this study, their authorship order is interchangeable, and they are listed in alphabetical order. D.Z. and S-J.P. designed and performed experiments, analyzed data, organized figures and wrote the paper. D.Z. developed the patient-derived and genome-edited iPSC-CMs and performed cell cluster assays, with help from Y.Q., P.Y., S.X., X.L., and F.L. S-J. P. developed the opto-MTF platform and analysis methods, performed experiments on opto-MTFs, and analyzed data. Y.L performed the western blot experiments. J.G. and A.E.R. recruited the CPVT patient. V.J.B. contributed to iPSC-CM characterization and with D.J.A. provided clinical data and insights. K.Y.L. conducted immunostaining. S.L.K. assisted with opto-MTF fabrication and analysis. K.Y.L. and F.S.P. optimized gelatin substrates and provided insights in experimental design and analysis. P.H.C. performed NRVM harvest for MTF optimization. A.G.K. provided mechanistic insights and edited the manuscript. W.T.P. conceived of and organized the project and co-wrote the manuscript. K.K.P. supervised the project, developed the opto-MTF platform, and edited the manuscript.
Sources of Funding
WTP was funded by NIH grant U01 HL100401, the Boston Children’s Translational Investigator Service, and the Boston Children’s Heart Center. The Boston Children’s Hospital Inherited Cardiac Arrhythmia Program (DJA, WTP) is funded by generous support from the Mannion and Roberts families. KKP was funded by the John A. Paulson School of Engineering and Applied Sciences at Harvard, the Wyss Institute for Biologically Inspired Engineering at Harvard, NIH National Center for Advancing Translational Sciences grant UH3TR000522, and National Science Foundation Materials Research Science and Engineering Center grant DMR-1420570. KKP and WTP were sponsored by NIH NCATS grant 1-UG3-HL-141798-01. KKP, WTP, and DJA were supported by a collaborative research grant from the American Heart Association (16CSA28750006). DZ was funded by the Ministry of Science and Technology of China (National Science and Technology Major Project, Grant No. 2018YFA0109100) and the National Natural Science Foundation of China (Grants 31871496 and 31741090). The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the NIH, the Defense Advanced Research Projects Agency, the U.S. Government, or the Chinese. This work was performed in part at the Center for Nanoscale Systems, a member of the National Nanotechnology Infrastructure Network, which is supported by NSF under Award No. ECS-0335765. Center for Nanoscale Systems is part of Harvard University. Certain aspects of the paper are described in U.S. patent 8,492,150 and 9,669,141, and U.S. patent cooperation treaty PCT/US2015/051818.
Footnotes
Conflict of Interest Disclosures
None
References
- 1.Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–575. [DOI] [PubMed] [Google Scholar]
- 2.Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, Chen SRW, Yan N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science [Internet]. 2016;354:AAH5324. [DOI] [PubMed] [Google Scholar]
- 3.Priori SG, Chen SRW. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu N, Ruan Y, Denegri M, Bachetti T, Li Y, Colombi B, Napolitano C, Coetzee WA, Priori SG. Calmodulin kinase II inhibition prevents arrhythmias in RyR2(R4496C+/−) mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol. 2011;50:214–222. [DOI] [PubMed] [Google Scholar]
- 5.Di Pasquale E, Lodola F, Miragoli M, Denegri M, Avelino-Cruz JE, Buonocore M, Nakahama H, Portararo P, Bloise R, Napolitano C, Condorelli G, Priori SG. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013;4:e843–e843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerrone M, Noujaim SF, Tolkacheva EG, Talkachou A, O’Connell R, Berenfeld O, Anumonwo J, Pandit SV, Vikstrom K, Napolitano C, Priori SG, Jalife J. Arrhythmogenic mechanisms in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2007;101:1039–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bers DM. Calcium Cycling and Signaling in Cardiac Myocytes. Annu Rev Physiol. 2008;70:23–49. [DOI] [PubMed] [Google Scholar]
- 8.Flores DJ, Duong T, Brandenberger LO, Mitra A, Shirali A, Johnson JC, Springer D, Noguchi A, Yu Z-X, Ebert SN, Ludwig A, Knollmann BC, Levin MD, Pfeifer K. Conditional ablation and conditional rescue models for Casq2 elucidate the role of development and of cell-type specific expression of Casq2 in the CPVT2 phenotype. Hum Mol Genet. 2018;27:1533–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bezzerides VJ, Zhang D, Pu WT. Modeling Inherited Arrhythmia Disorders Using Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ J. 2016;81:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jung CB, Moretti A, Schnitzler MM y., Iop L, Storch U, Bellin M, Dorn T, Ruppenthal S, Pfeiffer S, Goedel A, Dirschinger RJ, Seyfarth M, Lam JT, Sinnecker D, Gudermann T, Lipp P, Laugwitz K-L. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol Med. 2012;4:180–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Preininger MK, Jha R, Maxwell JT, Wu Q, Singh M, Wang B, Dalal A, Mceachin ZT, Rossoll W, Hales CM, Fischbach PS, Wagner MB, Xu C. A human pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia recapitulates patient-specific drug responses. Dis Model Mech. 2016;9:927–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fatima A, Xu G, Shao K, Papadopoulos S, Lehmann M, Arnáiz-Cot JJ, Rosa AO, Nguemo F, Matzkies M, Dittmann S, Stone SL, Linke M, Zechner U, Beyer V, Hennies HC, Rosenkranz S, Klauke B, Parwani AS, Haverkamp W, Pfitzer G, Farr M, Cleemann L, Morad M, Milting H, Hescheler J, Saric T. In vitro modeling of ryanodine receptor 2 dysfunction using human induced pluripotent stem cells. Cell Physiol Biochem. 2011;28:579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itzhaki I, Maizels L, Huber I, Gepstein A, Arbel G, Caspi O, Miller L, Belhassen B, Nof E, Glikson M, Gepstein L. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J Am Coll Cardiol. 2012;60:990–1000. [DOI] [PubMed] [Google Scholar]
- 14.Qu Z, Weiss JN. Mechanisms of ventricular arrhythmias: from molecular fluctuations to electrical turbulence. Annu Rev Physiol. 2015;77:29–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feinberg AW, Feigel A, Shevkoplyas SS, Sheehy S, Whitesides GM, Parker KK. Muscular thin films for building actuators and powering devices. Science. 2007;317:1366–1370. [DOI] [PubMed] [Google Scholar]
- 16.Wang G, McCain ML, Yang L, He A, Pasqualini FS, Agarwal A, Yuan H, Jiang D, Zhang D, Zangi L, Geva J, Roberts AE, Ma Q, Ding J, Chen J, Wang D-Z, Li K, Wang J, Wanders RJA, Kulik W, Vaz FM, Laflamme MA, Murry CE, Chien KR, Kelley RI, Church GM, Parker KK, Pu WT. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat Med. 2014;20:616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCain ML, Agarwal A, Nesmith HW, Nesmith AP, Parker KK. Micromolded gelatin hydrogels for extended culture of engineered cardiac tissues. Biomaterials. 2014;35:5462–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinberger F, Mannhardt I, Eschenhagen T. Engineering Cardiac Muscle Tissue: A Maturating Field of Research. Circ Res. 2017;120:1487–1500. [DOI] [PubMed] [Google Scholar]
- 19.Wang G, Yang L, Grishin D, Rios X, Ye LY, Hu Y, Li K, Zhang D, Church GM, Pu WT. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat Protoc. 2017;12:88–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ran FA, Ann Ran F, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park S-J, Gazzola M, Park KS, Park S, Di Santo V, Blevins EL, Lind JU, Campbell PH, Dauth S, Capulli AK, Pasqualini FS, Ahn S, Cho A, Yuan H, Maoz BM, Vijaykumar R, Choi J-W, Deisseroth K, Lauder GV, Mahadevan L, Parker KK. Phototactic guidance of a tissue-engineered soft-robotic ray. Science. 2016;353:158–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW, Helmchen F, Denk W, Brecht M, Osten P. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci U S A. 2004;101:18206–18211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 24.Baader A Real time, confocal imaging of Ca2 waves in arterially perfused rat hearts. Cardiovasc Res. 2002;53:105–115. [DOI] [PubMed] [Google Scholar]
- 25.Grosberg A, Alford PW, McCain ML, Parker KK. Ensembles of engineered cardiac tissues for physiological and pharmacological study: heart on a chip. Lab Chip. 2011;11:4165–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q, Ni RR, Hong H, Goh KY, Rossi M, Fast VG, Zhou L. Electrophysiological Properties and Viability of Neonatal Rat Ventricular Myocyte Cultures with Inducible ChR2 Expression. Sci Rep. 2017;7:1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penttinen K, Swan H, Vanninen S, Paavola J, Lahtinen AM, Kontula K, Aalto-Setälä K. Antiarrhythmic Effects of Dantrolene in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia and Replication of the Responses Using iPSC Models. PLoS One. 2015;10:e0125366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi S, Yano M, Uchinoumi H, Suetomi T, Susa T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Yamamoto T, Matsuzaki M. Dantrolene, a Therapeutic Agent for Malignant Hyperthermia, Inhibits Catecholaminergic Polymorphic Ventricular Tachycardia in a RyR2R2474S/Knock-In Mouse Model. Circ J. 2010;74:2579–2584. [DOI] [PubMed] [Google Scholar]
- 29.Peracchia C Chemical gating of gap junction channels; roles of calcium, pH and calmodulin. Biochim Biophys Acta. 2004;1662:61–80. [DOI] [PubMed] [Google Scholar]
- 30.Liu MB, de Lange E, Garfinkel A, Weiss JN, Qu Z. Delayed afterdepolarizations generate both triggers and a vulnerable substrate promoting reentry in cardiac tissue. Heart Rhythm. 2015;12:2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Glass DB, Cheng HC, Mende-Mueller L, Reed J, Walsh DA. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J Biol Chem. 1989;264:8802–8810. [PubMed] [Google Scholar]
- 32.Ji Y, Li B, Reed TD, Lorenz JN, Kaetzel MA, Dedman JR. Targeted inhibition of Ca2+/calmodulin-dependent protein kinase II in cardiac longitudinal sarcoplasmic reticulum results in decreased phospholamban phosphorylation at threonine 17. J Biol Chem. 2003;278:25063–25071. [DOI] [PubMed] [Google Scholar]
- 33.Pereira L, Cheng H, Lao DH, Na L, van Oort RJ, Brown JH, Wehrens XHT, Chen J, Bers DM. Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation. 2013;127:913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wehrens XHT, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. [DOI] [PubMed] [Google Scholar]
- 36.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, Wang Q, De Almeida AC, Skapura DG, Anderson ME, Bers DM, Wehrens XHT. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wehrens XHT, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen B-X, Marks AR. Role of chronic ryanodine receptor phosphorylation in heart failure and β-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sedej S, Heinzel FR, Walther S, Dybkova N, Wakula P, Groborz J, Gronau P, Maier LS, Vos MA, Lai FA, Napolitano C, Priori SG, Kockskämper J, Pieske B. Na+-dependent SR Ca2+ overload induces arrhythmogenic events in mouse cardiomyocytes with a human CPVT mutation. Cardiovasc Res. 2010;87:50–59. [DOI] [PubMed] [Google Scholar]
- 40.Zaglia T, Pianca N, Borile G, Da Broi F, Richter C, Campione M, Lehnart SE, Luther S, Corrado D, Miquerol L, Mongillo M. Optogenetic determination of the myocardial requirements for extrasystoles by cell type-specific targeting of ChannelRhodopsin-2. Proc Natl Acad Sci U S A. 2015;112:E4495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res. 2014;114:511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.