

Abstract
Prior to statistical analysis of mass spectrometry (MS) data, quality control (QC) of the identified biomolecule peak intensities is imperative for reducing process-based sources of variation and extreme biological outliers. Without this step, statistical results can be biased. Additionally, liquid chromatography–MS proteomics data present inherent challenges due to large amounts of missing data that require special consideration during statistical analysis. While a number of R packages exist to address these challenges individually, there is no single R package that addresses all of them. We present pmartR, an open-source R package, for QC (filtering and normalization), exploratory data analysis (EDA), visualization, and statistical analysis robust to missing data. Example analysis using proteomics data from a mouse study comparing smoke exposure to control demonstrates the core functionality of the package and highlights the capabilities for handling missing data. In particular, using a combined quantitative and qualitative statistical test, 19 proteins whose statistical significance would have been missed by a quantitative test alone were identified. The pmartR package provides a single software tool for QC, EDA, and statistical comparisons of MS data that is robust to missing data and includes numerous visualization capabilities.
Keywords: mass spectrometry, quality control, normalization, quantification, visualization, statistics, R package
Introduction
High-throughput mass spectrometry (MS)-based analyses generate large and complex data sets measuring hundreds to thousands of biomolecules (e.g., peptides, metabolites, and lipids). The term quality control (QC) can hold different meanings depending on the field. Here, QC refers to reducing process-based sources of variation and extreme biological outliers. The goal of this is to reduce bias in downstream statistical results and biological inference. QC processing of the MS-quantified biomolecule peak intensities is essential for removing outliers and other random effects arising from the mapping of raw mass spectra to identified biomolecules with observed values. Only after QC processing, including normalization, should statistical analysis be performed. Given the missing data issues inherent to liquid chromatography (LC)–MS-based proteomics data1,2 and the poor or inconsistent results of imputation to fill in missing data,1,3,4 QC and statistical methods that are robust to missing data are needed.
At the time of this writing, on Bioconductor5 there are a large number of R6 packages for processing MS data under the Proteomics, Metabolomics, and Lipidomics categories. The most comprehensive QC processing packages for proteomics in R focus on the raw data,7−10 which although necessary and valuable are not sufficient for removing process-based errors that will bias downstream statistical analysis. Several R packages are available for statistical analysis of proteomics data11,12 and have excellent functionality for statistics but minimal QC.12,13 Other packages support individualized analyses of single samples or biomolecules but rely on the user to add the functionality to statistically evaluate the data14 or have limited functionality for filtering biomolecules and performing statistical analysis.15 Additional packages16−22 cover extensive capabilities for QC and normalization, but none are focused on robust QC and statistics of mass spectrometry peak intensity data or specifically designed to be robust to the complex missing data of global proteomics. These missing data arise from a combination of left-censoring, instrument error, and physicochemical properties of the peptides, making imputation difficult.2,4 This is the overall gap that pmartR fills through end-to-end data processing capabilities.
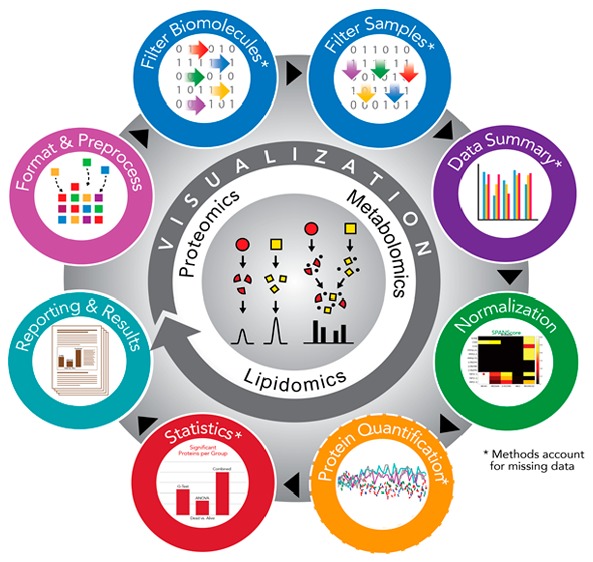
The pmartR R package provides QC, data pre-processing, and statistical analysis functionality that is both robust to missing values in analyses and uses the missing data structure to identify qualitative biomarker candidates. This is the primary capability that distinguishes pmartR from other mass spectrometry R packages, which rely on imputation of data. Many of the pmartR functions use attributes to capture information about the data set and the analyses that have been performed, allowing the user to query the data object and obtain a record of the analysis steps. pmartR has the added benefit of being applicable to multiple MS-quantified omics data types, including proteomics, metabolomics, and lipidomics gas chromatography (GC-) or LC-MS data. Finally, pmartR provides visualization and summary methods that allow the user to understand the effect of their filtering, normalization, and other analysis choices as they move through the QC, EDA, and statistics pipeline.
Software Design and Functionalities
The pmartR software package is implemented in R; the open source package is available for download at http://github.com/pmartR/pmartR. pmartR provides capabilities for preparing MS data for, and for performing, statistical analysis using simple function calls. Data objects are automatically updated with information about the analysis as it is performed, streamlining the data exploration, QC, and statistical analysis process for the user. Below, we provide brief descriptions of the capabilities of pmartR, and more details can be found on the github page and in the R package vignettes.
Data Format and Pre-Processing
Functions in the pmartR package operate primarily on an S3 data object defined at the beginning of an analysis by the user. There are separate S3 classes for peptide data (either unlabeled or labeled with an isobaric tag), protein data, metabolite data, and lipid data (Table 1), and the data object is created using the R function corresponding to the object type [as.pepData(), as.isobaricpepData(), as.proData(), as.metabData(), or as.lipidData()]. In R, S3 data objects have predefined structures and properties. Methods specific to a given data type can then be written by the developer and called generically by a user. For example, calling summary() on a vector of numbers returns the minimum, maximum, etc., of the vector’s values, while calling summary() on a pepData object returns quantities such as the number of samples, the number of biomolecules, and the amount of missing data.
Table 1. Types of Data Supported by pmartR and Their Corresponding S3 Object Classesa.
| type of data | S3 object class |
|---|---|
| peptide (unlabeled) | pepData |
| peptide (labeled) | isobaricpepData, pepData |
| protein | proData |
| metabolite | metabData |
| lipid | lipidData |
Note that an isobaricpepData object is also a pepData object, but the converse is not true.
The starting point for analysis using pmartR is the quantified peak intensities of identified biomolecules. More specifically, there are two required data tables and one optional data table for the creation of the pmartR S3 data object. Each component corresponds to a data.frame in R, which can be imported using the base functionality of R. (1) e_data is required and contains the expression data. This is a p × (n + 1) data.frame, where p is the number of biomolecules and n is the number of samples. Each row corresponds to a biomolecule, and each column corresponds to a sample except for one column, which gives a unique biomolecule identifier. (2) f_data is required and contains feature data about the samples. This is a data.frame with n rows, one for each sample, and one column for sample names with additional columns for information about the samples (e.g., treatment group or other experimental information). (3) e_meta is optional and contains metadata about the biomolecules. This is a data.frame with one column for the biomolecule identifier and the remaining columns for additional biomolecule information (e.g., for peptide data, e_meta might include a peptide-to-protein mapping). Supporting Information file S1 contains the e_data, f_data, and e_meta data tables corresponding to the data set discussed in Experimental Methods. Plot and summary methods for these data objects provide a visualization of the samples in the data set (as boxplots) and a quick overview of the data set properties (number of samples, number of biomolecules, and amount of missing data), respectively.
Several publications have shown that more reliable statistical results are obtained using methods that are robust to missing data to analyze mass spectrometry data.1 Thus, pmartR allows a user to analyze their data without imputation of missing values. However, there are some instances where it may be appropriate or desirable to impute missing values. Numerous R packages exist to perform imputation; therefore, pmartR does not attempt to recreate any of this functionality. A user should impute their data before using pmartR, if desired, and take care in choosing an imputation method as several studies have compared, contrasted, and demonstrated the differences in data generated by different imputation methodologies for mass spectrometry data.1,4
Functions for data value replacement [e.g., replace 0s with NA’s to represent missing values; edata_replace()] and transformation [e.g., to a log scale; edata_transform()] are provided to prepare data for statistical analyses. Statistical analysis with pmartR allows the user to make comparisons between two or more groups; we therefore provide a function for associating group membership with each sample, and this information is saved as part of the data object and used by some of the downstream filtering methods and functions[group_designation()].
For compatibility with the widely used MSnbase package,7 which contains an array of functionality for processing raw proteomics data, the pmartR package includes a function to transform data objects back and forth between the MsnSet class from MSnbase and the pepData class from pmartR. This allows users to take advantage of the raw data processing from MSnbase as well as the robust QC and statistical processing from pmartR without having to manually configure the data objects.
Data QC: Data Summary and Filtering
QC is crucial for accurate and unbiased downstream statistical analyses; therefore, pmartR offers a number of different functions to aid in this process, including multiple filter types and visualizations. Summary and plot methods are available for the data set S3 objects, to describe basic characteristics of the data and creating a boxplot for each sample; for the filter S3 objects, to summarize the effects of applying the filter and aid the selection of a threshold for the filter; for the normalization S3 object, to see boxplots before and after normalization; and for the summary S3 objects, to display information about the summary and graph the output. See Supporting Information file S2 for example code.
Data Summary and Visualizations
pmartR provides functions for generating numeric summaries (in the form of data.frames), graphical summaries to learn about the presence and patterns of missing values, a correlation heatmap, and probabilistic principal component analysis (PPCA).23 The correlation matrix is computed using Pearson correlation on pairwise complete observations in base R,24 via cor_result(), which creates an object of class corRes. This object can be displayed in a heatmap using the plot() command. PPCA is a PCA algorithm that can be used in the presence of missing data. The principal components are calculated using projection pursuit estimation, which implements an expectation–maximization algorithm when data are missing. We implement PPCA from the pcaMethods package in R23 using a wrapper function for dimension reduction, dim_reduction(), which creates an object of class dimRes. This object can be used to create a PCA scores plot via the plot() command.
Filters
pmartR allows the user to filter both biomolecules and samples based on objective or subjective characteristics of the data. Biomolecules can be filtered according to the number of observed values [molecule_filter()] or the variability according to the coefficient of variation [cv_filter()] across samples. They can also be filtered in anticipation of specific statistical tests that will be run subsequently [imdanova_filter()] via the removal of biomolecules for which no statistical test can be performed.25 A robust Mahalanobis distance (rMd) filter is used to identify potential outlying samples where each sample is assigned a p-value as described previously.26 The rMD filter [rmd_filter()] is based on any user-defined subset of the following five metrics: correlation, proportion of missing values, median absolute deviation, skew, and kurtosis. The rmd_filter() function uses a default p-value threshold of 0.0001; however, we recommend using the additional visualization capability for this method in combination with expert knowledge prior to removing outlying samples. User-specified biomolecules (e.g., contaminants and biomolecules of particular interest) or samples can be either removed from or retained in the data set using the custom_filter(). Each filter has an associated summary and plot method (except for the custom_filter(), which does not have a plot method) that can assist in making decisions about filtering thresholds and provide additional insight into the data set.
Normalization
A variety of data normalization options are available in pmartR. For labeled data, normalization to a reference sample is supported [normalize_isobaric()]. This can be followed by any of the other normalization approaches available in pmartR, which include quantile normalization [normalize_quantile()] and loess normalization27,28 [normalize_loess()] that operate on a biomolecule by biomolecule basis and a host of global normalization types [normalize_global()] that operate on a sample by sample basis.
For global normalization, one may choose a normalization strategy from the available options, or for proteomics data, the statistical procedure for the analyses of peptide abundance normalization strategies29 (SPANS) can be used to aid in the selection of a normalization strategy. The global normalization strategies consist of two parts, a subset function and a normalization function. The subset specifies the biomolecules (presented below in the context of peptide-level data) with which to generate the normalization factors using the normalization function. The normalization factors are then applied to the full data set. Currently available subset functions include the following. (1) All: Use all peptides to compute normalization factor(s). (2) Percentage of peptides present (PPP): Subset the data to peptides present in at least p percent of samples. (3) Rank invariant peptides (RIP): First, subset peptides to those present in every sample (e.g., complete peptides). Next, subject each peptide to a Kruskal–Wallis test on group, and those peptides not significant at a given p-value threshold are retained as invariant. (4) PPP-RIP: Rank invariant peptides among peptides present in a given percentage of samples. (5) Top “l” order statistics (LOS): The peptides with intensities in the top “l” order statistics are retained.
The currently available normalization functions (presented below in the context of log2-transformed peptide-level data) include the following, where missing values are ignored when computing means, medians, or standard deviations and data are on a log scale. (1) Median centering: The sample-wise median (median of all peptides in a given sample) is subtracted from each peptide in the corresponding sample. (2) Mean centering: The sample-wise mean (mean of all peptides in a given sample) is subtracted from each peptide in the corresponding sample. (3) Z-score transformation: The sample-wise mean (mean of all peptides in a given sample) is subtracted from each peptide, and the result is divided by the sample-wise standard deviation (standard deviation of all peptides in a given sample) in the corresponding sample. (4) Median absolute deviation (MAD) transformation: The sample-wise median (median of all peptides in a given sample) is subtracted from each peptide, and the result is divided by the sample-wise MAD (e.g., the MAD of all peptides in a given sample) in the corresponding sample.
Protein Quantification
For proteomics data, a number of algorithms for protein rollup are available in pmartR via the protein_quant() function, some of which account for different isoforms of the proteins. For any type of rollup, the user must specify a method for estimating protein abundances from observed peptides (“rollup”, “Rrollup”, “Qrollup”, or “Zrollup”)30 and a function to use for combining the peptide-level data (either “mean” or “median”). More information about the rollup methods can be found in the pmartR vignette. To account for protein isoforms, Bayesian Proteoform Quantification31 (BP-Quant) can be used.
Statistics
The pmartR package includes functions to analyze omics data for both quantitative and qualitative differences in abundance data between two or more groups [imd_anova()] using the IMD-ANOVA method.25 This functionality can handle up to two grouping factors (or “main effects”) with or without additional covariate information. Differences between the groups of main effects can be tested, adjusting for the covariates. The data can be paired or not.
A quantitative test for differential abundance in the data is accomplished via an analysis of variance (ANOVA) for each biomolecule. The Rcpp package is used to accelerate computation,33 and different ANOVA implementations are applied depending on the complexity of the supplied data. For example, if only two groups are supplied, then ANOVA reduces to a two-sample t-test that assumes equal variance for the two groups. Alternatively, if two groups are present and a covariate correction is required, then the effect of the covariates is removed using a reduced maximum likelihood approach. After the covariate correction is applied, a two-factor ANOVA is used to detect the difference between all combinations of groups or between main effects as appropriate.
In the event that there are not enough data to test for a quantitative difference in abundance between groups, one can still test for a qualitative difference in groups using the independence of missing data (IMD) test.25 This is often the case for proteomics data where a number of peptides or proteins could have missing data for one of several reasons. The idea is to assess if there are more missing values than expected by random chance in one group compared to another. If there are an adequate number of nonmissing data available, then the χ2 test of independence can be used.34 This assumption often fails, however, so a modified version of the χ2 test, called the g-test,25 should be used. The availability of both quantitative and qualitative statistical models and tests is one of the features that distinguish this package from other frequently used packages in R for mass spectrometry data.
Reporting and Results
The objects returned by imd_anova() are of the class statRes. Special functions are available for objects of this class, including print(), summary(), and plot(). The summary() function prints the type of test that was run, any adjustments that were made, the p-value threshold used to define significance, and a table that summarizes the number of significant biomolecules (up or down relative to the reference group) for each comparison.
The plot() function can be used to produce any of the following four plots. 1) A bar plot shows the number of significant biomolecules grouped by comparisons and indicates the direction of change. 2) A volcano plot is a plot of the ANOVA p-value by the fold-change estimate for each biomolecule, differentiated by test and faceted by comparison; 3) for the g-test, the resulting plot represents each biomolecule by the number of observations in each group as (x, y) coordinates. 4) A heatmap illustrates the fold changes for the statistically significant biomolecules (available only if comparisons among more than two groups are made).
Experimental Methods
We use a subset of samples from a larger study examining the effect of inhalation endotoxin exposure and obesity on lung inflammation in mice.35 The full experimental design and description of proteomics data generation have been presented previously.35 This identified and quantified example data set is available as Supporting Information file S1, and the R code used to analyze it is available as Supporting Information file S2. The data set contains 16 samples, eight belonging to the cigarette smoke exposure group and eight controls, with 5244 peptides corresponding to 3646 unique proteins.
Data processing begins with replacing values of 0 with NA and log2 transforming the data. We then apply the molecule filter to remove any peptides observed in fewer than two samples, which removes 679 peptides and results in 4565 peptides mapping to 3150 proteins. A proteomics filter to remove peptides mapping to more than one protein results in 1738 peptides mapping to 508 proteins. Finally, the IMD-ANOVA filter is applied to remove any peptides for which we will be unable to perform either the quantitative or qualitative statistical comparisons between the smoking and control samples. After this filtering, we are left with 1513 peptides mapping to 436 proteins. EDA functionality is demonstrated by boxplots for each sample (Figure 1A), PPCA plots of the peptide data (Figure 1B), a bar graph showing the number of missing values per sample (Figure 1C), and a Pearson correlation heatmap (Figure 1D).
Figure 1.

(A) Boxplots for each log2 sample prior to QC. (B) The PPCA plot of the log2 peptide data without imputation of missing values shows minimal clustering of the treatment groups. (C) A bar graph of the number of missing values per sample does not reveal anything systematically different between the two groups or the individual samples. (D) The Pearson correlation heatmap among the log2-transformed samples shows some variation in correlation across the samples but nothing to indicate a potential outlying sample. (E) The rMd plot does not identify any potential sample outliers. (F) The values for each of the metrics included in the rMd calculation are indicated on boxplots for sample U54_SMOKEmp_119, which is the control sample having the highest log2 rMd score.
Potential sample outliers are identified using a combination of the EDA results and the rMd filter based on correlation, the proportion of missing values, MAD, and skewness. To focus on extreme outliers, a p-value threshold of 0.0001 is used, but this can be easily modified to fit the needs of the analysis. For this data set, no potential outliers are identified as having a log2 rMd score exceeding the log2 rMd threshold associated with a p-value of 0.001 (Figure 1E). From Figure 1F, we can see where the metrics of correlation, proportion of missing data, MAD, and skewness for the sample with the highest rMd score fall relative to those of the other samples. These bar plots are particularly helpful in determining which samples, if any, to remove when there are samples that are identified as potential outliers. In this analysis, no samples were removed.
SPANS29 is used to select a normalization approach, where a heatmap of the SPANS scores for each combination of subset and normalization method (Figure 2A) indicates the more recommended approaches in lighter colors (yellows). For this data set, the SPANS algorithm identifies the MAD based on the top 5% of order statistics (LOS 0.05) as the ideal normalization approach, which is based on 121 peptides. The second choice for the normalization approach is the MAD based on the top 20% of order statistics (LOS 0.2), which is based on 508 peptides. Because the second choice is based on a larger number of peptides and has a SPANS score only 0.0035 below the top choice, we proceed using the second choice (Figure 2B).
Figure 2.

(A) Heatmap of the SPANS scores for each combination of data subset and normalization method that shows the use of the MAD of the LOS 0.05 peptides to be the top choice for the normalization approach, followed by the MAD of the LOS 0.2 peptides. (B) Boxplots for each sample that show the distributions of normalized log2 peptide abundance, where normalization was performed using the MAD of the LOS 0.2 peptides.
Protein quantification is performed using Rrollup,30 which first scales all peptides that map to a given protein by a reference peptide (the peptide, out of all peptides mapping to the given protein, with the most observations) and then takes the median of the scaled peptides to obtain the relative protein abundance. This results in 436 proteins. Statistical analysis at the protein level is performed using both quantitative and qualitative tests25 (IMD-ANOVA) on a protein by protein basis to compare the samples from the smoking group to the samples from the control group. A Holm p-value adjustment is used for the p values from both tests.
Results and Discussion
Of the 436 proteins tested, 29 showed significant differences between the smoking and control groups (based on Holm adjusted p-values of <0.05). Both the t-test and the g-test identified similar numbers of significant proteins that had a lower level of expression in the smoking group than in the control group. However, more proteins with a higher level of expression in the smoking group compared to the control group were identified as statistically significant by the g-test than by the t-test. There were 19 proteins that were found to be significant by the g-test and for which the t-test was not able to be run due to either the smoking or the control group having too few observed values. A graphical representation of the statistical results (Figure 3) was generated using the plot() command on the statRes object and includes a bar plot for the number of significant proteins, a volcano plot for the results of the t-test, and a plot showing the number of observations per group for the g-test results.
Figure 3.

Graphical summary of the statistical results that includes (A) a bar graph of the number of significant proteins, both in total and broken out by statistical test, and (B) a volcano plot for the t-test (ANOVA) results (top) and a plot of the number of observations per group for the g-test results (bottom).
Conclusions
The pmartR package is a collection of R functions that enable QC, EDA, and statistical processing of mass spectrometry data, without the necessity of imputing missing values. pmartR takes two to three data.frames, converts these to an S3 data object, and offers data cleaning, processing, summarizing, and statistical analysis capabilities. The pmartR package functions include automated tracking of the characteristics of the data [e.g., data scale (log2, log10, natural log, and abundance), whether and how the data were normalized, what biomolecules or samples were filtered, group membership for each sample, etc.], which streamlines analysis and reporting for the user. We provide sample data sets and R code as Supporting Information as well as additional example data sets in the pmartRdata package available on github that can be used for demonstration purposes. Therein, we demonstrate pmartR’s capabilities on a proteomics data set and present the results.
Acknowledgments
This work was supported by the National Cancer Institute of the National Institutes of Health via Grant U01 CA184783-02, by the U.S. Department of Energy (DOE), Office of Biological and Environmental Research, under Contract FWP 71226, and by the Microbiomes in Transition Initiative Laboratory Directed Research and Development Program at Pacific Northwest National Laboratory (PNNL). PNNL is a multiprogram national laboratory operated by Battelle for the U.S. DOE under Contract DE-AC05-76RL01830.
Glossary
Abbreviations
- MS
mass spectrometry
- QC
quality control
- EDA
exploratory data analysis
- LC
liquid chromatography
- GC
gas chromatography
- NA
not applicable
- PPCA
PCA probabilistic principal components
- rMd
robust Mahalanobis distance
- SPANS
statistical procedure for the analysis of peptide abundance normalization strategies
- PPP
percentage of peptides present
- RIP
rank invariant peptides
- LOS
top l order statistics
- MAD
median absolute deviation
- BP-Quant
Bayesian proteoform quantification
- ANOVA
analysis of variance
- IMD
independence of missing data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00760.
The authors declare no competing financial interest.
Notes
The pmartR package, including vignettes, is available at http://github.com/pmartR/pmartR. The pmartRdata package is available at http://github.com/pmartR/pmartRdata.
Supplementary Material
References
- Webb-Robertson B. J.; Wiberg H. K.; Matzke M. M.; Brown J. N.; Wang J.; McDermott J. E.; Smith R. D.; Rodland K. D.; Metz T. O.; Pounds J. G.; Waters K. M. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J. Proteome Res. 2015, 14 (5), 1993–2001. 10.1021/pr501138h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpievitch Y. V.; Dabney A. R.; Smith R. D. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinf. 2012, 13 (Suppl. 16), S5. 10.1186/1471-2105-13-S16-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Li L.; Chen T.; Ma J.; Zhu Y.; Zhuang J.; Chang C. In-depth method assessments of differentially expressed protein detection for shotgun proteomics data with missing values. Sci. Rep. 2017, 7 (1), 3367. 10.1038/s41598-017-03650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar C.; Gatto L.; Ferro M.; Bruley C.; Burger T. Accounting for the Multiple Natures of Missing Values in Label-Free Quantitative Proteomics Data Sets to Compare Imputation Strategies. J. Proteome Res. 2016, 15 (4), 1116–25. 10.1021/acs.jproteome.5b00981. [DOI] [PubMed] [Google Scholar]
- Huber W.; Carey V. J.; Gentleman R.; Anders S.; Carlson M.; Carvalho B. S.; Bravo H. C.; Davis S.; Gatto L.; Girke T.; Gottardo R.; Hahne F.; Hansen K. D.; Irizarry R. A.; Lawrence M.; Love M. I.; MacDonald J.; Obenchain V.; Oles A. K.; Pages H.; Reyes A.; Shannon P.; Smyth G. K.; Tenenbaum D.; Waldron L.; Morgan M. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12 (2), 115–21. 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R: A language and environment for statistical computing; R Foundation for Statistical Computing: Vienna, 2017.
- Gatto L.; Lilley K. S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2012, 28 (2), 288–9. 10.1093/bioinformatics/btr645. [DOI] [PubMed] [Google Scholar]
- Gatto L.; Christoforou A. Using R and Bioconductor for proteomics data analysis. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 42–51. 10.1016/j.bbapap.2013.04.032. [DOI] [PubMed] [Google Scholar]
- Gatto L.; Breckels L. M.; Naake T.; Gibb S. Visualization of proteomics data using R and bioconductor. Proteomics 2015, 15 (8), 1375–89. 10.1002/pmic.201400392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto L.; Hansen K. D.; Hoopmann M. R.; Hermjakob H.; Kohlbacher O.; Beyer A. Testing and Validation of Computational Methods for Mass Spectrometry. J. Proteome Res. 2016, 15 (3), 809–14. 10.1021/acs.jproteome.5b00852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M.; Chang C. Y.; Clough T.; Broudy D.; Killeen T.; MacLean B.; Vitek O. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30 (17), 2524–6. 10.1093/bioinformatics/btu305. [DOI] [PubMed] [Google Scholar]
- Wieczorek S.; Combes F.; Lazar C.; Giai Gianetto Q.; Gatto L.; Dorffer A.; Hesse A. M.; Coute Y.; Ferro M.; Bruley C.; Burger T. DAPAR & ProStaR: software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 2017, 33 (1), 135–136. 10.1093/bioinformatics/btw580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohart F.; Gautier B.; Singh A.; Le Cao K. A. mixOmics: An R package for ’omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13 (11), e1005752 10.1371/journal.pcbi.1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverner T.; Karpievitch Y. V.; Polpitiya A. D.; Brown J. N.; Dabney A. R.; Anderson G. A.; Smith R. D. DanteR: an extensible R-based tool for quantitative analysis of -omics data. Bioinformatics 2012, 28 (18), 2404–6. 10.1093/bioinformatics/bts449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggio R.; Villas-Boas S. G.; Ruggiero K. Metab: an R package for high-throughput analysis of metabolomics data generated by GC-MS. Bioinformatics 2011, 27 (16), 2316–8. 10.1093/bioinformatics/btr379. [DOI] [PubMed] [Google Scholar]
- Wen B.; Mei Z.; Zeng C.; Liu S. metaX: a flexible and comprehensive software for processing metabolomics data. BMC Bioinf. 2017, 18 (1), 183. 10.1186/s12859-017-1579-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C.; Maraschin M.; Rocha M.; Cardoso S.; Afonso T.; Beleites C.; Hao J.; Jacob D.. specmine: Metabolomics and Spectral Data Analysis and Mining, version 2.0.3; https://CRAN.R-project.org/package=specmine (accessed November 2018).
- Dorscheidt T.MetStaT: Statistical metabolomics tools, version 1.0; https://CRAN.R-project.org/package=MetStaT (accessed November 2018).
- Gaude E.; Chignola F.; Spiliotopoulos D.; Mari S.; Spitaleri A.; Ghitti M.. muma: Metabolomics Univariate and Multivariate Analysis, version 1.4; https://CRAN.R-project.org/package=muma (accessed November 2018).
- DeLivera A. M.metabolomics: Analysis of Metabolomics Data, version 0.1.4; https://CRAN.R-project.org/package=metabolomics (accessed November 2018).
- DeLivera A. M.MetNorm: Statistical Methods for Normalizing Metabolomics Data, version 0.1; https://CRAN.R-project.org/package=MetNorm (accessed November 2018).
- Möbius T. W. D.proteomics: Statistical Analysis of High Throughput Proteomics Data, version 0.2; https://CRAN.R-project.org/package=proteomics (accessed November 2018).
- Stacklies W.; Redestig H.; Scholz M.; Selbig J.; Walther D. pcaMethods: A Bioconductor package providing PCA methods for incomplete data. Bioinformatics 2007, 23 (9), 1164–7. 10.1093/bioinformatics/btm069. [DOI] [PubMed] [Google Scholar]
- R Core Team . R: A Language and Environment for Statistical Computing, version 3.5.0; https://www.R-project.org (accessed May 2018).
- Webb-Robertson B. J.; McCue L. A.; Waters K. M.; Matzke M. M.; Jacobs J. M.; Metz T. O.; Varnum S. M.; Pounds J. G. Combined statistical analyses of peptide intensities and peptide occurrences improves identification of significant peptides from MS-based proteomics data. J. Proteome Res. 2010, 9 (11), 5748–56. 10.1021/pr1005247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke M. M.; Waters K. M.; Metz T. O.; Jacobs J. M.; Sims A. C.; Baric R. S.; Pounds J. G.; Webb-Robertson B. J. Improved quality control processing of peptide-centric LC-MS proteomics data. Bioinformatics 2011, 27 (20), 2866–72. 10.1093/bioinformatics/btr479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad B. M.; Irizarry R. A.; Astrand M.; Speed T. P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19 (2), 185–93. 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Ballman K. V.; Grill D. E.; Oberg A. L.; Therneau T. M. Faster cyclic loess: normalizing RNA arrays via linear models. Bioinformatics 2004, 20 (16), 2778–86. 10.1093/bioinformatics/bth327. [DOI] [PubMed] [Google Scholar]
- Webb-Robertson B. J.; Matzke M. M.; Jacobs J. M.; Pounds J. G.; Waters K. M. A statistical selection strategy for normalization procedures in LC-MS proteomics experiments through dataset-dependent ranking of normalization scaling factors. Proteomics 2011, 11 (24), 4736–41. 10.1002/pmic.201100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polpitiya A. D.; Qian W. J.; Jaitly N.; Petyuk V. A.; Adkins J. N.; Camp D. G.; Anderson G. A.; Smith R. D. DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 2008, 24 (13), 1556–1558. 10.1093/bioinformatics/btn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb-Robertson B. J.; Matzke M. M.; Datta S.; Payne S. H.; Kang J.; Bramer L. M.; Nicora C. D.; Shukla A. K.; Metz T. O.; Rodland K. D.; Smith R. D.; Tardiff M. F.; McDermott J. E.; Pounds J. G.; Waters K. M. Bayesian Proteoform Modeling Improves Protein Quantification of Global Proteomic Measurements. Mol. Cell. Proteomics 2014, 13, 3639–3646. 10.1074/mcp.M113.030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddelbuettel D.Seamless R and C++ Integration with Rcpp; Springer: New York, 2013; pp 65–74. [Google Scholar]
- Ott R. L.; Longnecker M. T.. An Introduction to Statistical Methods and Data Analysis, 6th ed.; Brooks/Cole: Belmont, 2010; pp 503–504. [Google Scholar]
- Tilton S. C.; Waters K. M.; Karin N. J.; Webb-Robertson B. J.; Zangar R. C.; Lee K. M.; Bigelow D. J.; Pounds J. G.; Corley R. A. Diet-induced obesity reprograms the inflammatory response of the murine lung to inhaled endotoxin. Toxicol. Appl. Pharmacol. 2013, 267 (2), 137–48. 10.1016/j.taap.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.