

Abstract
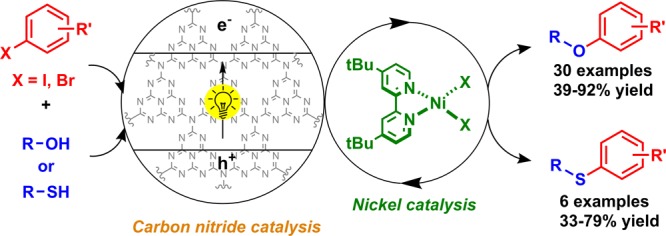
A carbon nitride material can be combined with homogeneous nickel catalysts for light-mediated cross-couplings of aryl bromides with alcohols under mild conditions. The metal-free heterogeneous semiconductor is fully recyclable and couples a broad range of electron-poor aryl bromides with primary and secondary alcohols as well as water. The application for intramolecular reactions and the synthesis of active pharmaceutical ingredients was demonstrated. The catalytic protocol is applicable for the coupling of aryl iodides with thiols as well.
Alkyl aryl ethers are a common structural motif in many active pharmaceutical ingredients (APIs), such as Fluoxetine, Ketoconazole, Raloxifene, and Flecainide (Figure 1).1,2
Figure 1.

Examples of APIs containing the alkyl aryl ether motif.
Nucleophilic substitution of alkyl halides with phenolates (Williamson ether synthesis) traditionally used to synthesize these scaffolds, suffers from functional group incompatibilities due to the harsh conditions it requires. Catalytic strategies for the synthesis of ethers were developed to overcome these drawbacks. Copper can catalyze the coupling of phenols and aryl halides (Ullmann-type reaction),3,4 as well as aryl boronic acids and alcohols (Chan–Evans–Lam coupling).5 Palladium-catalyzed O-arylations of alcohols via Buchwald–Hartwig-type cross-coupling reactions broaden the substrate scope.6,7 Strongly basic conditions and well-designed biaryl phosphine ligands are required for efficient cross-coupling reactions.8
Nickel catalysis gained increasing interest due to its significantly higher abundance compared to noble metals.9,10 The low electronegativity of Ni enables facile oxidative addition into carbon–halide bonds, whereas reductive elimination, especially in case of C–O couplings, is difficult.11−13 Thermolysis of Ni(II) oxametallacycles, for example, results in β-hydride elimination of undesired carbonyl compounds, whereas oxidation to Ni(III) complexes via single electron transfer (SET) with stoichiometric oxidants can induce reductive elimination resulting in C–O bond formation.14 Combining nickel with photoredox catalysis,15−17 enables the coupling of aryl bromides with alcohols18,19 or water20 without stoichiometric SET oxidants.
Photoredox catalysis is dominated by expensive, homogeneous iridium, and ruthenium complexes. Noble-metal-free, homogeneous photoredox catalysts, such as boron-dipyrromethene derivatives (BODIPY),20 require tedious purification procedures and are prone to degradation.21 Heterogeneous semiconductors are a recyclable alternative to common homogeneous photoredox catalysts.22−25 CdSe quantum dots26 and CdS27 were used for carbon–heteroatom couplings via the dual nickel/photoredox catalytic approach. Cadmium, however, is among the most toxic elements28 and strictly regulated.29 Its application in the synthesis of APIs is therefore not desirable.
Carbon nitride (CN) materials, a class of stable and metal-free semiconductors with low toxicity30 that can be easily made from commodity chemicals, are able to activate Ni complexes via photosensitization.31 Here, we show that these materials are also able to catalyze alkyl aryl ether synthesis, likely by triggering reductive elimination via SET modification of the oxidation state of Ni complexes.32
The etherification of methyl 4-bromobenzoate with 1-hexanol served as a model reaction for initial studies (Table 1). A careful optimization of all reaction parameters showed that a carbon nitride material prepared by polymerization of urea and oxamide (CN-OA-m)33 in combination with catalytic amounts of NiBr2·3H2O and di-tert-butylbipyridyl (dtbbpy) results in the selective synthesis of the desired ether (1) after 48 h irradiation with white LEDs in acetonitrile under mildly basic conditions (entry 1–2). The only side-products were small amounts of the corresponding phenol (2) from the cross-coupling with water and dehalogenated methyl benzoate (3). In homogeneous dual catalysis, the addition of catalytic amounts of quinuclidine was reported to accelerate the reaction.18,19 For similar reasons, an amine-modified organonickel complex was used in combination with photoredox catalysis for the synthesis of phenols from aryl halides and water.20 The semiheterogeneous protocol did not result in significant rate enhancement when 10 mol % quinuclidine was added (entries 3–4). The utilization of 6,6′-diamino-2,2′-bipyridyl instead of dtbbpy drastically reduced the efficacy of the C–O coupling in our model system (see Table S4). A reaction with methyl 4-chlorobenzoate as substrate resulted in very low amounts of the desired ether product under optimized conditions (Table 1, entry 5). No reaction was detected in the case of the mesyltate, triflate, or tosylate derivatives (see Table S10). Control experiments proved that light, CN-OA-m, NiBr2·3H2O, dtbbpy, N-tert-butylisopropylamine (BIPA), and oxygen-free conditions are essential for successful C–O cross-couplings (entries 6–11).
Table 1. Optimized Conditions and Control Experimentsa.

| entry | conditions | 1 (%)b | 2 (%)b | 3 (%)b |
|---|---|---|---|---|
| 1 | as shown | 96 | 3 | 1 |
| 2 | CN-OA-m (1.66 mg mL–1) | 90 | 6 | n.d. |
| 3 | 24 h | 55 | 3 | n.d. |
| 4 | 24 h with 10 mol % quinuclidine | 61 | 2 | n.d. |
| 5 | methyl 4-chlorobenzoate | 4 | n.d. | n.d. |
| 6 | no light | n.d. | n.d. | n.d. |
| 7 | no CN-OA-m | n.d. | n.d. | n.d. |
| 8 | no NiBr2·3H2O | n.d. | n.d. | n.d. |
| 9 | no dtbbpy | n.d. | n.d. | n.d. |
| 10 | no BIPA | n.d. | n.d. | n.d. |
| 11 | no degassing | n.d. | n.d. | n.d. |
Reaction conditions: methyl 4-bromobenzoate (0.3 mmol), 1-hexanol (0.6 mmol), CN-OA-m (10 mg), NiBr2·3H2O (30 μmol), dtbbpy (30 μmol), BIPA (1.5 mmol), MeCN (3.0 mL), white LEDs at 40 °C for 48 h.
Determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. n.d. = not detected.
With the optimized conditions in hand, the versatility of the semiheterogeneous cross-coupling was investigated (Scheme 1). Aryl bromides substituted with electron withdrawing groups in para-position were generally isolated in good to excellent yields. A broad range of functional groups including esters (1), nitriles (4), aldehydes (5), ketones (6, 15), phenylboronic acid pinacol esters (14), chlorides (10), and trifluoromethyl- (13) as well as methylsulfonyl-groups (16) were tolerated. Substrates with electron withdrawing meta-substituents (7, 8) did also yield the desired products, although with lower efficiency. Ortho-substituted aryl bromides (11, 12) resulted in a drastically decreased reactivity. Coupling of 1,4-dibromobenzene with 1-hexanol gave a selective monoetherification as the resulting aryl alkyl ether (9) deactivates the second bromide functionality. Heteroaryl bromides (17, 18) were successfully coupled under these conditions. Substrates lacking a strong electron withdrawing group gave very low amounts of the desired C–O coupling products (20, 21) within 48 h.
Scheme 1. Scope and Limitations of the Semiheterogeneous Etherification.

Reaction conditions: aryl bromide (1.2 mmol), alcohol (2.4–4.8 mmol), CN-OA-m (20 mg), NiBr2·3H2O (120 μmol), dtbbpy (120 μmol), BIPA (6.0 mmol), MeCN (6.0 mL), white LEDs at 40 °C.
Determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard.
DMF was used as solvent.
Electronic effects dictate the reactivity of para- and meta-substituted aryl bromides, whereas steric effects influence the reactivity of ortho-substituted analogs and are responsible for the scope and limitations of aliphatic alcohols (Scheme 1). Coupling of methyl 4-bromobenzoate with methanol (22) was completed within 24 h under standard conditions and within 8 h when MeOH was used as solvent (see the Supporting Information). The semiheterogeneous methodology provides an effective method to prepare deuterium-labeled anisoles (23) in excellent yield. Primary alcohols with benzyl (24), allyl (26), nitrile (29), trifluoromethyl (25), and tertiary amine (30) groups were coupled with high selectivity. The secondary alcohols isopropanol (31), cyclohexanol (32), and 1-phenylethanol (33) reacted efficiently as well. Sterically encumbered secondary (34, 35) and tertiary (36) alcohols resulted in low amounts of the desired ether products within 48 h. Formation of diaryl ethers from the reaction of aryl bromides and phenols was not observed, presumably due to the low nucleophilicity of aromatic alcohols.
Coupling of methyl 4-bromobenzoate with water as nucleophile gave phenol 2 in moderate isolated yield by switching to DMF as a solvent (for details, see Table S12). The ortho-substituted, electron-rich aryl bromide 2-(2-bromophenyl)ethanol did undergo an intramolecular C–O coupling, resulting in 2,3-dihydrobenzofurane (37).34 An analogous preparation of chromane and 1,4-benzodioxane was not feasible (see Table S11). The reason for this remains unclear, especially because chromanes were previously synthesized by reductive elimination from the corresponding nickel(II) oxametallacycles.14 The semiheterogeneous dual catalytic reaction of 1,4-dibromobenzene and isopropylideneglycerol afforded 38, a potential intermediate for the preparation of ketoconazole (Figure 1), itraconazole, terconazole, and their derivatives.35 The antidepressant Fluoxetine can be synthesized using this method. N-Protected 3-methylamino-1-phenylpropanol reacted with 1-bromo-4-(trifluoromethyl)benzene resulting in N-acetyl fluoxetine (39) in 66%.
The same catalytic system was evaluated for the coupling of thiols with aryl halides (Scheme 2).36−40 The reaction of aryl bromides and thiols usually requires strongly reducing photoredox catalysts,39 whereas aryl iodides can be successfully coupled using weaker reductants.38 When the optimized semiheterogeneous protocol (the conduction band minimum of CN-OA-m was reported to be at −1.6 V vs Ag/AgCl33) was applied on the reaction of methyl 4-bromobenzoate with methyl 3-mercaptopropionate, only 4% of the desired thioether were formed after 48 h (Table S15). The analogous reaction using methyl 4-iodobenzoate went to completion within 48 h resulting in 79% isolated yield of the desired thioether (40). When 2-mercaptoethanol was used, a selective C–S bond formation (41), with no detectable amount of the corresponding etherification product was obtained. In contrast to the C–O coupling, the semiheterogeneous C–S bond formation is not limited to primary and secondary thiols (40–42). Tertiary (43) and aromatic thiols (44, 45) also gave the desired thioethers in moderate isolated yields. This finding can be rationalized by the formation of highly reactive thiyl radicals, which are proposed to add to Ni(I) intermediates in the dual catalytic thioether synthesis.38,39
Scheme 2. Scope of Semiheterogeneous Thioetherification.

Reaction conditions: methyl 4-iodobenzoate (1.2 mmol), thiol (2.4 mmol), CN-OA-m (20 mg), NiBr2·3H2O (120 μmol), dtbbpy (120 μmol), BIPA (6.0 mmol), MeCN (6.0 mL), white LEDs at 40 °C.
The reaction rate was significantly increased by using blue LEDs with higher light intensity (for details, see Supporting Information). The coupling of methyl 4-bromobenzoate and methanol, for example, was complete after 16 h instead of 24 using a modified setup. These intensified conditions were used for studying the recyclability of the heterogeneous carbon nitride material (Figure 2). CN-OA-m was recycled six times, without losing its catalytic activity (Figure 2), proofing its high potential for sustainable photocatalysis.
Figure 2.

Recycling of CN-OA-m in the semiheterogeneous etherification.
Analysis of the recycled photocatalyst showed Ni deposition (see the Supporting Information). Attempts to use the recycled photocatalyst in absence of additional Ni(II) salts showed that the immobilized Ni species is not catalytically active in the model reaction.
In conclusion, a dual Ni/photocatalytic C–O coupling was developed using a carbon nitride semiconductor as recyclable photocatalyst with low toxicity. The semiheterogeneous nickel/carbon nitride catalysis is an inexpensive, sustainable alternative to homogeneous protocols. The method selectively couples a broad range of electron-poor aryl bromides with primary and secondary alcohols as well as water in good to excellent isolated yields. The application of this protocol for intramolecular reactions and API synthesis was demonstrated. The same catalytic protocol can be utilized to couple aryl iodides with thiols.
Acknowledgments
We gratefully acknowledge the Max-Planck Society for generous financial support. B.P. acknowledges financial support by a Liebig Fellowship of the German Chemical Industry Fund (Fonds der Chemischen Industrie, FCI). A.M. and B.P. acknowledge the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy–EXC 2008/1 (UniSysCat)–390540038 for financial support. We thank our colleagues Xuan Pham (FU Berlin) and Katharina ten Brummelhuis (MPIKG) for analytical support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.9b01957.
Experimental procedures and characterization data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
A version of this research was previously posted to ChemRxiv: Cavedon, Cristian; Madani, Amiera; Seeberger, Peter H.; Pieber, Bartholomäus (2019): Semi-Heterogeneous Dual Nickel/Photocatalytic (Thio)Etherification using Carbon Nitrides. ChemRxiv. Preprint. June 6, 2019. https://doi.org/10.26434/chemrxiv.8231144.v1
Supplementary Material
References
- Evano G.; Wang J.; Nitelet A. Org. Chem. Front. 2017, 4, 2480–2499. 10.1039/C7QO00671C. [DOI] [Google Scholar]
- McGrath N. A.; Brichacek M.; Njardarson J. T. J. Chem. Educ. 2010, 87, 1348–1349. 10.1021/ed1003806. [DOI] [Google Scholar]
- Bhunia S.; Pawar G. G.; Kumar S. V.; Jiang Y.; Ma D. Angew. Chem., Int. Ed. 2017, 56, 16136–16179. 10.1002/anie.201701690. [DOI] [PubMed] [Google Scholar]
- Monnier F.; Taillefer M. Angew. Chem., Int. Ed. 2009, 48, 6954–6971. 10.1002/anie.200804497. [DOI] [PubMed] [Google Scholar]
- Allen S. E.; Walvoord R. R.; Padilla-Salinas R.; Kozlowski M. C. Chem. Rev. 2013, 113, 6234–6458. 10.1021/cr300527g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enthaler S.; Company A. Chem. Soc. Rev. 2011, 40, 4912–4924. 10.1039/c1cs15085e. [DOI] [PubMed] [Google Scholar]
- Schlummer B.; Scholz U. Adv. Synth. Catal. 2004, 346, 1599–1626. 10.1002/adsc.200404216. [DOI] [Google Scholar]
- Zhang H.; Ruiz-Castillo P.; Buchwald S. L. Org. Lett. 2018, 20, 1580–1583. 10.1021/acs.orglett.8b00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Chem. Rev. 2011, 111, 1346–1416. 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker S. Z.; Standley E. A.; Jamison T. F. Nature 2014, 509, 299. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga P. T.; Mavropoulos J. C.; Hillhouse G. L. Polyhedron 1995, 14, 175–185. 10.1016/0277-5387(94)00330-H. [DOI] [Google Scholar]
- Koo K.; Hillhouse G. L. Organometallics 1995, 14, 4421–4423. 10.1021/om00009a054. [DOI] [Google Scholar]
- Matsunaga P. T.; Hillhouse G. L.; Rheingold A. L. J. Am. Chem. Soc. 1993, 115, 2075–2077. 10.1021/ja00058a085. [DOI] [Google Scholar]
- Han R.; Hillhouse G. L. J. Am. Chem. Soc. 1997, 119, 8135–8136. 10.1021/ja9714999. [DOI] [Google Scholar]
- Twilton J.; Le C.; Zhang P.; Shaw M. H.; Evans R. W.; MacMillan D. W. C. Nat. Rev. Chem. 2017, 1, 0052. 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- Skubi K. L.; Blum T. R.; Yoon T. P. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan J. A.; Phelan J. P.; Badir S. O.; Molander G. A. Angew. Chem., Int. Ed. 2019, 58, 6152–6163. 10.1002/anie.201809431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrett J. A.; Cuthbertson J. D.; Shurtleff V. W.; MacMillan D. W. C. Nature 2015, 524, 330–334. 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B.; Yan L.-K.; Geng Y.; Ren H.; Guan W.; Su Z.-M. Chem. Commun. 2018, 54, 5968–5971. 10.1039/C8CC03550D. [DOI] [PubMed] [Google Scholar]
- Yang L.; Huang Z.; Li G.; Zhang W.; Cao R.; Wang C.; Xiao J.; Xue D. Angew. Chem., Int. Ed. 2018, 57, 1968–1972. 10.1002/anie.201710698. [DOI] [PubMed] [Google Scholar]
- Yang L.; Simionescu R.; Lough A.; Yan H. Dyes Pigm. 2011, 91, 264–267. 10.1016/j.dyepig.2011.03.027. [DOI] [Google Scholar]
- Savateev A.; Ghosh I.; König B.; Antonietti M. Angew. Chem., Int. Ed. 2018, 57, 15936–15947. 10.1002/anie.201802472. [DOI] [PubMed] [Google Scholar]
- Lang X.; Chen X.; Zhao J. Chem. Soc. Rev. 2014, 43, 473–486. 10.1039/C3CS60188A. [DOI] [PubMed] [Google Scholar]
- Chen J.; Cen J.; Xu X.; Li X. Catal. Sci. Technol. 2016, 6, 349–362. 10.1039/C5CY01289A. [DOI] [Google Scholar]
- Friedmann D.; Hakki A.; Kim H.; Choi W.; Bahnemann D. Green Chem. 2016, 18, 5391–5411. 10.1039/C6GC01582D. [DOI] [Google Scholar]
- Caputo J. A.; Frenette L. C.; Zhao N.; Sowers K. L.; Krauss T. D.; Weix D. J. J. Am. Chem. Soc. 2017, 139, 4250–4253. 10.1021/jacs.6b13379. [DOI] [PubMed] [Google Scholar]
- Liu Y.-Y.; Liang D.; Lu L.-Q.; Xiao W.-J. Chem. Commun. 2019, 55, 4853–4856. 10.1039/C9CC00987F. [DOI] [PubMed] [Google Scholar]
- Godt J.; Scheidig F.; Grosse-Siestrup C.; Esche V.; Brandenburg P.; Reich A.; Groneberg D. A. J. Occup. Med. Toxicol. 2006, 1, 22–22. 10.1186/1745-6673-1-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Directive 2011/65/EU on the restriction of the use of certain hazardous substances in electrical and electronic equipment. 2011,Official Journal of the European Union, L174, 88–110. [Google Scholar]
- Dong Q.; Mohamad Latiff N.; Mazánek V.; Rosli N. F.; Chia H. L.; Sofer Z.; Pumera M. ACS Appl. Nano Mater. 2018, 1, 4442–4449. 10.1021/acsanm.8b00708. [DOI] [Google Scholar]
- Pieber B.; Malik J. A.; Cavedon C.; Gisbertz S.; Savateev A.; Cruz D.; Heil T.; Zhang G.; Seeberger P. H. Angew. Chem., Int. Ed. 2019, 10.1002/anie.201902785. [DOI] [PubMed] [Google Scholar]
- The etherification was proposed to proceed via SET oxidation/reduction of Ni intermediates (ref (18)). However, a photosensitization pathway cannot be excluded:Welin E. R.; Le C.; Arias-Rotondo D. M.; McCusker J. K.; MacMillan D. W. Science 2017, 355, 380–385. 10.1126/science.aal2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Li G.; Lan Z.-A.; Lin L.; Savateev A.; Heil T.; Zafeiratos S.; Wang X.; Antonietti M. Angew. Chem. 2017, 129, 13630–13634. 10.1002/ange.201706870. [DOI] [PubMed] [Google Scholar]
- During the preparation of this manuscript, an intramolecular dual nickel/photoredox C–O bond formation was described:Lee H.; Boyer N. C.; Deng Q.; Kim H.-Y.; Sawyer T. K.; Sciammetta N. Chem. Sci. 2019, 10, 5073–5078. 10.1039/C9SC00694J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Liu Z.; Cao X.; Liu X.; He H.; Yang Y. Bioorg. Med. Chem. Lett. 2011, 21, 4779–4783. 10.1016/j.bmcl.2011.06.062. [DOI] [PubMed] [Google Scholar]
- Marzo L.; Pagire S. K.; Reiser O.; König B. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. 10.1002/anie.201709766. [DOI] [PubMed] [Google Scholar]
- Jouffroy M.; Kelly C. B.; Molander G. A. Org. Lett. 2016, 18, 876–879. 10.1021/acs.orglett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oderinde M. S.; Frenette M.; Robbins D. W.; Aquila B.; Johannes J. W. J. Am. Chem. Soc. 2016, 138, 1760–1763. 10.1021/jacs.5b11244. [DOI] [PubMed] [Google Scholar]
- Du Y.; Pearson R. M.; Lim C.-H.; Sartor S. M.; Ryan M. D.; Yang H.; Damrauer N. H.; Miyake G. M. Chem. - Eur. J. 2017, 23, 10962–10968. 10.1002/chem.201702926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santandrea J.; Minozzi C.; Cruché C.; Collins S. K. Angew. Chem., Int. Ed. 2017, 56, 12255–12259. 10.1002/anie.201705903. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.