

Abstract

The development of bifunctional imaging probes can often be challenging with difficult and time-consuming solution phase chemistry protocols and purification techniques. A solid phase synthetic protocol was therefore utilized to produce a functionalized derivative of a potent bismacrocyclic calcium-responsive contrast agent for magnetic resonance imaging. Through a convenient building block approach, the applicability of this methodology in the preparation and simple future development of multifunctional imaging probes was demonstrated.
Bifunctional chelators based on polyazamacrocycles are often utilized in the development of imaging probes.1 The macrocyclic component acts as a metal chelator while the second function can be exploited for further synthetic modifications, conjugation to biomolecules, targeting, or the detection of specific analytes. As such, they are heavily used in various molecular imaging techniques, such as positron emission tomography (PET), single photon emission computed tomography (SPECT), or magnetic resonance imaging (MRI). Among these, MRI is a preferred technique utilized to obtain three-dimensional anatomical images of soft tissue with excellent spatial resolution. Moreover, the use of MRI contrast agents increases its specificity, while expanding the scope and utilization potential. For instance, its functional variant, which aims to observe processes on the molecular level (molecular fMRI), has progressively gained in importance lately,2 as its application aids the visualization of biochemical events in a dynamic manner. Subsequently, understanding various biological and pathological states becomes a challenging but highly rewarding task using this technique and the appropriate biomarkers. Thus, the application of bioresponsive or “smart” contrast agents (SCAs) in the development of functional MRI has become an increasingly investigated research area over recent years.3−5 In order to observe alterations that are coupled to the execution of particular biological processes, dedicated SCAs are employed, as they are capable of displaying a change in their MR behavior upon a physical or chemical change in their microenvironment. To achieve this, SCAs, which mostly consist of bifunctional systems, have been developed to detect enzymatic activity, pH changes, or various metal ion concentrations. However, the development of such systems includes arduous synthetic procedures. Moreover, limitations in synthetic modifications or further addition of functionalities to provide more diverse derivatives frequently lead to complications and challenges that cause limitations in structural diversity, hence the broader utilization of SCAs.
To this end, solid-phase synthetic (SPS) methods have been frequently used as an alternative to conventional solution-phase techniques, eventually finding its application in the development of MRI probes.6−11 Introduced by Bruce Merrifield in the 1960s, SPS has been typically used in the area of peptide synthesis, as it allows for the building of a molecule in a stepwise fashion on an insoluble solid support.12−14
Significantly, the use of an insoluble solid support gave significant advantages over standard solution-phase chemistry techniques,15 specifically: (i) the ability to drive reactions to completion through the use of excess reagents; (ii) the minimization of physical losses; (iii) quick washing steps after each reaction instead of tedious solution-phase workups and purification; (iv) to allow for the synthesis of more complex molecules and (v) the possibility to fully automate the synthetic procedure.
Such advantages could be useful for circumventing tedious and demanding preparation procedures of MRI chelates, as mentioned above. Yet, the potential of SPS has not been fully exploited in this field, especially on the preparation of SCAs. Being bifunctional chelators by default, SCAs are very suitable targets for convenient techniques such as SPS. Moreover, the establishment of a reliable SPS methodology to produce SCAs can enable additional modifications toward the incorporation of further functionalities (e.g., targeting vector, second probe suitable for additional imaging modality, etc.), thus expanding the scope of SCA use.
Hence, aiming to access more diverse bioresponsive probes, we embarked on developing an SPS synthetic protocol which would enable the production of such probes in a simple and convenient manner. Of particular importance was the preparation of a bismacrocyclic SCA, which generates a strong response in the presence of Ca2+, and in cellular model systems.16,17 The previously developed solution-phase procedure allowed for its convenient preparation; however, the symmetric nature did not allow for the incorporation of additional functionalities without damaging the response toward Ca2+.
We have therefore established a novel SPS approach based on the Fmoc-SPPS strategy15 to develop the desired multifunctional bismacrocyclic probe. The approach consisted of five stages (Figure 1): (1) the synthesis of a peptide unit capable of providing an orthogonal functionality; (2–4) the building of the bismacrocylic component with a series of smaller building blocks (BB1–BB3, Figure 2); (5) the incorporation of a functional molecule to the existing SCA. Beside the commercially available amino acids necessary for step 1, the exploration of this SPS methodology required the preparation of specific building blocks in solution prior to their introduction in steps 2–4.
Figure 1.

Schematic representation of the synthetic approach followed in this study.
Figure 2.

Chemical structures of the building blocks used.
The building blocks BB1–3 were selected as the best candidates for the assembly of the bismacrocyclic system. Importantly, these BBs included protecting groups that could be selectively deprotected and were compatible with the resin used. Furthermore, through the use of BB2 with the Fmoc protecting group, we were able to employ a strategy of alkylation in the final building block assembly step, which is a rather unconventional approach to SPS (vide infra).
BB1 and BB2 were prepared in a few straightforward synthetic steps (Scheme 1), whereas BB3 was synthesized in accordance with a reported literature procedure.16 The starting material for the synthesis of BB1 was secondary amine 1 (Scheme 1A). Its N-alkylation with N-(3-bromopropyl)phthalamide gave 2, which was reduced to give the BB1. Hence, this approach resulted in a DO2A-based macrocyclic component with one functional group (an aromatic amine) available for coupling to the peptide scaffold and another (phthalamide-protected amine) which can be selectively deprotected and used for further synthetic transformations.
Scheme 1. Synthetic Route towards Building Blocks BB1 and BB2.

(A) (i) N-(3-Bromopropyl)phthalamide, K2CO3, MeCN, 16 h, 70 °C, 66%; (ii) H2, Pd/C (20%wt), EtOH, rt, 78%. (B) (i) 2-Phenylpropan-2-yl 2-bromoacetate, DCC, DMAP, MeCN, 2 days, rt, 44%; (ii) FmocCl, Na2CO3, dioxane/water, 16 h, rt, 70%; (iii) 3% TFA/CH2Cl2, 1 h, rt, quant.
The design of the Ca2+ chelator BB2, derived from ethylene glycol tetraacetic acid (EGTA), required differentiation from the “standard” derivatives routinely used in our group. Typically, the EGTA-derived block used for the synthesis of this type of SCAs contains benzyl esters as orthogonal protecting groups, which are incompatible with SPS due to the use of a heterogeneous catalyst under a reducing hydrogen atmosphere. Thus, the alternative derivative bears an Fmoc protecting group and a phenyl isopropanol ester. The conditions of deprotection for this acid labile ester were orthogonal to that of Fmoc, tert-butyl esters, and the resin.
The synthesis of BB2 started via the monoalkylation of secondary diamine 3 with 2-phenylpropan-2-yl 2-bromoacetate in acetonitrile to give 4 (Scheme 1B). Acylation of 4 with FmocCl afforded 5. To yield the desired BB2, cleavage of the phenylisopropanol ester was carried out in a solution of 3% TFA in dichloromethane.
Once the BBs were prepared, the assembly of the multifunctional SCA was conducted using standard conjugation procedures (Scheme 2).15 Initially, we prepared the peptide sequence Lys-Gly-Gly between the rink-amide resin and the rest of the SCA to allow for the inclusion of an additional functional molecule at the final stage of conjugate preparation (vide infra). Consequently, Lys bearing a 4-methyltrityl (Mtt) orthogonal protection of the ω-NH2 was used as the first amino acid in the SPS protocol.
Scheme 2. Synthesis of Gd2L.

(i) BB1, HATU, DIPEA, DMF; (ii) ethylendiamine, iPrOH; (iii) BB2, HATU, DIPEA, DMF; (iv) 40% piperidine/DMF; (v) BB3, DIPEA, DMF; (vi) TFA/triisopropylsilane/CH2Cl2 (3:3:94), 4 × 2 min; (vii) biotin, HATU, HOBt, DIPEA, DMF; (viii) TFA/triisopropylsilane/H2O (95:2.5:2.5); (ix) GdCl3·6H2O, pH 7.
Furthermore, reaction of the terminal amine of the Lys-Gly-Gly backbone with succinic anhydride provided 6, which acted as a spacer unit and as a method to convert the N-terminal functionality to a carboxylic acid suitable for conjugation to BB1. Using the more reactive HATU coupling agent, the coupling of BB1 to the peptidyl resin was achieved to give 7. Here, phthalimide deprotection of 7 was carried out with 10 equiv of ethylene diamine in isopropanol at room temperature to yield amine 8. Interestingly, phthalimide is an amino protecting group that is not frequently employed in SPS. If utilized, the most popular deprotection conditions involve hydrazine.18 On the other hand, ethylene diamine is one attractive alternative due to its less harsh nature, increased reactivity, and overall safer use.19 Procedures describing this method typically employ an excess of ethylene diamine in butanol or isopropanol at reflux.19,20 Here we demonstrate the use of ethylene diamine as an efficient reagent for phthalimide deprotection on solid phase at room temperature, proceeding smoothly to provide conversion to the amino function. Importantly, the mild deprotection conditions described here can allow for increased flexibility in terms of future preparations involving hydrazine sensitive protecting groups.
Conjugation of amine 8 to BB2 was performed using HATU to afford 9. Subsequent Fmoc deprotection of 9 was carried out with 40% piperidine in DMF. The resulting amine was N-alkylated with BB3 to give 10. In total, 4.8 equiv of BB3 were added in two separate portions to drive the reaction to completion. Finally, with the SCA assembled on the resin, the incorporation of a functional molecule could be carried out. For this proof of concept study, we selected biotin, as its interaction with avidin is well characterized.21 However, in principle this site could be reserved for a number of various functional molecules such as antibodies, targeting moieties, or functional dyes depending on the desired application for the specific SCA.
Prior to biotin coupling, the orthogonal Mtt protecting group of lysine was removed with a solution of TFA/triisopropylsilane/dichloromethane (3:3:94) to afford the primary amine. This was then coupled to biotin with HATU yielding 11. Cleavage of the final biotinylated-SCA, 11, from the resin was achieved with the cleavage cocktail TFA/triisopropylsilane/H2O (95:2.5:2.5) to yield ligand L, which was purified by reversed-phase HPLC. Subsequent complexation with Gd3+ was carried out in H2O at room temperature and neutral pH to afford the final SCA Gd2L with an overall yield of 24% across the whole synthetic procedure.
To assess the potency of the multifunctional SCA toward Ca2+, its relaxometric characterization was performed with proton longitudinal and transverse relaxometric titrations. The response observed for Gd2L was excellent and even higher than the previously reported bismacrocyclic analogue.16 The relaxometric titrations show an increase of longitudinal r1 relaxivity upon the addition of Ca2+ from 3.47 to 7.58 mM–1 s–1, which is almost a 118% enhancement relative to the initial value in the absence of Ca2+. Concurrently, the transverse r2 relaxivity increased from 5.40 to 13.57 mM–1 s–1, or around 150% (Figure 3a). The obtained results indicate that the effective response of the SCA has been retained despite the change in molecular design. Moreover, the intrinsic property of Gd2L to trigger concurrent and massive changes on two paramagnetic Gd3+ ions upon interaction with a single Ca2+ ion is very advantageous, and could now be more frequently exploited with this new preparation methodology.
Figure 3.
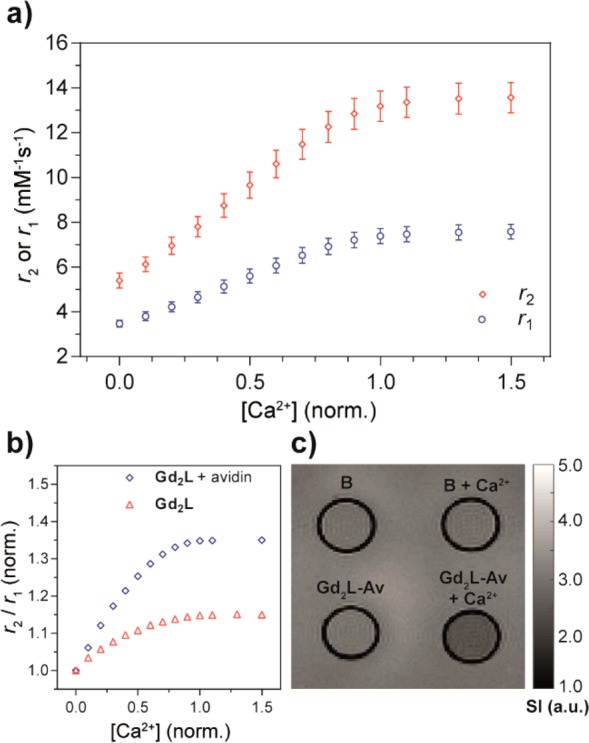
Relaxometric and MRI phantom experiments for Gd2L and Gd2L + avidin with Ca2+ ([Gd3+] = 1 mM, pH 7.4, 50 mM HEPES, 25 °C, 7 T). (a) Relaxometric titration of Gd2L with Ca2+; the r1 and r2 values are represented as means ± SD of three independent measurements. (b) Ratio of r2 over r1 from relaxometric titrations of Gd2L and Gd2L + avidin with Ca2+. (c) T2/T1 ratiometric MR phantom images of the buffer (B) or Gd2L + avidin (Gd2L-Av) sample tubes in presence and absence of Ca2+ (1 equiv).
With the inclusion of biotin as a functional moiety, we also proceeded to demonstrate its binding with avidin. The number of biotin binding sites available for Gd2L was determined through an HABA assay revealing three sites (Figure S1 in Supporting Information).22 Following this assessment, 3 equiv of Gd2L were added to avidin in HEPES buffer (50 mM) and incubated for 2 h. Relaxometric titrations and T2/T1-weighted ratiometric MR images were recorded showing an increase in r1 and r2 upon Ca2+ addition and consequently a change in MRI signal (Figures 3b,c and S2 in Supporting Information). The enhancement in r1 was lower than on Gd2L alone, which was previously observed after the attachment of “small-size” SCAs to nanosized systems.23 Furthermore, the unexplored interactions of Gd2L with the protein surface could also play a role. On the other hand, the values recorded for r2 were typical for nanosized-type SCAs, indicating the possibility to use the Gd2L avidin conjugate in combination with the fast T2/T1-weighted imaging protocol.24
In summary, we have developed a solid-phase synthetic protocol for the preparation of functionalized and bioresponsive bismacrocyclic SCAs. We diversified our building block synthesis to incorporate SPS-friendly functional groups such as phthalimide, which we successfully deprotected using mild conditions. Furthermore, through the use of a peptide backbone, significant flexibility can be achieved in terms of designing a molecule which can be highly specific, biocompatible and possess a range of functional molecules. The results described here offer an exciting range of possibilities for the development of various bifunctional bioresponsive probes ranging from multifunctional molecules to multimeric species.
Acknowledgments
The authors thank Ms. Vera F. C. Ferreira for assistance in initial SPS procedures and Mrs. Tanja Savić for performing MRI phantom experiments. The financial support of the German Research Foundation (DFG, grant AN 716/7-1) and COST TD1004 Action for the short term scientific mission of L.C. is gratefully acknowledged. J.D.G.C. thanks the Fundação para a Ciência e Tecnologia, Portugal, for financial support through Projects UID/Multi/04349/2019 and PTDC/QUI-NUC/30147/2017.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.9b01341.
Synthetic procedures for preparation of BB1, BB2, and Gd2L; purification procedure for Gd2L; HABA assay; additional relaxometric titration experiments; parameters for MRI phantom measurements; NMR and HR mass spectra; LC-MS and HPLC data (PDF)
Author Present Address
∥ Department of Chemical Science and Engineering, School of Engineering, Kathmandu University, Dhulikhel, Nepal.
The authors declare no competing financial interest.
Supplementary Material
References
- Lattuada L.; Barge A.; Cravotto G.; Giovenzana G. B.; Tei L. The synthesis and application of polyamino polycarboxylic bifunctional chelating agents. Chem. Soc. Rev. 2011, 40, 3019–3049. 10.1039/c0cs00199f. [DOI] [PubMed] [Google Scholar]
- Bartelle B. B.; Barandov A.; Jasanoff A. Molecular fMRI. J. Neurosci. 2016, 36, 4139–4148. 10.1523/JNEUROSCI.4050-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffern M. C.; Matosziuk L. M.; Meade T. J. Lanthanide Probes for Bioresponsive Imaging. Chem. Rev. 2014, 114, 4496–4539. 10.1021/cr400477t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelovski G. What We Can Really Do with Bioresponsive MRI Contrast Agents. Angew. Chem., Int. Ed. 2016, 55, 7038–7046. 10.1002/anie.201510956. [DOI] [PubMed] [Google Scholar]
- Carril M. Activatable probes for diagnosis and biomarker detection by MRI. J. Mater. Chem. B 2017, 5, 4332–4347. 10.1039/C7TB00093F. [DOI] [PubMed] [Google Scholar]
- Yoo B.; Sheth V. R.; Pagel M. D. An amine-derivatized, DOTA-loaded polymeric support for Fmoc solid phase peptide synthesis. Tetrahedron Lett. 2009, 50, 4459–4462. 10.1016/j.tetlet.2009.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo B.; Pagel M. D. Peptidyl molecular imaging contrast agents using a new solid-phase peptide synthesis approach. Bioconjugate Chem. 2007, 18, 903–911. 10.1021/bc060250q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripepi M.; Capuana F.; Gianolio E.; Kock F. V. C.; Pagoto A.; Stefania R.; Digilio G.; Aime S. Synthesis of High Relaxivity Gadolinium AAZTA Tetramers as Building Blocks for Bioconjugation. Bioconjugate Chem. 2018, 29, 1428–1437. 10.1021/acs.bioconjchem.8b00120. [DOI] [PubMed] [Google Scholar]
- Jayapaul J.; Schroder L. Complete Generation of a 129Xe Biosensor on the Solid Support by Systematic Backbone Assembly. Bioconjugate Chem. 2018, 29, 4004–4011. 10.1021/acs.bioconjchem.8b00814. [DOI] [PubMed] [Google Scholar]
- Singh J.; Rustagi V.; Zhang S. R.; Sherry A. D.; Udugamasooriya D. G. On-bead combinatorial synthesis and imaging of europium(III)-based paraCEST agents aids in identification of chemical features that enhance CEST sensitivity. Magn. Reson. Chem. 2017, 55, 747–753. 10.1002/mrc.4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano R.; Soesbe T. C.; De Leon-Rodriguez L. M.; Sherry A. D.; Udugamasooriya D. G. On-Bead Combinatorial Synthesis and Imaging of Chemical Exchange Saturation Transfer Magnetic Resonance Imaging Agents To Identify Factors That Influence Water Exchange. J. Am. Chem. Soc. 2011, 133, 13023–13030. 10.1021/ja201123f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield R. B. Solid Phase Peptide Synthesis. I. Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. 10.1021/ja00897a025. [DOI] [Google Scholar]
- Jaradat D. M. M. Thirteen decades of peptide synthesis: key developments in solid phase peptide synthesis and amide bond formation utilized in peptide ligation. Amino Acids 2018, 50, 39–68. 10.1007/s00726-017-2516-0. [DOI] [PubMed] [Google Scholar]
- Merrifield R. B. Solid-Phase Synthesis (Nobel Lecture). Angew. Chem., Int. Ed. Engl. 1985, 24, 799–810. 10.1002/anie.198507993. [DOI] [Google Scholar]
- Chan W. C.; White P. D.. Fmoc solid phase peptide synthesis: a practical approach; Oxford University Press: Oxford, 2000. [Google Scholar]
- Angelovski G.; Fouskova P.; Mamedov I.; Canals S.; Toth E.; Logothetis N. K. Smart magnetic resonance imaging agents that sense extracellular calcium fluctuations. ChemBioChem 2008, 9, 1729–1734. 10.1002/cbic.200800165. [DOI] [PubMed] [Google Scholar]
- Angelovski G.; Gottschalk S.; Milošević M.; Engelmann J.; Hagberg G. E.; Kadjane P.; Andjus P.; Logothetis N. K. Investigation of a Calcium-Responsive Contrast Agent in Cellular Model Systems: Feasibility for Use as a Smart Molecular Probe in Functional MRI. ACS Chem. Neurosci. 2014, 5, 360–369. 10.1021/cn500049n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cros E.; Planas M.; Barany G.; Bardaji E. N-tetrachlorophthaloyl (TCP) protection for solid-phase peptide synthesis. Eur. J. Org. Chem. 2004, 2004, 3633–3642. 10.1002/ejoc.200400244. [DOI] [Google Scholar]
- Murai N.; Miyano M.; Yonaga M.; Tanaka K. One-Pot Primary Aminomethylation of Aryl and Heteroaryl Halides with Sodium Phthalimidomethyltrifluoroborate. Org. Lett. 2012, 14, 2818–2821. 10.1021/ol301037s. [DOI] [PubMed] [Google Scholar]
- Kanie O.; Crawley S. C.; Palcic M. M.; Hindsgaul O. Acceptor-substrate recognition by N-acetylglucosaminyltransferase-V: Critical role of the 4″-hydroxyl group in β-D-GlcpNAc-(1 → 2)-α-D-Manp(1 → 6)-β-D-Glcp-OR. Carbohydr. Res. 1993, 243, 139–164. 10.1016/0008-6215(93)84087-M. [DOI] [PubMed] [Google Scholar]
- Wilchek M.; Bayer E. A. Introduction to Avidin-Biotin Technology. Methods Enzymol. 1990, 184, 5–13. 10.1016/0076-6879(90)84256-G. [DOI] [PubMed] [Google Scholar]
- Green N. M.[74] Spectrophotometric determination of avidin and biotin. Methods in Enzymology; Academic Press: 1970; Vol. 18, pp 418–424. [Google Scholar]
- Gündüz S.; Nitta N.; Vibhute S.; Shibata S.; Mayer M. E.; Logothetis N. K.; Aoki I.; Angelovski G. Dendrimeric calcium-responsive MRI contrast agents with slow in vivo diffusion. Chem. Commun. 2015, 51, 2782–2785. 10.1039/C4CC07540D. [DOI] [PubMed] [Google Scholar]
- Gündüz S.; Savić T.; Pohmann R.; Logothetis N. K.; Scheffler K.; Angelovski G. Ratiometric Method for Rapid Monitoring of Biological Processes Using Bioresponsive MRI Contrast Agents. ACS Sens. 2016, 1, 483–487. 10.1021/acssensors.6b00011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.