

SUMMARY
Anti-CRISPR proteins Acrs targeting CRISPR-Cas9 systems represent natural “off-switches” for Cas9-based applications. Recently, AcrIIC1, AcrIIC2, and AcrIIC3 proteins were found to inhibit Neisseria meningitidis Cas9 (NmeCas9) activity in bacterial and human cells. Here we report biochemical and structural data that suggest molecular mechanisms of AcrIIC2- and AcrIIC3-mediated Cas9 inhibition. AcrIIC2 dimer interacts with the bridge helix of Cas9, interferes with RNA binding, and prevents DNA loading into Cas9. AcrIIC3 blocks the DNA loading step through binding to a non-conserved surface of the HNH domain of Cas9. AcrIIC3 also forms additional interactions with the REC lobe of Cas9 and induces the dimerization of the AcrIIC3-Cas9 complex. While AcrIIC2 targets Cas9 orthologs from different subtypes albeit with different efficiency, AcrIIC3 specifically inhibits NmeCas9. Structure-guided changes in NmeCas9 orthologs convert them into anti-CRISPR-sensitive proteins. Our studies provide insights into anti-CRISPR-mediated suppression mechanisms and guidelines for designing regulatory tools in Cas9-based applications.
Graphical Abstract
eTOC Blurb
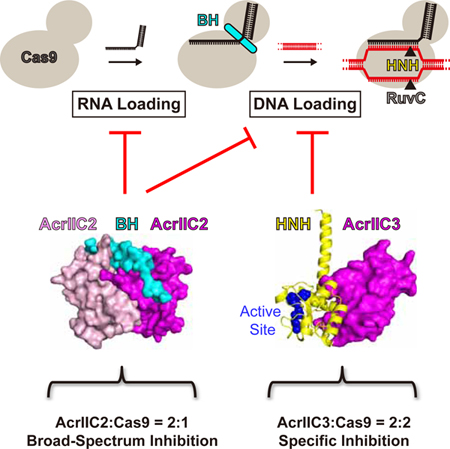
Zhu et al. report biochemical and structural data that suggest molecular mechanisms of AcrIIC2-and AcrIIC3-mediated inhibition of Cas9. The two inhibitors employ distinct means to block Cas9 activity that include binding to different regions, targeting distinct steps of catalysis, and inhibiting different scopes of Cas9 orthologs.
INTRODUCTION
The evolutionary arms race between bacteria and phages has led to evolving sophisticated antiphage defense systems in bacterial cells. Unique among them are the CRISPR-Cas systems, which provide bacteria with adaptive immunity against foreign nucleic acids (van der Oost et al., 2014). According to the updated phylogenetic classification, CRISPR-Cas systems are grouped into two classes, six types, and more than 20 subtypes (Koonin et al., 2017). Class 2 systems (comprising type II, V, and VI subtypes) represent the streamlined versions that require only a single protein to target and cleave foreign nucleic acids (Koonin et al., 2017; van der Oost et al., 2014). Notably, the type II CRISPR-Cas9 system, including subtypes IIA, IIB, and IIC, has been widely adapted for genome editing and other biotechnological applications (Hsu et al., 2014; Wang et al., 2016a). The cleavage activity of Cas9 requires either a pair of RNA molecules, namely crRNA (CRISPR-derived RNA) and tracrRNA (trans-activating crRNA), or a synthetic single-guide RNA (sgRNA) covalently linking the 3’ end of crRNA to the 5’ end of tracrRNA (Deltcheva et al., 2011; Jinek et al., 2012).
In response to development of CRISPR-Cas systems, phages have evolved anti-CRISPR proteins (Acrs) that directly bind to and inactivate CRISPR-Cas machinery (Maxwell, 2017). Recent studies have shown broad distribution of Acrs and suggested their critical role in the evolution of CRISPR-Cas systems (Gophna et al., 2015; van Houte et al., 2016). More than 30 unique Acr families have been described against type I (Bondy-Denomy et al., 2013; Marino et al., 2018; Pawluk et al., 2014; Pawluk et al., 2016b), type II (Hynes et al., 2017; Pawluk et al., 2016a; Rauch et al., 2017), and type V (Doron et al., 2018; Marino et al., 2018) CRISPR-Cas systems. Specifically, three Acrs (AcrIIC1, 2, and 3) that inhibit the type IIC Cas9 from Neisseria meningitidis (NmeCas9) have been identified along with five (AcrIIA1 through 5) that target select type IIA Cas9 orthologs. Given the extensive use of CRISPR-Cas9 in genome editing applications, the discovery of type II Acrs has provided the important prospect of introducing specific genetically encodable ‘‘off-switch” tools for modulating Cas9 activity. Acrs may also prove to be a useful addition to phage therapy protocols for treatment of bacterial infections.
Although the number of identified Acrs is quickly growing, the suppression mechanisms of only a few Acrs have been characterized in detail (Bondy-Denomy et al., 2015; Chowdhury et al., 2017; Dong et al., 2017; Guo et al., 2017; Harrington et al., 2017; Jiang et al., 2018; Liu et al., 2018; Peng et al., 2017; Shin et al., 2017; Wang et al., 2016b; Wang et al., 2016c; Yang and Patel, 2017). The complexity of the problem arises from the fact that Acrs can potentially inhibit several steps of CRSPR-Cas, including spacer acquisition, Cas protein expression, crRNA processing, crRNA assembly, target DNA binding, and target DNA cleavage. The CRISPR inhibition mechanisms determined in previous studies can be grouped into two general strategies aimed to disrupt DNA binding (AcrF1, AcrF2, AcrIIA2, AcrIIA4, and AcrIIC3) or inhibit target sequence cleavage (AcrF3 and AcrIIC1) (Maxwell, 2017).
The structural basis of inhibition of type II Acrs has been determined for AcrIIA2 (Jiang et al., 2018; Liu et al., 2018), AcrIIA4 (Dong et al., 2017; Shin et al., 2017; Yang and Patel, 2017), and AcrIIC1 (Harrington et al., 2017). Both AcrIIA2 (Jiang et al., 2018; Liu et al., 2018) and AcrIIA4 (Dong et al., 2017; Shin et al., 2017; Yang and Patel, 2017) binds to the Cas9-sgRNA complex and occupies the protospacer adjacent motif (PAM)-interacting site, thereby sterically blocking double-stranded DNA (dsDNA) binding. AcrIIC1 binds to the conserved HNH catalytic domain of Cas9 and inhibits DNA cleavage by trapping the complex in the sgRNA- and DNA-bound state (Harrington et al., 2017). Given distinct sequences of AcrIIC proteins, the published studies have not explained the suppression mechanisms employed by AcrIIC2 and AcrIIC3 while exposed differences from the reported ones (Harrington et al., 2017; Pawluk et al., 2016a).
Here, we investigate the mechanisms of inactivation of Cas9 by AcrIIC2 and AcrIIC3. The biochemical and structural data revealed that the two inhibitors employ distinct means to block Cas9 activity that include binding to various regions, targeting different steps of catalysis, and inhibiting different scopes of Cas9 orthologs. Therefore, our work provides critical molecular insights on the diverse mechanisms used by type II Acrs to disable Cas9 that will be essential for developing Cas9 modulators for biotechnological applications and understanding evolutionarily pressures that Acrs impose on CRISPR systems.
RESULTS
Mapping the Interacting Regions between NmeCas9 and AcrIIC2/3
To understand the molecular basis of Cas9 suppression by AcrIIC2 and AcrIIC3, we first determined which regions of Cas9 interact with the inhibitors. Instead of a thermostable NmeCas9 homolog from Geobacillus stearothermophilus (GeoCas9) utilized in the previous study for superior solubility of the protein constructs (Harrington et al., 2017), we used NmeCas9 to generate soluble truncated variants and tested their ability to bind AcrIIC2 and AcrIIC3 by size-exclusion chromatography (SEC) (Figures 1 and S1). In parallel, as a positive control, we conducted same experiments with the well-characterized AcrIIC1 known to bind to the isolated HNH domain of type IIC Cas9s (Harrington et al., 2017). Although the full-length NmeCas9 is able to interact with all three inhibitors, the variant lacking the HNH domain (∆HNH) can associate with AcrIIC2 but not with AcrIIC1 or AcrIIC3 (Figures 1A, 1B, and S1). Further SEC experiments with AcrIIC1 and AcrIIC3 showed that both proteins interact with the isolated HNH domain (Figures 1B–1D), suggesting that the primary binding region for AcrIIC3 is the same HNH domain that was shown earlier to bind AcrIIC1 (Harrington et al., 2017).
Figure 1. Mapping the interacting regions between NmeCas9 and AcrIIC1/2/3.

(A) NmeCas9 constructs used to map the interacting regions of NmeCas9 and AcrIIC1/2/3. NmeCas9 proteins were incubated with anti-CRISPR proteins, the mixtures were analyzed by size-exclusion chromatography (SEC) on Superdex 200 10/300 or Superdex 75 10/300 columns (void volumes 7.5 mL and 7.0 mL, respectively), and peak fractions were analyzed by SDS-PAGE. FL, full-length protein. (+) and (−) indicate Acr-Cas9 binding or no binding, respectively.
(B, E) SDS-PAGE of the peak fractions from SEC runs (Figures S1, 1C, 1D and 1F).
(C, D, F) Representative elution profiles of SEC runs on Superdex 75 10/300 column that show formation of Acr-Cas9 complexes. Black asterisks indicate the fractions analyzed in (B) and (E).
See also Figure S1.
To map the AcrIIC2 binding region, we tested binding of AcrIIC2 to additional truncations of NmeCas9 that omit either the recognition (REC) or nuclease (NUC) lobes. SEC indicated that interaction of AcrIIC2 with NmeCas9 requires the REC lobe (Figures 1E and S1C). To further delineate the interacting region, we split the REC lobe into its two respective domains and found that the bridge helix (BH)-REC1 region (aa 51–241) is sufficient for AcrIIC2 interaction (Figures 1E, 1F, and S1C).
AcrIIC2 Dimerization is Critical for NmeCas9 Inhibition
We noticed that AcrIIC2 is eluted significantly earlier than AcrIIC1 and AcrIIC3 on the size-exclusion column (Figure S2A), although the three proteins have similar molecular weights. Analytical ultracentrifugation revealed that AcrIIC2 forms a dimer (Figure S2B), in contrast to AcrIIC1 and AcrIIC3 (Figures S2C and S2D), which were previously shown to be monomeric (Harrington et al., 2017). To understand the structural basis of AcrIIC2 dimerization, we determined the crystal structure of AcrIIC2 at 2.2 Å resolution (Figures 2A, S2E and Table 1). Although there is only one AcrIIC2 molecule in the asymmetric unit, a dimer with extensive interface (2,064 Å2) is formed through the crystallographic symmetry (Figure 2A). AcrIIC2 adopts an α1β1–6α2 fold, with the C-terminal long α-helix inserted into the half-barrel structure formed by the six β-strands (Figure 2A). A Dali server search revealed that AcrIIC2 monomer shares some structural similarity with the tubular lipid binding proteins (Figure S2F) (Wong and Levine, 2017); however, the AcrIIC2 dimer represents a novel fold with no reported homologous structures. The dimerization of AcrIIC2 is mediated by α1, β1, β2, β3 and adjacent loops of both monomers through the formation of a compact hydrophobic core (Figures 2A and 2B). The intermolecular interactions are further strengthened by several hydrogen bonds (Figure S2G), including the antiparallel β-sheet formed by the β1 strand from each monomer. Mutations in the dimerization interface (L36D or Y11A/I15D/R20A) decrease the strength of intermolecular interactions between AcrIIC2 and NmeCas9 (Figure S2H) and diminish the inhibiting ability of AcrIIC2 (Figure 2C), highlighting the critical role of dimerization for the function of AcrIIC2. Remarkably, top and bottom faces of the AcrIIC2 dimer form surfaces with opposite electrostatic potential charges, with an “Acidic Groove” on top and an “Alkaline Platform” in the bottom (Figure S2I).
Figure 2. Structures of AcrIIC2 in apo and Cas9-bound forms.

(A) Apo structure of AcrIIC2 dimer with monomers colored in purple and light orange.
(B) Details of hydrophobic interactions between AcrIIC2 monomers.
(C) DNA cleavage assays conducted using NmeCas9 to test the inhibitory activity of AcrIIC2 variants that contain mutations in the dimerization interface.
(D) Overall structure of the AcrIIC2 dimer bound to BH (cyan ribbon).
(E) Top view of (D) depicting binding of BH in the “Acidic Groove” of AcrIIC2 dimer shown in the electrostatic potential surface representation. Arginines of BH are shown as sticks.
(F) Hydrogen bonding between BH and AcrIIC2 dimer. Same color code as in (D).
(G) DNA cleavage by NmeCas9 to test the inhibitory activity of the AcrIIC2 variants that contain mutations in the BH-binding surface.
(H) Isothermal titration calorimetry (ITC) assays to test binding of the NmeCas9-BH mutants to AcrIIC2. Left, representative binding curves. Right, binding affinities. KDs are mean±SE (n=2). NB, no binding.
See also Figures S2, S3 and Table 1.
Table 1.
Data Collection and Refinement Statistics
| Crystal | Apo_AcrIIC2 | NmeBH+AcrIIC2 | FnoBH+AcrIIC2 | NmeHNH+AcrIIC3 |
|---|---|---|---|---|
| Beam line | SSRF-18U | SSRF-18U | SSRF-18U | SSRF-18U |
| Wavelength (Å) | 0.9785 | 0.9785 | 0.9785 | 0.9785 |
| Space group | P6322 | P21 | P212121 | P64 |
| Unit Cell | ||||
| a, b, c (Å) | 72.0, 72.0, 105.2 | 46.4, 82.0, 76.1 | 49.7, 62.5, 88.8 | 129.1, 129.1, 35.2 |
| α, β, γ (°) | 90, 90, 120 | 90, 96.4, 90 | 90, 90, 90 | 90, 90, 12 0 |
| Resolution (Å) | 50–2.23 (2.30–2.23)a | 50–2.39 (2.43–2.39)a | 50–1.78 (1.81–1.78)a | 50–2.60 (2.66–2.60)a |
| Rmerge | 0.094 (4.078) | 0.097 (0.274) | 0.088 (0.398) | 0.126 (1.319) |
| I/σ(I) | 28.9 (1.0) | 29.6 (6.2) | 26.4 (4.5) | 26.2 (1.7) |
| Completeness (%) | 98.3 (81.3) | 99.8 (99.9) | 99.6 (96.2) | 99.9 (100.0) |
| Redundancy | 35.2 (25.6) | 6.7 (6.6) | 12.7 (11.0) | 17.4 (16.6) |
| Number of unique reflections | 8,250 | 22,256 | 27,092 | 10,567 |
| Rwork/Rfree (%) | 20.6/24.3 | 17.9/22.2 | 17.7/22.3 | 20.5/22.6 |
| Number of non-H atoms | ||||
| Protein | 954 | 4,110 | 2,075 | 2,207 |
| Water | 40 | 93 | 211 | 4 |
| Average B factor (Å2) | ||||
| Protein | 75.0 | 54.3 | 27.4 | 75.0 |
| Water | 74.9 | 44.7 | 36.4 | 63.5 |
| Ramachandran (%) | ||||
| Favored | 100 | 99.2 | 98.7 | 95.8 |
| Allowed | 0 | 0.8 | 1.3 | 4.2 |
| Outliers | 0 | 0 | 0 | 0 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.010 | 0.006 | 0.005 | 0.003 |
| Bond angles (°) | 1.213 | 1.185 | 0.923 | 0.640 |
| Methods to solve the phase | SAD | MR | MR | SAD |
| Estimated Coordinate Error (Maximum-Likelihood Based) | 0.30 | 0.28 | 0.08 | 0.27 |
Highest resolution shell (in Å) shown in parentheses.
Bridge Helix of NmeCas9 Binds in the Acidic Groove of AcrIIC2
To better understand how AcrIIC2 recognizes NmeCas9, we conducted extensive crystallization trials to determine the structure of AcrIIC2 bound to the BH-REC1 region of NmeCas9 (aa 51–241). These efforts yielded crystals with poor diffraction quality grown in the span of several months. SDS-PAGE and MALDI-TOF mass spectrometry of these crystals revealed the full-length AcrIIC2 and a spontaneously cleaved small fragment of BH-REC1 that contains an intact BH (Figures S3A and S3B). Based on these results, we crystallized the complex of AcrIIC2 and a shorter NmeCas9 construct (aa 51–123) and determined its structure at 2.4 Å resolution (Figure 2D and Table 1). The structure showed that the arginine-rich α-helix of BH (residues 56–79) tightly binds to the Acidic Groove of the AcrIIC2 dimer across the face formed by both monomers (Figure 2E), with the N-terminal non-helical tail of the motif (V51-G55) positioned down along the side of one monomer. The interface between BH and the AcrIIC2 dimer is not large (1,171 Å2), suggesting that twice smaller interacting surface for the monomeric AcrIIC2 may be insufficient for strong interactions and that dimerization represents a solution to enlarge the interacting surface. No electron density map was observed for residues 80–123 of NmeCas9 likely due to the lack of interactions with AcrIIC2 (Figure S3C).
Most of the intermolecular interactions in the AcrIIC2-BH complex are mediated by salt bridges and hydrogen bonds between arginine residues (R62, R63, R66, R69, R70, R73, and R74) of BH and acidic residues from both AcrIIC2 monomers (Figure 2F). The N-terminal tail (V51-G55) of BH forms several additional hydrogen bonds and Van-der-Vaal’s interactions with AcrIIC2, further strengthening the complex. Alanine substitutions (E18A and N23A/D24A/E25A) or deletion (∆109–124) of the residues in the Acidic Groove that contact BH abolish the inhibiting activity of AcrIIC2 (Figures 2F and 2G). Alanine substitutions for D42 and D43, which locate at the ends of the Acidic Groove and do not form stable contacts with Cas9, only slightly reduce the inhibition (Figure 2G). In line with the DNA cleavage results, the isothermal titration calorimetry (ITC) results reveal that E18A, N23A/D24A/E25A, and ∆109–124, but not D42A/D43A, dramatically reduce binding affinity between AcrIIC2 and BH (Figure S3D). Reciprocally, AcrIIC2 binding affinity is reduced for BH mutants T54A/D56A, R62A, R63A, R70A, and R73A, but not for R66A, R69A, and R74A (Figures 2H and S3E). A combined mutant R69A/R70A/R73A/R74A abolishes interactions (Figure 2H). Interestingly, R66, R69 and R74 are conserved and form extensive interactions with residues of AcrIIC2 but are not essential for complex formation (Figure S3F). In contrast, mutations of non-conserved T54 and D56 are most detrimental for binding affinity. As anticipated from the structure, mutations in the Alkaline Platform of AcrIIC2 did not affect inhibition by AcrIIC2 (Figure S3G).
AcrIIC2 Targets Diverse Cas9 Orthologs
Given that almost all Cas9 orthologs contain the arginine-rich BH, AcrIIC2 may be able to interact with other Cas9 proteins in addition to NmeCas9. However, residues of BH are moderately conserved and our data revealed that some highly conserved amino acids do not significantly contribute to the binding affinity of NmeCas9 for AcrIIC2 thereby questioning feasibility of AcrIIC2 binding to NmeCas9 orthologs. Indeed, an earlier study did not detect robust inhibition by AcrIIC2 in the DNA cleavage assays with GeoCas9, Campylobacter jejuni Cas9 (CjeCas9), and Streptococcus pyogenes Cas9 (SpyCas9) (Harrington et al., 2017). This study however did not reveal whether the lack of inhibition is caused by the inability of AcrIIC2 to bind to various Cas9s or by the failure to interfere with catalysis.
To assess whether Cas9s from different bacteria can interact with AcrIIC2, we conducted SEC of the mixtures between AcrIIC2 and Cas9s. Remarkably, AcrIIC2 formed stable complexes with SpyCas9, Staphylococcus aureus Cas9 (SauCas9), Francisella novicida Cas9 (FnoCas9), and CjeCas9 (Figures 3A and S3H). We further determined binding affinity between AcrIIC2 and BHs of these four Cas9 proteins using ITC (Figure 3B). While SauCas9 shared similar binding ability to NmeCas9, weaker binding was measured for SpyCas9, FnoCas9, and CjeCas9 (Figure 3B). Although SauCas9 and NmeCas9 have similar binding ability for AcrIIC2, SauCas9 lacks T54 and D56, which are important for binding to NmeCas9. These data suggest that AcrIIC2 forms non-identical contacts with SauCas9 and NmeCas9 and that different sets of residues contribute to binding affinity.
Figure 3. AcrIIC2 interacts with Cas9 orthologs from different subtypes.

(A) SDS-PAGE of SEC runs (Figure S3H) to test binding of MBP-tagged AcrIIC2 to Cas9 orthologs.
(B) Isothermal titration calorimetry assays for binding of AcrIIC2 to the BH motifs from different Cas9 proteins. KDs are mean±SE (n=2).
(C) Time course of the DNA cleavage by SpyCas9 in the presence or absence of AcrIIC2. The molar ratio of Cas9:AcrIIC2 was 1:4.
(D) The DNA cleavage by SpyCas9 in the presence of either WT or mutant AcrIIC2 that contains the Y11A/I15D/R20A mutation in the dimerization interface. The reaction time was 30 s.
(E) The BH motifs from both FnoCas9 (green) and NmeCas9 (cyan) bind within the same Acidic Groove of AcrIIC2 dimer.
(F) Hydrogen bonding interactions between the FnoCas9-BH motif (green) and the AcrIIC2 dimer (purple and light orange).
To check whether AcrIIC2 can inhibit the enzyme activity of other Cas9 proteins, we performed an in vitro DNA cleavage assay for SpyCas9. SpyCas9 shows a medium binding affinity for AcrIIC2 among all tested proteins (Figure 3B) and is the mostly used Cas9 in genome editing. Our kinetic analysis showed that AcrIIC2 efficiently inhibits SpyCas9 within 30 sec of the reaction but, because of lower binding affinity, inhibition decreases under prolonged incubation (Figure 3C). This result explains previously observed lack of the AcrIIC2-meidated inhibition of Cas9 orthologs in the 15 min assays (Harrington et al., 2017). As a negative control, the AcrIIC2 dimerization mutant Y11A/I15D/R20A did not show inhibition for SpyCas9 under the same conditions (Figure 3D). These data confirm that AcrIIC2 can bind to a Cas9 protein from a different bacterium and inhibit its activity albeit with much lower efficiency.
To visualize interactions between AcrIIC2 and orthologs of NmeCas9, we determined the crystal structure of AcrIIC2 in complex with BH of FnoCas9, which shows the weakest affinity for AcrIIC2 among all tested Cas9 orthologs (Figure 3B). The 1.8 Å resolution structure of AcrIIC2 bound to FnoCas9-BH reveals that the intermolecular interactions are mainly mediated by the arginine residues of BH and the charged residues in the Acidic Groove of AcrIIC2 (Figures 3E, 3F and Table 1), reminiscent but not identical to the interactions formed between AcrIIC2 and NmeCas9-BH. Although practically all amino acids of AcrIIC2 interacting with FnoCas9-BH, such as E18, E25, D109, and N113, were also found participating in the AcrIIC2-NmeCas9-BH complex formation, most of the interactions are formed with amino acids of FnoCas9-BH that are not equivalent to residues of NmeCas9-BH (Figures 2F and 3F). Some interactions, for example, equivalent to hydrogen bonding to T54 and D56, do not exist because the amino acids are not present in FnoCas9-BH. In addition, compared to NmeCas9-BH, FnoCas9-BH is slightly displaced to maximize interactions with the Acidic Groove (Figure 3E), indicating that AcrIIC2 can accommodate and tolerate diverse BHs from different Cas9 proteins. Interestingly, AcrIIC2 binding turns the non-helical S44-M52 region of FnoCas9 into a helical structure (Figure S3I), suggesting an induced conformational change of this region. Together, our structural and biochemical results demonstrate that AcrIIC2 can interact with diverse Cas9 orthologs from all three subtypes using similar but not identical binding patterns.
AcrIIC2 interferes with assembly of Cas9-sgRNA and Cas9-sgRNA-DNA complexes
BH is threaded through the center of Cas9 between tracrRNA and a guide region of crRNA in the RNA-DNA hybrid and, therefore, is essential for sgRNA-DNA recognition (Anders et al., 2014; Jiang et al., 2016; Nishimasu et al., 2014). Half of BH residues participate in sgRNA binding and mutations of several such amino acids, for example, R66, R70 and R74 of SpyCas9, resulted in decreased DNA cleavage (Nishimasu et al., 2014). These amino acids are conserved in BHs and mediate intermolecular interactions in our structures of the AcrIIC2-BH complexes. Thus, binding of AcrIIC2 likely prevents interactions of BH with two regions of sgRNA and negatively impacts sgRNA-DNA recognition and DNA cleavage.
To understand whether AcrIIC2 binding can prevent loading of sgRNA onto NmeCas9 or vice versa, we performed two types of experiments. In the first experiment, we preformed the AcrIIC2-NmeCas9 complex and then added sgRNA. In the second experiment, we preformed the sgRNA-NmeCas9 complex and then added AcrIIC2. In parallel, we conducted same experiments with AcrIIC1 and AcrIIC3. We analyzed formation of the complexes by SEC (Figures 4A, S4A and S4B) and SDS-PAGE (Figure 4B) and assessed activity of Cas9 by the DNA cleavage assay (Figure 4C). The first series of experiments showed that sgRNA can bind to all three preformed Acr-NmeCas9 complexes and that DNA cleavage is inhibited by all three Acrs. These results demonstrate that neither Acr, not even AcrIIC2, prevents sgRNA loading onto Cas9. The second series of experiments demonstrated that binding of AcrIIC2 to sgRNA-NmeCas9 complex is practically eliminated, AcrIIC1 binding is reduced, and AcrIIC3 binding is not affected. Concurrently, AcrIIC2 does not inhibit, AcrIIC1 inhibits with reduced efficiency, and AcrIIC3 completely inhibits DNA cleavage. These results suggest that AcrIIC2 should bind to Cas9 prior to sgRNA in order to be an effective inhibitor. In contrast, the order of binding is not important for AcrC1- and AcrC3-mediated inhibition.
Figure 4. Mechanism of Cas9 inhibition by AcrIIC2.

(A) Elution profiles of SEC runs to test binding of sgRNA to complexes formed with NmeCas9 and AcrIIC1 (left), AcrIIC2 (center), and AcrIIC3 (right).
(B) SDS-PAGE of peak fractions from SEC experiments (Figures 4A, S4A and S4B) to test binding of sgRNA (R) and AcrIICs to complexes formed with NmeCas9. Inhibitors were AcrIIC1 (C1, left), AcrIIC2 (C2, center), and AcrIIC3 (C3, right). Acrs were added to the reaction mixtures prior to or after adding sgRNA. Preformed complexes are shown in parentheses.
(C) DNA cleavage by NmeCas9 in the presence of AcrIIC1 (left), AcrIIC2 (center), and AcrIIC3 (right). Acrs were added into the reaction mixtures prior to or after adding sgRNA.
(D) Determination of binding affinity between sgRNA and NmeCas9 in the absence (black) and presence (blue) of AcrIIC2 using DRaCALA (Roelofs et al., 2011; Vasilyev et al., 2015). For comparisons of two data sets, plateaus of the binding curves were set to the bound RNA fraction of 1.0. KDs are 1.66±0.18 and 44.06±1.74 nM (mean±SE, n=2) in the absence and presence of AcrIIC2, respectively.
(E) Determination of binding affinity between NmeCas9-sgRNA and dsDNA in the absence (black) and presence (red) of AcrIIC2 using fluorescence polarization. KD is 2.08±0.17 μM (mean±SE, n=3) in the absence of AcrIIC2. KD in the presence of AcrIIC2 cannot be reliably determined.
(F, G) Superposition of the AcrIIC2-BH complex structure on the structures of the binary sgRNA-SpyCas9 complex (PDB ID: 4ZT0) (Jiang et al., 2015) (F) and ternary sgRNA-DNA-SpyCas9 complex with the HNH domain in the undocked conformation (PDB ID: 4UN3) (Anders et al., 2014) (G). All-atom superpositions were centered on BHs.
See also Figure S4.
To determine the effect of AcrIIC2 on sgRNA binding to Cas9, we conducted equilibrium binding assays with simultaneous addition of AcrIIC2 and sgRNA to NmeCas9 (Figure 4D). The data showed that sgRNA binding to NmeCas9 was significantly reduced (≈27-fold) in the presence of AcrIIC2. Accordingly, binding of dsDNA to the preformed sgRNA-NmeCas9-AcrIIC2 was practically abolished (Figure 4E).
To get insights into the molecular mechanisms of inhibition, we superposed the structure of the AcrIIC2-BH complex with the structures of SpyCas9 determined in different states, including apo SpyCas9 (PDB ID: 4CMP), the binary sgRNA-SpyCas9 (PDB ID: 4ZT0), and two ternary sgRNA-DNA-SpyCas9 (PDB ID: 4UN3 and 5F9R) complexes. Superimpositions revealed steric clashes of dimeric AcrIIC2 with the REC lobe, PI and RuvC domains in the apo Cas9, and with sgRNA and REC lobe in the binary and ternary complexes (Figures 4F, 4G, S4C and S4D). Thus, to bind AcrIIC2, Cas9 has to adopt a conformation not captured in available Cas9 structures. Regardless what this conformation could be, it is clear that the AcrIIC2 dimer provides major impediment for sgRNA and DNA binding in the vicinity of the BH of Cas9.
Together, biochemical and structural data demonstrate that AcrIIC2 weakens sgRNA-Cas9 interactions, prevents the correct assembly of the sgRNA-Cas9 complex, and, consecutively, dramatically affects formation of the sgRNA-DNA hybrid bound to Cas9. Because no Acrs have been shown to interfere directly with the sgRNA binding of Cas9, the AcrIIC2-mediated sequestration of the RNA binding site represents previously unobserved mechanism of Cas9 inhibition.
AcrIIC3 Binds NmeCas9 HNH Domain opposite to AcrIIC1
Although both AcrIIC1 and AcrIIC3 bind to the HNH domain of NmeCas9, the inhibition mechanisms are different for these two proteins (Harrington et al., 2017). To understand the molecular basis of the AcrIIC3-mediated inhibition, we determined the 2.6 Å crystal structure of the AcrIIC3-HNH complex and analyzed the binding interface (Figures 5, S5 and Table 1). AcrIIC3 is an α+β protein with the central four antiparallel β-strands surrounded by six α -helices (Figures 5A and S5A). A Dali server search revealed that AcrIIC3 has a similar fold to the CRISPR-associated protein Cas2 (Figure S5B). The two proteins also have rather high (~17%) identity, intriguingly suggesting that a phage may have co-opted a bacterial protein.
Figure 5. Interactions between AcrIIC3 and Cas9.

(A) Overall structure of AcrIIC3 (purple) in complex with the HNH domain of NmeCas9 (yellow). The catalytic residues of the HNH domain are shown in space-filling representation and orange color.
(B, C) Hydrogen bonding between AcrIIC3 and α 1 (B) and α 2 (C) helixes of the HNH domain. Same color code as in (A).
(D) DNA cleavage by NmeCas9 to test mutations in the HNH-binding surface of AcrIIC3.
(E) Pull-down analysis of AcrIIC3-NmeCas9 interactions. GST-tagged AcrIIC3 was incubated with different NmeCas9 constructs and the complexes were pulled down and analyzed by SDS-PAGE.
(F) Elution profiles of SEC runs on Superdex 200 10/300 column to test binding of AcrIIC3 to the MBP-tagged full-length (FL) NmeCas9.
(G) Elution profiles of SEC runs of the MBP-tagged NUC lobe of NmeCas9 complexed with AcrIIC3. Note that only FL but not the NUC lobe has a large shift in elution volume upon binding to AcrIIC3. Black asterisks indicate the fractions analyzed by SDS-PAGE.
(H) SDS-PAGE analysis (left panel) and quantitative measurement (right panel) for the purified AcrIIC3-Cas9 complex and the standard samples with 1:2 and 2:2 molar ratios of AcrIIC3 and Cas9, respectively.
(I) Elution profiles of SEC runs for MBP-AcrIIC3 alone and mixtures of AcrIIC3 and Cas9 at the 1:2 and 2:2 molar ratio.
See also Figures S5, S6 and Table 1.
In contrast to AcrIIC1, AcrIIC3 binds to the surface opposite to the active site of HNH (Figures 5A and S5C). Structure superimposition of AcrIIC1-HNH and AcrIIC3-HNH complexes shows that the two inhibitors can simultaneously interact with the HNH domain without steric clashes (Figure S5C). Furthermore, a SEC experiment confirmed that a ternary complex can be formed by HNH, AcrIIC1, and AcrIIC3 (Figure S5D). Despite having a potential dimerization interface of Cas2, AcrIIC3 is a monomer in solution (Harrington et al., 2017, and Figures S2A and S2D). Most likely, AcrIIC3 does not dimerize because the α5-Loop-α6 motif covers the Cas2-like dimerization surface of AcrIIC3 and prevents potential dimerization (Figures S5B and S5E). The AcrIIC3-HNH complex involves 2,212 Å2 interface mostly formed between two N-terminal α-helixes of HNH and several structural elements of AcrIIC3 (Figures 5A–5C and S5F). Intermolecular interactions are primarily mediated by hydrogen bonds and hydrophobic contacts with contribution of ionic interactions.
The first set of most prominent interactions involves the C-terminal region of α1 of HNH (K532-Y540), which forms multiple hydrogen bonds predominantly using side chains of charged residues (K532, K536, and E539) located on one side of the helix (Figure 5B). These amino acids and neighboring Y540 interact with residues of AcrIIC3 that belong to the end of β2 (L58 and N60), tip of α1 (R33), and the adjacent loop (V34 and D38). Although these multipoint interactions appear to provide substantial stability to the complex according to the structure of the complex, the triple mutant R33A/D38A/Y40A only moderately reduced inhibition of DNA cleavage and complex formation while a single N60A practically did not have impact on inhibition and minimally reduced complex formation (Figures 5D and S5G).
The second set of interactions involves the C-terminal half of α2 of HNH (R557-H563) and multiple residues of AcrIIC3 scattered on the entire binding interface (Figure 5C). These interactions mostly include hydrogen bonds between side chains of both proteins as well as cation-π interactions between R643 and Y40. Interestingly, aliphatic parts of some charged residues involved into intermolecular interactions (R5, E92, and R103) are embedded into the core of AcrIIC3 and their substitutions by alanines reduced formation of the complex two-fold (Figure S5G) and abolished inhibition of Cas9 (Figure 5D).
Lastly, the complex is locked up by hydrophobic interactions involving residues of both α1 and α2 of HNH and four residues of AcrIIC3 located in the middle of the interface (Figure S5F). Disruption of these interactions by mutations in AcrIIC3 (M1/F3/L58S) abolished the inhibitory effect of the protein and complex formation (Figures 5D and S5G). Thus, this set of interactions is most critical for the AcrIIC3-HNH complex formation.
The AcrIIC3-HNH binding interface is not conserved among Cas9 orthologs (Figure S6A) and contains only one highly conserved amino acid I553, which is involved in the formation of hydrophobic interactions. Therefore, AcrIIC3 may specifically bind only NmeCas9. SEC assays confirm that AcrIIC3 cannot bind other Cas9 proteins (Figures S6B and S6C), thus explaining the published DNA cleavage results that showed the lack of inhibition of CjeCas9, GeoCas9 and SpyCas9 by AcrIIC3 (Harrington et al., 2017).
REC Lobe is Critical for AcrIIC3-induced NmeCas9 Dimerization
Previous SEC, small-angle X-ray scattering and negative-stain electron microscopy data showed dimerization of NmeCas9 in the complex with AcrIIC3 (Harrington et al., 2017). However, both AcrIIC3 alone (Harrington et al., 2017, and Figure S2D) and the AcrIIC3-HNH complex (Figure S6D) are monomeric in solution, indicating that the AcrIIC3-HNH interaction is not sufficient for NmeCas9 dimerization.
These data suggest that NmeCas9 contain other AcrIIC3-interacting regions which contribute to NmeCas9 dimerization. Given the SEC can only identify strong interactions (Figures 1 and S1), we performed pull-down experiments to identify weaker interactions between GST-tagged AcrIIC3 and different NmeCas9 truncations. These experiments revealed that GST-AcrIIC3 binds to the isolated REC lobe (Figure 5E). Follow-up SEC analysis showed that the NmeCas9 variant lacking the REC lobe does not dimerize upon binding to AcrIIC3 (Figures 5F and 5G). To further confirm the critical role of the REC lobe for NmeCas9 dimerization, we generated hybrid proteins by replacing the HNH domains of SpyCas9, SauCas9, and CjeCas9 with the NmeCas9 counterpart and conducted SEC experiments. Although AcrIIC3 is able to bind to all hybrid proteins, it cannot induce dimerization (Figures S7A–S7D), indicating that interaction of AcrIIC3 with the REC lobe is species-specific. Indeed, the REC lobe has been shown to be one of the least conserved regions across all three Cas9 subtypes (Nishimasu et al., 2014). Together, these data demonstrate that both the HNH domain and REC lobe of NmeCas9 participate in interactions with AcrIIC3 and that the REC lobe is instrumental in inducing NmeCas9 dimerization.
To check whether Cas9 dimerization involves one or two AcrIIC3 molecules, we first performed SDS-PAGE experiment to quantitatively measure the stoichiometry of the SEC-purified AcrIIC3-Cas9 complex by comparing it with the standard samples prepared by mixing AcrIIC3 and Cas9 at 1:2 and 1:1 (equivalent to 2:2) molar ratios. The results clearly show that the purified AcrIIC3-Cas9 complex has a 2:2 stoichiometry (Figure 5H). We further conducted SEC experiments for both the 1:2 and 2:2 (AcrIIC3:Cas9) protein mixtures. As expected, the 1:2 sample still has a portion of monomeric Cas9 and the 2:2 sample entirely forms the dimerization complex (Figure 5I), confirming that the AcrIIC3-induced Cas9 dimerization involves two AcrIIC3 molecules.
Superpositions of AcrIIC3 onto Cas9
To better understand the molecular mechanism of AcrIIC3 inhibition, we superposed the structure of AcrIIC3-HNH complex with the structures of SpyCas9 determined in different states (Figures S7E–S7H). Superpositions with the HNH-AcrIIC3 structure placed AcrIIC3 on periphery in the apo Cas9, binary complex and ternary undocked complex (Figures S7E–S7G). In the binary complex, AcrIIC3 contacts the REC lobe while it is moved into the REC lobe producing multiple clashes in the ternary undocked complex. In the ternary docked complex, AcrIIC3 clashes with two other parts of the REC lobe (Figure S7H). Therefore, Cas9-bound AcrIIC3 must be positioned as in the binary complex or between its locations observed in the binary and ternary complexes. Given that AcrIIC3 was shown to negatively affect DNA binding and promote Cas9 dimerization (Harrington et al., 2017), we propose that each AcrIIC3 molecule binds the HNH domain positioned as in the binary complex and either reaches to the REC lobe of another Cas9 or binds to the REC lobe of the same molecule, causing dimerization of the Cas9-AcrIIC3 complex and closing the openings in Cas9s for DNA binding.
AcrIIC1/2/3 Simultaneously Interact with NmeCas9
Since AcrIIC1/2/3 interact with non-overlapping regions of Cas9, we wondered whether Cas9 can adopt a conformation compatible with binding to the three AcrIICs. While structure superposition suggest that such arrangement might be possible but requires Cas9 conformation not captured in the available Cas9 structures (Figures 6A and 6B), SEC and SDS-PAGE confirmed that AcrIIC1/2/3 can simultaneously interact with NmeCas9 (Figure 6C).
Figure 6. AcrIIC1/2/3 simultaneously interact with NmeCas9.

(A, B) Superpositions of the structures of the AcrIIC1-HNH, AcrIIC2-BH and AcrIIC3-HNH complexes with apo SpyCas9 (PDB ID: 4CMP) (Jinek et al., 2014) to show distinct location of bound Acrs on Cas9. All-atom superpositions were centered on Cas9 regions.
(C) Elution profiles of SEC runs on a Superdex 200 10/300 column to show simultaneous binding of AcrIIC1, AcrIIC2, and AcrIIC3 to NmeCas9. Black asterisk indicates the fractions analyzed by SDS-PAGE.
AcrIIC1 and AcrIIC3 Target HNH-switched Cas9 Protein
Given that the hybrid Cas9 proteins do not undergo dimerization upon binding to AcrIIC3, one can use these proteins to evaluate the effect of dimerization on Cas9 inhibition. To make sure that a hybrid Cas9 protein that contains the HNH domain of NmeCas9 is functional, we tested activity of a representative chimera, SpyCas9NmeHNH. The DNA cleavage assay conducted using end-labeled substrates showed that the SpyCas9NmeHNH hybrid protein can cut both the target and non-target DNA strands albeit with reduced efficiency than the WT SpyCas9 (Figures 7A, 7B, and S7I). We incubated AcrIIC3 with SpyCas9NmeHNH and found that AcrIIC3 cannot fully inhibit the DNA cleavage activity as compared with NmeCas9 under the same conditions (Figure 7C). Thus, although the AcrIIC3-HNH interaction is maintained, the lack of interactions with the REC lobe and consecutively the lack of SpyCas9NmeHNH dimerization negatively affect the inhibiting activity of AcrIIC3.
Figure 7. AcrIIC1/2/3 target hybrid SpyCas9 proteins.

(A) Schematic of the SpyCas9NmeHNH-mediated DNA cleavage. The hybrid Cas9 enzyme, sgRNA, target strand, non-target strand, and PAM sequence are colored in gray, black, blue, yellow, and red, respectively. The RuvC and HNH domains of NmeCas9 cleave the non-target and target strands (black triangles), respectively.
(B) End-labelled DNA cleavage assays conducted using the hybrid SpyCas9NmeHNH protein to assess the cleavage on the target and non-target strands. The lanes for a given condition correspond to 1 and 2 h of reaction. Red stars depict fluorescein labels placed on the ends of DNA strands.
(C) DNA cleavage assays conducted using SpyCas9NmeHNH or NmeCas9 with or without AcrIIC3.
(D) DNA cleavage assays conducted using SpyCas9NmeHNH with or without AcrIIC1.
(E) End-labelled DNA cleavage assays conducted using the hybrid. The lanes for a given condition correspond to 1 and 2 h of reaction.
(F) DNA cleavage assays conducted using WT SpyCas9 or SpyCas9NmeBH with or without AcrIIC2.
See also Figure S7.
In contrast to AcrIIC3, AcrIIC1 fully inhibits the DNA cleavage by SpyCas9NmeHNH (Figure 7D), indicating that the inhibiting activity of AcrIIC1 is mainly mediated through its interaction with the HNH domain. Based on this feature of AcrIIC1, one can convert the AcrIIC1-insensitive Cas9 orthologs into AcrIIC1-sensitive versions by simply swapping the HNH domains.
AcrIIC2 Targets BH-switched Cas9 Protein
Although AcrIIC2 was shown to be a broad-spectrum Cas9 binder (Figures 3A and S3H), both the binding affinity and inhibiting activity were reduced for Cas9 orthologs, such as SpyCas9 (Figures 3B–3D). Given that BH appears to be a sole determinant for AcrIIC2 binding, we reasoned that sensitivity of Cas9 orthologs to AcrIIC2 can be enhanced by introducing BH from NmeCas9. To prove our hypothesis, we prepared a hybrid SpyCas9NmeBH protein, in which the AcrIIC2-binding region of BH (D54-T73) of SpyCas9 was replaced by the NmeCas9 counterpart (T54-R73). In vitro cleavage results confirmed that SpyCas9NmeBH can cut both the target and non-target DNA strands with the efficiency similar to WT SpyCas9 (Figures 7E, 7F, and S7I). To assess whether AcrIIC2 inhibits SpyCas9NmeBH stronger, we measured the inhibitory effect with WT and hybrid SpyCas9 proteins by the in vitro DNA cleavage assay. While AcrIIC2 does not repress activity of WT SpyCas9 during long incubation (Figures 3C and 7F), it significantly represses activity of the SpyCas9NmeBH chimera (Figure 7F). Thus, AcrIIC2-insensitive or weakly sensitive Cas9 orthologs can be converted into AcrIIC2-sensitive variants by simply swapping small regions in BHs.
DISCUSSION
As more new anti-CRISPR protein families are identified, determination of their inhibitory mechanisms will be critical for understanding the basic principles of the bacteria-phage co-evolution and developing anti-CRISPR-based technological innovations. We now provide structural evidence for the proposed inhibitory mechanisms of AcrIIC3 and report a molecular mechanism of Cas9 inhibition employed by AcrIIC2.
To date, structural data were obtained only for a single member of the AcrIIC family, AcrIIC1. This protein interacts with the HNH domain of NmeCas9 and, while allowing binding of both sgRNA and dsDNA, impedes access to the active site and restricts conformational changes required for DNA cleavage (Harrington et al., 2017). Our work revealed that AcrIIC3 also binds to the HNH domain but to its opposite less conserved side. We also showed that AcrIIC3 makes additional weak interactions with the REC lobe that promote dimerization of Cas9, earlier proposed to reduce dsDNA binding affinity (Harrington et al., 2017). These interactions are species-specific and involve two AcrIIC3 molecules. An intriguing possibility is that the dimerization would be mediated by the Cas2-like surfaces of AcrIIC3s, which become accessible after binding to Cas9. However, testing this hypothesis is difficult because deletion or mutation of α5 and α6 cause aggregation of AcrIIC3.
Comparison with other Acrs further highlights distinct features employed by AcrIIC3 for inhibiting Cas9. Although two other type II Acrs, AcrIIA2 and AcrIIA4, also interfere with DNA binding step, they bind to the preformed Cas9-RNA complex, do not interact with the apo form of Cas9, and do not induce Cas9 dimerization (Dong et al., 2017; Jiang et al., 2018; Liu et al., 2018; Shin et al., 2017; Yang and Patel, 2017). Inhibitory mechanisms of AcrIIC3 and type I Acrs, AcrF1 and AcrF2, is even less similar. As AcrIIC3, AcrF1 and AcrF2 compete with target DNA for crucial interactions (Bondy-Denomy et al., 2015; Chowdhury et al., 2017; Guo et al., 2017; Peng et al., 2017) but they bind to two or more subunits of the multisubunit Cascade complex (Chowdhury et al., 2017; Guo et al., 2017; Peng et al., 2017).
We showed that, in contrast to AcrIIC1 and AcrIIC3, AcrIIC2 dimerizes and interacts with the bridge helix of Cas9, which is required for binding to tracrRNA and crRNA and formation of the RNA-DNA hybrid. Our data demonstrated that AcrIIC2 significantly weakens interaction with sgRNA, which comprises tracrRNA and crRNA, and strongly reduces dsDNA binding. Interestingly, AcrIIC2 does not block but only decreases sgRNA binding. Indeed, sgRNA has 10,840 Å2 binding interface with SpyCas9 that is much more than the ~1,170 Å2 interface between AcrIIC2 and NmeCas9. Thus, AcrIIC2 maximizes effectiveness of its Cas9 binding by sequestering the region of Cas9 that interact with the functionally essential region of sgRNA while not preventing loading of the sgRNA onto Cas9.
As a consequence of direct competition for binding to the same region of Cas9, preformation of the sgRNA-Cas9 complex impedes AcrIIC2 binding and obliterates the inhibitory effect of AcrIIC2 but not AcrIIC1 or AcrIIC3, which can bind to the preformed sgRNA-Cas9 complexes and efficiently inhibit Cas9 activity. Therefore, a full inhibition of NmeCas9 by AcrIIC2 in human cells required more AcrIIC2 than AcrIIC1 or AcrIIC3 likely to obtain higher AcrIIC2 concentration to compete with sgRNA (Pawluk et al., 2016a). This feature of inhibition also raises an intriguing question on how AcrIIC2 can block CRISPR immunity in the context of phage infection. Indeed, constitutively expressed Cas9, crRNA, and tracrRNA should preform the surveillance ternary complex prior to the phage infection. Therefore, we speculate that AcrIIC2 has to be at higher concentration than host RNA molecules to overcome ~50-fold lower affinity of Cas9 for sgRNA than for AcrIIC2 and inhibit activity of Cas9. Alternatively, AcrIIC2 might compete more effectively with individual tracrRNA and crRNA than with the sgRNA used in our study. One may also imagine that AcrIIC2 functions in concert with other proteins from the same or another phage (Borges et al., 2018; Landsberger et al., 2018).
It is particularly interesting that AcrIIC1 and AcrIIC2 can target a broad spectrum of Cas9 orthologs from different subtypes by binding to the moderately conserved HNH and BH domains, respectively. This feature will be important for designing AcrIIC1- and AcrIIC2-based molecular tools for biotechnological applications. In contrast, the application of AcrIIC3 may be restricted to the NmeCas9-related research because of lower conservation of targeted regions. The scope of inhibition of a particular Acr can be improved by the structure-based rational engineering of both Cas9 and Acr. We substantially increased sensitivity of SpyCas9, the enzyme most used for gene-editing applications, to inhibition by AcrIIC1 and AcrIIC2 using simple modification of the HNH and BH regions of SpyCas9, respectively. Through this strategy, one can potentially manipulate the SpyCas9 activity at different steps, including sgRNA binding (by AcrIIC2), dsDNA binding (by AcrIIC2 and AcrIIA4), as well as DNA cleavage (by AcrIIC1). Although further research is required to optimize the efficiency of catalysis and inhibition of the modified SpyCas9, our proof-of-principle work provides the first insights on the rational design of anti-CRISPR-sensitive Cas9 enzymes.
In summary, our work has revealed diverse molecular mechanisms of AcrIIC2/3-mediated inhibition of Cas9 activity and identified a previously unrecognized strategy, interference with both sgRNA and dsDNA binding steps and blockage of the sgRNA-dsDNA pairing, employed by an Acr to inhibit Cas9. Our study also provided insights into design of broad spectrum Cas9 inhibitors and may serve as a reference in engineering molecular tools with potentially far-reaching implications. Remarkably, the three type IIC inhibitors can simultaneously interact with NmeCas9, suggesting a potential synergistic inhibiting effect and combinatorial approaches for effective silencing of Cas9 activity in future studies. Finally, the diverse inhibition mechanisms of AcrIIC1/2/3 and their high sequence diversity support the independent evolution of different Acrs.
STAR METHODS TEXT
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Pu Gao (gaopu@ibp.ac.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Top10 Chemically Competent Cells were used for molecular cloning. Rosetta (DE3) Chemically Competent Cells were used for protein expression. Both of them were grown in LB medium culture at 37°C. After induction, Rosetta (DE3) cel ls were transferred to 20°C.
METHOD DETAILS
Plasmid construction
The genes encoding the NmeCas9, SauCas9, CjeCas9, AcrIIC1, AcrIIC2 and AcrIIC3 were synthesized and codon-optimized for expression in E. coli. The genes encoding SpyCas9 and FnoCas9 were amplified from PX459 pSpCas9(BB)-2A-Puro (Addgene: #62988) and PX408 Francisella tularensis subsp. novicida Cas9 (Addgene: #68705), respectively, using standard PCR method. All constructs were inserted into modified pRSF-Duet-1 vector (Novagen) with the N-terminal His6-SUMO tag followed by an ubiquitin-like protease (ULP1) cleavage site. In addition, NmeCas9, AcrIIC2, and AcrIIC3 were inserted into modified pET-28a vector (Novagen) with the N-terminal His6-MBP tag; and AcrIIC3 was inserted into pGEX-6p-1 vector bearing the N-terminal GST tag. All point mutants, truncations and chimeras were prepared from the full-length constructs using PCR.
Protein expression and purification
The recombinant proteins were overexpressed in Rosetta (DE3) strain (Novagen) in Lysogeny broth (LB) medium. The cells were grown at 37°C unt il OD600=0.8 and induced with 0.4 mM isopropyl b-D-1-thiogalactopyranoside at 20°C for 1 8 h. Cell pellets were resuspended in buffer A (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10% glycerol, 20 mM imidazole, 5 mM 2-hydroxy-1-ethanethiol) and cells were lysed by French Press. Lysates were cleared by centrifugation.
For the recombinant proteins with N-terminal His6-SUMO tag:
The supernatant was loaded onto the Ni column prepared with Ni-Sepharose 6 FF beads (GE Healthcare) and the protein sample was eluted with buffer B (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 500 mM imidazole, 10% glycerol, 5 mM 2-hydroxy-1-ethanethiol).
For Cas9 proteins, eluates were treated with ULP1 and loaded on HiTrap™ Heparin HP column (GE Healthcare). Proteins were eluted with a linear 0.25–1.0 M gradient of NaCl, followed by gel filtration on 16/60 G200 Superdex column (GE Healthcare) equilibrated with buffer C (20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 2 mM MgCl2, 1 mM DTT).
For AcrIIC1, the ULP1-treated sample was loaded onto Ni column to remove the His6-SUMO tag. The flowthrough was concentrated and purified on 16/60 G200 Superdex column equilibrated with buffer C.
For AcrIIC2 and AcrIIC3, the ULP1-cleaved tags were removed on the Ni column. The protein sample was purified on HiTrap™ Heparin HP column with a linear 0.25–1.0 M gradient of NaCl, followed by gel filtration on a 16/60 G200 Superdex column equilibrated with the buffer C.
For NmeBH (aa 51–123), FnoBH (aa 45–90), SauBH (aa 36–81) and CjeBH (aa 37–82), His6-SUM0-tagged proteins were loaded onto HiTrap™ Heparin HP column and eluted with linear 0.25–1.0 M gradient of NaCl. Samples were then treated with ULP1 and the tag was removed on Ni column.
For the recombinant proteins with N-terminal His6-MBP tag:
The supernatant was loaded onto Ni column and the protein was eluted with buffer B (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 500 mM imidazole, 10% glycerol, 5 mM 2-hydroxy-1-ethanethiol). The protein sample was loaded onto HiTrap™ Heparin HP column (MBP-AcrIIC2) or HiTrap™ SP Sepharose FF column (GE Healthcare Life Sciences) with a linear 0.25–1.0 M gradient of NaCl, followed by gel filtration on 16/60 G200 Superdex column equilibrated with buffer C.
For GST-AcrIIC3:
The supernatant was loaded onto GST column prepared with Glutathione-Sepharose 4 Fast Flow beads (GE Healthcare) and the protein sample was eluted with buffer D (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10 mM glutathione, 10% glycerol, 5 mM 2-hydroxy-1-ethanethiol). The protein was further purified on HiTrap™ Heparin HP column with a linear 0.25–1.0 M gradient of NaCl, followed by gel filtration on a 16/60 G200 Superdex column equilibrated with buffer C.
All truncated constructs and mutants were cloned and mutated proteins were purified as described for wild type proteins. The Se-methionine-substituted protein was produced in Se-methionine-containing medium and purified as described for the wild-type protein. The final sample was concentrated and stored at −80°C before use.
Complex formation and crystallization
Complex formation:
To prepare the HNH-AcrIIC3 complex, the HNH domain (~15 mg/mL) was incubated with 3-fold excess of AcrIIC3 in buffer C on ice for 30 min and the samples were loaded on 16/60 G200 Superdex column equilibrated with buffer C. The purified complex was concentrated to 10–15 mg/mL and stored at −80°C before use.
The complex between AcrIIC2 and Nme-BH-REC1 (aa 51–123) was prepared by mixing AcrIIC2 (~15 mg/mL) with Nme-BH-REC1 at a molar ratio of 2:1.5 in buffer C and incubating on ice for 30 min. The complex was purified on 16/60 G200 Superdex column, concentrated and stored at −80°C before use.
The complex between AcrIIC2 and Fno-BH (aa 45–90) was prepared by mixing AcrIIC2 (~15 mg/mL) with Fno-BH peptide at a molar ratio of 2:1.5 in buffer C and incubating on ice for 30 min. The complex was directly used for crystallization.
Crystallization:
Crystals of the complex between NmeHNH (aa 508–667) and AcrIIC3, were grown by hanging drop vapor diffusion method at 20°C using drops con taining 1 µL protein sample and 1 µL reservoir solution (0.1 M sodium citrate, pH 5.9, 48% (v/v) PEG200). Crystals of Se-methionine-substituted protein were grown using different reservoir solution (0.2 M NaCl, 0.1 M HEPES, pH 7.5, 20% (v/v) PEG400).
Crystals of AcrIIC2 were grown by sitting drop vapor diffusion method at 20°C using drops containing 1 µL protein solution and 1 µL reservoir solution (0.1 M Tris, pH 8.5 20% (v/v) ethanol). The crystals of Se-methionine-substituted protein were grown under the same conditions.
Crystals of the complex between NmeBH (aa 51–123) and AcrIIC2 were grown by sitting drop vapor diffusion method at 20°C using drops con taining 1 µL protein sample and 1 µL reservoir solution (0.1 M CHES, pH 9.5, 19% (v/v) ethanol).
Crystals of the complex between FnoBH (aa 45–90) and AcrIIC2 were grown by sitting drop vapor diffusion method at 20°C using drops con taining 1 µL protein solution and 1 µL reservoir solution (0.1 M MES, pH 5.6 5% (v/v) PEG3000, 28% (v/v) PEG200).
Structure determination
Diffraction datasets were collected at the BL18U1 and BL19U1 beamlines of Shanghai Synchrotron Radiation Facility (SSRF) and were indexed, integrated, and scaled using the HKL suite (Otwinowski and Minor, 1997). The structure of the HNH-AcrIIC3 complexes and apo AcrIIC2 were solved using single-wavelength anomalous dispersion (SAD) method using PHENIX (Adams et al., 2010). The figures of merit values of the SAD phasing calculations prior to density modification for HNH-AcrIIC3 and apo AcrIIC2 are 0.29 and 0.3, respectively. The structures of the NmeBH-AcrIIC2 and FnoBH-AcrIIC2 complexes were solved by molecular replacement method in PHENIX (Adams et al., 2010), using the structure of apo AcrIIC2 as the search model. Model building and structure refinement were carried out using COOT (Emsley et al., 2010) and PHENIX (Adams et al., 2010), respectively. Statistics of data collection and refinement are shown in Table 1.
In vitro transcription and purification of sgRNA
The construct for in vitro transcription of sgRNA, followed by the hammerhead ribozyme, was cloned into pUT7 vector bearing a promoter for T7 RNA polymerase (Peselis et al., 2016). Large scale transcription reaction (20 mL) was performed in the buffer containing 100 mM Tris-HCl, pH 7.9, 30 mM DTT, 15 mM MgCl2, 2 mM spermidine, 4 mM each NTP, 50 mg/ml DNA template, and 2.5 mg home-made T7 RNA polymerase. The mixture was incubated at 37°C for 3 h, supplemented by MgCl2 to final concentration of 50 mM, and incubated for 30 min. The transcribed sgRNA was purified on 10% polyacrylamide gel (PAG) containing 1x Tris/Borate/EDTA (TBE) buffer and 8M urea, extracted from the gel by electroelution using Elutrap (GE Healthcare), and further purified by ion-exchange chromatography using HiTrap DEAE Fastflow Sepharose column (GE Healthcare) pre-equilibrated with 20 mM Tris-HCl, pH 7.0 (Peselis et al., 2016). sgRNA was eluted by a linear gradient of 1 M NaCl in 20 column volumes. The RNA was denatured at 65°C for 5 min an d slowly cooled to room temperature.
Template of NmeCas9 sgRNA used for cleavage assay:
GGCTCCTCAGATGTAGTATTCAGAGTTGTAGCTCCCTTTCTCATTTCGGAAACGAAATGAG AACCGTTGCTACAATAAGGCCGTCTGAAAAGATGTGCCGCAACGCTCTGCCCCTTAAAGCC TCTGCTTTAAGGGGCATCGTTTC
Template of NmeCas9 sgRNA used for binding assay:
GGCTCCTCAGATGTAGTATTCAGAGTTGTAGCTCCCTTTCTGAAAAGAACCGTTGCTACAAT AAGGCCGTCTGAAAAGATGTGCCGCAACGCTC
Template of SpyCas9 sgRNA:
GGAAATTAGGTGCGCTTGGCGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCC GTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCT
In vitro DNA cleavage assay
The 660 and 654 bp linear target DNAs were synthesized and amplified by PCR; the 61 bp 5’-FAM fluorescein-labeled linear target DNA was synthesized by Zixi (Beijing, China).
To test activity of NmeCas9, the cleavage reaction was performed by mixing NmeCas9, Acr, sgRNA and target DNA (660bp) at the molar ratio of 4:12:4:1. For the assays including AcrIIC1 or AcrIIC3, the cleavage buffer E1 contained 20 mM Tris-HCl, pH 7.5, 100 mM KCl, 5 mM MgCl2, 5 mM DTT, and 5% glycerol. For the assays including AcrIIC2, the cleavage buffer E2 contained 20 mM Tris-HCl, pH 7.5, 300 mM KCl, 5 mM MgCl2, 5 mM DTT, and 5% glycerol. The reaction was initiated by incubation of NmeCas9 with Acr or sgRNA on ice for 10 min, followed by simultaneous addition of other components. The reaction was incubated at 37°C for 15 min and stopped by adding 2xTBE/8M urea gel loading buffer and heating at 95°C for 5 min.
To test activity of SpyCas9, the cleavage reaction was performed by mixing SpyCas9, AcrIIC2, sgRNA and target DNA (654 bp) at the molar ratio of 2:8:2:1 (Figures 5E and 5F) in the cleavage buffer E1. To initiate the reaction, SpyCas9 was pre-incubated with AcrIIC2 for 10 min on ice and other components were added to the reaction system simultaneously. The reaction was incubated at 37°C for 30 s (Figure 5F) or 20 s, 30 s, 40 s, and 15 min (Figure 5E) and quenched as described above.
To test activity of SpyCas9NmeHNH hybrid protein, the cleavage reaction was performed by mixing SpyCas9NmeHNH, Acr, sgRNA and target DNA (654 bp) at the molar ratio of 4:20:4:1 in the cleavage buffer E1 and incubating at 37°C for 1 h; or by incubating SpyCasNmeHNH, sgRNA and 5’-FAM labeled target DNA (61 bp) mixed at the molar ratio of 8:8:1 in the same buffer at 37°C for 1 or 2 h. The reaction was quenched as described above.
To test activity of SpyCas9NmeBH hybrid protein, the cleavage reaction was performed by mixing SpyCas9NmeBH, AcrIIC2, sgRNA and target DNA (654 bp) at the molar ratio of 2:8: 2:1 in the cleavage buffer E1 and incubating at 37°C for 1 5 min; or by mixing SpyCasNmeBH, sgRNA and 5’-FAM labeled target DNA (61 bp) at the molar ratio of 2:2:1 in the same buffer and incubating at 37°C for 1 or 2 h. The reaction was q uenched as described above. Control reactions with NmeCas9 and SpyCas9 were conducted as described for the SpyCas9NmeBH hybrid protein.
All cleavage products were analyzed by electrophoresis on 15% PAG TBE-urea gel and visualized by ethidium bromide staining or fluorescence scanning.
The DNA sequences used in this study are listed below (PAM is highlighted in bold and the target sequence is in italics):
NmeCas9 Target DNA sequence (660 bp):
TCCTGGATGGCATTGCCTGTGCCTGGGTTGGTGCCCGCGTTCCGTGGAGTGAGAAGTATG TGCAGGCAACCATGAGCTTTGAACCGCCGGGTGCCTGCCGTGTTATCGGTTATGGCCAGA AGCTGGGTCCTGTTGCAGCCGCCATGACCAATAGTGCATTCATCCAGGCAACCGAGCTGG TCCTCAGATGTAGTATTCAGAATATGATTATGACTACCATAGCGAGGCACCGCTGCACAGC GCAAGTATTGTGCTGCCTGCCGTGTTCGCAGCAAGTGAAGTGCTGGCCGAACAAGGCAAA ACCATTAGCGGCATCGACGTGATCCTGGCCGCCATTGTTGGCTTTGAAAGCGGTCCGCGT ATTGGCAAAGCCATCTACGGTAGCGATCTGCTGAATAACGGTTGGCATTGCGGTGCCGTTT ACGGTGCACCTGCCGGCGCATTAGCCACCGGCAAACTGCTGGGTCTGACCCCGGACAGC ATGGAGGATGCCCTGGGTATTGCATGCACCCAAGCCTGCGGTCTGATGAGCGCCCAGTAT GGCGGTATGGTTAAACGCGTGCAGCATGGCTTCGCCGCACGCAATGGTCTGCTGGGTGG TCTGCTGGCACATGGCGGCTATGAGGCCATGAAAGGCGTGCTGGAACGCAGCTATGGTG G
SpyCas9 Target DNA sequence (654 bp):
TCCTGGATGGCATTGCCTGTGCCTGGGTTGGTGCCCGCGTTCCGTGGAGTGAGAAGTATG TGCAGGCAACCATGAGCTTTGAACCGCCGGGTGCCTGCCGTGTTATCGGTTATGGCCAGA AGCTGGGTCCTGTTGCAGCCGCCATGACCAATAGTGCATTCATCCAGGCAACCGAGCTGG GGAAATTAGGTGCGCTTGGCTGGATGACTACCATAGCGAGGCACCGCTGCACAGCGCAAG TATTGTGCTGCCTGCCGTGTTCGCAGCAAGTGAAGTGCTGGCCGAACAAGGCAAAACCAT TAGCGGCATCGACGTGATCCTGGCCGCCATTGTTGGCTTTGAAAGCGGTCCGCGTATTGG CAAAGCCATCTACGGTAGCGATCTGCTGAATAACGGTTGGCATTGCGGTGCCGTTTACGG TGCACCTGCCGGCGCATTAGCCACCGGCAAACTGCTGGGTCTGACCCCGGACAGCATGG AGGATGCCCTGGGTATTGCATGCACCCAAGCCTGCGGTCTGATGAGCGCCCAGTATGGCG GTATGGTTAAACGCGTGCAGCATGGCTTCGCCGCACGCAATGGTCTGCTGGGTGGTCTGC TGGCACATGGCGGCTATGAGGCCATGAAAGGCGTGCTGGAACGCAGCTATGGTGG
SpyCas9NmeHNH and SpyCas9NmeBH Non-Target DNA sequence (60 bp; 5’-FAM fluorescein labeled):
CATTCATCCAGGCAACCGAGCTGGGGAAATTAGGTGCGCTTGGCTGGATGACTACCATAG C
SpyCas9NmeHNH and SpyCas9NmeBH Target DNA sequence (60 bp; 5’-FAM fluorescein labeled):
GCTATGGTAGTCATCCAGCCAAGCGCACCTAATTTCCCCAGCTCGGTTGCCTGGATGAATG
Size-exclusion binding assays
To test complexes formation between Acrs and NmeCas9 or its truncations, mixtures were prepared by incubating each Acr with NmeCas9 at the 1:3 molar ratio or with NmeCas9+sgRNA at the 1:10 ratio in the size exclusion buffer C (20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 2 mM MgCl2, and 1 mM DTT) on ice for 1 h.
Mixtures between Cas9 orthologs (SaCas9, SpyCas9, FnoCas9 and CjeCas9) with AcrIIC3 were prepared by mixing Cas9 and inhibitor at the molar ratio of 1:3 in the size exclusion buffer C; or with MBP-AcrIIC2 at the molar ratio of 1:6 in the size exclusion buffer F (20 mM Tris-HCl, pH 7.5, 100 mM NaCl, 2 mM MgCl2, 1 mM DTT) and incubating on ice for 1 h.
Mixtures between hybrid proteins SauCas9NmeHNH, SpyCas9NmeHNH and CjeCas9NmeHNH were prepared by mixing each hybrid Cas9 protein with AcrIIC3 at the 1:3 molar ratio in the size exclusion buffer C and incubating on ice for 1 h.
Protein mixtures were analyzed on either a Superdex 200 10/300 or a Superdex 75 10/300 column. Fractions were analyzed using SDS-polyacrylamide gel electrophoresis (PAGE) and visualized by Coomassie blue staining.
Pull-down assays
GST pull-down:
Free GST and the GST-AcrIIC3 complex were incubated with 20 µl glutathione-Sepharose 4B beads (GE Healthcare) at 10°C for 30 min and washed three times with the binding buffer C (20 mM Tris-HCl, pH 7.5, 300 mM NaCl, 2 mM MgCl2, 1 mM DTT). The purified NmeCas9, NmeCas9-∆HNH, and REC lobe proteins were incubated with the protein-coated beads at 10°C for 30 min. After incubation, the beads were washed three times with the same binding buffer. The bound proteins were eluted using the binding buffer containing 10 mM glutathione and analyzed by SDS-PAGE. The experiment was repeated three times.
MBP pull-down:
MBP-NmeCas9 was incubated with 50 µl Dextrin-Sepharose Hight Performance (GE Healthcare) beads at 10°C for 30 min and washed three times wit h the binding buffer C. The purified AcrIIC2, AcrIIC3 and their mutants were incubated with the protein-coated beads at 10°C for 30 min. After incubation, the beads were washed three times with the same binding buffer. The bound proteins were eluted using the binding buffer containing 20 mM maltose and analyzed by SDS-PAGE. The experiment was repeated three times.
Isothermal titration calorimetry binding assay
The dissociation constants of binding reactions of AcrIIC2 or AcrIIC2 mutants with BHs were determined by isothermal titration calorimetry (ITC) using a MicroCal ITC200 calorimeter. Proteins were dialyzed overnight against the working buffer (20 mM Tris-HCl, pH 7.5, 300 mM NaCl). The titration was carried out with 19 successive injections of 2 µL BH peptides at the 0.21–0.34 mM concentration, spaced 120 s apart, into the sample cell containing the AcrIIC2 or its mutants at the 0.03 mM concentration at 25°C. T he Origin software was used for baseline correction, integration, and curve fitting to a single site binding model.
Analytical ultracentrifugation
Analytical sedimentation velocity experiments were performed using a Proteome Lab XL-I analytical ultracentrifuge system (Beckman Coulter) at 20°C. Proteins were diluted with centrifugation buffer (20 mM Tris-HCl, pH 7.5, 300 mM NaCl) to a concentration of about 1 mg/ml. Samples were loaded onto a conventional double-sector quartz cell, mounted in a Beckman four-hole An-60 Ti rotor, and centrifuged at 50,000 rpm. Migration of the protein was monitored by absorbance at 280 nm. Data were calculated and analyzed using the SEDFIT software (http://www.analyticalultracentrifugation.com).
RNA binding assay
The affinity of sgRNA-Cas9 interactions in the absence and presence of AcrIIC2 was determined by DRaCALA (Roelofs et al., 2011; Vasilyev et al., 2015). Uniformly labelled radioactive 94-nt sgRNA was prepared from HindIII-linearized plasmid by in vitro transcription with phage T7 RNA polymerase and [α32P]-UTP. The labelled RNA was purified on 8% PAG with 8M urea, extracted from the gel by crash-and-soak method overnight, additionally purified by phenol-chloroform and chloroform extractions, precipitated by ethanol, and dissolved in water. Labeled RNA (500 cpm per experimental point) was diluted in 10 mM Tris-HCl, pH 7.5, 250 mM KCl, 5 mM MgCl2, 5 mM DTT, and 10 μg/mL bovine serum albumin. RNA was folded by heating at 42 °C for 15 min and cooling down on ice. AcrIIC 2 at 20 μM was added to the RNA samples when necessary. 9-µL RNA samples were mixed with 1-µL aliquot of Cas9 to yield final protein concentrations 2.5×10−11 to 1.5×10−6 M. After 60-min incubation on ice, 4-µL aliquot from each sample was spotted onto nitrocellulose membrane (Bio-Rad) and allowed to diffuse and dry on air for 30 min before exposing on the phosphorimager screen and scanning on Typhoon scanner (GE Healthcare). Radioactive signal was measured using Image J. The bound RNA was quantified as described in (Roelofs et al., 2011), and the binding curves were fitted to a one specific binding site model with Prism (GraphPad Software).
DNA binding assay
The 48 bp 5’-FAM fluorescein labeled linear target DNA was synthesized by Zixi. Binding reactions were conducted in the binding buffer (20 mM Tris-HCl, pH 7.5, 250 mM KCl, 5 mM MgCl2, 5 mM DTT, 5% glycerol). Variable amounts (2.0×10−9 to 1.0×10−4 M) of catalytically inactive NmeCas9D16/N611A and constant amount of AcrIIC2 (100 μM) were incubated on ice for 30 min. NmeCas9D16/N611A or NmeCas9D16/N611A-AcrIIC2 complex were incubated with 94 nt sgRNA for 30 min on ice at the molar ratio of 1:1. After sgRNA loading, 10 nM FAM-labeled DNA was added into the mixture and the binding reaction continued at 37°C for 30 min. Fluorescent polarization signal was recorded on a PerkinElmer EnSpire Multimode Plate Reader.
The sequence of the DNA oligonucleotide used in this study is listed below (PAM is in bold and the target sequence is in in italics):
NmeCas9 Non-Target DNA sequence (48 bp; 5’-FAM fluorescein labeled):
GGCAACCGAGCTGGTCCTCAGATGTAGTATTCAGAATATGATTATGAC
Peptide mass fingerprinting
Gel plugs were destained ultrasonically in 50 mM ammonium bicarbonate containing 40% (v/v) acetonitrile for 5 min, dehydrated in 200 mL 100% acetonitrile for 10 min, and dried in Speedvac (Labconco) for approximately 15 min. Proteins were reduced in 10 mM DTT (1 h, 56°C) and alkylated in 55 mM iodoacetamide (45 min, room temperature, in darkness). The gel plugs were washed twice with 50% acetonitrile in 25 mM ammonium bicarbonate, dried in Speedvac, and digested overnight (37°C) with 10 mL of 0.1 mg sequ ence-grade modified trypsin (Promega, v5113) in 25 mM ammonium bicarbonate. Five microliters of 10% trifluoroacetic acid were added to terminate the enzymatic reaction. The peptide mixture was desalted with Ziptip C18 column (Millipor). MALDI-TOF/TOF-MS analysis was performed on the New UltrafleXtreme mass spectrometer controlled by FlexControl 3.4 software package (Bruker Daltonics). The instrument was externally calibrated using the Bruker peptides calibration kit. The spectra were acquired in the positive ion reflection mode over the m/z range from 700 to 3500. MS data were analyzed on website http://www.matrixscience.com/, searching against the NCBI (or Swissprot) protein database.
QUANTIFICATION AND STATISTICAL ANALYSIS
Radioactive and fluorescent polarization data in Figures 4D and 4E are analyzed with Graphpad Prism software. ITC data in Figures 2H, 3B, S3D and S3E are analyzed with Origin software. Data are represented as mean ± SEM.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the coordinates and structure factors reported in this paper are PDB: 6J9K (Apo_AcrIIC2), 6J9M (NmeBH+AcrIIC2), 6J9L (FnoBH+AcrIIC2) and 6J9N (NmeHNH+AcrIIC3).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Top10 Chemically Competent Cell | TransGen Biotech | Cat#CD101-01 |
| Rosetta (DE3) Chemically Competent Cell | TransGen Biotech | Cat#CD801-01 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ULP1 | Homemade | N/A |
| T7 RNA polymerase | Homemade | N/A |
| MES | Sigma-Aldrich | Cat#M8250-250G |
| PEG 3000 | Sigma-Aldrich | Cat#81227-1KG |
| ethanol | Sigma-Aldrich | Cat#459836-1L |
| CHES | Sigma-Aldrich | Cat#C2885-100G |
| Sodium citrate | Hampton | Cat#HR2-235 |
| PEG 400 | QIAGEN | Cat#133086 |
| PEG 200 | Hampton | Cat#HR2-601 |
| 30% acrylamide and bis-acrylamide solution, 29:1 | LABLEAD | Cat#A3291-500ML |
| Critical Commercial Assays | ||
| JCSG Core Suite I | QIAGEN | Cat#130724 |
| JCSG Core Suite II | QIAGEN | Cat#130725 |
| JCSG Core Suite III | QIAGEN | Cat#130726 |
| JCSG Core Suite IV | QIAGEN | Cat#130727 |
| Crystal Screen | Hampton | Cat#HR2-110/112 |
| Index | Hampton | Cat#HR2-144 |
| Deposited Data | ||
| Structure of Apo_AcrIIC2 | This paper | PDB: 6J9K |
| Structure of NmeBH+AcrIIC2 | This paper | PDB: 6J9M |
| Structure of FnoBH+AcrIIC2 | This paper | PDB: 6J9L |
| Structure of NmeHNH+AcrIIC3 | This paper | PDB: 6J9N |
| Recombinant DNA | ||
| pRSF-Duet-1-His6-SUMO-NmeCas9 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas9_HNH508-667 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-455 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-241 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas9249-455 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas9Δ51-455 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas9ΔHNH | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-123 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W T54/D56A | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W R62A | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W R63A | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W R66A | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W R69A | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W R70A | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W R73A | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W R74A | This study | N/A |
| pRSF-Duet-1-His6-SUMO-NmeCas951-96W R69/R70/R73/R74A | This study | N/A |
| pRSF-Duet-1-His6-SUMO-SpyCas9 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-SpyCas951-96W | This study | N/A |
| pRSF-Duet-1-His6-SUMO-SpyCas9Nme-HNH | This study | N/A |
| pRSF-Duet-1-His6-SUMO-SaCas9 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-SaCas936-81W | This study | N/A |
| pRSF-Duet-1-His6-SUMO-SaCas9 Nme-HNH | This study | N/A |
| pRSF-Duet-1-His6-SUMO-CjeCas9 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-CjeCas937-82W | This study | N/A |
| pRSF-Duet-1-His6-SUMO-CjeCas9 Nme-HNH | This study | N/A |
| pRSF-Duet-1-His6-SUMO-FnoCas9 | This study | N/A |
| pRSF-Duet-1-His6-SUMO-FnoCas945-90W | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC1 | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC2 | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC2L36D | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC2Y11/I15/R20A | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC2E18A | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC2N23/D24/E25A | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC2D42/D43A | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC2Δ109-C | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC2K4/K10/R65/R66A | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC3 | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC3R5A | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC3R33A/D38A/Y40A | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC3N60A | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC3E92A/R103A | This study | N/A |
| pRSF-Duet-1-His6-SUMO- AcrIIC3M1S/F3S/L58S | This study | N/A |
| pET-28a-His6-MBP- NmeCas9 | This study | N/A |
| pET-28a-His6-MBP- NmeCas9Δ51-455 | This study | N/A |
| pET-28a-His6-MBP- AcrIIC2 | This study | N/A |
| pGEX-6P-1- AcrIIC3 | This study | N/A |
| Software and Algorithms | ||
| HKL2000 | Otwinowski and Minor, 1997 | http://www.hkl-xray.com/ |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org/ |
| Coot | Emsley et al., 2010 | http://www2.mrc-lmb.cam.ac.uk/Personal/pemsley/coot/ |
| Pymol | The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. | https://pymol.org/ |
| OriginPro 8 | OriginPro software | N/A |
| ImageJ | ImageJ software | N/A |
| Graphpad Prism | GraphPad software | N/A |
| Other | ||
| Amicon concentrators (3K) | Millipore | Cat#UFC900324 |
| Amicon concentrators (30K) | Millipore | Cat#UFC90302 |
| Ni Sepharose™ 6 FF beads | GE Healthcare | Cat#17-5318-03 |
| Glutathione Sepharose™ 4 Fast Flow beads | GE Healthcare | Cat#17-5132-02 |
| HiLoad™ 16/600 Superdex™ 200 pg | GE Healthcare | Cat#28-9893-35 |
| Superdex™ 75 10/300 GL | GE Healthcare | Cat#17-5174-01 |
| Superdex 200 10/300 GL | GE Healthcare | Cat#17-5175-01 |
| HiTrap™ Heparin HP (5 mL) | GE Healthcare | Cat#17-0407-01 |
| HiTrap™ SP Sepharose FF (5 mL) | GE Healthcare | Cat#17-5157-01 |
HIGHLIGHTS.
Crystal structures of Cas9-bound AcrIICs suggest distinct inhibitory mechanisms
AcrIIC2 interferes with RNA- and DNA-loading steps through binding to Cas9 BH motif
AcrIIC3 induces Cas9 dimerization by interacting with the HNH domain and REC lobe
Cas9 enzymes can be reengineered to become susceptible to AcrIIC’s inhibition
ACKNOWLEDGMENTS
We thank the staff from the BL18U1 and BL19U1 beamlines of National Facility for Protein Science in Shanghai (NFPS) at Shanghai Synchrotron Radiation Facility (SSRF) for assistance during data collection. We thank Dr. Fuquan Yang and Dr. Yuanyuan Chen for their support with the mass spectrometry and ITC assays. This research was supported by funds from the National Key R&D Program of China (2018YFA0508000 and 2018YFA0507203), the National Natural Science Foundation of China (91753133 and 31670903), and the CAS Pilot Strategic Science and Technology Projects B (XDB08020204) to P.G., and by funds from the NIH (R01GM112940) to A.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C, Niewoehner O, Duerst A, and Jinek M (2014). Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Garcia B, Strum S, Du MJ, Rollins MF, Hidalgo-Reyes Y, Wiedenheft B, Maxwell KL, and Davidson AR (2015). Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 526, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL, and Davidson AR (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–U181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AL, Zhang JY, Rollins MF, Osuna BA, Wiedenheft B, and Bondy-Denomy J (2018). Bacteriophage cooperation suppresses CRISPR-Cas3 and Cas9 immunity. Cell 174, 917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, Hoffmann C, Nosaka L, Bondy-Denomy J, Maxwell KL, Davidson AR, et al. (2017). Structure reveals mechanisms of viral suppressors that intercept a CRISPR RNA-guided surveillance complex. Cell 169, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, and Charpentier E (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong D, Guo MH, Wang SH, Zhu YW, Wang S, Xiong Z, Yang JZ, Xu ZL, and Huang ZW (2017). Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature 546, 436–439. [DOI] [PubMed] [Google Scholar]
- Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, and Sorek R (2018). Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, eaar4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gophna U, Kristensen DM, Wolf YI, Popa O, Drevet C, and Koonin EV (2015). No evidence of inhibition of horizontal gene transfer by CRISPR-Cas on evolutionary timescales. Isme J 9, 2021–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, Merk A, Eng ET, Raczkowski AM, Fox T, Earl LA, et al. (2017). Cryo-EM structures reveal mechanism and inhibition of DNA targeting by a CRISPR-Cas surveillance complex. Cell 171, 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LB, Doxzen KW, Ma E, Liu JJ, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, et al. (2017). A Broad-spectrum inhibitor of CRISPR-Cas9. Cell 170, 1224–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, and Zhang F (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes AP, Rousseau GM, Lemay ML, Horvath P, Romero DA, Fremaux C, and Moineau S (2017). An anti-CRISPR from a virulent streptococcal phage inhibits Streptococcus pyogenes Cas9. Nat. Microbiol 2, 1374–1380. [DOI] [PubMed] [Google Scholar]
- Jeong H, Park J, Jun Y, and Lee C (2017). Crystal structures of Mmm1 and Mdm12-Mmm1 reveal mechanistic insight into phospholipid trafficking at ER-mitochondria contact sites. Proc. Natl. Acad. Sci. USA 114, E9502–E9511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Liu JJ, Osuna BA, Xu M, Berry JD, Rauch BJ, Nogales E, Bondy-Denomy J, and Doudna JA (2018). Temperature-responsive competitive inhibition of CRISPR-Cas9. Mol. Cell, 10.1016/j.molcel.2018.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Taylor DW, Chen JS, Kornfeld JE, Zhou K, Thompson AJ, Nogales E, and Doudna JA (2016). Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang FG, Zhou KH, Ma LL, Gressel S, and Doudna JA (2015). A Cas9-guide RNA complex preorganized for target DNA recognition. Science 348, 1477–1481. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, et al. (2014). Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsberger M, Gandon S, Meaden S, Rollie C, Chevallereau A, Chabas H, Buckling A, Westra ER, and van Houte S (2018). Anti-CRISPR phages cooperate to overcome CRISPR-Cas immunity. Cell 174, 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yin M, Wang M, and Wang Y (2018). Phage AcrIIA2 DNA mimicry: structural basis of the CRISPR and Anti-CRISPR arms race. Mol. Cell, 10.1016/j.molcel.2018.11.016 [DOI] [PubMed] [Google Scholar]
- Marino ND, Zhang JY, Borges AL, Sousa AA, Leon LM, Rauch BJ, Walton RT, Berry JD, Joung JK, Kleinstiver BP, et al. (2018). Discovery of widespread Type I and Type V CRISPR-Cas inhibitors. Science 362, 240–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell KL (2017). The anti-CRISPR story: a battle for survival. Mol. Cell 68, 8–14. [DOI] [PubMed] [Google Scholar]
- Nishimasu H, Cong L, Yan WX, Ran FA, Zetsche B, Li Y, Kurabayashi A, Ishitani R, Zhang F, and Nureki O (2015). Crystal structure of Staphylococcus aureus Cas9. Cell 162, 1113–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, and Nureki O (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee JY, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, et al. (2016a). Naturally occurring off-switches for CRISPR-Cas9. Cell 167, 1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Bondy-Denomy J, Cheung VHW, Maxwell KL, and Davidson AR (2014). A new group of phage anti-CRISPR genes inhibits the Type I-E CRISPR-Cas system of Pseudomonas aeruginosa. Mbio 5, e00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Staals RHJ, Taylor C, Watson BNJ, Saha S, Fineran PC, Maxwell KL, and Davidson AR (2016b). Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat. Microbiol 1, 16085. [DOI] [PubMed] [Google Scholar]
- Peng RC, Xu Y, Zhu TF, Li NN, Qi JX, Chai Y, Wu M, Zhang XZ, Shi Y, Wang PY, et al. (2017). Alternate binding modes of anti-CRISPR viral suppressors AcrF1/2 to Csy surveillance complex revealed by cryo-EM structures. Cell Res 27, 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peselis A, Gao A, and Serganov A (2016). Preparation and crystallization of riboswitches. Methods Mol. Biol 1320, 21–36. [DOI] [PubMed] [Google Scholar]
- Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, and Bondy-Denomy J (2017). Inhibition of CRISPR-Cas9 with bacteriophage Proteins. Cell 168, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofs KG, Wang J, Sintim HO, and Lee VT (2011). Differential radial capillary action of ligand assay for high-throughput detection of protein-metabolite interactions. Proc. Natl. Acad. Sci. USA 108, 15528–15533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Jiang FG, Liu JJ, Bray NL, Rauch BJ, Baik SH, Nogales E, Bondy-Denomy J, Corn JE, and Doudna JA (2017). Disabling Cas9 by an anti-CRISPR DNA mimic. Sci. Adv 3, e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, LaFrance B, Kaplan M, and Doudna JA (2015). Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527, 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilyev N, Polonskaia A, Darnell JC, Darnell RB, Patel DJ, and Serganov A (2015). Crystal structure reveals specific recognition of a G-quadruplex RNA by a beta-turn in the RGG motif of FMRP. Proc. Natl. Acad. Sci. USA 112, E5391–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Oost J, Westra ER, Jackson RN, and Wiedenheft B (2014). Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol 12, 479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Houte S, Ekroth AKE, Broniewski JM, Chabas H, Ashby B, Bondy-Denomy J, Gandon S, Boots M, Paterson S, Buckling A, et al. (2016). The diversity-generating benefits of a prokaryotic adaptive immune system. Nature 532, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, La Russa M, and Qi LS (2016a). CRISPR/Cas9 in genome editing and beyond. Annu. Rev. Biochem 85, 227–264. [DOI] [PubMed] [Google Scholar]
- Wang JY, Ma J, Cheng Z, Meng X, You LL, Wang M, Zhang XZ, and Wang YL (2016b). A CRISPR evolutionary arms race: structural insights into viral anti-CRISPR/Cas responses. Cell Res 26, 1165–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XF, Yao DQ, Xu JG, Li AR, Xu JP, Fu PH, Zhou Y, and Zhu YQ (2016c). Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat. Struct. Mol. Biol 23, 868–870. [DOI] [PubMed] [Google Scholar]
- Wong LH, and Levine TP (2017). Tubular lipid binding proteins (TULIPs) growing everywhere. Bba-Mol. Cell Res 1864, 1439–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, and Patel DJ (2017). Inhibition mechanism of an anti-CRISPR suppressor AcrIIA4 targeting SpyCas9. Mol. Cell 67, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the coordinates and structure factors reported in this paper are PDB: 6J9K (Apo_AcrIIC2), 6J9M (NmeBH+AcrIIC2), 6J9L (FnoBH+AcrIIC2) and 6J9N (NmeHNH+AcrIIC3).