

We identified unique molecular mechanisms of resistance to CDK4/6 inhibitors, an area of intense biomedical investigation.
Abstract
The cyclin-dependent kinase 4/6 (CDK4/6) kinase is dysregulated in melanoma, highlighting it as a potential therapeutic target. CDK4/6 inhibitors are being evaluated in trials for melanoma and additional cancers. While beneficial, resistance to therapy is a concern, and the molecular mechanisms of such resistance remain undefined. We demonstrate that reactivation of mammalian target of rapamycin 1 (mTORC1) signaling through increased expression of the amino acid transporter, solute carrier family 36 member 1 (SLC36A1), drives resistance to CDK4/6 inhibitors. Increased expression of SLC36A1 reflects two distinct mechanisms: (i) Rb loss, which drives SLC36A1 via reduced suppression of E2f; (ii) fragile X mental retardation syndrome–associated protein 1 overexpression, which promotes SLC36A1 translation and subsequently mTORC1. Last, we demonstrate that a combination of a CDK4/6 inhibitor with an mTORC1 inhibitor has increased therapeutic efficacy in vivo, providing an important avenue for improved therapeutic intervention in aggressive melanoma.
INTRODUCTION
Dysregulation of p16INK4a–cyclin D1–CDK4/6-Rb pathway frequently occurs in melanoma (1, 2). p16INK4a, an allosteric inhibitor of cyclin-dependent kinase 4/6 (CDK4/6), is inactivated in 70% of melanomas (3); cyclin D1 is amplified in 44.4% of acral melanoma, 10.5% of lentigo maligna melanoma, and 5.6% of superficial spreading melanoma; cyclin D1 is overexpressed in an additional 30% of melanomas without amplification (4). Further support for the importance of dysregulation of cyclin D1-CDK4/6 stems from the demonstration that inactivation of p16INK4a or deletion of Fbxo4, the specificity component of the E3 ligase that directs cyclin D1 degradation, cooperates with BrafV600E to induce metastatic melanoma, while reduced cyclin D1 gene dosage abrogates melanoma development in mouse models (5), implicating inhibition of cyclin D1–CDK4/6 as a potential strategy for melanoma therapy.
Palbociclib is an orally available U.S. Food and Drug Administration (FDA)–approved CDK4/6 inhibitor (CDK4/6i) that specifically inhibits CDK4/6 (6). Palbociclib exposure triggers early G1 cell cycle arrest in normal and tumor cells, including those derived from melanoma, breast, colon, lung, and myeloid leukemia cells. Arrest reflects inhibition of CDK4/6-dependent phosphorylation of Rb; consequently, CDK4/6i or CKD4/6i’s such as palbociclib are being tested in a variety of clinical trials (7–9). Given that a number of patients exposed to a therapeutic modality will develop resistance and consequently require additional therapies, dissecting the mechanisms of acquired resistance to CDK4/6i is of clinically necessity.
With regard to melanoma, melanoma-derived cells are responsive to CDK4/6 inhibition with CDK4/6i (10), reflecting, in part, maintenance of wild-type Rb in a majority of melanomas (>95%). Prolonged exposure of CDK4/6i induces senescence in vitro and in vivo, which is associated with inhibition of mammalian target of rapamycin (mTOR) (10). The latter result suggested that combining mTORC1 inhibitors with CDK4/6 inhibition in melanoma could have important clinical implications. It further implies the potential for reactivation of mTOR as a potential mechanism of acquired resistance (10). In addition, recent studies demonstrated that inhibition of mitogen-activated protein kinase (MAPK) or phosphatidylinositol 3-kinase (PI3K) pathways enhances the efficacy of CDK4/6i in esophageal squamous cell carcinoma and human epidermal growth factor receptor 2 (HER2)–positive breast cancer, respectively (11, 12). However, critical factors that directly drive the acquired resistance to CDK4/6i are undefined.
We performed whole-exome sequencing and RNA sequencing (RNA-seq) analyses to dissect the molecular mechanisms of CDK4/6i-induced senescence and resistance to CDK4/6i and demonstrate that mTORC1 signaling activation is needed for the resistance. None of the known substrates for the CDK4/6 kinase, which include forkhead box protein M1 (FOXM1), protein arginine N-methyltransferase 5 (PRMT5), and histone acetyltransferase GCN5 (GCN5), regulate mTORC1 signaling. Here, we identify solute carrier family 36 member 1 (SLC36A1) (13–15), a neutral amino acid/proton symporter that has a pH-dependent electronic transport activity for small amino acids such as alanine, glycine, and proline, as a driver that drives mTORC1 signaling, contributing CDK4/6i resistance in melanoma. Furthermore, we identify fragile X mental retardation syndrome–associated protein 1 (FXR1), an RNA binding protein with putative oncogenic potential in head and neck carcinomas, overexpression as a resistance mechanism. FXR1 overexpression is associated with senescence bypass (16) and is linked with cyclin D1 through a common regulatory E3 ligase (17). We demonstrate a novel mechanism where FXR1 promotes SLC36A1-mTORC1 signaling and drives resistance to CDK4/6i. Last, we demonstrate the therapeutic efficacy of CDK4/6i and an mTORC1 inhibitor in vivo, providing support for a new therapeutic strategy in melanoma.
RESULTS
E2f targets and mTOR signaling are dysregulated in CDK4/6i-resistant cells
CDK4/6i-dependent senescence in melanoma correlates with mTORC1 inhibition; conversely, mTORC1 reactivation correlates with resistance to CDK4/6i (10). However, in the absence of the molecular mechanisms of mTORC1 regulation in cells treated with CDK4/6i, the biological significance of the relationship remains uncertain. We therefore established CDK4/6i-resistant (CR) cells via long-term culture with palbociclib using either melanoma 1205Lu (1205CR1, 1205CR2, 1205CR6, and 1205CR7)– or esophageal TE7 (TE7CR)–derived cells. We verified resistance by proliferation in 1 μM palbociclib (fig. S1A). Clones (1205CR1, 1205CR2, 1205CR6, and 1205CR7) are cross-resistant to ribociclib (LEE011) (fig. S1A) and are referred to as CR. To circumvent issues of mechanistic heterogeneity associated with “populations” of resistant cells, we isolated clonal lines, allowing us to precisely identify unique resistance mechanisms. All CR cells were continuously maintained in the CDK4/6i-containing medium. Critically, we confirmed that everolimus, an mTORC1 inhibitor, reversed CDK4/6i resistance in all of CR cells, consistent with previous work that CDK4/6i resistance correlates with mTORC1 activation (fig. S1, B to D) (10).
We first assessed potential alterations of DNA and mRNA expression in control versus palbociclib-treated cells [1-day treatment for cell cycle arrest and 8-day treatment for senescence (10)] and CR clones (1205CR1, 1205CR2, 1205CR6, 1205CR7, and TE7CR) by whole-exome sequencing and RNA-seq (Fig. 1A) to dissect the molecular mechanisms of how mTORC1 signaling is regulated by CDK4/6i. While exome sequencing revealed no additional genomic alterations in the CR cells compared to control cells, RNA-seq revealed substantial changes when comparing control cells with either senescent or resistant cells (Fig. 1B). Differentially regulated transcripts in each group are highlighted by a Venn diagram (>1.5-fold, q < 0.1; Fig. 1C). The top ranked biological pathways from systems-level analysis of the RNA-seq data revealed that senescence-associated pathways such as cytokine-cytokine receptor pathway, cell adhesion molecules pathway, tumor necrosis factor signaling pathway, and DNA replication pathway that is primarily controlled by E2f transcription factors are significantly different between senescent and CR cells, supporting our previous work that CDK4/6i resulted in cell cycle arrest and senescence (fig. S1E). The PI3K-Akt pathway is also significantly suppressed in CDK4/6i-induced senescent cells and reactivated in 1205CR1, 1205CR2, 1205CR6, and 1205CR7 cells, suggesting a link between E2f and PI3K-Akt pathways (fig. S1E). A heat map comparing 1205Lu parental cells, CDK4/6i-treated cells, CR cells, and Gene Set Enrichment Analysis (GSEA; www.broadinstitute.org/GSEA) using CDK4/6i-treated cells and CR cells revealed that CDK4/6i significantly inhibits the expression of E2f targets and mTOR-dependent signaling (Fig. 1, D and E). To verify the differential expression of E2f targets, we assessed mRNA and protein accumulation of CDK1, Flap endonuclease 1 (FEN1), and proliferating cell nuclear antigen (PCNA), well-known E2f1 targets, and demonstrated that their expression is significantly reduced in CDK4/6i-treated cells (Fig. 1, F and G).
Fig. 1. Comprehensive analyses of CR cells.
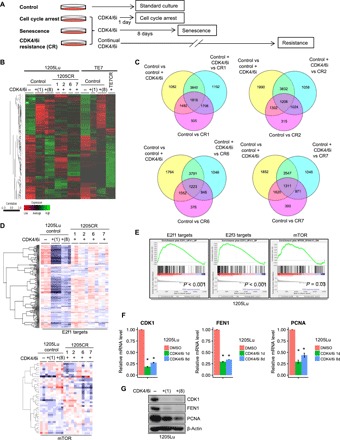
(A) Scheme for exposure of melanoma-derived cell lines to palbociclib and outcome. (B) Heat map with hierarchical clustering of samples from 1205Lu cells (control), 1205Lu cells treated with palbociclib (1 μM) for 1 or 8 days, and CR clones (1205CR1, 1205CR2, 1205CR6, and 1205CR7) (left) and TE7 cells treated with palbociclib (1 μM) for 1 or 8 days and CR cells (TE7CR) (right). (C) Venn diagrams of differentially expressed genes of samples [dimethyl sulfoxide (DMSO) versus palbociclib treatment (1 μM) for 8 days (control versus control + CDK4/6i), palbociclib treatment (1 μM) for 8 days versus 1205CR1, 1205CR2, 1205CR6, or 1205CR7 (CDK4/6i versus CR1, CR2, CR6, or CR7), control + palbociclib (CDK4/6i) versus CR1, CR2, CR6, or CR7 (q < 0.1)]. (D) Heat map of 1205Lu cells from (B) for expression of E2f target genes and mTOR signaling. (E) GSEA analysis of CR cells compared to senescent cells [palbociclib treatment (1 μM) for 8 days] for E2f1 targets (P < 0.001), E2f3 targets (P < 0.001), and mTOR signaling (P = 0.03). (F and G) Samples from 1205Lu cells with or without treatment of palbociclib (1 μM) for 24 hours or 8 days were prepared. (F) Quantitative polymerase chain reaction (qPCR) analysis using sets of primers for CDK1, FEN1, and PCNA. Data were normalized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and represent means ± SD. *P < 0.01 (one-sample two-tailed Student’s t test; n = 3). (G) Western blot analysis using antibodies to CDK1, FEN1, PCNA, and β-actin.
CDK2 overexpression contributes but does not drive CDK4/6i resistance
Given that bioinformatic analysis highlighted reactivation of E2f1-dependent target genes in all CR cell lines analyzed, consistent with cell cycle reentry (Fig. 1, D and E), we speculated that loss of the key CDK4/6-cyclin D1 substrate and upstream inhibitor of E2f, Rb, might contribute to resistance. Consistently, Rb was undetectable in 1205CR1-2; conversely, Rb was readily detectable in 1205CR6-7, highlighting different mechanisms of resistance (Fig. 2A). We also established CDK4/6i-resistant cells in WM3918 melanoma cells [3918CR1, 3918CR2, and 3918CR4; resistance was confirmed by 5-bromo-2′-deoxyuridine (BrdU) incorporation and expression of E2f targets; fig. S2, A and B] and observed Rb retention in 3918CR1, 3918CR2, and 3918CR4 cells (fig. S2C). Because Rb loss eliminates cellular dependence on CDK4/6 kinase function (18), its loss at a detectable frequency is expected. Enforced expression of Rb resensitized 1205CR1-2 cells to CDK4/6i treatment (Fig. 2, B to D, and fig. S3A), analogous with parental cells demonstrating arrest by showing that CDK4/6i is Rb dependent (10).
Fig. 2. Different resistance mechanisms between 1205CR1-2 and 1205CR6-7 cells.
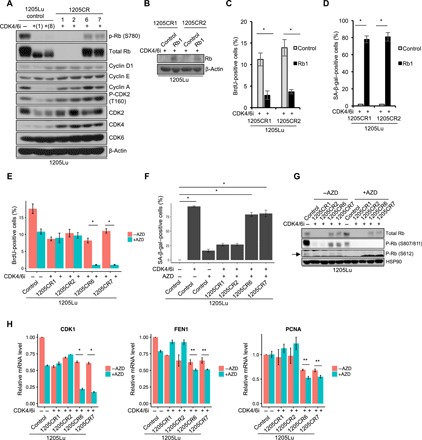
(A) Western blot analysis of lysates from 1205Lu cells treated with or without palbociclib (1 μM) for 1 or 8 days, and CR clones proliferating with palbociclib (1 μM) (1205CR1, 1205CR2, 1205CR6, and 1205CR7) using antibodies indicated on the right of the panel. (B) Western blot analysis of lysates from 1205CR1-2 cells overexpressing vehicle (control) or Rb1 using antibodies to Rb1 and β-actin. (C) 1205CR1-2 cells were subjected to BrdU incorporation for 45 min following introduction of Rb1 for 48 hours. BrdU-positive cells were determined by fluorescence-activated cell sorting (FACS) analysis. Data represent means ± SD. *P < 0.01 (two-tailed Student’s t test; n = 3). (D) Quantification of senescence-associated β-galactosidase (SA-β-gal)–positive cells in 1205CR1-2 with or without Rb1. Data represent means ± SD. *P < 0.01 (two-tailed Student’s t test; n = 3). (E) 1205Lu, 1205CR1, 1205CR2, 1205CR6, and 1205CR7 were subjected to a 45-min BrdU pulse following exposure to AZD5438 (AZD) treatment (0.5 μM). BrdU-positive cells were determined by FACS analysis and quantified; data represent means ± SD, *P < 0.01 (two-tailed Student’s t test; n = 3). (F) Quantification of SA-β-gal–positive cells in 1205Lu, 1205CR1, 1205CR2, 1205CR6, and 1205CR7 cells treated with AZD5438 (0.5 μM) or palbociclib (1 μM) for 8 days; data represent means ± SD, *P < 0.01 (two-tailed Student’s t test; n = 3). (G) Western blot analysis of lysates from 1205Lu, 1205CR1, 1205CR2, 1205CR6, and 1205CR7 cells after treatment of AZD5438 (0.5 μM) for 24 hours using antibodies indicated on the right of the panel. (H) qPCR analysis of samples from (G) using sets of primers for CDK1, FEN1, and PCNA. Data were normalized by GAPDH and represent means ± SD, *P < 0.01 (two-tailed Student’s t test; n = 3) and **P < 0.05 (two-tailed Student’s t test; n = 3).
The retention of Rb in 1205CR6-7, 3918CR1, 3918CR2, and 3918CR4 versus its loss in 1205CR1-2, TE7CR, and TE10CR (CR cells derived from esophageal TE7 and TE10) reveals differing mechanisms of resistance to CDK4/6i (Fig. 2A and fig. S2, C and D). When considering potential mechanisms, we noted that Rb is phosphorylated in the presence of CDK4/6i (Fig. 2A), suggesting retention of CDK function and potential CDK redundancy. Although CDK2, CDK4, and CDK6 levels are relatively stable when comparing 1205CR1 and 1205CR2 with 1205CR6 and 1205CR7 (Fig. 2A), we considered whether cells remained dependent on CDK2 activity. We treated CR cells with AZD5438, a CDK2 inhibitor (The half maximal inhibitory concentration: 6 nM toward CDK2, 16 nM toward CDK1, and 20 nM toward CDK9) (19). AZD5438 treatment triggered G1 cell cycle arrest after 1 day and senescence by 8 days following treatment in 1205CR6-7; conversely, 1205CR1-2, where Rb is lost, are refractory to AZD5438 (Fig. 2, E and F, and fig. S3, B and C). AZD5438 exposure suppressed CDK2-mediated Rb phosphorylation at serine-612 and serine-807/811, sites associated with CDK2 activity (Fig. 2G) (20, 21). Consistently, AZD5438 also suppressed E2f targets such as CDK1, FEN1, and PCNA, indicating that E2f1 activity is suppressed by AZD5438 (Fig. 2H). To ensure that AZD5438-induced G1 cell cycle arrest and senescence in 1205CR6-7 cells reflect specific inhibition of CDK2, we performed CDK2 knockdown by siCDK2. BrdU incorporation and senescence-associated β-galactosidase (SA-β-gal) assay revealed that CDK2 knockdown induced G1 cell cycle arrest after 2 days and senescence by 8 days in 1205CR6-7 cells (fig. S3, D to F). We confirmed reduced CDK2 expression following siCDK2 transfection, while CDK1 expression is not affected (fig. S3G). Consistently, CDK2 knockdown also suppressed E2f1 transcriptional activity in 1205CR6-7 cells (fig. S3H).
The above results demonstrate that 1205CR6-7 cells remain dependent on CDK2 activity once resistance is established but do not address whether increased CDK2 can override CDK4/6i-induced senescence and drive resistance. We used an overexpression approach to determining whether increased CDK2 can drive resistance to CDK4/6i-induced senescence. We also assessed CDK4 and CDK6, since CDK6 overexpression is thought to be a potential mechanism of resistance to LY2835219, a unique CDK4/6i (22). While overexpression of CDK2 had little effect on cell cycle distribution, overexpression of either CDK4 or CDK6 increased S phase and decreased G1 phase as previously reported (fig. S4, A and B) (23). Notably, cells overexpressing CDK2, CDK4, or CDK6 arrested in G1 phase within 24 hours of CDK4/6i treatment (fig. S4, A and B) and entered into a senescent state as determined by SA-β-gal staining and clonogenic growth assays by 8 days of treatment (fig. S4, C to E). Collectively, these data suggest that while CR cells that retain Rb remain dependent on CDK2 activity and CDK2i might be a promising target with CDK4/6i in CR cells, overexpression of individual CDKs does not drive CDK4/6i resistance.
SLC36A1 drives resistance to CDK4/6i-induced senescence in melanoma
While Rb loss results in cells that proliferate independent of CDK4/6, our data suggest that retention of Rb expression results from events upstream or parallel to cyclin D-CDK4/6. Given that the cyclin D-CDK4/6 kinase is acutely responsive to a multitude of signal transduction pathways, we took note of the mTORC1 signature highlighted by the GSEA (Fig. 1E). Because mTOR activity is regulated by amino acid availability, we considered whether increasing amino acid availability would contribute to reactivation of mTORC1. Supplementation of medium with small neutral amino acids did not activate phosphorylated S6 (p-S6), a downstream target of mTORC1 signaling (fig. S5A), and did not override CDK4/6i-induced senescence (fig. S5, B and C), suggesting loss of components that regulate amino acid transport.
We turned our attention to amino acid transporters, which, upon overexpression, have the potential to drive reactivation of mTOR (24, 25). To gain a broad insight into expression of transporters from our RNA-seq data, we generated a heat map for SLC (SLC) transporters, which revealed differential expression of select SLC transporters that regulate uptake of nutrients, amino acids, absorption of drugs, and metabolism, across CR cell lines (Fig. 3A). We focused our attention on SLC family members known to function in the transport of amino acids linked with mTOR activation and assessed mRNA abundance of select SLC members in parental, parental + palbociclib and CR cells (fig. S5D). We identified SLC36A1, a neutral amino acid/proton symporter that has a pH-dependent electronic transport activity for small amino acids such as alanine, glycine, and proline. Critically, SLC36A1 localizes on the lysosome where it has been linked with the regulation of mTORC1 signaling (14, 26, 27). SLC36A1 was significantly suppressed by CDK4/6i but back to or slightly higher than basal expression in CR cells (Fig. 3, B and C). SLC36A1 is also highly expressed in 3918 CR cells compared to control (fig. S2C). We also assessed the effect of CDK4/6i on the regulation of SLC36A1 expression in vivo. We performed immunohistochemistry (IHC) staining for SLC36A1 with tumors generated following injection of 983B melanoma cells into the flanks of severe combined immunodeficient (SCID) mice and treated with or without CDK4/6i (10). IHC staining revealed that CDK4/6i treatment reduced SLC36A1 in responsive tumors, but as cells acquired resistance to CDK4/6i, SLC36A1 expression was restored (Fig. 3D).
Fig. 3. SLC36A1 overrides CDK4/6i-induced senescence.
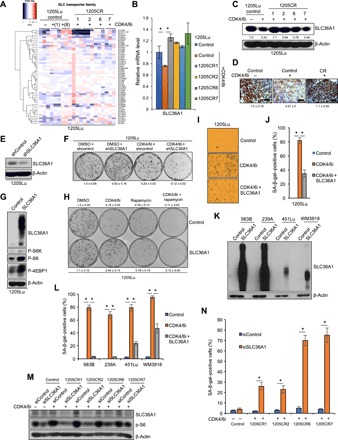
(A) Clustering of SLC transporters from 1205Lu cells treated with or without palbociclib (1 μM) for 1 day and 1205CR1, 1205CR2, 1205CR6, and 1205CR7 cells proliferating with palbociclib (1 μM) for 8 days. (B) qPCR analysis of samples from 1205Lu cells treated with or without palbociclib (1 μM) for 8 days or in 1205CR1, 1205CR2, 1205CR6, and 1205CR7 cells proliferating with palbociclib (1 μM) using a set of primers for SLC36A1. Data were normalized by GAPDH and represent means ± SD. *P < 0.01 (two-tailed Student’s t test; n = 3). (C) Western blot analysis of lysates from (B) for SLC36A1 and β-actin. The numbers indicate quantifications of SLC36A1 determined by SLC36A1/β-actin ratio. (D) Representative images of IHC staining of sections from xenograft tumors treated with either vehicle or palbociclib (1 μM) or tumors resistance to CDK4/6i for SLC36A1. Scale bars, 100 μm. The numbers indicate quantification of SLC36A1 intensity determined by IHC scoring (see Materials and Methods) from three independent experiments. (E) Western blot analysis of lysates from 1205Lu cells introduced with shcontrol or shSLC36A1 using antibodies to SLC36A1 and β-actin. (F) Clonogenic colony formation assay of cells from (E) treated with or without palbociclib (1 μM) for 8 days. The numbers indicate quantification of colonies from three independent experiments. (G) Western blot analysis of lysates from 1205Lu cells infected with empty vector (control) or SLC36A1 using antibodies to SLC36A1, p-S6K, and β-actin. (H) Clonogenic colony formation assay of cells from (G) treated with rapamycin (50 nM), palbociclib (1 μM), or rapamycin (50 nM) + palbociclib (1 μM) for 8 days. The numbers indicate quantification of colonies from three independent experiments. (I) Representative images of SA-β-gal staining from (G) treated with palbociclib (1 μM) for 8 days. (J) Quantification of SA-β-gal–positive cells from (I). Data represent means ± SD. *P < 0.01 (two-tailed Student’s t test; n = 3). (K) Western blot analysis of lysates from WM983B, WM239A, 451Lu, and WM3918 cells infected with empty vector (control) or SLC36A1 using antibodies to SLC36A1 and β-actin. (L) Quantification of SA-β-gal–positive cells from (K) and parental cells treated with or without palbociclib (1 μM) for 8 days. Data represent means ± SD. *P < 0.01 (two-tailed Student’s t test; n = 3). (M) Western blot analysis of lysates from 1205Lu, 1205CR1, 1205CR2, 1205CR6, and 1205CR7 cells transfected with siControl or siSLC36A1 using antibodies against SLC36A1, p-S6, and β-actin. (N) Quantification of SA-β-gal–positive cells after 8 days after transfection; data represent means ± SD, *P < 0.01 (two-tailed Student’s t test; n = 3).
We reasoned that if SLC36A1 contributed to CDK4/6i resistance, then SLC36A1 knockdown should facilitate CDK4/6i-induced senescence. Clonogenic colony assay revealed that combined shSLC36A1 + CDK4/6i was more effective in reducing colony outgrowth, a surrogate for stable senescence (10), than CDK4/6i alone (Fig. 3, E and F). We obtained consistent results when different short hairpin RNAs to knockdown SLC36A1 were used (fig. S5, E and F). Conversely, enforced expression of SLC36A1 should bypass CDK4/6i-induced senescence. 1205Lu cells stably expressing SLC36A1 were generated with successful activation of mTORC1 signaling (Fig. 3G). Note that when amino acid transporters are stably overexpressed in cells, they migrated as a smear, indicating the expression of glycosylated forms of amino acid transporters including SLC36A1 and SLC38A9 (28, 29). Recent work demonstrated that a glycosylation-deficient mutant of SLC36A1 is unstable and degraded mainly via the endoplasmic reticulum–associated degradation pathway (30). Clonogenic colony outgrowth and SA-β-gal assay revealed that SLC36A1 overexpression resulted in cells refractory to CDK4/6i-induced senescence (Fig. 3, H to J), while cells overexpressing SLC36A1 still arrest following CDK4/6i treatment (fig. S5G). We restored senescence by treatment with rapamycin, an mTORC1 inhibitor, consistent with mTORC1 dependence (Fig. 3H). To ensure that SLC36A1 universally overrides CDK4/6i-induced senescence in melanoma but is not limited to 1205Lu cell line, SLC36A1 was overexpressed in different melanoma cell lines (Fig. 3K). SA-β-gal staining confirmed that SLC36A1 is critical for bypass of CDK4/6i-induced senescence in all of melanoma cell lines tested (Fig. 3L and fig. S5H). We note that SLC36A1 completely overcomes CDK4/6i-induced senescence in 983B and 239A (Fig. 3L), which might reflect extremely high expression of SLC36A1 (Fig. 3K). Last, we assessed whether loss of SLC36A1 resensitizes CR cells to CDK4/6i. We used knockdown approach to eliminating the expression of SLC36A1 since SLC36A1 inhibitors are not commercially available. Knockdown of SLC36A1 mitigated mTORC1 signaling as assessed by p-S6 (Fig. 3M) and reduced S phase population in both 1205Lu parental cells and CR cells (fig. S5I). SA-β-gal staining revealed that acute knockdown of SLC36A1 resensitized CR cells to CDK4/6i-induced senescence, while parental cells did not undergo senescence by knockdown of SLC36A1 (Fig. 3N and fig. S5J), consistent with the mTORC1 inhibitor everolimus treatment, reversing CDK4/6i resistance (fig. S1, B to D) and suggesting that increased SLC36A1-driven CDK4/6i resistance is elicited through mTORC1 signaling. We noted that 1205CR6-7 cells, where Rb is retained, are more sensitive to acute knockdown of SLC36A1 than 1205CR1-2 cells, where Rb is lost (Fig. 3N). We reasoned that 1205CR6-7 cells are more dependent on SLC36A1-mTORC1 signaling to maintain CDK4/6i resistance, while Rb loss can directly drive CDK4/6i resistance through E2F1 targets regulating cell division, and up-regulation of SLC36A1 is a mechanism to CDK4/6i in 1205CR1-2 cells. Together, these results indicate that suppression of SLC36A1 is essential for CDK4/6i-dependent inhibition of mTORC1 signaling and promotion of senescence. Furthermore, it suggests that reexpression of SLC36A1 drives resistance to CDK4/6i-induced senescence.
SLC36A1 is an E2f target gene
E2f target genes and mTORC1 signaling (including SLC36A1) are repressed in CDK4/6i-induced senescent cells and reactivated in CR cells, implying a direct connection between E2f1 and SLC36A1-mTORC1. Further support stems from data demonstrating that E2f1 confers anticancer drug resistance by targeting the ATP-binding cassette (ABC) transporter family members and B cell lymphoma 2 (Bcl-2) via the p73/DNp73-miR205 circuitry (31). We hypothesized that E2f1 (or related E2f2/3) directly regulates SLC36A1 expression and thereby regulates mTORC1 activity. The SLC36A1 promoter contains putative binding sites for E2f proximal to the transcription start site (TSS) of SCL36A1 (Fig. 4A). We cloned a 1.3-kb genomic fragment upstream of the TSS of SLC36A1, which includes two potential E2f consensus binding sites, into a luciferase reporter plasmid. Expression of this reporter is increased in concert with exogenous E2f1 (Fig. 4B). We observed similar activation when coexpressed with either E2f2 or E2f3 but not expression of E2f1 (E132), a DNA binding–deficient mutant (Fig. 4B). We hampered E2f1-dependent SLC36A1 activation when a SLC36A1 promoter construct lacking of putative E2f biding sites was used, demonstrating the specificity of E2f1-dependent activation for SLC36A1 (Fig. 4C). In addition, E2f increased the SLC36A1 promoter activity in a dose-dependent manner, indicating that E2f binding sites of SLC36A1 promoter are responsible for E2f expression (Fig. 4D).
Fig. 4. SLC36A1 is an E2f target gene.
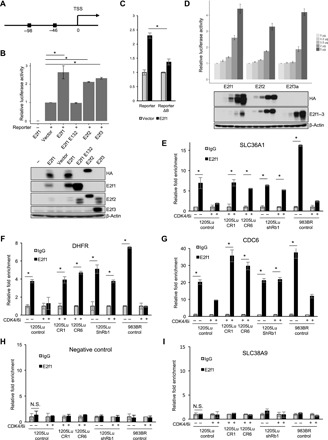
(A) Schematic of the SLC36A1 promoter. Boxes indicate the positions of putative E2f1 binding motifs. (B) Luciferase assays of lysates from HEK293T cells transfected with luciferase reporter construct containing SLC36A1 promoter (Reporter) and secreted alkaline phosphatase (SEAP) together with either empty vector, E2f1, E2f1 E132 (E2f1m), E2f2, or E2f3a. Data were normalized by SEAP for each sample (top). Data were normalized by SEAP and represent means ± SD, *P < 0.01 (one-sample two-tailed Student’s t test; n = 3). Western blot analysis of samples from (B) (top) using antibodies indicated on the right of the panel (bottom). (C) Luciferase assays of lysates from HEK293T cells transfected with Reporter or Reporter lacking of putative E2F1 binding sites (Reporter ΔB) and SEAP together with either empty vector or E2f1. Data were normalized by SEAP and represent means ± SD. *P < 0.01 (one-sample two-tailed Student’s t test; n = 3). (D) Luciferase assays of lysates from HEK293T cells transfected with Reporter and SEAP together with different amount plasmids of E2f1, E2f2, or E2f3a (0, 0.2, 0.5, 2, and 5 μg). Data were normalized by SEAP for each sample (top). Western blot analysis of samples from (C) (top) using antibodies indicated on the right of the panel (bottom). (E to I) The fixed chromatin of 1205Lu cells with or without treatment of palbociclib (1 μM) for 24 hours, 1205CR1 proliferating with palbociclib (1 μM), 1205CR6 proliferating with palbociclib (1 μM), 1205Lu cells introduced shRb with or without treatment of palbociclib (1 μM) for 24 hours, and 983BR cells with or without treatment of palbociclib (1 μM) for 24 hours was analyzed by chromatin immunoprecipitation (ChIP) assay using specific antibodies to control immunoglobulin G (IgG) (gray bars) or E2f1 (black bars). Precipitated DNA was subjected to qPCR analysis using sets of primers recognizing sequences that flank the E2f1 putative binding sites (SLC36A1) (E) or sequences that do not contain E2f motifs (negative control) (H). E2f binding to dihydrofolate reductase (DHFR) (F), CDC6 (G), or SLC38A9 (I) served as controls. Data were normalized by IgG and represent means ± SD, *P < 0.01 (two-tailed Student’s t test; n = 3). N.S., not significant (n = 3).
To determine whether E2f directly binds to the SLC36A1 promoter, we performed chromatin immunoprecipitation (ChIP). Critically, E2f1 is the most abundant E2f family member in melanoma cells and is overexpressed when compared with normal melanocytes (32). E2f1 was significantly enriched on the promoter of SLC36A1, and this binding was abolished following CDK4/6i treatment, same as dihydrofolate reductase and CDC6, known E2f1 targets (Fig. 4, E to G). We obtained consistent results in 983BR, a vemurafenib-resistant melanoma cell line (Fig. 4, E to G). To assess specificity, we designed sets of primers against sequences in the SLC36A1 promoter that do not include consensus E2f1 binding sites and also for the promoter of SLC38A9, an amino acid transporter that regulates mTOR (29, 33), but whose expression is independent of E2f1 (fig. S4D), as negative controls. We observed no E2f1 binding in the SLC36A1 promoter that does not include binding sites of E2f1 and in the SLC38A9 promoter (Fig. 4, H and I), indicating that E2f1 specifically binds to consensus binding sites in the promoter of SLC36A1 to regulate SLC36A1 expression.
FXR1 regulates SLC36A1 translation
The data above provide a plausible mechanism whereby Rb loss–mediated E2f activation increases SLC36A1, which then contributes to resistance to CDK4/6i once cells reenter the cell cycle in 1205CR1-2 cells (Fig. 4). However, this model does not address the initial event that triggers increased SLC36A1 expression, which is necessary to provide the initial reactivation of mTORC1 in 1205CR6-7, 3918CR1, 3918CR2, and 3918CR4 cells, where Rb is retained. We next focused our attention on the FXR1; FXR1 is an RNA binding protein frequently overexpressed in both melanoma and head and neck carcinomas. FXR1 expression is associated with senescence bypass (16) and linked with cyclin D1 through a common regulatory E3 ligase (17). Notably, FXR1 is highly expressed in CR cells [1205CR6-7 (Fig. 5A) and 3918CR1, 3918CR2, and 3918CR4 (fig. S2C)] relative to control and senescent cells (8-day treatment of CDK4/6i); however, no significant difference in mRNA levels was observed across the samples, indicating that FXR1 overexpression reflects a posttranscriptional mechanism (Fig. 5A and fig. S6A). Consistently, IHC staining from xenograft tumors (Fig. 3D) revealed that CDK4/6i treatment reduced FXR1, but FXR1 expression was restored or highly expressed in CR cells in vivo (Fig. 5B). To determine the potential contribution of FXR1 to CDK4/6i resistance, we knocked down FXR1. FXR1 knockdown resulted in markedly reduced SLC36A1 protein levels in 1205CR6-7 cells (Fig. 5C). We obtained similar results in human embryonic kidney (HEK293T) cells (fig. S6B). We also observed increased p27, p21, and p53 consistent with reports linking FXR1 with these targets (Fig. 5C) (16). FXR1 knockdown had little effect on mRNA levels of SLC36A1 (fig. S6C). In contrast, p53, phosphatase and tensin homolog (Pten), and p21 mRNAs were induced by FXR1 knockdown, consistent with FXR1 targeting p53, Pten, and p21 mRNAs for degradation (fig. S6C) (34). While FXR1 can suppress gene expression through destabilization of mRNA, as with p21, it can also increase protein accumulation by increasing mRNA on active polysomes (17, 35). We reasoned that the FXR1-dependent increase in SLC36A1 likely reflects FXR1-dependent regulation of SLC36A1 protein synthesis. We performed polysome fractionations to determine the impact of FXR1 on SLC36A1 protein synthesis. Lysates from HEK293T cells transfected with or without shFXR1 were subjected to polysome fractionation. Polysome profiles of 40S, 60S, 80S and polysome were determined by assessing 18S and 28S from each fraction. shFXR1 reduced SLC36A1 mRNA in the polysomes and increased in monosome fractions compared to control, indicating that knockdown of FXR1 attenuates translation of SLC36A1 (Fig. 5D, top). Ribosomal protein RPS18S mRNA was unchanged with knockdown of FXR1 (Fig. 5D, bottom). Conversely, overexpression of FXR1 resulted in a modest but significant increase in SLC36A1 (figs. S6B and S7D). To further investigate the molecular mechanisms by which FXR1 regulates SLC36A1, we examined whether FXR1 directly binds to the mRNA of SLC36A1. Since FXR1 targets G-quadraduplex (G4) RNA structure (36), we searched for G4 RNA structure within mRNA of SLC36A1. Quadruplex forming G-rich sequences (QGRS) mapper software determined high G score enriched in mRNA of SLC36A1, specifically 3′ untranslated region (3′UTR) (fig. S6D), fitting our hypothesis that FXR1 could bind to 3′UTR of SLC36A1. Supporting this hypothesis, SLC36A1 mRNA was copurified with FXR1 following RNA IP (RIP) using a specific FXR1 antibody in 1205Lu melanoma cells (Fig. 5, E and F). p21 and p27 were used as a positive control and a negative control for FXR1 RIP experiment, respectively (Fig. 5E) (16). Together, these results indicate that FXR1 regulates translation of SLC36A1 by its direct binding to the mRNA.
Fig. 5. FXR1 overrides CDK4/6i-induced senescence through SLC36A1.
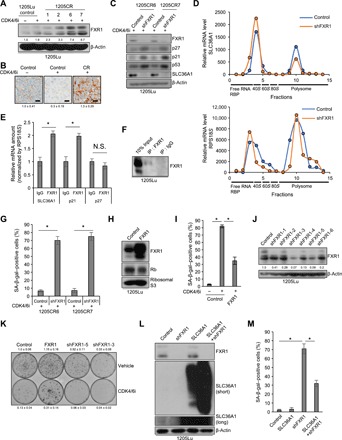
(A) Western blot analysis of lysates from 1205Lu cells treated with or without palbociclib (1 μM) for 8 days and 1205CR1, 1205CR2, 1205CR6, and 1205CR7 cells using antibodies to FXR1 and β-actin. The numbers indicate quantifications of FXR1 determined by FXR1/β-actin ratio. (B) Representative images of IHC staining of sections from xenograft tumors treated with either vehicle or palbociclib (1 μM) or tumors resistance to CDK4/6i for FXR1. Scale bars, 50 μm. The numbers indicate quantification of FXR1 intensity determined by IHC scoring (see Materials and Methods) from three independent experiments. (C) Western blot analysis of lysates from 1205CR6-7 cells with or without shFXR1 using antibodies indicated on the right of the panels. (D) Fractions from HEK293T cells with or without shFXR1 were collected by density gradient fractionation. Purified RNA was subjected to qRT-PCR analysis using sets of primers for SLC36A1 (top) and RPS18S (bottom). This experiment was performed three times, and representative data were shown. RBP, ribose-binding protein. (E) Lysates of 1205Lu cells were analyzed by RIP assay using antibodies to control IgG or FXR1 and subjected to qRT-PCR using sets of primers for SLC36A1, p21, and p27. Data were normalized by IgG and represent means ± SD, *P < 0.01 (one-sample two-tailed Student’s t test; n = 3). (F) Western blot analysis of the lysates from (E) using antibodies to FXR1. (G) Quantification of SA-β-gal–positive cells in 1205CR6-7 cells introduced with shcontrol (control) or shFXR1. Data represent means ± SD, *P < 0.01 (two-tailed Student’s t test; n = 3). (H) Western blot analysis of lysates from 1205Lu cells introduced with empty vector (control) or FXR1 using antibodies to FXR1, Rb, and ribosomal S3. (I) Quantification of SA-β-gal–positive cells in 1205Lu cells introduced with vehicle (control) or FXR1 with or without palbociclib treatment (1 μM) for 8 days. Data represent means ± SD, *P < 0.01 (two-tailed Student’s t test; n = 3). (J) Western blot analysis of lysates from 1205Lu cells with shcontrol or shFXR1 using antibodies to FXR1 and β-actin. The numbers indicate quantifications of FXR1 determined by FXR1/β-actin ratio. (K) Clonogenic colony formation assay of cells from (H) and (J) treated with or without palbociclib (1 μM) for 8 days. The numbers indicate quantification of colonies from three independent experiments. (L) Western blot analysis of lysates from 1205Lu cells infected with shcontrol, shFXR1, SLC36A1, or shFXR1 + SLC36A1 using antibodies to FXR1, SLC36A1, and β-actin. (M) Quantification of SA-β-gal–positive cells from (L). Data represent means ± SD, *P < 0.01 (two-tailed Student’s t test; n = 3).
FXR1 overrides CDK4/6i-induced senescence through SLC36A1
If resistance to CDK4/6i in 1205CR6-7, 3918CR1, 3918CR2, and 3918CR4 cells, where Rb is retained, is driven by FXR1-dependent regulation of SLC36A1, FXR1 overexpression should be essential for resistance. To test this, we knocked down FXR1 in 1205CR6-7 cells. CDK4/6i treatment along with FXR1 knockdown in 1205CR6-7 cells resulted in greater than 70% of the cells entering senescence as judged by SA-β-gal positivity (Fig. 5G and fig. S7A). Rb is maintained in cells overexpressing FXR1, ensuring that resistance to senescence does not reflect Rb loss (Fig. 5H). We also determined whether FXR1 overexpression is sufficient to drive resistance to CDK4/6i-dependent senescence. Consistent with our hypothesis, FXR1 overexpression effectively inhibited cells from entering into a senescent state as determined by SA-β-gal positivity and long-term colony formation through activation of SLC36A1-mTORC1 signaling (Fig. 5, I and K, and fig. S7, B and D). FXR1 overexpression did not prevent CDK4/6i-induced G1 arrest (fig. S7C), demonstrating that CDK4/6 inhibition is sufficient for cell cycle arrest, but coordinate inhibition of mTOR is necessary for senescence. We obtained consistent results in 983B melanoma cells, suggesting that acquired resistance mechanisms to CDK4/6i are not limited to 1205Lu cell lines (fig. S7, C to F). Consistent with a role for FXR1 in acquisition of a senescent phenotype, knockdown of FXR1 with two independent hairpins enhanced CDK4/6i-induced senescence (Fig. 5, J and K). Last, we determined whether SLC36A1 overexpression could rescue knockdown of FXR1-mediated senescence induction. Knockdown of FXR1 induced senescence, but SLC36A1 overexpression inhibited senescence induction by knockdown of FXR1, indicating that knockdown of FXR1-induced senescence is mediated through SLC36A1 (Fig. 5, L and M, and fig. S7G).
SLC36A1-mTORC1 signaling regulates CDK2 expression
The fact that cells overexpressing FXR1 still arrest (fig. S7C) suggests that mTORC1 activation is sufficient to override senescence, but a more direct intersection with CDK activation is necessary for cell cycle reentry. We therefore assessed whether mTORC1 activation, by overexpression of Raptor or SLC36A1, increases CDK expression. Critically, CDK2 expression is increased along with proper activation of E2f targets (fig. S8, A and B), suggesting that CDK2 up-regulation in resistant cells is a consequence of activation of SLC36A1-mTORC1 signaling by FXR1, followed by an increase in key CDK-E2f functions needed for S phase entry. In summary, while Rb loss–mediated E2f activation increases the expression of cell cycle regulators and SLC36A1-mTORC1 signaling activation drives resistance to CDK4/6i in 1205CR1-2, FXR1 overexpression reactivates mTORC1 through SLC36A1, which, in turn, increases CDK2 expression thereby promoting E2f-dependent cell cycle progression and SLC36A1 activation as a positive feedback loop in 1205CR6-7 cells.
Cooperative effects of a CDK4/6i and an mTORC1 inhibitor
The current study reveals that acquired resistance to CDK4/6i correlates with an initial reactivation of mTORC1 signaling, a direct result of increased expression of the SLC36A1 transporter. Clinically, this could be targeted through coordinate treatment with palbociclib (6) and ribociclib, both of which are highly selective for CDK4 and CDK6 along with an mTORC1 inhibitor, such as everolimus (37). Combination treatment is expected to induce senescence at a lower dose of CDK4/6i and achieve a more durable response. Palbociclib + rapamycin exhibited increased efficacy with regard to senescence induction than palbociclib single treatment (10). Since treatment with palbociclib results in severe neutropenia (8), a combination therapy wherein reduced dosage of individual inhibitors is used to achieve the same outcome will be highly beneficial to the patient.
To assess the potential efficacy of coordinate inhibition of CDK4/6 and mTORC1 in melanoma, we assessed the impact of drug treatment on cell proliferation using different concentrations of palbociclib or ribociclib + everolimus. We treated 1205Lu melanoma cells seeded on 96-well plates with CDK4/6i (either palbociclib or ribociclib) ± everolimus for 4 days, and we assessed cell proliferation. While single treatment of either CDK4/6i or everolimus has an inhibitory effect on proliferation, a combination of palbociclib + everolimus has the same impact at lower concentrations of CDK4/6i (Fig. 6A). To test the combined efficacy of palbociclib + everolimus in vivo, we subcutaneously injected 1205Lu melanoma cells into SCID mice. When tumors reached 2 mm in diameter, mice were administered palbociclib (90 mg/kg), everolimus (10 mg/kg), or a combination (palbociclib, 45 mg/kg; everolimus, 10 mg/kg) once daily (oral gavage for palbociclib, intraperitoneal injection for everolimus) for 8 days based on our previous findings that 8 days of CDK4/6i resulted in maximal induction of senescence, which is associated with phosphorylated Rb reduction (10). While all groups of tumors with single treatment exhibited growth suppression as summarized previously (8, 38), combination treatment of half dosage of palbociclib (45 mg/kg) + everolimus (10 mg/kg) has a more durable effect on tumor growth than palbociclib alone (90 mg/kg) or everolimus (10 mg/kg) alone (Fig. 6B). We further assessed the efficacy of this drug combination by ex vivo tumor explants using primary melanoma from patient. Freshly isolated primary melanoma was obtained, and approximately 1 to 10 mm3 of tissue slices was cultured on dental sponge in melanoma medium treated with or without palbociclib and/or rapamycin to assess whether palbociclib induces senescence. Palbociclib + rapamycin induced a robust induction of senescence compared to palbociclib alone (Fig. 6, C and D). These results indicate that combined treatment of a CDK4/6i and an mTORC1 inhibitor will have better efficacy than single agents and induce a more durable senescence in vivo and primary human melanoma ex vivo.
Fig. 6. Synergistic effects of a CDK4/6i and an mTORC1 inhibitor.
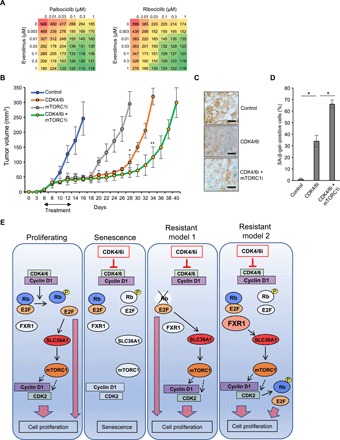
(A) Cell proliferation assay of 1205Lu cells treated with palbociclib (left) or ribociclib (right) at varying concentrations (0, 0.01, 0.03, 0.1, 0.3, and 1 μM) and everolimus at varying concentrations (0, 0.003, 0.01, 0.03, 0.1, 0.3, and 1 μM) for 4 days. Red indicates high cell number, and green color indicates low cell number. (B) A total of 2 × 106 cells of 1205Lu cells were subcutaneously injected into 6-week-old SCID mice. Treatments (palbociclib; 90 mg/kg) by oral gavage, everolimus (10 mg/kg) by intraperitoneal injection, or palbociclib (45 mg/kg) + everolimus (10 mg/kg) were initiated when tumors reached to 2 mm of diameter as indicated with arrows. Each group, 10 tumors; tumor volumes were measured every 2 days. Data represent means ± SD from n = 10 tumors per group, *P < 0.001 (two-tailed Student’s t-test; control versus palbociclib or everolimus, n = 10) and **P < 0.001 (two-tailed Student’s t-test; palbociclib versus palbociclib + everolimus, n = 10). (C) Representative images of SA-β-gal staining in fresh primary tumors culture on the dental sponges in cell culture medium with palbociclib (1 μM), rapamycin (50 nM), or palbociclib (1 μM) + rapamycin (50 nM) for 8 days (n = 3). Scale bars, 50 μm. (D) Quantification of SA-β-gal–positive cells from (C). Data represent means ± SD, *P < 0.01 (two-tailed Student’s t test; n = 3). (E) Model of CDK4/6i-induced senescence and its acquired resistance in melanoma.
DISCUSSION
Inhibition of CDK4/6 has emerged as a powerful anticancer therapeutic (7–9). Selective CDK4/6i or CDK4/6i’s such as palbociclib and ribociclib are approved by the FDA and are currently being evaluated in phase 3 clinical trials for estrogen receptor (ER+)/HER2− breast cancer and phase 1 for metastatic melanoma harboring Neuroblastoma RAS Viral Oncogene Homolog (NRAS) mutation or CDKN2A loss (8). We previously demonstrated that palbociclib treatment induced tumor senescence in vemurafenib-resistant melanoma, providing support for the use of palbociclib as a second line therapy. While CDK4/6i is expected to benefit patients, including those whom harbor vemurafenib resistance, patients are likely to develop resistance to palbociclib/ribociclib, emphasizing the importance for additional treatment options. Reactivation of MAPK or mTOR signaling can contribute to resistance to CDK4/6i or CKD4/6i’s (10, 39). However, the precise molecular mechanism driving mTOR activation and resistance to CDK4/6i remained unclear. We have explored the mechanisms of acquired resistance to CDK4/6i in melanoma and demonstrate that SLC36A1-mTORC1 activation drives resistance. We identified two molecular mechanisms whereby SLC36A1 is activated in CR cells: (i) Rb loss leads to mTORC1 activation through E2f-dependent induction of SLC36A1; (ii) FXR1 accumulation, which results in activation of mTORC1 through direct regulation of SLC36A1 protein synthesis. As a consequence of mTORC1 reactivation, the expression of cell cycle regulators, such as CDK2, is restored/increased, thereby triggering cell division through E2f-dependent activation (Fig. 6E). Given that CDK2 expression is necessary but not sufficient to drive resistance to CDK4/6i, it emphasizes that, while there is dependence on core cell cycle regulators, resistance is driven by signals extrinsic to the cell cycle.
To test the generality of SLC36A1 as a driver of CDK4/6i resistance in melanoma, we overexpressed SLC36A1 in a variety of melanoma cell lines and confirmed that SLC36A1 overrides CDK4/6i-induced senescence in all of melanoma cell lines tested. To further assess whether our findings are universal in cells spontaneously developed CDK4/6i resistance, we established another CDK4/6i-resistant cells derived from the WM3918 melanoma cell line (3918CR1, 3918CR2, and 3918CR4). We first confirmed resistance to CDK4/6i by BrdU incorporation and E2f1 activation judged by increased expression of its targets. Last, we observed Rb retention, increased FXR1, SLC36A1, p-S6, and CDK2 in 3918CR1, 3918CR2, and 3918CR4 cells compared to CDK4/6i exposure for 8 days in WM3918 cells, which is consistent with our model that FXR1 overexpression contributes to resistance to CDK4/6i through SLC36A1 (Fig. 6E).
To assess the tissue specificity of resistance mechanisms to CDK4/6i, we also established CR cells using esophageal squamous carcinoma cell lines TE7 and TE10. We found Rb loss in both TE7CR and TE10CR cells, suggesting that loss of Rb expression is likely a common mechanism to acquired resistance to CDK4/6i (Fig. 6E). Exome sequencing revealed no mutations on whole genome including at the Rb locus in the CR cells (both melanoma and esophageal squamous carcinoma) compared to control cells. Furthermore, Rb expression can be restored following culture of cells in the absence of CDK4/6i (10); however, the precise mechanisms contributing to Rb loss require additional investigation.
FXR1 is a driver of CDK4/6i resistance in cases where Rb expression is maintained; while increased protein levels were observed in resistant cells, no alteration in FXR1 mRNA or alteration at the FXR1 gene locus was observed, suggesting that dysregulated FXR1 is posttranscriptional. FXR1 is known to regulate many targets, but regulation of FXR1 expression is poorly characterized. We recently demonstrated that Fbxo4 is a component of an ubiquitin ligase that determines FXR1 accumulation (17). However, 1205Lu melanoma cells harbor an Fbxo4 mutation (I377M) that is refractory to substrate binding including FXR1 (5, 17). To further assess FXR1 regulation by Fbxo4 in the context of CDK4/6i resistance, we established CR cells using WM3918 cells that retain wild-type Fbxo4 (3918CR1, 3918CR2, and 3918CR4 cells) and observed reduced FXR1 protein in 3918CR cells (fig. S2C). However, there is no evidence that CDK4/6 regulates FXR1 expression. Critically, FXR1 drives reexpression of SLC36A1 in CR cells where it contributes to reactivation of mTOR; SLC36A1 is both necessary and sufficient for resistance to CDK4/6i. The importance of SLC36A1 is emphasized by the observation that addition of amino acids to culture medium is not sufficient to drive CDK4/6i resistance in the absence of SLC36A1 overexpression.
Last, concurrent treatment of palbociclib + everolimus induced a more durable impact of tumor progression to palbociclib alone, which reflects clinical significance. Notably, ribociclib + everolimus (ClinicalTrials.gov identifier: NCT03114527) is being evaluated in a phase 2 clinical trial in advanced dedifferentiated liposarcoma and leiomyosarcoma; palbociclib + PI3K/mTOR inhibitor gedatolisib (PF-05212384) (ClinicalTrials.gov identifier: NCT03065062) is also being tested in a phase 1 clinical trial for advanced squamous cell lung, pancreatic, head and neck, and other solid tumors. In addition, ribociclib + everolimus + exemestane (ClinicalTrials.gov identifier: NCT01857193) is being tested in a phase 1b clinical trial in hormone receptor–positive Her2-negative advanced breast cancer. While this combination is exciting, it should be noted that SLC36A1 overexpression is one mechanism of resistance. This is highlighted by our observation that SLC36A1 expression is down-regulated following CDK4/6i in the mouse mammary cancer cell line V720, the human ovarian cancer cell line HEY, and the esophageal adenocarcinoma cell line TE7 but not in the human breast cancer cell line T47D (fig. S8C). The contribution of SLC36A1-mTORC1 will be dependent on many contexts including cancer type, microenvironment, global/specific gene expression alterations (e.g., Rb), and genomic mutations (fig. S8C).
The lack of ongoing trials using CDK4/6i in melanoma precludes direct analysis of this resistance mechanism in primary human tissue at this time. However, mouse xenograft and three-dimensional culture with human melanoma experiments strongly support our conclusions. In summary, our work demonstrated the precise molecular mechanisms to acquired resistance to CDK4/6i in melanoma. Ultimately, our observations provide an important avenue for therapeutic intervention by targeting SLC36A1 to increase efficacy of CDK4/6i.
MATERIALS AND METHODS
Cell culture
Authenticated human melanoma cell lines (1205Lu, WM983B, WM239A, 451Lu, WM3918, and WM983BR) were obtained from the Wistar Institute collection (Philadelphia, PA) and cultured in Tu 2% media [80% MCDB153 medium supplemented with 20% Leibovitz L-15, 2% fetal bovine serum (FBS), and 1.68 mM CaCl2]. Authenticated HEK293T cells were purchased from American Type Culture Collection in 2014 and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). For viral production, viral expression plasmids were transfected into HEK293T cells with Lipofectamine together with packaging plasmid. Virus supernatants harvested 48 to 72 hours after transfection were used to infect melanoma cells with polybrene (10 μg/ml).
Flow cytometry
Cells were collected and stained with propidium iodide (10 μg/ml) containing ribonuclease A (100 μg/ml) and 0.5% FBS and fixed with 70% ethanol. For the BrdU detection, cells were cultured in medium containing 10 μM BrdU for 45 min, fixed with 70% ethanol, treated with 1.5 M HCl, and stained with an anti-BrdU mouse monoclonal antibody, followed by fluorescein isothiocyanate–linked mouse immunoglobulin G (IgG) staining. Cell cycle distribution and BrdU-positive cells were measured with BD LSRFortessa (BD Biosciences).
Western blot analysis
Cells were lysed in EBC buffer [50 mM tris (pH 8.0), 120 mM NaCl, 1 mM EDTA, and 0.5% NP-40] containing 1 mM phenylmethylsulfonyl fluoride, aprotinin (20 U/ml), 1 μM leupeptin, 1 mM dithiothreitol, 0.1 mM NaF, 0.1 mM sodium orthovanadate, and 10 mM β-glycerophosphate. Proteins were resolved by SDS–polyacrylamide gel electrophoresis, transferred to membrane, and immunoblotted with the indicated antibodies as follows: p-Rb (S780), cyclin E (D7T3U), cyclin A (B-8), p-CDK2 (M2), CDK4 (H-303), CDK6 (C-21), E2f1 (C-20), E2f2 (C-20), E2f3 (C-18), p53 (DO-1), and p21 (C-19) were obtained from Santa Cruz Biotechnology. p-Rb (S807/811) (D20B12), p-CDK2 (T160), p27 (D69C12), p-S6K (Thr389), p-S6 (Ser235/236) (2F9), hemagglutinin (HA) (C29F4), CDC2 (POH1), FEN1, PCNA (D3H8P), heat shock protein 90 (HSP90), and ribosomal protein S3 (D50G7) were purchased from Cell Signaling Technology. Raptor was purchased from Bethyl Laboratories. Cycin D1 (Ab-3) and FXR1 antibodies were purchased from Millipore. p-Rb (S612) and β-actin (AC-15) were purchased from Sigma-Aldrich. Rb (G3-245) and SLC36A1 were obtained from BD Pharmingen and Abcam, respectively. Membranes were incubated with horseradish peroxidase–conjugated anti-mouse or rabbit antibodies, and signals were developed with the enhanced chemiluminescence system (PerkinElmer) according to the manufacturer’s instructions.
Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted using the RNeasy Mini Kit (Qiagen) and reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad), and the data were normalized by glyceraldehyde-3-phosphate dehydrogenase (GADPH) or RPS18S.
Immunohistochemistry
Tissues from xenograft mice were fixed in 4% paraformaldehyde and deparaffinized and rehydrated in gradient ethanol. Sections were blocked with 10% goat serum and incubated with the primary antibodies against SLC36A1 that were purchased from Abcam. For IHC, sections were incubated with biotinylated rabbit antibodies and developed with an ABC substrate kit (Vector Laboratories) followed by 3,3’-diaminobenzidine (DAB) reaction (Vector Laboratories), counterstained with hematoxylin (Thermo Fisher Scientific), and mounted with Permount mounting media (Thermo Fisher Scientific). The IHC score in each experiment was defined by the following formula: Intensity = [staining positive population (1 to 3) × staining intensity (1 to 3)].
Senescence analysis
For SA-β-gal activity assay, cultured melanoma cells or primary melanoma tumors in Tissue-Tek O.C.T. compounds (Sakura Finetek) were fixed and incubated with X-gal staining solution (Sigma) according to the manufacturer’s instructions following exposure of 1 μM palbociclib for 8 days. For clonogenic colony formation assay, 2.5 × 103/60-mm plate plates were treated with 1 μM palbociclib with fresh palbociclib added daily for 8 days and cultured for an additional 14 days following removal of drug in complete growth media and colonies that were visualized by Giemsa staining.
Luciferase assay
The pEZX-LvPG04 vector (GeneCopoeia) inserted with fragments containing 1292 base pairs (bp) (from 190-bp downstream of transcription start sites (TSSs) of SLC36A1 to 1101-bp upstream of TSSs of SLC36A1) was used. HEK293T cells were transfected with the SLC36A1 promoter plasmid with E2f1, E2f2, E2f3a, or E2f1 E132 expression plasmids. Luciferase activity and secreted alkaline phosphatase (SEAP) activity as a control were determined using reporter gene assay kits (GeneCopoeia) according to the manufacturer’s instructions. The luciferase activity was normalized by SEAP for each sample.
Chromatin immunoprecipitation
Chromatin was prepared using the truChIP High Chromatin Shearing Kit (Covaris) and sheared into about 500- to 1000-bp fragments using Covaris S2. Immunoprecipitation was performed with the antibodies indicated as follows: E2f1 and normal IgG were obtained from Cell Signaling Technology. Genomic DNA was isolated from precipitated protein-DNA complex, and qPCR was performed as mentioned above using sets of primers for specific promoters. Data were normalized to IgG.
RNA immunoprecipitation
Cell lysates were prepared from growing cells, and equal amounts of protein (1000 μg) were subjected to IP reaction using FXR1 antibody (Millipore) or isotype control IgG in an RIP kit (Millipore) according to the manufacturer’s instructions. Total RNA from each IP was extracted with phenol and chloroform and precipitated by isopropanol in the presence of glycogen. Complementary DNA (cDNA) was synthesized as described above for qRT-PCR reaction. The data were normalized to RPS18S.
Polysome fractionation
Cell lysates were extracted in TMK100 buffer [10 mM tris-HCl (pH 7.4), 5 mM MgCl2, 1% (v/v) Triton X-100, 0.5% deoxycholate, 2 mM dithiothreitol, and cycloheximide (100 μg/ml)], followed by centrifugation with 14,000 rpm for 10 min. Supernatants were layered onto sucrose gradients (10 to 50%) and centrifuged at 35,000 rpm in a SW40Ti rotor for 3 hours at 4°C. Fractions were collected using a density gradient fractionation system (Teledyne ISCO), and then, RNAs in each fraction were isolated using QIAzol (Qiagen). Monosomal and polysomal fractions were determined by analysis of 18S and 28S ribosomal RNA levels using denaturing agarose gel electrophoresis. For analysis of SLC36A1 translation, cDNA was synthesized from RNA in each fraction, followed by qPCR analysis using sets of primers for SLC36A1 and RPS18S as a control.
Cell proliferation assay
A total of 2500 cells were fed on 96-well plates and treated with CDK4/6i or CKD4/6i’s (either palbociclib or ribociclib) and everolimus for 4 days. Medium was changed every 2 days. Cell proliferation was assessed by the CyQUANT NF Cell Proliferation Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Xenograft mouse model
A total of 2 × 106 melanoma cells were subcutaneously injected into 6-week-old SCID mice with Matrigel (BD Biosciences). Mice were treated with vehicle, everolimus (10 mg/kg) by IP injection, or palbociclib (90 mg/kg) by oral gavage for single treatment and everolimus (10 mg/kg) and palbociclib (45 mg/kg) for concomitant treatment for 8 days daily when tumors developed 2 mm in diameter after injection. Tumor volumes were measured by caliper every 2 days and calculated by the following formula: V = (length × width × height)/2. Mice were euthanized, and the tumor size was measured when tumors reached 10 mm of diameter. Care of experimental animals was performed in accordance with the institutional guidelines.
Human primary samples
Fresh primary tumor tissue was obtained from patients with metastatic melanoma at the Hollings Cancer Center at the Medical University of South Carolina (MUSC) in accordance with Institutional Review Board standards and in compliance with federal regulations governing research on specimens and/or clinical data (45CFR46). Tumors were dissected by a clinical pathologist and subdivided into approximately 1- to 10-mm3 pieces and placed on presoaked 1-cm3 dental sponges (Novartis; Vetspon) submerged in 2 ml of culture melanoma media with or without of treatment of palbociclib and/or rapamycin for 8 days. Treatments were refreshed daily, and tumors were harvested after 8 days and fixed in Tissue-Tek O.C.T. compounds (Sakura Finetek) for SA-β-gal assay.
Exome sequencing, RNA-seq, and analysis
Exome capture was carried out using the Nextera Rapid Capture Expanded Exome Kit (Illumina), and samples were sequenced on the HiSeq 2500 to a minimum depth of 4 to 5 Gb of sequence per exome. Data were subjected to Illumina quality control (QC) procedures (>80% of data must yield a Phred score of 30 and trimming of read bases below the Phred quality score of 20 from the 3′ end of reads). Secondary analysis was carried out the OnRamp Genomics Research Platform (GRP). Data were analyzed using the variant analysis pipeline implemented at the MUSC. This leverages the OnRamp Bioinformatics GRP, a Hadoop solution stack configured for high-throughput genomic analysis. The GRP was used to align raw fastq reads to the human genome 19 (hg19) (NCBI build 36.1) reference genome with the Burrow-Wheeler Aligner (BWA). The Genome Analysis Tool Kit was used for base quality score recalibration, indel realignment, duplicate removal, and computation of a false discovery rate (FDR) for each variant. Single-nucleotide variant (SNV) and insertion/deletion (InDel) discovery and genotyping were performed simultaneously across all samples using standard hard filtering parameters or variant quality score recalibration. SNVs were called using SAMTools (Sequence Alignment/Map Tools). The mapped reads of all SNVs were reviewed and filtered, and the number of total and amino acid changing variants was calculated for each sample. For each gene, we determined the number of samples that contain any amino acid changing (nonsynonymous) mutations. Last, we exploited the GEnome MINIng (GEMINI) framework to further annotate each variant by comparing it to several genome annotations from multiple sources including Encyclopedia of DNA Elements (ENCODE) tracks, University of California, Santa Cruz (UCSC) tracks, Online Mendelian Inheritance in Man (OMIM), The Single Nucleotide Polymorphism Database (dbSNP), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Human Protein Reference Database (HPRD). The dataset was deposited in Gene Expression Omnibus (GEO) repository under the accession number GSE109350.
A total of 100 to 200 ng of total RNA was used to prepare RNA-seq libraries using the TruSeq RNA Sample Prep Kit V2 (Illumina, San Diego, CA), following the protocol described by the manufacturer. High-throughput sequencing (HTSeq) was performed using an Illumina HiSeq 2500 with each sample sequenced to a minimum depth of ~50 million reads. A paired-end 2X125 cycle sequencing strategy was used. Data were subjected to Illumina QC procedures (>80% of the data yielded a Phred score of 30). Secondary analysis was carried out on an OnRamp Bioinformatics Genomics Research Platform (OnRamp Bioinformatics, San Diego, CA). OnRamp’s advanced Genomics Analysis Engine used an automated RNA-seq workflow to process the data, including (i) data validation and QC, (ii) read alignment to the human genome (hg19) using TopHat2, which revealed >90% mapping of the paired-end reads, (iii) generation of gene-level count data with HTSeq, and (iv) differential expression analysis with DEseq2, which enabled the inference of differential signals with robust statistical power (Genomics Research Platform with RNA-seq workflow v1.0.1, including FastQValidator v0.1.1a, Fastqc v0.11.3, Bowtie2 v2.1.0, TopHat2 v2.0.9, HTSeq v0.6.0, and DEseq v1.8.0). The resulting SAM files were sorted and inputted into the Python package HTSeq to generate count data for gene-level differential expression analyses. To infer differential signal within the datasets with robust statistical power, we used DEseq2. Transcript count data from DEseq2 analysis of the samples were sorted according to their adjusted P value or q value, which is the smallest FDR at which a transcript is called significant. FDR is the expected fraction of false-positive tests among significant tests and was calculated using the Benjamini-Hochberg multiple testing adjustment procedure. Statistical analysis of pathways and gene ontology terms was carried out using this sorted transcript list as described previously and Advaita iPathwayGuide. The datasets were deposited in the GEO database (GEO accession: GSM3101838). Extracted data were used for heat map generation by R and GSEA analysis by javaGSEA application. Gene sets from Broad Institute were used for heat map generation and GSEA analysis.
Statistical analysis
The normality and equal variance of data were evaluated by Shapiro-Wilk test and F test, respectively. When two groups were normally distributed, the parametric test was applied such as Student’s t test for equal variance and Welch’s t test for unequal variance. When group was not normally distributed, nonparametric test such as Mann-Whitney U test was applied. For xenograft experiment, unpaired two-tailed Student’s t test was applied after data were transformed to log values. The error bars represent SD of the mean, and significance was defined as a P value of less than 0.05 with two-tailed, unless otherwise specified. N.S., not significant. Details and significant values are described in the figure legends.
Supplementary Material
Acknowledgments
We thank M. Herlyn (The Wistar Institute) for providing melanoma cell lines and K. Armeson (MUSC) for assistance of statistical analysis. We also thank assistance from the Division of Laboratory Animal Resource (DLAR), Flow Cytometry Facility, and Genomics Shared Resource in the MUSC. Funding: This work was supported by grants from the NIH [R01CA093237 and P01098101 (to J.A.D.) and R01CA176401 and R01CA217329 (to K.E.K.)], Prostate Cancer Foundation Young Investigator Award (to R.d.L.), Outrun the Sun Foundation (to A.Y.), and Novartis (to J.A.D. and K.E.K.). G.H. acknowledges support from 1U01DA045300-01A1. This work was supported, in part, by a grant from Novartis. Author contributions: A.Y. and J.A.D. designed experiments, analyzed data, and wrote the manuscript. A.Y. and Y.B. performed experiments. J.W. and E.R.C. provided human melanoma samples. Y.B., E.S.H., and G.H. performed and analyzed whole-exome sequencing and RNA-seq. S.Q. provided materials. R.d.L. and K.E.K. provided intellectual input. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax6352/DC1
Fig. S1. Palbociclib-resistant melanoma cells retain broad resistance to CDK4/6i or CKD4/6i’s.
Fig. S2. Analyses of WM3918, TE7, and TE10 cells with CDK4/6i resistance.
Fig. S3. Different acquired resistance mechanisms between 1205CR1-2 and 1205CR6-7 cells.
Fig. S4. Overexpression of CDKs does not override CDK4/6i-induced senescence in melanoma.
Fig. S5. The impacts of adding amino acids and SLC36A1 in the context of CDK4/6i-induced senescence.
Fig. S6. FXR1 regulates SLC36A1 protein expression.
Fig. S7. FXR1 overrides CDK4/6i-induced senescence.
Fig. S8. RNA-seq analysis of additional cancers with CDK4/6i resistance.
REFERENCES AND NOTES
- 1.Choi Y. J., Li X., Hydbring P., Sanda T., Stefano J., Christie A. L., Signoretti S., Look A. T., Kung A. L., von Boehmer H., Sicinski P., The requirement for cyclin D function in tumor maintenance. Cancer Cell 22, 438–451 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackermann J., Frutschi M., Kaloulis K., McKee T., Trumpp A., Beermann F., Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res. 65, 4005–4011 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Ibrahim N., Haluska F. G., Molecular pathogenesis of cutaneous melanocytic neoplasms. Annu. Rev. Pathol. 4, 551–579 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Sauter E. R., Yeo U.-C., von Stemm A., Zhu W., Litwin S., Tichansky D. S., Pistritto G., Nesbit M., Pinkel D., Herlyn M., Bastian B. C., Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res. 62, 3200–3206 (2002). [PubMed] [Google Scholar]
- 5.Lee E. K., Lian Z., D’Andrea K., Letrero R., Sheng W. Q., Liu S., Diehl J. N., Pytel D., Barbash O., Schuchter L., Amaravaradi R., Xu X., Herlyn M., Nathanson K. L., Diehl J. A., The FBXO4 tumor suppressor functions as a barrier to BRAFV600E-dependent metastatic melanoma. Mol. Cell. Biol. 33, 4422–4433 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fry D. W., Harvey P. J., Keller P. R., Elliott W. L., Meade M., Trachet E., Albassam M., Zheng X., Leopold W. R., Pryer N. K., Toogood P. L., Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol. Cancer Ther. 3, 1427–1438 (2004). [PubMed] [Google Scholar]
- 7.Knudsen E. S., Witkiewicz A. K., The strange case of CDK4/6 inhibitors: Mechanisms, resistance, and combination strategies. Trends Cancer 3, 39–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sherr C. J., Beach D., Shapiro G. I., Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 6, 353–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Leary B., Finn R. S., Turner N. C., Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 13, 417–430 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Yoshida A., Lee E. K., Diehl J. A., Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 76, 2990–3002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou J., Wu Z., Wong G., Pectasides E., Nagaraja A., Stachler M., Zhang H., Chen T., Zhang H., Liu J. B., Xu X., Sicinska E., Sanchez-Vega F., Rustgi A. K., Diehl J. A., Wong K.-K., Bass A. J., CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in oesophageal squamous cell carcinoma. Nat. Commun. 8, 13897 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goel S., Wang Q., Watt A. C., Tolaney S. M., Dillon D. A., Li W., Ramm S., Palmer A. C., Yuzugullu H., Varadan V., Tuck D., Harris L. N., Wong K.-K., Liu X. S., Sicinski P., Winer E. P., Krop I. E., Zhao J. J., Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell 29, 255–269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boll M., Foltz M., Rubio-Aliaga I., Kottra G., Daniel H., Functional characterization of two novel mammalian electrogenic proton-dependent amino acid cotransporters. J. Biol. Chem. 277, 22966–22973 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Heublein S., Kazi S., Ögmundsdóttir M. H., Attwood E. V., Kala S., Boyd C. A. R., Wilson C., Goberdhan D. C. I., Proton-assisted amino-acid transporters are conserved regulators of proliferation and amino-acid-dependent mTORC1 activation. Oncogene 29, 4068–4079 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sagné C., Agulhon C., Ravassard P., Darmon M., Hamon M., El Mestikawy S., Gasnier B., Giros B., Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc. Natl. Acad. Sci. U.S.A. 98, 7206–7211 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Majumder M., House R., Palanisamy N., Qie S., Day T. A., Neskey D., Diehl J. A., Palanisamy V., RNA-binding protein FXR1 regulates p21 and TERC RNA to bypass p53-mediated cellular senescence in OSCC. PLOS Genet. 12, e1006306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qie S., Majumder M., Mackiewicz K., Howley B. V., Peterson Y. K., Howe P. H., Palanisamy V., Diehl J. A., Fbxo4-mediated degradation of Fxr1 suppresses tumorigenesis in head and neck squamous cell carcinoma. Nat. Commun. 8, 1534 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukas J., Bartkova J., Rohde M., Strauss M., Bartek J., Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol. Cell. Biol. 15, 2600–2611 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byth K. F., Thomas A., Hughes G., Forder C., McGregor A., Geh C., Oakes S., Green C., Walker M., Newcombe N., Green S., Growcott J., Barker A., Wilkinson R. W., AZD5438, a potent oral inhibitor of cyclin-dependent kinases 1, 2, and 9, leads to pharmacodynamic changes and potent antitumor effects in human tumor xenografts. Mol. Cancer Ther. 8, 1856–1866 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Gu Y., Rosenblatt J., Morgan D. O., Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 11, 3995–4005 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitagawa M., Higashi H., Jung H. K., Suzuki-Takahashi I., Ikeda M., Tamai K., Kato J., Segawa K., Yoshida E., Nishimura S., Taya Y., The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 15, 7060–7069 (1996). [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C., Li Z., Bhatt T., Dickler M., Giri D., Scaltriti M., Baselga J., Rosen N., Chandarlapaty S., Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 36, 2255–2264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quelle D. E., Ashmun R. A., Shurtleff S. A., Kato J. Y., Bar-Sagi D., Roussel M. F., Sherr C. J., Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 7, 1559–1571 (1993). [DOI] [PubMed] [Google Scholar]
- 24.Glavinas H., Krajcsi P., Cserepes J., Sarkadi B., The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 1, 27–42 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Dean M., ABC transporters, drug resistance, and cancer stem cells. J. Mammary Gland Biol. Neoplasia 14, 3–9 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Ögmundsdóttir M. H., Heublein S., Kazi S., Reynolds B., Visvalingam S. M., Shaw M. K., Goberdhan D. C., Proton-assisted amino acid transporter PAT1 complexes with Rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PLOS ONE 7, e36616 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goberdhan D. C., Wilson C., Harris A. L., Amino acid sensing by mTORC1: Intracellular transporters mark the spot. Cell Metab. 23, 580–589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorn M., Jaehme M., Weiwad M., Markwardt F., Rudolph R., Brandsch M., Bosse-Doenecke E., The role of N-glycosylation in transport function and surface targeting of the human solute carrier PAT1. FEBS Lett. 583, 1631–1636 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Wang S., Tsun Z.-Y., Wolfson R. L., Shen K., Wyant G. A., Plovanich M. E., Yuan E. D., Jones T. D., Chantranupong L., Comb W., Wang T., Bar-Peled L., Zoncu R., Straub C., Kim C., Park J., Sabatini B. L., Sabatini D. M., Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347, 188–194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo H., Zhao L., Ji X., Zhang X., Jin Y., Liu W., Glycosylation affects the stability and subcellular distribution of human PAT1 protein. FEBS Lett. 591, 613–623 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Alla V., Kowtharapu B. S., Engelmann D., Emmrich S., Schmitz U., Steder M., Pützer B. M., E2F1 confers anticancer drug resistance by targeting ABC transporter family members and Bcl-2 via the p73/DNp73-miR-205 circuitry. Cell Cycle 11, 3067–3078 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halaban R., Cheng E., Smicun Y., Germino J., Deregulated E2F transcriptional activity in autonomously growing melanoma cells. J. Exp. Med. 191, 1005–1016 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rebsamen M., Pochini L., Stasyk T., de Araújo M. E., Galluccio M., Kandasamy R. K., Snijder B., Fauster A., Rudashevskaya E. L., Bruckner M., Scorzoni S., Filipek P. A., Huber K. V., Bigenzahn J. W., Heinz L. X., Kraft C., Bennett K. L., Indiveri C., Huber L. A., Superti-Furga G., SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 519, 477–481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davidovic L., Durand N., Khalfallah O., Tabet R., Barbry P., Mari B., Sacconi S., Moine H., Bardoni B., A novel role for the RNA–binding protein FXR1P in myoblasts cell-cycle progression by modulating p21/Cdkn1a/Cip1/Waf1 mRNA stability. PLOS Genet. 9, e1003367 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vasudevan S., Steitz J. A., AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell 128, 1105–1118 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaeffer C., Bardoni B., Mandel J. L., Ehresmann B., Ehresmann C., Moine H., The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J. 20, 4803–4813 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lane H. A., Wood J. M., McSheehy P. M. J., Allegrini P. R., Boulay A., Brueggen J., Littlewood-Evans A., Maira S.-M., Martiny-Baron G., Schnell C. R., Sini P., O’Reilly T., mTOR inhibitor RAD001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin. Cancer Res. 15, 1612–1622 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Laplante M., Sabatini D. M., mTOR signaling in growth control and disease. Cell 149, 274–293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Leeuw R., McNair C., Schiewer M. J., Neupane N. P., Brand L. J., Augello M. A., Li Z., Cheng L. C., Yoshida A., Courtney S. M., Hazard E. S., Hardiman G., Hussain M. H., Diehl J. A., Drake J. M., Kelly W. K., Knudsen K. E., MAPK reliance via acquired CDK4/6 inhibitor resistance in cancer. Clin. Cancer Res. 24, 4201–4214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax6352/DC1
Fig. S1. Palbociclib-resistant melanoma cells retain broad resistance to CDK4/6i or CKD4/6i’s.
Fig. S2. Analyses of WM3918, TE7, and TE10 cells with CDK4/6i resistance.
Fig. S3. Different acquired resistance mechanisms between 1205CR1-2 and 1205CR6-7 cells.
Fig. S4. Overexpression of CDKs does not override CDK4/6i-induced senescence in melanoma.
Fig. S5. The impacts of adding amino acids and SLC36A1 in the context of CDK4/6i-induced senescence.
Fig. S6. FXR1 regulates SLC36A1 protein expression.
Fig. S7. FXR1 overrides CDK4/6i-induced senescence.
Fig. S8. RNA-seq analysis of additional cancers with CDK4/6i resistance.