

Directly disrupting the Hsp90-Cdc37 complex can selectively down-regulate kinase clients of Hsp90 and achieve cell cycle arrest.
Abstract
Disrupting the interactions between Hsp90 and Cdc37 is emerging as an alternative and specific way to regulate the Hsp90 chaperone cycle in a manner not involving adenosine triphosphatase inhibition. Here, we identified DDO-5936 as a small-molecule inhibitor of the Hsp90-Cdc37 protein-protein interaction (PPI) in colorectal cancer. DDO-5936 disrupted the Hsp90-Cdc37 PPI both in vitro and in vivo via binding to a previously unknown site on Hsp90 involving Glu47, one of the binding determinants for the Hsp90-Cdc37 PPI, leading to selective down-regulation of Hsp90 kinase clients in HCT116 cells. In addition, inhibition of Hsp90-Cdc37 complex formation by DDO-5936 resulted in a remarkable cyclin-dependent kinase 4 decrease and consequent inhibition of cell proliferation through Cdc37-dependent cell cycle arrest. Together, our results demonstrated DDO-5936 as an identified specific small-molecule inhibitor of the Hsp90-Cdc37 PPI that could be used to comprehensively investigate alternative approaches targeting Hsp90 chaperone cycles for cancer therapy.
INTRODUCTION
Heat shock protein 90 (Hsp90) is a well-known adenosine 5′-triphosphate (ATP)–dependent protein chaperone that achieves cellular protein homeostasis (1). A remarkable number of Hsp90 client proteins play important roles in the growth and proliferation of cancer cells (2). Beyond their effects on all the clients, kinase levels are highly correlated with the Hsp90 chaperone cycle, which makes Hsp90 an attractive anticancer therapeutic target (3). At present, the main focus of research targeting Hsp90 is competitively inhibiting the adenosine triphosphatase (ATPase) binding site on the Hsp90 N-terminal domain, leading to 17 small-molecule inhibitors with diverse structure types entered into clinical trials. However, current Hsp90 inhibitors exert a variety of toxicities (such as cardiotoxicity, gastrointestinal toxicity, and ocular toxicity) and ineluctable heat shock responses with limited clinical validity, which become the major obstacles restricting their approval to the market (4, 5). During the Hsp90 chaperone cycle, the toxicity and heat shock response of Hsp90 inhibitors might be induced by direct binding to the ATPase pocket, which ultimately prevents the vital step of ATP hydrolysis, inevitably impairing all Hsp90 clients (5). Current results provide evidence that entirely inhibiting Hsp90 or directly targeting the Hsp90 ATPase binding site may not be an optimal choice to achieve cancer therapy. Therefore, it is imperative to find new strategies or previously unknown sites to achieve anticancer potency and functional specificity through an alternative Hsp90 chaperone regulation approach.
The Hsp90 chaperone cycle works through a dynamic process that includes many protein-protein interactions (PPIs) with diverse cochaperones. Quantitative analysis of Hsp90-cochaperone interactions has been reported to reveal specificity in substrate recognition (6). Different cochaperones, including Hsp70, Hsp40, Hsp70/Hsp90-organizing protein, Cdc37, C terminus of heat shock cognate protein 70 (HSC70)–interacting protein, small glutamine-rich tetratricopeptide repeat (TPR)–containing protein α, TPR-containing protein associated with Hsp90 (TAH1), p23, protein phosphatase 5, and activator of Hsp90 ATPase protein 1, are indispensable in achieving different functions and selectivity through direct PPIs with Hsp90. Among them, cochaperone Cdc37 is responsible for recruiting kinases to Hsp90, and it has been assumed to be a specifically kinase-targeting subunit for the Hsp90 chaperone system (7). These kinases contain receptor tyrosine kinases, nonreceptor tyrosine kinases, lymphocyte-specific protein tyrosine kinases, and intracellular serine/threonine kinases, indicating an extensive degree of dependency of the cancer cell kinome on Cdc37 expression (8). As a specific kinase-targeting cochaperone, Cdc37 selectively recognizes and combines unfolded kinase clients before binding open-state Hsp90. When Hsp90-Cdc37-kinase complexes are established, Hsp90 integrates ATP and adopts a closed state to accomplish a regular kinase folding process (9). Hsp90-Cdc37 synergistically acts as a facilitator of oncogenesis to help the correct folding of a wide range of overexpressed or mutated oncogenic proteins, accelerating tumorigenic influence (10).
To date, two cocrystal structures of Hsp90-Cdc37 with different truncations have been reported, and these structures revealed a dynamic recruitment cycle in Hsp90-Cdc37-kinase recognition and folding mechanism (9, 11). Cdc37 contains an N-terminal client kinase interaction domain involving a key phosphorylated residue serine (S13), a central middle domain required for Hsp90 binding, and a C-terminal domain of unknown functions. The binding interface between Hsp90 and Cdc37 appears to be discrete and dynamic, involving a ~1056-Å2 molecular surface and limited critical interactions to reveal a micromolar binding affinity in vitro (11). Although some mutagenesis studies were performed to explore the important residues in Hsp90-Cdc37, during dynamic processes, the precise recognition interactions and potential small-molecule binding sites remained unclear (12, 13). Without a deep binding pocket, constrained secondary conformation, and definite action site, the superficial binding mode makes it challenging to find specific small molecules interacting with critical residues of Hsp90-Cdc37. The expression level of Hsp90 is similar in both normal cells and malignant cells, while Cdc37 functions as an overexpressed oncogene to force cells to transform and become proliferative, resulting in tumors (14). Cdc37 exhibits increasing levels in proliferating tissues and is prominently expressed in multiple cancers, while most normal tissues, without malignant proliferation, do not require Cdc37 (15). The formation of the Hsp90-Cdc37-kinase polycomplex ultimately facilitates kinases integrating into the Hsp90 chaperone machine to result in a mature state (9). Thus, these advantages indicate that inhibiting the Hsp90-Cdc37 PPI without impairing Hsp90 ATPase activity might achieve a promising therapeutic window that is completely different from direct inhibition of Hsp90. On the basis of the above mechanism, specifically recognizing pivotal residues and selectively disrupting the interactions between Hsp90 and Cdc37 might precisely block the folding of kinase clients to achieve anticancer therapeutic effects.
Until now, no small molecule has been reported to specifically bind to Hsp90 or Cdc37 with the potency to disrupt Hsp90-Cdc37 at the cellular level by blocking critical recognition residues, resulting in a lack of evidence for identifying the specific binding site and explicit inhibition mechanism, although several natural products were reported to exhibit anticancer activity via a mechanism of Hsp90-Cdc37 inhibition (16–19). Here, we are the first to describe a complete process from critical residue discovery on a protein-protein binding interface to the identification of a small-molecule binding site. Through molecular dynamics (MD) simulations, we focused on residues that contribute greatly to the Hsp90-Cdc37 binding interface and made further identifications by mutagenesis data; the results suggested an interaction between E47 and Q133 on Hsp90 and R167 on Cdc37 as a binding determinant for the Hsp90-Cdc37 PPI. To disrupt the most important interactions between Hsp90 and Cdc37, we designed a screening workflow that identified DDO-5936 as a cellularly active inhibitor that disrupted the Hsp90-Cdc37 interaction. Nuclear magnetic resonance (NMR) characterization and binding assays with different mutants confirmed that DDO-5636 selectively bound to a previously unknown site on the Hsp90 N terminus and exhibited almost no ATPase inhibition. As expected, DDO-5936 selectively down-regulated kinases without effects on other nonkinase clients of Hsp90, exhibited antiproliferative potency with a high correlation to the expression level of Hsp90-Cdc37, arrested the cell cycle via a cyclin-dependent kinase 4 (CDK4) decrease in HCT116 cells, and exhibited in vivo potency in a xenograft model. Collectively, these results indicate that the discovery of DDO-5936 might have identified a previously unknown binding site on the Hsp90 N terminus that disrupts its interaction with Cdc37, which might lead to an advanced understanding of Hsp90-Cdc37 function as well as a promising lead compound for alternative drug discovery strategies through regulation of Hsp90 with its cochaperone cycles.
RESULTS
Discovery of small-molecule inhibitors to block determinant interactions of Hsp90-Cdc37
Considering the complexity of the Hsp90-Cdc37 interface, the lack of positive compounds with well-defined binding sites and high-throughput screening methods makes it challenging to find small-molecule inhibitors targeting the Hsp90-Cdc37 PPI. Although few studies have revealed possible key residues on the Hsp90-Cdc37 binding interface, an explicit small-molecule binding site to disrupt Hsp90-Cdc37 remains unknown (12, 20, 21). Given the difficulties in disrupting the Hsp90-Cdc37 interaction, we first searched the binding interface to find the critical residues on both Hsp90 and Cdc37. It has been reported that a small number of pivotal contacts in Hsp90-Cdc37 lead to a moderate binding affinity (Kd = 1.46 μM) in vitro (11). To determine the potential binding sites on the Hsp90-Cdc37 binding interface, we calculated the per-residue effective binding energy based on a 50-ns MD simulation trajectory to provide the per-residue energy contribution to the Hsp90-Cdc37 binding (fig. S1A). The residues contributing more than −3 kcal/mol of binding energy were labeled, including R46, E47, Y61, and Q133 on Hsp90 and R167, Q208, and R246 on Cdc37 (fig. S1C). The root mean square fluctuations (RMSFs) of Hsp90 also revealed two additional stabilized regions when bound to Cdc37, correlating with per-residue energy contributions (fig. S1B). Each of the highly contributing residues was further separately mutated to alanine for mutated molecular mechanics/generalized Born surface area (MM/GBSA) calculations and experiments. The binding affinity between wild-type (WT) Hsp90 and Cdc37 was 2.17 μM as determined by isothermal titration calorimetry (ITC), which was comparable to the reported affinity, suggesting that both Hsp90 and Cdc37 proteins were in their active form (11). Mutants including R167A on Cdc37 or E47A or Q133A on Hsp90 resulted in a complete loss of binding affinity, while mutants of Q208A or R246A on Cdc37 or R46A or Y61A on Hsp90 retained binding affinity with Kd values of 3.51, 6.02, 8.85, and 6.85 μM, respectively (Fig. 1A, table S1, and fig. S2C). An in vitro pulldown assay showed that E47A and Q133A mutations on Hsp90 disrupted the Hsp90-Cdc37 interaction (fig. S2A), which might indicate a promising small-molecule binding site. In addition, four mutants of Hsp90 exhibited little effect on Hsp90 ATPase activity (fig. S2B). Thus, through MD simulation calculations of binding energy and determinations of mutant affinities, we identified polar interactions between R167 on Cdc37 and E47 and Q133 on Hsp90 as the most important binding determinants in recognition during the dynamic cycle of the Hsp90-Cdc37 PPI (fig. S2D).
Fig. 1. Discovery of DDO-5936, an Hsp90-Cdc37 PPI inhibitor without ATPase inhibition, based on a site screening strategy involving critical residues identified at the binding interface.
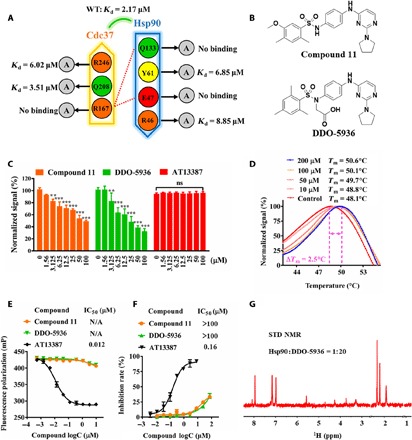
(A) Binding affinities of the WT Hsp90-Cdc37 complex and complexes with key residues mutated, determined by ITC. Hydrogen bonds are presented as red dotted lines. Data are from three independent experiments. (B) Chemical structures of compound 11 and DDO-5936. (C) Dose-dependent inhibition of the Hsp90-Cdc37 interaction by compound 11, DDO-5936, and AT13387. Twofold diluted compounds with seven concentrations were simultaneously tested by an Hsp90-Cdc37 homogeneous time-resolved fluorescence (HTRF) assay. The results are shown as the means ± SD, n = 3 wells, from three independent experiments. P values were calculated by pairwise comparisons to the dimethyl sulfoxide (DMSO) control (**P<0.01, ***P<0.001, Student’s t test). ns, not significant. (D) Thermostability of Hsp90 treated with 0, 10, 50, 100, and 200 μM DDO-5936, determined by a standard thermo shift assay. ΔTm results were obtained from the mean values of three independent assays (n = 6 wells). (E) Dose-dependent competitive binding of compound 11, DDO-5936, and AT13387 to the Hsp90 ATP binding site, determined by fluorescence polarization (FP) assays using a fluorescein isothiocyanate–geldanamycin probe. Data are presented as the means ± SD, n = 3 wells, from three independent experiments. N/A, not applicable. (F) Dose dependence of compound 11, DDO-5936, and AT13387 inhibition of Hsp90 ATPase activity. Data are presented as the means ± SD, n = 3 wells, from three independent experiments. (G) 1H saturation transfer difference (STD) NMR spectrum of 100 μM DDO-5936 binding to 5 μM Hsp90. ppm, parts per million.
On the basis of the binding determinants of the Hsp90-Cdc37 interface, we designed a hierarchical strategy that combines cascade docking, experimental assays, and chemical optimization to screen and find potential Hsp90-Cdc37 PPI inhibitors (fig. S3A). First, to interfere with the critical interactions formed between R167 (Cdc37) and E47 and Q133 (Hsp90), we assumed that Hsp90-Cdc37 PPI inhibitors might contain charged or alkaline groups, including amidines, amines, imidazoles, imines, or NH-imidazoles, to interact with a previously unknown site involving E47 or Q133 on Hsp90. To obtain a focused compound database, we performed a ligand filtering process from a commercial database (containing more than 1,500,000 compounds) to establish a focused library (120,537 compounds). We next virtually docked the compounds into a binding site predicted using critical residues (E47 and Q133) on the Hsp90 interface. After the MM/GBSA rescore and binding interaction filter, we lastly selected 33 compounds based on the top-ranked binding models (fig. S3B). To determine the ability of the compounds to disrupt the Hsp90-Cdc37 PPI, we developed a homogeneous time-resolved fluorescence (HTRF) assay using glutathione S-transferase (GST)–tagged Cdc37, His-tagged Hsp90, and the corresponding anti-GST and anti-His antibodies (fig. S3C). Among the 33 compounds, compounds 3 and 11 decreased the normalized signal by more than 50% at 200 μM (fig. S3D). Subsequently, we used biolayer interferometry (Octet) assays to measure the binding of hits to a chip-immobilized Hsp90. In a concentration dependency test, compound 11 exhibited a moderate binding affinity to Hsp90, with a KD value of 21.1 μM, while compound 3 showed weak binding, with a KD value of 568 μM (fig. S3E). The above results demonstrated that compound 11 exhibited Hsp90-Cdc37 PPI inhibition ability through a direct binding mode.
We then synthesized a series of derivatives of compound 11 to obtain DDO-5936 as a more potent inhibitor with increased solubility for further structural biology and functional assays (Fig. 1B and fig. S4A). HTRF assays showed that both compound 11 and DDO-5936 dose-dependently inhibit Hsp90-Cdc37 binding at a half-maximal inhibitory concentration (IC50) in a micromolar range, while Hsp90 ATPase inhibitor (AT13387, a phase 2 drug candidate targeting the Hsp90 ATPase pocket) exhibited no activity under the same experimental conditions (Fig. 1C). Furthermore, we performed an ATPase assay and fluorescence polarization (FP) assay (a classic approach to screening Hsp90 inhibitors targeting the ATP pocket) to determine whether DDO-5936 affects the Hsp90 ATPase function (22). With AT13387 (whose determined IC50 was 160 nM in the ATPase assay and 12 nM in the FP assay) as a positive control, compound 11 and DDO-5936 exhibited little effect on Hsp90 ATPase activity, with IC50 values greater than 100 μM in the ATPase assay and almost no activity in the FP competitive assay (Fig. 1, E and F). Thermal unfolding assays showed that DDO-5936 increased the melting temperature (Tm) of Hsp90 in a dose-dependent manner. The presence of 200 μM DDO-5936 increased the thermal stability of Hsp90 by 2.5°C (ΔTm) (Fig. 1D). In addition, we detected positive saturation transfer difference (STD) signals in STD spectra, which confirmed that DDO-5936 directly bound Hsp90 (Fig. 1G). Together, on the basis of a previously unknown site involving E47 and Q133 on Hsp90, compound screening and in vitro assays confirmed that DDO-5936 inhibits Hsp90-Cdc37 through directly binding Hsp90 without interfering with Hsp90 ATPase activity.
DDO-5936 targets a surface binding site on Hsp90 to inhibit the Hsp90-Cdc37 PPI
To elucidate the potential binding site and molecular interactions between DDO-5936 and Hsp90, we sought to map residues influenced by DDO-5936 using structural biology and binding assays with mutants of key residues. We attempted to obtain the cocrystal structure of DDO-5936–Hsp90 but unfortunately failed. We reasoned that the flexibility of the potential binding site might cause structural heterogeneity that precluded cocrystallization. Therefore, we obtained a 1H-15N transverse relaxation optimized spectroscopy (TROSY) heteronuclear single-quantum coherence (HSQC) spectrum, which was sensitive to weak and transient interactions, to display and map the residues of Hsp90 with or without treatment with DDO-5936. Sequential assignment of the Hsp90 N terminus complexed with DDO-5936 was achieved as previously reported following a minimum deviation principle in chemical shift perturbations (fig. S4B) (23–25). As shown in Fig. 2A, the 1H-15N TROSY spectrum of the critical residues (especially E47 and Q133) of the Cdc37 binding domain in Hsp90 exhibited gradual shifts in NMR resonance positions upon titration with increasing concentrations of DDO-5936. Per-residue chemical shift perturbations (Δω) were calculated as the distance between the peaks in the free and bound protein spectra. Chemical shift perturbations of more than 15 Hz were considered notable and were mapped onto the structure of Hsp90, with E47 and Q133 exhibiting the largest chemical shift perturbations and the residues in the Hsp90 ATPase domain experiencing relatively minor changes (fig. S4D). The changes in peak intensity (difference between DDO-5936–free and DDO-5936–bound states of Hsp90) were calculated as ΔI. Residues that lost over 80% of their free peak intensity were highlighted, identifying E47 and Q133 by them both having Δω values more than 15 Hz and ΔI values indicating a loss of over 80% of the free peak intensity with treatment with DDO-5936 (fig. S4C). The above structural results revealed that DDO-5936 might occupy a Cdc37-binding site with determinant residues, including E47 and Q133, entirely distinct from the ATP-binding pocket to disrupt the Hsp90-Cdc37 PPI, consistent with docking screening predictions (Fig. 2B).
Fig. 2. DDO-5936 binds to a critical residue on the Hsp90-Cdc37 PPI interface.
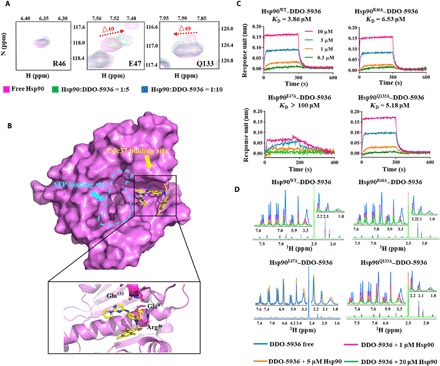
(A) HSQC spectra (three residues, including R46, E47, and Q133) of 50 μM 15N-labeled Hsp90 N terminus in the absence (magenta) and presence of 250 μM (green) or 500 μM DDO-5936 (blue). (B) Overview of the DDO-5936 binding site on Hsp90. Top: Overall structure with the surface of Hsp90 colored magenta. Inset: Detailed binding region of DDO-5936. Hsp90 is represented as a magenta cartoon, and DDO-5936 is shown as yellow sticks. The residues Arg46, Glu47, and Gln133 are highlighted within the stick model. Hydrogen bonds are displayed as green dotted lines. (C) Binding affinities of WT or mutant Hsp90 proteins for DDO-5936, determined by biolayer interferometry (ForteBio Octet) assay. Data are representative of three independent experiments. (D) Dose-dependent T1ρ NMR spectra for 200 μM DDO-5936 (blue) in the presence of 1 μM Hsp90 (magenta), 5 μM Hsp90 (orange), and 20 μM Hsp90 (green). Data are representative of three independent experiments.
To validate the binding site of DDO-5936, we separately mutated the residues that might be involved in binding in the Hsp90–DDO-5936 interaction site. Direct binding affinities revealed that the KD values for binding DDO-5936 were remarkably decreased for the E47A mutant (KD > 100 μM) and moderately lower for the R46A mutant (KD = 6.53 μM) and Q133A mutant (KD = 5.18 μM) than those for WT Hsp90 (KD = 3.86 μM), indicating direct binding of DDO-5936 to a previously unknown site involving E47 on Hsp90 (Fig. 2C). Consistent results were observed in the T1ρ NMR spectra, indicating that the WT Hsp90 protein and R46A and Q133A mutants interfered with the binding of DDO-5936 in a dose-dependent manner. However, no interference was observed in the E47A mutant by its NMR signals, which additionally supported DDO-5936 binding the key determinant site E47 (Fig. 2D). Together, our observations of the DDO-5936 binding mode via NMR detection and mutagenesis validation demonstrated that DDO-5936 specifically bound to a previously unknown site on Hsp90 involving E47 to block the critical interactions in the Hsp90-Cdc37 PPI.
DDO-5936 disrupts the Hsp90-Cdc37 PPI in cells
To further verify the interactions between DDO-5936 and the Hsp90 protein, we carried out a cellular thermal shift assay (CETSA) to determine the affinity, cellular uptake, and target engagement in vivo. The ability of DDO-5936 to thermally stabilize Hsp90 in HCT116 cells was determined, and representative Western blots for analyzing the stabilization of Hsp90 are shown in fig. S5A. The detectable Hsp90 exhibited a clear difference between being untreated and treated with 25 μM DDO-5936 for 12 hours in cells, with denaturation temperatures ranging from 51° to 61°C, indicating that DDO-5936 entered cells and directly bound the Hsp90 protein. To determine the Hsp90-Cdc37 PPI inhibition efficiency in vivo, we performed coimmunoprecipitation (co-IP) experiments after treatment with DDO-5936 at different concentrations. To precisely assess the Hsp90-Cdc37 PPI disruption activity of DDO-5936, we immunoprecipitated Hsp90 and Cdc37 to detect the bait proteins. It was distinctly observed that DDO-5936 disrupted the Hsp90-Cdc37 interaction in HCT116 cells in a dose-dependent manner whenever Hsp90 or Cdc37 immunoprecipitated (Fig. 3A). Recently, CDK4, one of the most well-studied kinase clients of Hsp90-Cdc37, and a crystal structure of the Hsp90-Cdc37-CDK4 kinase complex (9) were used as a model to explain how Hsp90 and Cdc37 stabilize and activate human kinases. Thus, different consequences might be observed by detecting CDK4 when immunoprecipitating either Hsp90 or Cdc37 in co-IP assays. As expected, CDK4 decreased when Hsp90 was immunoprecipitated, while no variant could be observed when Cdc37 was immunoprecipitated, indicating a direct binding of CDK4 to Cdc37 and a specific inhibition mechanism for DDO-5936.
Fig. 3. DDO-5936 disrupts the Hsp90-Cdc37 interaction, represses cell proliferation through a strong correlation with the Hsp90-Cdc37 expression level, and selectively down-regulates kinase clients of Hsp90.
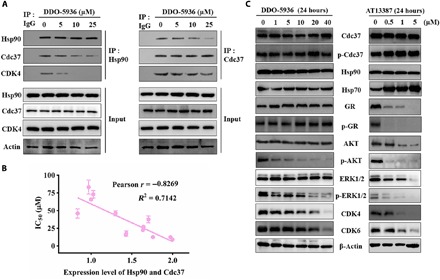
(A) Co-IP in HCT116 cells after treatment with DMSO and increasing concentrations of DDO-5936 (5, 10, and 25 μM) for 24 hours. Western blots were performed with anti-Hsp90, anti-Cdc37, or anti-CDK4 in each experiment. Data are representative of three independent experiments. (B) Correlation between the protein expression level of Hsp90 and Cdc37 in diverse cell lines and antiproliferative activities (IC50 values). The Pearson correlation coefficient (r) was calculated by GraphPad Prism 6.0 software. Data are presented as the means ± SD, n = 6 wells, from three independent experiments. (C) Western blot analysis of Cdc37, p (phosphorylated)–Cdc37, Hsp90, Hsp70, GR (glucocorticoid receptor), p-GR, AKT, p-AKT, ERK1/2 (extracellular signal–regulated kinase 1/2), p-ERK1/2, CDK4, and CDK6 protein expression levels in HCT116 cells after treatment with 0, 1, 5, 10, 20, and 40 μM DDO-5936 or 0, 0.5, 1, and 5 μM AT13387 for 24 hours. β-Actin was used as a loading control. Data are representative of three independent experiments.
Increased levels of Hsp90-Cdc37 complexes regulate the folding and stabilization of protein kinases required for proliferation or recycle rounds of kinase reactivation (26, 27). Thus, the correlations between the expression levels of Hsp90-Cdc37 and the antiproliferative activity of DDO-5936 could be determined to elucidate the specific inhibition mechanism used by DDO-5936 in cells. Given that DDO-5936 disrupted the Hsp90-Cdc37 PPI, we next determined its activity in a panel of human cancer and normal cell lines. Hsp90 expressed in similar levels in 13 tested cell lines, while Cdc37 expression was variable among them (fig. S5C). Notably, DDO-5936 exhibited diverse antiproliferative IC50 values, and its IC50 values were highly correlated with the expression levels of Hsp90 and Cdc37. We calculated the correlation coefficient based on the IC50 values and expression levels of both Hsp90 and Cdc37, indicating a strong correlation with a Pearson r value of −0.8451 (Fig. 3B). Among these cell lines, the Hsp90-Cdc37 complex exhibited the highest expression, and DDO-5936 achieved the most potent antiproliferative activity (an IC50 of 8.99 ± 1.21 μM) in HCT116 cells, indicating a reasonable target engagement of DDO-5936 (fig. S5D).
To further assess the effect of DDO-5936 on downstream Hsp90 client proteins, we detected representative kinases and nonkinase clients of Hsp90-Cdc37 in a dose-dependent and time-dependent manner (Fig. 3C and fig. S5B). As expected, DDO-5936 selectively affected kinases but exhibited no influence on the expression of both the glucocorticoid receptor (GR) and its phosphorylated form (p-GR), a nonkinase Hsp90 client, indicating a preferable specificity over AT13387 to achieve selective inhibition of Hsp90 clients. In contrast to AT13387, DDO-5936 treatment resulted in no increase in Hsp70, indicating no involvement of DDO-5936 in the heat shock response. Both CDK4 and CDK6 also decreased in a concentration- and time-dependent manner. Smith et al. (28) reported that the knockdown of Cdc37 in HCT116 cells significantly reduced the phosphorylation of AKT and extracellular signal–regulated kinase 1/2 (ERK1/2) without obvious effects on the protein levels of both AKT and ERK1/2. Consistent with their study, we found that the block of the interaction between Hsp90 and Cdc37 by DDO-5936 decreased the phosphorylated states of AKT and ERK1/2 (p-AKT and p-ERK1/2, respectively) to reduce their activities with no change in their protein levels. To further confirm the mechanism, using Cdc37–knockout (KO) HCT116 cell line, we revealed that p-AKT and p-ERK1/2 were markedly reduced, and the total AKT and ERK1/2 remained constant compared to those in the Cdc37-WT HCT116 cell line (Fig. 4A), in agreement with the effect of DDO-5936. Thus, the inhibitor DDO-5936 through the block of Hsp90-Cdc37 interaction will provide a chemical tool to unravel novel insights into the signal transduction of Hsp90.
Fig. 4. DDO-5936 arrests the cell cycle in HCT116 cells.
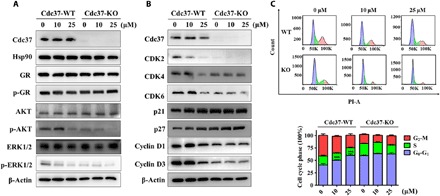
(A) Western blot analysis of Cdc37, Hsp90, GR, p-GR, AKT, p-AKT, ERK1/2, and p-ERK1/2 protein expression levels in Cdc37-WT HCT116 cells and corresponding Cdc37-KO HCT116 cells after treatment with 0, 10, or 25 μM DDO-5936 for 24 hours. β-Actin was used as a loading control. Data are representative of three independent experiments. (B) Western blot analysis of Cdc37, CDK2, CDK4, CDK6, p21, p27, cyclin D1, and cyclin D3 protein expression levels in Cdc37-WT HCT116 cells and corresponding Cdc37-KO HCT116 cells after treatment with 0, 10, or 25 μM DDO-5936 for 24 hours. β-Actin was used as a loading control. Data are representative of three independent experiments. (C) Top: Cell cycle distribution measured by propidium iodide (PI) staining of Cdc37-WT HCT116 cells and corresponding Cdc37-KO cells after treatment with 0, 10, or 25 μM DDO-5936 for 24 hours. Bottom: The cell cycle distribution is represented as a graphic histogram. Data are representative of three independent experiments as the means ± SD (***P < 0.001).
Recently, it has been proposed that binding of CDK4/6 to Cdc37 could be blocked by their ATP competitive inhibitors (29). Considering the possibility of direct inhibition effects of DDO-5936 on kinases, especially cell cycle–related kinases, we screened 20 kinases using DDO-5936. As expected, DDO-5936 exhibited no inhibition effects on ATPase activity of the selected kinases (IC50 > 100 μM), leading to exclude the possibility of DDO-5936 as a kinase inhibitor (table S2).
DDO-5936 causes cell cycle arrest in HCT116 cells in a Cdc37-dependent manner
To demonstrate the specificity and functional effects of DDO-5936 on Hsp90-Cdc37 complex inhibition in HCT116 cells, we generated a Cdc37-KO cell line by the CRISPR-Cas9 system. In agreement with the above results, DDO-5936 selectively down-regulated p-AKT and p-ERK1/2, a behavior similar to that in the Cdc37-KO cell line, consistent with previous reports (Fig. 4A) (28). Notably, DDO-5936 had attenuated effects on Cdc37-KO HCT116 cells, exhibiting a weakened dose dependence and indicating a valid and specific activity of DDO-5936.
As mentioned previously, the correct folding of CDK4 is highly correlated with Hsp90-Cdc37 and plays a key role in the cell cycle and related regulatory events. Thus, we determined the levels of related regulators of the cell cycle after DDO-5936 treatment. A detectable decrease in CDK2, CDK4, CDK6, cyclin D1, and cyclin D3 and an increase in p21 and p27 were observed in HCT116 cells, suggesting consistent results with Cdc37-KO cell lines (Fig. 4B). Treatment of Cdc37-KO cells with different concentrations of DDO-5936 had negligible effects on all the regulators through quantification results (fig. S6). We next performed a cell cycle distribution analysis based on a standard flow cytometry method by staining DNA with propidium iodide (PI) 24 hours after DDO-5936 treatment of both Cdc37-WT and Cdc37-KO HCT116 cells. DDO-5936 dose-dependently increased the percentages of HCT116 cells in G0-G1 phase, suggesting a manner of G0-G1 phase arrest that inhibits cell cycle progression (Fig. 4C). In comparison to DDO-5936–induced G0-G1 phase cell cycle arrest in Cdc37-WT HCT116 cells, Cdc37-KO HCT116 cells exhibited high percentages of G0-G1 phase arrest similar to the levels when Cdc37-WT HCT116 cells were treated with 25 μM DDO-5936. Treatment of Cdc37-KO HCT116 cells with different concentrations of DDO-5936 had negligible effects on the percentages of G0-G1 phase. Thus, DDO-5936 specifically induced cell cycle arrest at the G0-G1 phase and consequently inhibited HCT116 cell proliferation in a Cdc37-dependent manner.
DDO-5936 exhibits potency in vivo
To evaluate the antitumor efficiency of DDO-5936 in vivo, we administered different doses of inhibitors into nude mice bearing subcutaneous HCT116 tumor cell xenografts by intraperitoneal injection once per day for 21 days. We monitored tumor growth and body weight with and without inhibitor treatment. Two dosages (50 and 100 mg/kg per day) for treatment were set for in vivo assays. As shown in Fig. 5A, DDO-5936 had negligible effects on body weight in mice injected with these two dosages of compound. A dose-dependent reduction in HCT116 tumor growth and decreased tumor weight were observed in mice treated with DDO-5936 compared with vehicle-injected controls (Fig. 5, B and C). We performed histopathological analysis using hematoxylin and eosin (H&E) staining of tumor tissue and multiple organs from mice injected intraperitoneally (ip) with DDO-5936 for 21 days. The results revealed that compared with the vehicle group, the DDO-5936–treated groups showed no significant variations in multiple organs, indicating a low toxicity of DDO-5936 (fig. S7A). In contrast, H&E staining of tumor tissue exhibited a remarkable change upon treatment, suggesting the efficiency and specificity of DDO-5936 in vivo. We next checked the effects of DDO-5936 on Hsp90-Cdc37 signaling in xenograft tumors. Western blots of tumor tissues showed that phosphorylated forms of AKT and ERK1/2 were less abundant in the DDO-5936–treated group than in the vehicle group. CDK4 and CDK6 were also down-regulated dose-dependently. Hsp90, Hsp70, GR, and p-GR levels remained relatively constant, indicating consistent results with the observations in the HCT116 cell experiments (Fig. 5D). We then progressed DDO-5936 to a single-dose (100 mg/kg, ip) pharmacokinetic/pharmacodynamic (PK/PD) experiment in HCT116 xenografts to investigate whether the in vitro and PK profiles translated into sustained inhibition of the PD biomarkers in vivo (fig. S7, B and C). We measured DDO-5936 concentration in both plasma and tumor tissue. The plasma exposure of DDO-5936 (area under the curve) was 52.20 hours·μg/ml, the clearance was moderate (CL = 1794.95 ml/hour per kilogram), and the volume of distribution was large (Vz_F = 15.90 liters/kg). A moderate uptake of DDO-5936 in tumor tissue was observed, associated with a reasonable half-life (t1/2 = 4.05 hours), compared to 6.06 hours in plasma. In PD evaluation, DDO-5936 led to a reduction of CDK4 after 4 hours, which was consistent with the tmax value determined in tumor tissue. All these results revealed that DDO-5936 exhibited antiproliferative activity both in vitro and in vivo through an Hsp90-Cdc37 PPI inhibition mechanism (Fig. 5E).
Fig. 5. DDO-5936 dose-dependently impairs the growth of xenografted HCT116 cells in nude mice.
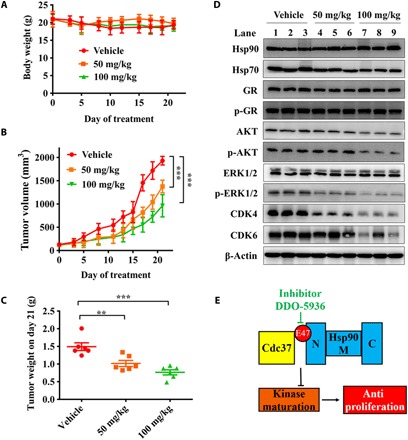
(A) Body weights of nude mice treated with either vehicle or DDO-5936 are shown. Body weight is plotted as the means ± SEM (n = 6 mice for each group). (B) Volume measurements of HCT116 xenograft tumors treated with vehicle, DDO-5936 (50 mg/kg per day), or DDO-5936 (100 mg/kg) for 21 days. DDO-5936 was administered by intraperitoneal injection once a day. Tumor volume is plotted as the means ± SEM (n = 6 mice for each group) (**P < 0.01, ***P < 0.001, paired Student’s t test, two-tailed). (C) Volume distribution of HCT116 xenograft tumors treated with vehicle or DDO-5936 at 21 days. (D) Western blot analysis of Hsp90, Hsp70, GR, p-GR, AKT, p-AKT, ERK1/2, p-ERK1/2, CDK4, and CDK6 in three representative xenograft tumor tissue samples from each group. β-Actin was used as a loading control. Data are representative of three technical replicates. (E) Schematic model of the process in which DDO-5936 modulates the Hsp90-Cdc37 chaperone cycle. DDO-5936 inhibited the Hsp90-Cdc37 PPI through binding to a previously unknown pocket on Hsp90 involving E47, a binding determinant of the Hsp90-Cdc37 complex, inhibiting the maturation of the kinase client to implement the antiproliferation effects of HCT116 cells.
DISCUSSION
Currently, most known Hsp90 inhibitors exert antitumor activity by being specific for ATP binding sites. However, feedback regulation of the heat shock response and high toxicity have limited the progress of those inhibitors in clinical trials. To obtain a full view of the Hsp90 chaperone system, we focused on the global regulation process, which indicated a key role of multichaperone Hsp90 complexes formed with different cochaperones by diverse PPIs (30). Because of the clear functional mechanism and evidence of structural biology, the dynamic regulation and specific kinase-associated features of the Hsp90-Cdc37 PPI gradually become distinct and appealing. Although direct inhibition of Hsp90 ATPase activity emerged as a potent and effective antitumor therapy, specific modulation of certain functional interactions may achieve therapeutic effects that avoid the heat shock response and high toxicity caused by wide-spectrum inhibition of Hsp90 clients.
The Hsp90-Cdc37 complex might be an attractive target because it meets the following criteria: (i) Cdc37 is expressed more in cancer cells than in normal cells to provide a potential therapeutic window (27); (ii) Cdc37 is verified to be a kinase-specific cochaperone of Hsp90, and disrupting the interactions between Hsp90 and Cdc37 or directly inhibiting Cdc37 can achieve specific inhibition of Hsp90 kinase clients; and (iii) Cdc37 interacts with Hsp90 through specific binding sites and certain motifs without affecting other cochaperone binding, indicating a potential specific modulation mechanism to avoid unnecessary toxicity. Thus, the development of small molecules targeting the Hsp90-Cdc37 PPI represents an alternative and effective strategy to implement cancer therapy.
Currently, because of the large and dynamic binding surface of Hsp90-Cdc37 PPI, the biggest challenge to developing specific Hsp90-Cdc37 PPI inhibitors is the uncertainty of accurate binding site for small molecules. Although DCZ3112 and many other natural products (such as celastrol) could inhibit Hsp90-Cdc37 PPI at the cellular level, the accurate binding site and regulation mechanism remained unclear (18, 19). In summary, our work first revealed the binding determinants of the Hsp90-Cdc37 complex and identified them by a specific small molecule to provide further insights into the modulation of interactions between Hsp90 and Cdc37 or other cochaperones in cancer therapy.
METHODS
MD simulation and analysis of MD trajectories
The currently used structures of the Hsp90-Cdc37 complex were all obtained from the Protein Data Bank (PDB). All structures were corrected and cleaned using the clean protein tool in the Discovery Studio 3.0 package (Accelrys Inc., San Diego, CA, USA). All calculations were conducted using a Dawning TC2600 cluster. Except when otherwise mentioned, parameters were set at their default values. Two known complexes (PDB ID: 1US7 and 2K5B) were obtained from the PDB for further analysis. All crystallographic water molecules were removed from the coordinate set. MD simulations of the Hsp90-Cdc37 complex were performed using the particle mesh Ewald MD module of AMBER 12 combined with the ff99SB modifications of Simmerling et al. (31) and Darian and Gannett (32). The complex was solvated using TIP3P water molecules, which extended at least 15 Å from the protein. To keep the whole system neutral, counterions were added to the solvent (four Na+ ions for 2K5B and nine Na+ ions for 1US7). Before performing the MD simulation, the whole system was minimized in two steps. Minimization 1 included refining the water molecules through 5000 steps of steepest descent, followed by 5000 conjugate gradient steps. The whole protein was kept fixed with a constraint of 3.0 kcal/mol per angstrom squared. Minimization 2 aimed to relax the complexes by 10,000 cycles of minimization, including 5000 cycles of steepest descent and 5000 cycles of conjugate gradient minimization. In the whole simulation process, the particle mesh Ewald method was used to calculate long-range electrostatic interactions (33). All covalent bonds involving hydrogen atoms were constrained to allow a time step of 2 fs using the SHAKE method. Meanwhile, a 12-Å cutoff value was used for nonbonded interactions. Then, the whole system was heated from 0 to 300 K while running 50-ps MD simulations with position restraints at constant volume. For pressure relaxation, isothermal-isobaric ensemble (NPT)–MD was subsequently carried out for 1000 ps with a time constant of 1.0 ps to adjust the solvent density. All receptors and proteins were applied with harmonic restraints with force constants of 2.0 kcal/mol per angstrom squared in this step. To relax the system without constraints, an extra 500 ps of unconstrained NPT-MD at 300 K with a time constant of 2.0 ps was applied. Last, 50 ns of production dynamics at constant pressure was used for further analysis, in which the snapshots were saved at 20-ps intervals.
The “ptraj” tool in AMBER 12 was applied to analyze the root mean square deviation and RMSF of the backbone atoms for studying the stability of the complexes during the simulation and for ensuring the basic analysis of trajectories of the sampling method. In addition, the hydrogen bonds were determined by a distance cutoff value of 3.2 Å and an angle cutoff value of 120°. Hydrogen bonds were retained only if their occupancies were >80% (the percentage of the whole simulation time that the hydrogen bonds were formed).
Free energy calculations and the investigation of the per-residue energy contributions were conducted using the MM-PBSA method, implemented in the AMBER program (34). The binding free energy can be obtained through calculation of the end points of the thermodynamic cycle of ligand binding, which is the basic mechanism of the MM-PBSA method. All binding free energies were averaged over the ensemble of conformers. An energy decomposition scheme was conducted by MM-PBSA to explore the detailed contributions of individual residues as introduced by Gohlke et al. (35, 36), which might provide critical insights into hotspots.
Virtual screening
On the basis of the crystal structure of the Hsp90-Cdc37 complex (PDB ID: 2K5B and 1US7), we used critical residues, including E47 and Q133, on Hsp90 to predict the potential binding site. Then, grid-based ligand docking was performed using energetics (Glide) software (Schrödinger suite 2009, v5.5) through cascade docking, including standard precision docking, extra precision docking, and MM/GBSA rescore. The 33 compounds with the top-ranked scores and ideal binding features were purchased from a commercial compound database for experiments.
Cloning, expression, and purification of recombinant WT and mutant Hsp90 and Cdc37
The DNA encoding Hsp90 (both the full-length and N-terminal truncated forms, i.e., residues 9 to 236), Cdc37, and site-directed mutants (R46A, E47A, Y61A, and Q133A in Hsp90 and R167A, Q208A, and R246A in Cdc37) was cloned into pET28a. The Escherichia coli BL21 (DE3) strain was transfected with recombinant plasmids and grown at 37°C in lysogeny broth medium in the presence of ampicillin (100 μg/ml). After the optical density (OD) values reached 0.6 to 0.8, protein expression was then induced by isopropyl β-d-1-thiogalactopyranoside at a final concentration of 1 mM for an additional 20 hours at 16°C. The cells were harvested by centrifugation at 5000 rpm for 15 min and stored at −80°C for use.
Protein purification procedures included the following steps: first, the cells were suspended in 50 ml of lysis buffer [20 mM tris-HCl (pH 7.4), 200 mM NaCl, and 1 mM dithiothreitol], lysed by sonication, and further centrifuged at 12,000 rpm for 30 min at 4°C. Next, the supernatant was filtered using 0.45-μm syringe filters, purified with a nickel column, and concentrated via centrifugal filtration (Millipore) with a molecular weight cutoff of 3000. Last, the proteins were subjected to gel filtration on HiLoad 60 Superdex 200 columns (GE Healthcare), and the eluted proteins were analyzed by SDS–polyacrylamide gel electrophoresis (PAGE). All proteins were concentrated and stored in phosphate-buffered saline (PBS) buffer at −80°C.
ITC assay
The binding affinities between Hsp90 and Cdc37 as well as those of all mutants were determined by ITC (MicroCal iTC200). Proteins in titration cells were prepared at a concentration of 50 μM in assay buffer [20 mM tris-HCl and 150 mM NaCl (pH 7.4)], and proteins in syringes were prepared at a concentration of 300 μM in the same assay buffer. Intervals of 180 s and a stirring speed of 1000 rpm were used for the entire injection procedure, which involved a total of 19 injections. In addition, the first 0.5 μl of the ligand solution was titrated to prevent initial interference. All the data obtained from the experiment were analyzed by the Origin software package to determine binding parameters, including the association constant (Ka = 1/Kd), enthalpy value (ΔH), and entropy value (ΔS).
Hsp90 ATPase assays
A DiscoveRx ADP Hunter Plus Assay Kit (DiscoveRx, Fremont, CA) was used to evaluate the Hsp90 ATPase activity. Following the standard protocols, all tests were performed in 384-well black plates at 37°C. Each well contained 20 μl of diluted compound (replaced with assay buffer when testing protein controls or the activity of mutants), 20 μl of Hsp90 protein (5 μM), and 20 μl of ATP (100 μM) for 1-hour incubations. Subsequently, 10 μl of detection reagent A and 20 μl of detection reagent B were added for another 30-min incubation at room temperature. Then, a Varioskan multimode microplate spectrophotometer (Thermo Scientific Varioskan Flash; 540-nm excitation and 620-nm emission) was used to examine the adenosine 5′-diphosphate generation. Meanwhile, the background values were determined without protein and compounds, while the negative control was determined with protein only and regarded as 100% ATPase activity.
In vitro pulldown assay
In vitro pulldown experiments were conducted using a Pierce GST Protein Interaction Pull-Down Kit (Thermo Fisher Scientific). GST-Cdc37 protein bound to GST Sepharose beads was then incubated with His-Hsp90 N terminus or Hsp90 mutants overnight at 4°C in the kit buffer. After being washed five times with washing buffer, the proteins were eluted from the beads with elution buffer and subjected to SDS-PAGE, followed by Western blots.
HTRF assays
HTRF assays were performed using a standard protocol (www.cisbio.com). A total volume of 8 μl of His-tag Hsp90 and GST-tag Cdc37 was premixed in PBS buffer containing an additional 200 mM KF (pH 7.4), with a final concentration of 80 nM. Then, 4 μl of prediluted solution with compounds at the indicated concentration was added and incubated for 1 hour at 37°C. Subsequently, 4 μl of anti-GST Cryptate (61GSTKLA, Cisbio) and 4 μl of anti-6HisXL665 (61HISXLA, Cisbio) detection reagent were added to reach a 20-μl volume for the experimental system, which was then incubated for another 30 min at room temperature. Time-resolved fluorescence intensities were measured using a Molecular Devices instrument (SpectraMax Paradigm; excitation, 320 nm; emission, 665 and 620 nm). The final HTRF ratio was calculated by taking the ratio of signals at two different wavelengths as follows: ratio = (Signal 665 nm/Signal 620 nm) × 10,000.
FP assay
In general, the ATP binding site competitive FP assay was performed as previously reported (22). FP values were determined on a SpectraMax multimode microplate reader (Molecular Devices SpectraMax Paradigm; excitation, 485 nm; emission, 535 nm). The final inhibition ratio was calculated by GraphPad Prism 6.0, and the values of the inhibition rate were determined as previously reported (37).
Biolayer interferometry assay
The dose-dependent binding affinities of compounds for WT or mutant Hsp90 were determined by a biolayer interferometry assay using Octet RED96 (ForteBio). All the proteins used in this assay were biotinylated by EZ-Link NHS-Biotin (20217, Thermo Fisher Scientific) at room temperature with a 1:1 incubation ratio according to the manufacturer’s instructions. Super Streptavidin (SSA) biosensor tips (ForteBio, Menlo Park, CA) were used to immobilize the biotinylated proteins after prewetting with kinetic buffer (PBS, 0.05%; bovine serum albumin, 0.01% Tween 20). The equilibrated SSA biosensors were loaded with WT or mutant Hsp90 (100 μg/ml). Background binding controls used a duplicate set of sensors that incubated in buffer without proteins. All assays were performed by a standard protocol in 96-well black plates with a total volume of 200 μl per well at 28°C. All the data were analyzed by Octet data analysis software. The signals were analyzed by a double reference subtraction protocol to deduce nonspecific and background signals and signal drifts caused by biosensor variability. A 1:1 binding model was used to fit the association and dissociation rates. Equilibrium dissociation constant (KD) values were calculated from the ratio of Koff to Kon.
Thermal shift assay
First, 10 μM N-terminal Hsp90 with or without the indicated concentrations (200, 100, 50, and 10 μM) of DDO-5936 was mixed in assay buffer [1× PBS (pH 8.0)], followed by the addition of 6× SYPRO Orange dye (Thermo Fisher Scientific, UK) to a final volume of 20 μl that was seeded in 96-well polymerase chain reaction (PCR) plates. Subsequently, the samples were heated in a PCR system (StepOnePlus, Applied Biosystems) from room temperature at a rate of 1°C/min for 74 cycles. Fluorescence intensities were monitored with 492-nm excitation and 610-nm emission. Control wells were used to compare the melting temperature (Tm) without compounds [replaced by the same amount of dimethyl sulfoxide (DMSO)]. Tm values were obtained from the maximum value of first derivative (dF/dT) plots of the unfolding protein curves and then analyzed by Protein Thermal Shift software v1.3 (Thermo Fisher Scientific). Experiments were performed in triplicate.
Kinase panel screening
The enzyme assays for 20 cell cycle–related kinases were conducted by the ChemPartner company (Shanghai, China) according to standard mobility shift assay protocols. Briefly, the diluted compounds were performed in prepared kinase buffer with ATP solution. Then, all samples were transferred to each well of a 384-well plate for incubation at room temperature for 10 min. The data were collected after adding stop buffer to stop reaction. The IC50 values were calculated by fitting the data points with the dose-response function in GraphPad Prism 6.0 (GraphPad Software) from three independent experiments.
NMR assays
Ligand-observed T1ρ and STD NMR experiments were used to verify compound-protein interactions. All NMR spectra were acquired at 25°C on a Bruker Avance III 600 MHz (proton frequency) spectrometer equipped with a cryogenically cooled probe (Bruker BioSpin, Germany). For ligand-observed T1ρ measurements, 200 μM compound with 0, 1, 5, and 20 μM Hsp90 were prepared as samples. For the STD experiments, 100 μM compound with or without 5 μM Hsp90 was prepared as samples. Compounds and proteins were dissolved in phosphate buffer [20 mM sodium phosphate (pH 7.4), 150 mM NaCl, and 5% DMSO] for NMR data acquisition.
Two-dimensional HSQC experiments were performed on a Bruker 800-MHz NMR spectrometer, and samples involving 50 μM 15N-labeled Hsp90 protein with or without the indicated concentration of compound (250 μM and 500 μM) were investigated in assay buffer containing 20 mM sodium phosphate (pH 7.4), 150 mM NaCl, and 5% DMSO. NMR data processing and analysis used the SPARKY (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco) program. Images of the structures were generated by PyMOL. Chemical shift changes were calculated using the combined shift changes of amide nitrogens and protons, Δω = (ΔωN2 + ΔωH2)1/2, where ΔωN and ΔωH are the differences in 15N and 1H frequencies, respectively, between WT protein and compound-bound protein in hertz.
Cell culture
HCT116, A549, H460, L02, HepG2, 786-0, HT1080, MCF-7, SKBR3, PC3, and HT29 cell lines were obtained from the Cell Resource Center of Shanghai Institute for Biological Sciences, Chinese Academy of Sciences. PANC-1 and SW480 were provided by J.Z. (Department of Pathophysiology, Key Laboratory of Cell Differentiation and Apoptosis of Ministry of Education, Shanghai Jiao Tong University School of Medicine). The MCF-7, SKBR3, and PC3 cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS). HCT116, A549, H460, L02, HepG2, 786-0, HT1080, HT29, NCM460, and CCD18C0 cell lines were maintained in RPMI 1640 medium supplemented with 10% FBS. All cells were authenticated by short tandem repeat profiling, examined for mycoplasma contamination, and cultured at 37°C in a humidified, 5% CO2-containing atmosphere incubator (Thermo Fisher Scientific).
Cellular thermal shift assay
CETSAs were performed by previously published standard protocols (38). HCT116 cells were seeded in 10-cm cell culture dishes to reach ~90% confluence. After treatment with 25 μM compound or the same concentration of DMSO, HCT116 cells were collected and washed three times with PBS buffer to exclude excess residual compound. Subsequently, cells were isolated by centrifugation (1200 rpm for 5 min at 4°C) and resuspended in PBS. Cell suspensions were equally distributed into 200-μl PCR tubes (approximately 1 million cells per tube). Then, the tubes were heated at the indicated temperature (37° to 61°C) to denature samples for 3 min, and the cells were freeze-thawed twice by liquid nitrogen. Last, the samples were centrifuged, and the supernatants were analyzed by Western blotting.
Co-IP assay
HCT116 cells were seeded in 10-cm cell culture dishes to reach ~90% confluence. Then, the cells were incubated with the indicated concentrations of compounds or the same amount of DMSO for 12 hours. After washing the cells twice with ice-cold PBS, radioimmunoprecipitation assay (RIPA) buffer [50 mM tris (pH 7.4), 150 mM NaCl, 1% NP-40, and protease inhibitor cocktail (Roche)] was used to lyse the cells for 1 hour on ice. Cell lysates were centrifuged at 12,000 rpm at 4°C for 15 min. Then, 1 mg of cell lysates was incubated with 5 μg of anti-Hsp90 (sc-13119, Santa Cruz Biotechnology), anti-Cdc37 (4793S, Cell Signaling Technology), or normal rabbit immunoglobulin G (IgG) (sc-2027, Santa Cruz Biotechnology) separately and overnight at 4°C on a vertical roller. Protein A/G Magnetic Agarose Beads (78609, Thermo Fisher Scientific) were added to lysates for another 3-hour incubation at 4°C. Last, the beads were washed five times with RIPA buffer and subjected to SDS-PAGE, followed by Western blot analysis.
Antiproliferative assay
Antiproliferative assays of the compounds against different cancer cell lines were performed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) assays. Cells were seeded into 96-well plates at 3000 to 6000 cells per well and incubated for 24 hours. Subsequently, the cells were treated with the indicated concentration of compounds or the same amount of DMSO for 72 hours. MTT (5 mg/ml) was added and incubated at 37°C for 4 hours. Then, the solution was removed, and 150 μl of DMSO was added to dissolve MTT formazan crystals. Last, the OD values at 570 nm were determined by an ELx800 absorbance microplate reader (BioTek, Vermont, USA). The IC50 values were calculated by nonlinear fit curves using GraphPad Prism 6.0 software.
Immunoblotting and antibodies
For all Western blots, the cell lysates were prepared in 1× SDS loading buffer. Total protein was applied to SDS-PAGE and transferred to Immobilon-PSQ Transfer Membranes (Merck Millipore). The membranes were blocked in 5% nonfat milk for 1 hour at room temperature, followed by the addition of specific primary antibodies overnight at 4°C. Horseradish peroxidase (HRP)–linked anti-rabbit IgG (catalog no. 7074P2, lot no. 26, Cell Signaling Technology) or HRP-linked anti-mouse IgG (catalog no. 7076P2, lot no. 32, Cell Signaling Technology) was used as a secondary antibody, corresponding to different proteins. Protein bands were detected using an Immobilon Western Chemiluminescent HRP Substrate Kit (Merck Millipore).
CRISPR-Cas9 KO cell lines
Cdc37-KO cell lines were generated through a standard CRISPR-Cas9 protocol (39). Guide RNA (gRNA) was designed with guidance from the website of the Zhang Lab (http://crispr.mit.edu/). Then, gRNA targeting exon 2 was cloned into the vector lentiCRISPR v2 (Addgene plasmid no. 52961). LentiCRISPR v2 vectors involving gRNA, psPAX2, and pMD2·G were prepared and transfected by Lipofectamine 2000 (Thermo Fisher Scientific) into human embryonic kidney 293T cells. Lentiviruses were cultured for 2 days after transfection and subsequently collected. Then, the HCT116 cells were infected with viral supernatant with polybrene (8 μg/ml; Sigma-Aldrich). The infected cells were screened by puromycin selection (Sigma-Aldrich), and the indicated clones were collected from living cells by limited dilution methods. Last, the selected clones were examined by Western blots.
Cell cycle analysis
Cells were treated with the indicated concentrations of compounds for 24 h at 37°C in the incubator and were subsequently harvested and fixed with 75% ethanol overnight at 4°C. Ribonuclease I (50 μg/ml) was incubated with the cells in PBS at 37°C for 30 min, and then PI (50 μg/ml) was used to stain the cells for 15 min. Flow cytometry and FACSDiva software, v 6.2 (BD Biosciences), were used to perform cell cycle assays. The distribution of the cell cycle phases was analyzed by FlowJo software.
Tumor xenograft experiments
HCT116 cells (1 × 107) were mixed with Matrigel (BD Biosciences) in a 3:1 volume on ice and then injected into the flanks of 6-week-old BALB/c nude mice to generate xenografts. The mice were randomly divided into three groups (n = 6 or 8 per group), and treatment was started when the mean tumor volumes reached ~125 mm3. The mice received an intraperitoneal injection of 0.9% saline solution containing 5% DMSO (vehicle) and compound (50 or 100 mg/kg) daily for 21 days. Tumor volume was determined every other day by measuring the two perpendicular diameters of the tumors and using the formula V = length (mm) × width (mm)2/2, and the body weight was recorded every 3 days. After 21 days of treatment, the mice were sacrificed, and the tumors were dissected and weighed. Tumor tissues and experimental organs were collected for further studies. All animals used in this study were handled in accordance with federal and institutional guidelines under a protocol approved by the Institutional Animal Care and Use Committee at China Pharmaceutical University.
H&E staining
Tumor tissues and normal organs were fixed in 4% formaldehyde solution and paraffin-embedded. All sections were cut to 4-μm thickness, immobilized on a glass slide, and subsequently stained with H&E for imaging to detect the morphology.
Pharmacokinetics/pharmacodynamics
The xenograft model mice (n = 6 per group) were administered DDO-5936 intravenously at 100 mg/kg. Blood samples with a volume of 150 μl were collected into heparinized Eppendorf tubes at indicated time points (0, 0.25, 0.5, 1, 2, 8, 12, and 24 hours) and centrifuged at 3000 rpm for 10 min to obtain plasma samples (the samples were stored at −80°C until liquid chromatography–tandem mass spectrometry analysis). Tumor samples were collected at the same time points before homogenized. The concentration of the protein lysates of tumor tissue was determined by a bicinchoninic acid kit. Then, the protein lysates were analyzed by Western blots.
Statistics
GraphPad Prism 6.0 software was used to calculate statistical significance. Values are expressed as the means ± SD. The statistical significance of the data was analyzed from at least three independent assays using Student’s t test. Significant differences between two groups were examined statistically as indicated (*P < 0.05, **P < 0.01, ***P < 0.001).
Supplementary Material
Acknowledgments
We thank W. Fu and T. Hou (College of Pharmacy, Zhejiang University) for helping with the computational calculations and virtual screening, L. Feng (High-Field NMR Center, College of Chemistry and Chemical Engineering, Xiamen University) and F. Lian (Department of Analytical Chemistry, Shanghai Institute of Materia Medica, Chinese Academy of Sciences) for NMR assays, and Z. Qiu (School of Traditional Chinese Pharmacy, China Pharmaceutical University) for PK/PD assays. Funding: This study was supported by projects 81573346, 81773639, 81773581, and 81602948 of the National Natural Science Foundation of China; BK20160746 and BK20190559 of the Natural Science Foundation of Jiangsu Province of China; CPU2018GY02 of the Double First Class Innovation Team of China Pharmaceutical University; the National Science and Technology Major Project “Key New Drug Creation and Manufacturing Program,” China (nos. 2018ZX09711002, 2017ZX09302003, and 2015ZX09101032); the Open Project of State Key Laboratory of Natural Medicines (SKLNMZZCX201803); the China Postdoctoral Science Foundation (no. 2018 M642376); the Program for Outstanding Scientific and Technological Innovation Team of Jiangsu Higher Education; and the Young Elite Scientists Sponsorship Program by CAST. Innovation Program of Shanghai Municipal Education Commission (Innovation Program of Shanghai Municipal Education Commission). Author contributions: Q.Y. initiated and supervised the research. Q.Y., J.Z., and L.W. designed the experiments. L.W. and Z.J. conducted MD simulations, compound screening, and hit validation. L.W., L.Z., and L.L. performed the biological assays and compound synthesis. Z.Z. helped perform the NMR assays. J.S., C.W., X.X., Q.B., and W.C. helped perform cellular assays. Q.Y., J.Z., Z.J., and L.W. wrote the manuscript, and all other authors contributed specific parts of the manuscript. Q.Y. and J.Z. assume responsibility for the manuscript in its entirety. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax2277/DC1
Supplementary Materials and Methods
Fig. S1. Searching for critical residues on the Hsp90-Cdc37 binding interface through MD simulation calculation.
Fig. S2. Identification of critical residues on Hsp90-Cdc37 binding interface.
Fig. S3. The rational design and discovery of Hsp90-Cdc37 PPI inhibitors.
Fig. S4. Synthetic scheme and binding site characterization of DDO-5936.
Fig. S5. DDO-5936 bound to Hsp90 in cells and the antiproliferative activities of DDO-5936 were correlated with the protein expression level of Hsp90 and Cdc37.
Fig. S6. Quantification of protein expression in Fig. 4 (A and B).
Fig. S7. Histological morphology of H&E-stained tissues sections of representative nude mice and PK/PD studies of DDO-5936.
Fig. S8. 1H NMR spectrum and 13C NMR spectrum of DDO-5936.
Table S1. The change of Gibbs free energy from calculation and experimental results.
Table S2. ATPase inhibition of DDO-5936 on cell cycle–related kinases by enzyme assays.
REFERENCES AND NOTES
- 1.Chiosis G., Dickey C. A., Johnson J. L., A global view of Hsp90 functions. Nat. Struct. Mol. Biol. 20, 1–4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahalingam D., Swords R., Carew J. S., Nawrocki S. T., Bhalla K., Giles F. J., Targeting HSP90 for cancer therapy. Br. J. Cancer 100, 1523–1529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trepel J., Mollapour M., Giaccone G., Neckers L., Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neckers L., Workman P., Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 18, 64–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hong D. S., Banerji U., Tavana B., George G. C., Aaron J., Kurzrock R., Targeting the molecular chaperone heat shock protein 90 (HSP90): Lessons learned and future directions. Cancer Treat. Rev. 39, 375–387 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Taipale M., Krykbaeva I., Koeva M., Kayatekin C., Westover K. D., Karras G. I., Lindquist S., Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearl L. H., Hsp90 and Cdc37—A chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 15, 55–61 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Mandal A. K., Lee P., Chen J. A., Nillegoda N., Heller A., DiStasio S., Oen H., Victor J., Nair D. M., Brodsky J. L., Caplan A. J., Cdc37 has distinct roles in protein kinase quality control that protect nascent chains from degradation and promote posttranslational maturation. J. Cell Biol. 176, 319–328 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verba K. A., Wang R. Y., Arakawa A., Liu Y., Shirouzu M., Yokoyama S., Agard D. A., Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stepanova L., Yang G., DeMayo F., Wheeler T. M., Finegold M., Thompson T. C., Harper J. W., Induction of human Cdc37 in prostate cancer correlates with the ability of targeted Cdc37 expression to promote prostatic hyperplasia. Oncogene 19, 2186–2193 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Roe S. M., Ali M. M., Meyer P., Vaughan C. K., Panaretou B., Piper P. W., Prodromou C., Pearl L. H., The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 116, 87–98 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Jiang Y., Bernard D., Yu Y., Xie Y., Zhang T., Li Y., Burnett J. P., Fu X., Wang S., Sun D., Split Renilla luciferase protein fragment-assisted complementation (SRL-PFAC) to characterize Hsp90-Cdc37 complex and identify critical residues in protein/protein interactions. J. Biol. Chem. 285, 21023–21036 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He J., Niu X., Hu C., Zhang H., Guo Y., Ge Y., Wang G., Jiang Y., Expression and purification of recombinant NRL-Hsp90α and Cdc37-CRL proteins for in vitro Hsp90/Cdc37 inhibitors screening. Protein Expr. Purif. 92, 119–127 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Schwarze S. R., Fu V. X., Jarrard D. F., Cdc37 enhances proliferation and is necessary for normal human prostate epithelial cell survival. Cancer Res. 63, 4614–4619 (2003). [PubMed] [Google Scholar]
- 15.Gray P. J. Jr., Stevenson M. A., Calderwood S. K., Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 67, 11942–11950 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Wang L., Li L., Zhou Z. H., Jiang Z. Y., You Q. D., Xu X. L., Structure-based virtual screening and optimization of modulators targeting Hsp90-Cdc37 interaction. Eur. J. Med. Chem. 136, 63–73 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Huang W., Ye M., Zhang L. R., Wu Q. D., Zhang M., Xu J. H., Zheng W., FW-04-806 inhibits proliferation and induces apoptosis in human breast cancer cells by binding to N-terminus of Hsp90 and disrupting Hsp90-Cdc37 complex formation. Mol. Cancer 13, 150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang T., Hamza A., Cao X., Wang B., Yu S., Zhan C. G., Sun D., A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 7, 162–170 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Chen X., Liu P., Wang Q., Li Y., Fu L., Fu H., Zhu J., Chen Z., Zhu W., Xie C., Lou L., DCZ3112, a novel Hsp90 inhibitor, exerts potent antitumor activity against HER2-positive breast cancer through disruption of Hsp90-Cdc37 interaction. Cancer Lett. 434, 70–80 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Zhang T., Li Y., Yu Y., Zou P., Jiang Y., Sun D., Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J. Biol. Chem. 284, 35381–35389 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sreeramulu S., Jonker H. R., Langer T., Richter C., Lancaster C. R., Schwalbe H., The human Cdc37.Hsp90 complex studied by heteronuclear NMR spectroscopy. J. Biol. Chem. 284, 3885–3896 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Kim J., Felts S., Llauger L., He H., Huezo H., Rosen N., Chiosis G., Development of a fluorescence polarization assay for the molecular chaperone Hsp90. J. Biomol. Screen. 9, 375–381 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Zhang H., Zhou C., Chen W., Xu Y., Shi Y., Wen Y., Zhang N., A dynamic view of ATP-coupled functioning cycle of Hsp90 N-terminal domain. Sci. Rep. 5, 9542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobs D. M., Langer T., Elshorst B., Saxena K., Fiebig K. M., Vogtherr M., Schwalbe H., NMR backbone assignment of the N-terminal domain of human HSP90. J. Biomol. NMR 36, 52 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Dehner A., Furrer J., Richter K., Schuster I., Buchner J., Kessler H., NMR chemical shift perturbation study of the N-terminal domain of Hsp90 upon binding of ADP, AMP-PNP, geldanamycin, and radicicol. Chembiochem 4, 870–877 (2003). [DOI] [PubMed] [Google Scholar]
- 26.Vaughan C. K., Gohlke U., Sobott F., Good V. M., Ali M. M., Prodromou C., Robinson C. V., Saibil H. R., Pearl L. H., Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell 23, 697–707 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gray P. J. Jr., Prince T., Cheng J., Stevenson M. A., Calderwood S. K., Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer 8, 491–495 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith J. R., Clarke P. A., de Billy E., Workman P., Silencing the cochaperone CDC37 destabilizes kinase clients and sensitizes cancer cells to HSP90 inhibitors. Oncogene 28, 157–169 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hallett S. T., Pastok M. W., Morgan R. M. L., Wittner A., Blundell K., Felletar I., Wedge S. R., Prodromou C., Noble M. E. M., Pearl L. H., Endicott J. A., Differential regulation of G1 CDK complexes by the Hsp90-Cdc37 chaperone system. Cell Rep. 21, 1386–1398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J., Soroka J., Buchner J., The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 1823, 624–635 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Simmerling C., Strockbine B., Roitberg A. E., All-atom structure prediction and folding simulations of a stable protein. J. Am. Chem. Soc. 124, 11258–11259 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Darian E., Gannett P. M., Application of molecular dynamics simulations to spin-labeled oligonucleotides. J. Biomol. Struct. Dyn. 22, 579–593 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Cheatham T. E., Miller J. L., Fox T., Darden T. A., Kollman P. A., Molecular-dynamics simulations on solvated biomolecular systems: The particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc. 117, 4193–4194 (1995). [Google Scholar]
- 34.Kollman P. A., Massova I., Reyes C., Kuhn B., Huo S. H., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D. A., Cheatham T. E., Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Gohlke H., Kiel C., Case D. A., Insights into protein-protein binding by binding free energy calculation and free energy decomposition for the Ras-Raf and Ras–RalGDS complexes. J. Mol. Biol. 330, 891–913 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Gohlke H., Case D. A., Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 25, 238–250 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Jiang F., Wang H. J., Jin Y. H., Zhang Q., Wang Z. H., Jia J. M., Liu F., Wang L., Bao Q. C., Li D. D., You Q. D., Xu X. L., Novel tetrahydropyrido[4,3-d]pyrimidines as potent inhibitors of chaperone heat shock protein 90. J. Med. Chem. 59, 10498–10519 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Molina D. M., Jafari R., Ignatushchenko M., Seki T., Larsson E. A., Dan C., Sreekumar L., Cao Y. H., Nordlund P., Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Cong L., Ran F. A., Cox D., Lin S., Barretto R., Habib N., Hsu P. D., Wu X., Jiang W., Marraffini L. A., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/9/eaax2277/DC1
Supplementary Materials and Methods
Fig. S1. Searching for critical residues on the Hsp90-Cdc37 binding interface through MD simulation calculation.
Fig. S2. Identification of critical residues on Hsp90-Cdc37 binding interface.
Fig. S3. The rational design and discovery of Hsp90-Cdc37 PPI inhibitors.
Fig. S4. Synthetic scheme and binding site characterization of DDO-5936.
Fig. S5. DDO-5936 bound to Hsp90 in cells and the antiproliferative activities of DDO-5936 were correlated with the protein expression level of Hsp90 and Cdc37.
Fig. S6. Quantification of protein expression in Fig. 4 (A and B).
Fig. S7. Histological morphology of H&E-stained tissues sections of representative nude mice and PK/PD studies of DDO-5936.
Fig. S8. 1H NMR spectrum and 13C NMR spectrum of DDO-5936.
Table S1. The change of Gibbs free energy from calculation and experimental results.
Table S2. ATPase inhibition of DDO-5936 on cell cycle–related kinases by enzyme assays.