

Abstract

Metal-free graphitic carbon nitride (g-CN) has attracted significant attention recently due to its multiple applications, such as photocatalysis, energy storage and conversion, and biomaterials, albeit formation of g-CN films is challenging. Herein, a “grafting to” route to graft polymer brushes onto g-CN via visible-light irradiation is described. Afterward, g-CN/polymer films can be obtained through spin coating on glass substrates. As such, the present material provides an improved process toward further application of g-CN in thin films. Moreover, an improved dispersibility in organic solvent was realized after grafting and functional groups (such as epoxides) were introduced to g-CN. Subsequently, the epoxy groups were utilized for further functionalization to adjust the surface polarity.
Introduction
Recently, metal-free graphitic carbon nitride (g-CN) has been widely investigated,1 due to its promising properties for many applications.2,3 It consists of repeating tri-s-triazine rings and features a band gap of about 2.7 eV absorbing visible light. Generally studied as a heterogeneous photocatalyst, g-CN can be used in water splitting,4−6 CO2 reduction,3,7 and pollution photodegradation.8−10 The unique optical properties such as photoluminescence11−13 and biocompatibility14 make g-CN an ideal candidate for bioimaging15,16 and biomedical17 applications. Recently, two-dimensional g-CN films have been frequently studied18 and have broad applications in solar cells,19 ionic devices,20 and sensing.21 In the past, various approaches targeted for the fabrication of g-CN films via chemical vapor deposition (CVD),22 thermal vapor condensation,23 or magnetron sputtering24 were investigated, during which g-CN directly grows from the substrate under suitable conditions, and usually uniform films can be obtained after these processes. For example, Xu et al. fabricated continuous polymeric CN thin films for solar cell application,19 and Xiao et al. used CVD-fabricated CN for ion transport.20 However, problems such as being costly, complicated processing, and high time requirements are disadvantages of these technologies. Solution processing would be an useful method for film formation, albeit the weak dispersibility of g-CN in most solvents caused by strong van der Waals attractions is the main problem for the solution approach. Hence, it is challenging to employ solution-based common methods, like spin coating25 or drop casting26 to form uniform g-CN films. Moreover, g-CN shows limited selectivity toward CO2 photoreduction because of unfavorable surface properties, which require further modification.27,28
In general, several strategies were attempted to improve the physiochemical properties of g-CN, such as doping,29 morphology changing,2 precursor functionalization,30 copolymerization,31 and surface grafting.32,33 For example, Xu et al. grafted n-bromobutane onto mesoporous CN for heterogeneous catalysis.34 Bu et al. modified g-CN with hydroxylamine groups by treating with oxygen plasma, thus forming g-CN with excellent hydrophilicity.35 Very recently, our group investigated the photoinitiated surface modification of g-CN, e.g., to introduce hydrophilic or hydrophobic moieties.36,37 In this process, g-CN promoted radical formation under visible-light irradiation that was utilized to initiate polymerizations38,39 or hydrogel formation as well.40,41 Among the modification of g-CN, polymers obtained more and more attraction, due to their versatile properties (e.g., flexibility, easy functionalization, and affordability).42 For example, poly(glycidyl methacrylate) (PGMA) is widely utilized because of the epoxy group that can be easily functionalized in different ways, e.g., Abedul and co-workers employed PGMA as a tunable platform to modify microfluidic devices.43 Usually utilized as an economical alternative to polycarbonate, poly(methyl methacrylate) (PMMA) possesses moderate properties of easy handling and processing, while poly(isobornyl acrylate) (PIBA) is commonly applied on microfluidic devices44 due to the properties of inertness and transparency. Thus, the surface properties of g-CN might be facilely adjusted by functionalized polymers while preserving its inherent catalytic features.
However, to the best of our knowledge, grafting polymer brushes onto g-CN has not been reported before, which in turn might lead to an improvement of interface properties of g-CN. Polymer brushes are surface coatings that consist of polymers tethered to a substrate, which can be realized by physisorption or covalent attachment. There are two ways for the covalent attachment, one is “grafting from”, where a controlled polymerization45 is performed and the polymer chains grow via monomer propagation on the surface. Another approach is grafting to; in this process, an end-functionalized polymer is reacted with a substrate surface. Several factors have an effect on the final grafting density, such as polymer molecular weight, surface coverage, solvent quality, and steric hindrance.46−48 Grafting of polymer brushes is commonly used for fabrication of hybrid structures,49 where polymers are tethered on a substrate, e.g., inorganic materials such as silicon, gold, hexagonal boron nitride, or silica,50−52 so that organic–inorganic films can be patterned as well, which can be applied for biotechnology,53 microelectronics,54,55 microreaction vessels,56 and electrical devices.57 In this perspective, g-CN as a functional material might be an ideal option for polymer grafting, and a combination of polymer with g-CN might simplify the process for g-CN utilization in various applications.
In the present work, the grafting to method is utilized to graft ene-functional polymers onto g-CN. Ene-functional polymers with different side groups, such as PMMA, PGMA, and PIBA were synthesized by atom transfer radical polymerization (ATRP), and ene-modified initiator 10-undecenyl 2-bromopropionate (Un-EBrP) was used during polymerization, as depicted in Scheme 1. The very common and affordable CM (formed from cyanuric acid and melamine as precursors) was utilized as the substrate. Due to the formation of surface radicals under visible light, the CM surface was employed for grafting. The polymer-grafted CM (polymer/CM) was characterized via elemental analysis (EA), thermal gravimetric analysis (TGA), and Fourier transform infrared spectroscopy (FT-IR). Afterward, polymer/CM film materials can be facilely obtained via spin coating and analyzed with an atomic force microscope (AFM) and a scanning electron microscope (SEM). Moreover, PGMA-grafted CM can be utilized to tune the material via further modification. Surface properties after modification was subsequently determined by contact angle and SEM–energy-dispersive X-ray spectroscopy (SEM–EDX).
Scheme 1. Overview of Atom Transfer Radical Polymerization and Grafting of Polymer Brushes onto g-CN under Visible-Light Irradiation.

Experimental Section
Materials
Acetic acid (glacial, ≥99.9%, Sigma-Aldrich), Al2O3 basic (Sigma-Aldrich), 2-bromopropionyl bromide (98%, Sigma-Aldrich), cyanuric acid (98%, Sigma-Aldrich), deuterated chloroform (CDCl3, 99.96%, Sigma-Aldrich), ethanol (absolute, 95%, Sigma-Aldrich), ethyl acetate (99.8%, Sigma-Aldrich), hexane (95%, Sigma-Aldrich), melamine (99%, Sigma-Aldrich), ethyl 2-bromopropionate (EBrP, 98%, Sigma-Aldrich), pyridine (99.8%, Sigma-Aldrich), N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA, ≥98%, Sigma-Aldrich), 1H,1H,2H,2H-perfluorodecanethiol (pFDe, ≥97%, Sigma-Aldrich), sodium 2-mercaptoethanesulfonate (MESNA, ≥98%, Sigma-Aldrich), tetrahydrofuran (THF, ≥99.9%, inhibitor-free, Sigma-Aldrich), silica gel (for column chromatography, Sigma-Aldrich), methanol (≥99.9%, Sigma-Aldrich), hydrochloric acid (HCl, 37%, Sigma-Aldrich), and 10-undecen-1-ol (98%, Sigma-Aldrich) were used as received. Copper(I) bromide (Cu(I)Br, 98%, Sigma-Aldrich) was purified by stirring in glacial acetic acid overnight, filtered, and washed with absolute ethanol. Isobornyl acrylate (IBA, 98.5%, Sigma-Aldrich), glycidyl methacrylate (GMA, 97%, Sigma-Aldrich), and methyl methacrylate (MMA, 99%, Sigma-Aldrich) were passed through a basic alumina column prior to use. 50 W Light-emitting diode (LED) chips (Foxpic High Power 50 W LED Chip Bulb Light DIY White 3800LM 6500 K) were connected to a self-made circuit and cooling system. Sonication was performed in a sonication bath from Elma (Transsonic T310). The initiator 10-undecenyl 2-bromopropionate (Un-EBrP) was synthesized following the work of Matyjaszewski and co-workers,58 and the utilized g-CN (CM) was synthesized from the cyanuric acid–melamine complex according to the literature.59
ATRP Employing Un-EBrP as an Initiator
General Procedure
Cu(I)Br was added to a 25 mL round bottom flask in an ice bath, and the flask was sealed with a rubber septum and flushed with argon for 30 min. Subsequently, deoxygenated monomers and deoxygenated PMDETA were added via a syringe. After Cu(I)Br/PMDETA complex formation, the solution turned light green and the initiator Un-EBrP was added via a syringe, the ice bath was removed and the mixture was stirred at the noted temperature and time. Subsequently, the flask was removed from the oil bath and a sample of the raw products was subjected to 1H NMR for calculation of monomer conversion. The reaction mixture was diluted in THF and passed through an aluminum oxide column to remove the oxidized catalyst. The polymer was purified by precipitation into methanol, filtered, dried, and finally the polymer was obtained as a white solid. Reference polymerizations with EBrP were performed in the same way.
Polymerization of MMA
PMMA was obtained according to the general procedure with the following equivalents: MMA (3.76 g, 0.0376 mol, 376 equiv), Cu(I)Br (7.5 mg, 0.05 mmol, 0.5 equiv), PMDETA (8.6 mg, 0.05 mmol, 0.5 equiv), and Un-EBrP (32 mg, 0.1 mmol, 1 equiv). The reaction was carried out at 45 °C for 30 min (conversion = 65%). (Mn,SEC = 21.5 kg/mol, Đ = 1.21.)
Polymerization of GMA
PGMA was obtained according to the general procedure with the following equivalents: GMA (5.4 g, 0.04 mol, 200 equiv), Cu(I)Br (0.15 mg, 0.1 mmol, 0.5 equiv), PMDETA (17 mg, 0.1 mmol, 0.5 equiv), and Un-EBrP (64 mg, 0.2 mmol, 1 equiv). The reaction was carried out at 25 °C for 25 min (conversion = 33%). (Mn,SEC = 25.9 kg/mol, Đ = 1.75.)
Polymerization of IBA
PIBA was obtained according to the general procedure with the following equivalents: IBA (6.23 g, 0.03 mol, 150 equiv), Cu(I)Br (30 mg, 0.2 mmol, 1 equiv), PMDETA (34 mg, 0.2 mmol, 1 equiv), and Un-EBrP (64 mg, 0.2 mol, 1 equiv). The reaction was carried out at 65 °C for 90 min (conversion = 96%). (Mn,SEC = 21.3 kg/mol, Đ = 1.41.)
Grafting Polymer Brushes onto g-CN (Polymer/CM)
CM (30 mg) and 15 mL of THF were mixed in a round bottom flask and sonicated for 1 h to yield a CM dispersion. Afterward, 300 mg of the related polymer (PMMA, PGMA, or PIBA) were added to the CM dispersion; the flask was sealed with a rubber septum and flushed with argon for 30 min. The mixture was put between two 50 W LED daylight sources (20 cm distance between light sources) to initiate the grafting. After 2 days, the mixture was washed two times with THF and dried under vacuum.
The control samples were prepared by the same process but treated only with stirring for 2 days without light irradiation.
The study of the CM/PMMA weight ratio was performed with 75, 150, 200, and 300 mg of PMMA and CM (30 mg), respectively.
Preparation of the Films
Polymer/CM films were prepared via spin coating. At first, 4 mg of the as-prepared PIBA/CM was dispersed in 1 mL of THF to obtain a PIBA/CM dispersion. Glass slides were used as substrates. The initially stationary glass was flooded with PIBA/CM solution until the entire surface was covered, then accelerated to the desired rotation rate. Acceleration times were 10 s, total spin times were 60 s, and the spin speed was 3000 rpm. Afterward, the glass slides were placed under vacuum at ambient temperature for 2 h to obtain the final dry films.
Modification of PGMA/CM
MESNA–PGMA/CM
In a 15 mL glass vial, 2 mL of THF, 5 mg of PGMA/CM, and 50 mg of MESNA were mixed and cooled with an ice bath. Then, 3 mg of LiOH was added slowly, the cooling was removed, and the resulting reaction mixture was stirred at ambient temperature for 3 h. Afterward, the mixture was diluted with 2 mL of H2O three times and centrifuged. The precipitate was dried under vacuum, and finally, 4.5 mg of dry products were obtained.
pFDe–PGMA/CM
In a 15 mL glass vial, 2 mL of THF, 7 mg of PGMA/CM, and 300 μL of pFDe were mixed and cooled with an ice bath. Then, 3 mg of LiOH was added slowly, the cooling was removed, and the resulting reaction mixture was stirred at ambient temperature for 1 h. Afterward, the mixture was diluted with 3 mL of THF three times and centrifuged. The precipitate was dried under vacuum, and finally, 5 mg of dry products were obtained.
As a reference, PMMA/CM was utilized together with pFDe and MESNA in the same process as with PGMA/CM.
Characterization
Fourier transform infrared (FT-IR) spectra were measured on a Nicolet iS 5FT-IR spectrometer. Elemental analysis (EA) for measuring the polymer/CM sample was recorded via a Vario Micro device. Thermogravimetric analysis (TGA) was carried out from ambient temperature to 1000 °C at a heating rate of 10 °C/min using a thermos microbalance TG 209 F1 Libra (Netzsch, Selb, Germany) in an air atmosphere, and data was recorded and analyzed by the Proteus (6.1.0) software package. Proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were recorded at ambient temperature on a Bruker Ascend 400. Size exclusion chromatography (SEC) for PGMA, PIBA, and PMMA was conducted in THF with toluene as an internal standard using PSS 1260-Iso as a pump, a column system of PSS SDV column (8 × 300 mm2) with a PSS SDV precolumn (8 × 50 mm2), PSS-SECcurity-VWD, and PSS-SECcurity-RID as detectors and a calibration with PMMA standards from PSS. The theoretical number-averaged molecular weight was calculated according to the equation (Mn,theo = [monomer]0/[I]0 × conversion × Mmonomer). Scanning electron microscopy (SEM) was performed using JSM-7500F (JEOL) equipped with an Oxford Instruments X-MAX 80 mm2 detector for imaging of the film. The SEM–energy-dispersive X-ray spectroscopy (EDX) measurements were conducted by using a JEOL JED-2300 detector in a JEOL JSM-5600 at an accelerating voltage of 15 kV. Atomic force microscopy (AFM) was performed with a Nanoscope IIIa Scanning Probe Microscope Controller and NanoWorld Arrow-NCR probes from Digital Instruments. The size of polymer-grafted g-CN was determined by a Zeta Nanosizer instrument (Malvern Instruments, U.K.) at a fixed scattering angle of 90°. All measurements were repeated at least three times. The three-phase contact angle of the polymer-grafted g-CN films was recorded by sessile drop analysis (OCA instrument, Dataphysics ES, Germany); the polymer-grafted g-CN was placed at the bottom of the stage, and about 2 μL of water droplet was placed gently on the sample, and three different spots on the film were measured. X-ray diffraction (XRD) patterns of polymer/CM and CM powders were obtained using a Bruker D8 Advance X-ray diffractometer via Cu Kα radiation. Solid state ultraviolet–visible (UV–vis) spectra of polymer/CM and CM were recorded via a Cary 500 Scan spectrophotometer equipped with an integrating sphere.
Results and Discussion
Synthesis of Ene-Functionalized Polymer
To obtain ene end-functionalized polymers for g-CN modification, a double-bond-functionalized initiator Un-EBrP was prepared following the work of Matyjaszewski and co-workers,58 which was used for atom transfer reaction polymerization (ATRP) with Cu(I)Br as the catalyst and PMDETA as the ligand. 1H NMR and 13C NMR (Figure S1) characterized the successful synthesis of Un-EBrP, with signals from the double bond at about 5 ppm. Three different monomers, namely MMA, IBA, and GMA, were used in the polymerization. The obtained polymers, PMMA, PIBA, and PGMA were characterized via SEC (Table 1). According to PMMA calibration, Mn,SEC values of 21.3 kg/mol for PIBA, 25.9 kg/mol for PGMA, and 21.5 kg/mol for PMMA were obtained, which indicates the successful polymerization of PGMA, PIBA, and PMMA, with Un-EBrP as an initiator. Unimodal molecular mass distributions were obtained (Figure S2), yet the ene-functionalized polymers show relatively broad molecular mass distributions with Đ of 1.41 for PIBA, 1.75 for PGMA, and 1.21 for PMMA. The reason might be chain transfer events during the polymerization, although test polymerizations with EBrP led to similar results (Table S1). In addition, the obtained molecular masses were in reasonable agreement with Mn,theo. Moreover, the double-bond incorporation was confirmed via 1H NMR (Figure 1) as the signals from the ene end-group can be found around 5 ppm.
Table 1. SEC Results for Ene-Functionalized Polymers Obtained via ATRP.
| ene-polymer | monomer/initiator | aconversion (%) | bMn,theo (kg/mol) | cMn,SEC(kg/mol) | dĐ |
|---|---|---|---|---|---|
| PGMA | 200:1 | 33 | 16 | 25.9 | 1.75 |
| PIBA | 150:1 | 96 | 29 | 21.3 | 1.41 |
| PMMA | 376:1 | 65 | 24 | 21.5 | 1.21 |
Calculated via 1H NMR.
Calculated according to the Experimental Section.
Determined via SEC in THF based on PMMA calibration.
Đ = Mw/Mn.
Figure 1.

1H NMR of ene end-functionalized PGMA (a), PIBA (b), and PMMA (c) (peak a corresponds to CHCl3 and peak b to water).
Ene-Polymer-Grafted CM
After synthesis of ene-functionalized polymers, grafting of CM was attempted. CM is readily producing radicals under visible-light irradiation, which can be exploited for photoinitiation. Thus, the double bond at the end of the polymer can be effectively attacked by those radicals and grafting proceeds as expected from the literature-known reactivity of g-CN and ene-functional molecules.38,60 By these means, functional polymer brushes onto CM were installed. Bulk CM was dispersed in THF and sonicated in advance to increase the accessibility of CM sheet structures (Figure S3a). Afterward, the as-prepared ene-polymers PMMA, PIBA, and PGMA were added, respectively. Irradiation with visible lights led to radical formation. Thus, polymers can be introduced to CM via radical addition, namely PIBA/CM (Figure S3b), PGMA/CM (Figure S3c), and PMMA/CM (Figure S3d). The grafting was followed by FT-IR spectroscopy (Figure 2a). Compared to CM precursor spectra, PIBA/CM, PGMA/CM, and PMMA/CM IR spectra show an additional peak originating from C=O stretching at about 1730 cm–1, which corresponds to the carbonyl group in the methacrylic polymers. Moreover, the corresponding ene-polymer FT-IR spectra with a direct comparison (Figure S4), indicated the grafting of PIBA, PGMA, and PMMA on CM, respectively. Subsequently, elemental analysis (Figure 2b) results showed a significant increase of the C/N ratio compared to the CM precursor, whereas CM only had a C/N ratio of 0.57, a change up to 1.88 (PIBA/CM), 1.00 (PGMA/CM), and 3.75 (PMMA/CM) was observed after grafting (Table S2), demonstrating successful ene-polymer grafting. Notably, only minor changes were observed for the reference samples, which were prepared via the same process without light irradiation, i.e., C/N ratios of 0.61 (ref PIBA/CM), 0.71 (ref PGMA/CM), and 0.61 (ref PMMA/CM) were observed. Hence, it can be confirmed that only slight physical attachment was present during mixing of polymers and CM, while a chemical bond was formed under visible-light irradiation.
Figure 2.
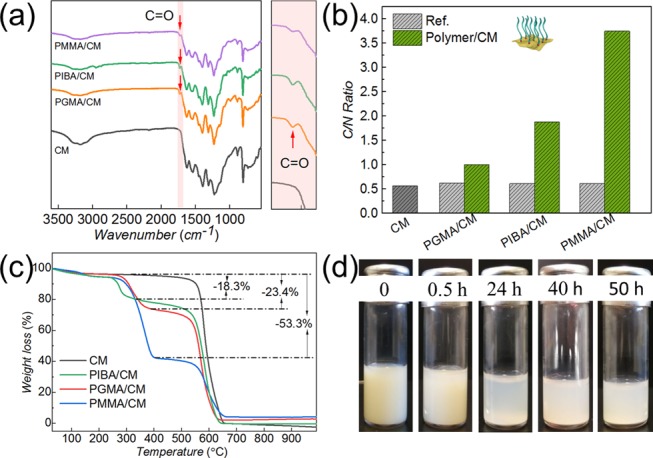
(a) FT-IR spectra of the CM precursor and ene-polymer-grafted CM. (b) C/N ratio of the CM precursor and ene-polymer-grafted CM obtained via elemental analysis. (c) TGA of the CM precursor and ene-polymer-grafted CM. (d) Photographs of PGMA/CM dispersed in THF over 2 days.
The amount of grafted polymers differs for the respective polymers, and thus thermogravimetric analysis (TGA, Figure 2c) was performed. The weight loss of polymers was analyzed during heating: 18.3% weight loss of PIBA/CM on heating at 220–300 °C, 23.4% loss of weight of PGMA/CM on heating at 250–380 °C, and 53.3% loss of weight of PMMA/CM on heating at 260–400 °C, respectively. Afterward, the amount of grafted ene-polymer grafting was calculated61 as shown specifically in Table S3. Therefore, a CM unit is defined consisting of seven nitrogen atoms and six carbon atoms. According to this calculation, PIBA- and PGMA-grafted CM show almost the same grafting density of 1 polymer chain grafted per 526 CM units (1.11 × 10–2 mmol/g) and 500 CM units (1.18 × 10–2 mmol/g), respectively. On the other hand, PMMA/CM shows a significantly higher grafting density of 4.46 × 10–2 mmol/g, meaning 1 PMMA chain per 132 units of CM. Demonstrating that even with almost the same molecular weight, a difference of the grafting density from different polymers is obtained. In the literature, it was stated that the cross-sectional area of a polymer chain with a similar Mn and in good solvent is around 170 Å2,62 which might be larger than the spaces between active species on the g-CN surface. Hence, only a limited amount of active radicals are utilized in the grafting to process. Moreover, steric hindrance differs according to the polymer sidechains, as PGMA with an epoxide group and PIBA with a cycloalkyl group cause larger steric hindrance than PMMA (only with methyl and methylcarboxylate groups), which also might result in higher PMMA grafting density.
To further elucidate the grafting process, utilization of different mass ratios of CM to PMMA was attempted (Figures S5 and S6). With a certain amount of CM (30 mg), simply adjusting the PMMA from 75 to 300 mg, an increased weight loss of PMMA with thermal treatment and increased C/N ratio from elemental analysis were found. Thus, it is indicated that with the introduction of increased amounts of PMMA, increased numbers of surface active species were utilized for grafting. In such a way, it can be concluded that the active sites on the surface are not saturated employing the initial ratios of PMMA and CM. Moreover, XRD (Figure S7a) and ultraviolet absorption spectrum (Figure S7b) show no significant changes compared to CM after grafting, which indicated that the inherent structural and absorption properties of CM were still preserved enabling further applications of the grafted materials.
Dispersibility and Film Fabrication
Commonly, g-CN has poor dispersibility, which limits its application, for example in solvent processing. Thus, further deposition is restricted, e.g., spin coating results in complicated processing for further applications as film materials, e.g., solar cells, fuel cells and organic light-emitting diodes. Therefore, the dispersibility of ene-polymer-grafted CM in organic media was investigated. As an example, PIBA/CM was dispersed in THF (Figures 2d and S8) over 2 days and chloroform (Figure S9) over 20 h, respectively, and only minor sedimentation was found. In comparison, the CM precursor does not form a stable dispersion at all, which confirms an improved stability of polymer/CM dispersions in organic solvent. Furthermore, dynamic light scattering (Table S4) in THF shows a significant decrease of CM hydrodynamic diameters after grafting, because CM is a sheetlike structure, and during the visible-light-inducing process, repelling force, and steric hindrance between two polymer chains causing the exfoliation of the CM layer structure, thus resulting in a smaller size after grafting.
Subsequently, the polymer-brush-grafted CM was utilized to form films via the solution-based deposition method of spin coating, e.g., a PIBA/CM film on a glass substrate was demonstrated (Figure 3a). A transparent film was obtained and compared to opaque CM films (Figure S10a), without an obvious change of the UV–vis absorption (Figure S11). The structure of the PIBA/CM film was characterized by scanning electron microscopy (SEM) and atomic force microscopy (AFM) images (Figure 3b,c), and a smooth thin film structure was confirmed, with a surface roughness of 1.49 nm (Figure S12). Besides, with the same condition for spin coating of CM precursors, no clear film structure was found via AFM investigation (Figure S10b,c). On the one hand, improved dispersibility in organic solvent promoted better film formation. On the other hand, PIBA possesses excellent adhesion63 and film-forming properties, which has been recognized as a commercial coating material,64 as accelerated film formation contributes to a better film structure. The thickness of the PIBA/CM film was characterized via section analysis (Figure 3d,e). The corresponding micrograph indicates a thickness of the PIBA/CM film of approximately 62 nm, and the thickness of the CM film (Figure S13) was approximately 20 nm. Thus, the difference in thickness between PIBA/CM and CM might be the thickness of polymer brushes as a rough assumption. Moreover, PIBA can be used as a polymer dielectric, and such thickness of thin films is appropriate for the electric gate function.65 As mentioned before, the structural and absorption properties of CM were still preserved after grafting. Hence, an improved process toward g-CN utilization as an energy conversion device, solar cell, and other film materials is provided.
Figure 3.
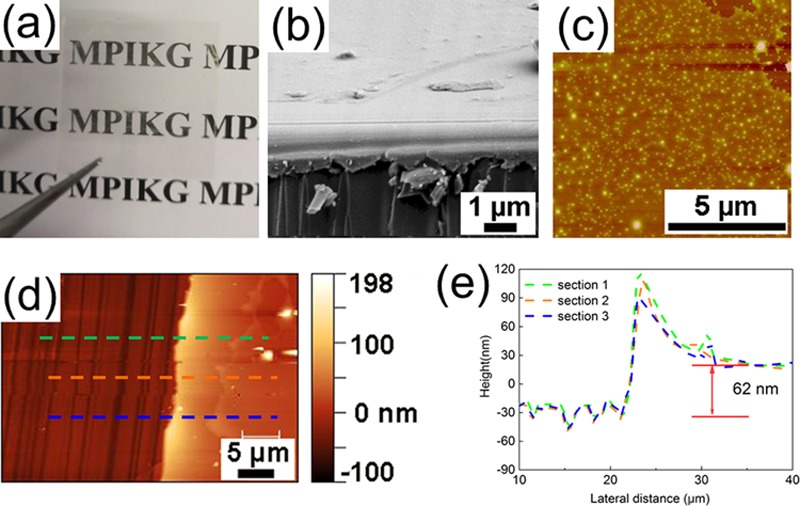
(a) Digital picture of the PIBA/CM film after spin coating on a glass substrate. (b) SEM image of the PIBA/CM film after spin coating. (c)–(e) show the AFM profile of spin-coated PIBA/CM film, thickness, and thickness distribution with three lines in detail, respectively.
PGMA/CM Surface Modification
In addition to the facile formation of thin films via spin coating, the functional groups such as epoxides in PGMA can be utilized for further modification. Hence, the surface properties of PGMA/CM brush materials can be readily adjusted. As a proof of concept, the thiol–epoxy addition reaction of PGMA/CM with sodium 2-mercaptoethanesulfonate (MESNA) and perfluorodecanethiol (pFDe) was attempted, respectively. The functionalization was carried out following the work of De and Khan,66 using LiOH as a catalyst at ambient temperature and reaction for 3 and 1 h, respectively (Figure 4a). The obtained MESNA–PGMA/CM and pFDe–PGMA/CM materials were characterized via several methods, e.g., FT-IR (Figure S14a). For example, compared to reference experiments with PMMA/CM (Figure S14b), C–F stretch and S=O stretch bands were found from the related FT-IR spectra, respectively. As MESNA is more hydrophilic and pFDe is more hydrophobic, PGMA/CM films (Figures S15 and S16) with opposite polarity were obtained (Figure 4b). The surface properties were characterized via contact angle measurements (Figures 4c and S17). MESNA–PGMA/CM was highly hydrophilic (contact angle 32°), and pFDe–PGMA/CM showed a highly hydrophobic property (contact angle 120°), whereas PGMA/CM showed a relatively hydrophilic property with a contact angle of 75° as expected. Further investigation was performed by SEM (Figure S18a,b), which showed no change in morphology after modification. Moreover, energy-dispersive X-ray (EDX) (Figure S18c,d) indicated the appearance of C, N, O, Na, and S throughout the surface of MESNA–PGMA/CM and C, N, O, F, and S throughout the surface of pFDe–PGMA/CM, indicating successful functionalization.
Figure 4.
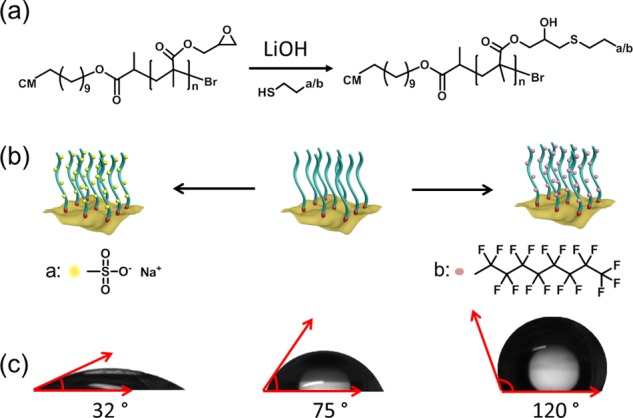
(a) Modification of PGMA/CM via thiol-addition-mediated epoxy ring-opening. (b) Illustration of PGMA/CM modified with sodium 2-mercaptoethanesulfonate (a, MESNA) and 1H,1H,2H,2H-perfluorodecanethiol (b, pFDe), respectively. (c) Contact angle of MESNA–PGMA/CM, PGMA/CM, and pFDe–PGMA/CM from left to right, respectively.
Conclusions
In conclusion, a grafting to method was utilized to realize the grafting of defined polymers on g-CN. Thus, polymer/g-CN hybrid materials were fabricated. Functional polymers were prepared via ATRP from an initiator with ene functionality, and visible light was used to perform photoinduced grafting. Thus, functional polymers namely PIBA, PGMA, and PMMA were grafted onto g-CN. However, varied grafting density was observed for the different polymers probably due to different steric hindrance. The as-prepared polymer/g-CN has improved dispersibility in organic solvent, which simplifies the further processing toward film fabrication. Compared to CM, PIBA/CM films on a glass substrate were facilely obtained via the spin coating deposition method. Film thicknesses in the range of 60 nm and surface roughness around 1.49 nm were observed, with a relatively smooth surface and ideal thickness toward further application as film materials. Moreover, the epoxide group of PGMA/CM was utilized for further modification. Thus, the surface properties of PGMA/CM brush materials were readily adjusted to be either hydrophilic or hydrophobic according to the attached species. Overall, polymer-grafted g-CN provides a novel way for combination of polymers with g-CN. On one hand, improvement of the process toward g-CN utilization in film materials was achieved, as such materials might be of interest for solar cells, electric devices, or photocatalysis. On the other hand, g-CN-containing films with adjustable hydrophobicity were provided, which might be a promising material for photocatalysis with improved selectivity.
Acknowledgments
The authors are grateful for funding from the Max-Planck Society. The authors acknowledge Marlies Graewert for SEC measurements, Ang Li for assistance with illustrations, Antje Voelkel for elemental analysis and TGA investigation, Reinhild Dünnebacke for AFM investigation, Tobias Heil for assisting with SEM and EDX measurements, and Irina Shekova and Jianrui Zhang for assistance with contact angle measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.9b00894.
Additional synthetic procedures; calculation; FT-IR; AFM; XRD; UV–vis; and DLS (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Oh J.; Lee J. M.; Yoo Y.; Kim J.; Hwang S.-J.; Park S. New insight of the photocatalytic behaviors of graphitic carbon nitrides for hydrogen evolution and their associations with grain size, porosity, and photophysical properties. Appl. Catal., B 2017, 218, 349–358. 10.1016/j.apcatb.2017.06.067. [DOI] [Google Scholar]
- Yan H. Soft-templating synthesis of mesoporous graphitic carbon nitride with enhanced photocatalytic H2 evolution under visible light. Chem. Commun. 2012, 48, 3430–3432. 10.1039/c2cc00001f. [DOI] [PubMed] [Google Scholar]
- Wang K.; Li Q.; Liu B.; Cheng B.; Ho W.; Yu J. Sulfur-doped g-C3N4 with enhanced photocatalytic CO2-reduction performance. Appl. Catal., B 2015, 176–177, 44–52. 10.1016/j.apcatb.2015.03.045. [DOI] [Google Scholar]
- Martin D. J.; Qiu K.; Shevlin S. A.; Handoko A. D.; Chen X.; Guo Z.; Tang J. Highly efficient photocatalytic H2 evolution from water using visible light and structure-controlled graphitic carbon nitride. Angew. Chem., Int. Ed. 2014, 53, 9240–9245. 10.1002/anie.201403375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Jiao Y.; Zhu Y.; Li L. H.; Han Y.; Chen Y.; Du A.; Jaroniec M.; Qiao S. Z. Hydrogen evolution by a metal-free electrocatalyst. Nat. Commun. 2014, 5, 3783 10.1038/ncomms4783. [DOI] [PubMed] [Google Scholar]
- Wang X.; Maeda K.; Thomas A.; Takanabe K.; Xin G.; Carlsson J. M.; Domen K.; Antonietti M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. 10.1038/nmat2317. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Wang Y.; Bi Y.; Jin J.; Ehsan M. F.; Fu M.; He T. Preparation of 2D hydroxyl-rich carbon nitride nanosheets for photocatalytic reduction of CO2. RSC Adv. 2015, 5, 33254–33261. 10.1039/C5RA04227E. [DOI] [Google Scholar]
- Ma J.; Wang C.; He H. Enhanced photocatalytic oxidation of NO over g-C3N4-TiO2 under UV and visible light. Appl. Catal., B 2016, 184, 28–34. 10.1016/j.apcatb.2015.11.013. [DOI] [Google Scholar]
- Sano T.; Tsutsui S.; Koike K.; Hirakawa T.; Teramoto Y.; Negishi N.; Takeuchi K. Activation of graphitic carbon nitride (g-C3N4) by alkaline hydrothermal treatment for photocatalytic NO oxidation in gas phase. J. Mater. Chem. A 2013, 1, 6489–6496. 10.1039/c3ta10472a. [DOI] [Google Scholar]
- Sun J.; Schmidt B. V.; Wang X.; Shalom M. Self-standing carbon nitride-based hydrogels with high photocatalytic activity. ACS Appl. Mater. Interfaces 2017, 9, 2029–2034. 10.1021/acsami.6b14879. [DOI] [PubMed] [Google Scholar]
- Cui Q.; Xu J.; Wang X.; Li L.; Antonietti M.; Shalom M. Phenyl-modified carbon nitride quantum dots with distinct photoluminescence behavior. Angew. Chem., Int. Ed. 2016, 55, 3672–3676. 10.1002/anie.201511217. [DOI] [PubMed] [Google Scholar]
- Shen J.; Zhu Y.; Yang X.; Li C. Graphene quantum dots: emergent nanolights for bioimaging, sensors, catalysis and photovoltaic devices. Chem. Commun. 2012, 48, 3686–3699. 10.1039/c2cc00110a. [DOI] [PubMed] [Google Scholar]
- Son E. J.; Lee S. H.; Kuk S. K.; Pesic M.; Choi D. S.; Ko J. W.; Kim K.; Hollmann F.; Park C. B. Carbon nanotub-graphitic carbon nitride hybrid films for flavoenzyme-catalyzed photoelectrochemical cells. Adv. Funct. Mater. 2018, 28, 1705232 10.1002/adfm.201705232. [DOI] [Google Scholar]
- Cao Q.; Cui Q.; Yang Y.; Xu J.; Han C.; Li L. Graphitic carbon nitride as a distinct solid stabilizer for emulsion polymerization. Chem. - Eur. J. 2018, 24, 2286–2291. 10.1002/chem.201705885. [DOI] [PubMed] [Google Scholar]
- Lu Y.-C.; Chen J.; Wang A.-J.; Bao N.; Feng J.-J.; Wang W.; Shao L. Facile synthesis of oxygen and sulfur co-doped graphitic carbon nitride fluorescent quantum dots and their application for mercury (II) detection and bioimaging. J. Mater. Chem. C 2015, 3, 73–78. 10.1039/C4TC02111H. [DOI] [Google Scholar]
- Kim J. K.; Park S.; Yoo R. J.; Jeong H. J.; Oh J.; Lee Y. J.; Park S.; Kim D. W. Thin PEGylated carbon nitrides: water-dispersible organic nanodots as bioimaging probes. Chem. - Eur. J. 2018, 24, 3506–3511. 10.1002/chem.201704761. [DOI] [PubMed] [Google Scholar]
- Dearnaley G.; Arps J. H. Biomedical applications of diamond-like carbon (DLC) coatings: A review. Surf. Coat. Technol. 2005, 200, 2518–2524. 10.1016/j.surfcoat.2005.07.077. [DOI] [Google Scholar]
- Hu Y.; Shim Y.; Oh J.; Park S.; Park S.; Ishii Y. Synthesis of 13C-,15N-labeled graphitic carbon nitrides and NMR-based evidence of hydrogen-bonding assisted two-dimensional assembly. Chem. Mater. 2017, 29, 5080–5089. 10.1021/acs.chemmater.7b00069. [DOI] [Google Scholar]
- Xu J.; Brenner T. J. K.; Chabanne L.; Neher D.; Antonietti M.; Shalom M. Liquid-based growth of polymeric carbon nitride layers and their use in a mesostructured polymer solar cell with Voc exceeding 1 V. J. Am. Chem. Soc. 2014, 136, 13486–13489. 10.1021/ja508329c. [DOI] [PubMed] [Google Scholar]
- Xiao K.; Giusto P.; Wen L.; Jiang L.; Antonietti M. Nanofluidic ion transport and energy conversion through ultrathin free-standing polymeric carbon nitride membranes. Angew. Chem. 2018, 130, 10280–10283. 10.1002/ange.201804299. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Wang W.; Lei J.; Xu N.; Gao F.; Ju H. Fluorescence quenching of carbon nitride nanosheet through its interaction with DNA for versatile fluorescence sensing. Anal. Chem. 2013, 85, 12182–12188. 10.1021/ac403646n. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhou Z.; Li H. Crystalline carbon nitride films formation by chemical vapor deposition. Appl. Phys. Lett. 1996, 68, 634–636. 10.1063/1.116492. [DOI] [Google Scholar]
- Bian J.; Li J.; Kalytchuk S.; Wang Y.; Li Q.; Lau T. C.; Niehaus T. A.; Rogach A. L.; Zhang R.-Q. Efficient emission facilitated by multiple energy level transitions in uniform graphitic carbon nitride films deposited by thermal vapor condensation. ChemPhysChem 2015, 16, 954–959. 10.1002/cphc.201402898. [DOI] [PubMed] [Google Scholar]
- Lejeune M.; Durand-Drouhin O.; Zellama K.; Benlahsen M. Structure and optical properties of carbon nitride films deposited by magnetron sputtering. Solid State Commun. 2001, 120, 337–342. 10.1016/S0038-1098(01)00411-2. [DOI] [Google Scholar]
- Safaei J.; Mohamed N. A.; Noh M. F. M.; Soh M. F.; Riza M. A.; Mustakim N. S. M.; Ludin N. A.; Ibrahim M. A.; Isahak W. N. R. W.; Teridi M. A. M. Facile fabrication of graphitic carbon nitride (g-C3N4) thin film. J. Alloys Compd. 2018, 769, 130–135. 10.1016/j.jallcom.2018.07.337. [DOI] [Google Scholar]
- Ge L.; Han C. Synthesis of MWNTs/g-C3N4 composite photocatalysts with efficient visible light photocatalytic hydrogen evolution activity. Appl. Catal., B 2012, 117–118, 268–274. 10.1016/j.apcatb.2012.01.021. [DOI] [Google Scholar]
- Raciti D.; Mao M.; Park J. H.; Wang C. Mass transfer effects in CO2 reduction on Cu nanowire electrocatalysts. Catal. Sci. Technol. 2018, 8, 2364–2369. 10.1039/C8CY00372F. [DOI] [Google Scholar]
- Li A.; Wang T.; Li C.; Huang Z.; Luo Z.; Gong J. Adjusting the reduction potential of electrons by quantum confinement for selective photoreduction of CO2 to methanol. Angew. Chem. 2019, 131, 3844–3848. 10.1002/ange.201812773. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Mori T.; Niu L.; Ye J. Non-covalent doping of graphitic carbon nitride polymer with graphene: controlled electronic structure and enhanced optoelectronic conversion. Energy Environ. Sci. 2011, 4, 4517–4521. 10.1039/c1ee01400e. [DOI] [Google Scholar]
- Zhang J.; Zhang G.; Chen X.; Lin S.; Möhlmann L.; Dołęga G.; Lipner G.; Antonietti M.; Blechert S.; Wang X. Co-monomer control of carbon nitride semiconductors to optimize hydrogen evolution with visible light. Angew. Chem., Int. Ed. 2012, 51, 3183–3187. 10.1002/anie.201106656. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chen X.; Takanabe K.; Maeda K.; Domen K.; Epping J. D.; Fu X.; Antonietti M.; Wang X. Synthesis of a carbon nitride structure for visible-light catalysis by copolymerization. Angew. Chem., Int. Ed. 2010, 49, 441–444. 10.1002/anie.200903886. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Li H.; Wang Y. Post-functionalization of graphitic carbon nitrides by grafting organic molecules: toward C-H bond oxidation using atmospheric oxygen. Chem. Commun. 2014, 50, 6312–6315. 10.1039/C4CC02676D. [DOI] [PubMed] [Google Scholar]
- Sun J.; Phatake R.; Azoulay A.; Peng G.; Han C.; Barrio J.; Xu J.; Wang X.; Shalom M. Covalent functionalization of carbon nitride frameworks through cross-coupling reactions. Chem. - Eur. J. 2018, 24, 14921–14927. 10.1002/chem.201803201. [DOI] [PubMed] [Google Scholar]
- Xu J.; Wu F.; Jiang Q.; Li Y.-X. Mesoporous carbon nitride grafted with n-bromobutane: a high-performance heterogeneous catalyst for the solvent-free cycloaddition of CO2 to propylene carbonate. Catal. Sci. Technol. 2015, 5, 447–454. 10.1039/C4CY00770K. [DOI] [Google Scholar]
- Bu X.; Li J.; Yang S.; Sun J.; Deng Y.; Yang Y.; Wang G.; Peng Z.; He P.; Wang X.; et al. Surface modification of C3N4 through oxygen-plasma treatment: a simple way toward excellent hydrophilicity. ACS Appl. Mater. Interfaces 2016, 8, 31419–31425. 10.1021/acsami.6b10516. [DOI] [PubMed] [Google Scholar]
- Kumru B.; Antonietti M.; Schmidt B. V. K. J. Enhanced dispersibility of graphitic carbon nitride particles in aqueous and organic media via a one-pot grafting approach. Langmuir 2017, 33, 9897–9906. 10.1021/acs.langmuir.7b02441. [DOI] [PubMed] [Google Scholar]
- Kumru B.; Cruz D.; Heil T.; Schmidt B. V. K. J.; Antonietti M. Electrostatic stabilization of carbon nitride colloids in organic solvents enables stable dispersions and transparent homogeneous CN-films for optoelectronics. J. Am. Chem. Soc. 2018, 140, 17532–17537. 10.1021/jacs.8b08956. [DOI] [PubMed] [Google Scholar]
- Fu Q.; Ruan Q.; McKenzie T. G.; Reyhani A.; Tang J.; Qiao G. G. Development of a robust PET-RAFT polymerization using graphitic carbon nitride (g-C3N4). Macromolecules 2017, 50, 7509–7516. 10.1021/acs.macromol.7b01651. [DOI] [Google Scholar]
- Dadashi-Silab S.; Tasdelen M. A.; Kiskan B.; Wang X.; Antonietti M.; Yagci Y. Photochemically mediated atom transfer radical polymerization using polymeric semiconductor mesoporous graphitic carbon nitride. Macromol. Chem. Phys. 2014, 215, 675–681. 10.1002/macp.201400063. [DOI] [Google Scholar]
- Kumru B.; Shalom M.; Antonietti M.; Schmidt B. V. K. J. Reinforced hydrogels via carbon nitride initiated polymerization. Macromolecules 2017, 50, 1862–1869. 10.1021/acs.macromol.6b02691. [DOI] [Google Scholar]
- Kumru B.; Molinari V.; Dunnebacke R.; Blank K. G.; Schmidt B. V. K. J. Extremely compressible hydrogel via incorporation of modified graphitic carbon nitride. Macromol. Rapid Commun. 2019, 40, 1800712 10.1002/marc.201800712. [DOI] [PubMed] [Google Scholar]
- Kumru B.; Barrio J.; Zhang J.; Antonietti M.; Shalom M.; Schmidt B. V. K. J. Robust carbon nitride-based thermoset coatings for surface modification and photochemistry. ACS Appl. Mater. Interfaces 2019, 11, 9462–9469. 10.1021/acsami.8b21670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez-Martos I.; Alonso-Bartolomé R.; Mulas Hernández V.; Anillo A.; Costa-García A.; García Alonso F. J.; Fernández-Abedul M. T. Poly(glycidyl methacrylate) as a tunable platform of modifiers for microfluidic devices. React. Funct. Polym. 2016, 100, 89–96. 10.1016/j.reactfunctpolym.2016.01.002. [DOI] [Google Scholar]
- López-García M. d. C.; Beebe D. J.; Crone W. C. Characterization of poly (isobornyl acrylate) as a construction material for microfluidic applications. J. Appl. Polym. Sci. 2007, 105, 1894–1902. 10.1002/app.26195. [DOI] [Google Scholar]
- Ejaz M.; Yamamoto S.; Ohno K.; Tsujii Y.; Fukuda T. Controlled graft polymerization of methyl methacrylate on silicon substrate by the combined use of the langmuir-blodgett and atom transfer radical polymerization techniques. Macromolecules 1998, 31, 5934–5936. 10.1021/ma980240n. [DOI] [Google Scholar]
- Devaux C.; Cousin F.; Beyou E.; Chapel J. P. Low swelling capacity of highly stretched polystyrene brushes. Macromolecules 2005, 38, 4296–4300. 10.1021/ma047478y. [DOI] [Google Scholar]
- Liao W.-P.; Elliott I. G.; Faller R.; Kuhl T. L. Normal and shear interactions between high grafting density polymer brushes grown by atom transfer radical polymerization. Soft Matter 2013, 9, 5753–5761. 10.1039/c3sm50261a. [DOI] [Google Scholar]
- Goujon F.; Malfreyt P.; Tildesley D. J. The compression of polymer brushes under shear: the friction coefficient as a function of compression, shear rate and the properties of the solvent. Mol. Phys. 2005, 103, 2675–2285. 10.1080/00268970500134706. [DOI] [Google Scholar]
- Senaratne W.; Andruzzi L.; Ober C. K. Self-assembled monolayers and polymer brushes in biotechnology: current applications and future perspectives. Biomacromolecules 2005, 6, 2427–2448. 10.1021/bm050180a. [DOI] [PubMed] [Google Scholar]
- Ostaci R.-V.; Damiron D.; Capponi S.; Vignaud G.; Léger L.; Grohens Y.; Drockenmuller E. Polymer brushes grafted to “passivated” silicon substrates using click chemistry. Langmuir 2008, 24, 2732–2739. 10.1021/la703086x. [DOI] [PubMed] [Google Scholar]
- Tokareva I.; Minko S.; Fendler J. H.; Hutter E. Nanosensors based on responsive polymer brushes and gold nanoparticle enhanced transmission surface plasmon resonance spectroscopy. J. Am. Chem. Soc. 2004, 126, 15950–15951. 10.1021/ja044575y. [DOI] [PubMed] [Google Scholar]
- Sheng W.; Amin I.; Neumann C.; Dong R.; Zhang T.; Wegener E.; Chen W. L.; Forster P.; Tran H. Q.; Loffler M.; Winter A.; Rodriguez R. D.; Zschech E.; Ober C. K.; Feng X.; Turchanin A.; Jordan R. Polymer Brushes on Hexagonal Boron Nitride. Small 2019, 15, 1805228 10.1002/smll.201805228. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Emmenegger C.; Preuss C. M.; Yameen B.; Pop-Georgievski O.; Bachmann M.; Mueller J. O.; Bruns M.; Goldmann A. S.; Bastmeyer M.; Barner-Kowollik C. Controlled cell adhesion on poly(dopamine) interfaces photopatterned with non-fouling brushes. Adv. Mater. 2013, 25, 6123–6127. 10.1002/adma.201302492. [DOI] [PubMed] [Google Scholar]
- Ionov L.; Bocharova V.; Diez S. Biotemplated synthesis of stimuli-responsive nanopatterned polymer brushes on microtubules. Soft Matter 2009, 5, 67–71. 10.1039/B813295J. [DOI] [Google Scholar]
- Tu H.; Heitzman C. E.; Braun P. V. Patterned poly (N-isopropylacrylamide) brushes on silica surfaces by microcontact printing followed by surface-initiated polymerization. Langmuir 2004, 20, 8313–8320. 10.1021/la049663a. [DOI] [PubMed] [Google Scholar]
- Costantini F.; Bula W. P.; Salvio R.; Huskens J.; Gardeniers H. J. G. E.; Reinhoudt D. N.; Verboom W. Nanostructure based on polymer brushes for efficient heterogeneous catalysis in microreactors. J. Am. Chem. Soc. 2009, 131, 1650–1651. 10.1021/ja807616z. [DOI] [PubMed] [Google Scholar]
- Ouyang H.; Xia Z.; Zhe J. Static and dynamic responses of polyelectrolyte brushes under external electric field. Nanotechnology 2009, 20, 195703 10.1088/0957-4484/20/19/195703. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski K.; Miller P. J.; Shukla N.; Immaraporn B.; Gelman A.; Luokala B. B.; Siclovan T. M.; Kickelbick G.; Vallant T.; Hoffmann H.; Pakula T. Polymers at interfaces: using atom transfer radical polymerization in the controlled growth of homopolymers and block copolymers from silicon surfaces in the absence of untethered sacrificial initiator. Macromolecules 1999, 32, 8716–8724. 10.1021/ma991146p. [DOI] [Google Scholar]
- Shalom M.; Inal S.; Fettkenhauer C.; Neher D.; Antonietti M. Improving carbon nitride photocatalysis by supramolecular preorganization of monomers. J. Am. Chem. Soc. 2013, 135, 7118–7121. 10.1021/ja402521s. [DOI] [PubMed] [Google Scholar]
- Mazzolini J.; Boyron O.; Monteil V.; Gigmes D.; Bertin D.; D’Agosto F.; Boisson C. Polyethylene end functionalization using radical-mediated thiol–ene chemistry: use of polyethylenes containing alkene end functionality. Macromolecules 2011, 44, 3381–3387. 10.1021/ma200342y. [DOI] [Google Scholar]
- Zeininger L.; Portilla L.; Halik M.; Hirsch A. Quantitative determination and comparison of the surface binding of phosphonic acid, carboxylic acid, and catechol ligands on TiO2 nanoparticles. Chem. - Eur. J. 2016, 22, 13506–13512. 10.1002/chem.201601920. [DOI] [PubMed] [Google Scholar]
- Jones D. M.; Brown A. A.; Huck W. T. J. L. Surface-initiated polymerizations in aqueous media: effect of initiator density. Langmuir 2002, 18, 1265–1269. 10.1021/la011365f. [DOI] [Google Scholar]
- Inui T.; Sato E.; Matsumoto A. Pressure-sensitive adhesion system using acrylate block copolymers in response to photoirradiation and postbaking as the dual external stimuli for on-demand dismantling. ACS Appl. Mater. Interfaces 2012, 4, 2124–2132. 10.1021/am300103c. [DOI] [PubMed] [Google Scholar]
- Shaw D. G.; Dawson E.; Cline D.; Langlois M.. Acrylate coating material. Google Patent US5440446, 1995.
- Seong H.; Pak K.; Joo M.; Choi J.; Im S. G. Vapor-phase deposited ultrathin polymer gate dielectrics for high-performance organic thin film transistors. Adv. Electron. Mater. 2016, 2, 1500209 10.1002/aelm.201500209. [DOI] [Google Scholar]
- De S.; Khan A. Efficient synthesis of multifunctional polymers via thiol-epoxy “click” chemistry. Chem. Commun. 2012, 48, 3130–3132. 10.1039/c2cc30434a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.