

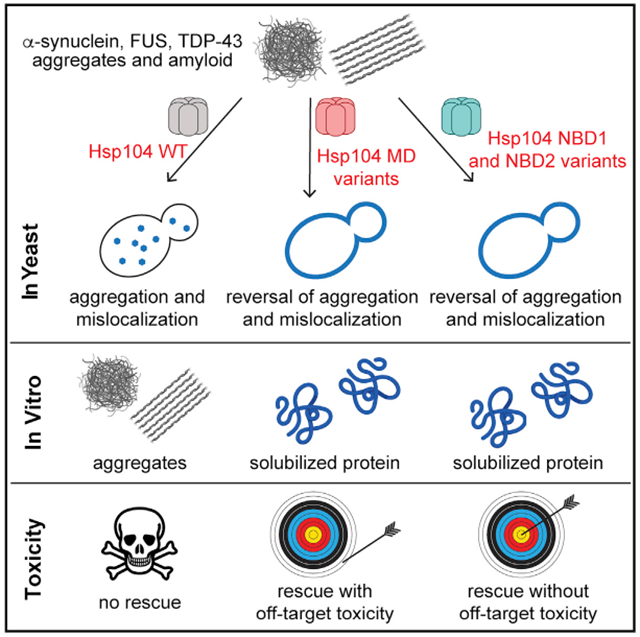
SUMMARY
Hsp104 is an AAA+ protein disaggregase, which can be potentiated via diverse mutations in its autoregulatory middle domain (MD) to mitigate toxic misfolding of TDP-43, FUS, and α-synuclein implicated in fatal neurodegenerative disorders. Problematically, potentiated MD variants can exhibit off-target toxicity. Here, we mine disaggregase sequence space to safely enhance Hsp104 activity via single mutations in nucleotide-binding domain 1 (NBD1) or NBD2. Like MD variants, NBD variants counter TDP-43, FUS, and α-synuclein toxicity and exhibit elevated ATPase and disaggregase activity. Unlike MD variants, non-toxic NBD1 and NBD2 variants emerge that rescue TDP-43, FUS, and α-synuclein toxicity. Potentiating substitutions alter NBD1 residues that contact ATP, ATP-binding residues, or the MD. Mutating the NBD2 protomer interface can also safely ameliorate Hsp104. Thus, we disambiguate allosteric regulation of Hsp104 by several tunable structural contacts, which can be engineered to spawn enhanced therapeutic disaggregases with minimal off-target toxicity.
In Brief
Tariq et al. disambiguate the allosteric regulation of Hsp104 by several tunable structural contacts, which can be engineered to spawn enhanced therapeutic disaggregases with minimal off-target toxicity. Non-toxic nucleotide-binding domain 1 (NBD1) and NBD2 variants emerge that rescue TDP-43, FUS, and α-synuclein toxicity connected to neurodegenerative disease.
Graphical Abstract
INTRODUCTION
Aberrant protein aggregation is linked with several neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and Parkinson’s disease (PD) (Chuang et al., 2018). In ALS-FTD, subsets of patients display cytoplasmic aggregation of TDP-43 or FUS in degenerating neurons (Robberecht and Philips, 2013). TDP-43 and FUS are RNA-binding proteins with prion-like domains, which shuttle between the nucleus and cytoplasm (Harrison and Shorter, 2017). However, cytoplasmic TDP-43 or FUS aggregation in disease restricts their transport back to the nucleus. Dissolution of cytoplasmic TDP-43 and FUS aggregates and restoration of functional TDP-43 and FUS to the nucleus is a major therapeutic goal in ALS-FTD (Guo et al., 2019). In PD, α-synuclein (α-syn) forms toxic soluble oligomers and amyloid fibrils that accumulate in cytoplasmic Lewy bodies and neurites in degenerating dopaminergic neurons (Abeliovich and Gitler, 2016). α-Syn is a lipid-binding protein that typically localizes to the plasma membrane but forms cytoplasmic inclusions in PD patients and in yeast (Outeiro and Lindquist, 2003). Inclusions of α-syn, TDP-43, and FUS are widely considered intractable. Moreover, the genes encoding TDP-43 and FUS are essential (Ling et al., 2013), and severe depletion of α-syn from nigrostriatal neurons might elicit degeneration (Collier et al., 2016). Thus, strategies that decrease expression or promote degradation of these proteins may be counterproductive, and reactivation of these proteins is an intriguing alternative approach (Guo et al., 2018, 2019; Jackrel et al., 2014a; Jackrel and Shorter, 2017; Shorter, 2016, 2017).
Hsp104 is a hexameric AAA+ protein disaggregase found in yeast, which harbors an N-terminal domain (NTD), nucleotide-binding domain 1 (NBD1), a middle domain (MD), NBD2, and a short C-terminal domain (CTD; Figure 1A). Hsp104 resolves diverse protein aggregates (Shorter and Southworth, 2019; Sweeny and Shorter, 2016). Following environmental stress, Hsp104 solubilizes aggregated proteins (Parsell et al., 1994; Wallace et al., 2015). Additionally, Hsp104 constructs and deconstructs yeast prions (infectious amyloids) for adaptive purposes (DeSantis et al., 2012; DeSantis and Shorter, 2012; Klaips et al., 2014; Newby and Lindquist, 2013; Park et al., 2014; Paushkin et al., 1996; Shorter and Lindquist, 2004, 2006, 2008; Sweeny et al., 2015; Zhao et al., 2017). The remarkable ability of Hsp104 to dissolve prions is intriguing, as prions are commonly perceived as ineradicable. We have suggested that Hsp104 could be applied to eliminate aggregated species and toxic, soluble oligomers linked to neurodegeneration (March et al., 2019; Shorter, 2008). Indeed, Hsp104 actively remodels amyloids and toxic soluble oligomers formed by several disease-linked proteins (Castellano et al., 2015; DeSantis et al., 2012; Liu et al., 2011; Lo Bianco et al., 2008; Michalska et al., 2019; Park et al., 2017). Importantly, Hsp104 antagonizes protein misfolding and neurodegeneration in animal models of neurodegenerative disease (Cushman-Nick et al., 2013; Lo Bianco et al., 2008; Satyal et al., 2000; Vacher et al., 2005). Nonetheless, Hsp104 activity against many amyloidogenic substrates has limits, and effective remodeling can require high Hsp104 concentrations (DeSantis et al., 2012; Lo Bianco et al., 2008). Moreover, Hsp104 displays limited activity against TDP-43 and FUS (Jackrel et al., 2014a). TDP-43, FUS, and α-syn do not have yeast homologs and are not natural Hsp104 substrates. Thus, we have engineered Hsp104 via diverse single missense mutations in its autoregulatory MD to potentiate activity against various disease-linked proteins (Castellano et al., 2015; Jackrel et al., 2014a, 2015; Jackrel and Shorter, 2014; Ryan et al., 2019; Tariq et al., 2018).
Figure 1. Hsp104 NBD1 Variants Suppress α-syn Toxicity, Aggregation, and Mislocalization.

(A) Domain map of Hsp104 shows the location of potentiating mutations in NBD1 (dark blue) and NBD2 (light blue). A503V is in the MD (green). Pink, NTD; brown, CTD.
(B) NBD1 variants suppress α-syn toxicity in yeast. W303aΔhsp104-pAG-303GAL-α-syn-YFP-304GALα-syn-YFP yeast were transformed with Hsp104 variants or vector. Strains were serially diluted 5-fold and spotted in duplicate onto glucose (non-inducing) and galactose (inducing) media.
(C) NBD1 variants do not reduce α-syn expression in yeast. Strains in (B) were induced for 8 h, lysed, and immunoblotted for Hsp104, α-syn, and 3-phosphoglycerate kinase (PGK; loading control).
(D) NBD1 variants suppress α-syn aggregation and mislocalization in yeast. Selected strains in (B) were induced for 8 h and prepared for fluorescence microscopy. Scale bar, 2.5 μm.
(E) α-syn aggregation and localization were quantified by calculating the proportion of cells exhibiting either cytoplasmic aggregates or plasma membrane localization. Values represent means ± SEM (n = 3).
See also Figures S1 and S2.
Potentiated Hsp104 MD variants display a therapeutic gain of function. They rescue TDP-43, FUS, and α-syn aggregation and toxicity in yeast under conditions in which wild-type Hsp104 is ineffective (Mack and Shorter, 2016). Certain enhanced MD variants suppress TDP-43, FUS and α-syn mislocalization and rescue dopaminergic neurodegeneration in a C. elegans PD model (Jackrel et al., 2014a; Yasuda et al., 2017). Potentiated MD variants disassemble TDP-43, FUS, and α-syn fibrils more effectively than Hsp104 (Jackrel et al., 2014a). Problematically, however, potentiated MD variants can exhibit off-target toxicity (Jackrel et al., 2014a). This off-target toxicity refers to adverse effects on targets other than those desired (i.e., TDP-43, FUS, or α-syn) because of limited substrate specificity.
The MD is just one domain of Hsp104, and vast tracts of Hsp104 sequence space remain unexplored and poorly understood. Here, we establish that Hsp104 can be more safely potentiated to counter ALS-FTD-linked and PD-linked proteotoxicity by single missense mutations in NBD1 or NBD2. To do so, we exploited yeast-based screens to hack disaggregase sequence space. We have isolated several non-toxic, potentiated Hsp104 variants with single missense mutations in NBD1 or NBD2. We thus expand the repertoire of mutations that enhance Hsp104 activity and illuminate several critical contacts that allosterically tune Hsp104 activity. Our studies open a gateway to synthetic, non-toxic disaggregases with therapeutic utility.
RESULTS
Specific Missense Mutations in NBD1 Potentiate Hsp104
Yeast models of α-syn, TDP-43, and FUS proteinopathy have been established in which the proteins are overexpressed through a galactose-inducible promoter. α-Syn, TDP-43, and FUS overexpression is toxic and the proteins accumulate in cytoplasmic inclusions, recapitulating phenotypes of PD and ALS-FTD patients (Johnson et al., 2008; Outeiro and Lindquist, 2003; Sun et al., 2011). However, Hsp104 cannot rescue this aggregation and toxicity, providing a platform to screen for Hsp104 variants with enhanced activity (Jackrel et al., 2014a). A key feature of this screen is that overtly toxic, hyperactivated Hsp104 variants will not survive the selection process. Rather, the screen selects for Hsp104 variants that are not toxic at 30°C and rescue α-syn, TDP-43, or FUS toxicity. We have used this strategy to isolate MD variants that suppress α-syn, TDP-43, and FUS toxicity (Jackrel et al., 2014b). We implemented a similar approach to uncover potentiated NBD1 or NBD2 variants. Thus, we used domain-specific error-prone PCR to randomly mutagenize NBD1 or NBD2 while leaving the remainder of the Hsp104 gene unperturbed. We achieved a mutation rate of approximately one mutation per gene. To isolate potentiated variants, we co-expressed these Hsp104 variant libraries with α-syn, TDP-43, or FUS in yeast. Thus, we identified a series of NBD1 variants that potently suppress α-syn, TDP-43, and FUS toxicity. We subsequently validated eight NBD1 variants: Hsp104I187F, Hsp104E190V, Hsp104I193L, Hsp104I193T, Hsp104I230N, Hsp104Q347L, Hsp104L355A, and Hsp104E360K (Figure 1A).
Potentiated NBD1 Variants Suppress α-Syn Toxicity, Aggregation, and Mislocalization
Not any mutation in NBD1 potentiates Hsp104 (Torrente et al., 2016). For example, Hsp104A178D and Hsp104T196S, like Hsp104, had no effect on α-syn toxicity (Figure 1B). In contrast, Hsp104I187F and Hsp104E360K very strongly suppressed α-syn toxicity, rescuing at a similar level as Hsp104A503V (Figures 1B and S1A), a potentiated variant bearing a mutation in MD helix L3 (Jackrel et al., 2014a). Hsp104I230N and Hsp104L355A strongly suppressed α-syn toxicity (Figures 1B and S1A). Hsp104Q347L and Hsp104I193T displayed a slightly weaker rescue, and Hsp104E190V and Hsp104I193L displayed the least rescue (Figures 1B). Rescue of α-syn toxicity was not due to altered expression levels, as α-syn and Hsp104 levels were roughly equal across the strains (Figures 1C and S1B).
We also tested whether Hsp104A330V could rescue α-syn toxicity, as the equivalent mutation in the E. coli Hsp104 homolog, ClpB, has been reported to hyperactivate ClpB (Franke et al., 2017). However, Hsp104A330V only very slightly reduced α-syn toxicity (Figure S1A). Thus, mutations that hyperactivate ClpB do not necessarily translate to Hsp104 variants able to potently suppress α-syn toxicity.
A potentiated MD variant, Hsp104A503V, suppresses formation of cytoplasmic α-syn foci and enables α-syn to localize to the plasma membrane, whereas Hsp104 is ineffective (Jackrel et al., 2014a). We tested the NBD1 variants and found that Hsp104I187F and Hsp104E360K, like Hsp104A503V, suppress formation of cytoplasmic foci and restore α-syn to the plasma membrane (Figures 1D and 1E). Upon overexpression of Hsp104I187F and Hsp104E360K, more than 70% of cells displayed α-syn solely localized to the plasma membrane, compared with ~30% of cells expressing Hsp104 (Figure 1E). In contrast, ~47% of cells expressing Hsp104I230N and ~54% of cells expressing Hsp104Q347L harbored cytoplasmic α-syn inclusions (Figure 1E). Thus, Hsp104 variants that provided the strongest rescue of α-syn toxicity also provided the greatest reduction in α-syn aggregation (Figures 1B and 1E).
Potentiated NBD1 Variants Suppress TDP-43 Toxicity, Aggregation, and Mislocalization
The same NBD1 variants that potently suppress α-syn toxicity also suppress TDP-43 toxicity but display slightly different trends. As with α-syn, Hsp104, Hsp104A178D, and Hsp104T196S were ineffective (Figure 2A). In contrast, Hsp104I230N, Hsp104E360K, and Hsp104L355A confer the strongest rescue of TDP-43 toxicity (Figures 2A and S1A). Hsp104I187F and Hsp104Q347L confer a weaker rescue, while Hsp104E190V, Hsp104I193T, and Hsp104I193L confer a very weak rescue (Figures 2A and S1A). Hsp104A330V failed to rescue TDP-43 toxicity (Figure S1A). As with α-syn, rescue of TDP-43 toxicity was not due to altered expression levels, as TDP-43 and Hsp104 levels were roughly equal across strains (Figures 2B and S1B).
Figure 2. Hsp104 NBD1 Variants Suppress TDP-43 Toxicity, Aggregation, and Mislocalization.

(A) NBD1 variants suppress TDP-43 toxicity in yeast. W303aΔhsp104-pAG-303GAL-TDP-43 yeast were transformed with Hsp104 variants or vector. Strains were serially diluted 5-fold and spotted in duplicate onto glucose (non-inducing) and galactose (inducing) media.
(B) NBD1 variants do not reduce TDP-43 expression in yeast. Strains in (A) were induced for 5 h, lysed, and immunoblotted for Hsp104, TDP-43, and PGK (loading control).
(C) NBD1 variants suppress TDP-43 aggregation and mislocalization in yeast. Fluorescence microscopy of cells coexpressing fluorescently tagged TDP-43 and Hsp104 variants. Strains were induced for 5 h in galactose, fixed, and stained with DAPI (blue) to visualize nuclei. Scale bar, 2.5 μm.
(D) TDP-43 localization was quantified by calculating the proportion of cells containing colocalized nuclear staining. Values represent means ± SEM (n = 3).
See also Figures S1 and S2.
TDP-43 shuttles between the nucleus and cytoplasm but aggregates and mislocalizes in the cytoplasm of degenerating neurons of ALS-FTD patients (Guo and Shorter, 2017). Expression of TDP-43 in yeast recapitulates this phenotype (Johnson et al., 2008). Hsp104A503V suppresses formation of cytoplasmic TDP-43 foci and restores TDP-43 to the nucleus (Jackrel et al., 2014a). The NBD1 variants Hsp104I187F, Hsp104I230N, Hsp104Q347L, and Hsp104E360K reduced cytoplasmic TDP-43 foci and increased nuclear localization (Figures 2C and 2D). Indeed, ~53% of cells expressing Hsp104I187F or ~50% of cells expressing Hsp104E360K displayed nuclear TDP-43, while only ~24% of cells expressing Hsp104 or ~31% of cells with the vector control displayed nuclear TDP-43 (Figures 2C and 2D). Yeast coexpressing Hsp104I230N or Hsp104Q347L displayed slightly fewer cells with nuclear TDP-43 (~49% for I230N and ~47% for Q347L) than the other potentiated variants (Figures 2C and 2D). Thus, Hsp104I187F, Hsp104I230N, Hsp104Q347L, and Hsp104E360K are potent suppressors of TDP-43 aggregation and toxicity. Clearing cytoplasmic TDP-43 aggregates and restoring TDP-43 to the nucleus is anticipated to be highly therapeutic for ALS-FTD (Gasset-Rosa et al., 2019; Mann et al., 2019; McGurk et al., 2018a, 2018b).
Potentiated NBD1 Variants Suppress FUS Toxicity and Aggregation
As with TDP-43, Hsp104I230N and Hsp104E360K conferred the strongest rescue of FUS toxicity, comparable with the rescue by Hsp104A503V, whereas Hsp104, Hsp104A178D, and Hsp104T196S were ineffective (Figures 3A and S1A). Hsp104I187F, Hsp104Q347L, Hsp104I193T, and Hsp104L355A were slightly less robust suppressors of FUS toxicity (Figures 3A and S1A). Hsp104E190V and Hsp104I193L displayed very mild rescue of FUS toxicity (Figure 3A). Hsp104A330V displayed minimal rescue of FUS toxicity (Figure S1A). Although some enhanced Hsp104 variants (e.g., Hsp104I187F) appeared to reduce FUS levels, in other cases enhanced Hsp104 variants (e.g., Hsp104I193T and Hsp104L355A) did not (Figures 3B and S1B). Thus, rescue of FUS toxicity by Hsp104 variants does not require reduced FUS protein levels (Figures 3B and S1B; Jackrel et al., 2014a; Jackrel and Shorter, 2014).
Figure 3. Hsp104 NBD1 Variants Suppress FUS Toxicity and Aggregation.

(A) NBD1 variants suppress FUS toxicity in yeast. W303aΔhsp104-pAG-303GAL-FUS yeast were transformed with Hsp104 variants or vector. Strains were serially diluted 5-fold and spotted in duplicate onto glucose (non-inducing) and galactose (inducing) media.
(B) NBD1 variants do not grossly reduce FUS expression in yeast. Strains in (A) were induced for 5 h, lysed, and immunoblotted for Hsp104, FUS, and PGK (loading control).
(C) NBD1 variants suppress FUS aggregation in yeast. Fluorescence microscopy of cells coexpressing FUS-GFP and Hsp104 variants. Strains were induced for 5 h in galactose and imaged. Scale bar, 2.5 μm.
(D) FUS aggregation was quantified by calculating the proportion of cells containing multiple foci, a single focus, or no foci. Values represent means ± SEM (n = 2–3).
(E) Potentiated NBD1 variants typically do not exhibit reduced growth at 37°C. Hsp104 variants were expressed in the 416GAL vector in Δhsp104 yeast in the absence of any disease protein. Strains were serially diluted 5-fold and spotted in duplicate onto glucose (non-inducing) and galactose (inducing) media and grown at 30°C or 37°C.
See also Figures S1 and S2.
Hsp104A503V disaggregates FUS inclusions (Jackrel et al., 2014a; Yasuda et al., 2017). Thus, we tested if NBD1 variants also antagonize FUS aggregation. Upon overexpression of FUS, ~75% of cells contain multiple foci, and Hsp104 had no effect on this phenotype (Figures 3C and 3D). Hsp104A503V reversed this phenotype, clearing foci entirely from ~73% of cells. In contrast, Hsp104I187F and Hsp104E360K decreased the percentage of cells containing multiple foci to ~18% while increasing the percentage of cells with a single focus to ~30% (Figures 3C and 3D). More than 50% of these cells were cleared of foci entirely (Figures 3C and 3D). Hsp104I230N and Hsp104Q347L increased the percentage of cells with a single focus even further to ~38% (Figures 3C and 3D). FUS foci were cleared entirely from ~40% of cells expressing Hsp104I230N and ~27% of cells expressing Hsp104Q347L (Figure 3C and Figure 3D). Unlike TDP-43, FUS is not returned to the nucleus, as the yeast nuclear-import machinery does not recognize the FUS PY-NLS (Guo et al., 2018; Ju et al., 2011; Sun et al., 2011). Nonetheless, Hsp104I187F, Hsp104I230N, Hsp104Q347L, and Hsp104E360K antagonize FUS aggregation and toxicity.
Potentiated NBD1 Variants Typically Do Not Exhibit Reduced Growth at 37°C
Potentiated Hsp104 MD variants often confer a temperature-sensitive growth phenotype in which yeast grow normally at 30°C but exhibit reduced growth at 37°C (Jackrel et al., 2014a). This off-target toxicity likely stems from promiscuous substrate recognition resulting in unfolding of essential proteins (Jackrel et al., 2014a; Jackrel and Shorter, 2014; Schirmer et al., 2004). Thus, we determined if the NBD1 variants also conferred this phenotype. Hsp104I193T, Hsp104I230N, Hsp104Q347L, and Hsp104L355A exhibited similar growth to Hsp104 at 37°C, whereas Hsp104E360K exhibited minimally reduced growth at 37°C (Figure 3E). In contrast, Hsp104I187F displayed a strong growth defect at 37°C that was more severe than the potentiated MD variant, Hsp104A503V (Figure 3E). Aside from Hsp104I187F and to a lesser extent Hsp104E360K, the potentiated NBD1 variants were more like Hsp104 and did not display a temperature-sensitive growth phenotype. Thus, Hsp104 activity can be potentiated via specific mutations in NBD1 without conferring undesirable toxicity, which contrasts with potentiated MD variants that typically exhibit toxicity in yeast at 37°C.
Hsp104A330V was an outlier, which we studied because the equivalent mutation hyperactivates ClpB (Franke et al., 2017). Hsp104A330V fails to rescue TDP-43 toxicity and confers negligible rescue of FUS and α-syn toxicity (Figure S1A). However, Hsp104A330V was subtly toxic to yeast at 30°C (unlike Hsp104I187F) and more toxic than Hsp104A503V at 37°C (Figure 3E). Thus, Hsp104A330V is likely hyperactive with severe off-target effects. However, this hyperactivity is not coupled to an ability to potently rescue TDP-43, FUS, and α-syn toxicity. Thus, we reserve the term “potentiated” (i.e. endowed with greater potency) for Hsp104 variants able to potently rescue TDP-43, FUS, or α-syn toxicity, whereas we reserve the term “hyperactive” (i.e. more active than desired) for Hsp104 variants that are toxic to yeast at 30°C and 37°C and have extremely limited ability to rescue TDP-43, FUS, or α-syn toxicity. Aside from Hsp104A330V, the only other hyperactive Hsp104 variants we have encountered are Hsp104E360P (see below) and Hsp104A503P (Jackrel et al., 2014a).
Potentiating Substitutions Alter NBD1 Residues that Contact ATP, ATP-Binding Residues, or the MD
We next mapped the potentiating mutations onto the structure of Hsp104 hexamers bound to AMP-PNP or ADP (Figures 4A–4F; Gates et al., 2017; Yokom et al., 2016). E190 and I193 are highly conserved residues (Figure 4G) that reside in helix B3 (Figures 4C and 4D). Interestingly, in the AMP-PNP-bound structure, E190 lies in close proximity to R419 in helix L1 of the MD of the counterclockwise protomer (Figure 4C, left; Yokom et al., 2016). The E190V mutation would likely alter this inter-protomer interaction with the MD. However, the R419V mutation did not potentiate Hsp104 (Figure S2A), and R419M inhibited Hsp104 activity (Wendler et al., 2007). Thus, the mechanism of potentiation is likely more complex than simply disrupting this NBD1:MD interaction. Indeed, I193 also interacts with E190 in the ADP-bound structure (Figure 4D, right). Thus, alteration of E190 or I193 may also affect their interaction within NBD1 and elicit enhanced activity.
Figure 4. Location of Potentiating NBD1 and NBD2 Mutations.

(A) Domain map of Hsp104 shows the location of potentiating mutations in NBD1 (dark blue) and NBD2 (light blue). A503V is located in the MD (green). Pink, NTD; brown, CTD.
(B) Location of residues in NBD1 and NBD2 that can be mutated to potentiate Hsp104 are shown on the hexameric structure of Hsp104 bound to AMP-PNP (left; PDB: 5KNE) and bound to ADP (right; PDB: 5VY8). Protomer3(P3) is colored to denote the different domains: pink, NTD; dark blue, NBD1; green, MD; and light blue, NBD2 (as in A). P1, P2, P4, P5, and P6 are in gray.
(C–F) Zooms to show the positions of residues that can be mutated to potentiate Hsp104. (C) Top views showing the positions of E190 and E360 in P3 of the Hsp104 hexamer in AMP-PNP (left; PDB: 5KNE) and ADP (right; PDB: 5VY8). (D) Sideviews showing the positions of I193 and E190 in P3 of the Hsp104 hexamer bound to AMP-PNP (left; PDB: 5KNE) and bound to ADP (right; PDB: 5VY8). (E) Topviews showing the positions of I187 and Q347 in P3 of the Hsp104 hexamer in AMP-PNP (left; PDB: 5KNE) and ADP (right; PDB: 5VY8). (F) Side views showing the positions of I230 and L355 in P3 of the Hsp104 hexamer in AMP-PNP (left; PDB: 5KNE) and ADP (right; PDB: 5VY8).
(G) Clustal Omega (Sievers and Higgins, 2018) alignment of portions (residues 185–195, 225–235, 342–352, and 355–365) of NBD1 from Saccharomyces cerevisiae Hsp104 with S. cerevisiae Hsp78, Schizosaccharomyces pombe Hsp104, Chlamydomonas reinhardtii Hsp104, Arabidopsis thaliana Hsp101, Monosiga brevicollis Hsp104, Thermus thermophilus ClpB, Escherichia coli ClpB, and E. coli ClpA. I187, E190, I193, I230, Q347, L355, and E360 are indicated with red arrowheads and highlighted in yellow. Asterisk denotes fully conserved residue, colon denotes conservation of residues with strong similarity, and period indicates conservation of residues with weak similarity.
I187 is a highly conserved residue (Figure 4G) that lies in a loop between helix B2 and B3 of the NBD1 large domain (Figure 4E). The carbonyl oxygen of the main chain at position I187 directly contacts nucleotide in the AMP-PNP- and ADP-bound structures (Figure 4E; Gates et al., 2017; Lee et al., 2003; Yokom et al., 2016). Thus, alteration of this side chain likely alters the architecture and regulation of the nucleotide-binding pocket.
I230 is a highly conserved residue in helix B4, which also carries the critical K218 Walker A residue at its N-terminal end (Figures 4F and 4G). I230 is in close proximity to R495 in the MD in the ADP-bound hexamer (Figure 4F, right). Hence, the I230N mutation may disrupt this contact and confer potentiation. Indeed, R495D, R495E, R495N, and R495M also potentiate Hsp104 (Figure S2A), and R495M elevates Hsp104 ATPase activity (Wendler et al., 2007). Thus, the I230N mutation likely perturbs an intra-protomer NBD1:MD interaction in the ADP-bound hexamer.
Q347 is a poorly conserved residue in helix C1 of the NBD1 small domain (Figures 4E and 4G). In fact, glutamate or aspartate is typically found at this position in Hsp104 homologs, indicating a divergent feature of S. cerevisiae Hsp104 (Figure 4G). Q347 contacts I351, an ATP-binding residue (Figure 4E; Gates et al., 2017; Lee et al., 2003; Yokom et al., 2016). Thus, alteration of the Q347 side chain may alter regulation of the nucleotide-binding pocket.
L355 is a highly conserved residue in helix C1 of the NBD1 small domain (Figures 4F and 4G). L355 helps confine nucleotide within the binding pocket (Figure 4F; Lee et al., 2003). Thus, the L355A mutation likely alters the architecture and regulation of the nucleotide-binding pocket.
E360 is a highly conserved residue also in helix C1 (Figures 4C and 4G). In the AMP-PNP-bound hexamer, E360 lies in proximity to R433 in the distal loop between helix L1 and L2 of the MD of the counterclockwise protomer (Figure 4C, left; Yokom et al., 2016). The E360K mutation would likely alter this inter-protomer NBD1:MD interaction. However, R433E, R433K, and R433Y did not potentiate Hsp104 (Figure S2A). Thus, disruption of the E360:R433 contact is likely not critical for enhanced activity. In the ADP-bound hexamer, E360 lies in proximity to K480 in MD helix L2 of the same protomer (Figure 4C, right; Gates et al., . Thus, the E360K mutation would disrupt this intra-protomer NBD1:MD contact. Importantly, K480E also enables Hsp104 to rescue α-syn, TDP-43, and FUS toxicity, although rescue of TDP-43 toxicity was not as strong as E360K, perhaps because of lower expression of Hsp104K480E compared to Hsp104E360K (Figures S2A and S2B; Jackrel et al., 2015). Breaking the intra-protomer NBD1:MD contact between E360 and K480 in the ADP-bound hexamer likely elicits potentiated Hsp104 activity.
Diverse Mutations at Certain NBD1 Positions Potentiate Hsp104
At several MD positions, Hsp104 can be mutated to diverse amino acids and confer potentiation. For example, A503 can be mutated to any amino acid, except proline to suppress α-syn, FUS, and TDP-43 toxicity (Jackrel et al., 2014a). We were curious if the NBD1 variants also displayed this degeneracy. Thus, we introduced conservative or non-conservative mutations at several of the potentiating NBD1 sites. We mutated Hsp104I187, Hsp104I230, Hsp104Q347, and Hsp104E360 to nine or ten different residues with different properties. Interestingly, none of these NBD1 positions displayed the same level of degeneracy as A503 to yield potentiated activity. Thus, more specific effects dictate which mutations in NBD1 enhance activity.
Hsp104I187X (where X represents any amino acid) displayed the strongest rescue of α-syn, TDP-43, and FUS toxicity when mutated to aromatic residues phenylalanine, tryptophan, or tyrosine (Figure 5A). Valine did not confer potentiation, possibly because this side chain is not sufficiently different from isoleucine (Figure 5A). Indeed, valine is often found at this position in Hsp104 homologs (data not shown). In contrast, Hsp104I187A and Hsp104I187S rescued α-syn, TDP-43, and FUS toxicity (Figures 5A). Mutation to asparagine enabled some rescue of α-syn and FUS toxicity but not TDP-43 toxicity (Figure 5A). Substitution with aspartate, lysine, or proline abolished rescue (Figure 5A). Thus, alteration of this nucleotide-binding residue can have diverse effects on Hsp104 activity. Charged substitutions at I187 do not potentiate activity, whereas aromatic substitutions broadly enhance activity. Moreover, mutation of I187 to alanine or serine mildly increases activity, whereas mutation of I187 to asparagine tunes Hsp104 to selectively antagonize α-syn and FUS toxicity. Notably, Hsp104I187F/Y/W exhibited off-target toxicity at 37°C, whereas Hsp104I187A/S/N did not (Figure S3A).
Figure 5. Diverse Mutations at Certain NBD1 Sites Potentiate Hsp104.

(A) Hsp104 variants mutated at the I187 position and controls were transformed into W303aΔhsp104 yeast harboring α-syn (left), TDP-43 (center), or FUS (right) genes. Strains were serially diluted 5-fold and spotted in duplicate onto glucose (non-inducing) or galactose (inducing) media.
(B–D) Hsp104 variants mutated at the I230 position (B), Q347 position (C), or E360 position (D) were assessed as in (A).
See also Figures S3.
Hsp104I230X displayed the strongest rescue of α-syn, TDP-43, and FUS toxicity when mutated to asparagine, serine, aspartate, glutamine, or phenylalanine (Figure 5B). Hsp104I230A and Hsp104I230K restricted rescue of toxicity to α-syn and FUS (Figure 5B). As with position I187, mutation of I230 to valine (which is often found at position 230 in Hsp104 homologs) or proline did not confer potentiation (Figures 4G and 5B). In contrast to I187, however, an aspartate substitution at I230 enhanced activity (Figure 5B). None of the Hsp104I230X variants exhibited off-target toxicity, with the exception of Hsp104I230D (Figure S3B). Thus, mutation of I230 is a safer way to elicit enhanced activity than mutation of I187.
Hsp104Q347X rescued α-syn, TDP-43, and FUS toxicity when mutated to leucine, threonine, or valine (Figure 5C). There was no rescue of α-syn, FUS, or TDP-43 toxicity when Q347 was mutated to alanine, asparagine, proline, phenylalanine, aspartate (a residue commonly found at this position in Hsp104 homologs), or lysine (Figures 4G and 5C). Thus, potentiation of Hsp104 via mutation of Q347 can be conferred through introduction of branched amino acids or polar uncharged side chains smaller than asparagine. Remarkably, Hsp104Q347X variants did not exhibit off-target toxicity, indicating multiple safe paths to Hsp104 potentiation (Figure S3C).
Hsp104E360X displayed the strongest rescue of α-syn, TDP-43, and FUS toxicity when mutated to lysine, arginine, or asparagine (Figure 5D). Hsp104E360F suppressed α-syn and FUS toxicity but not TDP-43 toxicity (Figure 5D). Phenylalanine is not found naturally at position 360 in any Hsp104 homolog. Only the charge-reversing mutations to lysine or arginine, or uncharged, polar asparagine conferred broad potentiated activity. Thus, there are fewer routes to potentiated Hsp104 activity via mutation of E360. Hsp104E360K exhibited minimally reduced growth at 37°C, whereas Hsp104E360F/N/R did not (Figure S3D). In contrast, Hsp104E360P, which failed to rescue α-syn, TDP-43, or FUS toxicity (Figure 5D) exhibited strong off-target toxicity even at 30°C (Figure S3D). Thus, Hsp104E360P is a toxic hyperactive Hsp104 variant, exhibiting even greater toxicity than Hsp104A330V. Mutation of E360 can therefore yield toxic hyperactive variants (e.g., E360P) as well as potentiated variants with minimal (e.g., E360K) or no toxicity (e.g., E360F).
Potentiated NBD1 Variants Are Enhanced ATPases, Disaggregases, and Unfoldases
We next purified Hsp104I187F, Hsp104I230N, Hsp104Q347L, and Hsp104E360R and assessed their ATPase, disaggregase, and unfoldase activity. The majority of potentiated MD variants display higher ATPase activity than Hsp104 (Jackrel et al., 2014a). Indeed, Hsp104A503V displays ~5-fold greater ATPase activity than Hsp104 (Jackrel et al., 2014a; Schirmer et al., 2004). The four NBD1 variants we tested also displayed higher ATPase activity than Hsp104 (Figure 6A). Hsp104I187F had the highest ATPase activity, ~10-fold higher than Hsp104 and ~2-fold higher than Hsp104A503V (Figure 6A). Hsp104I230N exhibited similar ATPase activity as Hsp104A503V (Figure 6A). The ATPase activity for Hsp104Q347L and Hsp104E360R was slightly less than that of Hsp104A503V and Hsp104I230N (Figure 6A). Thus, potentiating NBD1 mutations elevate ATPase activity, which may enable more work to be performed on substrate per unit time.
Figure 6. Potentiated NBD1 Variants Display Enhanced ATPase, Disaggregase, and Unfoldase Activity.

(A) NBD1 variants exhibit elevated ATPase activity. Values represent means ± SEM (n = 2–3).
(B) NBD1 variants exhibit elevated disaggregase activity. Luciferase aggregates were incubated with Hsp104 variant plus (gray bars) or minus (black bars) Hsc70 (0.167 μM) and Hdj2 (0.167 μM). Values represent means ± SEM (n = 2–7).
(C) NBD1 variants exhibit elevated unfoldase activity. RepA1–70-GFP was incubated with Hsp104 variant and GroELtrap plus ATP or ATP:ATPγS (1:1). GFP unfolding was measured by fluorescence. Representative data from three trials are shown.
See also Figures S4.
We next tested the protein disaggregation and reactivation activity of the NBD1 variants using denatured luciferase aggregates (Glover and Lindquist, 1998). Although Hsp104 requires Hsp70 and Hsp40 for luciferase reactivation (Figure 6B), potentiated MD variants do not (Jackrel et al., 2014a). Similar to enhanced MD variants, Hsp104I187F, Hsp104I230N, and Hsp104Q347L do not require Hsp70 or Hsp40 for luciferase reactivation (Figure 6B). In the absence of Hsp70 and Hsp40, Hsp104I187F, Hsp104I230N, and Hsp104Q347L were slightly less active than Hsp104 with Hsp70 and Hsp40 (Figure 6B). Hsp104E360R alone was also more active than Hsp104 without Hsp70 and Hsp40, but less active than Hsp104I187F, Hsp104I230N, and Hsp104Q347L (Figure 6B). The addition of Hsp70 and Hsp40 to the NBD1 variants increased luciferase reactivation substantially (Figure 6B). In the presence of Hsp70 and Hsp40, Hsp104I187F was ~3-fold more active, Hsp104I230N was ~4-fold more active, Hsp104Q347L was ~5-fold more active, and Hsp104E360R was ~1.5-fold more active than Hsp104 with Hsp70 and Hsp40 (Figure 6B). Thus, collaboration with Hsp70 and Hsp40 enables optimal disaggregase activity in vitro and also likely in vivo. Nonetheless, potentiating NBD1 mutations increase disaggregase activity in the absence or presence of Hsp70 and Hsp40.
We next analyzed unfoldase activity. We used the substrate RepA1–70-GFP, where RepA1–70 serves as an unfolded tag and upon translocation of RepA1–70, the appended GFP moiety is unfolded and loses fluorescence. To eliminate confounding effects of spontaneous GFP refolding, we used GroELtrap, which prevents GFP refolding (Doyle et al., 2007). Hsp104 unfolds RepA1–70-GFP in the presence of a 1:1 ratio of ATP:ATPγS but not in the presence of ATP alone (Figure 6C; Jackrel et al., 2014a). However, the potentiated NBD1 variants Hsp104I187F, Hsp104I230N, Hsp104Q347L, and Hsp104E360R, like the potentiated MD variants, unfold RepA1–70-GFP in the presence of only ATP (Figure 6C) (Jackrel et al., 2014a; Jackrel and Shorter, 2014). The potentiated NBD1 variants unfold RepA1–70-GFP more rapidly with ATP than Hsp104 with ATP and ATPγS (Figure 6C). Hsp104I187F and Hsp104I230N displayed similar unfoldase activity to Hsp104A503S (a potentiated MD variant), whereas Hsp104Q347L and Hsp104E360R were slightly less effective (Figure 6C). Thus, potentiating NBD1 mutations enhance unfoldase activity.
Potentiated NBD1 Variants Confer Thermotolerance but Not Resistance to Azetidine-2-Carboxylic Acid
We next assessed whether enhanced NBD1 variants generally rendered yeast less sensitive to protein aggregation in two ways. First, we assessed thermotolerance to 50°C, at which many proteins aggregate and must be solubilized and reactivated (but not degraded) to promote viability (Parsell et al., 1994). We found that enhanced Hsp104 variants conferred similar thermotolerance to Hsp104 (Figure S4A). There were some exceptions: Hsp104E360K, Hsp104E360F, Hsp104E360R, and Hsp104A330V displayed ~2-fold reduced thermotolerance (Figure S4A). In contrast, Hsp104I187S and Hsp104I187N conferred an ~60% increase in thermotolerance (Figure S4A). Overall, however, enhanced Hsp104 variants were similar to Hsp104 in conferring thermotolerance.
Second, we assessed growth on azetidine-2-carboxylic acid (AZC), a toxic proline analog. AZC is incorporated into proteins competitively with proline and elicits widespread protein aggregation (Trotter et al., 2001; Weids and Grant, 2014). We induced expression of the Hsp104 variant prior to plating on AZC. Neither Hsp104 nor enhanced variants rescued AZC toxicity (Figure S4B). Thus, enhanced Hsp104 variants do not generally render cells less sensitive to toxicity connected to widespread protein aggregation. Rather, they exhibit enhanced activity against the selection pressures screened for here (i.e., α-syn, TDP-43, or FUS toxicity). We suggest that enhanced Hsp104 variants are better equipped to combat aggregation and toxicity of a single dominant protein (e.g., α-syn, TDP-43, or FUS) but exhibit similar activity to wild-type when challenged with multiple aggregating proteins as in thermotolerance or growth on AZC. Nonetheless, Hsp104 variants that confer greater thermotolerance or AZC resistance can likely be found via screens using growth at 50°C or growth on AZC as the selection pressure.
Hsp104N566I Is a Potentiated NBD2 Variant
We also generated libraries of NBD2 variants and screened them for rescue of α-syn, TDP-43, or FUS toxicity. Remarkably, we uncovered only one potentiated variant, Hsp104N5661 (Figures 4A and 4B). Several NBD2 variants with a single missense mutation (e.g., H571Q, V581A, V581F, S597A, S618A, S618Y, K654N, G668S, T697S, L703S, N728A, N728K, V754I, H781L, K782T, E792K, E793R, R794D, L806V, L877W) were unable to rescue α-syn, FUS, and TDP-43 toxicity in yeast (Torrente et al., 2016). The difficulty in isolating potentiated NBD2 variants indicates that NBD2 may be less receptive to single amino acid changes that enhance Hsp104 activity. Moreover, when we aligned Hsp104 NBD1 to NBD2 and made analogous mutations in NBD2 equivalent to the potentiating mutations in NBD1, then we also did not observe enhanced activity. Thus, V580F (equivalent to I187F), I587T (equivalent to I193T), L631N (equivalent to I230N), H781L (equivalent to Q347L), and E793K (equivalent to E360K) did not potentiate Hsp104. These findings emphasize the non-equivalent roles that NBD1 and NBD2 play in Hsp104 disaggregase activity, and likely reflect that NBD1 and NBD2 are from different clades of the AAA+ family (Erzberger and Berger, 2006).
N566 is a poorly conserved residue in helix D2 of the NBD2 large domain (Figures 4A, 4B, and Figure 7A). Arginine or lysine is typically found at this position in Hsp104 homologs, indicating a divergent feature of S. cerevisiae Hsp104 (Figure 7A). Interestingly, N566 lies at the NBD2 protomer interface (Gates et al., 2017; Ye et al., 2019). N566 is in proximity to L845 of the neighboring protomer at the P2-P3, P3-P4, P4-P5, and P5-P6 interfaces in the extended state of substrate-bound Hsp104 in the presence of ATPγS (Figure 7B; Gates et al., 2017). In contrast, N566 is shifted away from L845 in most protomers in the closed state of substrate-bound Hsp104 in the presence of ATPγS and is only in proximity to L845 at the P6-P1 interface (Figure 7B; Gates et al., 2017). Thus, N566I could alter communication between adjacent Hsp104 protomers during polypeptide translocation.
Figure 7. Hsp104N566I Is a Potentiated NBD2 Variant.

(A) Clustal Omega (Sievers and Higgins, 2018) alignment of a portion (residues 561–571) of NBD2 from S. cerevisiae Hsp104 with S. cerevisiae Hsp78, S. pombe Hsp104, C. reinhardtii Hsp104, A. thaliana Hsp101, M. brevicollis Hsp104, T. thermophilus ClpB, E. coli ClpB, and E. coli ClpA. N566 is indicated with a red arrowhead and highlighted in yellow. Asterisk denotes fully conserved residue, colon denotes conservation of residues with strong similarity, and period indicates conservation of residues with weak similarity.
(B) Bottom-up view of Hsp104 hexamers bound to ATPγS and casein in the extended state (top panel; PDB: 5VYA) or closed state (bottom panel; PDB: 5VJH). Protomer 1 (P1) is shown in red, P2 in orange, P3 in green, P4 in cyan, P5 in blue, and P6 in purple. N566I lies in close proximityto L845 in the neighboring subunit at the P2-P3, P3-P4, P4-P5, and P5-P6 subunit interfaces in the extended state bound to casein (top panel). In contrast, N566I lies in close proximity to L845 in the neighboring subunit at the P6-P1 interface in the closed state (bottom panel).
(C) Hsp104N566I rescues α-syn, FUS, and TDP-43 toxicity in yeast. W303aΔhsp104 yeast were transformed with α-syn, FUS, orTDP-43 and Hsp104 variants or vector. Strains were serially diluted 5-fold and spotted in duplicate onto glucose (non-inducing) and galactose (inducing) media.
(D) Hsp104N566I does not grossly reduce α-syn, FUS, or TDP-43 expression in yeast. Strains in (A) were induced for 5 h (FUS and TDP-43) or 8h (α-syn), lysed, and immunoblotted. PGK serves as a loading control.
(E) Hsp104N566I does not exhibit reduced growth at 37°C. Hsp104 variants were expressed in the 416GAL vector in W303aΔhsp104 yeast in the absence of any disease protein. Strains were serially diluted 5-fold and spotted onto galactose (inducing) media and analyzed at 30°C or 37°C.
(F) Hsp104N566I exhibits elevated ATPase activity. Values represent means ± SEM (n = 3).
(G) Hsp104N566I exhibits elevated disaggregase activity in the absence of Hsp70 and Hsp40. Luciferase aggregates were incubated with Hsp104 variant plus (checkered bars) or minus (clear bars) Hsc70 (0.167 μM) and Hdj2 (0.167 μM). Values represent means ± SEM (n = 3).
See also Figures S5–S7.
Like enhanced NBD1 variants, Hsp104N566I conferred similar thermotolerance to Hsp104 and was unable to counter AZC toxicity (Figures S4A and S4B). Remarkably, Hsp104N566I effectively rescued α-syn, FUS, and TDP-43 toxicity in yeast without grossly affecting disease protein expression level and was almost as effective as Hsp104A503V (Figures 7C and 7D). Accordingly, Hsp104N566I reduced α-syn aggregation but not as potently as Hsp104A503V (Figure S5). Hsp104N566I also reduced the proportion of cells with multiple FUS foci but was not as effective as Hsp104A503V or potentiated NBD1 variants at eliminating cytoplasmic FUS foci (Figures 3C, 3D, and S6). Thus, the presence of multiple FUS foci may be particularly toxic. Unexpectedly, Hsp104N566I does not restore TDP-43 back to the nucleus like Hsp104A503V (Figure S7), despite rescuing TDP-43 toxicity without affecting TDP-43 levels (Figures 7C and 7D). Thus, Hsp104N566I likely rescues TDP-43 toxicity via another mechanism.
Importantly, Hsp104N566I exhibited similar growth to Hsp104 at 37°C (Figure 7E). Hsp104N566I was not toxic like Hsp104A503V at 37°C Figure 7E). Thus, Hsp104N566I is akin to non-toxic potentiated NBD1 variants (Figure 3E). Like Hsp104A503V, Hsp104N566I exhibited elevated ATPase activity (Figure 7F). However, Hsp104N566I luciferase disaggregase activity was not as potent as Hsp104A503V in the presence or absence of Hsp70 and Hsp40 (Figure 7G). Yet Hsp104N566I luciferase reactivation activity was higher than Hsp104 in the absence of Hsp70 and Hsp40 (Figure 7G). Thus, separation of Hsp104 disaggregase activity from Hsp70 and Hsp40 might enable rescue of α-syn, FUS, and TDP-43 toxicity (Jackrel et al., 2014a; Torrente et al., 2016).
DISCUSSION
Here, we define single missense mutations in NBD1 or NBD2 that potentiate Hsp104. Potentiated NBD1 and NBD2 variants suppress the toxicity of TDP-43 and FUS (implicated in ALS-FTD), as well as α-syn toxicity (implicated in PD), whereas Hsp104 is ineffective. Several NBD1 variants also suppressed aggregation of these disease-associated substrates. Previously, we isolated numerous missense mutations located at disparate positions throughout the Hsp104 MD that confer potentiation (Jackrel et al., 2014a, 2015). Several of these potentiating MD mutations likely disrupt inter-protomer MD contacts or intra-protomer NBD1:MD contacts in the ADP-bound hexamer (Gates et al., 2017; Heuck et al., 2016). The potentiating NBD1 mutations alter residues that contact ATP (I187, L355), contact ATP-binding residues (Q347), or reside at intra-protomer (I230, E360) or inter-protomer (E190, E360) NBD1:MD interfaces. Importantly, potentiated NBD1 variants do not typically exhibit off-target toxicity, unlike several potentiated MD variants, and thus provide a risk-averse mechanism to enhance Hsp104 activity.
We have expanded the repertoire of mutations that enhance Hsp104 activity into the NBD1 ATP-binding pocket. I187 directly contacts nucleotide, and L355 helps confine nucleotide in the nucleotide-binding pocket. Q347 contacts I351, which also directly binds nucleotide (Gates et al., 2017; Lee et al., 2003; Yokom et al., 2016). Alteration of these side chains likely affects NBD1 activity. Indeed, Hsp104I187F and Hsp104Q347L exhibited elevated ATPase activity that was coupled to enhanced disaggregase activity. Interestingly, the equivalent mutations in NBD2 did not potentiate activity, which likely reflects that NBD1 and NBD2 are from different clades of the AAA+ family and may operate differently (Sweeny and Shorter, 2016). This finding also reinforces functional differences between NBD1 and NBD2 in Hsp104 disaggregase activity (Doyle et al., 2007; Hattendorf and Lindquist, 2002b; Schirmer et al., 2001; Torrente et al., 2016).
E190 and E360 are in close contact with the MD of the counterclockwise protomer in the AMP-PNP-bound hexamer (Yokom et al., 2016). Thus, mutation of these residues may alter NBD1:MD communication between adjacent protomers. Curiously, mutation of several other residues that form salt bridges in this inter-protomer NBD1:MD contact in the AMP-PNP-bound hexamer inactivates Hsp104 (Gates et al., 2017). Thus, residues involved in this inter-protomer NBD1:MD contact area are sensitive to mutation, with some mutations inactivating Hsp104 (Gates et al., 2017) and other mutations enhancing Hsp104 (this study). However, mutating residues in the MD that contact E190 (R419) or E360 (R433) in the AMP-PNP-bound hexamer did not potentiate Hsp104. Neither Hsp104R419V nor Hsp104R433E rescued TDP-43, FUS, or α-syn toxicity. Thus, the mechanism of potentiation may not be as simple as disrupting this inter-protomer NBD1:MD interaction. Notably, another residue that can be altered to yield potentiated activity, I193, interacts with E190 in the ADP-bound hexamer. Thus, altered E190 or I193 contacts may enhance activity.
NBD1 residues I230 and E360 contact MD residues R495 and K480 respectively in the ADP-bound hexamer (Gates et al., 2017). I230N and E360K may perturb these interactions and enhance Hsp104 activity. Importantly, R495E and K480E mutations also potentiate Hsp104 (Jackrel et al., 2015).Thus, disrupting intra-protomer NBD1:MD interactions in the ADP-bound hexamer can enhance Hsp104 activity.
We also expand the landscape of potentiating mutations into NBD2. Far fewer potentiated Hsp104 variants emerged from our NBD2 screen compared with our NBD1 or MD screens. Thus, NBD2 may be less receptive to single missense mutations that enhance activity. Hsp104N566I was the only potentiated NBD2 variant to emerge. Intriguingly, N566 resides in the NBD2 protomer interface and could alter inter-protomer communication during substrate translocation (Gates et al., 2017). Thus, altering hexamer interfaces in NBD2 can yield enhanced activity, delineating another route to potentiated activity that does not appear to involve the MD.
Hsp104N566I rescued α-syn aggregation and toxicity. In contrast, Hsp104N566I rescued FUS and TDP-43 toxicity without drastically affecting their aggregation. Hsp104N566I reduced the proportion of cells with multiple cytoplasmic FUS foci, which may be critical to mitigate FUS toxicity. However, Hsp104N566I had no obvious effect on cytoplasmic TDP-43 foci and did not return TDP-43 to the nucleus, unlike enhanced NBD1 variants. Thus, Hsp104N566I may rescue TDP-43 toxicity via a distinct mechanism. Hsp104N566I may direct TDP-43 into less toxic aggregated structures or material states. Alternatively, Hsp104N566I might eliminate toxic, soluble TDP-43 oligomers, while leaving less toxic aggregated structures intact. Hsp104N566I might also extract essential proteins whose coaggregation with TDP-43 contributes to toxicity. These possible mechanisms are not mutually exclusive and may combine to rescue toxicity.
Loss of amino acid identity at some positions in the MD can potentiate Hsp104. Indeed, mutation of A503 to any residue except proline enabled Hsp104 to potently rescue α-syn, FUS, and TDP-43 toxicity (Jackrel et al., 2014a). This same level of degeneracy did not apply to our potentiated NBD1 variants. Tighter constraints on NBD1 appear to reduce the number of mutagenic routes to enhanced activity. Indeed, not any mutation would potentiate at the I187, I230, Q347, or E360 positions. The I187 and I230 positions were the most permissive as five residues we assessed at these positions enabled rescue of α-syn, FUS, and TDP-43 toxicity. Q347 and E360 were more restrictive with only three residues we tested conferring broad-spectrum potentiated activity. These findings refine our understanding of disaggregase sequence space and clarify mutagenic routes to potentiated activity. For example, there appear to be many possible routes to enhanced activity by mutating A503, fewer for I230, and fewer still for E360.
Our findings reveal deep insights into disaggregase mechanism, regulation, therapeutics, and evolution. We define how altering NBD1 or NBD2 can enhance Hsp104 activity. We have been concerned that the lack of substrate specificity of the variants may lead to the non-specific unfolding of proteins and off-target toxicity under some conditions, as with many potentiated MD variants (Jackrel and Shorter, 2015). However, the majority of potentiated NBD1 (I193T, I230N, Q347L, L355A, and E360R) and NBD2 (N566I) variants explored did not exhibit reduced growth at 37°C in yeast. Thus, potentiated NBD1 and NBD2 variants appear less prone to deleterious off-target effects.
Several distinct misfolded conformers of a single protein can contribute to toxicity in neurodegenerative disorders (Chuang et al., 2018; Liu et al., 2016; Rasmussen et al., 2017). Moreover, misfolded conformers of different proteins can promote neurodegeneration in the same disease (Aoyagi et al., 2019; Robinson et al., 2018). Ideally, we envisage engineering Hsp104 variants that selectively purge the entire cloud of diverse toxic conformers to remediate disease progression rather than focusing on a single conformer (Mack and Shorter, 2016).
The non-toxic, potentiated NBD1 and NBD2 variants uncovered here represent attractive variants to advance to animal models. Hsp104 variants could ultimately be developed as therapeutics (Jackrel and Shorter, 2015; Lo Bianco et al., 2008; Shorter, 2008, 2017). Several strategies can be conceived, including gene-, mRNA-, or protein-based therapies (Shorter, 2016, 2017). For example, Hsp104 variants could be delivered via neuron-specific adeno-associated viruses (AAVs) that selectively target degenerating neurons (Bedbrook et al., 2018; Chan et al., 2017; Deverman et al., 2016; Tervo et al., 2016). AAVs have key advantages including minimal immunogenicity and robust transgene expression in the CNS without integration into the host genome (Bedbrook et al., 2018; Hocquemiller et al., 2016). Game-changing AAV therapies are now U.S. Food and Drug Administration (FDA) approved for congenital blindness and spinal muscular atrophy (Apte, 2018; Mendell et al., 2017). Ultimately, we envision a therapy in which an inducible disaggregase transgene is delivered in a single AAV dose and serves as a long-lasting, lifelong treatment, which can be switched on or off as needed by an orally administered drug. This approach is attractive, as we desire to only transiently express a therapeutic disaggregase until protein misfolding is reversed. Once reversed, then the disaggregase would be silenced to minimize any off-target effects or immune response. Should disease resurface, the disaggregase would be switched back on. Rapid advances in AAV technology make this approach feasible (Bed-brook et al., 2018). Finally, lessons learned from tailoring therapeutic Hsp104 disaggregases will prove instructive for safely enhancing the activity of several human protein disaggregases, which might also be applied to treat neurodegenerative disorders (Guo et al., 2018, 2019; Shorter, 2011, 2016, 2017).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James Shorter (jshorter@pennmedicine.upenn.edu). Plasmids newly generated in this study will be made readily available to the scientific community. We will honor requests in a timely fashion. Material transfers will be made with no more restrictive terms than in the Simple Letter Agreement or the Uniform Biological Materials Transfer Agreement and without reach through requirements.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast Strains and Media
Yeast were WT W303a (MATa, can1-100, his3-11, 15, leu2-3, 112, trp1-1, ura3-1, ade2-1) or the isogenic strain W303aΔhsp104 (Jackrel et al., 2014a). The yeast strains W303aΔhsp104-pAG303GAL-α-syn-YFP-pAG304GAL-α-syn-YFP, W303aΔhsp104-pAG303GAL-FUS, W303aΔhsp104-pAG303GAL-TDP-43, W303aΔhsp104-pAG303GAL-TDP-43-GFPS11-pAG305GAL-GFPS1-10, and W303aΔhsp104-pAG303GAL-FUS-GFP have been previously described (Jackrel et al., 2014a, 2014b; Jackrel and Shorter, 2014). Yeast were grown in rich media (YPD) or synthetic dropout media, supplemented with 2% glucose, raffinose, or galactose.
METHOD DETAILS
Plasmids
QuikChange site-directed mutagenesis (Agilent) was used to create mutations in the pAG416GAL-Hsp104, pNOTAG-Hsp104, and pRS313-Hsp104 plasmids (Gates et al., 2017; Hattendorf and Lindquist, 2002a; Jackrel et al., 2014a). Mutations were confirmed by DNA sequencing and are detailed in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Hsp104 | Enzo | Cat#ADI-SPA-1040; RRID: AB_10631415 |
| Mouse monoclonal anti-PGK | Invitrogen | Cat#459250; RRID: AB_2532235 |
| Rabbit polyclonal anti-GFP | Sigma-Aldrich | Cat#G1544; RRID: AB_439690 |
| Rabbit polyclonal anti-TDP-43 | Proteintech | Cat#10782-2-AP; RRID: AB_615042 |
| Rabbit polyclonal anti-FUS | Bethyl Laboratories | Cat#A300-302A; RRID: AB_309445 |
| Goat anti-rabbit secondary antibody | Li-cor | Cat#926-68071; RRID: AB_10956166 |
| Goat anti-mouse secondary antibody | Li-cor | Cat#926-32210; RRID: AB_621842 |
| Bacterial and Virus Strains | ||
| BL21(DE3)RIL cells | Agilent | Cat#230245 |
| XL10-Gold cells | Agilent | Cat#200314 |
| One Shot TOP10 Chemically Competent E. coli | Invitrogen | Cat#C404010 |
| ElectroMAX DH5α-E Competent Cells | Invitrogen | Cat#11-319-019 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Affi-Gel Blue Gel | Bio-Rad | Cat#1537302 |
| Hsc70 | Enzo | Cat#ADI-SPP-751 |
| Hdj2 | Enzo | Cat#ADI-SPP-405 |
| Firefly luciferase | Sigma-Aldrich | Cat#L6876 Cat# SRE0045 |
| Luciferase Assay Reagent | Promega | Cat#E1483 |
| Creatine kinase | Roche | Cat# 10127566001 |
| Creatine phosphate | Roche | Cat# 10621722001 |
| Adenosine-5’-triphosphate disodium salt hydrate, 98% | Alfa Aesar | Cat# J61125 |
| Adenosine 5′-[ã-thio]triphosphate tetralithium salt | Roche | Cat#10102342001 |
| Hsp104 | (Jackrel et al., 2014a) | N/A |
| Hsp104A503V | (Jackrel et al., 2014a) | N/A |
| Hsp104I187F | This paper | N/A |
| Hsp104I230N | This paper | N/A |
| Hsp104Q347L | This paper | N/A |
| Hsp104E360R | This paper | N/A |
| Hsp104N566I | This paper | N/A |
| RepA11-70-GFP | (Jackrel et al., 2014a) | N/A |
| GroELtrap | (Jackrel et al., 2014a) | N/A |
| L-Azetidine-2-carboxylic acid | Bachem | Cat#4019045 |
| Vectashield mounting medium with DAPI | Vector Laboratories | Cat#H-1200 |
| 5-Fluoroorotic acid (5-FOA) | Research Products International | Cat#F10501 |
| StrataClean Resin | Agilent | Cat#400714 |
| DpnI | New England Biolabs | R0176S |
| PfuUltra II Fusion HS DNA Polymerase | Agilent | 600672 |
| Critical Commercial Assays | ||
| Pi ColorLock Gold Phosphate Detection System | Innova | Cat#303-0030 |
| GeneMorph II EZClone Domain Mutagenesis kit | Agilent | Cat#200552 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: W303a (MATa, can1-100, his3-11, 15, leu2-3, 112, trp1-1, ura3-1, ade2-1) | (Schirmer et al., 2004) | N/A |
| S. cerevisiae: W303aΔhsp104 (MATa, can1-100, his3-11, 15, leu2-3, 112, trp1-1, ura3-1, ade2-1, hsp104::KanMX) | (Schirmer et al., 2004) | A3224 |
| S. cerevisiae: W303aΔhsp104-pAG303GAL-α-syn-YFP-pAG304GAL-α-syn-YFP | (Jackrel et al., 2014a) | N/A |
| S. cerevisiae: W303aΔhsp104-pAG303GAL-TDP-43 | (Jackrel et al., 2014a) | N/A |
| S. cerevisiae: W303aΔhsp104-pAG303GAL-FUS | (Jackrel et al., 2014a) | N/A |
| S. cerevisiae: W303aΔhsp104-pAG303GAL-TDP-43-GFPS11-pAG305GAL-GFPS1-10 | (Jackrel et al., 2014a) | N/A |
| S. cerevisiae: W303aΔhsp104-pAG303GAL-FUS-GFP | (Jackrel et al., 2014a) | N/A |
| Recombinant DNA | ||
| pAG416Gal-CCDB | (Jackrel et al., 2014a) | N/A |
| pAG416Gal-Hsp104 | (Jackrel et al., 2014a) | N/A |
| pAG416Gal-Hsp104A503V | (Jackrel et al., 2014a) | N/A |
| pAG416Gal-Hsp104I187F | This paper | N/A |
| pAG416Gal-Hsp104I230N | This paper | N/A |
| pAG416Gal-Hsp104Q347L | This paper | N/A |
| pAG416Gal-Hsp104E360K | This paper | N/A |
| pAG416Gal-Hsp104A178D | This paper | N/A |
| pAG416Gal-Hsp104E190V | This paper | N/A |
| pAG416Gal-Hsp104I193L | This paper | N/A |
| pAG416Gal-Hsp104I193T | This paper | N/A |
| pAG416Gal-Hsp104T196S | This paper | N/A |
| pAG416Gal-Hsp104L355A | This paper | N/A |
| pAG416Gal-Hsp104A330V | This paper | N/A |
| pAG416Gal-Hsp104N566I | This paper | N/A |
| pAG416Gal-Hsp104R419V | This paper | N/A |
| pAG416Gal-Hsp104R495N | This paper | N/A |
| pAG416Gal-Hsp104R495E | This paper | N/A |
| pAG416Gal-Hsp104R433E | This paper | N/A |
| pAG416Gal-Hsp104K480E | (Jackrel et al., 2015) | N/A |
| pAG416Gal-Hsp104R433K | This paper | N/A |
| pAG416Gal-Hsp104R433Y | This paper | N/A |
| pAG416Gal-Hsp104R495D | This paper | N/A |
| pAG416Gal-Hsp104R495M | This paper | N/A |
| pNOTAG-Hsp104 | (Hattendorf and Lindquist, 2002a) | N/A |
| pNOTAG-Hsp104A503V | (Jackrel et al., 2014a) | N/A |
| pNOTAG-Hsp104I187F | This paper | N/A |
| pNOTAG-Hsp104I230N | This paper | N/A |
| pNOTAG-Hsp104Q347L | This paper | N/A |
| pNOTAG-Hsp104E360R | This paper | N/A |
| pNOTAG-Hsp104N566I | This paper | N/A |
| pAG416Gal-Hsp104I187A | This paper | N/A |
| pAG416Gal-Hsp104I187S | This paper | N/A |
| pAG416Gal-Hsp104I187V | This paper | N/A |
| pAG416Gal-Hsp104I187P | This paper | N/A |
| pAG416Gal-Hsp104I187Y | This paper | N/A |
| pAG416Gal-Hsp104I187W | This paper | N/A |
| pAG416Gal-Hsp104I187D | This paper | N/A |
| pAG416Gal-Hsp104I187N | This paper | N/A |
| pAG416Gal-Hsp104I187K | This paper | N/A |
| pAG416Gal-Hsp104I230A | This paper | N/A |
| pAG416Gal-Hsp104I230S | This paper | N/A |
| pAG416Gal-Hsp104I230V | This paper | N/A |
| pAG416Gal-Hsp104I230P | This paper | N/A |
| pAG416Gal-Hsp104I230F | This paper | N/A |
| pAG416Gal-Hsp104I230D | This paper | N/A |
| pAG416Gal-Hsp104I230Q | This paper | N/A |
| pAG416Gal-Hsp104I230K | This paper | N/A |
| pAG416Gal-Hsp104Q347A | This paper | N/A |
| pAG416Gal-Hsp104Q347T | This paper | N/A |
| pAG416Gal-Hsp104Q347V | This paper | N/A |
| pAG416Gal-Hsp104Q347P | This paper | N/A |
| pAG416Gal-Hsp104Q347F | This paper | N/A |
| pAG416Gal-Hsp104Q347D | This paper | N/A |
| pAG416Gal-Hsp104Q347N | This paper | N/A |
| pAG416Gal-Hsp104Q347K | This paper | N/A |
| pAG416Gal-Hsp104E360A | This paper | N/A |
| pAG416Gal-Hsp104E360T | This paper | N/A |
| pAG416Gal-Hsp104E360V | This paper | N/A |
| pAG416Gal-Hsp104E360P | This paper | N/A |
| pAG416Gal-Hsp104E360F | This paper | N/A |
| pAG416Gal-Hsp104E360D | This paper | N/A |
| pAG416Gal-Hsp104E360N | This paper | N/A |
| pAG416Gal-Hsp104E360R | This paper | N/A |
| pRS313-CCDB | (Gates et al., 2017) | N/A |
| pRS313-Hsp104 | (Gates et al., 2017) | N/A |
| pRS313-Hsp104A503V | This paper | N/A |
| pRS313-Hsp104I187F | This paper | N/A |
| pRS313-Hsp104I230N | This paper | N/A |
| pRS313-Hsp104Q347L | This paper | N/A |
| pRS313-Hsp104E360K | This paper | N/A |
| pRS313-Hsp104I193T | This paper | N/A |
| pRS313-Hsp104L355A | This paper | N/A |
| pRS313-Hsp104N566I | This paper | N/A |
| pRS313-Hsp104A330V | This paper | N/A |
| pRS313-Hsp104E360F | This paper | N/A |
| pRS313-Hsp104I187S | This paper | N/A |
| pRS313-Hsp104I187N | This paper | N/A |
| pRS313-Hsp104E360R | This paper | N/A |
| pRS416GAL-Hsp104 | (Jackrel et al., 2014a) | |
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| GraphPad Prism | GraphPad | Version 8.0 |
Yeast Transformation and Spotting Assays—Figures 1, 2, 3, 5, 7, S1, S2, S3, and S4
Yeast transformations were performed using standard polyethylene glycol and lithium acetate procedures (Gietz and Schiestl, 2007). For the spotting assays, yeast were grown to saturation in raffinose supplemented dropout media overnight at 30°C. The saturated overnight cultures were normalized to an OD600 of 2.0 (A600nm = 2.0) and serially diluted five-fold. A 96-bolt replicator tool (frogger) was used to spot the strains in duplicate onto both glucose and galactose dropout plates. These plates were grown at 30°C and imaged after 72h to assess suppression of disease toxicity. Each spotting assay shown is representative of at least two or three biological replicates.
Library Generation and Screening
Library generation and screening were performed as described previously (Jackrel et al., 2014a, 2014b). The GeneMorph II EZClone Domain Mutagenesis kit (Agilent) was used to create a library of NBD1 or NBD2 mutants, with modifications. NBD1 or NBD2 was amplified by PCR using Mutazyme. Gel extraction was used to purify the resulting PCR product, and this PCR product was used as the template in a PCR reaction containing pRS416GAL-Hsp104WT and PfuUltraII HS polymerase. DpnI restriction enzyme was used to digest the parental DNA product, and the product was purified using StrataClean resin (Agilent). The product was then ethanol precipitated and transformed by electroporation into ElectroMAX DH5α cells in quadruplicate to maintain library size. Representative clones were sequenced to confirm mutagenesis of NBD1 or NBD2.
The NBD1 or NBD2 library was transformed into yeast containing pAG303GAL-α-syn-YFP-pAG304GAL-α-syn-YFP, pAG303GAL-FUS, or pAG303GAL-TDP-43 and the yeast were harvested and pooled to form the final library. The library was grown overnight in raffinose-containing media and then plated onto galactose media. Individual yeast colonies were selected for further analysis. A secondary screen was performed to confirm that toxicity suppression was Hsp104-dependent. Here, Hsp104 variants were streaked onto 5-FOA media to counterselect for strains that lost the Hsp104 plasmids. Toxicity of the disease-substrate in the absence of Hsp104 was then assessed to validate strains as containing true hits (Jackrel et al., 2014a, 2014b). All missense mutations were then constructed freshly in the pAG416GAL-Hsp104 template plasmid using QuikChange mutagenesis for all additional experiments.
Western Blotting—Figures 1, 2, 3, 7, S1, S2, and S4
Transformed Hsp104 variants and controls were grown overnight in raffinose media. The overnight cultures were diluted to an OD600 of 0.3 (A600nm = 0.3) and grown in galactose-supplemented media at 30°C. α-synuclein samples were induced for 8h, while FUS and TDP-43 samples were induced for 5h. Samples were then normalized to an OD600 of 0.6 (A600nm = 0.6). The pelleted cells were resuspended in 0.1 M NaOHfor5 min and then pelleted again and resuspended in 1xSDS sample buffer. The samples were then boiled and separated by SDS-PAGE (4%–20% gradient, Bio-Rad) and transferred to a PVDF membrane. The following primary antibodies were used: anti-GFP polyclonal (Sigma-Aldrich), anti-FUS polyclonal (Bethyl Laboratories), anti-TDP-43 polyclonal (Proteintech), anti-Hsp104 polyclonal (Enzo Life Sciences), and anti-PGK monoclonal (Invitrogen). Two fluorescently labeled secondary antibodies were used: anti-rabbit (Li-Cor) and anti-mouse (Li-Cor). Blots were imaged using a LI-COR Odyssey FC Imaging system.
Toxicity Assay—Figures 3, 7, and S3
Hsp104 variants along with applicable controls were transformed into W303aΔhsp104 yeast. The strains were grown overnight in raffinose dropout media at 30°C with shaking. The saturated cultures were normalized to an OD600 of 2.0 (A600nm = 2.0) and spotted in duplicate onto two sets of SD-Ura and SGal-Ura plates. One set of plates was placed at 37°C and the other at 30°C. Both sets of plates were analyzed for toxicity after 72-96h.
Yeast Thermotolerance Assay—Figure S4
pRS313 plasmid encoding the indicated Hsp104 variant under the control of the HSP104 promoter or an empty vector control was transformed into W303aΔhsp104 yeast as described in the “Yeast Transformation and spotting assays” section and plated on Glucose-His-dropout (SD-His) plates. The transformants were then inoculated in SD-His media for 4 h at 30°C and normalized to OD600 of 0.6 (A600nm = 0.6). Yeast were then incubated at 37°C for 30min to induce Hsp104 expression, followed by a 0 or 20 min heat shock at 50°C. Yeast were then plated onto SD-His after a 1000-fold dilution. Yeast colonies were then counted after three days at 30°C using an aCOLyte colony counter (Synbiosis). The number of colonies after 20 min heat shock was divided by the number of colonies after no heat shock to enable calculation of the survival (%) conferred by each Hsp104 variant or the empty vector control.
Yeast L-Azetidine-2-carboxylic acid (AZC) tolerance Assay—Figure S4
pAG416Gal encoding the indicated Hsp104 variant or empty vector control was transformed into W303aΔhsp104 yeast as described in the “Yeast Transformation and spotting assays” section and plated on SD-Ura. The transformants were then inoculated in SRaff-Ura media and grown overnight until saturation at 30°C. Yeast were then inoculated into SGal-Ura media and grown for 6 h at 30°C to induce Hsp104 expression. The samples were then normalized to OD600 of 1 and spotted on SD-Ura or SGal-Ura plates containing 0mM, 2mM, or 5mM AZC as described in the “Spotting assay” section.
Fluorescence Microscopy—Figures 1, 2, 3, S5, S6, and S7
Microscopy samples were grown and induced as they were for immunoblotting. For TDP-43 samples (W303aΔhsp104-pAG303GAL-TDP-43-GFPS11-pAG305GAL-GFPS-10), cells were harvested, fixed in 1mL 70% ethanol, and immediately pelleted. The cells were then washed 3 times with cold PBS and resuspended in 15μL of Vectashield mounting medium with 4’,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). α-syn (W303aΔhsp104-pAG303GAL-α-syn-YFP-pAG304GAL-α-syn-YFP) and FUS (W303aΔhsp104-pAG303GAL-FUS-GFP) samples were imaged live. All cells were imaged at 100x magnification using a Leica-DMIRBE microscope. Analysis of cells was performed in ImageJ (Schneider et al., 2012). For each sample, 97-530 cells were quantified in two or three independent trials.
Protein Purification
Protein purification was performed as described previously (DeSantis et al., 2014; Jackrel et al., 2014a). Hsp104 proteins were expressed and purified as untagged proteins from E. coli. Proteins were overexpressed in BL21(DE3) RIL. Cells were harvested, lysed with lysis buffer (50mM Tris pH 8.0,10mM MgCl2, 2.5% glycerol, 2mM β-mercaptoethanol) supplemented with protease inhibitors, and the protein was purified using Affi-Gel Blue Gel (Bio-Rad). The protein was eluted with elution buffer (50mM Tris pH 8.0,1M KCl, 10mM MgCl2, 2.5% glycerol, 2mM β-mercaptoethanol). The eluate was buffer exchanged into high salt storage buffer (40mM HEPES-KOH pH 7.4, 500mM KCl, 20mM MgCl2,10% glycerol, 1mM DTT). The protein was then further purified by ResourceQ anion exchange chromatography using running buffer Q (20mM Tris pH 8.0, 0.5mM EDTA, 5mM MgCl2, 50mM NaCl) and eluted with a linear gradient of buffer Q+ (20mM Tris pH 8.0, 0.5mM EDTA, 5mM MgCl2, 1M NaCl). Immediately before loading the column, the protein was diluted to a final concentration of 10% in buffer Q supplemented to 150mM NaCl and loaded onto the column using a 50ml Superloop. The eluted protein was then concentrated and exchanged into high salt storage buffer, flash frozen in liquid N2, and stored at −80°C until use. Hsp104 concentrations refer to the hexamer concentration. RepA1–70-GFP was purified by expressing N-terminally His-tagged protein in E. coli and purifying from inclusion bodies in 6M urea. Urea was removed by dialysis and the protein was applied to Ni-NTA beads. The eluted protein was then used with the tag. Hsc70 and Hdj2 were from Enzo Life Sciences.
ATPase assay—Figure 6
Hsp104 (0.042μM hexamer) was incubated with ATP (1mM) for 5min at 25°C. ATPase activity was assessed by the release of inorganic phosphate, which was determined using a malachite green phosphate detection kit (Innova). Background hydrolysis was determined at time zero and subtracted.
Luciferase Reactivation assay—Figure 6
Luciferase reactivation was performed as described (DeSantis et al., 2012; Glover and Lindquist, 1998; Lo Bianco et al., 2008). Briefly, to assemble aggregates, firefly luciferase (50μM, Sigma) in luciferase-refolding buffer (LRB: 25mM HEPES-KOH pH 7.4, 150mM KAOc, 10mM MgAOc, 10mM DTT) plus 8M urea was incubated at 30°C for 30min. The sample was then rapidly diluted 100-fold into LRB. Aliquots were snap frozen and stored at −80°C until use. Aggregated luciferase (50nM) was incubated with Hsp104 (0.167μM hexamer) with ATP (5.1mM) and an ATP regeneration system (1mM creatine phosphate, 0.25μM creatine kinase) in the presence or absence of Hsc70 (0.167mM) and Hdj2 (0.167mM) for 90min at 25°C. At the end of the reaction, luciferase activity was assessed with a luciferase assay system (Promega). Recovered luminescence was monitored using a Tecan Infinite M1000 plate reader.
RepA1-70-GFP Unfolding Assay—Figure 6
RepA1–70-GFP unfolding was performed as described (Jackrel et al., 2014a). Unfolding assays were performed in buffer A (20mM Tris-HCl, pH 7.5, 100mM KCl, 0.1mM EDTA, 10% (v/v) glycerol, 20 mg/ml BSA, 0.005% (vol/vol) Triton X-100, 10mM MgCl2, and 5mM DTT) in the presence of 20mM creatine phosphate, 60 mg/ml creatine kinase and 4mM ATP. RepA1–70-GFP (0.7μM) was incubated with the indicated Hsp104 variant (2.1 μM) plus GroELtrap (2.5 μM). Fluorescence was monitored using a plate reader (TECAN) with an excitation wavelength at 395nm and emission wavelength at 51 nm every min for 60 min. F0 denotes the fluorescence of a sample at time = 0. F denotes fluoresce at any time T. F/F0 as a function of time was plotted.
QUANTIFICATION AND STATISTICAL ANALYSIS
As described in the figure legends, all data points in each graph are means ± standard error of the mean (SEM), unless otherwise indicated. For yeast experiments, n represents a biological replicate. For biochemical experiments, n represents an independent experimental trial. N is indicated in the figure legends. No tests of statistical significance were employed in this study.
DATA AND CODE AVAILABILITY
The original/source data in the paper have not been deposited in a public repository because for these types of data it is not mandatory. The original/source data in the paper are available from the corresponding author upon reasonable request. This study did not generate code.
Supplementary Material
Highlights.
Mining Hsp104 sequence space to safely enhance disaggregase activity
Non-toxic NBD1 and NBD2 variants counter TDP-43, FUS, and α-synuclein toxicity
Mutating NBD1 residues that engage ATP or ATP-binding residues potentiates activity
Mutating the NBD2 protomer interface can safely ameliorate Hsp104 activity
ACKNOWLEDGMENTS
We thank Zach March, Edward Chuang, and Ryan Cupo for feedback. We were supported by an Alzheimer’s Association Research Fellowship (J.L.), an American Heart Association (AHA) post-doctoral fellowship, a Target ALS Springboard Award (M.E.J), National Science Foundation (NSF) graduate research fellowship DGE-1321851 (K.L.M.), NIH training grant T32GM008275, an AHA predoctoral fellowship (E.A.S.), NIH grants DP2OD002177 and R01GM099836, a Muscular Dystrophy Association Research Award, an ALS Association Award, the Life Extension Foundation, a Linda Montague Pechenik Research Award, the Packard Center for ALS Research at Johns Hopkins University, and Target ALS (J. Shorter).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.069.
DECLARATION OF INTERESTS
M.E.J. and J. Shorter are inventors on U.S. Patent No. 9994625 (HSP104 variants and uses thereof).
REFERENCES
- Abeliovich A, and Gitler AD (2016). Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539, 207–216. [DOI] [PubMed] [Google Scholar]
- Aoyagi A, Condello C, Stohr J, Yue W, Rivera BM, Lee JC, Woerman AL, Halliday G, van Duinen S, Ingelsson M, et al. (2019). Ab and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Sci. Transl. Med 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte RS (2018). Gene Therapy for Retinal Degeneration. Cell 173, 5. [DOI] [PubMed] [Google Scholar]
- Bedbrook CN, Deverman BE, and Gradinaru V (2018). Viral strategies for targetingthecentral and peripheral nervoussystems. Annu. Rev. Neurosci 41, 323–348. [DOI] [PubMed] [Google Scholar]
- Castellano LM, Bart SM, Holmes VM, Weissman D, and Shorter J (2015). Repurposing Hsp104 to antagonize seminal amyloid and counter HIV infection. Chem. Biol 22, 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, Sanchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, and Gradinaru V (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci 20, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang E, Hori AM, Hesketh CD, and Shorter J (2018). Amyloid assembly and disassembly. J. Cell Sci 131, jcs189928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Redmond DE Jr., Steece-Collier K, Lipton JW, and Manfredsson FP (2016). Is alphα-synuclein loss-of-function a contributor to Parkinsonian pathology? Evidence from non-human primates. Front. Neurosci 10, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushman-Nick M, Bonini NM, and Shorter J (2013). Hsp104 suppresses polyglutamine-induced degeneration post onset in a Drosophila MJD/SCA3 model. PLoS Genet. 9, e1003781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis ME, and Shorter J (2012). Hsp104 drives “protein-only” positive selection of Sup35 prion strains encoding strong [PSI(+)]. Chem. Biol 19, 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis ME, Leung EH, Sweeny EA, Jackrel ME, Cushman-Nick M, Neuhaus-Follini A, Vashist S, Sochor MA, Knight MN, and Shorter J (2012). Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 151, 778–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis ME, Sweeny EA, Snead D, Leung EH, Go MS, Gupta K, Wendler P, and Shorter J (2014). Conserved distal loop residues in the Hsp104 and ClpB middle domain contact nucleotide-binding domain 2 and enable Hsp70-dependent protein disaggregation. J. Biol. Chem 289, 848–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deverman BE, Pravdo PL, Simpson BP, Kumar SR, Chan KY, Banerjee A, Wu WL, Yang B, Huber N, Pasca SP, and Gradinaru V (2016). Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol 34, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SM, Shorter J, Zolkiewski M, Hoskins JR, Lindquist S, and Wickner S (2007). Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat. Struct. Mol. Biol 14, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzberger JP, and Berger JM (2006). Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct 35, 93–114. [DOI] [PubMed] [Google Scholar]
- Franke KB, Bukau B, and Mogk A (2017). Mutant analysis reveals allosteric regulation of ClpB disaggregase. Front. Mol. Biosci 4, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, Shorter J, Da Cruz S, and Cleveland DW (2019). Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102, 339–357.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates SN, Yokom AL, Lin J, Jackrel ME, Rizo AN, Kendsersky NM, Buell CE, Sweeny EA, Mack KL, Chuang E, et al. (2017). Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, and Schiestl RH (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc 2, 31–34. [DOI] [PubMed] [Google Scholar]
- Glover JR, and Lindquist S (1998). Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82. [DOI] [PubMed] [Google Scholar]
- Guo L, and Shorter J (2017). Biology and pathobiology ofTDP-43 and emergent therapeutic strategies. Cold Spring Harb. Perspect. Med 7, a024554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, O’Donovan K, Fare CM, Diaz Z, Singh N, et al. (2018). Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 173, 677–692.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Fare CM, and Shorter J (2019). Therapeutic dissolution of aberrant phases by nuclear-import receptors. Trends Cell Biol. 29, 308–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison AF, and Shorter J (2017). RNA-binding proteins with prion-like domains in health and disease. Biochem. J 474, 1417–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattendorf DA, and Lindquist SL (2002a). Analysis of the AAA sensor-2 motif in the C-terminal ATPase domain of Hsp104 with a site-specific fluorescent probe of nucleotide binding. Proc. Natl. Acad. Sci. USA 99, 2732–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattendorf DA, and Lindquist SL (2002b). Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. EMBO J. 21, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuck A, Schitter-Sollner S, Suskiewicz MJ, Kurzbauer R, Kley J, Schleiffer A, Rombaut P, Herzog F, and Clausen T (2016). Structural basis for the disaggregase activity and regulation of Hsp104. eLife 5, e21516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocquemiller M, Giersch L, Audrain M, Parker S, and Cartier N (2016). Adeno-associated virus-based gene therapy for CNS diseases. Hum. Gene Ther 27, 478–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, and Shorter J (2014). Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins. Dis. Model. Mech 7, 1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, and Shorter J (2015). Engineering enhanced protein disaggregases for neurodegenerative disease. Prion 9, 90–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, and Shorter J (2017). Protein-remodeling factors as potential therapeutics for neurodegenerative disease. Front. Neurosci 11, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, DeSantis ME, Martinez BA, Castellano LM, Stewart RM, Caldwell KA, Caldwell GA, and Shorter J (2014a). Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell 156,170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, Tariq A, Yee K, Weitzman R, and Shorter J (2014b). Isolating potentiated Hsp104 variants using yeast proteinopathy models. J. Vis. Exp (93), e52089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackrel ME, Yee K, Tariq A, Chen AI, and Shorter J (2015). Disparate mutations confer therapeutic gain of Hsp104 function. ACS Chem. Biol 10, 2672–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, McCaffery JM, Lindquist S, and Gitler AD (2008). A yeast TDP-43 proteinopathy model: exploring the molecular determinants ofTDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 105, 6439–6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, Bosco DA, Hayward LJ, Brown RH Jr., Lindquist S, et al. (2011). A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 9, e1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaips CL, Hochstrasser ML, Langlois CR, and Serio TR (2014). Spatial quality control bypasses cell-based limitations on proteostasis to promote prion curing. eLife 3, e04288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sowa ME, Watanabe YH, Sigler PB, Chiu W, Yoshida M, and Tsai FT (2003). The structure of ClpB: a molecular chaperone that rescues proteins from an aggregated state. Cell 115, 229–240. [DOI] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, and Cleveland DW (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YH, Han YL, Song J, Wang Y, Jing YY, Shi Q, Tian C, Wang ZY, Li CP, Han J, and Dong XP (2011). Heat shock protein 104 inhibited the fibrillization of prion peptide 106-126 and disassembled prion peptide 106-126 fibrils in vitro. Int. J. Biochem. Cell Biol 43, 768–774. [DOI] [PubMed] [Google Scholar]
- Liu J, Costantino I, Venugopalan N, Fischetti RF, Hyman BT, Frosch MP, Gomez-Isla T, and Makowski L (2016). Amyloid structure exhibits polymorphism on multiple length scales in human brain tissue. Sci. Rep 6, 33079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Shorter J, Regulier E, Lashuel H, Iwatsubo T, Lindquist S, and Aebischer P (2008). Hsp104 antagonizes α-synuclein aggregation and reduces dopaminergic degeneration in a rat model of Parkinson disease. J. Clin. Invest 118, 3087–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack KL, and Shorter J (2016). Engineering and evolution of molecular chaperones and protein disaggregases with enhanced activity. Front. Mol. Biosci 3, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, Copley KE, Hurtle B, Portz B, Pyles NJ, et al. (2019). RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102, 321–338.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March ZM, Mack KL, and Shorter J (2019). AAA+ protein-based technologies to counter neurodegenerative disease. Biophys. J 116, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk L, Gomes E, Guo L, Mojsilovic-Petrovic J, Tran V, Kalb RG, Shorter J, and Bonini NM (2018a). Poly(ADP-ribose) prevents pathological phase separation ofTDP-43 by promoting liquid demixing and stress granule localization. Mol. Cell 71, 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGurk L, Mojsilovic-Petrovic J, Van Deerlin VM, Shorter J, Kalb RG, Lee VM, Trojanowski JQ, Lee EB, and Bonini NM (2018b). Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol. Commun 6, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, et al. (2017). Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med 377, 1713–1722. [DOI] [PubMed] [Google Scholar]
- Michalska K, Zhang K, March ZM, Hatzos-Skintges C, Pintilie G, Bigelow L, Castellano LM, Miles LJ, Jackrel ME, Chuang E, et al. (2019). Structure of Calcarisporiella thermophila Hsp104 disaggregase that antagonizes diverse proteotoxic misfolding events. Structure 27, 449–463.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newby GA, and Lindquist S (2013). Blessings in disguise: biological benefits of prion-like mechanisms. Trends Cell Biol. 23, 251–259. [DOI] [PubMed] [Google Scholar]
- Outeiro TF, and Lindquist S (2003). Yeast cells provide insight into alphasynuclein biology and pathobiology. Science 302, 1772–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YN, Zhao X, Yim YI, Todor H, Ellerbrock R, Reidy M, Eisenberg E, Masison DC, and Greene LE (2014). Hsp104 overexpression cures Saccharomyces cerevisiae [PSI+] by causing dissolution of the prion seeds. Eukaryot. Cell 13, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Jang HR, Lee IY, Oh HK, Choi EJ, Rhim H, and Kang S (2017). Amyotrophic lateral sclerosis-related mutant superoxide dismutase 1 aggregates inhibit 14-3-3-mediated cell survival by sequestration into the JUNQ compartment. Hum. Mol. Genet 26, 3615–3629. [DOI] [PubMed] [Google Scholar]
- Parsell DA, Kowal AS, Singer MA, and Lindquist S (1994). Protein disaggregation mediated by heat-shock protein Hsp104. Nature 372, 475–478. [DOI] [PubMed] [Google Scholar]
- Paushkin SV, Kushnirov VV, Smirnov VN, and Ter-Avanesyan MD (1996). Propagation of the yeast prion-like [psi+] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 15, 3127–3134. [PMC free article] [PubMed] [Google Scholar]
- Rasmussen J, Mahler J, Beschorner N, Kaeser SA, Hasler LM, Baumann F, Nystrom S, Portelius E, Blennow K, Lashley T, et al. (2017). Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 114, 13018–13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, and Philips T (2013). The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci 14, 248–264. [DOI] [PubMed] [Google Scholar]
- Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, Caswell C, Van Deerlin VM,Yan N, Yousef A, et al. (2018). Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141, 2181–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan JJ, Sprunger ML, Holthaus K, Shorter J, and Jackrel ME (2019). Engineered protein disaggregases mitigate toxicity of aberrant prion-like fusion proteins underlying sarcoma. J. Biol. Chem 294, 11286–11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyal SH, Schmidt E, Kitagawa K, Sondheimer N, Lindquist S, Kramer JM, and Morimoto RI (2000). Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U S A 97, 5750–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer EC, Ware DM, Queitsch C, Kowal AS, and Lindquist SL (2001). Subunit interactions influence the biochemical and biological properties of Hsp104. Proc. Natl. Acad. Sci. U S A 98, 914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer EC, Homann OR, Kowal AS, and Lindquist S (2004). Dominant gain-of-function mutations in Hsp104p reveal crucial roles for the middle region. Mol. Biol. Cell 15, 2061–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J (2008). Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals 16, 63–74. [DOI] [PubMed] [Google Scholar]
- Shorter J (2011). The mammalian disaggregase machinery: Hsp110 synergizes with Hsp70 and Hsp40 to catalyze protein disaggregation and reactivation in a cell-free system. PLoS ONE 6, e26319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J (2016). Engineering therapeutic protein disaggregases. Mol. Biol. Cell 27, 1556–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J (2017). Designer protein disaggregases to counter neurodegenerative disease. Curr. Opin. Genet. Dev 44, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, and Lindquist S (2004). Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 304, 1793–1797. [DOI] [PubMed] [Google Scholar]
- Shorter J, and Lindquist S (2006). Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Mol. Cell 23, 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, and Lindquist S (2008). Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination ofSup35 prions. EMBO J. 27, 2712–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, and Southworth DR (2019). Spiraling in Control: Structures and Mechanisms of the Hsp104 Disaggregase. Cold Spring Harb Perspect Biol. 11, a034033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, and Higgins DG (2018). Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 27, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, and Gitler AD (2011). Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 9, e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeny EA, and Shorter J (2016). Mechanistic and structural insights into the prion-disaggregase activity of Hsp104. J. Mol. Biol 428 (9 Pt B), 1870–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeny EA, Jackrel ME, Go MS, Sochor MA, Razzo BM, DeSantis ME, Gupta K, and Shorter J (2015). The Hsp104 N-terminal domain enables disaggregase plasticity and potentiation. Mol. Cell 57, 836–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariq A, Lin J, Noll MM, Torrente MP, Mack KL, Murillo OH, Jackrel ME, and Shorter J (2018). Potentiating Hsp104 activity via phosphomimetic mutations in the middle domain. FEMS Yeast Res. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervo DG, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente MP, Chuang E, Noll MM, Jackrel ME, Go MS, and Shorter J (2016). Mechanistic insights into Hsp104 potentiation. J. Biol. Chem 291, 5101–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter EW, Berenfeld L, Krause SA, Petsko GA, and Gray JV (2001). Protein misfolding and temperature up-shift cause G1 arrest via a common mechanism dependent on heat shock factor in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 98, 7313–7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacher C, Garcia-Oroz L, and Rubinsztein DC (2005). Overexpression of yeast hsp104 reduces polyglutamine aggregation and prolongs survival of a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet 14, 3425–3433. [DOI] [PubMed] [Google Scholar]
- Wallace EW, Kear-Scott JL, Pilipenko EV, Schwartz MH, Laskowski PR, Rojek AE, Katanski CD, Riback JA, Dion MF, Franks AM, et al. (2015). Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 162, 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weids AJ, and Grant CM (2014). The yeast peroxiredoxin Tsa1 protects against protein-aggregate-induced oxidative stress. J. Cell Sci 127, 1327–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendler P, Shorter J, Plisson C, Cashikar AG, Lindquist S, and Saibil HR (2007). Atypical AAA+ subunit packing creates an expanded cavity for disaggregation by the protein-remodeling factor Hsp104. Cell 131, 1366–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Clatterbuck-Soper SF, Jackrel ME, Shorter J, and Mili S (2017). FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J. Cell Biol 216, 1015–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Lin J, Mayne L, Shorter J, and Englander SW (2019). Hydrogen exchange reveals Hsp104 architecture, structural dynamics, and energetics in physiological solution. Proc. Natl. Acad. Sci. USA 116, 7333–7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokom AL, Gates SN, Jackrel ME, Mack KL, Su M, Shorter J, and Southworth DR (2016). Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat. Struct. Mol. Biol 23, 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Rodriguez R, Silberman RE,Ahearn JM, Saidha S, Cummins KC, Eisenberg E, and Greene LE (2017). Heat shock protein 104 (Hsp104)-mediated curing of [PSI+] yeast prions depends on both [PSI+] conformation and the properties of the Hsp104 homologs. J. Biol. Chem 292,8630–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original/source data in the paper have not been deposited in a public repository because for these types of data it is not mandatory. The original/source data in the paper are available from the corresponding author upon reasonable request. This study did not generate code.