

Summary
Recent experimental data, clinical, genetic and transcriptome evidence from patients converge to suggest a key role of interleukin-1β (IL-1β) in the pathogenesis of Kawasaki Disease (KD). However, the molecular mechanisms involved in the development of cardiovascular lesions during KD vasculitis are still unknown. Here, we investigated intestinal barrier function in KD vasculitis, and observed evidence of intestinal permeability and elevated circulating secretory IgA (sIgA) in KD patients, as well as elevated sIgA and IgA deposition in vascular tissues in a mouse model of KD vasculitis. Targeting intestinal permeability corrected gut permeability, prevented IgA deposition and ameliorated cardiovascular pathology in the mouse model. Using genetic and pharmacologic inhibition of IL-1β signaling, we demonstrated that IL-1β lay upstream of disrupted intestinal barrier function, subsequent IgA vasculitis development, and cardiac inflammation. Targeting mucosal barrier dysfunction and the IL-1β pathway may also be applicable to other IgA-related diseases including IgA vasculitis and IgA nephropathy.
Keywords: Kawasaki Disease, IgA, intestinal permeability, vasculitis, Complement, interleukin-1β
Graphical Abstract
eTOC
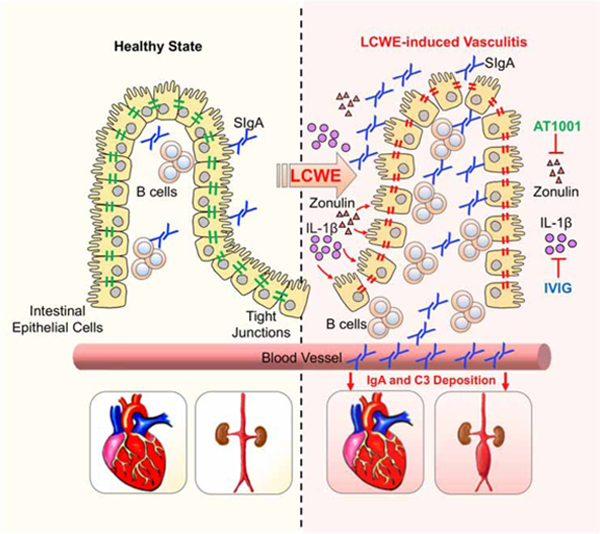
Kawasaki Disease (KD) is the leading cause of acquired heart disease among children and the etiology is unknown. Noval Rivas and colleagues demonstrate that a murine model of KD vasculitis is dependent on intestinal barrier dysfunction leading to secretory IgA leakage and IgA-C3 immune complex deposition in cardiovascular lesions.
Introduction
The critical role of intestinal barrier function in health and disease, and intestinal permeability is increasingly recognized as a pathogenic factor in many inflammatory diseases (Bischoff et al., 2014). Kawasaki Disease (KD) is an acute febrile childhood vasculitis of unknown etiology with increasing incidence, and is the leading cause of acquired heart disease among children (McCrindle et al., 2017). Coronary artery aneurysms (CAA) occur in approximately 30% of untreated pediatric patients, but this is reduced to 5% with high-dose intravenous immunoglobulin (IVIG) treatment (McCrindle et al., 2017). KD also affects the mucosal intestinal immune responses, and increased numbers of activated T cells and macrophages are present in the small intestine of KD patients (Nagata et al., 1993). A recent multicenter study of over 300 patients reveals that abdominal and gastrointestinal (GI) symptoms at KD onset, such as abdominal pain, vomiting, and diarrhea, complicate KD diagnosis, delay treatment and correlate with IVIG-resistance and severe CAA (Fabi et al., 2018). However, no studies have investigated the role of intestinal permeability in the development of KD vasculitis.
To investigate the role of intestinal barrier function in KD, we used the Lactobacillus casei cell wall extract (LCWE)-induced KD vasculitis murine model that is associated with systemic inflammation and interleukin-1β (IL-1β) production (Lee et al., 2012; Wakita et al., 2016), mimics features of human cardiovascular lesions (Noval Rivas et al., 2017) and responds to IVIG therapy (Myones et al., 1995). We show that LCWE-induced KD vasculitis is associated with increased gut permeability and secretory IgA (sIgA) leakage. We also observed IgA-Complement component 3 (C3) immune complex deposition in cardiovascular lesions and in the kidney, reminiscent of two other forms of vasculitis: IgA vasculitis (Henoch-Schönlein purpura) and IgA nephropathy (IgAN). These data indicate that KD vasculitis may be a form of IgA vasculitis and suggest that KD vasculitis as well as IgA-dependent vasculitis may share common pathological mechanisms involving a gut-vascular axis. Furthermore, we found that pharmacological blockade of intestinal gut permeability or inhibition of intestinal IL- 1β signaling on intestinal epithelial cells in mice abrogated the development of KD vasculitis, indicating that intestinal permeability and IL-1β could be targeted to treat not only the immune vasculitis of KD but also IgA-vasculitis and IgAN.
Results
LCWE-induced KD vasculitis is associated with a defective intestinal barrier.
LCWE-injected mice exhibited pathological features similar to those observed in human KD, including aortitis, coronary artery lesions (Figure 1A), and abdominal aortic aneurysms (AAA) (Figure 1B). Human KD is frequently accompanied by vague intestinal dysfunction in children (Fabi et al., 2018) and pathological studies reveal increased activation of immune cells in the small intestine (SI) lamina propria (LP) of KD patients (Nagata et al., 1993). Histological analysis of the SI and colon did not demonstrate apparent differences in architecture or evidence of inflammation between PBS and LCWE-injected KD mice (Figure S1A). However, we found that the intestinal barrier integrity was damaged in LCWE-injected KD mice as reflected by increased efflux of FITC-dextran and lipopolysaccharide (LPS) from the intestinal lumen into the circulation at 24 h and 1 wk post-LCWE injection, respectively (Figures 1C and 1D). We were unable to culture any anaerobic bacteria from the blood or spleens of LCWE-injected mice, indicating that the increased intestinal permeability was not associated with bacterial translocation into the blood or distant organs (data not shown).
Figure 1. Defective intestinal barrier and abnormal small intestinal TJ expression in LCWE-induced KD vasculitis.

(A) H&E staining of heart sections and heart vessels inflammation score of PBS or LCWE-injected WT mice (n=10 per group). Scale bars: 500μm. (B) Pictures of abdominal area, H&E staining of abdominal aorta cross-section and maximal abdominal aorta diameter of PBS or LCWE-injected WT mice (n=10 per group). Scale bars: 200μm. (C) FITC-dextran intestinal permeability assay in WT mice 24 h after PBS or LCWE-injection (n=18 per group). Data normalized to PBS controls. (D) Serum LPS concentrations of PBS or LCWE-injected WT mice at 24 h and 1 wk post-injection (n=8–17 per group). (E) SI expression of TJ components relative to β-actin 24 h after PBS or LCWE injection (n=10 per group). (F, G) ZO-1 (red, left panel) and OCLN (red, right panel) immunofluorescent staining (F) and immunofluorescence intensity quantification (G) in the SI of PBS or LCWE-injected WT mice 24 h after injection. Scale bars: 25μm. (H, I) PV-1 immunofluorescence (green, H) and immunofluorescence intensity (I) in SI sections of PBS or LCWE-injected WT mice 24 h post-injection (n=4 per group). Scale bars: 50μm. (J-L) MUC2 immunofluorescent staining (J), immunofluorescence intensity quantification (K) and mRNA expression (L) in the SI of PBS or LCWE-injected WT mice (n=2–10 per group). Scale bars: 50μm. Nuclei were stained with DAPI (blue) in all the immunofluorescent staining. Data are presented as mean ± s.e.m., *p<0.05, **p<0.01, ***p<0.001, ****p<0.001 by two-tailed unpaired t test or and 1-way ANOVA with Bonferroni post-test analysis (D). Data representative of a least 3 independent experiments (A, B) or compiled from 2 independent experiments (C-E). CA, coronary artery; Ao, aorta; AU, arbitrary units. See also Figure S1.
The formation and maintenance of tight junctions (TJ) between intestinal epithelial cells (IECs) is crucial to maintain barrier function and regulate intestinal permeability (Turner, 2009). Consistent with a deficit in intestinal barrier function and increased gut permeability, the mRNA expression of TJ protein-1 (Tjp1), Tjp2, occludin (Ocln), claudin-3 (Cldn3), Cldn7, Cldn12 and Cldn15 were significantly decreased in the SI of LCWE-injected KD mice 24 h post-injection (Figure 1E). In contrast, the expression of Cldn2, which indicates a leaky intestinal barrier (Zeissig et al., 2007), was upregulated in LCWE-injected KD mice (Figure 1E). Reduced expression of two TJ proteins, zonula occludens-1 (ZO-1) and occludin (OCLN), were also observed by immunofluorescence in the jejunum of KD mice 24 h after LCWE injection (Figures 1F and 1G). Except for Cldn15, decreased expression of TJ proteins was not observed in the colon of LCWE-injected mice, suggesting that LCWE specifically affects the SI barrier integrity (Figures S1B–S1D).
At steady-state, the gut-vascular barrier (GVB) blocks bacteria, bacterial antigens, and other luminal contents from systemic dissemination through the intestinal epithelial barrier. To determine whether LCWE-induced KD vasculitis was associated with a breach of the GVB, we evaluated the expression pattern of plasmalemma vesicle-associated protein-1 (PV-1), which is upregulated in intestinal lamina propria endothelial cells upon GVB dysfunction (Spadoni et al., 2015). At 24 h after LCWE injection, increased PV-1 signal was seen in the SI, but not the colon (Figures 1H,1I and Figure S1E). Intestinal bacteria are separated from the IECs by the mucus layer (Turner, 2009). In LCWE-injected KD mice, mucus thickness was decreased only in the SI as assessed by reduced Mucin 2 (MUC2) protein and Muc2 mRNA expression (Figures 1J–1L, Figure S1F) as well as decreased numbers of MUC2 producing goblet cells (Figures S1G and S1H). Overall, we found that LCWE-induced KD vasculitis was associated with increased gut permeability, decreased SI TJ expression, and a dysfunctional GVB resulting in the translocation of LPS into the bloodstream.
Pharmacological blockade of intestinal permeability improves LCWE-induced KD vasculitis.
Zonulin activates intestinal TJ protein phosphorylation, which opens the paracellular space and increases intestinal permeability (Fasano et al., 2000). To confirm further increased intestinal permeability and systemic inflammation in LCWE-induced KD vasculitis, we measured serum concentrations of zonulin and calprotectin (S100A8/A9 complexes), which is also increased in inflammatory disorders such as rheumatoid arthritis and inflammatory bowel disease (Brun et al., 1992; Kalla et al., 2016). Zonulin and calprotectin concentrations increased significantly in mice 24 h after LCWE-injection and dropped by 1 wk (Figure 2A). We also examined serum from KD children and febrile control subjects for evidence of increased intestinal permeability (Table S1). We observed signs of increased intestinal barrier dysfunction and systemic inflammation in KD children, as zonulin and calprotectin were elevated in the serum of KD patients pre-IVIG treatment (Figure 2B).
Figure 2. Intestinal permeability inhibitors significantly reduce LCWE-induced KD vasculitis.

(A) Zonulin (n=12 PBS; n=8 for 24 h and n=15 for 1 wk LCWE) and Calprotectin (n=9 PBS; n=10 for 24 h and n=10 for 1 wk LCWE) concentrations in the serum of PBS or LCWE-injected WT mice. (B) Zonulin (n=25 control patients; n=30 KD patients including 6 KD patients with coronary artery lesions (CAL) and Calprotectin (n=23 control patients; n=34 KD patients including 8 KD patients with CAL) concentrations measured in the serum of KD patients pre-IVIG treatment. Red circles indicate patients with CAL. (C) FITC-Dextran intestinal permeability assay in WT mice 24 h after injection of PBS (n=15), LCWE (n=20) or LCWE with either AT1001 (n=10) or ML-7 (n=10). Data normalized to PBS control mice. (D) Serum LPS concentrations in PBS (n=8), LCWE (n=10), or LCWE-injected WT mice treated with either AT1001 (n=7) or ML-7 (n=10). (E, F) Heart sections H&E staining (E) and heart vessels inflammation score (F) of PBS (n=10), LCWE (n=20) or LCWE-injected WT mice treated with either AT1001 (n=10) or ML-7 (n=10). Scale bars: 500μm. (G, H) Representative pictures of abdominal area, H&E staining of abdominal aorta cross-section (G) and maximal abdominal aorta diameter (H) of the mouse groups shown in (E, F). Scale bars: 250μm. Data are mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001, ****p<0.001 by two-tailed unpaired t test (B) or 1-way ANOVA with Bonferroni post-tests (A, C, D, F and H). Data compiled from 2 independent experiments. CA: coronary artery; Ao: aorta; CAL: coronary artery lesions.
To determine if increased intestinal permeability in LCWE-injected KD mice was a consequence of elevated concentrations of serum zonulin, and to assess if blocking intestinal permeability could reduce LCWE-induced KD vasculitis, we treated mice with the zonulin inhibitor AT1001 (Paterson et al., 2007) or ML-7, which inhibits myosin light chain kinase (MLCK). MLCK regulates intestinal permeability by contracting the IECs actin cytoskeleton, and its upregulation is associated with disruption of intestinal epithelium integrity and increased gut permeability (Ferrier et al., 2003). At 24 h post-LCWE injection, both inhibitors significantly reduced intestinal permeability and LPS translocation to amounts similar to those in control mice (Figures 2C and 2D). Furthermore, LCWE-injected KD mice treated with AT1001 or ML-7 had significantly reduced cardiovascular inflammation (Figures 2E and 2F) and abdominal aorta dilation (Figures 2G and 2H). Collectively, these results indicate that elevated concentrations of zonulin and calprotectin in KD patients represent a dysregulated intestinal barrier function phenotype that stimulates systemic inflammation. Furthermore, blocking pathways that promote intestinal permeability prevented the development of the cardiovascular lesions associated with LCWE-induced KD vasculitis.
Increased secretory IgA production and tissue deposition during LCWE-induced KD vasculitis.
IgA, the predominant antibody isotype produced in the GI tract, is translocated in mucosal secretions as polyreactive dimers, sIgA, and protects the intestinal epithelium from toxins and pathogenic microorganisms (Pabst, 2012). Compared with febrile control patients, KD subjects had increased concentrations of sIgA in their serum (Figure 3A). Similarly, sIgA concentrations were increased in the serum of LCWE-injected KD mice (Figure 3B). Blocking intestinal permeability with AT1001 or ML-7 decreased serum sIgA concentrations in LCWE-injected KD mice (Figure 3C). The amounts of intestinal luminal and fecal sIgA (Figures 3D and S2A), but not serum IgA (Figure S2A), were also increased in LCWE-injected KD mice as compared with control mice. In contrast, as reported (Lehman et al., 1985), serum concentrations of IgG were higher in LCWE-induced KD mice (Figure S2A). Because we observed a dysfunctional intestinal barrier and increased intestinal permeability in LCWE-injected KD mice, we reasoned that an unusual activation of the mucosal immune response may develop in these mice. This hypothesis is supported by the observation that acute KD patients display an increased proportion of IgA-producing B cells within inflammatory infiltrates in involved coronary arteries and other tissues (Rowley et al., 2000). We observed increased IgA and polymeric immunoglobulin receptor (pIgR) expression in the SI of LCWE-injected KD mice compared with controls (Figures S2B–S2E). We next examined the frequency of germinal center (GC) B cells and IgA-positive B cells, and found that LCWE-injected KD mice also displayed increased numbers of GC B cells and CD19+ IgA+ class-switched B cells in the spleen, abdominal aorta draining lymph nodes (AA LN), Peyer’s Patches (PP) and in the SI lamina propria (LP) (Figures 3E–H).
Figure 3. Increased IgA production during LCWE-induced KD vasculitis.

(A) sIgA concentrations measured in the serum of KD patients pre-IVIG treatment (n=22 control patients; n=34 KD patients including 8 KD patients with CAL). Red circles indicate patients with CAL. (B) sIgA concentrations in the serum of PBS or LCWE-injected WT mice (n=10 per group). (C) sIgA concentrations in the serum of PBS, LCWE-injected and LCWE-injected WT mice treated with AT1001 or ML7 (n=8–10 per group). (D) Intestinal luminal concentrations of IgA, IgM and IgG in PBS or LCWE-injected WT mice (n=10–15 per group). (E, F) Flow cytometric analysis (E), enumerated frequencies and absolute numbers (F) of GC B cells (viable CD3− CD19+ CD95+ GL7+ cells) present in the spleen, AA LN, MLN, PP, SI and colon LP of PBS or LCWE-injected WT mice (n=6–10 mice per group). (G, H) Flow cytometric analysis (G), enumerated frequencies and absolute numbers (H) of IgA+ B cells (defined as viable CD3− CD19+ IgA+ cells) present in the spleen, AA LN, MLN, PP, SI and colon LP of PBS or LCWE-injected WT mice (n=6–10 mice per group). Data are presented as mean ± s.e.m., *p<0.05, **p<0.01, ***p<0.001 by two-tailed unpaired t test and 1-way ANOVA with Bonferroni post-tests analysis. Data compiled from 2 to 3 independent experiments. See also Figure S2.
Pathological changes in the IgA compartment are associated with several autoimmune conditions, such as IgA nephropathy (IgAN), wherein IgA-immune complexes deposit in the renal glomeruli resulting in inflammation and renal failure (Floege and Feehally, 2016). Therefore, we assessed if increased intestinal permeability could also result in IgA deposition in LCWE-injected KD mice. Compared with control mice, IgA deposition was observed in the heart and abdominal aorta lesions of KD vasculitis mice (Figures 4A, 4B and S3A). Blocking intestinal permeability with AT1001 strongly reduced IgA deposition in those tissues (Figures 4A, 4B and S3A). Because sIgA is transported across the intestinal epithelium into the lumen by pIgR (Kaetzel et al., 1991), we confirmed the intestinal origin of IgA deposition by staining the abdominal aorta of LCWE-injected mice for pIgR and observed a similar deposition pattern as IgA (Figure S3B). We also observed C3 deposition and colocalization with IgA in the heart and abdominal aorta of LCWE-injected WT mice (Figures 4C, 4D and S3C). Periodic acid-Schiff (PAS) staining of kidney tissues revealed no remarkable cellular inflammatory infiltrates, although glomerular mesangial proliferation and expansion were observed in LCWE-injected KD mice (Figure 4E).
Figure 4. IgA-C3 immune complex deposition in vascular tissues and kidneys during vasculitis.

(A, B) Immunostaining of CD31 (green) and IgA (red) in heart (A) and abdominal aorta sections (B) of PBS, LCWE and LCWE-injected mice treated with AT1001. Scale bars: 100μm (A) and 25μm (B). (C, D) Immunostaining of CD31 (green), C3 (teal) and IgA (red) in heart (C) and abdominal aorta sections (D) of PBS or LCWE-injected mice. Scale bars: 50μm (C) and 25μm (D). (E) PAS staining and glomeruli mesangial cell proliferation and expansion score observed in the kidneys of PBS (n=10) or LCWE-injected mice (n=7); magnification 400x. (F) Immunostaining of CD31 (green), C3 (teal) and IgA (red) in kidney sections of PBS or LCWE-injected mice. White arrows indicate C3 and IgA colocalization. Scale bars: 50μm. Data in E are mean ± s.e.m., *p<0.05, **p<0.01, ***p<0.001 by two-tailed unpaired t test. Immunofluorescence images are representative of n= 4–8 per group and nuclei were stained with DAPI (blue). Data representative of 2 independent experiments. See also Figure S3.
In kidney sections, we observed fine granular deposition of IgA and C3 in glomerular mesangial areas only in the LCWE-injected KD mice (Figures 4F and S3D), reminiscent of the phenotype observed in IgAN (Floege and Feehally, 2016). Transmission electron microscopy also identified electron dense deposits corresponding to IgA deposition in the mesangial areas of LCWE-injected mice (Figures S3E and S3F). Overall, these results support the involvement of the mucosal immune response, sIgA, and complement in KD pathology and demonstrate that glomerular capillaries in kidneys may also be affected by this vasculitis.
IgA is required for the development of LCWE-induced KD vasculitis.
To explore the requirement for IgA in LCWE-induced KD vasculitis lesions, Igha−/− mice as well as mice deficient for the IgA and IgM Fc receptor, Fcamr−/− mice, were injected with PBS or LCWE. Absence of IgA signaling, or IgA production, resulted in a significant reduction in cardiovascular inflammation in the aortic root, coronary arteries, and the abdominal aorta (Figures 5A, 5B and Figures S4A–S4F). As expected, IgA deposition was not seen in the heart and abdominal aortic lesions of LCWE-injected Igha−/− mice (Figures 5C, 5D and Figure S3A). IgM concentrations were higher in their serum and intestinal mucus of LCWE-injected Igha−/− mice (Figures S4G and S4H), indicating that IgM does not participate in the development of LCWE-induced KD vasculitis. To specifically determine the pathogenic role of IgA and its vascular deposition during murine KD vasculitis, we sorted CD19+ IgA+ B cells from the PP of WT CD45.1+ mice and adoptively transferred them into CD45.2+ Igha−/− mice. Recipient mice were then injected with LCWE, and 2 weeks later we evaluated the presence of adoptively transferred CD19+ IgA+ B cells, serum and SI IgA concentrations, and KD vasculitis (Figure S5A). CD19+ IgA+ B cells were detected by flow cytometry in the PP, the AA LN and the mesenteric lymph nodes (MLN) of Igha−/− recipient mice (Figures S5B and S5C). Transfer of IgA+ B cells into Igha−/− mice resulted in significantly increased intestinal and serum IgA concentrations without impacting IgM and IgG concentrations (Figures S5D and S5E). Transfer of CD19+ IgA+ B cells significantly increased cardiovascular inflammation in the aortic root, coronary arteries, and abdominal aorta in LCWE-injected Igha−/− mice (Figures 5E–5H). Furthermore, adoptive transfer of CD19+ IgA+ B cells into Igha−/− mice also restored IgA deposition in the heart and abdominal aorta lesions (Figures 5I – 5L).
Figure 5. IgA promotes coronary vasculitis and abdominal aorta dilation.

(A) Heart vessels inflammation score and maximal abdominal diameter of WT and Fcamr−/− mice 1 wk post-LCWE injection (n=10 per group). (B) Heart vessels inflammation score and maximal abdominal diameter of WT and Igha−/− mice 1 wk post-LCWE injection (n=10–11 per group). (C, D) H&E and immunofluorescent staining for CD31 (green) and IgA (red) in heart sections (C) and abdominal aorta (D) of PBS or LCWE-injected WT mice. Scale bars: 200μm (C) and 50μm (D). (E, F) Representative H&E staining of heart sections (E) and heart vessels inflammation score (F) of LCWE-injected Igha−/− and LCWE-injected Igha−/− mice that received CD45.1+ CD19+ IgA+ B cells (n=9–10 per group). Scale bars: 500μm. (G, H) Representative pictures of abdominal area, H&E staining of abdominal aorta cross-section (G) and maximal abdominal aorta diameter (H) of Igha−/− and Igha−/− mice that received CD45.1+ CD19+ IgA+ B cells injected with LCWE (n=9–10 per group). Scale bars: 250μm. (I-L) Representative H&E sections and immunostaining of CD31 (green) and IgA (red) and IgA immunofluorescence quantification in heart (I, J) and abdominal aorta sections (K, L) of Igha−/− and Igha−/− mice that received CD45.1+ CD19+ IgA+ B cells and were injected with LCWE (n=4 per group). Scale bars: 100μm (I) and 50μm (K). Data are presented as mean ± s.e.m. **p<0.01, ***p<0.001 by two-tailed unpaired t test. Nuclei were stained with DAPI (blue) in all the immunofluorescent staining. Data pooled from 2 to 3 independent experiments. AU; arbitrary units. See also Figures S4, S5 and S6.
As a complementary approach, to restore IgA concentrations in Igha−/− mice, an enriched SI IgA fraction was prepared from the SI mucus of WT mice by discarding the bacterial pellet and depleting IgG from the remaining supernatant (Figure S6A and S6B). A SI control fraction lacking IgA was similarly prepared from the SI mucus of Igha−/− mice (Figure S6B). We found that WT enriched SI IgA fraction contained mostly free IgA, which was not bound to luminal bacteria (Figure S6C–S6E). The WT SI IgA enriched fraction and the Igha−/− SI control fraction were transferred by oral gavage into Igha−/− recipients (Figures S6F). We then injected recipient Igha−/− mice with LCWE, and examined the serum and intestinal IgA concentrations and KD vasculitis development 2 wks later. Oral gavage with the WT enriched SI IgA fraction, but not the Igha−/− SI control fraction, resulted in detectable IgA concentrations in the serum and SI of the recipient mice (Figure S6G) and exacerbated KD-associated cardiovascular lesions (Figures S6H, S6I). Reconstitution of with the WT enriched SI IgA fraction was also associated with IgA deposition in heart and abdominal aorta tissues (Figures S6J–S6L). These results indicate that IgA production, signaling and deposition are key events in the development of LCWE-induced KD vasculitis.
IVIG treatment blocks intestinal permeability and prevents the development of LCWE-induced KD vasculitis.
The mechanisms by which IVIG downregulates cardiovascular tissue inflammation and prevents coronary artery damages in acute KD are not completely understood. IVIG may act by blocking Fc receptors, decreasing complement-mediated tissue damage, neutralizing autoantibodies, or modulating cytokine production (Kazatchkine and Kaveri, 2001). In acute human KD, IVIG administration leads to decreased IL-1β production (Leung et al., 1989) and increased IL-1 receptor antagonist (IL-1Ra) production (Aukrust et al., 1994). To analyze the possible role of IVIG in modulating intestinal permeability, WT mice were treated with IVIG and injected with LCWE. IVIG treatment decreased intestinal permeability 24 h post LCWE injection, as assessed by FITC-dextran translocation across the intestinal epithelium (Figure 6A). IVIG treatment strongly reduced the cardiovascular lesions in LCWE-injected KD mice (Figures 6B and 6C). Abdominal aorta dilation and aneurysm development were also decreased in IVIG-treated LCWE-injected mice (Figures 6D–6F). Importantly, IVIG treatment was associated with reduced SI IgA concentrations after LCWE-injection (Figure 6G) and diminished IgA deposition in heart and abdominal aorta tissues (Figures 6H and 6I). Taken together, these results demonstrate that a possible therapeutic effect of IVIG during KD vasculitis is to prevent intestinal barrier dysfunction and, in turn, decrease the development of the cardiovascular lesions associated with KD vasculitis.
Figure 6. IVIG treatment blocks intestinal permeability and IgA tissue deposition in LCWE-injected mice.

(A) FITC-dextran intestinal permeability assay in WT mice 24 h after PBS (n=10), LCWE (n=12) and LCWE injection with IVIG treatment (n=10). Data normalized to PBS controls. (B, C) H&E staining of heart sections (B) and heart vessels inflammation score (C) of PBS, LCWE-injected and LCWE-injected IVIG-treated WT mice 1 wk post-LCWE injection (n=10 per group). Scale bars: 500μm. (D-F) Representative pictures of abdominal area (D), H&E staining of abdominal aorta cross-sections (E) and maximal abdominal aorta diameter (F) of the mouse groups in (B, C). Scale bars: 250μm. (G) SI IgA concentrations in PBS, LCWE-injected and LCWE-injected IVIG-treated WT mice 1 wk post-LCWE injection (n=5 per group). (H) H&E staining of heart coronary artery, abdominal aorta and immunostaining of CD31 (green) and IgA (red) in the heart and abdominal aorta sections of WT mice injected with PBS, LCWE and LCWE-injected IVIG-treated mice (n=4 per group). Scale bars: 25μm. (I) IgA immunofluorescence intensity quantification in the heart and abdominal aorta of WT mice injected with PBS, LCWE and LCWE-injected IVIG-treated mice. Data are presented as mean ± s.e.m., *p<0.05, ***p<0.001 and ****p<0.0001 by 1-way ANOVA with Bonferroni post-tests. Data in A, C and F are compiled from two independent experiments and data in G are representative of 2 independent experiments. Immunofluorescence images are representative of n=4 mice per group and nuclei were stained with DAPI (blue). CA: Coronary Artery; Ao: Aorta; AU: Arbitrary Units.
IL-1β signaling on IEC is required for KD murine vasculitis and IgA tissue deposition.
Proinflammatory cytokines, such as IFN-γ and IL-1β modulate intestinal barrier permeability by reducing TJ protein expression, altering their distribution, and rearranging the IECs actin cytoskeleton (Al-Sadi et al., 2009). Because IL-1β is known to be required for the development of LCWE-induced KD vasculitis (Lee et al., 2012; Wakita et al., 2016), we interrogated the role of intestinal IL-1β signaling in this effect. For this, we generated IEC-specific IL1 receptor (IL1R; I1r1) deficient mice by crossing mice expressing Cre recombinase under the control of the Vil1 promoter (Vil1Cre) with mice harboring a floxed Il1r1 allele (Il1r1fl/fl mice) (Figure 7A). The resulting mice, Vil1CreIl1r1Δ/Δ, had deletion of IL1R specifically in IECs (Figure 7B). Vil1CreIl1r1Δ/Δ and control littermate Il1r1fl/fl mice were injected with LCWE. Compared with LCWE-injected Il1r1fl/fl mice, intestinal permeability was decreased 24 h after LCWE injection in Vil1CreIl1r1Δ/Δ mice (Figure 7C). Furthermore, absence of IL-1β signaling in IECs also led to reduced heart inflammation (Figures 7D and 7E) and abdominal aorta dilation (Figures 7F and 7G). LCWE-injected Vil1CreIl1r1Δ/Δ mice had decreased IgA deposition in heart and abdominal aorta lesions (Figures 7H–7J). These results suggest that IL-1β signaling in IECs promotes intestinal barrier dysfunction in experimental KD vasculitis.
Figure 7. IL-1β signaling in intestinal epithelial cells is required for LCWE-induced KD vasculitis.

(A) Scheme for the generation of Vil1CreIl1r1Δ/Δ mice. (B) Immunofluorescence analysis showing absence of IL-1R1 expression (red) in SI sections of Vil1CreIl1r1Δ/Δ as compared with Il1r1fl/fl mice. (C) FITC-dextran intestinal permeability assay in PBS-injected Il1r1fl/fl, LCWE-injected Il1r1fl/fl and LCWE-injected Vil1CreIl1r1Δ/Δ 24 h post-injection (n=7–10 per group). Data normalized to PBS controls. (D, E) Representative H&E staining of heart sections (D) and heart vessels inflammation score (E) of LCWE-injected Il1r1fl/fl and Vil1CreIl1r1Δ/Δ (n=14 per group). Scale bars: 500μm. (F) Representative pictures of abdominal area and H&E staining of abdominal aorta cross-section from LCWE-injected Il1r1fl/fl and Vil1CreIl1r1Δ/Δ. Scale bars: 200μm. (G) Measurement of maximal abdominal diameter of LCWE-injected Il1rl1fl/fl and Vil1CreIl1r1Δ/Δ (n=12–14 per group). (H) Representative H&E sections and immunostaining of CD31 (green) and IgA (red) in the heart coronary artery of LCWE-injected Il1r1fl/fl (1; non-obstructed and 2; obstructed coronary) and Vil1CreIl1r1Δ/Δ. Scale bars: 25μm. (I) H&E abdominal aorta cross sections staining and immunostaining of CD31 (green) and IgA (red) in the abdominal aorta of LCWE-injected Il1r1fl/fl. Scale bars: 10μm. (J) IgA immunofluorescence quantification in the heart and abdominal aorta of LCWE-injected Il1rl1fl/fl and Vil1CreIl1r1Δ/Δ (n=5 per group). Data are presented as mean ± s.e.m. *p<0.05,**p<0.01, ***p<0.001 by 1-way ANOVA with Bonferroni post-test (C) or two-tailed unpaired t test (E, G and J) and pooled from 2 to 4 independent experiments.
Discussion.
Dysfunction of the intestinal barrier function, manifested by increased flux across TJ-dependent pores or through a TJ-independent pathway, is a risk factor for a variety of disorders (Turner, 2009). KD patients exhibit multiple signs of disrupted intestinal barrier, including increased serum concentrations of anti-Lipid A antibodies (Takeshita et al., 2002), sIgA (Ohshio et al., 1987), IgA anti-cardiolipin antibodies (Gupta et al., 2002), and increased numbers of IgA+ plasma cells at mucosal surfaces, coronary arteries, and kidneys (Rowley et al., 2000). However, data linking these observations to the development of the cardiovascular pathologies associated with KD have been lacking. Our results provide strong evidence that intestinal IgA production is increased during the acute phase of KD in human patients and in a murine model of KD vasculitis, and that intestinal epithelial barrier dysfunction in this model results in sIgA leakage and sIgA-C3 complex deposition into vascular tissues and glomeruli. Taken together, these data suggest that KD may be a form of IgA vasculitis.
Intestinal permeability can be altered by a range of factors, including viral infection, mucus barrier degradation and epithelial damage or change in the microbiome composition (Bischoff et al., 2014). KD patients presenting with intestinal symptoms are at higher risk of developing coronary aneurysms, and IVIG-resistance (Fabi et al., 2018). In KD mice, we found that intestinal permeability increased rapidly after LCWE injection, well before the development of coronary artery inflammation and abdominal aorta dilatation, suggesting this defective intestinal barrier is more likely a trigger than a result of vasculitis development.
Although the exact mechanisms are not completely understood, IVIG reduces the incidence of CAA in KD patients from 25% to approximately 4% (McCrindle et al., 2017). IVIG likely acts in part via its regulation of IL-1β; IVIG inhibits IL-1β production from in vitro stimulated macrophages and induces secretion of IL-1Ra (Iwata et al., 1987; Ruiz de Souza et al., 1995). Accordingly, IL-1β is elevated in the serum of acute KD patients (Leung et al., 1989) and IVIG treatment reduces IL-1β and upregulates IL-1Ra (Aukrust et al., 1994). Furthermore, an IL1B gene related signature is associated with the acute phase of KD and IVIG-resistance (Fury et al., 2010). Pro-inflammatory cytokines such as IL-1β increase intestinal barrier dysfunction by disrupting intestinal TJs (Al-Sadi et al., 2009). Intestinal epithelial-derived IL-1β is the primary trigger of intestinal barrier dysfunction in a murine model of mucositis (Kanarek et al., 2014). Furthermore, IL-1β plays a critical role in many autoinflammatory and chronic inflammatory diseases (Gabay et al., 2010), human KD (Leung et al., 1989) and LCWE-induced KD vasculitis (Lee et al., 2012; Wakita et al., 2016). Here, we found that IL-1 signaling in IECs increased intestinal permeability, as deleting IL-1R specifically in those cells not only prevented increased permeability but also reduced heart inflammation and abdominal aortic aneurysm development in LCWE-injected KD mice.
However, IL-1β production, which contributes to fever induction, is not sufficient for the development of KD vasculitis and coronary arteritis, which specifically requires a currently unidentified disease-inducing agent. Because KD occurs in geographical clusters and seasonal patterns similar to those observed for respiratory infections (McCrindle et al., 2017), it has been suggested that the disease is triggered in genetically predisposed children by an infectious agent entering the mucosal surfaces either through the lungs or the GI tract (Rowley, 2018). Increased numbers of IgA+ plasma cells at the proximal respiratory tract mucosal surfaces, coronary arteries, and kidney support a respiratory portal of entry (Rowley et al., 2000). Our results showing that LCWE-induced KD mice have increased intestinal permeability and that KD patients have elevated serum concentrations of calprotectin (Abe et al., 2005) also support this infectious agent hypothesis. Furthermore, using intestinal permeability inhibitors such as AT1001, which is currently in clinical trials to treat celiac disease (Paterson et al., 2007), we were able to abrogate the cardiovascular lesions at the aortic root, coronary arteries, and abdominal aorta, and block sIgA deposition in the LCWE-induced KD mice, highlighting the key role of intestinal epithelium disruption as an early pathogenic event.
The majority of intestinal IgA production is microbiota-reactive and polyreactive (Bunker et al., 2017). However, the role of the intestinal microbiome in modulating intestinal permeability during LCWE-induced KD vasculitis is unknown and will need to be further investigated. Human sIgA is known to bind self-antigens such as actin, myosin and tubulin (Quan et al., 1997), and compared with febrile controls, acute phase KD patients have increased serum concentration of IgA-containing circulating immune complexes and sIgA (Ohshio et al., 1987). We found that in LCWE-injected mice, C3 and IgA form an immune complex that deposits in the aortic root, coronary artery, and abdominal aorta. Similar findings have been described in an independent KD vasculitis model, in which C3d deposits together with mannose binding lectin (MBL) in aortic root lesions (Nakamura et al., 2014). Several studies suggest that the anti-inflammatory effect of high dose IVIG in KD and other autoimmune diseases might be explained, at least partially, by attenuation of complement induced amplification (Lutz and Späth, 2005). While IgA is a weak activator of classical complement pathways, human polymeric serum IgA can bind to MBL, which activates the alternative lectin complement pathway (Roos et al., 2001). Furthermore, a polymorphism in MBL2 is an age-related risk factor for coronary artery lesions in KD patients (Biezeveld et al., 2003; Sato et al., 2009) and complement breakdown products C4d and C3d are upregulated in KD patients (Kohsaka et al., 1994). The finding that C3 deposits with IgA in LCWE-injected KD mice may indicate that these immune complexes contribute to inflammation-driven tissue injury and vessel damage.
We also observed C3 and IgA deposits in the kidney glomeruli of LCWE-induced KD vasculitis mice. Several reports describe the development of acute kidney injury involving immune complex deposition of IgA and C3 and immune complex-mediated glomerulonephritis with KD (Watanabe, 2018). Autopsy findings revealed that 73% of KD patients had renal artery involvement (Asachi, 1989). These findings and our observations are reminiscent of two other human vasculitis diseases: IgA vasculitis and IgA nephropathy (IgAN), which are both characterized by IgA immune-complexes with C3 deposition in the kidney. The commonality of kidney involvement points to the gut-kidney axis, which has been highlighted by recent studies identifying genetic, microbial and dietary factors which affect intestinal mucosal immunity and promote both IgAN (Coppo, 2018a) and IgA vasculitis in pediatric and adult patients (Moja et al., 1998). There are additional indications of common pathophysiology of IgA vasculitis, IgAN and KD vasculitis. IgAN patients exhibit signs of subclinical inflammation associated with the activation of inflammatory cells in the small intestinal mucosa, increased intestinal permeability, and in a subpopulation, sIgA colocalizes with complement in the glomerular mesangium (Floege and Feehally, 2016; Oortwijn et al., 2006). In addition, a polymorphism in the promoter of the LPS receptor CD14 (CD14/159) associated with coronary artery abnormalities in KD patients (Nishimura et al., 2003) has been linked to progression of IgAN patients to more severe renal disease (Yoon et al., 2003). Furthermore, children with KD (Stagi et al., 2006) and those with IgAN (Coppo, 2018b) have increased prevalence of coeliac disease. A recent case report of a child with IgA vasculitis who developed dilated coronary arteries and responded to IVIG and infliximab therapy further supports an overlap in IgA vasculitis and KD pathogenesis (Bloom et al., 2018). Finally, similar to the important role that IL-1β plays in KD vasculitis (Burns et al., 2017), this master cytokine has been implicated in the pathogenesis and renal complications of both IgA vasculitis (Besbas et al., 1997; Boyer et al., 2011) and IgAN (Atkins, 1995; Chun et al., 2016). Several recent studies suggest that targeting intestinal mucosal immunity is likely to be beneficial for patients with IgAN (Coppo, 2018a), and our findings suggest that this approach may be broadly applicable to IgA-related diseases, including KD.
Overall, our findings provide a mechanistic link between the gastrointestinal symptoms observed in acute KD vasculitis and intestinal barrier dysfunction and demonstrate that targeting intestinal permeability in a murine model of KD vasculitis improves intestinal barrier dysfunction and strongly decreases cardiovascular tissue inflammation. We propose the transformative concept that KD may be a form of IgA vasculitis, characterized by increased gut permeability with leakage of sIgA, and by IgA-C3 immune complex deposition in cardiovascular lesions and the kidney, which promote further vascular inflammation. Therefore, therapies aiming to decrease intestinal permeability, such as TJ-targeted therapeutic interventions, intestinal permeability inhibitors, IVIG, or drugs targeting cytokines known to regulate and open TJs, such as TNF-a or IL-1, should be evaluated as additional treatments for KD vasculitis. Recently, IgG endopeptidase treatment has been successfully used to block donor pathogenic IgG and complement activation in sensitized renal transplant patients (Jordan et al., 2017). Our results suggest that comparable therapeutic strategies targeting IgA, such as IgA1 protease that prevents mesangial IgA deposits in humanized murine model of IgAN (Lamm et al., 2008), could also be developed to treat IVIG-resistant KD patients. More importantly, our data identify systemic IL-1β production as an upstream disruptor of intestinal barrier function and mediator of IgA and C3 deposition for vasculitis. Therefore, diseases such as IgA vasculitis and IgAN may also benefit from the anti-IL-1β treatment approaches that proved to be beneficial in this immune vasculitis mouse model, and which led to the current Phase II clinical trials of Anakinra in IVIG-unresponsive KD vasculitis patients.
STAR METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Moshe Arditi (Moshe.Arditi@cshs.org).
EXPERIMENTAL MODEL AND SUBJECTS DETAILS
Experimental Animals
WT congenic mice CD45.1+ (B6.SJL-Ptprca Pepcb/BoyJ ) and C57BL/6J CD45.2+ were obtained from the Jackson Laboratory. Igha−/− mice were a gift from Dr. Masayuki Fukata (Cedars-Sinai Medical Center). Fcamr−/− (Shibuya et al., 2000) experiments were performed at Tsubuka University Japan in collaboration with Dr. Akira Shibuya. Experimental Igha−/− mice were obtained from homozygous breeding and age-matched WT C57BL/6J from our internal colony were used as controls. The Il1r1fl/fl mice on the C57BL/6 background were produced by blastocyst injection of conditional-ready embryonic stem cells clone Il1r1tm1a(EUCOMM)Hmgu obtained from the European conditional mouse mutagenesis project (EUCOMM)(Skarnes et al., 2011). The Il1r1 allele which contains loxP sites flanking exons 2 and a FRT-lacZ-loxP-neo-FRT cassette introduced between exons 1 and 2. For initial screening of Il1r1fl/lf we used the following primers; Il1r1 Forward: GAGGTATGGACGGGGAGAGGAAGC and loxP Reverse: TGAACTGATGGCGAGCTCAGACC. Chimeric mice were then crossed with C57BL/6 and the offspring with germ line transmission of the recombined allele were crossed with mice expressing a FLP1 recombinase (B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/RainJ – Jackson Laboratory) to excise the FRT flanked lacZ-neo cassette to obtain Il1rfl/fl mice. Il1r1fl/fl mice were subsequently bred with B6.Cg-Tg(Vil1-cre)997Gum/J (Jackson Laboratory) to specifically delete IL1R in intestinal epithelial cells and generate Vil1CreIl1r1Δ/Δ. Age-matched Il1r1fl/fl mice littermate controls were used in the study of Vil1CreIl1r1Δ/Δ mice. Only 5 week old male animals were used in this study as it has been shown that LCWE-injection induces stronger and more consistent coronary vasculitis lesions and abdominal aorta aneurysms in male compared with female mice (Wakita et al., 2016). All mutant mouse strains were backcrossed 8–10 generations on C57BL/6 background. Mice were housed under specific pathogen-free conditions and used according to the guidelines of the Cedars Sinai Medical Center institutional committee.
Human Study population
Human sera from febrile control patients and KD subjects were previously collected and biobanked at 2 different sites (Northwestern University; Chicago and Nippon Medical School; Tokyo) under a protocol approved by the respective institutional boards. The study population was composed of 2 groups. Group (1) contains 25 febrile controls and group (2) contains 34 acute phase KD patients. The demographics and clinical characteristics of those samples are shown in Table S1. The febrile control subjects included 9 boys and 16 girls, ranging in age from 1 month to 9 years. The febrile controls were age-matched and diagnosed with joint infection, lower or upper respiratory tract infection or viral illness. The KD patient group is composed of 23 boys and 11 girls, ranging from 4 months to 15 years. Group 2 also contained sera from 8 KD patients with coronary artery lesions (CAL), defined by a Z score >2.5 in the left anterior descending coronary artery or the right coronary artery (McCrindle et al., 2017). The KD sera used was collected during KD acute phase pre-IVIG treatment.
LCWE-induced KD vasculitis murine model
Lactobacillus casei (ATCC 11578) cell wall extract was prepared as previously published (Lee et al., 2012). 5 week old male mice were injected i.p. with either 500μg of LCWE or PBS. One- to 2-weeks post-injection, depending of LCWE batch efficiency, mice were euthanized, hearts and kidneys were removed and embedded in Optimal Cutting Temperature (OCT) compound for histological examination. Abdominal aorta were also collected, photographed to determine the maximal abdominal aorta diameter in Image J, then embedded on OCT.
METHOD DETAILS
Tissue Fixation, Staining and histopathology
Serial cryosections (7μm) of heart tissue and abdominal aorta were H&E stained and histopathological scoring of coronary arteritis, aortic root vasculitis, and myocarditis were performed by a pathologist blinded to the experimental set up as previously described (Lee et al., 2012; 2015; Wakita et al., 2016). Kidney, small and large intestines were collected, fixed in formaldehyde (4% vol/vol) and embedded in paraffin blocks. Periodic acid-Schiff (PAS) staining was performed on kidney sections (4μm thickness) to semi-quantitatively score mesangial cell proliferation and expansion with the previously published scoring system (Suzuki et al., 2005): score 0, 0%; score 1, 1 to 24%; score 2, 25 to 49%; and score 3, Š50% of all glomeruli. Cryosections of kidney tissues (4μm thickness) were also obtained to perform immunofluorescence staining for IgA and complement C3. Jejunum and colon tissues were fixed in Carnoy’s buffer and embedded in paraffin. Jejunum and colon sections (5μm thickness) were used for intestinal histological analysis by H&E staining and PV-1, OCLN, ZO-1, pIgR and IgA expression quantification by immunofluorescent staining. Goblet cell numbers were assessed by counting Alcian blue (American MasterTech) positive cell number per crypt unit, with a minimum of 20 crypt units assessed for each tissue samples. All images were acquired either with a Biorevo BZ-9000 or BZ-X710 (Keyence), and were further analyzed with ImageJ software.
Electron microscopy.
Kidneys were collected from PBS and LCWE-injected mice, the capsule was removed, and the tissues were sliced into small pieces (approximately 2 mm3). Kidney tissues were then fixed with 3% glutaraldehyde in 0.1 M cacodylate buffer at 4°C for 24 hrs. As previously described (Eriguchi et al., 2018), ultrathin sections (at 70 nm thickness) of plastic-embedded tissue stained with uranyl acetate and lead citrate were examined in a JEOL 100CX transmission electron microscope (JEOL Ltd., Tokyo, Japan).
Antibodies and Immunofluorescence.
OCT frozen 7μm tissue sections from heart, abdominal aorta and kidneys were fixed in acetone, and after washing with PBS 0.05% Tween, were stained overnight with the following antibodies: IgA (A90–103P; Bethyl Laboratories), CD31 (390; eBioscience or abcam ab28364), pIgR (AF2800; R&D systems) and C3 (11H9; Abcam). Jejunum and colon tissues were fixed in 10% formalin, and processed for paraffin sectioning. Five μm sections were deparaffinized in xylene and hydrated by serial immersion in ethanol and PBS. Sections were stained overnight with the following antibodies: ZO1 (40–2200; Thermo Fisher Scientific), OCLN (40–4700; Thermo Fisher Scientific), PV-1 (MECA-2; BD Biosciences), CD19 (6OMP31, Thermo Fisher Scientific), IgA (A90–103P; Bethyl Laboratories), IL-1RI (RM0013–11B7; Novus Biological) and MUC2 (Gene Tex). Isotype controls were used as negative controls and sections were incubated appropriately with one of the following fluorophore-conjugated secondary antibodies: Donkey anti-goat Alexa Fluor 647 (Thermo Fisher Scientific, A-21447), Donkey anti-goat Alexa Fluor 594 (Thermo Fisher Scientific, A-11058), Donkey anti-rat Alexa Fluor 555 (Abcam, ab150154), Donkey anti-rat Alexa Fluor 488 (Thermo Fisher, A-21208) and Donkey anti-Rabbit Alexa Fluor 488 (Abcam, ab150065). Before imaging, the nuclei were counterstained with ProLong Gold Antifade Reagent containing DAPI (Invitrogen). Images were obtained using a Biorevo BZ-9000 (Keyence) fluorescent microscope and were further analyzed with ImageJ software.
In vivo Intestinal Permeability Assay.
To asses intestinal permeability in vivo, we performed a FITC-dextran intestinal permeability. At 16 hours post-PBS or LCWE injection, mice were fasted for 4 hours. At the end of the fasting, mice received FITC-dextran (4 kDA; FD4; 2mg/kg of body weight, Sigma Aldrich) by oral gavage. Four hours later, serum were collected and read for fluorescence intensity at 521 nm using a SpectraMax M2 spectrometer (Molecular Device).
Modulation of Intestinal Permeability.
To block intestinal permeability, mice were treated with either the Zonulin antagonist AT-1001 (500μg i.p.; daily), the Myosin Light Chain Kinase inhibitor, ML-7 (40μg i.p. twice a day, Sigma) (Al-Sadi et al., 2012) or Human IVIG (2.5kg/kg i.p. 1 day before LCWE injection; Gamunex-C; Grifols)(Myones et al., 1995). Mice were injected i.p. with LCWE, intestinal permeability and KD vasculitis were assessed as described above at 24 h and 1 wk post-LCWE injection respectively.
Antibodies and Flow cytometry.
Cell suspensions were obtained from the spleen, the PP, the MLN and the AA LN (lumbar and renal lymph nodes). Jejunum and colon sections from PBS and LCWE-injected mice were harvested and dissociated into single-cell suspensions with a gentleMACS™ Octo Dissociator (Miltenyi Biotec) and a mouse Lamina Propria Dissociation kit (Miltenyi Biotec) following the complete protocol from the manufacturer. The following antibodies against the respective murine antigens were used: IgA (mA-6E1,Thermo Fisher Scientific), CD19 (eBio1D3, Thermo Fisher Scientific), CD4 (RM4–5, Tonbo Biosciences), CD3 (145–2C11, BioLegend and Tonbo Biosciences), CD45.1 (A20, Thermo Fisher Scientific), CD45.2 (104, BioLegend), CD95 (SA367H8, BioLegend), GL-7 (GL-7, BioLegend). Dead cells were routinely excluded based on the staining of Fixable Viability dye (FVD) eFluor 506 (Thermo Fisher Scientific). Cell numbers were calculated by flow cytometry with the CountBright Absolute Counting Beads (Thermo Fisher Scientific). Stained cells were analyzed on a LSRII (BD Biosciences) and the data were processed using Flowjo (Tree Star Inc.).
Intestinal IgA purification.
Small intestines were removed from naïve WT and Igha−/− mice, cut open longitudinally and mucus was scrapped out and suspended in PBS. Luminal content was filtered and spun twice at 8000g for 5 minutes, the bacterial pellets discarded, and the supernatant collected was further processed on a Nab Protein A/G Spin kit (Thermo Fisher Scientific) according to the manufacturer’s instruction. The concentration of IgA in the eluted fraction, depleted of IgG, was then measured by ELISA (Bethyl). Igha−/− recipient mice received by oral gavage 100μg of purified IgA the day before and day 3 after LCWE i.p. injection.
Detection of Fecal and SI mucus Bacteria by Flow Cytometry.
50 mg of fecal pellet and SI scraped mucus from WT and Igha−/− mice were homogenized in 1ml of sterile cold PBS and centrifuged at 40g for 10 minutes at 4°C to remove large particles. Supernatant was collected, filtered through a 70μm strainer and centrifuged at 8000 g for 5 minutes to pellet the bacteria. The supernatant from the SI mucus preparation was kept for bacterial flow staining and the bacterial fecal pellets and bacterial SI mucus pellets were washed twice with 1ml of sterile PBS and incubated on ice for 15 minutes with blocking buffer (1% FCS in PBS). Samples were further centrifuged at 8000g for 5 minutes and subsequently stained with 5μM SYTO-BC (Thermo Fisher Scientific) along with anti-mouse IgA (clone mA-6E1, Thermo Fisher Scientific). Samples were then washed 3 times with 1 ml of PBS and fluorescent Countbright Absolute Counting beads (Thermo Fisher Scientific) were added to the samples to calculate absolute cell numbers. Flow cytometric analysis was performed on a BD LSR Fortessa cell analyzer (BD Biosciences), and data processed using FlowJo (Tree Star Inc.).
Adoptive transfer of CD45.1+ IgA+ B cells.
PP cells of CD45.1+ WT congenic donor mice were isolated, stained for CD3 (145–2C11, BioLegend), CD19 (eBio1D3, Thermo Fisher Scientific) and IgA (mA-6E1, Thermo Fisher Scientific). CD3− CD19+ IgA+ B cells were isolated by cell-sorting on a BD FACSAria III (BD Biosciences). After isolation, 0.2–0.3 x105 viable CD3− CD19+ IgA+ B cells were immediately transferred i.p. into CD45.2+ Igha−/− recipient mice as previously described (Wang et al., 2015). One day after, non-reconstituted and reconstituted Igha−/− mice were injected with LCWE. KD vasculitis and heart pathology was assessed as previously described (Noval Rivas et al., 2017).
ELISAs.
To quantify luminal Igs, small intestines and colon were cut open longitudinally and mucus and feces were scrapped out and suspended into 1ml of PBS. Luminal content were spun at 8000g for 5 minutes and the supernatant collected and stored at −80°C. LPS (MyBiosource), sIgA (Cloud-Clone Corp.), IgA (Bethyl), IgM (Bethyl), IgG (Bethyl) and Calprotectin (S100A8/S100A9 Heterodimer Duoset; R&D systems) concentrations were measured by ELISAs in the serum or luminal content according to the manufacturer instructions. For luminal Igs measurement, concentration was normalized to the mucus weight. Murine serum Zonulin concentrations were measured by using a mouse Zonulin ELISA kit (MyBioSource).
Quantitative Real-Time PCR analysis.
Jejunum and colon sections were flushed with PBS, snap-frozen in liquid nitrogen and homogenized in Trizol (Thermo Fisher Scientific). RNA was extracted from the homogenized tissues by using the RNeasy kit (Qiagen). Quantitative PCR analysis were performed using the following primers (bellow) and SYBR Green Premix Ex Taq (Takara) on a CFX96 Real Time system (Biorad). β-actin was used as an endogenous control and WT mouse RNA as the exogenous control. Samples were run in triplicate and the relative expression was calculated using the ΔΔCt method. qPCR was performed for β-actin, Tjp1, Tjp2, Ocln, Cldn2, Cldn3, Cldn4, Cldn7, Cldn12, Cldn15 and Muc2 with primers listed in the Key Resource Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD3ε – APC/Cy7 (145–2C11) | BioLegend | Cat# 100222, RRID:AB_2242784 |
| Anti-mouse CD3ε – FITC (145–2C11) | Tonbo Biosciences | Cat# 35–0031, RRID:AB_2621659 |
| Anti-mouse CD3ε – PerCP/Cy5.5 (145–2C11) | BioLegend | Cat# 100330, RRID:AB_1877170 |
| Anti-mouse CD4 – PerCP Cy5.5 (RM4–5) | Tonbo Biosciences | Cat # 65–0042, RRID:AB_2621876 |
| Anti-mouse CD19 – eFluor 450 (eBio1D3) | Thermo Fisher Scientific | Cat# 48–0193-82, RRID:AB_2734905 |
| Anti-mouse CD45.1 – APC/Cy7 (A20) | Tonbo Biosciences | Cat# 25–0453, RRID:AB_2621629 |
| Anti-mouse CD45.2 – FITC (104) | Tonbo Biosciences | Cat# 35–0454, RRID:AB_2621692 |
| Anti-mouse CD95 (Fas) – FITC (SA367H8) | BioLegend | Cat# 152605, RRID:AB_2632900 |
| Anti-mouse/human GL7 – PerCP/Cy5.5 (GL-7) | Biolegend | Cat# 144609, RRID:AB_2562978 |
| Anti-mouse IgA – PE (mA-6E1) | Thermo Fisher Scientific | Cat# 12–4204-82, RRID:AB_465917 |
| Anti-MUC2 antibody (C3) | GeneTex | Cat# GTX100664 |
| OCLN Polyclonal Antibody | Thermo Fisher Scientific | Cat # 40–4700 RRID:AB_2533468 |
| ZO-1 Polyclonal Antibody | Thermo Fisher Scientific | Cat# 402200 RRID:AB_2533456 |
| Anti-CD31 antibody | Abcam | Cat# ab28364 RRID:AB_726362 |
| Anti-mouse CD31 (PECAM-1; 390) | Thermo Fisher Scientific | Cat# 16–0311-81 RRID:AB_468931 |
| Mouse IgA Antibody | Bethyl | Cat# A90–103P, RRID:AB_67140 |
| Panendothelial Cell Antigen Antibody (PV1; MECA-32) | Novus | Cat# NB100–77668 RRID:AB_2276108 |
| Mouse pIgR Antibody | R&D Systems | Cat# AF2800, RRID:AB_2283871 |
| C3 antibody (11H9) | Abcam | Cat# 11862 RRID:AB_2066623 |
| CD19 Monoclonal Antibody (6OMP31) | Thermo Fisher Scientific | Cat# 11–0194-80 RRID:AB_2637170 |
| Rat Anti-Murine IL1R-I Monoclonal, Unconjugated (m0013–11b7) | Novus | Cat# NB110–85471 RRID:AB_1201285 |
| Donkey Anti-Goat IgG (H+L) Antibody – Alexa Fluor 647 | Molecular Probes | Cat# A-21447 RRID:AB_141844 |
| Donkey Anti-Goat IgG (H+L) Antibody – Alexa Fluor 594 | Molecular Probes | Cat# A-11058 RRID:AB_142540 |
| Donkey Anti-Rat IgG (H+L) – Alexa Fluor 488 | Molecular Probes | Cat# A-21208 RRID:AB_141709 |
| Donkey Anti-Rabbit IgG (H+L) – Alexa Fluor 488 | Abcam | Cat# ab150065 |
| Donkey Anti-Rat IgG (H+L) – Alexa Fluor 555 | Abcam | Cat# ab150154 |
| Bacterial strains | ||
| Lactobacillus casei subsp. casei (OrlaJensen) Hansen and Lessel | ATCC | ATCC #11578 |
| Biological Samples | ||
| Healthy and KD patient sera | This Study | N/A |
| Murine Abdominal Aorta | This Study | N/A |
| Murine Blood | This Study | N/A |
| Murine Heart | This Study | N/A |
| Murine Small Intestine | This Study | N/A |
| Murine Colon | This Study | N/A |
| Murine Mesenteric Lymph Nodes | This Study | N/A |
| Murine Abdominal Aorta Lymph Nodes | This Study | N/A |
| Murine Kidneys | This Study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AT1001 | Dr. Alessio Fasano | N/A |
| ML-7 | Sigma-Aldrich | Cat# I2764 |
| Fixable Viability Dye (FVD) – eFluor 506 | Thermo Fisher Scientific | Cat# 65–0866-14 |
| CountBright Absolute Counting Beads, for flow cytometry | Thermo Fisher Scientific | Cat# 675269 |
| Fluorescein isothiocyanate-dextran FD4 | Sigma-Aldrich | Cat# FD4–250MG |
| Sodium dodecyl sulfate solution | Sigma-Aldrich | Cat# 05030 |
| Eosin Y solution | Sigma-Aldrich | Cat# HT110132 |
| Hematoxylin Solution, Harris Modified | Sigma-Aldrich | Cat# HHS32 |
| MRS Broth | Millipore Sigma | Cat# 110661 |
| ProLong Gold Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# P36931 |
| Gamunex® -C | Grifols | N/A |
| SYTO™ BC Green Fluorescent Nucleic Acid Stain | Thermo Fisher Scientific | Cat# S34855 |
| TRIzol™ Reagent | Thermo Fisher Scientific | Cat# 15596018 |
| RNAlater RNA Stabilization Reagent | Qiagen | Cat# 76104 |
| Critical Commercial Assays | ||
| Mouse Secretory Immunoglobulin A (sIgA) ELISA Kit, Cloud-Clone. | ARP American Research Products | Cat# SEA641Mu |
| Human Secretory Immunoglobulin A (sIgA) ELISA Kit, Cloud-Clone. | ARP American Research Products | Cat# SEA641Hu |
| Mouse IgA ELISA Quantification Set | Bethyl | Cat# E90–103 |
| Mouse IgG ELISA Quantification Set | Bethyl | Cat# E90–131 |
| Mouse IgM ELISA Quantification Set | Bethyl | Cat# E90–101 |
| Mouse Zonulin ELISA Kit | MyBioSource | Cat# MBS748504 |
| Mouse Endotoxin ELISA Kit | MyBioSource | Cat# MBS722939 |
| Human Zonulin Serum ELISA | Alpco | Cat# 30-ZONSHU-E01 |
| Mouse S100A8/S100A9 Heterodimer DuoSet ELISA | R&D systems | Cat# DY8596–05 |
| Human S100A8/S100A9 Heterodimer Quantikine ELISA Kit | R&D systems | Cat# DS8900 |
| Lamina Propria Dissociation Kit, mouse | Miltenyi Biotec | Cat# 130–097-410 |
| Alcian Blue -P.A.S Stain Kit | American MasterTech | Cat# KTAPA |
| Periodic Acid Schiff (PAS) Stain kit | Abcam | Cat# ab150680 |
| SDS-out SDS precipitation Kit | Thermo Fisher Scientific | Cat# 20308 |
| NAb™ Protein A/G Spin Kit, 1 mL | Thermo Fisher Scientific | Cat# 89980 |
| RNeasy Midi Kit | Qiagen | Cat# 75144 |
| TB Green Premix Ex Taq II (Tli RNase H Plus) | Takara Bio | Cat# RR820A |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | The Jackson Laboratory | Stock 000664 |
| B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | Stock 002014 |
| B6.Cg-Tg(Vil1-cre)997Gum/J | The Jackson Laboratory | Stock 004586 |
| B6.129S4-Gt(ROSA)26Sortm1(FLP1)Dym/RainJ | The Jackson Laboratory | Stock 009086 |
| Il1r1fl/fl | Cedars Sinai Medical Center, Los Angeles, CA | Dr. Moshe Arditi |
| Igha−/− | Cedars Sinai Medical Center, Los Angeles, CA | Dr. Masayuki Fukata |
| Fcα/μR−/− | University of Tsubuka, Ibaraki, Japan | Dr. Akira Shibuya |
| Oligonucleotides | ||
|
Il1r1fwd:GAGGTATGGACGGGGAGAGGAAGC LoxP rev: TGAACTGATGGCGAGCTCAGACC |
This Study | N/A |
|
Actb fwd: CTTCTTTGCAGCTCCTTCGTT Actb rev: TTCTGACCCATTCCCACCA |
This Study | N/A |
|
Tjp1 fwd: GCGCGGAGAGAGACAAGATG Tjp1 rev: CTGTGAAGCGTCACTGTGTG |
This Study | N/A |
|
Tjp2 fwd: GCCTACGAGAAGGTTCTGCT Tjp2 rev: AGATCCGGCATCTTTGGGTT |
This Study | N/A |
|
Ocln fwd: GTGAGCACCTTGGGATTCCG Ocln rev: TTCAAAAGGCCTCACGGACA |
This Study | N/A |
|
Cldn2 fwd: CTCTTCGAAAGGACGGCTCC Cldn2 rev: CAGTGTCTCTGGCAAGCTGA |
This Study | N/A |
|
Cldn3 fwd: GACCGTACCGTCACCACTAC Cldn3 rev: CAGCCTAGCAAGCAGACTGT |
This Study | N/A |
|
Cldn4 fwd: ACACGTTACTCCAGCGCTAC Cldn4 rev: CTCTCAATGGCCCCTCAGTC |
This Study | N/A |
|
Cldn7 fwd: AATGTACGACTCGGTGCTCG Cldn7 rev: GTGTGCACTTCATGCCCATC |
This Study | N/A |
|
Cldn8 fwd: GCAGTGCAAGGTCTACGACT Cldn8 rev: CATTCCGAGGATGGCTGTCA |
This Study | N/A |
|
Cldn12 fwd: CAAGGTATTCCCGAGCGGAG Cldn12 rev: GCACATCTGCACACAAAGCA |
This Study | N/A |
|
Cldn15 fwd: CGTGGGCAACATGGATCTCT Cldn15 rev: CCACGAGATAGCCACCATCC |
This Study | N/A |
|
MUC2 fwd: ATGCCCACCTCCTCAAAGAC MUC2 rev: GTAGTTTCCGTTGGAACAGTGAA |
This Study | N/A |
| Recombinant DNA | ||
| N/A | ||
| Software and Algorithms | ||
| FlowJo 10 | FlowJo LLC | https://www.flowjo.com/ |
| GraphPad Prism 7 | GraphPad Software, Inc. | https://www.graphpad.com/ |
| Image J | Image J | https://imagej.nih.gov/ij/ |
| Keyence BZ-analyzer | Keyence | https://www.keyence.com |
| Gimp 2.8.10 | GIMP | https://www.gimp.org |
| Other | ||
| 75 mm Hematocrit Tubes | Fisher Scientific | Cat # 21–176-6 |
| Fisherbrand™ Superfrost™ Plus Microscope Slides, Superfrost Plus | Fisher Scientific | Cat# 12–550-15 |
| Tissue-Tek® O.C.T. Compound, Sakura ® Finetek | VWR | Cat# 25608–930 |
| Parablast® embedding agent for histology | VWR | Cat# 15159–409 |
ELISA on KD and febrile childhood control sera.
Human sera Zonulin (Alpco), sIgA (Cloud-Clone Corp.) and Calprotectin (R&D systems) concentrations were measured by ELISAs according to the manufacturer’s instructions. Those measurements were only performed when enough volume of human serum sample was available to perform the assay.
Statistical Analysis.
Statistical analyses of data are described in figure legends. Statistics were done with Prism software (GraphPad). A p-value of p<0.05 was considered statistically significant. *: p<0.05, **: p<0.01, ***: p<0.001 and ****p<0.0001. Results are reported as mean ± SEM.
Supplementary Material
Highlights.
IL-1β-driven increased intestinal permeability is observed in murine KD model
Blocking intestinal permeability or IgA production decreases murine KD vasculitis
IgA and IgA-C3 are observed in cardiovascular lesions of murine KD model
KD vasculitis may be a form of IgA vasculitis involving a gut-vascular axis
Acknowledgements.
We thank Ganghua Huang, Wenxuan Zhang, Malcolm Lane and Debbie Moreira for their technical support and the lab members for their helpful discussions. We thank the EUCOMM consortium for providing the ES cells and the Cedars-Sinai Genomic core for generating the IL1r1fl/fl mice. We thank Dr. Fukata for giving us the Igha−/− mice, Dr. Rowley, Dr. Shulman and Dr. Fukazawa for kindly sharing human serum samples. We would like to thank Dr. Waters at CSMC for the histological analysis of the murine intestinal sections and Daniel N. Leal, BS, CEMT, for assistance in electron microscopy study. Research was supported by grants from the NIH R01 AI072726 to M.AR01 HL139766 and AHA award 17SDG33671141 to M.N.R. T.T.C was supported by the CAPES - PDSE 88881.133324/2016-01 grant. M.Y. is supported by NCATS UCLA CTSI KL2 grant (KL2TR001882) and Cedars-Sinai CTSI Clinical Scholar Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: A.F. is co-founder and stock holder of Alba Therapeutics, a company licensing patents for the therapeutic use of AT1001. All other authors declare no competing interests.
References
- Abe J, Jibiki T, Noma S, Nakajima T, Saito H, and Terai M (2005). Gene Expression Profiling of the Effect of High-Dose Intravenous Ig in Patients with Kawasaki Disease. J. Immunol 174, 5837–5845. [DOI] [PubMed] [Google Scholar]
- Al-Sadi R, Boivin M, and Ma T (2009). Mechanism of cytokine modulation of epithelial tight junction barrier. Front Biosci 14, 2765–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sadi R, Guo S, Dokladny K, Smith MA, Ye D, Kaza A, Watterson DM, and Ma TY (2012). Mechanism of interleukin-1β induced-increase in mouse intestinal permeability in vivo. J. Interferon Cytokine Res 32, 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asachi S (1989). Pathomorphological Study of Renal Lesions in Kawasaki Disease. The Journal of Japanese College of Angiology 29, 453–460. [Google Scholar]
- Atkins RC (1995). Interleukin-1 in crescentic glomerulonephritis. Kidney Int 48, 576–586. [DOI] [PubMed] [Google Scholar]
- Aukrust P, Frøland SS, Liabakk NB, Müller F, Nordøy I, Haug C, and Espevik T (1994). Release of cytokines, soluble cytokine receptors, and interleukin-1 receptor antagonist after intravenous immunoglobulin administration in vivo. Blood 84, 2136–2143. [PubMed] [Google Scholar]
- Besbas N, Saatci U, Ruacan S, Ozen S, Sungur A, Bakkaloglu A, and Elnahas AM (1997). The role of cytokines in Henoch Schonlein purpura. Scand. J. Rheumatol 26, 456–460. [DOI] [PubMed] [Google Scholar]
- Biezeveld MH, Kuipers IM, Geissler J, Lam J, Ottenkamp JJ, Hack CE, and Kuijpers TW (2003). Association of mannose-binding lectin genotype with cardiovascular abnormalities in Kawasaki disease. The Lancet 361, 1268–1270. [DOI] [PubMed] [Google Scholar]
- Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A, and Wells JM (2014). Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol 14, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JL, Darst JR, Prok L, and Soep JB (2018). A case of Henoch-Schonlein Purpura with dilated coronary arteries. Pediatric Rheumatology 16, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer EM, Turman M, and O’Neil KM (2011). Partial response to anakinra in life-threatening Henoch-Schönlein purpura: case report. Pediatric Rheumatology 9, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun JG, Haga HJ, Boe E, Kallay I, Lekven C, Berntzen HB, and Fagerhol MK (1992). Calprotectin in patients with rheumatoid arthritis: relation to clinical and laboratory variables of disease activity. J. Rheumatol 19, 859–862. [PubMed] [Google Scholar]
- Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, Meisel M, Jabri B, Antonopoulos DA, Wilson PC, and Bendelac A (2017). Natural polyreactive IgA antibodies coat the intestinal microbiota. Science 358, eaan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JC, Kone-Paut I, Kuijpers T, Shimizu C, Tremoulet A, and Arditi M (2017). Review: Found in Translation: International Initiatives Pursuing Interleukin-1 Blockade for Treatment of Acute Kawasaki Disease. Arthritis Rheumatol 69, 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Chung H, Wang X, Barry R, Taheri ZM, Platnich JM, Ahmed SB, Trpkov K, Hemmelgarn B, Benediktsson H, et al. (2016). NLRP3 Localizes to the Tubular Epithelium in Human Kidney and Correlates With Outcome in IgA Nephropathy. Sci. Rep 6, 24667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppo R (2018a). The gut-kidney axis in IgA nephropathy: role of microbiota and diet on genetic predisposition. Pediatr Nephrol 33, 53–61. [DOI] [PubMed] [Google Scholar]
- Coppo R (2018b). The Gut-Renal Connection in IgA Nephropathy. Semin. Nephrol 38, 504–512. [DOI] [PubMed] [Google Scholar]
- Eriguchi M, Lin M, Yamashita M, Zhao TV, Khan Z, Bernstein EA, Gurley SB, Gonzalez-Villalobos RA, Bernstein KE, and Giani JF (2018). Renal tubular ACE-mediated tubular injury is the major contributor to microalbuminuria in early diabetic nephropathy 314, F531–F542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabi M, Corinaldesi E, Pierantoni L, Mazzoni E, Landini C, Bigucci B, Ancora G, Malaigia L, Bodnar T, Di Fazzio G, et al. (2018). Gastrointestinal presentation of Kawasaki disease: A red flag for severe disease? PLoS ONE 13, e0202658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano A, Not T, Wang W, Uzzau S, Berti I, Tommasini A, and Goldblum SE (2000). Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. The Lancet 355, 1518–1519. [DOI] [PubMed] [Google Scholar]
- Ferrier L, Mazelin L, Cenac N, Desreumaux P, Janin A, Emilie D, Colombel J-F, Garcia-Villar R, Fioramonti J, and Bueno L (2003). Stress-induced disruption of colonic epithelial barrier: role of interferon-gamma and myosin light chain kinase in mice. Gastroenterology 125, 795–804. [DOI] [PubMed] [Google Scholar]
- Floege J, and Feehally J (2016). The mucosa-kidney axis in IgA nephropathy. Nat Rev Nephrol 12, 147–156. [DOI] [PubMed] [Google Scholar]
- Fury W, Tremoulet AH, Watson VE, Best BM, Shimizu C, Hamilton J, Kanegaye JT, Wei Y, Kao C, Mellis S, et al. (2010). Transcript abundance patterns in Kawasaki disease patients with intravenous immunoglobulin resistance. Hum Immunol 71, 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabay C, Lamacchia C, and Palmer G (2010). IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol 6, 232–241. [DOI] [PubMed] [Google Scholar]
- Gupta M, Johann-Liang R, Bussel JB, Gersony WM, and Lehman TJA (2002). Elevated IgA and IgM anticardiolipin antibodies in acute Kawasaki disease. Cardiology 97, 180–182. [DOI] [PubMed] [Google Scholar]
- Iwata M, Shimozato T, Tokiwa H, and Tsubura E (1987). Antipyretic activity of a human immunoglobulin preparation for intravenous use in an experimental model of fever in rabbits. Infect. Immun 55, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan SC, Lorant T, Choi J, Kjellman C, Winstedt L, Bengtsson M, Zhang X, Eich T, Toyoda M, Eriksson B-M, et al. (2017). IgG Endopeptidase in Highly Sensitized Patients Undergoing Transplantation. N Engl J Med 377, 442–453. [DOI] [PubMed] [Google Scholar]
- Kaetzel CS, Robinson JK, Chintalacharuvu KR, Vaerman JP, and Lamm ME (1991). The polymeric immunoglobulin receptor (secretory component) mediates transport of immune complexes across epithelial cells: a local defense function for IgA. P Natl Acad Sci USA 88, 8796–8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalla R, Kennedy NA, Ventham NT, Boyapati RK, Adams AT, Nimmo ER, Visconti MR, Drummond H, Ho G-T, Pattenden RJ, et al. (2016). Serum Calprotectin: A Novel Diagnostic and Prognostic Marker in Inflammatory Bowel Diseases. Am. J. Gastroenterol 111, 1796–1805. [DOI] [PubMed] [Google Scholar]
- Kanarek N, Grivennikov SI, Leshets M, Lasry A, Alkalay I, Horwitz E, Shaul YD, Stachler M, Voronov E, Apte RN, et al. (2014). Critical role for IL-1β in DNA damage-induced mucositis. Proc. Natl. Acad. Sci. U.S.a 111, E702–E711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazatchkine MD, and Kaveri SV (2001). Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med 345, 747–755. [DOI] [PubMed] [Google Scholar]
- Kohsaka T, Abe J, Asahina T, and Kobayashi N (1994). Classical pathway complement activation in Kawasaki syndrome. The Journal of Allergy and Clinical Immunology 93, 520–525. [DOI] [PubMed] [Google Scholar]
- Lamm ME, Emancipator SN, Robinson JK, Yamashita M, Fujioka H, Qiu J, and Plaut AG (2008). Microbial IgA protease removes IgA immune complexes from mouse glomeruli in vivo: potential therapy for IgA nephropathy. Am. J. Pathol 172, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Schulte DJ, Shimada K, Chen S, Crother TR, Chiba N, Fishbein MC, Lehman TJ, and Arditi M (2012). Interleukin-1beta is crucial for the induction of coronary artery inflammation in a mouse model of Kawasaki disease. Circulation 125, 1542–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Wakita D, Dagvadorj J, Shimada K, Chen S, Huang G, Lehman TJA, Fishbein MC, Hoffman HM, Crother TR, et al. (2015). IL-1 Signaling Is Critically Required in Stromal Cells in Kawasaki Disease Vasculitis Mouse Model: Role of Both IL-1a and IL-1β. Arterioscler. Thromb. Vasc. Biol 35, 2605–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman TJ, Walker SM, Mahnovski V, and McCurdy D (1985). Coronary arteritis in mice following the systemic injection of group B Lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum 28, 652–659. [DOI] [PubMed] [Google Scholar]
- Leung DY, Cotran RS, Kurt-Jones E, Burns JC, Newburger JW, and Pober JS (1989). Endothelial cell activation and high interleukin-1 secretion in the pathogenesis of acute Kawasaki disease. The Lancet 2, 1298–1302. [DOI] [PubMed] [Google Scholar]
- Lutz HU, and Späth PJ (2005). Anti-inflammatory effect of intravenous immunoglobulin mediated through modulation of complement activation. Clin Rev Allergy Immunol 29, 207–212. [DOI] [PubMed] [Google Scholar]
- McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, Baker AL, Jackson MA, Takahashi M, Shah PB, et al. (2017). Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals From the American Heart Association. Circulation 135, e927–e999. [DOI] [PubMed] [Google Scholar]
- Moja P, Quesnel A, Resseguier V, Lambert C, Freycon F, Berthoux F, and Genin C (1998). Is there IgA from gut mucosal origin in the serum of children with Henoch-Schönlein purpura? Clin. Immunol. Immunopathol 86, 290–297. [DOI] [PubMed] [Google Scholar]
- Myones BL, Bathoria B, Lehman L, and Shulman S (1995). Human IVIG inhibits Lactobacillus casei-inducible coronary arteritis in a murine model. Proceedings of the V International Kawasaki Disease Symposium 252–256.
- Nagata S, Yamashiro Y, Maeda M, Ohtsuka Y, and Yabuta K (1993). Immunohistochemical studies on small intestinal mucosa in Kawasaki disease. Pediatr. Res 33, 557–563. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Okigaki M, Miura N, Suzuki C, Ohno N, Kametani F, and Hamaoka K (2014). Involvement of mannose-binding lectin in the pathogenesis of Kawasaki disease-like murine vasculitis. Clinical Immunology 153, 64–72. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Zaitsu M, Hara M, Yokota G, Watanabe M, Ueda Y, Imayoshi M, Ishii E, Tasaki H, and Hamasaki Y (2003). A polymorphism in the promoter of the CD14 gene (CD14/-159) is associated with the development of coronary artery lesions in patients with Kawasaki disease. J. Pediatr 143, 357–362. [DOI] [PubMed] [Google Scholar]
- Noval Rivas M, Lee Y, Wakita D, Chiba N, Dagvadorj J, Shimada K, Chen S, Fishbein MC, Lehman TJ, Crother TR, et al. (2017). CD8+ T Cells Contribute to the Development of Coronary Arteritis in the Lactobacillus casei Cell Wall Extract-Induced Murine Model of Kawasaki Disease. Arthritis Rheumatol 69, 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshio G, Furukawa F, Khine M, Yoshioka H, Kudo H, and Hamashima Y (1987). High levels of IgA-containing circulating immune complex and secretory IgA in Kawasaki disease. Microbiol. Immunol 31, 891–898. [DOI] [PubMed] [Google Scholar]
- Oortwijn BD, van der Boog PJM, Roos A, van der Geest RN, de Fijter JW, Daha MR, and van Kooten C (2006). A pathogenic role for secretory IgA in IgA nephropathy. Kidney Int 69, 1131–1138. [DOI] [PubMed] [Google Scholar]
- Pabst O (2012). New concepts in the generation and functions of IgA. Acta Pharmacologica Sinica 12, 821–832. [DOI] [PubMed] [Google Scholar]
- Paterson BM, Lammers KM, Arrieta MC, Fasano A, and Meddings JB (2007). The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther 26, 757–766. [DOI] [PubMed] [Google Scholar]
- Quan CP, Berneman A, Pires R, Avrameas S, and Bouvet JP (1997). Natural polyreactive secretory immunoglobulin A autoantibodies as a possible barrier to infection in humans. Infect. Immun 65, 3997–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, and Daha MR (2001). Human IgA activates the complement system via the mannan-binding lectin pathway. J. Immunol 167, 2861–2868. [DOI] [PubMed] [Google Scholar]
- Rowley AH, Shulman ST, Mask CA, Finn LS, Terai M, Baker SC, Galliani CA, Takahashi K, Naoe S, Kalelkar MB, et al. (2000). IgA plasma cell infiltration of proximal respiratory tract, pancreas, kidney, and coronary artery in acute Kawasaki disease. Jid 182, 1183–1191. [DOI] [PubMed] [Google Scholar]
- Rowley AH (2018). Is Kawasaki disease an infectious disorder? Int J Rheum Dis 21, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Souza V, Carreno MP, Kaveri SV, Ledur A, Sadeghi H, Cavaillon JM, Kazatchkine MD, and Haeffner-Cavaillon N (1995). Selective induction of interleukin-1 receptor antagonist and interleukin-8 in human monocytes by normal polyspecific IgG (intravenous immunoglobulin). Eur. J. Immunol 25, 1267–1273. [DOI] [PubMed] [Google Scholar]
- Sato S, Kawashima H, Kashiwagi Y, Fujioka T, Takekuma K, and Hoshika A (2009). Association of mannose-binding lectin gene polymorphisms with Kawasaki disease in the Japanese. Int J Rheum Dis 12, 307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya A, Sakamoto N, Shimizu Y, Shibuya K, Osawa M, Hiroyama T, Eyre HJ, Sutherland GR, Endo Y, Fujita T, et al. (2000). Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat Immunol 1, 441–446. [DOI] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, et al. (2011). A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474, 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, Caprioli F, Bottiglieri L, Oldani A, Viale G, et al. (2015). A gut-vascular barrier controls the systemic dissemination of bacteria. Science 350, 830–834. [DOI] [PubMed] [Google Scholar]
- Stagi S, Simonini G, Ricci L, de Martino M, and Falcini F (2006). Coeliac disease in patients with Kawasaki disease. Is there a link? Rheumatology 45, 847–850. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Suzuki Y, Yamanaka T, Hirose S, Nishimura H, Toei J, Horikoshi S, and Tomino Y (2005). Genome-wide scan in a novel IgA nephropathy model identifies a susceptibility locus on murine chromosome 10, in a region syntenic to human IGAN1 on chromosome 6q22–23 16, 1289–1299. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Kawase H, Shimizu T, Yoshida M, and Sekine I (2002). Increased production of serum IgA-class antibody to lipid A in Kawasaki disease. Pediatr Int 44, 5–11. [DOI] [PubMed] [Google Scholar]
- Turner JR (2009). Intestinal mucosal barrier function in health and disease. Acta Pharmacologica Sinica 9, 799–809. [DOI] [PubMed] [Google Scholar]
- Wakita D, Kurashima Y, Crother TR, Noval Rivas M, Lee Y, Chen S, Fury W, Bai Y, Wagner S, Li D, et al. (2016). Role of Interleukin-1 Signaling in a Mouse Model of Kawasaki Disease-Associated Abdominal Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol 36, 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Charbonnier L-M, Noval Rivas M, Georgiev P, Li N, Gerber G, Bry L, and Chatila TA (2015). MyD88 Adaptor-Dependent Microbial Sensing by Regulatory T Cells Promotes Mucosal Tolerance and Enforces Commensalism. Immunity 43, 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T (2018). Clinical features of acute kidney injury in patients with Kawasaki disease. World J Clin Pediatr 7, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H-J, Shin JH, Yang SH, Chae D-W, Kim H, Lee D-S, Kim HL, Kim S, Lee JS, and Kim YS (2003). Association of the CD14 gene -159C polymorphism with progression of IgA nephropathy. J. Med. Genet 40, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, and Schulzke JD (2007). Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.