

Antimicrobial resistance in bacteria represents one of the most consequential problems in modern medicine, and its emergence and spread threaten to compromise central advances in the treatment of infectious diseases. Ceftazidime-avibactam (CZA) belongs to a new class of broad-spectrum beta-lactam/beta-lactamase inhibitor combinations designed to treat infections caused by multidrug-resistant bacteria. Understanding the emergence of resistance to this important new drug class is of critical importance. In this work, we demonstrate that evolved mismatch repair deficiency in P. aeruginosa, an important pathogen responsible for significant morbidity and mortality among hospitalized patients, may facilitate rapid acquisition of resistance to CZA in the context of acute infection. These findings are relevant for both diagnosis and treatment of antimicrobial resistance emerging in acute infection in the hypermutator background and additionally have implications for the emergence of more virulent phenotypes.
KEYWORDS: Pseudomonas aeruginosa, antimicrobial agents, bacterial evolution, ceftazidime-avibactam, host-pathogen interactions, hypermutator, microbial genomics, mismatch repair
ABSTRACT
Strains of Pseudomonas aeruginosa with deficiencies in DNA mismatch repair have been studied in the context of chronic infection, where elevated mutational rates (“hypermutation”) may facilitate the acquisition of antimicrobial resistance. Whether P. aeruginosa hypermutation can also play an adaptive role in the more dynamic context of acute infection remains unclear. In this work, we demonstrate that evolved mismatch repair deficiencies may be exploited by P. aeruginosa to facilitate rapid acquisition of antimicrobial resistance in acute infection, and we directly document rapid clonal succession by such a hypermutating lineage in a patient. Whole-genome sequencing (WGS) was performed on nine serially cultured blood and respiratory isolates from a patient in whom ceftazidime-avibactam (CZA) resistance emerged in vivo over the course of days. The CZA-resistant clone was differentiated by 14 mutations, including a gain-of-function G183D substitution in the PDC-5 chromosomal AmpC cephalosporinase conferring CZA resistance. This lineage also contained a substitution (R656H) at a conserved position in the ATPase domain of the MutS mismatch repair (MMR) protein, and elevated mutational rates were confirmed by mutational accumulation experiments with WGS of evolved lineages in conjunction with rifampin resistance assays. To test whether MMR-deficient hypermutation could facilitate rapid acquisition of CZA resistance, in vitro adaptive evolution experiments were performed with a mutS-deficient strain. These experiments demonstrated rapid hypermutation-facilitated acquisition of CZA resistance compared with the isogenic wild-type strain. Our results suggest a possibly underappreciated role for evolved MMR deficiency in facilitating rapid adaptive evolution of P. aeruginosa in the context of acute infection.
INTRODUCTION
In general, the maintenance of high-fidelity DNA replication and the reduction of spontaneous mutation rates are beneficial to organisms. Previous work has demonstrated that even moderately elevated mutational rates may be substantially deleterious and that exceeding certain threshold mutational rates results in the irrecoverable loss of essential wild-type sequences and population collapse (1–10). Replication fidelity in bacteria is maintained by elaborate machinery that includes the proofreading exonucleases in replicative DNA polymerases and an array of DNA repair proteins broadly organized into homologous DNA recombination, base and nucleotide excision repair pathways, and methyl-directed mismatch repair (MMR) (11–13). While in general beneficial, very low mutational rates can reduce the ability to adapt in the face of a changing microhabitat. As a result, optimal mutation rates can differ between these high and low extremes and may be dynamically modified by selection, particularly when increases in mutational rate may be beneficial for adapting bacterial populations (6, 14–18). Supporting this hypothesis, moderately increased mutational rates are shown to be advantageous in both in vitro and in vivo competition experiments under conditions of selective bottlenecks in which sharp reductions in population numbers occur (6, 7, 19–22).
Increased spontaneous mutation rates are extensively documented among natural and human clinical bacterial isolates (15, 23–28). Elevated mutational rates may result from the induction of error-prone polymerases or from defects in DNA repair systems (11–13, 16, 17, 29–33). Depending on the exact mutation and gene target, DNA repair defects may result in increases in mutational rates from 10-fold to greater than 1,000-fold, resulting in hypermutator phenotypes (27, 29, 30). Hypermutation carries with it many consequences for both genome plasticity and population structure under selection (7, 14). While increasing the ability to adapt, DNA repair lesions are likely to be associated with a negative fitness cost due to a secondary mutational load. Nevertheless, hypermutators appear to arise with high frequency in chronic respiratory colonization and infection in cystic fibrosis (25, 34). These studies have demonstrated that hypermutators may fix in populations through coselection in vivo with beneficial mutations such as antimicrobial resistance in the context of infection (25, 27, 28). In vitro selection experiments have demonstrated the increased evolvability of antimicrobial resistance in strains with elevated mutational rates, and others have demonstrated that antibiotic exposure itself promotes mutagenesis (21, 26–28, 33, 35–38). Elevated mutational rates may additionally facilitate the discovery of secondary mutations that mitigate fitness costs often associated with primary antimicrobial resistance mutations (26, 39).
Phenotypic screens for hypermutators among commensal and pathogenic clinical isolates have demonstrated a wide range of prevalence, from ∼1% when only strong hypermutators were included to 14% when mild hypermutating phenotypes were included (23, 24, 26). Thus, hypermutators occur broadly among clinical isolates. However, the question of whether selection for hypermutation may play a role in facilitating intrahost evolution in the context of acute clinical infection has not been extensively evaluated in longitudinal clinical studies. In this work, we used whole-genome sequencing (WGS) of serially collected clinical isolates and in vitro adaptive evolution experiments to demonstrate that evolved de novo MMR deficiency may be exploited by Pseudomonas aeruginosa to facilitate the rapid emergence of resistance to ceftazidime-avibactam (CZA), a new broad-spectrum beta-lactam/beta-lactamase inhibitor combination antibiotic, in acute infection, and we directly documented rapid clonal succession by such a hypermutating lineage in a patient.
RESULTS
Clinical case.
A patient undergoing treatment at the NIH Clinical Center for refractory T-cell lymphoblastic leukemia developed fever in the context of neutropenia. Gram-negative empirical antimicrobial therapy during the patient’s febrile course included trimethoprim-sulfamethoxazole, levofloxacin, meropenem, tigecycline, gentamicin, and colistimethate. After the patient’s transfer to the intensive care unit (ICU), blood cultures grew P. aeruginosa (day 1) (Fig. 1 and Table 1). Ceftazidime (CAZ) and amikacin were substituted for meropenem and gentamicin, and the patient became afebrile after 4 days. A chest computed tomography (CT) scan from day 4 revealed a large, dense consolidation that occupied most of the left lower lobe, and a sputum culture from day 5 grew P. aeruginosa with susceptibilities identical to those of the blood isolate. Gram-negative antibacterial therapy was further adjusted to ceftazidime-avibactam (CZA) and amikacin, and amikacin was discontinued on day 8 after several days of negative blood cultures. On day 10, hypotension and fever recurred, and cultures drawn on day 9 grew P. aeruginosa that was resistant to all agents tested except colistin, notably with a CZA MIC of >256 μg/ml. Subsequent testing demonstrated that these isolates had simultaneously acquired high-level resistance (MIC > 256 μg/ml) to ceftolozane-tazobactam (C/T), although this antimicrobial was not used in treatment. Amikacin and colistimethate were restarted, but the patient developed respiratory failure and hypotension and expired.
FIG 1.

Summary of antibiotic treatment and isolate collection during the course of infection. Bars at the top indicate Gram-negative antimicrobial treatment. Time of culture collection and positivity are noted with circles connected by lines. Dashed lines indicate approximate positivity time, as respiratory cultures were not continuously monitored for growth.
TABLE 1.
Summary of antimicrobial susceptibility testing for all 9 isolates recovered from the patient
| Antimicrobiala | MIC for indicated isolateb
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Paer_1 (blood, day 0)c |
Paer_2 (blood, day 0) |
Paer_3 (sputum, day 5) |
Paer_4 (blood, day 8) |
Paer_5 (blood, day 10) |
Paer_6 (tracheal aspirate, day 11) |
Paer_7 (blood, day 11) |
Paer_8 (blood, day 11) |
Paer_9 (lung, day 13) |
|
| Amikacin | ≤8 (S) | ≤8 (S) | ≤8 (S) | 16 (S) | 16 (S) | 32 (I) | 32 (I) | 32 (I) | 16 (S) |
| Aztreonam | ≤4 (S) | ≤4 (S) | ≤4 (S) | >16 (R) | >16 (R) | >16 (R) | >16 (R) | >16 (R) | >16 (R) |
| Cefepime | 8 (S) | 8 (S) | 8 (S) | >16 (R) | >16 (R) | >16 (R) | >16 (R) | >16 (R) | >16 (R) |
| Ceftazidime | 8 (S) | 8 (S) | 8 (S) | >128 (R) | >128 (R) | >128 (R) | >128 (R) | >128 (R) | >128 (R) |
| CZA | 1 (S) | 2 (S) | 2 (S) | >256 (R) | >256 (R) | >256 (R) | >256 (R) | >256 (R) | >256 (R) |
| Ciprofloxacin | >2 (R) | >2 (R) | >2 (R) | >2 (R) | >2 (R) | >2 (R) | >2 (R) | >2 (R) | >2 (R) |
| Doripenem | 4 (I) | 4 (I) | 4 (I) | >4 (R) | 4 (I) | 4 (I) | 4 (I) | 4 (I) | 4 (I) |
| Gentamicin | >8 (R) | >8 (R) | >8 (R) | >8 (R) | >8 (R) | >8 (R) | >8 (R) | ||
| Imipenem | >8 (R) | >8 (R) | >8 (R) | >8 (R) | 4 (I) | 4 (I) | 4 (I) | 4 (I) | 4 (I) |
| Levofloxacin | >4 (R) | >4 (R) | >4 (R) | >4 (R) | >4 (R) | >4 (R) | >4 (R) | ||
| Meropenem | 4 (I) | 4 (I) | 4 (I) | >8 (R) | 8 (R) | 8 (R) | 8 (R) | 8 (R) | 8 (R) |
| Pip/Tazo | 32/4 (I) | 64/4 (I) | 32/4 (I) | >64/4 (R) | >64/4 (R) | >64/4 (R) | >64/4 (R) | 64/4 (I) | 64/4 (I) |
| Tobramycin | >8 (R) | >8 (R) | >8 (R) | >8 (R) | >8 (R) | >8 (R) | >8 (R) | >8 (R) | >8 (R) |
| C/T | 1 (S) | >256 (R) | >256 (R) | ||||||
CZA, ceftazidime-avibactam; Pip/Tazo, piperacillin-tazobactam; C/T, ceftolozane-tazobactam.
Values are MICs (μg/ml) measured by automated broth microdilution, except those for CZA and C/T, which were measured by Etest. Interpretive breakpoint criteria are included from CLSI M100, 27th ed. R, resistant; S, susceptible; I, intermediate.
The source of the isolate and the day of collection are given in parentheses.
Emergence of a clonal CZA-resistant lineage during infection.
During the course of acute illness, eight serial isolates were cultured from blood and respiratory sources, and a final isolate was cultured postmortem from lung tissue (Fig. 1). To understand the genetic basis of the rapid emergence of CZA resistance, we performed WGS. Individual isolates were sequenced to 41× to 123× depth using an Illumina MiSeq system (see Table S1 in the supplemental material), and de novo genome assemblies were generated. All of the isolates were closely related and demonstrated an ∼6.8-Mb genome belonging to sequence type 155 (ST155), an internationally distributed strain of P. aeruginosa that has been recovered from patients with and without cystic fibrosis, animals (dogs and horses), and environmental sources (40–43). Additionally, multidrug-resistant ST155 strains have been associated with outbreaks (41–43). Notably, the genome assembly contains two beta-lactamases (OXA-50 and PDC-5 AmpC cephalosporinase).
Isolate sequencing summary. Download Table S1, DOCX file, 0.01 MB (13.6KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Using the de novo draft genome assembly of the early CZA-susceptible isolate Paer_3 (nomenclature defined in Fig. 1 and Table 1) as a reference, we identified 31 substitutions and 6 indels in the set of sequenced isolates (Fig. 2 and Table 2). The three early-course, CZA-susceptible isolates were found to be indistinguishable, and all variants were confined to the late-course, CZA-resistant isolates. Out of the 37 sequence variants, 14 single nucleotide substitutions comprising 2 intergenic, 3 synonymous, and 9 nonsynonymous substitutions were shared by all CZA-resistant isolates, and each of the remaining 23 variants occurred in exactly one isolate each. These mutations thus divide the isolates into two lineages: the initial clonal CZA-susceptible isolate and the CZA-resistant isolate containing the 14 shared mutations. We subsequently focused our analysis on the 9 nonsynonymous substitutions (Table 2).
FIG 2.
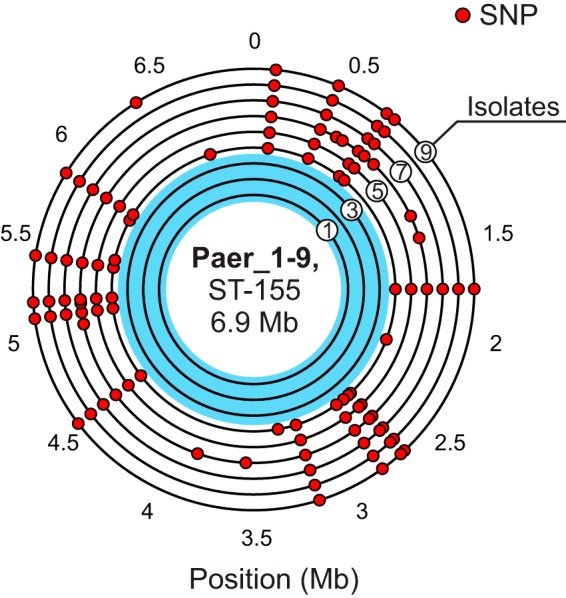
Genomic distribution of 31 detected SNPs in 9 isolates. All isolate genomes are arranged in concentric circles according to time of collection (isolate 1 to isolate 9), with the earliest isolate closest to the center. Position is given relative to the genome assembly of the Paer_3 isolate.
TABLE 2.
Summary of mutations present in late-course isolates and their effects on annotated coding sequencesa
| Position | Reference | Alternate | Starting position |
Ending position |
Strand | Geneb | Annotation | AA change |
|---|---|---|---|---|---|---|---|---|
| 112902 | G | A | 112341 | 114170 | − | ftsH | ATP-dependent zinc metallo-peptidase | G→G |
| 440224 | C | T | 438791 | 441085 | − | bglX | Periplasmic beta-glucosi-dase | G→S |
| 721564 | C | T | 720960 | 723527 | − | mutS | Mismatch repair protein | R→H |
| 771283 | A | G | 770709 | 771347 | − | bepR | Transcriptional repressor | C→R |
| 1747408 | A | G | NA | Intergenic region | ||||
| 2661379 | A | G | 2660819 | 2661772 | − | gshB | Glutathione synthetase | F→L |
| 2686555 | C | T | 2685579 | 2688719 | + | mexB | Multidrug resistance protein | P→L |
| 2805500 | A | G | 2805376 | 2805930 | + | puuR | Transcriptional regulator | Q→R |
| 3161857 | C | T | NA | Intergenic region | ||||
| 4523157 | A | G | 4523030 | 4523299 | + | fliQ | Flagellar biosynthesis | Q→R |
| 5102104 | G | T | 5101420 | 5102391 | − | hprA | Glycerate dehydrogenase | V→V |
| 5184929 | C | T | 5184345 | 5187176 | + | ileS | Isoleucine-tRNA ligase | A→A |
| 5422370 | T | C | 5421685 | 5423961 | − | clpA | ATP-dependent Clp protease | E→G |
| 5869250 | C | T | 5868604 | 5869797 | − | blaPDC-5 | AmpC beta-lactamase | G→D |
Nucleotide and amino acid (AA) positions are given along with strand position.
NA, not applicable.
G183D substitution in PDC-5 confers CZA resistance.
Mutations in beta-lactamase genes can confer resistance to CZA. One of the 14 mutations shared by all of the CZA-resistant isolates was a nonsynonymous G548A nucleotide substitution in a chromosomal AmpC cephalosporinase gene, blaPDC-5 (44). Importantly, the resulting G183D amino acid substitution has been previously described in PDC beta-lactamases in P. aeruginosa and has been shown to confer resistance to both CZA and C/T (45–47).
Late-course isolates contain a defective MutS mismatch repair protein and display a hypermutator phenotype.
In addition to the PDC-5 G183D substitution, all of the CZA-resistant isolates contained an R656H substitution in the MutS mismatch repair protein. R656H is located in a conserved position in the ATPase domain of the protein (Fig. 3) that is critically required for its function. R656 is a part of the “palm” region within the AAA family ATPase domain and is immediately adjacent to the strongly conserved MutS “signature” motif (48). Mutations within this same domain in the human MutS homologue underlie the hypermutation phenotype and microsatellite instability seen in hereditary nonpolyposis colorectal cancer (49, 50). Thus, we expected that the R656H substitution may inactivate MutS, resulting in MMR deficiency and a hypermutator phenotype with an elevated spontaneous mutation rate and a mutation spectrum dominated by transitions. Consistent with MutS MMR-deficient mutagenesis, 30 of the 31 single nucleotide substitutions in the late-course CZA-resistant isolates were transition mutations (Table S2).
FIG 3.

The R656H substitution is located in a critical domain of the MutS protein. (A) Crystal structure of the Escherichia coli MutS protein (PDB accession no. 1E3M) with the highlighted R656 amino acid and ADP in space-fill representation. (B) Domain structure of MutS family of proteins. (C) Multiple sequence alignment of MutS-related proteins around position 656. R656 is universally conserved across a range of homologous proteins from bacteria to humans.
Mutation classes found in late-course CZA-resistant isolates. Download Table S2, DOCX file, 0.01 MB (13.5KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
To determine whether the apparent MutS defect results in an increase in the spontaneous mutation rate, we first used a rifampin reversion assay (51). We found the substitution frequency as measured by this assay to be >100-fold higher in the R656H Paer_6 strain than in the wild-type Paer_3 strain (Table S3). Next, to provide an independent measure of mutational rates and spectrum, we performed mutation accumulation experiments, followed by WGS. In this approach, initially isogenic lineages were serially propagated through single-colony subcultures without selection, conditions under which genetic drift dominates and a high percentage of mutations accumulate in an effectively neutral fashion (52). We propagated one of the early, CZA-susceptible isolates containing the intact, wild-type MutS sequence and one of the late, CZA-resistant isolates containing the MutS R656H substitution for approximately 150 generations. WGS of progeny revealed only one newly acquired mutation in the isolate with intact wild-type MutS protein, compared to 18 substitutions (all transitions) and 5 small indels in the late MutS R656H isolate (Fig. S1 and Table S4). These results demonstrate that the R656H substitution significantly increases the mutation rate in the late-course CZA-resistant isolates with a transition mutation spectrum. Taken together, these data demonstrate an acquired hypermutation phenotype of MutS R656H in the late-course isolates.
In vitro mutation accumulation in MutS-deficient isolates. (A) Design and passaging scheme of a mutation accumulation experiment. Early-course CZA-susceptible isolate with WT MutS (Paer_3) and late-course, CZA-resistant isolate with MutS R656H substitution (Paer_6) were passaged for ∼150 generations in parallel and then sequenced. (B) Genomic distribution of SNPs detected by WGS following mutation accumulation. Each genome is represented as a large concentric circle, and positions with mutations are indicated with small blue (reference) and red (mutant) circles. During the course of the experiment, isolates with intact MutS (wt) acquired a single mutation, whereas 18 substitutions (all transitions) and 5 small indels (not shown) accumulated in isolates with the R656H variant of MutS. Download FIG S1, EPS file, 0.4 MB (434.4KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Rifampin reversion frequencies for Paer_3, Paer_6, and Paer_9. Results are shown for two experiments (n = 3 replicates). Download Table S3, DOCX file, 0.01 MB (14KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Mutation classes found in the mutation accumulation experiment in a late-course, CZA-resistant isolate with the MutS R656H substitution (Paer_6) following ∼150 generations. All observed mutations are transitions. Download Table S4, DOCX file, 0.01 MB (13.6KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Other mutations present in the late isolates.
Among the remaining 12 mutations in the CZA-resistant isolates, three other genes known to be associated with the development of antimicrobial resistance—mexB, bepR, and clpA—contained single nonsynonymous substitutions (53, 54). MexB is an MDR family efflux pump that, when overexpressed, can confer resistance to multiple classes of antimicrobials in P. aeruginosa, including CZA, and numerous studies have demonstrated the evolution of resistance in association with mutations in the mexB gene (53–56). A P326L substitution was present in the MexB protein in a position adjacent to the channel domain in the crystal structure (PDB accession no. 2V50) (57). Although the structure of the P326L MexB mutant is unknown, the proline is conserved and located at an alpha-helix-to-beta-strand transition point, such that a change to a leucine residue (with the ability to hydrogen bond, which the proline lacks) may have structural consequences. The effects of this mutation were not further explored, but the structure and conformation of the pore region control substrate specificity (58).
An A65G transition was present in the open reading frame coding for the BepR protein, an ortholog of NalD and a well-studied negative regulator of the MexAB-OprM operon in P. aeruginosa (59). BepR/NalD is a member of the TetR helix-turn-helix (HTH) family of transcription factors that contain an N-terminal DNA binding domain. The corresponding C22R amino acid substitution in the CZA-resistant isolates is located in the HTH domain, suggesting the possibility that DNA binding might be disrupted, resulting in transcriptional derepression of MexAB-OprM, as has been noted in other studies (53, 54).
An E531G substitution was observed in ClpA, an AAA family ATPase and a motor subunit of the ClpP protease that is involved in a broad variety of cellular functions in bacteria, including virulence and bacterial persistence (60). Mutations in Clp family proteins have been associated with cephalosporin resistance and have been observed to develop under monobactam, CAZ, and CZA selection (53, 54, 61). Although the exact impact of the observed substitution in ClpA is unclear, the above evidence suggests the potential involvement in CZA resistance observed in this case.
MutS deficiency facilitates rapid acquisition of CZA resistance in vitro.
Based on the observation that CZA resistance in the late-course isolates emerged rapidly and was mediated by a single transition mutation (G548A) within the mutational spectrum of the MMR-deficient hypermutator, we hypothesized that this phenotype might have facilitated the rapid acquisition of CZA resistance in this clinical case. To test the degree to which an MMR-deficient hypermutation could increase the speed of acquisition of CZA resistance, we performed in vitro adaptive evolution experiments under CZA selection using a pair of strains derived from laboratory reference P. aeruginosa PAO1, one containing a mutS gene interrupted by a transposon insertion (MPAO1 mutS) and the isogenic wild type (MPAO1 WT) (C. Manoil laboratory, University of Washington) (62, 63). In agreement with the gross defects in mismatch repair expected in the absence of an intact MutS protein, rifampin reversion assay testing demonstrated that the spontaneous mutational rate of the MPAO1 mutS transposon knockout strain was 98-fold greater than that observed in wild-type MPAO1 (reversion frequencies of 3.8 × 10−6 versus 3.9 × 10−8; single experiment). Four independent wild-type MPAO1 and MPAO1 mutS lineages were passaged over a range of escalating concentrations of ceftazidime (0.25 to 512 μg/ml) and a fixed concentration of avibactam (4 μg/ml). Following each passage, the population growing at the highest ceftazidime concentration was transferred into the next round (Fig. 4) (see Materials and Methods for details of the passaging experiment design).
FIG 4.

Serial passaging of MPAO1 and the MMR-deficient MPAO1 MutS– derivative under CZA selection. (A) Average highest CZA concentration (mg/liter of ceftazidime) for which growth was detected versus passage number. Four independent lineages were passaged in parallel for MMR-proficient MPAO1 and MMR-deficient MPAO1 MutS– isolates over a gradient of increasing CZA concentrations (see Fig. S2 in the supplemental material) (see also Materials and Methods for details of the experimental design). Averages and standard deviations of maximum CZA concentrations at which growth was still detected are plotted on the graph. (B) Number of passages required to achieve population growth at a CZA concentration of ≥16 mg/liter. MPAO1 MutS– achieves resistance to CZA significantly faster than the wild type (P = 0.006).
Serial passaging of the WT MPAO1 and MMR-deficient MPAO1 derivative under CZA selection (full data for Fig. 4). Heatmap of maximum CZA concentration (μg/ml of ceftazidime) for which growth was detected versus passage number. Four independent lineages (A to D) were passaged in parallel for WT MPAO1 and MPAO1 MutS– isolates over a gradient of increasing CZA concentrations (see Materials and Methods for details of experimental design). Download FIG S2, EPS file, 0.2 MB (243.1KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Strikingly, we observed that the MMR-deficient MPAO1 mutS lineages acquired high-level resistance to CZA within 4 to 5 passages, much more rapidly (P = 0.006) than did the wild-type MPAO1 strain, which took 12 to 18 passages to reach the same levels of resistance (Fig. 4 and Fig. S2). This observation is perhaps not surprising, given the high spontaneous mutation rate of MutS-deficient strains. An estimated mutational rate of MMR-deficient isolates was measured in this work and by others as 10−8 to 10−7 per position per generation (0.7 to 0.07 mutations per genome per division) (64, 65). To a rough first approximation, the population would need to undergo x cell divisions to sample all possible single mutations, where x is on the order of (mutation rate per position per generation)−1. If replication can proceed unimpeded, this requires y generations, where y = −log2(mutation rate per position per generation). In the case of the isolates considered here, this number is 7 · log2(10) to 8 · log2(10) or 23 to 27 generations. Given that there are multiple pathways to CZA resistance based on single mutations (46), and that the starting population is likely to be greater in size than one cell, CZA resistance could potentially be found in even fewer generations. These theoretical considerations, taken in combination with the experimental results described above, demonstrate that MMR deficiency can facilitate rapid acquisition of CZA resistance under conditions of in vitro selection.
MutS inactivation likely preceded PDC-5 mutation.
While independent clinical isolates that separate MutS and PDC-5 mutations were not cultured from this patient—all late isolates share 14 common mutations—there are several distinct features of the evolutionary process in this case that provide insight into the sequence of mutation acquisition. Base pair substitutions in MMR-deficient P. aeruginosa are extremely biased toward transitions relative to those in the wild type. While in wild-type strains transitions account for two-thirds of all base pair substitutions (65), they account for nearly all substitutions in MMR-deficient strains, with estimates ranging from 98% under neutral conditions (65) to 99.5% under conditions of antibiotic selection (66) and 97% in our data. We thus asked how likely it is to acquire only one transversion as a function of the timing of the MutS inactivation step. We performed numeric simulation using the following scenario. Mutations are acquired according to a neutral model (66% transitions) until MutS inactivation, after which point mutations accumulate following an MMR-deficient mutation spectrum (98% transitions) (Fig. S3A). This simulation demonstrated that it is likely that MutS was inactivated on steps 1 to 5 rather than steps 6 to 14 (proportional risk = 1.6) (Fig. S3B and S3C).
Mutation acquisition as a function of MutS inactivation step. (A) Schematic representation of the simulation approach. Mutations are acquired using the wild-type mutation spectrum until MutS is inactivated, after which point mutations accumulate following an MMR-deficient mutation spectrum. Mutations are assumed to be independent. (B) The probability of observing one transversion out of 14 mutations depending on MutS inactivation step. (C) Fifth to 95th percentile ranges of numbers of substitutions for each of the evolutionary scenarios. Blue squares indicates median numbers of transversions. Download FIG S3, EPS file, 0.6 MB (673KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Additional independent evidence suggests that PDC-5 G183D occurred as a later mutation in the series. Three of the seven nonsynonymous mutations that occurred along with mutS and blaPDC-5 involved genes associated with cephalosporin resistance: mexB, nalD/bepR, and clpA. We find it unlikely (P < 0.01%) that the mutations in these genes occurred by chance, based on the fact that fewer than 100 genes out of a total of >5,000 genes have been described to be involved in CAZ and CZA resistance in P. aeruginosa. If an allele conferring high-level resistance to a given antibiotic is found during stepwise adaptation, subsequent mutations are unlikely to provide additional adaptive benefit and evolution should proceed in a neutral manner via random drift. Since it is known that PDC-5 G183D is a strongly protective variant conferring a high MIC to CZA (45–47), we believe the most likely scenario is that the discovery of the PDC-5 G183D substitution was preceded by a series of beneficial but less protective mutations in mexB, nalD/bepR, and clpA, in a process similar to what has been described for selection of antimicrobial resistance genes under conditions of lower antibiotic concentrations (67, 68). We then can combine this observation with the probability distribution of MutS inactivation as a function of a given step (Fig. S2B) to estimate the likelihood of the PDC-5 mutation preceding MutS inactivation. Using the simplest assumption that the PDC-5 mutation was acquired with uniform probability among the last 10 steps, we find the probability that the PDC-5 G183D mutation occurred prior to the MutS mutation, P(PDC < MutS), is 0.14. This is, however, a very conservative estimate. In this clinical case, the population likely underwent a selective sweep, and the founder clone with 14 mutations present in all late isolates became dominant. Since random drift following acquisition of a strong protective mutation is slow, it is likely that the strongly protective G183D variant in PDC-5 was found much later in the process, among the last few steps.
DISCUSSION
Here, we report the dynamic emergence of MMR deficiency and resistance to CZA in P. aeruginosa in acute systemic infection. Using whole-genome sequencing of serial clinical isolates, we directly documented clonal succession and fixation of an inactivating mutation in the mutS gene and a blaPDC-5 mutation conferring CZA resistance in isolates from two sites (blood and respiratory). We provide corroborating evidence from in vitro adaptive evolution experiments that hypermutation due to MutS inactivation can facilitate the rapid emergence of CZA resistance in a period of days. In the clinical case studied here, the speed with which CZA resistance emerged may have been facilitated by the fact that the substitution in the blaPDC-5 chromosomal AmpC cephalosporinase gene is a transition, a class of substitutions that is predominant in the MutS mutational spectrum, accounting for 30 of 31 mutations in the clinical isolates containing the R656H MutS and 18 of 18 mutations in the MPAO1 mutS isolate.
In our analysis of the evolutionary process in this case, we are limited by the isolates actually recovered from clinical culture and cannot determine with certainty the relative order in which all individual mutations arose. However, there are a number of lines of evidence that suggest that the inactivating mutS mutation occurred early in the evolutionary sequence. If MMR was inactivated early and thus mediated subsequent mutagenesis, one would expect to observe a substantially higher rate of mutation acquisition than that of wild-type, nonhypermutator isolates. Although the literature on genomic data for serial isolates from acute infections is relatively limited compared to that for chronic infections, two recent works (69, 70) provide data on mutation accumulation during acute infections. In these works, between 3 and 8 isolates were serially cultured from individual patients with ventilator-associated pneumonia within a period of 14 to 78 days (see Table S5 in the supplemental material). Persyn et al. reported the accumulation of between 2 and 4 mutations following 14 to 48 days in patients with ventilator-associated pneumonia (70), and Wang et al. reported similar numbers of substitutions (69) resulting in mutation acquisition rates not exceeding 0.1 to 0.3 mutations per day in 35 out of 36 isolates. In the isolates studied in the present work, the mutation acquisition rate is ∼3.5 mutations per day. We thus believe that mutation acquisition for a nonhypermutator phenotype in this clinical case is substantially more rapid than would be expected, providing corroborating evidence that the mutS inactivating mutation occurred early in the sequence.
Literature data summary of P. aeruginosa mutation accumulation during the course of acute infection in 8 patients. Download Table S5, DOCX file, 0.02 MB (20.5KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Another distinct feature of the late-course isolates is a striking enrichment for mutations in genes potentially mediating ceftazidime resistance, including mexB, bepR, and clpA, in addition to blaPDC-5. These targets accounted for 4 of 9 total nonsynonymous mutations and are a likely consequence of strong selective pressures on P. aeruginosa isolates due to treatment with CAZ and CZA during the course of infection. It is possible that the mutations in mexB, bepR, and clpA individually confer low-level resistance to CAZ or CZA and represent early steps along the evolutionary path to high-level resistance conferred by PDC-5 G183D. In addition, the large number of functionally relevant mutations observed in the late-course isolates, which are almost completely skewed toward transition mutations, argues strongly for the role of inactivation of mismatch repair in the evolution of CZA resistance. To test the hypothesis that MMR deficiency alone could facilitate the rapid development of CZA resistance, we performed in vitro evolution experiments using isogenic P. aeruginosa laboratory strains with a mutS gene disabled by a transposon insertion. These experiments demonstrated that resistance to CZA developed within 4 to 5 passages under selection in all four mutS MMR-deficient lineages in comparison to 12 to 18 passages in the MMR-proficient lineages. This is perhaps not surprising due to the high mutation rate in the MMR-deficient strain. If there is at least one pathway to CZA resistance based on a single transition, we hypothesize that it will be rapidly discovered once an inactivating MutS MMR lesion has evolved. Under the assumption of ∼1 substitution per genome per 10 generations, we estimate that in as few as 27 generations, a MutS hypermutator may discover any particular transition mutation, such as blaPDC-5 G548A. This estimate is conservative, as several different point mutations have been associated with CZA resistance in both PDC beta-lactamases and KPC carbapenemases in P. aeruginosa and Enterobacteriaceae, and the development of resistance may be further facilitated by mutations in auxiliary genes (46, 71–75).
The presence of antibiotic may indeed substantially reduce the viability of the wild type and most mutants and may slow the division time and substantially change the dynamics of the search for CZA resistance. In the hypothetical setting in which antibiotic treatment is completely effective, cell division does not take place to allow discovery of CZA mutants. However, it is difficult to know these parameters with any degree of precision in the relevant in vivo context. Under the conditions used in our in vitro adaptive evolution experiments with fixed 24-h interval passages, mutations conferring CZA resistance are reliably found within days, well within the range of our theoretical estimates. Furthermore, empirical observation of CZA development within days in the clinical case suggests the plausibility of these estimates, at least for certain ranges of conditions.
Significant work has been done to study how the balance between adaptive and deleterious mutations in hypermutating strains determines evolutionary outcomes (1, 5–7, 12, 32, 76, 77). Both theoretical and experimental investigations suggest that hypermutators of various strengths will fix by hitchhiking with the adaptive mutations they facilitate under certain conditions (6, 14, 21, 22). This effect has been demonstrated both in vitro and in vivo and is also consistent with the observations of the cooccurrence of hypermutators and drug resistance mutations in chronic infections in humans (23–26, 30, 35–37). It is possible that secondary suppressor mutations, reversion mutations, or recombination (if wild-type copies remain) could reverse hypermutator phenotypes and restore fidelity of replication and may be subsequently selected, producing lineages that retain the selectively advantageous mutations discovered by hypermutation. Alternatively, favorable mutations discovered by mutators may be transferred to nonmutators by recombination (76).
The host constitutes an important part of the adaptive evolutionary equation. The patient discussed in this case had clinical features which almost certainly contributed to the rapid evolution of resistance documented here. Profound and prolonged neutropenia, resulting in a host dependent almost entirely on the action of antimicrobials, in combination with pneumonia, a disease with a high microorganism burden, likely favored the events described. Notably, it has been demonstrated in vitro that avibactam may impede the evolution of AmpC-dependent resistance to ceftazidime in P. aeruginosa (78). However, in the case presented here, the combination of host factors and increased mutational rate conferred by MMR deficiency appears to have overcome any such impediment.
Our work also highlights challenges in the study of serial clinical isolates in which the relative order in which mutations arose is not known with certainty, requiring indirect analysis and inferences regarding evolutionary trajectories and distinguishing causative versus secondary mutations. In the case analyzed here, the exact sequence of mutation acquisition is supported by indirect evidence and should be considered within these limitations. Aggregation of a large number of individual cases and broader surveillance in future studies may provide more power to detect similarities between cases and strengthen the conclusions.
Although hypermutators have been studied in the context of chronic colonization and infection, the question of whether they may also play a role in facilitating intrahost evolution in the context of acute clinical infection has been less well studied to date. Based on our findings, we suggest that the role of P. aeruginosa hypermutation in acute infection and the emergence of antimicrobial resistance may be underappreciated. Diagnostic assessment for hypermutation genotypes in clinical isolates by WGS may become more feasible as the cost and difficulty of sequencing continue to decrease. Clearly, these may have therapeutic implications. However, it is important to note that at this time, routine testing for hypermutating P. aeruginosa strains and specific strategies for managing infections caused by these strains are not included in clinical practice guidelines. In conclusion, our results suggest an underappreciated role for evolved MMR deficiency in facilitating rapid adaptive evolution of P. aeruginosa in the context of acute infection, with potentially important general consequences for the diagnosis and treatment of infections with these organisms.
MATERIALS AND METHODS
Ethical approval.
This study was excluded from NIH Institutional Review Board review, under the Office for Human Research Protections ID no. 19-NIAID-00814, on the basis of the fact that it was a study of a single case of a deceased individual, involving only sequencing and analysis of bacterial isolates.
Clinical isolates.
Blood cultures were performed using an automated BD Bactec FX blood culture instrument (Becton Dickinson, Franklin Lakes, NJ). Respiratory specimens were plated onto standard microbiologic culture media, including blood agar plates (Remel, Lenexa, KS). Isolates from all sources were subcultured prior to identification, sequencing, and antimicrobial susceptibility testing. Identification was performed using a Bruker matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometer (Bruker Daltonics, Billerica, MA) in accordance with the manufacturer’s instructions.
Antimicrobial susceptibility testing.
Susceptibility testing of isolates was performed by automated broth microdilution methods using a Trek Sensititre instrument (ThermoFisher Scientific, Waltham MA) with custom susceptibility panels, with the exception of CZA and C/T, which were tested by Etest. Interpretive breakpoints were applied per guidelines from the CLSI (79).
DNA purification.
Genomic DNA was extracted from 1 ml liquid culture using the DNeasy blood and tissue kit (Qiagen, Valencia, CA) per the manufacturer’s instructions. DNA yields were quantified with a Qubit 2.0 fluorometer (Invitrogen, Carlsbad, CA), and DNA quality was assessed by migration through a 0.8% agarose gel (Lonza, Rockland, ME).
Illumina MiSeq library preparation and sequencing of clinical isolates.
Extracted genomic DNA samples were fragmented using a Covaris M220 instrument (Covaris, Inc., Woburn, MA) to ∼450 bp. Whole-genome libraries were prepared using a TruSeq Nano DNA kit with the Illumina NeoPrep system (Illumina, San Diego, CA), by following the kit instructions. Samples were sequenced using an Illumina MiSeq in 2 × 300 bp mode with 600-cycle V3 sequencing kits to a depth of ∼40× to 120× (see Table S1 in the supplemental material).
Computational methods.
Whole-genome assembly was performed using SPAdes v.3.9.1, and obtained contigs were polished with Pilon v.1.22. Draft assemblies were annotated using prokka v.1.11. Sequence variant identification was performed using FreeBayes v1.0.2, followed by filtering using VCFtools v.0.1.14 to retain only high-quality single nucleotide polymorphisms (SNPs) in long (>5-kb), nonrepeated (mean coverage, 0.5× to 1.5× of single-copy coverage) contigs.
Mutational accumulation experiments.
The overall design is as detailed in Fig. S1. Isolates Paer_3 (wild-type MutS) and Paer_6 (R656H MutS) were streaked onto two blood agar plates each from frozen stocks. After 23 h of incubation at 37°C in 6% CO2, isolated 1-mm-diameter colonies from each of the four plates (one colony per plate) were streaked individually onto fresh blood agar plates (passage 1, ∼30 generations), and these four plates were incubated under the same conditions. After 23 h of incubation, 1-mm-diameter colonies were selected from each of the four plates. Each of these colonies was split and streaked onto two blood agar plates, yielding 8 plates total, with a split of each lineage (passage 2). Portions of these colonies were additionally inoculated into liquid broth cultures (one culture from each of four plates) for DNA extraction and sequencing as described below. These plates were incubated under the same conditions as the other plates. Simultaneously, 1-mm-diameter colonies were picked from all four initial plates and diluted into broth for counting and CFU estimation. Following incubation of the eight plates described above, 1-mm colonies were serially passaged two more times at 23-h intervals and then inoculated into 5 ml LB broth and incubated overnight. To prepare DNA for sequencing, cultures were spun at 5,000 × g for 10 min. Then, the supernatants were discarded and DNA was extracted using Qiagen DNeasy kits per kit instructions. DNA concentrations were measured by Qubit.
Sequencing of isolates from mutational accumulation experiments.
Six isolates each from Paer_3 and Paer_6 lineages were sequenced as follows. Libraries for Illumina MiSeq sequencing were prepared using the Nextera XT library prep kit (Illumina, San Diego, CA) by following the manufacturer’s instructions. Prepared libraries were normalized to 4 nM and sequenced on the Illumina MiSeq instrument in a paired-end 300-cycle mode using a 600-cycle reagent kit v3 (Illumina, San Diego, CA).
Rifampin reversion experiments.
One bacterial colony resuspended in 20 ml of Mueller-Hinton broth (MHB) was cultured overnight at 35°C. Bacterial cells were pelleted and resuspended in 1 ml of normal saline, and 10-fold serial dilutions were prepared in normal saline. Volumes of 100 μl of each dilution were plated on Mueller-Hinton agar (MHA) with and without 300 μg of rifampin/ml. After 48 h of aerobic incubation at 35°C, colonies were counted and the frequency of mutants was estimated. For mutS rifampin reversion experiments, MPAO1 MutS– (two allele transposon library strain PW7149 with genotype mutS-D02::ISlacZ/hah; purchased from the Colin Manoil laboratory, University of Washington) was used.
In vitro adaptive evolution experiments.
Serial CZA passaging was performed in 96-well plates with LB broth containing a concentration of ceftazidime in 2-fold serial dilutions ranging from 0.5 to 512 μg/ml with a fixed avibactam concentration of 4 μg/ml. The plates were initially inoculated with ∼4 × 106 cells each from the MPAO1 wild type (MPAO1 WT) and MPAO1 MutS– in quadruplicate in parallel. After 18 to 24 h of aerobic incubation at 35°C, cells from each population with the highest concentration well with growth (defined as an optical density at 600 nm [OD600] of >0.2 after background subtraction) were diluted 100-fold and reinoculated into fresh medium as before, containing the full range of CZA concentrations. This serial passaging was repeated for a total of 20 iterations. The P value for the MPAO1 WT and MPAO1 MutS– comparison presented in Fig. 4B was calculated using a t test.
Simulation of mutational acquisition as a function of MutS inactivation step.
A simulation was performed using the following scenario. Mutations are acquired according to a neutral model (66% transitions) until MutS inactivation, after which point mutations accumulate following an MMR-deficient mutation spectrum (98% transitions) (Fig. S3A). If mutations are acquired independently, acquisition of transversions can be approximated as the sum of two binomial distributions:
| (1) |
In equation 1, k is the MutS inactivation step, Pk(Tv = x) is the probability of observing x transversions, and TvWT and TvMutS are transversion proportions in wild-type and MMR-deficient strains, respectively. Since, in general, the sum of two binomial distributions does not have a simple analytical solution, we ran a numerical simulation over the full range of possible MutS inactivation steps (1 to 14). After simulation, we calculated the probability of observing a single transversion out of 14 mutations depending on the MutS inactivation step (Fig. S3B) and the 5th to 95th percentile range for the number of transversions that are expected to be observed depending on the MutS inactivation step (Fig. S3C).
Next we performed a calculation assuming that the blaPDC-5 mutation is preceded by three less-protective mutations in mexB, nalD/bepR, and clpA and therefore happened no earlier than on the 4th step (see Results). We can then approximate the probability of the blaPDC-5 mutation preceding MutS inactivation by summing over the previously derived probability of MutS inactivation on a given step:
| (2) |
In equation 2, k is the MutS inactivation step, P(PDC < MutS) is the probability of the blaPDC-5 mutation preceding MutS inactivation, and [k(MutS) is the probability of MutS inactivation on the kth step conditioned to have one transversion over the evolutionary course. This probability is derived from the probability distribution in Fig. S3B; P(pdc < k) is the probability of finding the PDC-5 mutation prior to the kth step. Under the simplest assumption that the blaPDC-5 mutation is acquired randomly with uniform probability, P(pdc < k) is equal to (k − 4)/10. By performing calculations over the range of 10 possible values of k, and using as before 98% and 66% transition proportions for MutS– and wild-type P. aeruginosa, respectively, we determine that the probability of the blaPDC-5 mutation preceding MutS inactivation is P(PDC < MutS) = 0.14.
Data availability.
Sequencing data have been deposited in the NCBI Sequence Read Archive under accession number PRJNA562735.
ACKNOWLEDGMENTS
This work was supported in part by the Intramural Research Programs of the National Institutes of Health Clinical Center (P.P.K., A.D.C., J.H., J.H.-Y., J.K.L., J.G.-B., K.M.F., J.P.D.) and National Institute of Allergy and Infectious Disease (P.P.K., A.D.C., J.P.D.). Research reported in this publication was supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers R01AI100560, R01AI063517, and R01AI072219 to R.A.B. This study was supported in part by funds and/or facilities provided by the Cleveland Department of Veterans Affairs, award number 1I01BX001974, to R.A.B. and from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development and the Geriatric Research Education and Clinical Center VISN 10 to R.A.B. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract no. HHSN261200800001E (M.P.).
The content of this publication is solely the responsibility of the authors and does not necessarily reflect the official views or policies of the Department of Health and Human Services, National Institutes of Health, or the Department of Veterans Affairs, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
The P. aeruginosa two allele transposon library strains used in this work were purchased from the laboratory of Colin Manoil at the University of Washington and produced there with funding from grant no. NIH P30 DK089507. We thank the staff of the NIH Microbiology Service, Department of Laboratory Medicine and Clinical Center, for their contributions to this study. This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov).
J. Lemon is presently employed by and receives a salary from OpGen, Inc. OpGen, Inc., played no role in this study. R. Bonomo reports current or prior research support from Roche, Shionogi, Merck, Achaogen, Allecra, Entasis, and Wockhardt, none of which funded or played any role in this study. The other authors declare no conflicts of interest.
Footnotes
Citation Khil PP, Chiang AD, Ho J, Youn J-H, Lemon JK, Gea-Banacloche J, Frank KM, Parta M, Bonomo RA, Dekker JP. 2019. Dynamic emergence of mismatch repair deficiency facilitates rapid evolution of ceftazidime-avibactam resistance in Pseudomonas aeruginosa acute infection. mBio 10:e01822-19. https://doi.org/10.1128/mBio.01822-19.
REFERENCES
- 1.Kimura M. 1967. On the evolutionary adjustment of spontaneous mutation rates. Genet Res 9:23–34. doi: 10.1017/S0016672300010284. [DOI] [Google Scholar]
- 2.Eigen M. 1971. Molecular self-organization and the early stages of evolution. Q Rev Biophys 4:149–212. doi: 10.1017/S0033583500000627. [DOI] [PubMed] [Google Scholar]
- 3.Drake JW. 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci U S A 88:7160–7164. doi: 10.1073/pnas.88.16.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drake JW, Charlesworth B, Charlesworth D, Crow JF. 1998. Rates of spontaneous mutation. Genetics 148:1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funchain P, Yeung A, Stewart JL, Lin R, Slupska MM, Miller JH. 2000. The consequences of growth of a mutator strain of Escherichia coli as measured by loss of function among multiple gene targets and loss of fitness. Genetics 154:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giraud A, Matic I, Tenaillon O, Clara A, Radman M, Fons M, Taddei F. 2001. Costs and benefits of high mutation rates: adaptive evolution of bacteria in the mouse gut. Science 291:2606–2608. doi: 10.1126/science.1056421. [DOI] [PubMed] [Google Scholar]
- 7.de Visser JA. 2002. The fate of microbial mutators. Microbiology 148:1247–1252. doi: 10.1099/00221287-148-5-1247. [DOI] [PubMed] [Google Scholar]
- 8.Eigen M. 2002. Error catastrophe and antiviral strategy. Proc Natl Acad Sci U S A 99:13374–13376. doi: 10.1073/pnas.212514799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biebricher CK, Eigen M. 2005. The error threshold. Virus Res 107:117–127. doi: 10.1016/j.virusres.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Sprouffske K, Aguilar-Rodriguez J, Sniegowski P, Wagner A. 2018. High mutation rates limit evolutionary adaptation in Escherichia coli. PLoS Genet 14:e1007324. doi: 10.1371/journal.pgen.1007324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radman M, Taddei F, Matic I. 2000. DNA repair systems and bacterial evolution. Cold Spring Harbor Symp Quant Biol 65:11–19. doi: 10.1101/sqb.2000.65.11. [DOI] [PubMed] [Google Scholar]
- 12.Bridges BA. 2001. Hypermutation in bacteria and other cellular systems. Philos Trans R Soc Lond B Biol Sci 356:29–39. doi: 10.1098/rstb.2000.0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang W. 2008. Structure and mechanism for DNA lesion recognition. Cell Res 18:184–197. doi: 10.1038/cr.2007.116. [DOI] [PubMed] [Google Scholar]
- 14.Taddei F, Radman M, Maynard-Smith J, Toupance B, Gouyon PH, Godelle B. 1997. Role of mutator alleles in adaptive evolution. Nature 387:700–702. doi: 10.1038/42696. [DOI] [PubMed] [Google Scholar]
- 15.Giraud A, Radman M, Matic I, Taddei F. 2001. The rise and fall of mutator bacteria. Curr Opin Microbiol 4:582–585. doi: 10.1016/S1369-5274(00)00254-X. [DOI] [PubMed] [Google Scholar]
- 16.Bjedov I, Tenaillon O, Gerard B, Souza V, Denamur E, Radman M, Taddei F, Matic I. 2003. Stress-induced mutagenesis in bacteria. Science 300:1404–1409. doi: 10.1126/science.1082240. [DOI] [PubMed] [Google Scholar]
- 17.Boshoff HI, Reed MB, Barry CE III, Mizrahi V. 2003. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 113:183–193. doi: 10.1016/S0092-8674(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 18.Weigand MR, Sundin GW. 2012. General and inducible hypermutation facilitate parallel adaptation in Pseudomonas aeruginosa despite divergent mutation spectra. Proc Natl Acad Sci U S A 109:13680–13685. doi: 10.1073/pnas.1205357109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chao L, Cox EC. 1983. Competition between high and low mutating strains of Escherichia coli. Evolution 37:125–134. doi: 10.1111/j.1558-5646.1983.tb05521.x. [DOI] [PubMed] [Google Scholar]
- 20.Mao EF, Lane L, Lee J, Miller JH. 1997. Proliferation of mutators in A cell population. J Bacteriol 179:417–422. doi: 10.1128/jb.179.2.417-422.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller JH, Suthar A, Tai J, Yeung A, Truong C, Stewart JL. 1999. Direct selection for mutators in Escherichia coli. J Bacteriol 181:1576–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gentile CF, Yu SC, Serrano SA, Gerrish PJ, Sniegowski PD. 2011. Competition between high- and higher-mutating strains of Escherichia coli. Biol Lett 7:422–424. doi: 10.1098/rsbl.2010.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeClerc JE, Li B, Payne WL, Cebula TA. 1996. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science 274:1208–1211. doi: 10.1126/science.274.5290.1208. [DOI] [PubMed] [Google Scholar]
- 24.Matic I, Radman M, Taddei F, Picard B, Doit C, Bingen E, Denamur E, Elion J. 1997. Highly variable mutation rates in commensal and pathogenic Escherichia coli. Science 277:1833–1834. doi: 10.1126/science.277.5333.1833. [DOI] [PubMed] [Google Scholar]
- 25.Oliver A, Canton R, Campo P, Baquero F, Blazquez J. 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 26.Denamur E, Bonacorsi S, Giraud A, Duriez P, Hilali F, Amorin C, Bingen E, Andremont A, Picard B, Taddei F, Matic I. 2002. High frequency of mutator strains among human uropathogenic Escherichia coli isolates. J Bacteriol 184:605–609. doi: 10.1128/jb.184.2.605-609.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chopra I, O’Neill AJ, Miller K. 2003. The role of mutators in the emergence of antibiotic-resistant bacteria. Drug Resist Updat 6:137–145. doi: 10.1016/S1368-7646(03)00041-4. [DOI] [PubMed] [Google Scholar]
- 28.Blazquez J. 2003. Hypermutation as a factor contributing to the acquisition of antimicrobial resistance. Clin Infect Dis 37:1201–1209. doi: 10.1086/378810. [DOI] [PubMed] [Google Scholar]
- 29.Horst JP, Wu TH, Marinus MG. 1999. Escherichia coli mutator genes. Trends Microbiol 7:29–36. doi: 10.1016/S0966-842X(98)01424-3. [DOI] [PubMed] [Google Scholar]
- 30.Boe L, Danielsen M, Knudsen S, Petersen JB, Maymann J, Jensen PR. 2000. The frequency of mutators in populations of Escherichia coli. Mutat Res 448:47–55. doi: 10.1016/s0027-5107(99)00239-0. [DOI] [PubMed] [Google Scholar]
- 31.Friedberg EC, Wagner R, Radman M. 2002. Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science 296:1627–1630. doi: 10.1126/science.1070236. [DOI] [PubMed] [Google Scholar]
- 32.Matic I, Taddei F, Radman M. 2004. Survival versus maintenance of genetic stability: a conflict of priorities during stress. Res Microbiol 155:337–341. doi: 10.1016/j.resmic.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 33.Gutierrez A, Laureti L, Crussard S, Abida H, Rodriguez-Rojas A, Blazquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, Matic I. 2013. beta-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat Commun 4:1610. doi: 10.1038/ncomms2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oliver A, Mena A. 2010. Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin Microbiol Infect 16:798–808. doi: 10.1111/j.1469-0691.2010.03250.x. [DOI] [PubMed] [Google Scholar]
- 35.Alonso A, Campanario E, Martínez JL. 1999. Emergence of multidrug-resistant mutants is increased under antibiotic selective pressure in Pseudomonas aeruginosa. Microbiology 145:2857–2862. doi: 10.1099/00221287-145-10-2857. [DOI] [PubMed] [Google Scholar]
- 36.Giraud A, Matic I, Radman M, Fons M, Taddei F. 2002. Mutator bacteria as a risk factor in treatment of infectious diseases. Antimicrob Agents Chemother 46:863–865. doi: 10.1128/aac.46.3.863-865.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller K, O’Neill AJ, Chopra I. 2002. Response of Escherichia coli hypermutators to selection pressure with antimicrobial agents from different classes. J Antimicrob Chemother 49:925–934. doi: 10.1093/jac/dkf044. [DOI] [PubMed] [Google Scholar]
- 38.Blázquez J, Rodríguez-Beltrán J, Matic I. 2018. Antibiotic-induced genetic variation: how it arises and how it can be prevented. Annu Rev Microbiol 72:209–230. doi: 10.1146/annurev-micro-090817-062139. [DOI] [PubMed] [Google Scholar]
- 39.Bjorkman J, Nagaev I, Berg OG, Hughes D, Andersson DI. 2000. Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance. Science 287:1479–1482. doi: 10.1126/science.287.5457.1479. [DOI] [PubMed] [Google Scholar]
- 40.Kidd TJ, Ritchie SR, Ramsay KA, Grimwood K, Bell SC, Rainey PB. 2012. Pseudomonas aeruginosa exhibits frequent recombination, but only a limited association between genotype and ecological setting. PLoS One 7:e44199. doi: 10.1371/journal.pone.0044199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Witney AA, Gould KA, Pope CF, Bolt F, Stoker NG, Cubbon MD, Bradley CR, Fraise A, Breathnach AS, Butcher PD, Planche TD, Hinds J. 2014. Genome sequencing and characterization of an extensively drug-resistant sequence type 111 serotype O12 hospital outbreak strain of Pseudomonas aeruginosa. Clin Microbiol Infect 20:O609–18. doi: 10.1111/1469-0691.12528. [DOI] [PubMed] [Google Scholar]
- 42.Kidd TJ, Ramsay KA, Vidmar S, Carlin JB, Bell SC, Wainwright CE, Grimwood K, AFCBAL Study Investigators. 2015. Pseudomonas aeruginosa genotypes acquired by children with cystic fibrosis by age 5-years. J Cyst Fibros 14:361–369. doi: 10.1016/j.jcf.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 43.Hammond JH, Hebert WP, Naimie A, Ray K, Van Gelder RD, DiGiandomenico A, Lalitha P, Srinivasan M, Acharya NR, Lietman T, Hogan DA, Zegans ME. 2016. Environmentally endemic Pseudomonas aeruginosa strains with mutations in lasR are associated with increased disease severity in corneal ulcers. mSphere 1:e00140-16. doi: 10.1128/mSphere.00140-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rodriguez-Martinez JM, Poirel L, Nordmann P. 2009. Extended-spectrum cephalosporinases in Pseudomonas aeruginosa. Antimicrob Agents Chemother 53:1766–1771. doi: 10.1128/AAC.01410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cabot G, Bruchmann S, Mulet X, Zamorano L, Moya B, Juan C, Haussler S, Oliver A. 2014. Pseudomonas aeruginosa ceftolozane-tazobactam resistance development requires multiple mutations leading to overexpression and structural modification of AmpC. Antimicrob Agents Chemother 58:3091–3099. doi: 10.1128/AAC.02462-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lahiri SD, Walkup GK, Whiteaker JD, Palmer T, McCormack K, Tanudra MA, Nash TJ, Thresher J, Johnstone MR, Hajec L, Livchak S, McLaughlin RE, Alm RA. 2015. Selection and molecular characterization of ceftazidime/avibactam-resistant mutants in Pseudomonas aeruginosa strains containing derepressed AmpC. J Antimicrob Chemother 70:1650–1658. doi: 10.1093/jac/dkv004. [DOI] [PubMed] [Google Scholar]
- 47.MacVane SH, Pandey R, Steed LL, Kreiswirth BN, Chen L. 2017. Emergence of ceftolozane-tazobactam-resistant Pseudomonas aeruginosa during treatment is mediated by a single AmpC structural mutation. Antimicrob Agents Chemother 61:e01183-17. doi: 10.1128/AAC.01183-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Acharya S, Patterson K. 2010. Mutations in the conserved glycine and serine of the MutS ABC signature motif affect nucleotide exchange, kinetics of sliding clamp release of mismatch and mismatch repair. Mutat Res 684:56–65. doi: 10.1016/j.mrfmmm.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 49.Martín-López JV, Fishel R. 2013. The mechanism of mismatch repair and the functional analysis of mismatch repair defects in Lynch syndrome. Fam Cancer 12:159–168. doi: 10.1007/s10689-013-9635-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nielsen SV, Stein A, Dinitzen AB, Papaleo E, Tatham MH, Poulsen EG, Kassem MM, Rasmussen LJ, Lindorff-Larsen K, Hartmann-Petersen R. 2017. Predicting the impact of Lynch syndrome-causing missense mutations from structural calculations. PLoS Genet 13:e1006739. doi: 10.1371/journal.pgen.1006739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garibyan L, Huang T, Kim M, Wolff E, Nguyen A, Nguyen T, Diep A, Hu K, Iverson A, Yang H, Miller JH. 2003. Use of the rpoB gene to determine the specificity of base substitution mutations on the Escherichia coli chromosome. DNA Repair (Amst) 2:593–608. doi: 10.1016/S1568-7864(03)00024-7. [DOI] [PubMed] [Google Scholar]
- 52.Lynch M, Ackerman MS, Gout JF, Long H, Sung W, Thomas WK, Foster PL. 2016. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet 17:704–714. doi: 10.1038/nrg.2016.104. [DOI] [PubMed] [Google Scholar]
- 53.Jorth P, McLean K, Ratjen A, Secor PR, Bautista GE, Ravishankar S, Rezayat A, Garudathri J, Harrison JJ, Harwood RA, Penewit K, Waalkes A, Singh PK, Salipante SJ. 2017. Evolved aztreonam resistance is multifactorial and can produce hypervirulence in Pseudomonas aeruginosa. mBio 8:e00517-17. doi: 10.1128/mBio.00517-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanz-Garcia F, Hernando-Amado S, Martinez JL. 2018. Mutation-driven evolution of Pseudomonas aeruginosa in the presence of either ceftazidime or ceftazidime-avibactam. Antimicrob Agents Chemother 62:e01379-18. doi: 10.1128/AAC.01379-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chalhoub H, Saenz Y, Nichols WW, Tulkens PM, Van Bambeke F. 2018. Loss of activity of ceftazidime-avibactam due to MexAB-OprM efflux and overproduction of AmpC cephalosporinase in Pseudomonas aeruginosa isolated from patients suffering from cystic fibrosis. Int J Antimicrob Agents 52:697–701. doi: 10.1016/j.ijantimicag.2018.07.027. [DOI] [PubMed] [Google Scholar]
- 56.Du D, Wang-Kan X, Neuberger A, van Veen HW, Pos KM, Piddock LJV, Luisi BF. 2018. Multidrug efflux pumps: structure, function and regulation. Nat Rev Microbiol 16:523–539. doi: 10.1038/s41579-018-0048-6. [DOI] [PubMed] [Google Scholar]
- 57.Sennhauser G, Bukowska MA, Briand C, Grutter MG. 2009. Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J Mol Biol 389:134–145. doi: 10.1016/j.jmb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 58.Sun J, Deng Z, Yan A. 2014. Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochem Biophys Res Commun 453:254–267. doi: 10.1016/j.bbrc.2014.05.090. [DOI] [PubMed] [Google Scholar]
- 59.Sobel ML, Hocquet D, Cao L, Plesiat P, Poole K. 2005. Mutations in PA3574 (nalD) lead to increased MexAB-OprM expression and multidrug resistance in laboratory and clinical isolates of Pseudomonas aeruginosa. Antimicrob Agents Chemother 49:1782–1786. doi: 10.1128/AAC.49.5.1782-1786.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Culp E, Wright GD. 2017. Bacterial proteases, untapped antimicrobial drug targets. J Antibiot (Tokyo) 70:366–377. doi: 10.1038/ja.2016.138. [DOI] [PubMed] [Google Scholar]
- 61.Fernandez L, Breidenstein EB, Song D, Hancock RE. 2012. Role of intracellular proteases in the antibiotic resistance, motility, and biofilm formation of Pseudomonas aeruginosa. Antimicrob Agents Chemother 56:1128–1132. doi: 10.1128/AAC.05336-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Held K, Ramage E, Jacobs M, Gallagher L, Manoil C. 2012. Sequence-verified two-allele transposon mutant library for Pseudomonas aeruginosa PAO1. J Bacteriol 194:6387–6389. doi: 10.1128/JB.01479-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee H, Popodi E, Tang H, Foster PL. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A 109:E2774–E2783. doi: 10.1073/pnas.1210309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dettman JR, Sztepanacz JL, Kassen R. 2016. The properties of spontaneous mutations in the opportunistic pathogen Pseudomonas aeruginosa. BMC Genomics 17:27. doi: 10.1186/s12864-015-2244-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cabot G, Zamorano L, Moya B, Juan C, Navas A, Blazquez J, Oliver A. 2016. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrob Agents Chemother 60:1767–1778. doi: 10.1128/AAC.02676-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Andersson DI, Hughes D. 2014. Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol 12:465–478. doi: 10.1038/nrmicro3270. [DOI] [PubMed] [Google Scholar]
- 68.Hughes D, Andersson DI. 2017. Evolutionary trajectories to antibiotic resistance. Annu Rev Microbiol 71:579–596. doi: 10.1146/annurev-micro-090816-093813. [DOI] [PubMed] [Google Scholar]
- 69.Wang K, Chen YQ, Salido MM, Kohli GS, Kong JL, Liang HJ, Yao ZT, Xie YT, Wu HY, Cai SQ, Drautz-Moses DI, Darling AE, Schuster SC, Yang L, Ding Y. 2017. The rapid in vivo evolution of Pseudomonas aeruginosa in ventilator-associated pneumonia patients leads to attenuated virulence. Open Biol 7:170029. doi: 10.1098/rsob.170029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Persyn E, Sassi M, Aubry M, Broly M, Delanou S, Asehnoune K, Caroff N, Cremet L. 2019. Rapid genetic and phenotypic changes in Pseudomonas aeruginosa clinical strains during ventilator-associated pneumonia. Sci Rep 9:4720. doi: 10.1038/s41598-019-41201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Livermore DM, Warner M, Jamrozy D, Mushtaq S, Nichols WW, Mustafa N, Woodford N. 2015. In vitro selection of ceftazidime-avibactam resistance in Enterobacteriaceae with KPC-3 carbapenemase. Antimicrob Agents Chemother 59:5324–5330. doi: 10.1128/AAC.00678-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Humphries RM, Yang S, Hemarajata P, Ward KW, Hindler JA, Miller SA, Gregson A. 2015. First report of ceftazidime-avibactam resistance in a KPC-3-expressing Klebsiella pneumoniae isolate. Antimicrob Agents Chemother 59:6605–6607. doi: 10.1128/AAC.01165-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shields RK, Nguyen MH, Press EG, Chen L, Kreiswirth BN, Clancy CJ. 2017. In vitro selection of meropenem resistance among ceftazidime-avibactam-resistant, meropenem-susceptible Klebsiella pneumoniae isolates with variant KPC-3 carbapenemases. Antimicrob Agents Chemother 61:e00079-17. doi: 10.1128/AAC.00079-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giddins MJ, Macesic N, Annavajhala MK, Stump S, Khan S, McConville TH, Mehta M, Gomez-Simmonds A, Uhlemann AC. 2018. Successive emergence of ceftazidime-avibactam resistance through distinct genomic adaptations in blaKPC-2-harboring Klebsiella pneumoniae sequence type 307 isolates. Antimicrob Agents Chemother 62:e02101-17. doi: 10.1128/AAC.02101-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gaibani P, Campoli C, Lewis RE, Volpe SL, Scaltriti E, Giannella M, Pongolini S, Berlingeri A, Cristini F, Bartoletti M, Tedeschi S, Ambretti S. 2018. In vivo evolution of resistant subpopulations of KPC-producing Klebsiella pneumoniae during ceftazidime/avibactam treatment. J Antimicrob Chemother 73:1525–1529. doi: 10.1093/jac/dky082. [DOI] [PubMed] [Google Scholar]
- 76.Radman M, Taddei F, Matic I. 2000. Evolution-driving genes. Res Microbiol 151:91–95. doi: 10.1016/S0923-2508(00)00122-4. [DOI] [PubMed] [Google Scholar]
- 77.Lynch M. 2010. Evolution of the mutation rate. Trends Genet 26:345–352. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gifford DR, Furio V, Papkou A, Vogwill T, Oliver A, MacLean RC. 2018. Identifying and exploiting genes that potentiate the evolution of antibiotic resistance. Nat Ecol Evol 2:1033–1039. doi: 10.1038/s41559-018-0547-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.CLSI. 2017. Performance standards for antimicrobial susceptibility testing, 27th ed Supplement M100 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Isolate sequencing summary. Download Table S1, DOCX file, 0.01 MB (13.6KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Mutation classes found in late-course CZA-resistant isolates. Download Table S2, DOCX file, 0.01 MB (13.5KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
In vitro mutation accumulation in MutS-deficient isolates. (A) Design and passaging scheme of a mutation accumulation experiment. Early-course CZA-susceptible isolate with WT MutS (Paer_3) and late-course, CZA-resistant isolate with MutS R656H substitution (Paer_6) were passaged for ∼150 generations in parallel and then sequenced. (B) Genomic distribution of SNPs detected by WGS following mutation accumulation. Each genome is represented as a large concentric circle, and positions with mutations are indicated with small blue (reference) and red (mutant) circles. During the course of the experiment, isolates with intact MutS (wt) acquired a single mutation, whereas 18 substitutions (all transitions) and 5 small indels (not shown) accumulated in isolates with the R656H variant of MutS. Download FIG S1, EPS file, 0.4 MB (434.4KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Rifampin reversion frequencies for Paer_3, Paer_6, and Paer_9. Results are shown for two experiments (n = 3 replicates). Download Table S3, DOCX file, 0.01 MB (14KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Mutation classes found in the mutation accumulation experiment in a late-course, CZA-resistant isolate with the MutS R656H substitution (Paer_6) following ∼150 generations. All observed mutations are transitions. Download Table S4, DOCX file, 0.01 MB (13.6KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Serial passaging of the WT MPAO1 and MMR-deficient MPAO1 derivative under CZA selection (full data for Fig. 4). Heatmap of maximum CZA concentration (μg/ml of ceftazidime) for which growth was detected versus passage number. Four independent lineages (A to D) were passaged in parallel for WT MPAO1 and MPAO1 MutS– isolates over a gradient of increasing CZA concentrations (see Materials and Methods for details of experimental design). Download FIG S2, EPS file, 0.2 MB (243.1KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Mutation acquisition as a function of MutS inactivation step. (A) Schematic representation of the simulation approach. Mutations are acquired using the wild-type mutation spectrum until MutS is inactivated, after which point mutations accumulate following an MMR-deficient mutation spectrum. Mutations are assumed to be independent. (B) The probability of observing one transversion out of 14 mutations depending on MutS inactivation step. (C) Fifth to 95th percentile ranges of numbers of substitutions for each of the evolutionary scenarios. Blue squares indicates median numbers of transversions. Download FIG S3, EPS file, 0.6 MB (673KB, eps) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Literature data summary of P. aeruginosa mutation accumulation during the course of acute infection in 8 patients. Download Table S5, DOCX file, 0.02 MB (20.5KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data Availability Statement
Sequencing data have been deposited in the NCBI Sequence Read Archive under accession number PRJNA562735.