

Separation of early to late Golgi cisternae shows the sequential localization of resident proteins and the sequence features that guide transmembrane proteins within the Golgi stack.
Abstract
The order of enzymatic activity across Golgi cisternae is essential for complex molecule biosynthesis. However, an inability to separate Golgi cisternae has meant that the cisternal distribution of most resident proteins, and their underlying localization mechanisms, are unknown. Here, we exploit differences in surface charge of intact cisternae to perform separation of early to late Golgi subcompartments. We determine protein and glycan abundance profiles across the Golgi; over 390 resident proteins are identified, including 136 new additions, with over 180 cisternal assignments. These assignments provide a means to better understand the functional roles of Golgi proteins and how they operate sequentially. Protein and glycan distributions are validated in vivo using high-resolution microscopy. Results reveal distinct functional compartmentalization among resident Golgi proteins. Analysis of transmembrane proteins shows several sequence-based characteristics relating to pI, hydrophobicity, Ser abundance, and Phe bilayer asymmetry that change across the Golgi. Overall, our results suggest that a continuum of transmembrane features, rather than discrete rules, guide proteins to earlier or later locations within the Golgi stack.
INTRODUCTION
The Golgi is an ancient organelle, common to all eukaryotic lineages (Klute et al., 2011), consisting of a stack of flattened, membranous discs, or cisternae, in which protein and lipid cargoes are modified in a progressive manner and substituted with complex glycan side chains (Ito et al., 2014; Strasser, 2016; van de Meene et al., 2017). The Golgi is the hub of the secretory pathway, trafficking cargo-containing vesicles to and from the endoplasmic reticulum (ER) at the cis face (Brandizzi and Barlowe, 2013) and to other cellular destinations at the trans face (Gendre et al., 2015). There have been important advances in understanding trafficking processes from the trans-Golgi network (TGN) to post-TGN destinations (Xiang et al., 2013; Robinson and Pimpl, 2014; Heard et al., 2015), and many regulatory components of ER to cis-Golgi traffic have been determined (Brandizzi and Barlowe, 2013; Hawes et al., 2015). However, our understanding of the trafficking pathways within the Golgi stack itself, and the mechanisms underlying spatial partitioning of proteins within stacks, is still somewhat limited.

Studying secretory organelle organization not only contributes to a general understanding of biochemical pathways and how protein localization is specified but also gives us the capacity to better control the complex, sequential biochemistry and trafficking processes of cellular secretion. Although understanding of how sequence characteristics localize proteins to organelles has advanced (Sharpe et al., 2010), no general sequence-based determinants of Golgi cisternal membrane localization are known (Banfield, 2011). Transmembrane (TM) span length, retrieval, and retention motifs (Saint-Jore-Dupas et al., 2006; Schoberer and Strasser, 2011; Gao et al., 2014; Woo et al., 2015) cannot sufficiently explain the distribution of resident proteins within the Golgi, implicating undiscovered factors governing intra-Golgi protein localization. Cutting-edge microscopy has localized a limited number of Golgi proteins (Tie et al., 2016), although tagging membrane proteins can increase aberrant localization (Stadler et al., 2013). Consequently, too few proteins have been accurately localized within the Golgi to identify cisternal targeting sequences or map intra-Golgi trafficking pathways.
Modern mass spectrometry, using multiple separation stages and peptide mass fingerprinting, provides a way of simultaneously detecting and quantifying the occurrence of thousands of proteins in purified and enriched samples. This has allowed the compilation of proteome sets for subcellular compartments. Generally, these comparative proteomic analyses, which have proved essential to our understanding of vesicular trafficking (Gilchrist et al., 2006; Heard et al., 2015), depend on some degree of physical separation of compartments. Here, the localization of organelle proteins by isotope tagging (LOPIT) technique, using density gradient centrifugation, has become the gold standard for subcellular proteome discovery (Mulvey et al., 2017) and has provided ER, Golgi, and TGN proteomes in Arabidopsis (Arabidopsis thaliana; Dunkley et al., 2006; Nikolovski et al., 2012; Groen et al., 2014). However, to date, only electrophoresis techniques have delivered adequate separation of Golgi cisternae. Free-flow electrophoresis (FFE) has been shown to separate vesicles according to small differences in surface charge (Barkla et al., 2007; Islinger et al., 2010). Although early attempts to separate the ER, Golgi cisternae, and TGN using FFE were promising (Morré and Mollenhauer, 2009), contemporary technical limitations prevented proper follow-up and validation. In this study, we separate the Golgi subcompartments in an endomembrane-enriched sample from an Arabidopsis cell-suspension culture using FFE.
Plant suspension-culture cells are an attractive option for studying the endomembrane, as they generate large quantities of intact Golgi cisternae (Parsons et al., 2012). Centrifugation and gentle manipulation under negative pressure efficiently unstack cisternae, which can be enriched on a simple step gradient. A gradient of surface charge, likely resulting from flipping of negatively charged phospholipids to the outer leaflet, exists between the ER, Golgi, TGN, and plasma membrane (PM; Morré and Mollenhauer, 2009; Parsons et al., 2012) and appears to exist across Golgi cisternae, which facilitates electrophoretic separation.
Here, we combine gentle electrophoretic fractionation of largely intact endomembranes with high-throughput mass spectrometry, bioinformatics, and imaging techniques to create one of the largest experimental data sets in this field to date. We use both LOPIT and FFE abundance profiles to determine the localization of hundreds of resident proteins, protein cargo, and glycan cargo through the secretory pathway at sub-Golgi resolution. Our approach is validated in several ways, including using glycan immunogold transmission electron microscopy and protein fluorescence microscopy. We show sub-Golgi categorizations that are consistent with the progressive glycosylation functions of the Golgi. This then allows us to bioinformatically analyze sub-Golgi-specific protein sequences to discover any trends or rules that may contribute to cisternal localization.
RESULTS
Experimental Inputs
Using FFE, we separated an endomembrane-enriched homogenate into 96 fractions according to surface charge. For each replicate sample, ∼45 fractions with significant endomembrane protein content were selected in each case and analyzed using shotgun proteomic mass spectrometry to gauge the identity and relative amount of each protein in each fraction. A schematic representation of our approach, using gentle separation of intact membrane samples, mass spectrometric proteomic identification, and subsequent abundance profile generation, is illustrated in Figure 1.
Figure 1.

Schematic Overview of Electrophoretic Separation Profile Analysis of Endomembrane Proteins.
(A) Samples from Arabidopsis cell-suspension cultures, enriched in intact endomembranes, were separated by voltage under laminar flow (i.e., using FFE). This provided gentle separation of membrane-bound compartments, according to their surface charges, and resulted in 96 separately collected fractions, ordered along the voltage axis.
(B) Total protein content of FFE fractions was determined via absorption at 280 nm to identify the range of fractions with major endomembrane protein enrichment. These and adjacent fractions were then taken forward for more detailed analysis. Nonmembrane components from the samples peaked in early fractions outside this range.
(C) Endomembrane fractions were primarily investigated using shotgun proteomics to measure the relative amounts of the different proteins contained therein. Here, proteins were identified via the mass fingerprints of trypic digest peptides searched against the most recent Arabidopsis proteome using MASCOT software.
(D) Average FFE abundance profiles for resident proteins from Golgi, ER, and other organelles using independent subcellular localizations derived from LOPIT analysis (Supplemental Data Set 1). Protein abundance values from multiple replicate FFE runs, in the form of spectral intensities, were combined (see Methods for the fraction-matching procedure), generating 25 merged, consensus endomembrane fractions. Combined data are shown for totals of 200 ER, 204 Golgi, and 1290 other organelle proteins.
Preliminary investigations with two biological replicate samples (R1 and R2), performed with an TripleTOF 5600 System (ABSciex), identified over 1500 proteins and established the basic utility of our approach (and R1, which contained more material than R2, was later used for glycan/carbohydrate analysis). This was then followed up with three high-sensitivity replicates (R3, R4, and R5) using anOrbitrap Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific), which detected over 2700 proteins and formed the basis of our main analysis.
Establishing Updated Subproteomes for the Golgi and Other Organelles
Before we could begin to dissect any cisternal separation of Golgi proteins, our first task was to establish updated protein sets of resident proteins for the Golgi and other membrane-bound compartments within our cell line. Current plant protein annotations sometimes contain contradictory locational information, often with no indication of which proteins are organelle residents or localize to multiple organelles. This is problematic when analyzing the Golgi, as distinguishing between cisternal residents, cargo, and vesicular proteins is essential. It was especially important to generate accurate, updated ER and TGN proteomes; the ER showed the closest degree of FFE fraction overlap with the Golgi (Figure 2A), and dual-localized ER-Golgi proteins were expected. Electrophoretic migration of the TGN was difficult to distinguish from the Golgi, as TGN proteins are both trafficked through and exchanged with the Golgi. Hence, updating the TGN proteome enabled TGN cargo to be distinguished from Golgi residents.
Figure 2.

Primary Determination of Organelle Subproteomes.
(A) PCA analysis of a single LOPIT experiment. Protein abundance profiles from density-based separation are presented by projection onto their two principal, orthogonal axes, representing most interprotein variance. Each point represents a single protein, which is colored according to its organelle classification. Organelle clusters were distinguished using multiple-class SVM on complete abundance profiles and used existing annotations for classification (see Methods).
(B) Presentation of the same LOPIT data and classifications shown in (A), presented as a 2D t-SNE plot. This visualization attempts to preserve the proximity of similar profiles, and the separation of distinct profiles, over all data dimensions (whole profiles). This is unlike PCA, which shows (dis)similarity along the selected projection axes.
(C) Average FFE profiles, across 25 merged fractions from replicates R3 to R5, are shown for organelle groups classified using LOPIT data. Plotted values represent the mean abundance for each fraction in each organelle class from per-protein normalized profiles (see Methods). Error bars represent the se. Data are shown separately for the ER and Golgi (upper plot), which peak as a class in central fractions, and the distinct profiles for other organelles/compartments (lower plot). ER and Golgi proteomes have been subdivided as either those belonging to the initial organelle markers or those newly classified as organelle residents, demonstrating the accuracy with which new residents were assigned.
(D) Hierarchical clustering of secretory (ER, Golgi, TGN, and PM) protein FFE profiles. Merged abundance profiles from proteins identified in high-quality replicates R3 to R5 were clustered using Ward’s method and presented as a dendrogram with the corresponding, underlying abundance profiles shown beneath as a color density plot, together with primary organelle classifications derived from LOPIT. The three major clusters that separated profiles generally into Golgi/TGN, ER, and PM were further separated into eight smaller clusters, labeled A to H. Here, a threshold was chosen so that each major ER and Golgi cluster contained three minor clusters.
To date, the only proteomics technique capable of distinguishing resident and cargo proteins is LOPIT. In LOPIT, organelles are separated on a linear density gradient, fractions of which are labeled using isobaric tags. Tagging enables very accurate quantitation of protein abundances along the gradients. Proteins from the same organelle have similar abundance profiles, so when, for example, principal component analysis (PCA) is applied to quantitation data, organelle residents form distinct clusters and multilocalized proteins do not. LOPIT was originally developed and validated using Arabidopsis over a decade ago (Dunkley et al., 2006). Thorough cross-validation using immunoblots and imaging, as well as technical and bioinformatic updates, have led to LOPIT becoming the technique of choice for high-accuracy, whole-cell proteomics analysis (Gatto et al., 2014; Breckels et al., 2016b; Christoforou et al., 2016; Mulvey et al., 2017; Thul et al., 2017), but it has never been reapplied to a whole-cell analysis of Arabidopsis. Updating resident organelle proteomes was therefore an essential first step in this study.
Multiple-class support vector machine (SVM)-based methods are frequently combined with LOPIT to classify proteins according to their location (Breckels et al., 2013, 2016a; Mulvey et al., 2017). Here, we used proteins with clearly annotated localizations derived from the subcellular localization database for Arabidopsis proteins (SUBA; Hooper et al., 2017a), and from Groen et al. (2014) as the initial classification inputs (Supplemental Data Set 1). This created organelle-specific clusters by partitioning the LOPIT profile data (i.e., density centrifugation profiles) according to the consensus of the initial markers. Classification parameters (see Methods) were set such that organelle clusters remained tight and were therefore most likely to contain only resident proteins. When compared against fluorescent protein localization records housed in SUBA, <5% of proteins showed conflicting localizations. Given that the Golgi has been subjected to relatively few proteomic studies, it was desirable to increase the number of known Golgi-resident proteins. Hence, the SVM classification parameters were relaxed to permit <2% conflicts. This did not affect the tightness of the Golgi cluster, meaning that accuracy was not compromised. For all organelles, proteins were only selected if present in two or more replicates.
PCA revealed tight, distinct clusters for all subcellular compartments (Figure 2A). The compartments could be largely, but not entirely, separated by projection onto only two principle components. Hence, results were also visualized using t-distributed stochastic neighbor embedding (t-SNE), which attempts to combine data from all dimensions to a two-dimensional (2D) plot (Van der Maaten and Hinton, 2008). t-SNE confirmed that clusters overlapping in Figure 2A, including the ER, Golgi, and TGN, were indeed separate (Figure 2B). Importantly, for our later analyses, the TGN group was entirely distinct from the Golgi.
LOPIT resulted in the identification of 345 ER-, 46 TGN-, and 397 Golgi-resident proteins in three spatially distinct clusters, along with comprehensive lists of resident protein markers for all other organelles (Supplemental Figure 1; Supplemental Data Set 1). The currently annotated Arabidopsis Golgi proteome (covering all cell types) is estimated at ∼530 proteins (Hooper et al., 2017b), suggesting that we identified a large majority of resident Golgi proteins present in our cell line.
Organelle FFE Protein Abundance Profiles
Having established updated, resident proteomes for all major subcellular compartments, we then used these to analyze FFE data. After merging high-sensitivity proteomic data from replicates R3 to R5 (see Methods), the combined, average FFE profiles of proteins previously known to reside in the ER and Golgi are illustrated in Figure 2C, alongside profiles for the newly assigned ER and Golgi sets from LOPIT; the newly assigned proteins had remarkably similar profiles to those of established residents. Additionally, the combined FFE profiles for all other LOPIT subcompartment classifications (Figure 2C) show that these data can be used to categorize nonendomembrane proteins as either nonsecretory contaminants or cargo. Contaminants (e.g., peroxisome, plus most chloroplast and PM proteins) had electrophoretic profiles similar to those observed in previous electrophoresis separations (Eubel et al., 2007; de Michele et al., 2016). Interestingly, some chloroplast, PM, vacuole, and mitochondrial proteins had flat profiles, which did not correspond to previous observations for those organelles (Barkla et al., 2007; Eubel et al., 2007). The subpopulation of proteins from these organelles with flat profiles was disproportionately enriched in features consistent with cargo subjected to posttranslational modifications in the Golgi. Over 40% of non-Golgi proteins identified in Golgi-enriched fractions had been found previously in vesicular trafficking proteomes (Heard et al., 2015), were S-acylated (Heard et al., 2015), contained a high-confidence N-glycosylation site (Zielinska et al., 2012), or had an experimentally determined glycosylphosphatidylinositol anchor (Yeats et al., 2018).
Overall, Golgi proteins were detected across the entire region of the selected membrane fractions and did not obviously separate into discrete surface-charge regions (e.g., corresponding to different cisternae). However, even with separate sub-Golgi proteomes, we would expect a somewhat overlapped situation here, given that resident proteins transit through, and possibly recycle via, adjacent compartments. Additionally, we are studying a superposition of different cellular and vesicular states (i.e., with varying surface charge).
As illustrated in Figure 2D for high-sensitivity replicates R3 to R5, protein profiles were hierarchically clustered according to the pattern of their merged FFE abundance along the separated fractions. This clustering effectively pairs the most similar abundance profiles, in a progressive manner, and allowed us to visualize any innate groups that may occur within the FFE data (i.e., which may correspond to different organelles and subcompartments). Given that Golgi cisternae remained largely intact during the FFE separation, we did not directly separate Golgi residents from trafficking cargo, even if we might expect resident and cargo proteins to have different, characteristic FFE profiles. Hence, to objectively assign organelle residents to FFE profile clusters with highest confidence, we used only resident proteins from Figures 2A and 2B and Supplemental Data Set 1.
When proteins with existing organelle annotations are compared by hierarchical clustering (Figure 2D; Supplemental Figure 2B), the grouped profiles clearly correspond to three major clusters: Golgi/TGN, ER and PM, which have peak abundances in different regions of the FFE profile. Overall, the Golgi/TGN proteins tend to peak in early fractions (nearer anode), ER residents come in the middle, and PM proteins come later. These features were also obvious in hierarchical clustering of the individual FFE replicate data sets R3 to R5 (Supplemental Figure 3), although they are clearest in the combined data, as expected. Also, looking within the large Golgi/TGN cluster, we can see that TGN annotations largely group together and Golgi subclusters are present. Although the TGN FFE profiles are similar to, and hence cluster with, those from the Golgi, this presents no problems for our analysis, as these compartments are entirely separate in the density centrifugation (LOPIT) analysis.
Dissecting the clustering further into minor subclusters that we label A to H, we can see that ER proteins were distributed over a larger cluster E and a smaller, higher-variance cluster F (Figure 2D). Cluster D contained ER and Golgi proteins with profiles intermediate to most ER and Golgi proteins, possibly indicating a dual-localized group. Golgi proteins could be grouped into three main clusters that appeared to form a continuum along the electrophoretic gradient. The Golgi cluster with peak abundance closest to the anode (cluster A) exhibited a zone of main protein abundance that was focused over a smaller number of fractions compared with, for example, cluster C, which was wider and peaked closer to the cathode. Clusters G and H comprised mainly PM proteins and migrated farthest toward the cathode. This is consistent with previous reports that PM vesicles come out farther toward that cathode than other endomembrane compartments (de Michele et al., 2016).
Evidence for Sub-Golgi Separation in FFE Profiles
To investigate whether the Golgi subclusters found in the FFE profiles had any correspondence with Golgi cisternae, we performed an analysis of glycans in the FFE fractions that was coupled to electron microscopy of individual cisternae and also looked at proteins with well-established cisternal identity.
Cisternal Polysaccharide Distribution
Using immunogold transmission electron microscopy (TEM), we performed an in situ analysis of glycan epitopes. These epitopes represented polysaccharides with different structural complexities, as would be found across the range of Golgi cisternae (see Supplemental Data Set 2 for details). Using TEM on samples with gold-labeled antibodies, we localized the glycans to individual cisternae with high spatial resolution.
As expected from previous analyses, glycan epitopes showed specific localizations for different Golgi membranes, with more structurally complex polysaccharides being associated with later cisternae (Figure 3A; summarized in Supplemental Data Set 2). The overall TEM results (Figures 3A and 3B) are summarized as follows. (1) Anti-extensin antibody LM1 was detected in the cis-Golgi; extensins have protein backbones, which provide a substrate for modification immediately after entering the Golgi. (2) Anti-mannan antibody LM21 was detected over cis and medial cisternae. (3) Antibodies for LM19, which recognizes partially methyl-esterified homogalacturonan, and LM15, which recognizes a simply branched, xylose-substituted epitope of xyloglucan (XG), occur early but overall have a medial distribution and peak before XG epitopes with longer side chains. (4) Anti-xyloglucan M87, which recognizes XG epitopes with medium-length side chains (xylose and galactose), was bound at late, trans cisternae. (5) Antibodies against long XG side chains, containing xylose, galactose, and fucose (M1 and M39), were also found in late cisternae. Of those polysaccharide epitopes that had been previously imaged within the Golgi, cisternal localization results matched earlier findings (Smallwood et al., 1994; Marcus et al., 2008; Viotti et al., 2010; Driouich et al., 2012).
Figure 3.

Establishing Characteristics of Early and Late Golgi FFE Profiles.
(A) Example negative-stain TEM images showing the in vivo distributions of several glycan epitopes, with varying structural complexity, across the Golgi stack. Glycans were localized using monoclonal antibodies linked to gold particles. All stacks are depicted with cis at the bottom and trans as the top, as indicated.
(B) Violin plots showing the overall data from the immunogold TEM localization of glycan epitopes, as illustrated in (A). The relative Golgi stack positions of gold particles represent the fraction of the particle distance to the outer cis face as a proportion of the total cis-trans thickness.
(C) FFE abundance profiles for four classes of glycan epitope, with varying structural complexity. Class members and epitope structures are detailed in Supplemental Data Set 2. Data are shown for detergent-extracted samples from FFE replicate R1 that were printed onto nitrocellulose microarrays and probed via alkaline phosphatase-linked monoclonal antibodies. Error bars show se for n = 3 antibodies (group 4), n = 9 (group 3), n = 2 (group 2), and n = 5 (group 1).
(D) Exemplar FFE protein abundance profiles as detected by high-throughput shotgun proteomics. Example proteins were selected on the basis of previously established sub-Golgi, ER, and transitional ER-Golgi localization relating to well-known biomolecular functions in the secretory pathway.
(E) FEE abundance profiles of selected proteins detected via high-sensitivity, targeted proteomics. Proteins (Supplemental Data Set 2) were chosen given an established function and localization specific to Golgi cisternae or the ER. Two independent peptides per protein were measured for n = 7 (ER), n = 1 (cis-Golgi), n = 5 (medial-Golgi), and n = 3 (trans-Golgi) proteins. Solid lines indicate mean abundance over all proteins in the class, and error bars represent se.
Following on from the TEM imaging, the FFE fractions (from R1) were analyzed for the same classes of polysaccharide, using carbohydrate antibody arrays immobilized on nitrocellulose membranes, which has been successfully applied to endomembrane enrichments (Okekeogbu et al., 2019) and post-Golgi compartments (Wilkop et al., 2019). Here, we were able to probe an expanded number of polysaccharide epitopes compared with TEM due to the high-throughput nature of the array assays. Where possible, antibodies were chosen against epitopes with a known, or likely, sub-Golgi distribution either from previous publications or from Figure 3A. Polysaccharide epitopes were placed into four groups (details in Supplemental Data Set 2) with correspondence to the TEM probes. It is notable that the rhamnogalacturonan class was not covered in the TEM analysis but localized to cis/medial cisternae, as described previously (Ralet et al., 2010), and so was grouped with homogalacturonan and XGs with shorter side chains. As shown in Figure 3C, the combined FFE profiles from the carbohydrate analysis show distinct distributions for the four epitope groups, with each peaking in the following anode-to-cathode order: complex and medium-branched XG (late), homogalacturonan, XG with shorter branching and rhamnogalacturonan (medial), mannans (early), and extensins (very early). Hence, the appearance of the polysaccharide epitopes along the FEE profile has a distinct cisternal bias in the order of trans- to medial- to cis-Golgi (i.e., going from glycans with more complex or longer to less complex or shorter branching as the fraction number increases toward the cathode).
Cisternal Protein Distribution
Next, the overall protein FFE profiles were examined for any evidence of ordering to proteins along the electrophoretic gradient, which might also correspond to different Golgi cisternae. An initial, approximate gauge was obtained by examining the distribution of N-glycosylation enzymes where ER or cisternal localization, and hence secretion pathway order, had been established previously (Nilsson et al., 2009; Schoberer and Strasser, 2011). FUT13, the trans-Golgi N-glycosylation marker, was not present in all replicates, so two alternative biosynthesis enzymes of known trans-Golgi location (Gao et al., 2008; Chevalier et al., 2010) were included. As illustrated in Figure 3D, the peak protein abundance was again observed to approximate the late/early/ER sequential order (i.e., with proteins from the medial and trans cisternae more abundant in earlier fractions [closer to the anode]). The COPII-associated proteins p24δ2 and p24δ5 were also included for comparison. As anticipated, these profiles were similar to ER and cis-Golgi proteins.
A second, more in-depth protein analysis was conducted using targeted proteomics for proteins previously localized at sub-Golgi resolution (Figure 3E). The notion here was that a higher-sensitivity but lower-bandwidth technique could be used to validate and complement the high-throughput shotgun proteomics mass spectrometry technique we were using in the main (Picotti et al., 2009, 2010). The proteins of known localization that were used as sub-Golgi markers for targeted proteomics are listed in Supplemental Data Set 2 and include N-glycosylation markers from Figure 3D. Profiles obtained using targeted (Figure 3E) and shotgun (Figure 3D) proteomics were comparable, and again, a cis-medial-trans-Golgi trend toward the anode was evident. Together with carbohydrate data, this analysis further corroborated that FFE can separate Golgi cisternae, with earlier cisternae migrating farther toward the cathode during separation.
Subcluster Discrimination
After establishing the general, peak cisternal ordering along the FFE gradient, we returned to analysis of the minor sub-Golgi FFE clusters. Following on from the initial hierarchical clustering of protein-abundance profiles, we next generated a more robust set of clusters using a bootstrapping approach, as detailed in Methods and illustrated in Figure 4A, which randomly omitted 20% of the proteins during repeat hierarchical clustering to generate consensus groups and a measure of uncertainty. This more general, consensus clustering generated clusters numbered 1 to 8 (Figure 4B). Consistent with the observation that more anodic clusters contained later Golgi proteins, proteins previously localized to the late Golgi (FUT12, XYLT, FUT1, and QUA2; Figures 3D and 3E; Supplemental Data Set 2) were found in clusters 1 and 2, and proteins previously localized to the early Golgi (GMII and MNS2; Figures 3D and 3E; Supplemental Data Set 2) were found in clusters 3 and 4. Given this, together with the general cisternal separation, we tentatively assigned clusters as follows: 1, trans-Golgi; 2, medial-Golgi and TGN; 3, cis-Golgi; 4, cis-Golgi and ER; 5 and 6, ER; and 7 and 8, PM.
Figure 4.

Classification of Sub-Golgi Compartments.
(A) Robust clustering of secretory protein FFE profiles via bootstrapping. Abundance profiles (second from top) were reclustered using Ward’s method 120 times, each time omitting 20% of the proteins. The resulting clusters were assigned to the corresponding initial clusters A to H (see Figure 2) by similarity to the cluster medioids. These clusters are shown as a color map (third panel), where each row corresponds to a different, random subset of proteins, and is presented in the initial hierarchical cluster column order (as used in Figure 2). The robust, consensus clusters (lower panel) were defined as the most common cluster identity for each protein over all the bootstrap trials.
(B) FFE profiles for each of the eight consensus groups were separately reclustered (Ward’s method) to clearly visualize profile characteristics of each group. The groups were relabeled 1 to 8 to discriminate them from the initial clusters A to H, which have (slightly) different memberships. These were then used for tentative assignment of particular groups (1–4) to sub-Golgi compartments using trends presented in Figure 3. Abundance profiles are presented as a color density map, as in (A), but in a new intragroup order.
(C) Merged FFE profile data, for proteins present in replicates R3 to R5, plotted as a 2D PCA projection and labeled according to the bootstrap consensus clusters 1 to 8, as illustrated in (B).
(D) Merged FFE profile data, for all secretory proteins detected in any of the replicates R1 to R5, presented as a 2D PCA projection. Multiple-class SVM was used to classify proteins (on whole FFE profiles, not the 2D map) into three sub-Golgi groups and an ER group. The group labels used in the classification came from LOPIT to provide distinction between resident ER and Golgi proteins (and to exclude TGN ones), given that profiles overlap, to a degree, in the FFE data but not in the LOPIT data. The consensus FFE subclusters (as in [C]) were then used to classify the three sub-Golgi groups from among the larger Golgi proteome. Consensus subclusters and final proteomes are detailed in Supplemental Data Set 3.
(E) Re-presentation of a section of the LOPIT PCA map shown in Figure 2A, now colored according to ER and sub-Golgi classes presented in (D).
(F) Re-presentation of a section of the 2D t-SNE map shown in Figure 2B, now colored according to ER and sub-Golgi classes presented in (D).
To visualize these clusters on a 2D map, and thus to better illustrate group relationships, PCA was performed on the merged FFE protein profiles (R3–R5) using robust clusters 1 to 8 as labels (Figure 3C). Here, Golgi clusters 1, 2, and 3 (trans, medial, and cis) formed a somewhat continuous grouping, while Golgi cluster 4 was peripheral to the ER group. Cluster 1, the largest Golgi cluster, appeared to be more diffuse at its outer edge, but this peripheral group did not obviously correspond to any subcluster, so cluster 1 was not further divided. It is notable that two medial-localized N-glycosylation enzymes, XYLT and FUT12, were consistently identified in the peripheral region of cluster 1. The dispersed, distal end of cluster 1 might correspond to Golgi residents in a specific trafficking pathway. Although cluster 4 was proximal to the ER cluster, the earlier LOPIT analysis had confirmed that cluster 4 members were resident Golgi proteins. This proximity to the ER suggests a similarity in compartment surface charge, hinting that cluster 4 may be either an intermediate compartment or a Golgi subcompartment that accepts ER vesicles.
To generate final proteome lists, the robust clusters 1 to 8 were used as labels for training data in a multidimensional SVM-based classification. This was used to further classify data from R3 to R5, this time considering proteins only detected in single replicates. These additional proteins clustered consistently and so were incorporated into an expanded training set, which was then used in a second round of SVM to classify merged data from all replicates R1 to R5 (see Figure 3D for 2D PCA projection). In the end, this yielded compartment proteomes of the following sizes: ER, 181; cis-Golgi, 41; medial-Golgi, 56; and trans-Golgi, 84 proteins (Supplemental Data Set 3).
Golgi cisternae were not expected to differ sufficiently in density to be separable on a density gradient, and LOPIT proteome maps were therefore not expected to reflect clustering observed in FFE data. Nevertheless, for comparison, subproteomes were plotted onto LOPIT data (Figure 4E). This revealed separate partitioning from the ER and, unexpectedly, some partial separation of Golgi cisternae proteins. The proposed proteomes largely separated along an ER-cis-medial-trans axis, indicating that classifications from electrophoretic separations were correct.
Validating Golgi Cisternae Separation
Super-Resolution Imaging of Protein Distributions
Next, we validated our observations by testing whether members of the sub-Golgi proteomes showed their proposed in vivo localizations. Using structured illumination microscopy (SIM; Heintzmann and Huser, 2017) of transiently transformed tobacco (Nicotiana benthamiana) leaves, we resolved RFP- and GFP-tagged protein pairs for cis/cis, medial/medial, trans/trans, cis/medial, medial/trans, and cis/trans locations (Figure 5A). Proteins were selected based on their functional association with cisternae or relevance to products localized in Figure 4. A visual overview of protein localization is provided in Figure 5A by showing protein localization in individual Golgi stacks. We sampled a large number of Golgi stacks from multiple images (Supplemental Data Set 4) to generate a statistically robust analysis of protein pair localization.
Figure 5.

Validation of Sub-Golgi Protein Localization.
(A) Example images of SIM of validatory protein pairs representative of cis- (C), medial- (M), and trans- (T) Golgi sublocalizations. Sub-Golgi locations of PUBQ10-driven, C-terminally tagged GFP and RFP fusion proteins (Grefen et al., 2010) were assayed to provide pairwise comparisons by using transient expression in N. benthamiana. For each protein pair, localization data were collected from nine regions (Supplemental Data Set 4), incorporating three image stacks from at least two leaves per plant. Localizations were visualized in a single Golgi body from each of the three image stacks. The gene identifiers for the proteins were as follows: AT2G20810.1 (C1), AT5G47780.1 (C2), AT2G43080.1 (C3), AT1G26850.1 (M1), AT3G62720.1 (M2), AT5G18480.1 (M3), AT1G19360.1 (M4), AT1G74380.1 (T1), AT1G08660.1 (T2), AT4G36890.1 (T4), AT2G35100.1 (T3), and AT5G11730.1 (T5). Bars = 400 nm.
(B) Three example histograms showing the distribution of distance transform values for image regions containing multiple Golgi stacks with spatially overlapping (top), partly overlapping (middle), and somewhat separate (bottom) labeled protein pairs (i.e., from red/green fluorescence microscopy illustrated in [A]). Channel signal overlap was quantified by thresholding intensities to generate ROIs, then summing the distance transform values for one channel’s ROIs within the ROI bounds of the other. Here, negative values indicate greater separation and positive values indicate overlap.
(C) The distribution of red/green channel overlap scores, over multiple image regions (n = 9), for the validatory protein pairs shown in (A), arranged in modal order. Overlap scores were calculated for each image region as the log2 ratio of mean absolute values on either side of zero distance (see blue and orange regions in [B]), with positive values indicating more overlap. Image regions are given in Supplemental Data Set 4.
(D) Occurrence of protein families and functional annotation in the secretory and sub-Golgi proteomes. Using ER, TGN, and PM localizations derived from LOPIT data and sub-Golgi localizations from FFE (Supplemental Data Set 3), proteins were grouped variously according to family, MapMan (Ramsak et al., 2014) functional categorization, and possession of the K/H/RDEL ER-retrieval motif. Groups with at least five members are presented here.
To give a measure of the overlap between the locations of the fluorescent proteins, we used a method based upon the distance transform (see Methods) to quantify how coincident the red and green signal intensities were in the Golgi image regions. From the values of the distance transform, we devised a simple log-ratio-based score to indicate whether the overall distribution of values for the two channels were generally overlapping (positive), partly overlapped (near zero), or separated (negative): examples of this are illustrated in Figure 5B. The results showed that values became more negative (more separated) when combinations were predicted to be more physically distant within the Golgi stack (Figure 5C). Results therefore confirm cis-/medial-/trans-Golgi separation using FFE and subsequent compilation of relevant subproteomes.
Distribution of Protein Function across Golgi Cisternae
The sub-Golgi proteomes were examined for evidence of functional differences associated with cisternae and were contrasted with the ER and PM. Proteins were grouped by subfamily where possible, given that functional categories such as “hemicellullose biosynthesis,” for example, were too broad for the high spatial resolution of Golgi biosynthetic processes. As summarized in Figure 5D for selected groups (see full descriptions in Supplemental Data Set 3), specific functions were clearly associated with cisternal subproteomes. There was little overlap of typical ER functions (or KDEL motif proteins) with the cis-Golgi and virtually no overlap of typical Golgi functions with the PM. Prolyl-4-hydroxylases were clearly cis-Golgi associated, as anticipated (Yuasa et al., 2005). The GT47 family was enriched in the trans-Golgi, as were glucuronic acid and xylose epimerases. A distinct cis/medial trend was observed in the galacturonosyltransferase (GAUT) and O-fucosyltransferase families.
Bioinformatics Analysis of Golgi and Sub-Golgi Trends
Paralogue TM Region Sequences
Having established proteomes for the sub-Golgi, we sought to identify features common to these subcompartments that might determine localization. We investigated proteins in our data set that are close paralogues (i.e., with highly similar amino acid sequences) but that have different cisternal localizations. We observed that the TM and near-TM regions of the paralogue sequences seemed somewhat variant (Figure 6). Although protein TM regions, because they form simple spanning helices, would be expected to vary somewhat during evolution (notwithstanding restraints on hydrophobicity), they are potentially ideal sites for specifying localization given that they can vary without affecting globular domains and are able to respond to a lipid membrane environment. Hence, we investigated the amino acid composition of the TM regions in detail to discern any compartment-specific patterns. As highlighted in Figure 6, an initial casual check on the sequences showed that the paralogues from later cisternae generally had more Phe residues on the exoplasmic/lumenal side of the TM/span and more Ser residues on the exoplasmic side after the TM span.
Figure 6.

Comparison of Type II TM Protein Paralogues with Different Sub-Golgi Classifications.
Alignments are shown for pairs of similar, homologous proteins from Arabidopsis that have different sub-Golgi localizations. TM span regions are indicated in boldface. The blue Arg/Lys at the cytoplasmic edge highlight the start of the TM span. Phe residues are colored either pink or cyan to indicate relative position in the TM span. Within 15 residues of the exoplasmic TM edge, Ser residues are colored yellow and three consecutive Ser residues are colored red.
Compartmental TM Region Logo Plots
To give a more general picture of TM region composition in the cisternae, and because differently localized paralogues are rare, we looked at the overall sequence properties of each localized subproteome group. Data sets for single TM span proteins were augmented using a similar approach to that of Sharpe et al. (2010), with only very close homologues selected and TM span edges determined from multiple alignments using a consistent, hydrophobicity-based informatics procedure (see Methods). We did this for all our localized single-span TM proteins using logo plots for visualization (Figure 7) and where we aligned different sequences according to the cytoplasmic edge or exoplasmic edge of their estimated TM span. This revealed several features that appeared to correlate with progression either through the Golgi stack or through the entire secretory pathway from ER to PM. From ER to PM there was an increased frequency of Arg/Lys at the cytoplasmic TM boundary. Also, the peak Arg/Lys abundance appears to be broader in the early Golgi compared with the ER. In the Golgi, there was increased Ser occurrence at the exoplasmic boundary, although a much weaker, diffuse Ser signal was present in TGN and PM proteins. From the cis- to trans-Golgi, Phe distribution became progressively more biased toward the exoplasmic half of the TM span and Val to the cytoplasmic half, but little change was seen in other hydrophobic residues within Golgi groups. In the PM, Ala, Val, Gly, and Ile were predominant in the exoplasmic TM half with Phe and Leu in the cytoplasmic half. Phe frequency was proportionally much lower in the TGN and PM TM span compared with the ER and Golgi. Hence, overall differences in amino acid distribution likely reflect organelle-specific changes in overall membrane composition and relative differences between the inner and outer membrane leaflets (Xu et al., 2013).
Figure 7.

TM Amino Acid Composition in Sub-Golgi and Secretory Compartments.
Logo plots of single-span TM proteins from secretory and sub-Golgi proteomes indicating the relative abundance of amino acids at and around aligned TM spans. Data are shown for the Arabidopsis proteins localized by LOPIT and FFE and their very close homologues. Different sequences were aligned at either the cytoplasmic (left column) or exoplasmic/luminal (right column) edge of the hydrophobic TM spans. (See Methods for details of gathering homologues and aligning TM sequences.) The different amino acids are color-coded according to their physiochemical properties, as indicated in the color key (bottom). Logo plots were generated after randomly sampling 1000 sequences for each data set from position-specific residue abundance probabilities calculated from dissimilarity weighted sequences. This was done to reduce the bias caused by the different sizes of protein families (i.e., which are informatically somewhat redundant).
Intraprotein Sequence Patterns
Although logo plots of aligned sequences provide a good illustration of amino acid composition, they only present an average picture and are agnostic as to residue correlations within individual sequences. Hence, we additionally analyzed single-span Arabidopsis TM proteins, at the TM spans and ±15 flanking residues, to look generally for patterns across the (sub)compartments that were hinted at when inspecting the logo plots together with example sequences.
First, we looked at trends that we would predict from the logo plots by investigating Arg, Lys, and Ser residues at TM edges (Figure 8Ai). Consistent with the logo plots, these showed some abundance differences for Arg/Lys at the cytoplasmic boundary and Ser at the exoplasmic boundary. However, overall, these trends were not especially discriminating for individual cisternae.
Figure 8.

Comparison of Protein Sequence Features in Organelle and Suborganelle Proteomes.
(A) Abundance of sequence features at and around the TM spans of single-span proteins in the secretory and sub-Golgi proteomes. Data are shown for 63 ER, 23 cis-, 37 medial-, and 54 trans-Golgi proteins, and 108 PM TM proteins. (i) The relative abundance of Lys or Arg at the cytoplasmic TM edge and Ser at the exoplasmic/luminal edge. Values were normalized relative to the maximum observation. (ii) Overall TM Phe content, as a proportion of TM span length, and the cytoplasmic-exoplasmic asymmetry of TM Phe; asymmetry was calculated as the difference in abundance between the two halves of each TM span. (iii) The relative abundance of Ser and presence of three or more consecutive Ser residues in the 15-residue exoplasmic region immediately flanking the TM spans. Values were normalized relative to the maximum observation. (iv) An overview of the results presented in (i) to (iii), but shown as a proportion of each subcellular proteome. Here, Phe asymmetry corresponded to positive values presented in (ii) and high Ser content corresponded to a count of at least five in the 15 flanking exoplasmic residues. For panels (i) to (iii), bar heights are mean values and error bars represent the se.
(B) Distributions of TM span properties for different subproteome groups. Data sets for localized single-span TM proteins from Arabidopsis were expanded through close homology searches (as used in Figure 7), where sequence contributions were weighted by dissimilarity and TM spans were edges defined, as detailed in Methods. TM span length (top), pI of the entire cytoplasmic region (middle), and pI of the entire exoplasmic region (bottom) are shown as violin plots for different secretory and sub-Golgi compartments (defined by LOPIT and FFE, respectively).
(C) Line plots of per-position TM hydrophobicity (top) and mean residue charge (bottom) for localized Arabidopsis and homologue over TM hydrophobic core and flanking regions (as in Figure 7). TM spans were anchored at their exoplasmic boundary. Plotted values represent the means at each TM aligned position over different, dissimilarity-weighted proteins. Error bars represent the se.
Next, we looked at Phe and Ser residues in more detail, given our initial observations on paralogues. Specifically, we measured the asymmetry of Phe composition by comparing the cytoplasmic and exoplasmic halves of the TM span sequences (Figure 8Aii). Notably, although overall TM Phe abundance was similar across compartments, Phe was more concentrated in the exoplasmic half of the medial- and trans-Golgi TM spans, while the PM, and to some degree the ER, showed the opposite tendency. When looking at Ser abundances (Figure 8Aiii), we saw that this increased through the secretory pathway, peaking in the trans-Golgi before dropping a little in the PM. However, a more striking observation was discovered when looking at the presence of three or more adjacent Ser residues (i.e., SSS in the sequence) on the exoplasmic side of the TM span; these only seemed to occur in the late Golgi to PM and peaked in the trans-Golgi.
An overview of these results is presented in Figure 8Aiv and expressed as a proportion of each subproteome, to illustrate the ubiquity of the trends. Overall, although each feature may not be present in all proteins of a given compartment, there is very clearly a fingerprint of characteristics for each. These measures are similar for the cis-Golgi and ER, the TM Phe asymmetry and exoplasmic Ser distinguish later cisternae, and cytosolic edge Arg/Lys (i.e., positive charges) are characteristic of trans-Golgi and PM. These features can potentially account for much of the residue intra-Golgi TM protein distribution. However, physical properties like hydrophobicity and exoplasmic and cytoplasmic pI, as we examine next, may also contribute.
TM Span Properties
When analyzing the derived, physical TM span properties, it was pertinent to investigate span length, as this is one of very few characteristics associated with increasing membrane thickness in later cisternae (Banfield, 2011), although the span-length variety in plant Golgi proteins (Schoberer and Strasser, 2011) implies the existence of other factors beyond those specific to protein families (Gao et al., 2014; Woo et al., 2015).
As shown in Figure 8B, the span length distributions for the cis-Golgi are similar to those of the ER, and then from the medial-Golgi onward the length tends to increase, on average, through the secretory pathway to the PM. The cytoplasmic pI distributions show analogous trends, albeit with the pI diminishing from the medial-Golgi to the PM. On the other side of the TM span, the exoplasmic pI is somewhat different between the cis-Golgi (lower) and later cisternae (higher), and both are distinct from the ER and PM. The per-residue hydrophobicity (relative to the TM edge) generally reflected the observed trends in TM span length. However, the most notable hydrophobicity differences occurred in the 10- to 15-residue segment flanking the exoplasmic TM boundary (Figure 8C). This increased in the Golgi from cis to trans but was appreciably lower in the TGN and PM. This was accompanied by a decrease in mean exoplasmic residue charge in the late Golgi, which also contrasted with the TGN and PM.
DISCUSSION
This study shows that the secretory pathway can be directionally separated from the ER to the trans-Golgi. We successfully performed a proteomic comparison of separated Golgi cisternae and elucidated a series of protein characteristics likely to affect protein location and longevity in different cisternae, along with a comprehensive Arabidopsis Golgi resident proteome. Our separation results were validated by comparing protein and glycan localization in vivo and post electrophoresis. Partial separation of cisternae by density gradient centrifugation provided additional independent validation of the cisternal proteomes.
The medial- and trans-Golgi are proposed to be the principal sites of polysaccharide synthesis (Driouich et al., 2012), and the complexity and length of side chains are known to increase from cis- to trans-Golgi (van de Meene et al., 2017). Our results agree overall but show considerable levels of polysaccharide synthesis in the early Golgi (Figures 3B and 3C). Consistently, fewer gold particles were detected in early compared with late Golgi compartments (Figure 3A), suggesting that polysaccharides are less readily detectable in the early Golgi using immunogold TEM. The signal from antibodies in the “very early” group was found to persist through Golgi-containing fractions (Figure 3C), even though the LM1 signal was restricted to the cis-Golgi in TEM images (Figure 3B). Some very early antibodies may exhibit some cross reactivity with arabinogalactan side chains (Pattathil et al., 2010), which may be present in the later Golgi. The overall increase in glycosyltransferase (GT) proteins in the trans-Golgi (Figure 3E) indicates that the diversity of glycosylation reactions is greatest in the trans-Golgi.
Functional analysis of cisternal proteomes supported the canonical view that molecular complexity of modified cargo increases through the Golgi and showed that our sub-Golgi categorization accurately reflects biological function. The GAUT family members that have been biochemically characterized are known to synthesize polysaccharide backbones (Atmodjo et al., 2011), while members of the GT47 family and core-2/I-branching β-1,6-GlcNAc transferase families transfer sugars to peripheral glycan branches (Zhong et al., 2005; Iwai et al., 2006; Jensen et al., 2008; Harholt et al., 2012; Knoch et al., 2013). As shown in Figure 5, the latter two families were found mainly in the trans-Golgi whereas GAUTs were in the cis/medial-Golgi. Several of the cis-Golgi-localized (Figure 5) P4H enzymes catalyze the first step in O-linked glycosylation and shuttle between the ER and the cis-Golgi (Yuasa et al., 2005; Velasquez et al., 2011), while the medial RRA3 (M4 in Figure 5A) catalyzes the subsequent arabinosylation of Hyp (Chen et al., 2015). Some S-adenosyl-methionine (SAM)-dependent methyltransferases have been associated with methyl esterification of substrates synthesized by GAUT1 and GAUT7 (Krupková et al., 2007; Miao et al., 2011). Consistently, these proteins localized subsequent to GAUT1 and GAUT7 (Figure 5), as did their reaction products (Figure 3). MUR3, a GT47 family member, was located in the trans-Golgi (Figure 5), along with its product, galactosylated xyloglucan (group 4 in Figure 3). Functional insight imparted by our results is demonstrated by analysis of DUF707 proteins, which are suggested to be a GT family (Nikolovski et al., 2012) but are otherwise unstudied. Of the 11 Arabidopsis family members, we identified 9 in the medial/trans-Golgi cluster in our LOPIT data. In electrophoretic data, 3 were identified, all exclusive to the trans-Golgi. Given their trans-Golgi association, and the family size, it seems likely that DUF707s make an important contribution to the diversity of terminal substitutions on glycan chains, possibly relating to the cell wall.
Nonfucosylated xyloglucan epitopes were not observed in the very latest Golgi cisternae (Figures 3A to 3C) but have been recorded in post-Golgi compartments and the cell wall (Wilkop et al., 2019). This suggests that their absence from the very late Golgi was not a consequence of further substitution preventing antibody binding. Possibly, epitopes not being further substituted pass through the very latest cisternae quickly and so are present at low concentrations, although it cannot be ruled out that select cargo may somehow bypass terminal cisternae.
A unique advantage of this study is that hundreds of cargo and resident proteins were tracked simultaneously through the secretory pathway. Profiles of these protein groups indicated distinct trafficking mechanisms; the flat profiles of cargo proteins (Supplemental Figure 2) were compatible with a uniform, nonselective mechanism of trafficking cargo from the cis- to trans-Golgi, such as cisternal maturation (Luini, 2011). Golgi residents accumulated above cargo abundance levels, which is most straightforwardly explained by recycling of resident proteins directionally opposite to the cargo flow, although anterograde trafficking mechanisms cannot be ruled out. Observations are therefore consistent with the current consensus model of combined cisternal maturation and retrograde vesicular trafficking (Glick and Luini, 2011; Luini, 2011; Donohoe et al., 2013). Interestingly, TGN proteins were somewhat more associated with medial- than trans-Golgi cisternae (Figure 2D). This could be a consequence of medial-Golgi receiving retrograde trafficked material in COPIb vesicles, as recently discussed (Schoberer et al., 2019).
The gradient of increasing electronegativity that appears to exist across the Golgi stack cannot be explained by bulk changes in the cytosolic pI of proteins (Figure 8B), so it must be attributed to lipid content. Phosphatidylserine is an endomembrane-associated monoacidic phospholipid whose concentration at the cytoplasmic leaflet is higher in the Golgi than in the ER (Leventis and Grinstein, 2010; Simon et al., 2014) due to the action of flippases (Poulsen et al., 2008). Our data indicate that cytoplasmic leaflet phosphatidylserine concentration increases from cis- to trans-Golgi. In this case, an extremely anodic migration of PM could have been expected, owing to the accumulation of cytoplasmic leaflet phosphatidylinositol-4-phosphate (Simon et al., 2014, 2016). The observed extremely cathodic migration (Supplemental Figure 2B) was therefore likely due to binding proteins, counterions, or most vesicles being in an exoplasmic-face-out orientation.
Phe asymmetry in the TM span, exoplasmic Ser concentration, multiple consecutive Ser residues, exoplasmic pI, and exoplasmic hydrophobicity were convincingly associated with the later Golgi (Figures 6 to 8). The changes in Phe asymmetry at the TGN and PM (Figure 7) suggest that this is an important identifier of Golgi residents. Phe stabilizes membrane proteins by inserting into the bilayer adjacent to ionic lipid-protein interactions (Bogdanov et al., 2014). Less asymmetric proteins could be progressively excluded if this feature confers stability in the late Golgi luminal environment. The luminal pH of plant secretory compartments decreases from the ER to the TGN and thereafter increases (Martinière et al., 2013). Total Golgi measurements in earlier studies suggest that this feature is not unique to plants (Sharpe et al., 2010; Quiroga et al., 2013). Exoplasmic Ser could further increase stability in tightly appressed trans-Golgi cisternae by facilitating hydrogen bonding and compact folding through its action as a flexible linker between the TM helix and catalytic domains (Sharpe et al., 2010). The increase in Arg/Lys at the cytoplasmic TM boundary from the ER to PM (Figure 7) may increase protein stability as outer-leaflet concentrations of negatively charged lipids increase throughout the entire secretory pathway. The observation that differences in these sequence features can be detected between differentially localized proteins of very high overall sequence similarity (Figure 6) lends weight to these features being important determining factors in sub-Golgi localization. Recently, Glu at the exoplasmic TM boundary was found to confer cis/medial-Golgi localization of GnTI (Schoberer et al., 2019). Exoplasmic anchoring of medial protein TM span sequences reveals a prominent Glu at this position in our data, suggesting that multiple medial-Golgi proteins are localized in this way. A single, cisternally specific amino acid at this location was not evident in cis or trans proteomes (Figure 7). If early to late cisternal localization is conferred by a gradient of preference for Ser and Phe, a specific central Golgi signal may add a further level of distinction. Alternatively, this may identify a specific retrieval pathway for medial proteins (Schoberer et al., 2019).
At the TGN, most resident proteins must be retained and recycled and relevant proteins selected for onward trafficking (Guo et al., 2014). The drop in exoplasmic pI and hydrophobicity at the TGN (Figure 8C) and loss of exoplasmic Ser (Figure 8A) indicate a sudden change in luminal environment, which could exclude Golgi residents from most TGN regions. Lipid zonation occurs within the TGN (Surma et al., 2012; Wattelet-Boyer et al., 2016); the decrease in TM Phe bias in TGN proteins indicates that TM span composition may exclude Golgi residents from certain TGN zones. Residue composition appears to play an important role in distinguishing PM proteins (Figure 7), as illustrated by the lack of Leu and prominence of Ile toward the exoplasmic TM edge, which is not observed in other membranes. Also, the strong, regular spacing of Gly residues toward the exterior of the TM span may indicate the presence of dimerization sites in these PM proteins (Teese and Langosch, 2015).
In summary, we have shown that the electrophoretic separation of Golgi cisternae is possible and provides a means to determine the order of proteins, and hence functions, within the secretory pathway and to discriminate residents from cargo. Through this separation, we have also uncovered a continuum of differences in TM amino acid sequences across the different Golgi cisternae. Our results provide a framework upon which the precise mechanisms of cisternal localization and longevity can be investigated and will contribute to an understanding of how the complex equilibrium of the Golgi is maintained.
METHODS
Preparation of Intact-Membrane Material
The Arabidopsis (Arabidopsis thaliana) cell-suspension culture line (ecotype Landsberg erecta) was maintained, homogenized, and enriched for endomembranes in a similar manner to that described previously (Parsons et al., 2012). For membrane separations, 60 to 80 g fresh weight (FFE separations) or 40 g fresh weight (LOPIT) of 7-d-old cells was protoplasted according to Eubel et al. (2008) and gently homogenized using six strokes of a glass-Teflon homogenizer in a 10 mM Na2HPO4, 3 mM EDTA, 2 mM DTT, protease inhibitor tablets (Roche), and 1% (w/v) dextran 200000 buffer (1:2 [w/v] ratio of fresh cell weight to buffer). The ensuing homogenate was clarified at 3000g for 15 min, then collected on a cushion of 1.4 M sucrose at 100,000g for 1.5 h. The cushion was overlaid with homogenization buffer containing 1.0 and 0.2 M sucrose, and endomembranes were collected at the 1.0/0.2 M interface after centrifugation for 100,000g for 1.5 h. Each biological replicate (FFE and LOPIT experiments) represented a separate preparation of homogenized cell-suspension culture, collected in different weeks, from different inoculations.
FFE
The electrophoresis was performed using continuous zone electrophoresis-FFE using an FFE System (BD Diagnostics) in the same manner as Parsons et al. (2012) on five separate biological replicates of endomembrane-enriched samples from Arabidopsis cell-suspension cultures (as above). Separation was by the tangential action of laminar flow and voltage using 700 V, which resulted in a current of 105 to 115 mA. The medium injection speed was 200 mL/h, and samples at 1500 μL/h. Fractions were collected and assessed for total protein content according to absorbance at 280 nm. Fractions corresponding to the main endomembrane separation zone (Figure 1) were analyzed using shotgun proteomics (all replicates) and further validated using targeted proteomics (replicate R4) and glycan epitope analysis (replicate R1) where material was available.
MS Analysis of Replicates R1 and R2
Proteins were reduced, alkylated, and digested with trypsin (1:10, w/w) overnight in 50% (v/v) acetonitrile and 10 mM Tris-HCl, pH 7.5. Peptides were injected onto a Pepmap100 μ-guard column on a Famos Autosampler (both Dionex-LC Packings) and washed for 10 min with buffer A (2% [v/v] acetonitrile and 0.1% [v/v] formic acid) flowing at 15 μL/min. Peptides were eluted onto an Acclaim Pepmap100 C18 column (75 μm × 150 mm, 300 nL/min flow rate; Dionex-LC Packings) and into the TripleTOF 5600 via a gradient of 5% buffer B (98% [v/v] acetonitrile and 0.1% [v/v] formic acid), increasing B to 35% B over 60 min. B was increased to 90% over 3 min and held for 15 min, followed by a ramp back down to 5% B over 3 min, where it was held for 15 min to reequilibrate the column. Peptides were introduced to the mass spectrometer using a Nanospray III source (AB Sciex) with a nanotip emitter (New Objective) in positive-ion mode (2400 V). Data were acquired with Analyst TF 1.5.1 operating in information-dependent acquisition mode. After a 250-ms scan, the 20 most intense ions (charge states 2–5) within 400 to 1600 mass-to-charge ratio (m/z) mass range above a threshold of 150 counts were selected for tandem mass spectrometry (MS/MS) analysis. MS/MS spectra were collected using time of flight resolution mode: high resolution with the quadrupole set to UNIT resolution and rolling collision energy to optimize fragmentation. MS/MS spectra were scanned from 100 to 1600 m/z and were collected for 50 ms. Selected precursor ions were excluded for 16 s following MS/MS acquisition.
MS Analysis of Replicates R3 to R5
Proteins were digested as above, and resulting peptides were injected onto a Q‐Exactive+ (Thermo Fisher Scientific) using a nanoACQUITY UltraPerformance LC system (Waters), incorporating a C18 reverse-phase column (Waters; 100 μm × 100 mm, 1.7-μm particle, BEH130C18, column temperature 40°C). Peptides were analyzed over a 150‐min gradient using buffer A and 5% buffer B. Buffer B was increased from 2 to 10% over 2 min, to 40% over 110 min, then to 85% over 1 min, maintained at 85% for 10 min, and equilibrated for 14 min with 2% buffer B. Peptides were eluted at a flow rate of 300 nL/min. A survey scan was obtained for the m/z range 300 to 1600. MS/MS spectra were acquired using a top 15 method, where the top 15 ions in the spectra were subjected to high‐energy collisional dissociation. An isolation mass window of 2.0 m/z was used for the precursor ion selection, and normalized collision energy of 27% was used for fragmentation. A duration of 5 s was used for the dynamic exclusion. An automatic gain control target of 1,000,000 for MS and 50,000 for MS/MS was used, while maximum injection time for MS was 30 ms and for MS/MS was 50 ms. The system employed a resolution of 70,000 for MS and 17,500 for MS/MS.
Label-Free Protein Quantitation Using the Normalized Spectral Index
Identification annotations were extracted from mzIdentML files. Spectra were clustered using the spectra-clusetr-cli version 1.0.3 (Griss et al., 2016), a precursor tolerance of 2 m/z, and a fragment tolerance of 0.1 m/z. All other settings were left at their defaults. The accuracy of label-free quantitation was improved using the id_transferer_cli tool to transfer identifications to unidentified spectra if these were part of a cluster with five or more identified spectra and at least 70% of these spectra identified the same peptide. This approach is comparable to a feature mapping based on precursor m/z and retention time but does not require complex retention time alignment to be performed between the different samples. Proteins were inferred from The Arabidopsis Information Resource 10 (Berardini et al., 2015), and the smallest number of proteins required to explain all observed peptides was retained. Peptides that could be assigned to more than one unambiguously identified protein/protein group were not taken into consideration for label-free quantitation.
Merged FFE Profile Generation
Fraction-separated spectral count data from different FFE replicates were merged into a single set of pseudo-fraction abundances prior to hierarchical clustering. Merging was achieved by progressive, pairwise aggregation of FFE profiles, using a scheme (see ‘fraction align-and-merge procedure') that aligns fraction data with the objective of maximizing the correlation between protein abundances in equivalent fractions. Alignment involved an exhaustive search of relative end offsets (and thus linear scaling) to pair-up overlapping/partially overlapping fractions from different experimental replicates. The open-source computer code that performed this operation is available atgithub.com/tjs23/ms_fraction_merge/.
Fraction Align-and-Merge Procedure
Data for each replicate, in terms of spectral counts for each protein in each fraction, were loaded from CSV files, and the later fractions, where total protein count was negligible, were discarded in each case (fractions 43, 50, 47, 37, and 44 for the replicates in this study). Missing abundance values from fractions not harvested after FFE were imputed by performing a linear interpolation of values from the closest fractions on either side that were recorded. Each protein’s abundance profile in each replicate was then normalized by subtracting its minimum value over all fractions (i.e., baseline correction for those proteins which do not have a zero-valued fraction) and dividing by the summation of counts; each protein had a fractional abundance profile that summed to 1.0. Each replicate fraction, containing proportional protein abundances, was than normalized by dividing by the fraction’s median value, thus centering each fraction irrespective of total protein content. Progressing from the most similar pair of replicates (or replicates combined in a previous round), fraction data were combined by an exhaustive search of relative offset of profile start and end, and hence also width scaling, to find the alignment with the best overall correlation in fraction protein abundances. Fraction offsets, which align the starts and ends of the replicate data, were sampled in the range of +/−5 in steps of 0.5 (original) fraction widths. Concomitantly, this also sampled fraction width scaling, to shrink or expand the fractions’ equivalent range in one data set relative to the other, where intermediate scale values are linearly interpolated. For each combination of start and end offset parameters, the similarity between two fractions from two different replicate experiments was calculated as the Pearson correlation in protein abundance (considering proteins common to both replicates) multiplied by the relative width of the overlap between fractions. The width-scaled overlap scores were then summed over all fractions to give an overall replicate-replicate similarity score for each particular combination of offsets. This score is maximized if the replicates are aligned to give equal abundances of each protein in equivalent fractions. The combination of alignment parameters that gave the highest score was then used to merge the replicate data. Merging was achieved by averaging the protein abundances in each pair of equivalent fractions over their region of overlap and generally resulted in merged pseudo-fractions with nonequal widths (i.e., partial overlap). Where merging was done with data that represent previously combined replicates, the protein abundances were scaled proportionality according to the number of original replicates in the combined data. After the first pair of replicates was merged, the next most similar replicate was then merged with the result of the previous merge, and the whole procedure was repeated until all replicates had been merged with the rest of the data. After the last merge, new pseudo-fractions were generated by imposing 25 equal-width bins on the final data, which averages the protein compositions of differently sized regions that result from the successive rounds of merging. The composition in each bin was simply the average of the protein abundances of the overlapping merge regions weighted according to the width of overlap.
LOPIT Analysis and Clustering
Organelle separation and fraction collection was performed according to Christoforou et al. (2016) with the following modifications: 20 g fresh weight of cells per gradient was protoplasted and homogenized as described above. Iodixanol was adjusted to the required concentrations using the above homogenization buffers without dextran. Membranes were collected on 25% iodixanol cushions, then adjusted to 25% iodixanol and loaded onto a gradient as described (Christoforou et al., 2016). Membranes were then fractionated according to their density by centrifuging at 100,000g for 8 h in an NVTi65 rotor (Beckman Coulter) using slow braking. Fractions (0.5 mL) were harvested top-down using an Auto Densi-flow collection device (Labconco).
Fractions were pelleted for 50 min at 100,000g in an SW55Ti rotor, then resuspended in 25 mM CaCO3 and shaken gently for 30 min at 4°C before repelleting. Membranes in fractions 1, 3, 6, 9, 11, 15, 18, and 20 were sonicated for 15 min in 10-s pulses and assayed for protein content. From each fraction, 80 µg of protein precipitated using chloroform:methanol:water (1:4:3), then resolubilized, and reduced in 50 μL of 8 M urea/100 mM HEPES (pH 7.8) containing 0.1% SDS and 7 mM DTT for 2 h (room temperature). Iodoacetamide was added to a final concentration of 15 mM for 2 h (dark, room temperature). Proteins were precipitated in 6 volumes of 80% acetone at −20°C, then pelleted at 16,000g for 10 min at 8°C and resuspended in 200 μL of 100 mM HEPES, pH 8.0. Proteins were digested with sequencing-grade trypsin (Promega) for 1 h with a 1:40 enzyme:protein ratio at 37°C. An additional aliquot of trypsin at 1:40 concentration was added and incubated overnight at 37°C. Trypsin digests were centrifuged for 10 min at 13,000g to remove any insoluble matter, then reduced to dryness by vacuum centrifugation. TMT 10 plex labeling, peptide desalting, and reverse-phase HPLC were conducted according to Christoforou et al. (2016) but using 100 mM HEPES and acetonitrile instead of triethylammonium bicabonate and isopropanol during peptide labeling.
MS, Raw Data Processing, and Quantification for LOPIT
All MS runs were performed on an Orbitrap Fusion Lumos Tribrid instrument coupled to a Dionex Ultimate 3000 RSLCnano system (Thermo Fisher Scientific) with parameters from Mulvey et al. (2017). Raw files were processed with Proteome Discoverer v1.4 (Thermo Fisher Scientific) using the MASCOT server v2.3.02 (Matrix Science), searched against the Arabidopsis proteome (canonical sequences, downloaded on April 2, 2017). Precursor and fragment mass tolerances were set to 10 ppm and 0.6 D, respectively. Trypsin was set as the enzyme of choice, and a maximum of two missed cleavages were allowed. Static modifications were carbamidomethyl (C), TMT6plex (N-term), and TMT6plex (K). Dynamic modifications were oxidation (M), TMT6plex(S), and TMT6plex(T). False discovery rate was assessed using percolator, and only high-confidence peptides were retained. Additional data reduction filters were peptide rank = 1 and ion score > 25. Quantification at the MS3 level was performed within the Proteome Discoverer workflow using the centroid sum method and an integration tolerance of 2 milli mass units. Isotope impurity correction factors were applied. Each raw peptide-spectrum match (PSM) reporter intensity was then divided by the sum of all intensities for that PSM (sum normalization). Protein grouping was performed according to the minimum parsimony principle, and the median of all sum-normalized PSM ratios belonging to each protein group was calculated as the protein group quantitation value. Only proteins with a full reporter ion series were retained.
Machine Learning and Establishment of Resident Organelle Proteomes Using LOPIT Data
Data analysis, including PCA, was performed using the R (R Core Team, 2013) Bioconductor (Gentleman et al., 2004) packages MSnbase (Gatto and Lilley, 2012) and pRoloc (Gatto et al., 2014) as described (Breckels et al., 2016b). t-SNE analysis was performed in the R programming environment using Rtsne, with the following parameters: theta=0, perplexity=80, and max_iter=800. Supervised machine learning using an SVM classifier with a radial basis function kernel was employed in order to predict the localization of unlabeled proteins. A training set of organelle markers specific to single subcellular compartments (PM, TGN, Golgi, ER, peroxisome, chloroplast, nucleus, mitochondria, cytosol, and vacuole) was compiled by selecting proteins whose combined historical data from confocal microscopy and organelle proteomics (Hooper et al., 2017a) showed a clear majority localization to any one compartment (Supplemental Data Set 1). Following the SVM protocol of Breckels et al. (2016b), 100 rounds of fivefold cross-validation were employed (creating five stratified test/train partitions) to estimate algorithmic performance. This protocol features an additional round of cross-validation on each training partition to optimize the free parameters of the SVM, sigma and cost, via a grid search. Based on the best F1 score (the harmonic mean of precision and recall), for each LOPIT data set the best sigma and cost were 0.01 and 16, respectively. Previously unclassified proteins with an SVM score greater or equal to the upper quartile value for each compartment were assigned as resident to that compartment if consistently classified in at least two of the four replicate LOPIT experiments. False assignment rates (FARs) were estimated by calculating conflicting microscopy data (Hooper et al., 2017a) in the new resident organelle proteomes. FARs were between 0.1 and 5% for all locations. The resident Golgi proteome was expanded by lowering the upper quartile threshold until the FAR was 2%. This did not result in the assignment of any new proteins beyond the main Golgi cluster in any replicates, so was deemed an appropriate method for expanding the number of Golgi-resident proteins. The final organelle resident proteomes are shown in Supplemental Data Set 1.
Hierarchical Clustering
Differences between protein abundance profiles were measured as Euclidean distances and grouped using Ward’s method for hierarchical clustering, considering the similarity of their merged abundance profiles, across (pseudo)fractions, from all experimental replicates. Initially, Scientific Python’s scipi.hierarchy implementation of Ward’s method was used to both create dendrograms and to set the order when plotting the abundance profiles as color matrices (Figures 2 and 4). A threshold was defined after inspection of the dendrogram so that proteins could be split into a useful number of groups (i.e., branches). In each case, the threshold was set so that the protein clusters were of roughly equal size and there were at least three predominantly Golgi-enriched groups (according to proteomes established by LOPIT). The output order of the initial clustering was also used to set the order of rows and columns in the corresponding correlation matrix shown in Supplemental Figure 2. This matrix contained the Pearson’s correlation coefficient between the (merged) profiles for each pair of proteins.
Immunogold Electron Microscopy
Arabidopsis roots were grown on one-half-strength Murashige and Skoog medium containing 1% sucrose under constant light. Three-day-old root tips were high-pressure frozen, freeze-substituted, embedded, sectioned, and immunolabeled according to McFarlane et al. (2008). Samples were cryofixed in B-type sample holders (Ted Pella) using a Leica HPM-100 high-pressure freezer with 1-hexadecene (Sigma-Aldrich) as a cryoprotectant. Samples were freeze-substituted for 5 d at −85°C in a Leica AFS2 in a solution of 0.25% gluteraldehyde, 0.1% uranyl acetate, and 8% 2,2-dimethoxypropane (Sigma-Aldrich) in acetone. Samples were then slowly warmed to room temperature over 2 d, infiltrated with LR White resin (London Resin) over 5 d, then resin was polymerized for 36 h at 70°C. Sections of ∼70 nm were cut with a DiATOME knife on a Leica UCS ultramicrotome, suspended on nickel grids (Gilder) with 0.3% formvar, blocked with 5% BSA in Tris-buffered saline with detergent (TBST; 10 mM Tris, 250 mM NaCl, and 0.1% [w/v] Tween 20, pH 7.4), and thoroughly washed with TBST before antibody application. Primary antibodies were CCRC-M1, CCRC-M39, CCRC-M87, CCRC-M89 (CarboSource Services), LM1, LM15, LM19, and LM21 (PlantProbes), all used at 1:10 dilution and applied for 1 h at room temperature, after which grids were thoroughly washed with TBST. Secondary antibodies were 1:100 goat anti-mouse conjugated to 18-nm gold (Jackson ImmunoResearch, 115-215-146) or 1:100 goat anti-rat conjugated to 18-nm gold (Abcam, ab105302), applied for 1 h at room temperature, after which grids were thoroughly washed with TBST and then water. Samples were poststained with 1% (w/v) aqueous uranyl acetate for 8 min and Reynolds’ lead citrate for 4 min. Grids were imaged using a Philips CM120 transmission electron microscope at 80 kV accelerating voltage coupled to a Gatan multiscan 791 CCD camera. The relative positions of gold particles were determined by measuring the thickness of each Golgi stack from cis to trans, then measuring the distance from the cis-most face for each gold particle.
Cis-to-trans polarity of the Golgi stacks was confidently determined by (1) the cis-to-trans decrease in cisternal lumen width, (2) the increase in cisternal lumen electron density (Staehelin et al., 1990), and (3) the location of a Golgi-associated TGN (where present). Under our fixation, embedding, and poststaining conditions, the electron density increased up to the trans-most cisternae, usually peaking in the penultimate cisterna. To avoid glancing sections through the margins of Golgi stacks, only Golgis with at least three clearly visible cisternae were imaged.
FFE Glycan Analysis
The distribution of glycans in replicate R1 after electrophoresis was quantified using carbohydrate microarrays. Here, polysaccharides were released from 20 µg of protein by digestion with 4 µg of Proteinase K for 4 h at 37°C, then dilution in an array-jet printing buffer (55.2% glycerol, 44% water, and 0.8% Triton X-100) in 0.8% Triton X-100. A twofold dilution series of four dilutions was loaded, printed onto nitrocellulose arrays, and quantified according to Pedersen et al. (2012) using anti-rat (for LM antibodies) or anti-mouse (for CCRC and BS-400-4 antibodies) secondary antibodies conjugated to alkaline phosphatase (Sigma-Aldrich). Primary antibodies were sources as above, with the addition of BS-400-4 (Australian Biosupplies). For each fraction, extracts equivalent to 0.1 or 0.05 µg of total protein were probed. Average antibody signal intensities from the dilution series of two technical replicates were normalized to the highest sample value per replicate.
Targeted Proteomics
Targeted proteomics of specific proteins was performed on replicate sample R4 by selected reaction monitoring (SRM). This was done on an Agilent 6460QQQ mass spectrometer according to Batth et al. (2014) using a 25-min method with the following gradient: 95% buffer A (2% acetonitrile and 0.1% formic acid), 5% buffer B (98% acetonitrile and 0.1% formic acid). Buffer B increased to 40% over 17 min, then to 80% B in 30 s, where it was held for 1 min, then ramped back down to 5% in 30 s, and equilibrated for 6 min prior to the next injection. Data analysis was performed using Skyline v2.6 (MacLean et al., 2010). Targets were selected according to the following criteria: confident identification by shotgun MS, appreciable increase in signal intensity after enrichment of endomembranes from whole-cell homogenates, and appreciable increase in signal intensity after focusing acquisition time around the anticipated retention time. Targets were identified using up to five transitions per peptide and at least two peptides per protein (Supplemental Figure 4). Peptide quantification was achieved by summing the integrated peak areas of two validated SRMs. Peptides were averaged for all proteins associated with subcompartments. Relative protein abundance was expressed as a percentage of the total for all fractions.
Robust Clustering of FFE Profiles by Bootstrapping
Initial hierarchical clustering of FFE profiles defined eight groups, and this number of groups was kept for a second, more robust round of clustering that was less sensitive to the inclusion of specific proteins. The secondary clustering also used Ward’s method (albeit via the sklearn.custering implementation) but was performed on the rows of the correlation matrix, rather than plain abundance profiles. The robustness of the clustering was assessed by bootstrapping, removing 20% of data each time, and randomly (and independently) resampling 120 times. The resulting clusters were colored according to the most similar cluster from the initial (dendrogram) clustering; taking the minimum Euclidean distance between mean correlation profiles, so that the correspondence between the initial and secondary clustering was obvious. The bootstrapping results were then used to estimate the variability in the cluster allocation of each protein (Supplemental Figure 2). The variability in the assignment of each protein to the clusters was simply measured as the fraction of the bootstrap samples that put the protein in its nonmodal cluster.
Machine Learning and Establishment of Resident Organelle Proteomes Using FFE Data
SVM was performed as described earlier for LOPIT data sets but using a best sigma and cost of 0.01 and 16, respectively. Results from the bootstrapped clustering described above provided the training input. Non-Golgi endosomal proteins could be confidently excluded from cis-, medial-, and trans-Golgi proteomes, as all endosomal organelles clustered distinctly in LOPIT analyses. Likewise, this approach was used to distinguish ER and cis-Golgi proteins in clusters 4 and 5. SVM training data and final ER and sub-Golgi proteomes are described in Supplemental Data Set 3.
SIM of Golgi Stacks
SIM was performed on the following representatives of cis (C), medial (M), and trans (T) proteins: AT2G20810.1 (C1), AT5G47780.1 (C2), AT2G43080.1 (C3), AT1G26850.1 (M1), AT3G62720.1 (M2), AT5G18480.1 (M3), AT1G19360.1 (M4), AT1G74380.1 (T1), AT1G08660.1 (T2), AT4G36890.1 (T4), AT2G35100.1 (T3), and AT5G11730.1 (T5). AT1G08660.1, AT2G20810.1, AT5G47780.1, AT5G18480.1, AT3G62720.1, AT5G11730.1, AT2G35100.1, AT1G74380.1, AT1G19360.1, and AT4G36890.1 in pDONR227 were a kind gift from Berit Ebert. Coding sequences for AT2G43080.1 and AT1G26850.1 were purchased from the ABRC and amplified using Gateway additions for C-terminal tagging using the following gene-specific primers: ATGGCTCCTGCCATGAAG (AT2G43080.1 Fwd), GTAGCTTTTTGCCTCATCC (AT2G43080.1 Rev), ATGGCGTTGAAGTCTAGTTCTG (AT2G26850.1 Fwd), and GTGAGTCGAGGTGGAGTTGG (AT2G26850.1 Rev), then recombined into pDONR227. All pDONR227 constructs were recombined into pUBC-GFP_Dest and pUBC_RFP_Dest vectors (Grefen et al., 2010). Sub-Golgi locations of P-UBQ10 driven, C-terminally tagged GFP and RFP fusion proteins were assayed by pairwise comparisons using transient expression in Nicotiana benthamiana according to Grefen et al. (2010). Localizations were visualized for the following pairs (Figure 5A): C1:C2, C2:C3, M1:M2, T3:T2, M2:T1, C2:M2, M2:M3, M4:T4, M3:C3, M1:C1, C1:T5, and C1:T3. For each protein pair, three images were taken from at least two leaves. From each image, three regions of >20 Golgi stacks were selected, giving nine regions per protein pair. Super-resolution images were acquired using a Deltavision OMX 3D-SIM System (https://ki.mit.edu/sbc/microscopy/instrumentation/omx) V3 BLAZE from Applied Precision (a GE Healthcare company) equipped with three sCMOS cameras, 405-, 488-, and 592.5-nm diode laser illumination, an Olympus Plan Apo N 60x 1.42 NA oil objective, and standard excitation and emission filter sets. Imaging of each channel was done sequentially using three angles and five phase shifts of the illumination pattern as described (Gustafsson et al., 2008). Sections were acquired at 0.125-μm z steps. Raw data were reconstructed and channel registered in SoftWoRx software version 6.5.2 (Applied Precision, a GE Healthcare company). Brightness/contrast was adjusted as necessary using Fiji (Schindelin et al., 2012).
Quantification of Microscopic Image Overlap
Analysis of all nine image regions per pair gave a statistically robust analysis of red/green channel overlap. Channel signal overlap was quantified by thresholding intensities to generate regions of interest (ROIs), then summing the distance transform values for one channel’s ROIs within the ROI bounds of the other. Voxelwise nearest-neighbor distances were measured for GFP signal relative to RFP signal using a custom script for Fiji (Schindelin et al., 2012) and a custom script in Supplemental Data Set 4. The latter maps signal volumes using Kapur’s maximum entropy thresholding method (Kapur et al., 1985) and measures distances using the exact signed 3D Euclidean distance transform with internal distances set to zero for display on the histogram. The distribution of distances was analyzed by using the log ratio of absolute values and comparing the average positive value with the average (absolute) negative value for each protein pair. Accordingly, log ratios larger than zero indicate overlap, values around zero represent partial overlap, and values less than zero indicate separation.
Alignments of Similar Golgi Protein Sequences from Different Cisternae
Pairwise sequence alignments were performed between proteins present in cis-, medial-, and trans-Golgi proteomes using the nwalign Python module (which implements the Needleman-Wunsch algorithm). Comparisons were ranked according to alignment bit score, and the eight most similar pairs of proteins, representing three protein families (GAUTs, GlcNAc transferases, and SAM-dependent methyltransferases) are shown in Figure 6.
Identification of TM Sequences in Localized Proteins and Close Homologues
Analysis of single-span TM protein sequences was performed in a similar manner to previous studies (Sharpe et al., 2010; Nikolovski et al., 2012), albeit with refinements. From the Arabidopsis organelle and sub-Golgi proteome lists, single-span TM proteins were identified by their UniProt database (UniProt Consortium, 2015) TM span annotation, where it exists, and otherwise by a combination of SignalP 4.0 (Petersen et al., 2011) and TMHMM (Krogh et al., 2001), taking predicted single TM spans and excluding those predicted to be signal peptides. Initial TM span edge positions and cytoplasm-exoplasm chain topology were taken from UniProt and otherwise from prediction by Phobius (Käll et al., 2004).
Arabidopsis protein sequences were augmented with sequence information from close homologues using BLAST+ (Camacho et al., 2009) searches of the UniProt reference proteomes within the Viridiplantae clade. All searches used an e-value cutoff of 10−20. Overlapping homologue families, from different initial queries, that had common members were separated by allocating each homologue to its most similar query. Resulting family groups all had a single, consistent organelle or subcompartment annotation that was derived from the Arabidopsis query protein.
Families of sequences were multiply aligned using Clustal Omega (Sievers and Higgins, 2014) with default parameters. TM span edge positions were further refined using the multiple alignment of each homologue family. First, the edges of the TM span (initially taken from UniProt annotations or Phobius) were adjusted within a region of five or more residues by selecting the point in the alignment with the maximum difference in GES-scale hydrophobicity (summed over all proteins in the alignment) between the adjacent five residues on the side of the TM span and the adjacent five residues on the opposite side. Next, the edge positions were trimmed or extended according to the average hydrophobicity over the whole alignment. If the mean hydrophobicity of the next residue exceeded 1.0 kcal/mol (Gly or more hydrophobic), the edge was extended. Similarly, if the mean hydrophobicity of an edge residue was below 1.0 kcal/mol, the edge was trimmed. Finally, individual protein adjustments were made, extending or trimming positions for each span sequence. Accordingly, individual TM span edges were trimmed if they ended in a gap or a hydrophilic residue (defined here as Arg, Lys, Asp, Glu, Gln, Asn, His, or Ser) or extended if the next residue was suitably hydrophobic (Phe, Met, Ile, Leu, Val, Cys, Trp, Ala, Thr, or Gly).
Next, families of proteins were multiply aligned again using Clustal Omega (Sievers and Higgins, 2014), and the following additional checks were made for a comparable TM span, comparing each BLAST+ hit with the query: (1) the length of the protein must not differ by more than 200 residues; (2) there must not be more than four gap insertions in the TM span region; (3) the separation from the TM span to the N terminus must not differ by more than 75 residues; and (4) there must be a cursory similarity between span sequences (mean, aligned regional BLOSUM62 score > 0.8).
Reduction of Protein Sequence Redundancy
Given that families contain different numbers of protein sequences with different degrees of similarity, each protein was weighted according to its dissimilarity to all other sequences in the whole data set. Dissimilarity weights for each protein (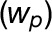 ) were obtained using a BLAST+ search of each sequence (maximum e-value 10−20) against a database of all the protein sequences and were calculated as:
) were obtained using a BLAST+ search of each sequence (maximum e-value 10−20) against a database of all the protein sequences and were calculated as:
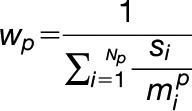 |
Here, 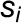 is the BLAST+ bit score of the aligned high-scoring database hit
is the BLAST+ bit score of the aligned high-scoring database hit 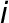 (from a total of
(from a total of 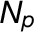 hits) and
hits) and 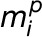 is the maximum possible bit score value, the bit score if the query were compared with itself over the same alignment region. Accordingly, the dissimilarity weight is 1.0 if the search only finds itself and approximately
is the maximum possible bit score value, the bit score if the query were compared with itself over the same alignment region. Accordingly, the dissimilarity weight is 1.0 if the search only finds itself and approximately 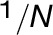 if it finds
if it finds 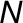 very similar sequences. This protects against large and/or well-conserved protein families having an undue influence on the measurement of general TM span properties.
very similar sequences. This protects against large and/or well-conserved protein families having an undue influence on the measurement of general TM span properties.
Protein Sequence Logo Plots
The frequency of residue occurrence in TM spans and flanking regions of cisternal proteins and their close homologues was visualized using logo plots. Logo plots were generated by specially written Python scripts (available at github.com/tjs23/logo_plot), after randomly sampling 1000 sequences for each data set, from position-specific residue abundance probabilities calculated from dissimilarity weighted sequences. The use of dissimilarity weights (as defined above) reduced the effect of redundant sequences (i.e., due to different sized homologous protein families). Different proteins within each subgroup were aligned by anchoring their sequences at the cytoplasmic or exoplasmic edge of the TM span, prior to the generation of logo plots (Figure 7).
Accession Numbers
Electrophoresis proteomics data are deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Vizcaíno et al., 2016) with identifier PXD004596. LOPIT proteomics data are deposited to the ProteomeXchange Consortium via the PRIDE partner repository with identifier PXD009978. SRM data are available from PASSEL, part of the PeptideAtlas repository (peptideatlas.org/passel/), with accession number PASS00908. The following proteins were localized to Golgi cisternae in Figure 5: AT2G20810.1, AT5G47780.1, AT2G43080.1, AT1G26850.1, AT3G62720.1, AT5G18480.1, AT1G19360.1, AT1G74380.1, AT1G08660.1, AT4G36890.1, AT2G35100.1, and AT5G11730.1.
Supplemental Data
Supplemental Figure 1. t-SNE plots of additional whole-cell Arabidopsis LOPIT experiments.
Supplemental Figure 2. Further details on hierarchical clustering of FFE profiles.
Supplemental Figure 3. Clustering and correlation of FEE profiles from individual replicates R3 to R5.
Supplemental Figure 4. Selection of SRM targets used in Figure 3.
Supplemental Data Set 1. Resident organelle proteomes from LOPIT experiments after SVM-based classification.
Supplemental Data Set 2. Additional information for monoclonal antibodies, polysaccharide epitopes, and protein targets featured in Figure 3.
Supplemental Data Set 3. Protein lists for sub-Golgi proteomes.
Supplemental Data Set 4. Complete suite of SIM images used in Figure 5.
Dive Curated Terms
The following phenotypic, genotypic, and functional terms are of significance to the work described in this paper:
ACKNOWLEDGMENTS
H.T.P. expresses thanks to Sean Munro, Nadine Muschalik, Stephen Fry, Janice Millar, Henrik Scheller, Yves Verhertbruggen, Michael Joo, and Purbasha Sarkar. Electron microscopy was performed at the Biosciences node of the Melbourne Advanced Microscopy Facility. This project was part of the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research (grant DE-AC02-05CH11231 to the Lawrence Berkeley National Laboratory and the U.S. Department of Energy). Additional support was provided by a European Commission Marie Curie Fellowship (PIEF-GA-2011-301401 to H.T.P.) and a Det Frie Forskningsral-Technologie og Produktion Fellowship (DFF-1337-00066 to H.T.P.). Support was provided by a European Molecular Biology Organization Long-Term Fellowship (ALTF 1246-2013 to H.E.M.) and an Australian Research Council Discovery Early Career Researcher Award (DE170100054 to H.E.M.). Funding was also provided by the Vienna Science and Technology Fund (LS11-045 to J.G.) and by the Medical Research Council (MC_U105178783 to T.J.S.). Generation of the CCRC series of monoclonal antibodies used in this work was supported by the National Science Foundation Plant Genome Program (grant DBI-0421683). Distribution of JIM and MAC antibodies used in this work was supported in part by the National Science Foundation (grants DBI-0421683 and RCN 009281).
AUTHOR CONTRIBUTIONS
H.T.P. conceptualized the project, prepared all samples, and performed all experimental procedures, except where otherwise stated below. H.T.P. and T.J.S. performed data analysis and wrote the article with assistance from K.S.L. T.J.S. performed programming and computational analyses. H.E.M. collected and analyzed electron microscopy data. J.G. performed label-free quantitation of protein abundances. N.L. and R.B. operated the fluorescence imaging microscope. W.G.T.W. and S.V.-M. collected carbohydrate epitope array data. C.J.P., M.M.L.S, and M.S. oversaw the collection of mass spectrometry data. K.S.L. and J.L.H. supervised the project and provided laboratory resources. All authors read and approved the final article.
Footnotes
Articles can be viewed without a subscription.
References
- Atmodjo M.A., Sakuragi Y., Zhu X., Burrell A.J., Mohanty S.S., Atwood J.A. III, Orlando R., Scheller H.V., Mohnen D. (2011). Galacturonosyltransferase (GAUT)1 and GAUT7 are the core of a plant cell wall pectin biosynthetic homogalacturonan:galacturonosyltransferase complex. Proc. Natl. Acad. Sci. USA 108: 20225–20230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield D.K. (2011). Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 3: a005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkla B.J., Vera-Estrella R., Pantoja O. (2007). Enhanced separation of membranes during free flow zonal electrophoresis in plants. Anal. Chem. 79: 5181–5187. [DOI] [PubMed] [Google Scholar]
- Batth T.S., Singh P., Ramakrishnan V.R., Sousa M.M.L., Chan L.J.G., Tran H.M., Luning E.G., Pan E.H.Y., Vuu K.M., Keasling J.D., Adams P.D., Petzold C.J. (2014). A targeted proteomics toolkit for high-throughput absolute quantification of Escherichia coli proteins. Metab. Eng. 26: 48–56. [DOI] [PubMed] [Google Scholar]
- Berardini T.Z., Reiser L., Li D., Mezheritsky Y., Muller R., Strait E., Huala E. (2015). The Arabidopsis Information Resource: Making and mining the “gold standard” annotated reference plant genome. Genesis 53: 474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov M., Dowhan W., Vitrac H. (2014). Lipids and topological rules governing membrane protein assembly. Biochim. Biophys. Acta 1843: 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandizzi F., Barlowe C. (2013). Organization of the ER-Golgi interface for membrane traffic control. Nat. Rev. Mol. Cell Biol. 14: 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckels L.M., Gatto L., Christoforou A., Groen A.J., Lilley K.S., Trotter M.W.B. (2013). The effect of organelle discovery upon sub-cellular protein localisation. J. Proteomics 88: 129–140. [DOI] [PubMed] [Google Scholar]
- Breckels L.M., Holden S.B., Wojnar D., Mulvey C.M., Christoforou A., Groen A., Trotter M.W.B., Kohlbacher O., Lilley K.S., Gatto L. (2016a). Learning from heterogeneous data sources: An application in spatial proteomics. PLOS Comput. Biol. 12: e1004920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckels L.M., Mulvey C.M., Lilley K.S., Gatto L. (2016b). A Bioconductor workflow for processing and analysing spatial proteomics data. F1000 Res. 5: 2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Dong W., Tan L., Held M.A., Kieliszewski M.J. (2015). Arabinosylation plays a crucial role in extensin cross-linking in vitro. Biochem. Insights 8 (suppl. 2): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier L., Bernard S., Ramdani Y., Lamour R., Bardor M., Lerouge P., Follet-Gueye M.-L., Driouich A. (2010). Subcompartment localization of the side chain xyloglucan-synthesizing enzymes within Golgi stacks of tobacco suspension-cultured cells. Plant J. 64: 977–989. [DOI] [PubMed] [Google Scholar]
- Christoforou A., Mulvey C.M., Breckels L.M., Geladaki A., Hurrell T., Hayward P.C., Naake T., Gatto L., Viner R., Martinez Arias A., Lilley K.S. (2016). A draft map of the mouse pluripotent stem cell spatial proteome. Nat. Commun. 7: 8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Michele R., McFarlane H.E., Parsons H.T., Meents M.J., Lao J., González Fernández-Niño S.M., Petzold C.J., Frommer W.B., Samuels A.L., Heazlewood J.L. (2016). Free-flow electrophoresis of plasma membrane vesicles enriched by two-phase partitioning enhances the quality of the proteome from Arabidopsis seedlings. J. Proteome Res. 15: 900–913. [DOI] [PubMed] [Google Scholar]
- Donohoe B.S., Kang B.-H., Gerl M.J., Gergely Z.R., McMichael C.M., Bednarek S.Y., Staehelin L.A. (2013). Cis-Golgi cisternal assembly and biosynthetic activation occur sequentially in plants and algae. Traffic 14: 551–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driouich A., Follet-Gueye M.-L., Bernard S., Kousar S., Chevalier L., Vicré-Gibouin M., Lerouxel O. (2012). Golgi-mediated synthesis and secretion of matrix polysaccharides of the primary cell wall of higher plants. Front. Plant Sci. 3: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkley T.P.J., et al. (2006). Mapping the Arabidopsis organelle proteome. Proc. Natl. Acad. Sci. USA 103: 6518–6523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eubel H., Lee C.P., Kuo J., Meyer E.H., Taylor N.L., Millar A.H. (2007). Free-flow electrophoresis for purification of plant mitochondria by surface charge. Plant J. 52: 583–594. [DOI] [PubMed] [Google Scholar]
- Eubel H., Meyer E.H., Taylor N.L., Bussell J.D., O’Toole N., Heazlewood J.L., Castleden I., Small I.D., Smith S.M., Millar A.H. (2008). Novel proteins, putative membrane transporters, and an integrated metabolic network are revealed by quantitative proteomic analysis of Arabidopsis cell culture peroxisomes. Plant Physiol. 148: 1809–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C., Cai Y., Wang Y., Kang B.-H., Aniento F., Robinson D.G., Jiang L. (2014). Retention mechanisms for ER and Golgi membrane proteins. Trends Plant Sci. 19: 508–515. [DOI] [PubMed] [Google Scholar]
- Gao P., Xin Z., Zheng Z.-L. (2008). The OSU1/QUA2/TSD2-encoded putative methyltransferase is a critical modulator of carbon and nitrogen nutrient balance response in Arabidopsis. PLoS One 3: e1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto L., Lilley K.S. (2012). Msnbase: An R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 28: 288–289. [DOI] [PubMed] [Google Scholar]
- Gatto L., Breckels L.M., Wieczorek S., Burger T., Lilley K.S. (2014). Mass-spectrometry-based spatial proteomics data analysis using pRoloc and pRolocdata. Bioinformatics 30: 1322–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendre D., Jonsson K., Boutté Y., Bhalerao R.P. (2015). Journey to the cell surface: The central role of the trans-Golgi network in plants. Protoplasma 252: 385–398. [DOI] [PubMed] [Google Scholar]
- Gentleman R.C., et al. (2004). Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist A., et al. (2006). Quantitative proteomics analysis of the secretory pathway. Cell 127: 1265–1281. [DOI] [PubMed] [Google Scholar]
- Glick B.S., Luini A. (2011). Models for Golgi traffic: A critical assessment. Cold Spring Harb. Perspect. Biol. 3: a005215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grefen C., Donald N., Hashimoto K., Kudla J., Schumacher K., Blatt M.R. (2010). A ubiquitin-10 promoter-based vector set for fluorescent protein tagging facilitates temporal stability and native protein distribution in transient and stable expression studies. Plant J. 64: 355–365. [DOI] [PubMed] [Google Scholar]
- Griss J., Perez-Riverol Y., Lewis S., Tabb D.L., Dianes J.A., Del-Toro N., Rurik M., Walzer M.W., Kohlbacher O., Hermjakob H., Wang R., Vizcaíno J.A. (2016). Recognizing millions of consistently unidentified spectra across hundreds of shotgun proteomics datasets. Nat. Methods 13: 651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen A.J., Sancho-Andrés G., Breckels L.M., Gatto L., Aniento F., Lilley K.S. (2014). Identification of trans-Golgi network proteins in Arabidopsis thaliana root tissue. J. Proteome Res. 13: 763–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Sirkis D.W., Schekman R. (2014). Protein sorting at the trans-Golgi network. Annu. Rev. Cell Dev. Biol. 30: 169–206. [DOI] [PubMed] [Google Scholar]
- Gustafsson M.G.L., Shao L., Carlton P.M., Wang C.J.R., Golubovskaya I.N., Cande W.Z., Agard D.A., Sedat J.W. (2008). Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 94: 4957–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harholt J., Jensen J.K., Verhertbruggen Y., Søgaard C., Bernard S., Nafisi M., Poulsen C.P., Geshi N., Sakuragi Y., Driouich A., Knox J.P., Scheller H.V. (2012). ARAD proteins associated with pectic arabinan biosynthesis form complexes when transiently overexpressed in planta. Planta 236: 115–128. [DOI] [PubMed] [Google Scholar]
- Hawes C., Kiviniemi P., Kriechbaumer V. (2015). The endoplasmic reticulum: A dynamic and well-connected organelle. J. Integr. Plant Biol. 57: 50–62. [DOI] [PubMed] [Google Scholar]
- Heard W., Sklenář J., Tomé D.F.A., Robatzek S., Jones A.M.E. (2015). Identification of regulatory and cargo proteins of endosomal and secretory pathways in Arabidopsis thaliana by proteomic dissection. Mol. Cell. Proteomics 14: 1796–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzmann R., Huser T. (2017). Super-resolution structured illumination microscopy. Chem. Rev. 117: 13890–13908. [DOI] [PubMed] [Google Scholar]
- Hooper C.M., et al. (2017b). Multiple marker abundance profiling: Combining selected reaction monitoring and data-dependent acquisition for rapid estimation of organelle abundance in subcellular samples. Plant J. 92: 1202–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper C.M., Castleden I.R., Tanz S.K., Aryamanesh N., Millar A.H. (2017a). SUBA4: The interactive data analysis centre for Arabidopsis subcellular protein locations. Nucleic Acids Res. 45: D1064–D1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islinger M., Eckerskorn C., Völkl A. (2010). Free-flow electrophoresis in the proteomic era: A technique in flux. Electrophoresis 31: 1754–1763. [DOI] [PubMed] [Google Scholar]
- Ito Y., Uemura T., Nakano A. (2014). Formation and maintenance of the Golgi apparatus in plant cells. Int. Rev. Cell Mol. Biol. 310: 221–287. [DOI] [PubMed] [Google Scholar]
- Iwai H., Hokura A., Oishi M., Chida H., Ishii T., Sakai S., Satoh S. (2006). The gene responsible for borate cross-linking of pectin rhamnogalacturonan-II is required for plant reproductive tissue development and fertilization. Proc. Natl. Acad. Sci. USA 103: 16592–16597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J.K., et al. (2008). Identification of a xylogalacturonan xylosyltransferase involved in pectin biosynthesis in Arabidopsis. Plant Cell 20: 1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L., Krogh A., Sonnhammer E.L.L. (2004). A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338: 1027–1036. [DOI] [PubMed] [Google Scholar]
- Kapur J.N., Sahoo P.K., Wong A.K.C. (1985). A new method for gray-level picture thresholding using the entropy of the histogram. Comput. Vis. Graph. Image Process. 29: 273–285. [Google Scholar]
- Klute M.J., Melançon P., Dacks J.B. (2011). Evolution and diversity of the Golgi. Cold Spring Harb. Perspect. Biol. 3: a007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoch E., et al. (2013). A β-glucuronosyltransferase from Arabidopsis thaliana involved in biosynthesis of type II arabinogalactan has a role in cell elongation during seedling growth. Plant J. 76: 1016–1029. [DOI] [PubMed] [Google Scholar]
- Krogh A., Larsson B., von Heijne G., Sonnhammer E.L.L. (2001). Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305: 567–580. [DOI] [PubMed] [Google Scholar]
- Krupková E., Immerzeel P., Pauly M., Schmülling T. (2007). The TUMOROUS SHOOT DEVELOPMENT2 gene of Arabidopsis encoding a putative methyltransferase is required for cell adhesion and co-ordinated plant development. Plant J. 50: 735–750. [DOI] [PubMed] [Google Scholar]
- Leventis P.A., Grinstein S. (2010). The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39: 407–427. [DOI] [PubMed] [Google Scholar]
- Luini A. (2011). A brief history of the cisternal progression-maturation model. Cell. Logist. 1: 6–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B., Tomazela D.M., Shulman N., Chambers M., Finney G.L., Frewen B., Kern R., Tabb D.L., Liebler D.C., MacCoss M.J. (2010). Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus S.E., Verhertbruggen Y., Hervé C., Ordaz-Ortiz J.J., Farkas V., Pedersen H.L., Willats W.G.T., Knox J.P. (2008). Pectic homogalacturonan masks abundant sets of xyloglucan epitopes in plant cell walls. BMC Plant Biol. 8: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinière A., Bassil E., Jublanc E., Alcon C., Reguera M., Sentenac H., Blumwald E., Paris N. (2013). In vivo intracellular pH measurements in tobacco and Arabidopsis reveal an unexpected pH gradient in the endomembrane system. Plant Cell 25: 4028–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane H.E., Young R.E., Wasteneys G.O., Samuels A.L. (2008). Cortical microtubules mark the mucilage secretion domain of the plasma membrane in Arabidopsis seed coat cells. Planta 227: 1363–1375. [DOI] [PubMed] [Google Scholar]
- Miao Y., Li H.-Y., Shen J., Wang J., Jiang L. (2011). QUASIMODO 3 (QUA3) is a putative homogalacturonan methyltransferase regulating cell wall biosynthesis in Arabidopsis suspension-cultured cells. J. Exp. Bot. 62: 5063–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morré D.J., Mollenhauer H.H. (2009). Isolation and subfractionation. In The Golgi Apparatus. (New York: Springer; ), pp. 39–61. [Google Scholar]
- Mulvey C.M., Breckels L.M., Geladaki A., Britovšek N.K., Nightingale D.J.H., Christoforou A., Elzek M., Deery M.J., Gatto L., Lilley K.S. (2017). Using hyperLOPIT to perform high-resolution mapping of the spatial proteome. Nat. Protoc. 12: 1110–1135. [DOI] [PubMed] [Google Scholar]
- Nikolovski N., Rubtsov D., Segura M.P., Miles G.P., Stevens T.J., Dunkley T.P.J., Munro S., Lilley K.S., Dupree P. (2012). Putative glycosyltransferases and other plant Golgi apparatus proteins are revealed by LOPIT proteomics. Plant Physiol. 160: 1037–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T., Au C.E., Bergeron J.J.M. (2009). Sorting out glycosylation enzymes in the Golgi apparatus. FEBS Lett. 583: 3764–3769. [DOI] [PubMed] [Google Scholar]
- Okekeogbu I.O., Pattathil S., González Fernández-Niño S.M., Aryal U.K., Penning B.W., Lao J., Heazlewood J.L., Hahn M.G., McCann M.C., Carpita N.C. (2019). Glycome and proteome components of Golgi membranes are common between two angiosperms with distinct cell-wall structures. Plant Cell 31: 1094–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons H.T., et al. (2012). Isolation and proteomic characterization of the Arabidopsis Golgi defines functional and novel components involved in plant cell wall biosynthesis. Plant Physiol. 159: 12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattathil S., et al. (2010). A comprehensive toolkit of plant cell wall glycan-directed monoclonal antibodies. Plant Physiol. 153: 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen H.L., et al. (2012). Versatile high resolution oligosaccharide microarrays for plant glycobiology and cell wall research. J. Biol. Chem. 287: 39429–39438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen T.N., Brunak S., von Heijne G., Nielsen H. (2011). SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 8: 785–786. [DOI] [PubMed] [Google Scholar]
- Picotti P., Bodenmiller B., Mueller L.N., Domon B., Aebersold R. (2009). Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138: 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P., Rinner O., Stallmach R., Dautel F., Farrah T., Domon B., Wenschuh H., Aebersold R. (2010). High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nat. Methods 7: 43–46. [DOI] [PubMed] [Google Scholar]
- Poulsen L.R., López-Marqués R.L., McDowell S.C., Okkeri J., Licht D., Schulz A., Pomorski T., Harper J.F., Palmgren M.G. (2008). The Arabidopsis P4-ATPase ALA3 localizes to the Golgi and requires a beta-subunit to function in lipid translocation and secretory vesicle formation. Plant Cell 20: 658–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroga R., Trenchi A., González Montoro A., Valdez Taubas J., Maccioni H.J.F. (2013). Short transmembrane domains with high-volume exoplasmic halves determine retention of type II membrane proteins in the Golgi complex. J. Cell Sci. 126: 5344–5349. [DOI] [PubMed] [Google Scholar]
- Ralet M.-C., Tranquet O., Poulain D., Moïse A., Guillon F. (2010). Monoclonal antibodies to rhamnogalacturonan I backbone. Planta 231: 1373–1383. [DOI] [PubMed] [Google Scholar]
- Ramsak Ž., Baebler Š., Rotter A., Korbar M., Mozetic I., Usadel B., Gruden K. (2014). GoMapMan: Integration, consolidation and visualization of plant gene annotations within the MapMan ontology. Nucleic Acids Res. 42: D1167–D1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A Language and Environment for Statistical Computing. (Vienna: R Foundation for Statistical Computing). https://www.R-project.org/. [Google Scholar]
- Robinson D.G., Pimpl P. (2014). Clathrin and post-Golgi trafficking: A very complicated issue. Trends Plant Sci. 19: 134–139. [DOI] [PubMed] [Google Scholar]
- Saint-Jore-Dupas C., Nebenführ A., Boulaflous A., Follet-Gueye M.-L., Plasson C., Hawes C., Driouich A., Faye L., Gomord V. (2006). Plant N-glycan processing enzymes employ different targeting mechanisms for their spatial arrangement along the secretory pathway. Plant Cell 18: 3182–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoberer J., Strasser R. (2011). Sub-compartmental organization of Golgi-resident N-glycan processing enzymes in plants. Mol. Plant 4: 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoberer J., Liebminger E., Vavra U., Veit C., Grünwald-Gruber C., Altmann F., Botchway S.W., Strasser R. (2019). The Golgi localization of GnTI requires a polar amino acid residue within its transmembrane domain. Plant Physiol. 180: 859–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe H.J., Stevens T.J., Munro S. (2010). A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142: 158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Higgins D.G. (2014). Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 1079: 105–116. [DOI] [PubMed] [Google Scholar]
- Simon M.L.A., Platre M.P., Assil S., van Wijk R., Chen W.Y., Chory J., Dreux M., Munnik T., Jaillais Y. (2014). A multi-colour/multi-affinity marker set to visualize phosphoinositide dynamics in Arabidopsis. Plant J. 77: 322–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M.L.A., Platre M.P., Marquès-Bueno M.M., Armengot L., Stanislas T., Bayle V., Caillaud M.-C., Jaillais Y. (2016). A PtdIns(4)P-driven electrostatic field controls cell membrane identity and signalling in plants. Nat. Plants 2: 16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood M., Beven A., Donovan N., Neill S.J., Peart J., Roberts K., Knox J.P. (1994). Localization of cell wall proteins in relation to the developmental anatomy of the carrot root apex. Plant J. 5: 237–246. [Google Scholar]
- Stadler C., Rexhepaj E., Singan V.R., Murphy R.F., Pepperkok R., Uhlén M., Simpson J.C., Lundberg E. (2013). Immunofluorescence and fluorescent-protein tagging show high correlation for protein localization in mammalian cells. Nat. Methods 10: 315–323. [DOI] [PubMed] [Google Scholar]
- Staehelin L.A., Giddings T.H. Jr., Kiss J.Z., Sack F.D. (1990). Macromolecular differentiation of Golgi stacks in root tips of Arabidopsis and Nicotiana seedlings as visualized in high pressure frozen and freeze-substituted samples. Protoplasma 157: 75–91. [DOI] [PubMed] [Google Scholar]
- Strasser R. (2016). Plant protein glycosylation. Glycobiology 26: 926–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surma M.A., Klose C., Simons K. (2012). Lipid-dependent protein sorting at the trans-Golgi network. Biochim. Biophys. Acta 1821: 1059–1067. [DOI] [PubMed] [Google Scholar]
- Teese M.G., Langosch D. (2015). Role of GxxxG motifs in transmembrane domain interactions. Biochemistry 54: 5125–5135. [DOI] [PubMed] [Google Scholar]
- Thul P.J., et al. (2017). A subcellular map of the human proteome. Science 356: 356. [DOI] [PubMed] [Google Scholar]
- Tie H.C., Mahajan D., Chen B., Cheng L., VanDongen A.M.J., Lu L. (2016). A novel imaging method for quantitative Golgi localization reveals differential intra-Golgi trafficking of secretory cargoes. Mol. Biol. Cell 27: 848–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium (2015). UniProt: A hub for protein information. Nucleic Acids Res. 43: D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Meene A.M.L., Doblin M.S., Bacic A. (2017). The plant secretory pathway seen through the lens of the cell wall. Protoplasma 254: 75–94. [DOI] [PubMed] [Google Scholar]
- Van der Maaten L., Hinton G. (2008). Visualizing data using t-SNE. J. Mach. Learn. Res. 9: 2579–2605. [Google Scholar]
- Velasquez S.M., et al. (2011). O-Glycosylated cell wall proteins are essential in root hair growth. Science 332: 1401–1403. [DOI] [PubMed] [Google Scholar]
- Viotti C., et al. (2010). Endocytic and secretory traffic in Arabidopsis merge in the trans-Golgi network/early endosome, an independent and highly dynamic organelle. Plant Cell 22: 1344–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J.A., et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44: D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattelet-Boyer V., Brocard L., Jonsson K., Esnay N., Joubès J., Domergue F., Mongrand S., Raikhel N., Bhalerao R.P., Moreau P., Boutté Y. (2016). Enrichment of hydroxylated C24- and C26-acyl-chain sphingolipids mediates PIN2 apical sorting at trans-Golgi network subdomains. Nat. Commun. 7: 12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkop T., Pattathil S., Ren G., Davis D.J., Bao W., Duan D., Peralta A.G., Domozych D.S., Hahn M.G., Drakakaki G. (2019). A hybrid approach enabling large-scale glycomic analysis of post-Golgi vesicles reveals a transport route for polysaccharides. Plant Cell 31: 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo C.H., Gao C., Yu P., Tu L., Meng Z., Banfield D.K., Yao X., Jiang L. (2015). Conserved function of the lysine-based KXD/E motif in Golgi retention for endomembrane proteins among different organisms. Mol. Biol. Cell 26: 4280–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang L., Etxeberria E., Van den Ende W. (2013). Vacuolar protein sorting mechanisms in plants. FEBS J. 280: 979–993. [DOI] [PubMed] [Google Scholar]
- Xu H., Su W., Cai M., Jiang J., Zeng X., Wang H. (2013). The asymmetrical structure of Golgi apparatus membranes revealed by in situ atomic force microscope. PLoS One 8: e61596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeats T.H., Bacic A., Johnson K.L. (2018). Plant glycosylphosphatidylinositol anchored proteins at the plasma membrane-cell wall nexus. J. Integr. Plant Biol. 60: 649–669. [DOI] [PubMed] [Google Scholar]
- Yuasa K., Toyooka K., Fukuda H., Matsuoka K. (2005). Membrane-anchored prolyl hydroxylase with an export signal from the endoplasmic reticulum. Plant J. 41: 81–94. [DOI] [PubMed] [Google Scholar]
- Zhong R., Peña M.J., Zhou G.-K., Nairn C.J., Wood-Jones A., Richardson E.A., Morrison W.H. III, Darvill A.G., York W.S., Ye Z.-H. (2005). Arabidopsis fragile fiber8, which encodes a putative glucuronyltransferase, is essential for normal secondary wall synthesis. Plant Cell 17: 3390–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinska D.F., Gnad F., Schropp K., Wiśniewski J.R., Mann M. (2012). Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol. Cell 46: 542–548. [DOI] [PubMed] [Google Scholar]