

Abstract
Background:
Death-Associated Protein Kinase 1 (DAPK1) plays an important role in apopto-sis, tumor suppression and neurodegeneration including Alzheimer’s Disease (AD).
Objective:
This review will describe the diverse roles of DAPK1 in the development of cancer and AD, and the current status of drug development targeting DAPK1-based therapies.
Methods:
Reports of DAPK1 regulation, function and substrates were analyzed using genetic DAPK1 manipulation and chemical DAPK1 modulators.
Results:
DAPK1 expression and activity are deregulated in cancer and AD. It is down-regulated and/or inactivated by multiple mechanisms in many human cancers, and elicits a protective effect to counteract numerous death stimuli in cancer, including activation of the master regulator Pin1. Moreover, loss of DAPK1 expression has correlated strongly with tumor recurrence and metastasis, suggesting that lack of sufficient functional DAPK1 might contribute to cancer. In contrast, DAPK1 is highly expressed in the brains of most human AD patients and has been identified as one of the genetic factors affecting suscepti-bility to late-onset AD. The absence of DAPK1 promotes efficient learning and better memory in mice and prevents the development of AD by acting on many key proteins including Pin1 and its downstream tar-gets tau and APP. Recent patents show that DAPK1 modulation might be used to treat both cancer and AD.
Conclusion:
DAPK1 plays a critical role in diverse physiological processes and importantly, its deregula-tion is implicated in the pathogenesis of either cancer or AD. Therefore, manipulating DAPK1 activity and/or expression may be a promising therapeutic option for cancer or AD.
Keyword: Alzheimer’s Disease (AD), Apoptosis, Cancer, Death-Associated Protein Kinase 1 (DAPK1), kinase modulator, Phosphorylation, Therapeutic target
1. INTRODUCTION
Phosphorylation of proteins on their Serine (Ser) or Threonine (Thr) residues plays important roles in controlling many diverse cellular processes under various conditions and its deregulation contributes to human pathological conditions, notably cancer and Alzheimer’s Disease (AD) [1-3]. In fact, many oncogenes and tumor suppressors are directly regulated by Ser/Thr phosphorylation and/or can trigger signaling pathways involving Ser/Thr phosphorylation [1, 2]. Moreover, Ser/Thr phosphorylation regulates cell survival by affecting many downstream events, such as coordination of apoptosis and autophagy, and its deregulation can result in resisting cell death in cancer or abnormal neuronal loss in AD [4, 5]. Another interesting feature is increased phosphorylation of certain proteins on their Ser/Thr motifs in degenerating neurons of AD brains [6-10]. Furthermore, many cell cycle-dependent kinases and stress-activated kinases are shown to be activated in AD brains [6-10]. In addition, a number of epidemiologic studies suggest that patients who develop neurodegenerative disease have a decreased risk of cancer [11-13]. This implies that two distinct diseases may have common signaling pathways that are deregulated in different directions.
Death-Associated Protein Kinase 1 (DAPK1) was first identified by a functional cloning strategy based on its involvement in interferon-γ induced apoptosis [14, 15]. DAPK1 is a 160 kDa calcium/calmodulin-regulated Ser/Thr kinase that mediates apoptosis induced by various stimuli [14]. Subsequent studies have shown that DAPK1 activation and/or overexpression promotes caspase-dependent or -independent apoptosis, autophagy, necrosis, and anoikis-like cell death [5, 14, 16, 17]. In addition to a pro-cell-death role, DAPK1 plays important roles in cell growth, tumorigenesis, inflammation, and neurodegeneration [14, 18-22]. These functions are coordinated with interactions between DAPK1 and its binding partners and/or DAPK1-mediated phosphorylation of target substrates. Furthermore, the findings of the opposite effects of DAPK1 in cancer and AD have been supported by epidemiological and genetic association studies [13, 23-29]. This review will focus on the role of DAPK1 in cancer and AD although DAPK1 plays important functions in apoptosis, autophagy and other neurological diseases. Recent reviews provide an up-to-date overview of the role of DAPK1 in apoptosis, autophagy, and DAPK1-related diseases, and present an in-depth discussion of DAPK1 regulation in various neurological disorders [30, 31]. Therefore, the broad aspects of DAPK1 will not be focused in this review. In addition, not all DAPK1 functions and its related diseases can be covered in a single review. Instead, this review provides a major impact of DAPK1 deregulation on cancer and AD development, discusses the development of DAPK1 activators and inhibitors on diseases, and summarizes recently selected patents and therapeutic interventions targeting DAPK1.
2. DAPK1 STRUCTURE
DAPK1 is one of five human DAPK family members including DAPK1-Related Protein 1 (DRP-1 or DAPK2), Zipper-Interacting Protein Kinase (ZIPK or DAPK3), DAPK1-Related Apoptosis-Inducing Protein Kinase 1 (DRAK1), and DRAK2 (Fig. 1) [14, 16, 31]. Although DAPK family members share common functions such as cell death due to their homologous catalytic domain, each DAPK family member has different cellular localization and distinct functions because they have different extra-catalytic domains [16]. DAPK1 contains multiple distinct domains such as an N-terminal kinase domain, a calcium/calmodulin (Ca2+ / CaM)-regulatory domain, eight ankyrin repeats, two putative P-loops, a Ras of Complex (ROC) domain, a C-terminal of ROC (COR) domain, a Death Domain (DD), and a C-terminal serine-rich tail [16]. Each DAPK1 domain plays a unique role in its catalytic activity, localization, stability, and interaction with its substrates or binding partners. The catalytic domain of DAPK1 (residues 13-267) consists of 11 subdomains and is highly conserved with all other DAPK family members [32-34]. A lysine residue (Lys42) is a part of the ATP binding site and mediates the DAPK1 kinase activity [34]. Mutation of this lysine to an alanine residue abolished DAPK1 catalytic activity and function [14, 34]. The Ca2+ / CaM-autoregulatory domain (residues 278-320) is critical for regulating DAPK1 activity in two ways [14, 16]. Ca2+-activated CaM binding to the autoregulatory/CaM binding domain pulls it out from the catalytic cleft and enables substrate phosphorylation [16, 35]. The autophosphorylation of Ser308 of DAPK1 stabilizes the contact of the autoregulatory domain and the catalytic cleft and decreases the affinity for CaM, thereby inhibiting the catalytic activity of DAPK1 [36, 37]. A deletional mutation of the CaM binding domain has a constitutive activation effect on its kinase activity. The phosphatase(s) responsible for the dephosphorylation of Ser308 have long been studied, and PP2A phosphatase has been identified as a strong candidate [38-40]. The phosphatases dephosphorylating DAPK1 at Ser308 might be tissue- or stress-specific under different cellular conditions. For example, Ca2+ activated calcineurin has been identified as another Ser308 phosphatase in response to ischemic insults in the brain [41]. More work needs to be done to fully identify phosphatases in different tissues. DAPK1 contains eight ankyrin repeats (residues 365-629) that mediate actin localization, interact with other proteins, and are implicated in DAPK1 degradation via the ubiquitin-proteasome pathway [35, 42-44]. The ROC-COR domain (residues 667-1,288) was previously identified as a cytoskeleton localization region binding to actin microfilaments [38]. DAPK1 is associated with GTP with a P-loop motif and mediates GTP hydrolysis in the ROC domain [38]. GTP binding inhibits DAPK1 activity and function by promoting Ser308 phosphorylation independent of PP2A phosphatase [39]. This domain also mediates protein-protein interactions that are important functions of cancer and AD such as Peptidyl-Prolyl Cis-Trans Isomerase (PPIase), Pin1 and N-Myc Downstream-Regulated Gene 2 (NDRG2) [45-47]. The death domain (residues 1,312-1,396) mediates most DAPK1-interacting proteins that promote apoptosis or mediate other functions through interacting with different proteins including Fas, Tumor Necrosis Factor Receptor (TNFR), TNFR-Associated Death Domain (TRADD), Fas Associated Death Domain (FADD), Tuberous Sclerosis Complex 2 (TSC2), UNC5H2, Kelch-Like Protein 20 (KLHL20), Mitogen-Activated Protein Kinase 1/2 (MARK1/2), Extracellular Signal-Regulated Kinase (ERK), Pyruvate Kinase Isoenzyme M2 (PKM2), and Amyloid Precursor Protein (APP) [14, 16, 22, 48-54]. Finally, a C-terminal serine-rich tail contains 17 serine amino acids and regulates DAPK1 function without affecting its kinase activity, presumably by protein-protein interaction [43]. Although many crystal structures of DAPK1 were reported to date, none of them include the three-dimensional structure of full-length DAPK1. This information is critical for understanding the molecular mechanisms of DAPK1 function and designing drugs targeting DAPK1 to treat disease.
Fig. (1).
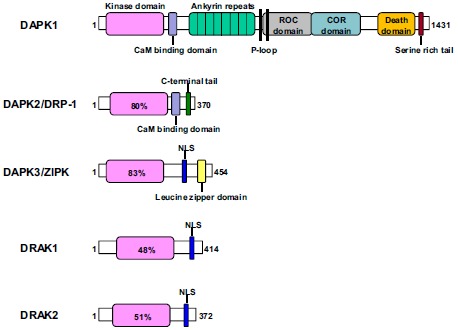
Schematic representation of the death-associated protein kinase family. All five members share a common kinase domain and vary significantly in their size and extra catalytic domains. The numbers of the proteins demarcate the amino acid residues. The percentage in the kinase domain indicates the degree of amino acid identity to the kinase domain of DAPK1. NLS, nuclear localization signal.
3. DAPK1 IN CANCER
Cancer is one of the leading causes of death worldwide. Currently, chemotherapeutics is the most commonly used treatment option for cancer patients. However, one of the major obstacles for successful cancer therapy using these chemotherapeutics is that patients often do not respond or later develop resistance to this chemotherapeutics after primary therapy [55]. Among the many reasons for drug resistance, loss or inactivation of tumor suppressor pathways is a major one. Tumor suppressor genes are frequently deregulated by mutations or epigenetic modifications in drug-resistant cancer and are also responsible for the initiation and progression of cancer, thereby composing an essential class of signaling molecules within the cell [56]. In addition to its death-promoting activity, a large number of studies indicate that DAPK1 plays an important role in tumor suppression and is downregulated or deactivated in a wide variety of cancers (Fig. 2) [14, 18, 20, 57].
Fig. (2).
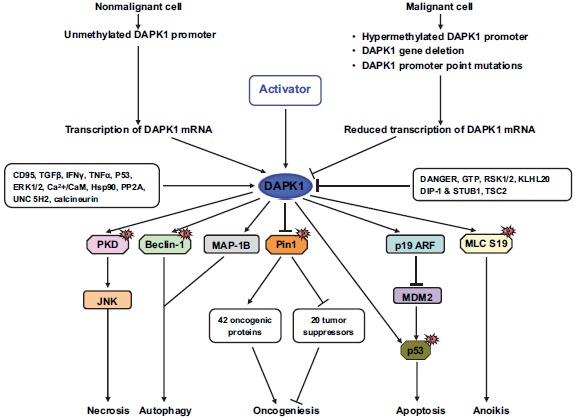
The molecular mechanisms by which DAPK1 suppresses cancer. DAPK1 function can be reduced at multiple levels by methylation and post-transcriptional or -translational modifications. DAPK1 controls two master regulators of many signaling pathways by activating p53 or inhibiting Pin1, resulting in growth arrest and apoptosis.
3.1. DAPK1 and its Regulation in Cancer
The connection between DAPK1 and cancer was first suggested when DAPK1 was found to be downregulated in human cancer tissues [20]. These observations have been subsequently confirmed by demonstrations that DAPK1 promoter regions are significantly methylated in 30 different human tumor types compared with their corresponding normal tissue [18, 20, 57-59]. Indeed, loss of DAPK1 expression is found in most prevalently encountered cancers including lung, colon, breast, head and neck, kidney, liver and B cell malignancies [18, 20, 57-59]. Comparison between DAPK1 promoter hypermethylation and clinical specimens of cancer patients shows a significant correlation. Hypermethylation of the DAPK1 promoter correlates with poor prognosis and advanced disease stage in lung cancer patients [60-62]. Moreover, lymph node involvement and DAPK1 promoter hypermethylation have been found to be correlated with gastric carcinoma and head and neck cancers [63-65]. Loss of DAPK1 expression has been shown to correlate strongly with recurrence and metastasis incidence in breast cancer patients [66]. DAPK1 promoter hypermethylation has been shown to be less sensitive to radiochemotherapy in esophageal squamous cell carcinoma and Non-Small Cell Lung Cancer (NSCLC) [60, 67]. Collectively, DAPK1 promoter hypermethylation is positively correlated with advanced stage and metastatic potential of specific cancers, suggesting that DAPK1 methylation may present a potential prognostic marker in selected human cancers. Indeed, restoration of physiological levels of DAPK1 in the highly metastatic Lewis carcinoma efficiently suppresses its ability to form metastases in mice [68]. Although hypermethylation of the DAPK1 promoter is frequently documented as an inactivation mechanism in cancers, DAPK1 protein can still be expressed in the presence of hypermethylation in primary tissues and NSCLC cell lines. Loss of DAPK1 expression in the absence of promoter hypermethylation has also been reported, suggesting the existence of an additional layer of DAPK1 regulation in tumors, including post-transcriptional regulation of DAPK1. In limited cases, homozygous deletions, allelic deletions, point mutations and single nucleotide polymorphism in the DAPK1 gene have been reported [18, 20, 59, 69]. Micro RNAs miR-103 and miR-107 bind to the 3-UTR of DAPK1 and suppress DAPK1 expression, thereby inactivating integrin B1 [70]. These two miRNAs mediate integrin B inactivation by targeting DAPK1, resulting in increased cell motility and cell-matrix adhesion and decreased cell-cell adhesion in colorectal cancer [70]. Post-translational modifications of DAPK1 such as phosphorylation and ubiquitination, can affect DAPK1 activity and stability. The Ser289, Ser308, Tyr491, Tyr492 and Ser735 residues in the DAPK1 protein are currently well-known phosphorylation sites. For example, oncogenic Src phosphorylates DAPK1 at Tyr491 and 492 in its ankyrin repeats, thereby inhibiting DAPK1 catalytic activity and biological function [71]. ERK binds to DAPK1 through the DAPK1 DD and directly phosphorylates it at Ser735 [49]. Phosphorylation of DAPK1 by ERK increases the catalytic activity of DAPK1 and promotes DAPK1 apoptotic activity [49]. In contrast, Ras-ERK activation through p90 ribosome S6 kinase phosphorylates DAPK1 at Ser289 and suppresses its apoptotic activity [72], suggesting that multiple oncoproteins and tumor suppressors might be involved in DAPK1 activity by influencing its function in cancer signaling. In addition, E3 ubiquitin ligases, carboxyl terminus of HSC70-Interacting Protein (CHIP), DAPK-Interacting Protein 1 (DIP1) and KLHL20 target DAPK1 and degrade it by the proteasome pathway, thereby regulating DAPK1 protein stability [44, 51, 73, 74]. However, the effect of DAPK1 ubiquitination in cancer has not been determined.
3.2. DAPK1 and its Substrates in Cancer
Several lines of evidence indicate that DAPK1 plays an important role in tumor suppression with interacting partners or downstream substrates such as tumor suppressor p53 and oncogenic Pin1 [18, 48]. First, DAPK1 was found to be capable of suppressing c-myc- and E2F-induced oncogenic transformation in a p53-dependent manner. DAPK1 activates p53 through induction of p19ARF, thereby binding to Mdm2 and increasing p53 protein stability [75]. DAPK1 increases p53 gene transcription and its responsive genes and induces apoptosis in the presence of p53 [75]. Moreover, DAPK1 inhibits integrin-mediated cell survival signals, thereby inactivating FAK and upregulating p53 [76]. Interestingly, since p53 can also increase DAPK1 transcriptional levels, a positive feedback regulation between DAPK1 and p53 has been suggested [77]. Recently, Brown and colleagues discovered that DAPK1 may increase cancer cell growth in p53-mutant triple-negative breast cancer via the mTOR/S6K pathway [78]. These results imply that DAPK1 may switch its apoptotic role to a proapoptotic role in certain cellular environments, suggesting a new therapeutic strategy for p53-mutant cancers.
In addition, DAPK1 inhibits the oncogenic ability of Pin1 to induce cell transformation and migration by suppressing Pin1 enzymatic activity via phosphorylation [3, 45, 46]. Pin1 is a unique peptidyl-prolyl cis-trans isomerase that binds only to phosphorylated Ser/Thr-Pro-motifs and induces conformational changes in proteins [79]. Pin1 has important roles in many cellular processes by regulating the functions of proline-directed phosphor proteins in the cell cycle, cell signaling, transcription and splicing, DNA damage responses, germ cell development and neuronal survival [79-81]. Pin1 activates more than 40 oncogenes or growth-promoting regulators and inhibits close to 20 tumor suppressor or growth-inhibiting regulators, indicating that Pin1 is a pivotal enzyme for tumorigenesis by turning on or off numerous oncogenes or tumor suppressors, respectively, at multiple steps at the same time [82-98]. Moreover, Pin1 overexpression in proliferative mammary glands leads to breast cancer [87], whereas Pin1 inhibition or Knockout (KO) prevents cancer induced by oncogenic Ras or Neu or by p53 KO in mouse models [90, 99-102]. Our group found that DAPK1 directly binds to the PPIase domain of Pin1 in contrast to all known Pin1-binding proteins at the WW domain of Pin1 and phosphorylates Pin1 at Ser71 in vitro and in vivo [45]. DAPK1-mediated Pin1 phosphorylation blocks Pin1 nuclear localization, inhibits the ability of Pin1 to activate transcription factors and stabilize proteins, and attenuates Pin1-induced centrosome amplification, chromosome instability and cell transformation [45]. Moreover, Ser71 phospho-mimicking mutations not only completely inactivate Pin1 activity, but also fully inhibit Pin1 function in cells, whereas Ser71 non-phosphorylation mutation is fully functional [45]. Finally, Pin1 Ser71 phosphorylation levels positively correlated with DAPK1 levels, but negatively correlated with centrosome amplification in breast cancer tissues [45]. These results indicate that the tumor suppressor DAPK1 is an important enzyme to suppress Pin1 oncogenic function and might eventually lead to more effective therapeutic strategies for cancer [3, 46]. DAPK1 can also affect cell mobility and cytoskeletal structure [18]. DAPK1 phosphorylates Myosin Light Chain II (MLCII) at Ser19, thereby promoting cell detachment from the extracellular matrix and membrane blebbing, resulting in cell death [42, 103]. Thus, accumulating evidence indicates that DAPK1 may have a critical role in tumor suppression.
4. DAPK1 IN ALZHEIMER’S DISEASE
Alzheimer’s Disease (AD) is the most prevalent neurodegenerative disease associated with a progressive loss of memory. AD is characterized by Neurofibrillary Tangles (NFTs) composed of hyperphosphorylated tau (p-tau) and senile plaques comprised of Amyloid-β Peptides (Aβ) derived from the APP [104-107]. Tau is a Microtubule (MT)-associated protein that normally stabilizes the MT cytoskeletal network that functions to maintain the unique neuronal structure and transports proteins and other molecules through the neurons [108-110]. Phosphorylation is a key regulatory mechanism, which disrupts the ability of tau to bind MTs and to promote their assembly [111]. In AD, tau is hyperphosphorylated and aggregated into abnormal conformations of filamentous Paired Helical Filaments (PHFs) that make up NFTs [112-114]. APP is a transmembrane protein processed by two different proteolytic processes, amyloidogenic or non-amyloidogenic pathways [107, 115]. In the amyloidogenic pathway, β-secretase (BACE1) cuts APP at the beginning of Aβ sequence, generating an extracellular soluble fragment called βAPPs and an intracellular COOH-terminal fragment called βCTF [107, 115]. Subsequently, γ-secretase cuts βCTF at residues 40/42/43 of the Aβ sequence, generating an intact insoluble Aβ species that is aggregated in senile plaques [107, 115]. Previous studies shown that Aβ exacerbates tangle formation in tau mutant mice and tau reduction blocks Aβ-mediated toxicity [116-120]. These results strongly indicate interactions between Aβ deposits and tau tangles, and a common molecular mechanism may influence both tangle and plaque formation, through the regulation of both tau and APP.
In addition to acting as a tumor suppressor, DAPK1 has been identified as a major common regulator of both tau and APP and might link both tangle and plaque pathologies in cell and animal AD models (Fig. 3) [21, 22]. Importantly, it was shown that DAPK1 regulates levels of phosphorylated tau as well as levels of Aβ, as gene knockout of DAPK1 protects against the age-dependent neurodegeneration of AD with decreased levels of phosphorylated tau and decreased pathogenic processing of APP and insoluble beta-amyloid peptide [21, 22]. In particular, DAPK1 expression is significantly upregulated in the hippocampal region of AD patients compared with age-matched normal subjects [21, 22]. These findings are particularly interesting because the hippocampal region of the brain is specifically affected by AD. Moreover, two Single-Nucleotide Polymorphisms (SNPs) that regulate DAPK1 allele-specific expression are strongly associated with late-onset AD [23-26]. In addition, DAPK1 kinase-activity-deficient mice are more efficient learners and have better spatial memory than Wild-Type (WT) mice [121, 122]. Taken together, these results indicate that DAPK1 plays an important role in neurodegenerative disorders including AD.
Fig. (3).
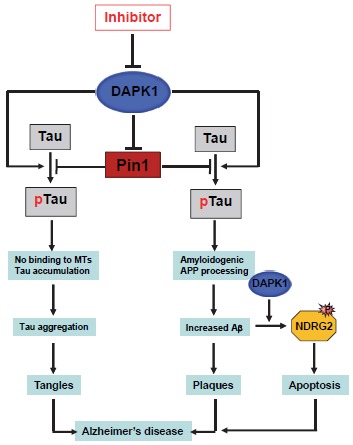
The regulation of tau function and APP processing by DAPK1 in AD neurons. DAPK1 expression leads to increased phosphorylation levels of tau, APP, Pin1, and NDRG2, thereby promoting tau-related pathology, amyloidogenic APP processing, neuronal cell death and AD.
4.1. DAPK1 and Tau Phosphorylation
Tau is hyperphosphorylated at more than 25 specific sites, aggregated into PHFs, and eventually forms NFTs in the human AD brain [112-114]. For example, Thr231 is phosphorylated at the beginning of sequential tau phosphoepitopes appearing during the early stages of AD pretangle formation [123]. Phosphorylation of tau on Ser262 facilitates the detachment of tau from MTs and Ser396 phosphorylation is found in insoluble PHFs [112-114]. Our group discovered that DAPK1 overexpression induces tau phosphorylation at sites including Thr231, Ser262, and Ser396 in neuronal cells [21]. However, DAPK1 kinase-deficient mutant (K42A) failed to increase tau phosphorylation [21], indicating that DAPK1 regulates tau phosphorylation by a kinase activity-dependent manner. Moreover, DAPK1 KO mice show decreased levels of tau phosphorylation in the brain in an age-dependent manner [21]. Furthermore, DAPK1 also increases tau protein stability by a kinase activity-dependent manner in neuronal cells and the mouse brain. Constitutive active DAPK1 mutant (ΔCaM), but not K42A, significantly enhances tau protein stability with cycloheximide treatment [21].
Interestingly, increased tau phosphorylation and stability is positively correlated with Pin1 Ser71 phosphorylation [21]. DAPK1 does not increase the stability of T231A tau mutant that is not affected by Pin1 and does not exert its effect on tau protein stability in Pin1 KO cells [21]. Moreover, the expression of phosphorylated Pin1 at Ser71 is significantly lower in DAPK1 KO mouse brain compared with WT mouse [21]. It has been reported that Pin1 restores the ability of p-tau to promote MT assembly, facilitates p-tau dephosphorylation, and promotes p-tau degradation by acting on the pThr231-Pro motif thereby isomerizing cis to trans conformation in tau [124-127]. Pin1 KO mice show age-dependent AD pathologies and neurodegeneration [125, 128]. Furthermore, Pin1 overexpression in mouse postnatal neurons promotes tau degradation and suppresses tauopathy induced by human tau [126]. These results indicate that Pin1 has a pivotal role against AD by regulating phosphorylated tau [3, 124, 125, 129-132], suggesting that DAPK1 might inhibit tau function and induce tau phosphorylation by inhibiting Pin1 function. Chen and colleagues found that DAPK1 increases tau toxicity by inhibiting microtubule assembly and polarizing neurite outgrowth through MARK1/2 phosphorylation [52]. Therefore, DAPK1 indirectly increases tau Ser262 phosphorylation by regulation of MARK1/2 activity. Recently, direct interaction of DAPK1 and tau was found to cause spinal damage and subsequent neuronal cell death in a stroke model [133]. DAPK1 directly phosphorylates tau at Ser262 and inhibition of DAPK1 activity or disruption of the DAPK1 and tau interaction protects against spinal damage in mice [133]. However, it has also been reported that DAPK1-induced tau phosphorylation is more resistant to apoptosis in human embryonic kidney cells, suggesting that tau hyperphosphorylation may have a neuroprotective effect in certain stages of AD progression [134]. This discrepancy might be due to the selected cell lines, different disease models or, in part, to differences in the sensitivity and specificity of the methods used. Therefore, the in vivo effects of DAPK1-mediated tau hyperphosphorylation in neuronal cell death in the brain remain to be determined.
4.2. DAPK1 and APP Processing
Increasing evidence suggests that APP processing and function is modulated by phosphorylation on the APP intracellular domain [135, 136]. Seven APP sites have been found to be phosphorylated in AD patients in a neuron-specific manner [137]. Among them, phosphorylation of APP at Thr668 is elevated in human AD brains and leads to the amyloidogenic processing of APP and increased Aβ formation [137]. It has been recently shown that DAPK1 significantly increases human Aβ secretion by a kinase activity-dependent manner in neuronal cell culture models [22]. Moreover, Knockdown (KD) of DAPK1 expression or inhibition of DAPK1 catalytic activity decreases both Aβ 40 and 42 secretions. In mice overexpressing APP, DAPK1 ablation has shifted APP processing toward a non-amyloidogenic pathway and decreases Aβ production [22]. DAPK1 DD is associated with APP and increases APP Thr668 phosphorylation [22]. Interestingly, DAPK1 activates brain-specific JNK3 under oxidative stress and increases APP phosphorylation and Aβ secretion [22]. In addition, our group found that the expression of DAPK1 is increased in hippocampal brain slices after Aβ or ceramide treatment and has identified N-myc Downstream-Regulated Gene 2 (NDRG2) as a novel DAPK1 substrate that is involved in neuronal cell death and AD under Aβ treatment [47]. DAPK1 ablation in APP overexpressing mice suppresses neuronal cell death in the brain [47]. Furthermore, NDRG2-mediated neuronal cell death by DAPK1 requires a caspase-dependent apoptosis pathway [47]. Since Pin1 also acts on the pThr668-Pro motif in APP to promote APP turnover and non-amyloidogenic APP processing, and reduce Aβ production, DAPK1 might regulate Aβ production by phosphorylating Pin1 on Ser71 [3, 128, 138]. Finally, a small molecule inhibitor of DAPK1 has been described for attenuating neuronal damage in a chronic infusion model of Aβ toxicity [139]. Together, these results strongly suggest that DAPK1 may be a critical regulator of APP processing and DAPK1 deregulation may contribute to AD progression. However, the upstream regulators of DAPK1 expression under Aβ treatment and whether modulating DAPK1 expression in vivo affects the development of AD remains unknown. Future studies should include the development of transgenic mice with inducible and brain-restricted DAPK1 expression and DAPK1 ablation or DAPK1-overexpression mice crossed with AD mouse models.
5. DAPK1 AS A POTENTIAL DIAGNOSTIC AND THERAPEUTIC TARGET
5.1. DAPK1 Activators
Because DAPK1 plays an important role in inhibiting neoplasmic transformation and suppressing metastasis, upregulation and/or activation of DAPK1 could be a viable strategy for cancer treatment if it could be limited to cancer tissues, due to its cell death potential in cells [29, 140]. The current approach to designing DAPK1-based therapy for cancer is to upregulate DAPK1 protein expression or inhibit Ser308 autophosphorylation of DAPK1 to activate its kinase activity. It has been reported that a total of seven reagents could regulate DAPK1 activity by post-translational modifications or regulate DAPK1 expression by transcription, translation and degradation (Table 1) [141-149]. The first FDA-approved pan-histone deacetylase (HDAC) inhibitor, panobinostat (LBH589), induces autophagy in the HCT116 colon cancer cell line through DAPK1 activation via induction of Ser308 dephosphorylation and DAPK1 protein levels [141]. Moreover, DAPK1 induces caspase-dependent and -independent apoptosis under panobinostat treatment in autophagy-deficient cells [141]. SB203580, a well-known p38 MAPK inhibitor is reported to mediate its anti-proliferative activity and pro-cell-death function through DAPK1 activation by dephosphorylation at Ser308, thus increasing autophagic cell death in human Hepatocellular Carcinoma (HCC) cells [142]. Another HDAC inhibitor, trichostatin A, which is isolated from Streptomyces hygroscopicus, induces chemosensitivity in ovarian cancer, gastric cancer, and erythroleukemia cells. Trichostatin A promotes apoptosis of A549 lung cancer cells and enhances the sensitivity of the cells to cisplatin, mediated by both upregulations of DAPK1 expression and downregulation of DAPK1 Ser308 [143]. Grifolin, a secondary metabolite of the mushroom Albatrellus confluens has been reported to suppress some cancer cell lines by promoting apoptosis. Grifolin induces DAPK1 mRNA and protein levels by increasing the phosphorylation of Ser392 and Ser20 in the p53 protein in breast cancer and colon cancer cell lines [144]. Since p53 is a positive transcription factor of DAPK1, grifolin may induce apoptosis in cancer cells via the p53-DAPK1 pathway. Another natural product, curcumin, which is a component of the perennial herb Curcuma longa increases levels of DAPK1 mRNA and protein in U251 cells [145]. The anti-proliferative and pro-apoptotic effects of curcumin are mediated by DAPK1 through STAT-3 and NF-kB inhibition and caspase-3 activation [145]. In addition, DNA methyltransferase inhibitors, Vidaza (5-azacytidine) and Decitabine (5-aza-2'-deoxycytidine), which were approved by the FDA, could restore DAPK1 expression from DAPK1 promoter hypermethylation, indicating an interesting therapeutic option for promoting re-expression of the silenced DAPK1 gene [146-149]. Since all DAPK1 activators mentioned in this section are not designed to directly target DAPK1 activity or expression, the development of DAPK1 activators are still in the early stages of evaluation.
Table 1. DAPK1 Activators.
| Name / Function | Effect on DAPK1 | DAPK1 Cellular Function | Reference |
|---|---|---|---|
| Panobinostat/LBH589 (Histone deacetylation inhibitor) |
mRNA & protein enhanced Ser308 dephosphorylation |
Induce autophagy | [141] |
| SB203580 (p38 MAPK inhibitor) | Ser308 dephosphorylation | Induce autophagy | [142] |
| Trichostatin A (Histone deacetylation inhibitor) |
Protein enhanced | Induce apoptosis | [143] |
| Grifolin (Albatrellus confluens extract) |
mRNA & protein enhanced Ser308 dephosphorylation |
Induce apoptosis | [144] |
| Curcumin (Curcuma longa extract) |
mRNA & protein enhanced | Induce apoptosis | [145] |
| Vidaza(5-azacytidine) (Demethylating agent) |
mRNA enhanced | Induce apoptosis | [146-149] |
| Decitabine (5-aza-2'-deoxycytidine) (Demethylating agent) |
mRNA enhanced | Induce apoptosis | [146-149] |
5.2. DAPK1 Inhibitors
DAPK1-specific inhibitors offer the potential of a novel therapeutic strategy for neurological disorders such as ischemia and AD because DAPK1 is essential for neuronal cell death and loss under various stimuli [5, 29, 30]. With the crystal structure of the catalytic domain of DAPK1, most DAPK1 inhibitors have been developed targeting its kinase activity [29, 140, 150] (Table 2; [151-158]). The first DAPK1 inhibitor, an alkylated 3-amino-6-phenylpyridazine was developed to target acute brain injury such as stroke [151, 152]. It has been shown to suppress DAPK1 kinase activity and decrease the loss of brain tissue after cerebral ischemia using both acute and sustained brain injury animal models after a single intraperitoneal injection, suggesting that DAPK1 inhibition may provide a new therapeutic approach to suppress early programmed cell death in acute brain injury [151]. Other DAPK1 inhibitors including (4Z)-4-(3-pyridylmethylene)-2-styryl-oxazol-5-one (Compound 6) have been synthesized using structure-based virtual screening [153, 154]. Compound 6 effectively and selectively inhibits DAPK1 activity (IC50 = 69nM) [153]. Our group discovered that compound 6 increases neurite outgrowth and microtubule polymerization in regulating tau function by affecting DAPK1 activity [21]. Compound 6 also reduces both Aβ40 and Aβ42 production in neuronal cell lines and primary cortical neurons, and significantly attenuates Aβ-induced neuronal cell death [22, 47]. A reversible peptide targeting DAPK1 has been shown to rapidly and specifically knock down DAPK1 through the lysosomal machinery [155]. The peptide consists of the DAPK1 binding domain of the NMDA receptor GluN2B subunit (GluN2B1242-1342) and Chaperone-Mediated Autophagy-Targeting Motif (CTM) (KFERQKILDQRFFE). DAPK1 activated by oxidative stress in neuronal cell culture or a focal ischemia model in rats is targeted for degradation through the lysosome by the peptide containing CTM in a dose- and time-dependent manner, indicating that a DAPK1-targeting peptide–based method might be an effective way to inhibit DAPK1. Other DAPK1 inhibitors in the table need more analysis to study DAPK1 specificity and effects on DAPK1-mediated cellular function [139, 151, 156-158]. Since DAPK1 inhibits the ability of Pin1 to promote cis/trans conformational changes of phosphorylated tau on Thr231 (pT231-tau) and cis pT231-tau is resistant to dephosphorylation and degradation, and lead to aggregation into NFTs, cis/trans conformation-specific antibodies might be an effective way to eliminate toxic p-tau [21, 132]. Indeed, our group developed monoclonal cis- and trans-specific pT231-tau antibodies (mAbs) and showed that cis mAb treatment could enter neurons, block cis-p-tau induction and prevent tau-related pathology development in mouse models [131, 159]. Thus, conformation-specific immunotherapies might offer a new therapeutic method against tau-related pathologies including AD [160-164]. It has also been reported that six FDA-approved drugs, sunitinib, ruxolitinib, baricitinib, dabrafenib, abemaciclib, alectinib can suppress DAPK1 activity, although all of these drugs target specific other kinases including c-Jun NH(2)-Terminal Protein Kinase (JAK), v-Raf murine sarcoma viral oncogene homolog-B1 (Braf), Epidermal Growth Factor Receptor (EGFR), Cyclin-Dependent Kinase (CDK) and Anaplastic Lymphoma Kinase (ALK) rather than DAPK1 [140]. However, FDA-approved drugs may be beneficial in developing more potent and selective DAPK1 inhibitors as a leading compound with the crystal structure for full-length DAPK1.
Table 2. DAPK1 Inhibitors.
| Name | Specificity on DAPK1 | DAPK1 Cellular Function | Reference |
|---|---|---|---|
| 3-Amino-6-phenylpyridazine derivative | IC50 = 13μM | Inhibit neuronal cell death | [151, 152] |
| (4Z)-4-(3-Pyridylmethylene)-2-styryl-oxazol-5-one | IC50 = 69nM | Promoter neuronal differentiation and inhibit DAPK1-induced Aβ production and neuronal cell death | [21, 22, 47, 153, 154] |
| 2-(3-Chlorophenyl)-4-(3-pyridylmethylene)-1,3-oxazolin-5-one | IC50 = 529.5nM | N/A | [154] |
| 2-(3-Methylphenyl)-4-(3-pyridylmethylene)-1,3-oxazolin-5-one | IC50 = 713.7nM | N/A | [154] |
| 2-(4-Bromo-3-methylphenyl)-4-(3-pyridylmethylene)-1,3-oxazolin-5-one | IC50 = 252.9nM | N/A | [154] |
| 2-Phenyl-4-(4-pyridinylmethylene)-5(4H)-oxazolone | IC50 = 583nM | N/A | [154] |
| (4Z)-2-(3,4-Difluorophenyl)-4-(3-pyridylmethylene) - 1,3-oxazolin-5-one | IC50 = 346.1nM | N/A | [154] |
| FK506 | N/A | Inhibit neuronal cell death | [156] |
| MK-801 | N/A | Inhibit neuronal cell death | [157] |
| Pyrazolo[3,4-d]pyrimidinone (HS38) | IC50 = 200nM | N/A | [157] |
| Morin | IC50 =15μM | N/A | [158] |
6. RECENT PATENTS TARGETING DAPK1 IN CANCER AND AD
Recent patents suggest that various compositions and methods for the modulation of DAPK1 activity and expression might give new therapeutic options for treating cancer and AD. Recent patents targeting DAPK1 in cancer therapy focus on diagnosis to detect DAPK1 expression status on early cancer stage and treatment to promote cancer cell death. New patents report methods and kits for the detection of DAPK1 methylation status in head and neck cancer, and ovarian cancer [165, 166]. DAPK1 promoter regions are highly methylated in more than 65% of head and neck cancer, and ovarian cancer patient samples [29]. The inventions provide high sensitivity and specificity for the detection of the methylation status in the targeted genes and regulatory regions of genes including DAPK1 in the saliva, serum, blood, and urine samples, suggesting that the test can be carried out at low-cost. Other recent patents suggest new methods for measuring DAPK1 expression status in different types of cancer. The inventions provide methods for detecting or diagnosing metastatic non-small cell lung cancer by promoter hypermethylation of DAPK1 in particular in the tumor, lymph nodes, bone marrow and blood [167]. Another patent provides a new non-invasive method for the early detection of gastric cancer [168]. Although the gastric cancer is the fourth most common cancer and the second leading cause of cancer related deaths in the world, the diagnosis of gastric cancer in the early stage is difficult because the majority of gastric cancer patient are asymptomatic until advanced stage. DAPK1 methylation is found in more than 80% of gastric cancer patient samples [29]. The inventions indicate that an alternative non-invasive method using patient plasma provides massive and rapid detection of the methylation status of twenty-four tumor suppressors including DAPK1. A recent patent provides combination therapy by activating DAPK1 may inhibit cancer cell growth in orthotopic spontaneous kidney cancer [169]. A pharmaceutical composition comprising sorafenib and GW5074 affects c-Raf-PP2A-DAPK1 signaling pathway in either in vitro or in preclinical animal model. The binding of c-Raf and GW5074 leads to conformational change that consequently increases the association of sorafenib and c-Raf, thereby increasing Ser308 dephosphorylation of DAPK1 by PP2A. The invention suggests a novel drug designed targeting c-Raf and DAPK1 protein complex and a new biomarker for phosphorylation status of DAPK1 for drug screening. A new patent provides a pharmaceutical composition comprising a therapeutically effective amount of DAPK1 activators or inhibitors in the treatment of cancer [170]. The inventions provide a specific design for DAPK1 activators and inhibitors for DAPK1 isoforms, DAPK1α and DAPK1β. DAPK1 is undergoing an alternative splicing leading to the production of two isoforms [171]. These two forms differ in ten amino acids at the C-terminus of the pre-mRNA and have antagonistic functions. DAPK1α possesses pro-apoptotic function whereas DAPK1β attenuates TNF-induced apoptosis [172], suggesting that activating DAPK1α and suppressing DAPK1β might be beneficial in the treatment of cancer. However, the exact mechanism of DAPK1 pre-mRNA alternative splicing remains unknown and the inventions were only performed in vitro, indicating that physiological consequences for the activation and inhibition of DAPK1 isoforms remain determined. A recent patent suggests novel compositions and methods for regulating DAPK1 activity by controlling ROC domain for identifying agents in cancer and AD [173]. DAPK1 ROC domain acts on a GTP binding site that suppresses DAPK1 activity by increasing Ser308 phosphorylation [38]. To screen agents for promoting DAPK1 activity, the invention provides a method that determines the ability of agents to negatively affect GTP binding to the ROC domain and to decrease Ser308 phosphorylation. The invention is also presented a method for the identification of agents to enhance GTP binding to the ROC domain, prevent GTP hydrolysis from the ROC domain and increase Ser308 phosphorylation for treating neurodegeneration including AD.
A recent patent provides novel chemical compounds, compositions and methods of making specific DAPK1 inhibition in stroke and AD [174]. The invention provides pyridazine compounds and related heterocyclic derivatives which specifically target DAPK1 kinase activity. The pyridazine compounds suppress neuronal cell death, Aβ-induced neuroinflammation and synaptic damage in mice. Since DAPK1 increases tau phosphorylation, Aβ production, and neuronal cell death, the invention might be useful for drug screening and therapeutic applications specifically targeting DAPK1. Several chemical formulas including peptides were developed for targeting DAPK1 and its related kinases [175-178]. The compounds bind to an ATP-binding site and a substrate-recognition site of DAPK1 simultaneously, thereby inhibiting DAPK1 catalytic activity. The inventions also provide various DAPK1 inhibiting compositions with other agents to suppress DAPK1 activity. Because the formulas suppress the activity of DAPK1 and other kinases which have similar ATP-binding pocket in the kinase domain, in vivo preclinical effect of DAPK1 remains elusive. A recent patent provides a peptide comprising a chaperone-mediated autophagy-targeting signal domain, a protein-binding domain that selectively binds to a target cytosolic protein, and a cell membrane penetrating domain [179]. The peptide (TNT-NR2BCTM) is observed to only target activated DAPK1 and cause DAPK1 protein degradation. The inventions show that the peptide treatment in mouse primary cortical neurons increases activated DAPK1 degradation after H2O2 treatment. Moreover, TAT-NR2BCTM specifically decreases DAPK1 levels in the ischemic brain areas and reduces ischemic neuronal damage in the rat middle cerebral artery occlusion focal ischemia model in vivo. Thus, the invention suggests that the peptide may selectively knockdown activated DAPK1 under certain pathological conditions such as AD.
CONCLUSION
DAPK1 plays an important role in regulating cell growth and death, which is critical to the survival of both proliferating and differentiated tissues. Deregulation in DAPK1 expression and/or activity might cause or promote the development of cancer and neurodegeneration. DAPK1 hypermethylation or inactivation promotes or causes cancer through anti-apoptotic pathways, and effective DAPK1 upregulation targeted only to cancer cells would be a powerful anti-cancer therapy. In contrast, DAPK1 is genetically associated with AD, highly overexpressed in the AD brain and promotes both tau and APP toxic pathways, thereby contributing to AD development.
Therefore, inhibiting DAPK1 targeting in degenerated neurons might be a novel therapeutic option for treating this devastating disease. However, development of DAPK1 modulators is still in the early stages and may be challenging due to specificity, selectivity issues, off-target effects, side effects, and the BBB. More research is needed to better understand how DAPK1 is regulated in certain tissues and diseases and how DAPK1 interplays with its binding partners.
CURRENT & FUTURE DEVELOPMENTS
The targeting of DAPK1 is an attractive drug candidate because DAPK1 has opposite effects in cancer and AD. Moreover, DAPK1 is an upstream negative regulator of Pin1 that activates numerous oncogenes and suppresses many tumor suppressors, suggesting that activating DAPK1 effectively inhibits multiple oncogenic signaling pathways. The biggest challenge of DAPK1 activating reagents to increase DAPK1 expression and activity is lack of specificity. Most DAPK1 activators inhibit DNA methyltransferase to increase DAPK1 transcriptional levels or general protein degradation pathways to stabilize DAPK1 protein [29]. It has also been reported that DAPK1 might be oncogenic on certain cellular condition such as p53 mutant cancers [78]. Therefore, more studies of DAPK1 regulatory mechanisms and specific DAPK1 activators are needed. DAPK1 could be reactivated by enhancing DAPK1 expression, DAPK1 activity, and DAPK1 protein delivery. Restoring of transcriptional or post-transcriptional activation of DAPK1 could be achieved by enhancing the activity of DAPK1 transactivators, inhibiting DAPK1-targeting miRNAs, and repressing epigenetic silencing. Targeting E3 ligases that promote DAPK1 degradation might be another option to restore DAPK1 protein expression levels. Recently, a new patent provides E3 ubiquitin ligases using gene mutation to promote DAPK1 protein degradation [180]. The invention only targets DAPK1 protein degradation via the ubiquitin proteasome pathway in a tissue specific manner, suggesting that this method might be effective in p53 mutant tumors. DAPK1 activity could be increased through various methods, including development of small molecules that can block the interaction of DAPK1 and negative binding partners, and or engineering DAPK1 variants with increased kinase activity [38, 181]. DAPK1 protein delivery and gene therapy to only cancer tissues that lose DAPK1 expression are another plausible DAPK1 reactivation strategy although it remains challenging due to degradation, low membrane permeability, and specificity.
The greatest challenge facing small molecule DAPK1 inhibitors treating AD is effective penetration of the Blood-Brain Barrier (BBB). The BBB is a highly specialized endothelial cell membrane and plays a key role in the generation of chronic inflammation during AD. It is estimated that 98% of small molecule drugs are unable to effectively cross the BBB [182]. Therefore, DAPK1 inhibitors should be systematically designed to penetrate BBB. For example, DAPK1 inhibitors could be conjugated with nano-particles such as a near-infrared, thereby visualizing and targeting DAPK1 inhibitors to brain [183]. Since DAPK1 is a well-known tumor suppressor and has an inverse association between cancer and AD, targeting DAPK1 for treating AD might lead to undesirable side effects such as tumorigenesis in other proliferative tissues. One of the solutions may be to design DAPK1 inhibitors that only target to damaged neurons. Because mice with whole-body DAPK1 KO do not produce any spontaneous tumors or have any defects compared with WT mice [14], long-term treatment of DAPK1 inhibitors may not have acute effects. Moreover, the inhibition of DAPK1 kinase activity has been shown to lead to better learning and memory in mice [121, 122], suggesting that DAPK1 inhibitors may be used to treat AD if they can be modified to be more selective with fewer off-target effects [150]. Therefore, the efficacy, potency and side effects should be evaluated in designing DAPK1-specific inhibitors.
ACKNOWLEDGEMENTS
This study was supported by the Fujian Medical University (XRCZX2017007) to Dongmei Chen and the Fujian Medical University (XRCZX2017019) and the Alzheimer’s Disease Research Program of the Alzheimer’s Association (AARG-17-528817) through research funds awarded to Tae Ho Lee.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are base of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
This study was supported by the Natural Science Foundation of Fujian Province (No. 2019J05072) to Dongmei Chen and the Natural Science Foundation of Fujian Province (No. 2019J01297) and the Alzheimer’s Disease Research Program of the Alzheimer’s Association (AARG-17-528817) to Tae Ho Lee.
CONFLICT OF INTEREST
Xiao Zhen Zhou is an inventor of Pin1 technology, which was licensed by Beth Israel Deaconess Medical Center (BIDMC) to Pinteon Therapeutics. Dr. Zhou owns equity in, and consult for, Pinteon. Her interests were reviewed and managed by BIDMC in accordance with its conflict of interest policy.
REFERENCES
- 1.Blume-Jensen P., Hunter T. Oncogenic kinase signalling. Nature. 2001;411(6835):355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 2.Pawson T., Scott J.D. Protein phosphorylation in signaling: 50 Years and counting. Trends Biochem. Sci. 2005;30(6):286–290. doi: 10.1016/j.tibs.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Lee T.H., Pastorino L., Lu K.P. Peptidyl-prolyl cis-trans isomerase Pin1 in ageing, cancer and Alzheimer disease. Expert Rev. Mol. Med. 2011;13:e21. doi: 10.1017/S1462399411001906. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D., Weinberg R.A. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Fujita Y., Yamashita T. Role of DAPK in neuronal cell death. Apoptosis. 2014;19(2):339–345. doi: 10.1007/s10495-013-0917-4. [DOI] [PubMed] [Google Scholar]
- 6.Yang Y., Geldmacher D.S., Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 2001;21(8):2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagy Z., Esiri M.M., Smith A.D. The cell division cycle and the pathophysiology of Alzheimer’s disease. Neuroscience. 1998;87(4):731–739. doi: 10.1016/s0306-4522(98)00293-0. [DOI] [PubMed] [Google Scholar]
- 8.Raina A.K., Monteiro M.J., McShea A., Smith M.A. The role of cell cycle-mediated events in Alzheimer’s disease. Int. J. Exp. Pathol. 1999;80(2):71–76. doi: 10.1046/j.1365-2613.1999.00106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gandy S.E., Caporaso G.L., Buxbaum J.D., De Cruz Silva O., Iverfeldt K., Nordstedt C., et al. Protein phosphorylation regulates relative utilization of processing pathways for Alzheimer beta/A4 amyloid precursor protein. Ann. N. Y. Acad. Sci. 1993;695:117–121. doi: 10.1111/j.1749-6632.1993.tb23038.x. [DOI] [PubMed] [Google Scholar]
- 10.Preuss U., Doring F., Illenberger S., Mandelkow E.M. Cell cycle-dependent phosphorylation and microtubule binding of tau protein stably transfected into Chinese hamster ovary cells. Mol. Biol. Cell. 1995;6(10):1397–1410. doi: 10.1091/mbc.6.10.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Musicco M., Adorni F., Di Santo S., Prinelli F., Pettenati C., Caltagirone C., et al. Inverse occurrence of cancer and Alzheimer disease: A population-based incidence study. Neurology. 2013;81(4):322–328. doi: 10.1212/WNL.0b013e31829c5ec1. [DOI] [PubMed] [Google Scholar]
- 12.Ou S.M., Lee Y.J., Hu Y.W., Liu C.J., Chen T.J., Fuh J.L., et al. Does Alzheimer’s disease protect against cancers? A nationwide population-based study. Neuroepidemiology. 2013;40(1):42–49. doi: 10.1159/000341411. [DOI] [PubMed] [Google Scholar]
- 13.Roe C.M., Behrens M.I., Xiong C., Miller J.P., Morris J.C. Alzheimer disease and cancer. Neurology. 2005;64(5):895–898. doi: 10.1212/01.WNL.0000152889.94785.51. [DOI] [PubMed] [Google Scholar]
- 14.Bialik S., Kimchi A. The death-associated protein kinases: Structure, function, and beyond. Annu. Rev. Biochem. 2006;75:189–210. doi: 10.1146/annurev.biochem.75.103004.142615. [DOI] [PubMed] [Google Scholar]
- 15.Deiss L.P., Feinstein E., Berissi H., Cohen O., Kimchi A. Identification of a novel serine/threonine kinase and a novel 15-kD protein as potential mediators of the gamma interferon-induced cell death. Genes Dev. 1995;9(1):15–30. doi: 10.1101/gad.9.1.15. [DOI] [PubMed] [Google Scholar]
- 16.Shiloh R., Bialik S., Kimchi A. The DAPK family: A structure-function analysis. Apoptosis. 2014;19(2):286–297. doi: 10.1007/s10495-013-0924-5. [DOI] [PubMed] [Google Scholar]
- 17.Levin-Salomon V., Bialik S., Kimchi A. DAP-kinase and autophagy. Apoptosis. 2014;19(2):346–356. doi: 10.1007/s10495-013-0918-3. [DOI] [PubMed] [Google Scholar]
- 18.Chen H.Y., Lee Y.R., Chen R.H. The functions and regulations of DAPK in cancer metastasis. Apoptosis. 2014;19(2):364–370. doi: 10.1007/s10495-013-0923-6. [DOI] [PubMed] [Google Scholar]
- 19.Lai M.Z., Chen R.H. Regulation of inflammation by DAPK. Apoptosis. 2014;19(2):357–363. doi: 10.1007/s10495-013-0933-4. [DOI] [PubMed] [Google Scholar]
- 20.Michie A.M., McCaig A.M., Nakagawa R., Vukovic M. Death-associated protein kinase (DAPK) and signal transduction: Regulation in cancer. FEBS J. 2010;277(1):74–80. doi: 10.1111/j.1742-4658.2009.07414.x. [DOI] [PubMed] [Google Scholar]
- 21.Kim B.M., You M-H., Chen C-H., Lee S., Hong Y., Hong Y., et al. Death-associated protein kinase 1 plays a critical role in aberrant tau protein regulation and function. Cell Death Dis. 2014;5:e1237. doi: 10.1038/cddis.2014.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim B.M., You M.H., Chen C.H., Suh J., Tanzi R.E., Lee T.H. Inhibition of death-associated protein kinase 1 attenuates the phosphorylation and amyloidogenic processing of amyloid precursor protein. Hum. Mol. Genet. 2016;25(12):2498–2513. doi: 10.1093/hmg/ddw114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y., Grupe A., Rowland C., Nowotny P., Kauwe J.S., Smemo S., et al. DAPK1 variants are associated with Alzheimer’s disease and allele-specific expression. Hum. Mol. Genet. 2006;15(17):2560–2568. doi: 10.1093/hmg/ddl178. [DOI] [PubMed] [Google Scholar]
- 24.Li H., Wetten S., Li L., St Jean P.L., Upmanyu R., Surh L., et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch. Neurol. 2008;65(1):45–53. doi: 10.1001/archneurol.2007.3. [DOI] [PubMed] [Google Scholar]
- 25.Laumet G., Chouraki V., Grenier-Boley B., Legry V., Heath S., Zelenika D., et al. Systematic analysis of candidate genes for Alzheimer’s disease in a French, genome-wide association study. J. Alzheimers Dis. 2010;20(4):1181–1188. doi: 10.3233/JAD-2010-100126. [DOI] [PubMed] [Google Scholar]
- 26.Gaj P., Paziewska A., Bik W., Dabrowska M., Baranowska-Bik A., Styczynska M., et al. Identification of a late onset Alzheimer’s disease candidate risk variant at 9q21.33 in Polish patients. J. Alzheimers Dis. 2012;32(1):157–168. doi: 10.3233/JAD-2012-120520. [DOI] [PubMed] [Google Scholar]
- 27.Kristensen L.S., Asmar F., Dimopoulos K., Nygaard M.K., Aslan D., Hansen J.W., et al. Hypermethylation of DAPK1 is an independent prognostic factor predicting survival in diffuse large B-cell lymphoma. Oncotarget. 2014;5(20):9798–9810. doi: 10.18632/oncotarget.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Behrens M.I., Lendon C., Roe C.M. A common biological mechanism in cancer and Alzheimer’s disease? Curr. Alzheimer Res. 2009;6(3):196–204. doi: 10.2174/156720509788486608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Y., Chen L., Guo L., Hupp T.R., Lin Y. Evaluating DAPK as a therapeutic target. Apoptosis. 2014;19(2):371–386. doi: 10.1007/s10495-013-0919-2. [DOI] [PubMed] [Google Scholar]
- 30.Xu L., Li B., Jia J. DAPK1: A Novel pathology and treatment target for Alzheimer’s Disease. Mol. Neurobiol. 2018;••• doi: 10.1007/s12035-018-1242-2. [DOI] [PubMed] [Google Scholar]
- 31.Singh P., Ravanan P., Talwar P. Death associated protein kinase 1 (DAPK1): A Regulator of apoptosis and autophagy. Front. Mol. Neurosci. 2016;9:46–52. doi: 10.3389/fnmol.2016.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tereshko V., Teplova M., Brunzelle J., Watterson D.M., Egli M. Crystal structures of the catalytic domain of human protein kinase associated with apoptosis and tumor suppression. Nat. Struct. Biol. 2001;8(10):899–907. doi: 10.1038/nsb1001-899. [DOI] [PubMed] [Google Scholar]
- 33.Zimmermann M., Atmanene C., Xu Q., Fouillen L., Van Dorsselaer A., Bonnet D., et al. Homodimerization of the death-associated protein kinase catalytic domain: Development of a new small molecule fluorescent reporter. PLoS One. 2010;5(11):e14120. doi: 10.1371/journal.pone.0014120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Velentza A.V., Schumacher A.M., Weiss C., Egli M., Watterson D.M. A protein kinase associated with apoptosis and tumor suppression: Structure, activity, and discovery of peptide substrates. J. Biol. Chem. 2001;276(42):38956–38965. doi: 10.1074/jbc.M104273200. [DOI] [PubMed] [Google Scholar]
- 35.Cohen O., Feinstein E., Kimchi A. DAP-kinase is a Ca2+/calmodulin-dependent, cytoskeletal-associated protein kinase, with cell death-inducing functions that depend on its catalytic activity. EMBO J. 1997;16(5):998–1008. doi: 10.1093/emboj/16.5.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shohat G., Spivak-Kroizman T., Cohen O., Bialik S., Shani G., Berrisi H., et al. The pro-apoptotic function of death-associated protein kinase is controlled by a unique inhibitory autophosphorylation-based mechanism. J. Biol. Chem. 2001;276(50):47460–47467. doi: 10.1074/jbc.M105133200. [DOI] [PubMed] [Google Scholar]
- 37.Shani G., Henis-Korenblit S., Jona G., Gileadi O., Eisenstein M., Ziv T., et al. Autophosphorylation restrains the apoptotic activity of DRP-1 kinase by controlling dimerization and calmodulin binding. EMBO J. 2001;20(5):1099–1113. doi: 10.1093/emboj/20.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carlessi R., Levin-Salomon V., Ciprut S., Bialik S., Berissi H., Albeck S., et al. GTP binding to the ROC domain of DAP-kinase regulates its function through intramolecular signalling. EMBO Rep. 2011;12(9):917–923. doi: 10.1038/embor.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gozuacik D., Bialik S., Raveh T., Mitou G., Shohat G., Sabanay H., et al. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008;15(12):1875–1886. doi: 10.1038/cdd.2008.121. [DOI] [PubMed] [Google Scholar]
- 40.Guenebeaud C., Goldschneider D., Castets M., Guix C., Chazot G., Delloye-Bourgeois C., et al. The dependence receptor UNC5H2/B triggers apoptosis via PP2A-mediated dephosphorylation of DAP kinase. Mol. Cell. 2010;40(6):863–876. doi: 10.1016/j.molcel.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 41.Tu W., Xu X., Peng L., Zhong X., Zhang W., Soundarapandian M.M., et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. 2010;140(2):222–234. doi: 10.1016/j.cell.2009.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bialik S., Bresnick A.R., Kimchi A. DAP-kinase-mediated morphological changes are localization dependent and involve myosin-II phosphorylation. Cell Death Differ. 2004;11(6):631–644. doi: 10.1038/sj.cdd.4401386. [DOI] [PubMed] [Google Scholar]
- 43.Raveh T., Berissi H., Eisenstein M., Spivak T., Kimchi A. A functional genetic screen identifies regions at the C-terminal tail and death-domain of death-associated protein kinase that are critical for its proapoptotic activity. Proc. Natl. Acad. Sci. USA. 2000;97(4):1572–1577. doi: 10.1073/pnas.020519497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin Y., Blue E.K., Dixon S., Shao Z., Gallagher P.J. A death-associated protein kinase (DAPK)-interacting protein, DIP-1, is an E3 ubiquitin ligase that promotes tumor necrosis factor-induced apoptosis and regulates the cellular levels of DAPK. J. Biol. Chem. 2002;277(49):46980–46986. doi: 10.1074/jbc.M208585200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee T.H., Chen C-H., Suizu F., Huang P., Schiene-Fischer C., Daum S., et al. Death associated protein kinase phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function. Mol. Cell. doi: 10.1016/j.molcel.2011.03.005. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bialik S., Kimchi A. Pin-pointing a new DAP kinase function: The peptidyl-proly isomerase Pin1 is negatively regulated by DAP kinase-mediated phosphorylation. Mol. Cell. 2011;42(2):139–141. doi: 10.1016/j.molcel.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 47.You M.H., Kim B.M., Chen C.H., Begley M.J., Cantley L.C., Lee T.H. Death-associated protein kinase 1 phosphorylates NDRG2 and induces neuronal cell death. Cell Death Differ. 2017;24(2):238–250. doi: 10.1038/cdd.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bialik S., Kimchi A. The DAP-kinase interactome. Apoptosis. 2014;19(2):316–328. doi: 10.1007/s10495-013-0926-3. [DOI] [PubMed] [Google Scholar]
- 49.Chen C.H., Wang W.J., Kuo J.C., Tsai H.C., Lin J.R., Chang Z.F., et al. Bidirectional signals transduced by DAPK-ERK interaction promote the apoptotic effect of DAPK. EMBO J. 2005;24(2):294–304. doi: 10.1038/sj.emboj.7600510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Llambi F., Lourenco F.C., Gozuacik D., Guix C., Pays L., Del Rio G., et al. The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. EMBO J. 2005;24(6):1192–1201. doi: 10.1038/sj.emboj.7600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee Y.R., Yuan W.C., Ho H.C., Chen C.H., Shih H.M., Chen R.H. The Cullin 3 substrate adaptor KLHL20 mediates DAPK ubiquitination to control interferon responses. EMBO J. 2010;29(10):1748–1761. doi: 10.1038/emboj.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu P.R., Tsai P.I., Chen G.C., Chou H.J., Huang Y.P., Chen Y.H., et al. DAPK activates MARK1/2 to regulate microtubule assembly, neuronal differentiation, and tau toxicity. Cell Death Differ. 2011;18(9):1507–1520. doi: 10.1038/cdd.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mor I., Carlessi R., Ast T., Feinstein E., Kimchi A. Death-associated protein kinase increases glycolytic rate through binding and activation of pyruvate kinase. Oncogene. 2012;31(6):683–693. doi: 10.1038/onc.2011.264. [DOI] [PubMed] [Google Scholar]
- 54.Stevens C., Lin Y., Harrison B., Burch L., Ridgway R.A., Sansom O., et al. Peptide combinatorial libraries identify TSC2 as a death-associated protein kinase (DAPK) death domain-binding protein and reveal a stimulatory role for DAPK in mTORC1 signaling. J. Biol. Chem. 2009;284(1):334–344. doi: 10.1074/jbc.M805165200. [DOI] [PubMed] [Google Scholar]
- 55.Fodale V., Pierobon M., Liotta L., Petricoin E. Mechanism of cell adaptation: When and how do cancer cells develop chemoresistance? Cancer J. 2011;17(2):89–95. doi: 10.1097/PPO.0b013e318212dd3d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morris L.G., Chan T.A. Therapeutic targeting of tumor suppressor genes. Cancer. 2015;121(9):1357–1368. doi: 10.1002/cncr.29140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bialik S., Kimchi A. DAP-kinase as a target for drug design in cancer and diseases associated with accelerated cell death. Semin. Cancer Biol. 2004;14(4):283–294. doi: 10.1016/j.semcancer.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 58.Gozuacik D., Kimchi A. DAPk protein family and cancer. Autophagy. 2006;2(2):74–79. doi: 10.4161/auto.2.2.2459. [DOI] [PubMed] [Google Scholar]
- 59.Benderska N., Schneider-Stock R. Transcription control of DAPK. Apoptosis. 2014;19(2):298–305. doi: 10.1007/s10495-013-0931-6. [DOI] [PubMed] [Google Scholar]
- 60.Tang X., Khuri F.R., Lee J.J., Kemp B.L., Liu D., Hong W.K., et al. Hypermethylation of the death-associated protein (DAP) kinase promoter and aggressiveness in stage I non-small-cell lung cancer. J. Natl. Cancer Inst. 2000;92(18):1511–1516. doi: 10.1093/jnci/92.18.1511. [DOI] [PubMed] [Google Scholar]
- 61.Kim D.H., Nelson H.H., Wiencke J.K., Christiani D.C., Wain J.C., Mark E.J., et al. Promoter methylation of DAP-kinase: Association with advanced stage in non-small cell lung cancer. Oncogene. 2001;20(14):1765–1770. doi: 10.1038/sj.onc.1204302. [DOI] [PubMed] [Google Scholar]
- 62.Harden S.V., Tokumaru Y., Westra W.H., Goodman S., Ahrendt S.A., Yang S.C., et al. Gene promoter hypermethylation in tumors and lymph nodes of stage I lung cancer patients. Clin. Cancer Res. 2003;9(4):1370–1375. [PubMed] [Google Scholar]
- 63.Hu S.L., Kong X.Y., Cheng Z.D., Sun Y.B., Shen G., Xu W.P., et al. Promoter methylation of p16, Runx3, DAPK and CHFR genes is frequent in gastric carcinoma. Tumori. 2010;96(5):726–733. doi: 10.1177/030089161009600515. [DOI] [PubMed] [Google Scholar]
- 64.Chan A.W., Chan M.W., Lee T.L., Ng E.K., Leung W.K., Lau J.Y., et al. Promoter hypermethylation of death-associated protein-kinase gene associated with advance stage gastric cancer. Oncol. Rep. 2005;13(5):937–941. [PubMed] [Google Scholar]
- 65.Sanchez-Cespedes M., Esteller M., Wu L., Nawroz-Danish H., Yoo G.H., Koch W.M., et al. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000;60(4):892–895. [PubMed] [Google Scholar]
- 66.Levy D, Plu-Bureau G, Decroix Y, Hugol D, Rostene W, Kimchi A, et al. Death-associated protein kinase loss of expression is a new marker for breast cancer prognosis. Clin Cancer Res 004. 10(9):3124–30. doi: 10.1158/1078-0432.ccr-03-0213. [DOI] [PubMed] [Google Scholar]
- 67.Brabender J., Arbab D., Huan X., Vallbohmer D., Grimminger P., Ling F., et al. Death-associated protein kinase (DAPK) promoter methylation and response to neoadjuvant radiochemotherapy in esophageal cancer. Ann. Surg. Oncol. 2009;16(5):1378–1383. doi: 10.1245/s10434-009-0356-1. [DOI] [PubMed] [Google Scholar]
- 68.Inbal B., Cohen O., Polak-Charcon S., Kopolovic J., Vadai E., Eisenbach L., et al. DAP kinase links the control of apoptosis to metastasis. Nature. 1997;390(6656):180–184. doi: 10.1038/36599. [DOI] [PubMed] [Google Scholar]
- 69.Raval A., Tanner S.M., Byrd J.C., Angerman E.B., Perko J.D., Chen S.S., et al. Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell. 2007;129(5):879–890. doi: 10.1016/j.cell.2007.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen H.Y., Lin Y.M., Chung H.C., Lang Y.D., Lin C.J., Huang J., et al. miR-103/107 promotes metastasis of colorectal cancer by targeting the metastasis suppressors DAPK and KLF4. Cancer Res. 2012;72(14):3631–3641. doi: 10.1158/0008-5472.CAN-12-0667. [DOI] [PubMed] [Google Scholar]
- 71.Wang W.J., Kuo J.C., Ku W., Lee Y.R., Lin F.C., Chang Y.L., et al. The tumor suppressor DAPK is reciprocally regulated by tyrosine kinase Src and phosphatase LAR. Mol. Cell. 2007;27(5):701–716. doi: 10.1016/j.molcel.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 72.Anjum R., Roux P.P., Ballif B.A., Gygi S.P., Blenis J. The tumor suppressor DAP kinase is a target of RSK-mediated survival signaling. Curr. Biol. 2005;15(19):1762–1767. doi: 10.1016/j.cub.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 73.Citri A., Harari D., Shohat G., Ramakrishnan P., Gan J., Lavi S., et al. Hsp90 recognizes a common surface on client kinases. J. Biol. Chem. 2006;281(20):14361–14369. doi: 10.1074/jbc.M512613200. [DOI] [PubMed] [Google Scholar]
- 74.Zhang L., Nephew K.P., Gallagher P.J. Regulation of death-associated protein kinase. Stabilization by HSP90 heterocomplexes. J. Biol. Chem. 2007;282(16):11795–11804. doi: 10.1074/jbc.M610430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Raveh T., Droguett G., Horwitz M.S., DePinho R.A., Kimchi A. DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat. Cell Biol. 2001;3(1):1–7. doi: 10.1038/35050500. [DOI] [PubMed] [Google Scholar]
- 76.Wang W.J., Kuo J.C., Yao C.C., Chen R.H. DAP-kinase induces apoptosis by suppressing integrin activity and disrupting matrix survival signals. J. Cell Biol. 2002;159(1):169–179. doi: 10.1083/jcb.200204050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martoriati A., Doumont G., Alcalay M., Bellefroid E., Pelicci P.G., Marine J.C. DAPK1, encoding an activator of a p19ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene. 2005;24(8):1461–1466. doi: 10.1038/sj.onc.1208256. [DOI] [PubMed] [Google Scholar]
- 78.Zhao J., Zhao D., Poage G.M., Mazumdar A., Zhang Y., Hill J.L., et al. Death-associated protein kinase 1 promotes growth of p53-mutant cancers. J. Clin. Invest. 2015;125(7):2707–2720. doi: 10.1172/JCI70805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu K.P., Zhou X.Z. The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 80.Lu K.P. Pinning down cell signaling, cancer and Alzheimer’s disease. Trends Biochem. Sci. 2004;29:200–209. doi: 10.1016/j.tibs.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 81.Lu K.P., Finn G., Lee T.H., Nicholson L.K. Prolyl cis-trans isomerization as a molecular timer. Nat. Chem. Biol. 2007;3(10):619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 82.Zhou X.Z., Lu K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer. 2016;16(7):463–478. doi: 10.1038/nrc.2016.49. [DOI] [PubMed] [Google Scholar]
- 83.Min S.H., Zhou X.Z., Lu K.P. The role of Pin1 in the development and treatment of cancer. Arch. Pharm. Res. 2016;39(12):1609–1620. doi: 10.1007/s12272-016-0821-x. [DOI] [PubMed] [Google Scholar]
- 84.Ryo A., Nakamura N., Wulf G., Liou Y.C., Lu K.P. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 2001;3:793–801. doi: 10.1038/ncb0901-793. [DOI] [PubMed] [Google Scholar]
- 85.Ryo A., Suizu F., Yoshida Y., Perrem K., Liou Y.C., Wulf G., et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell. 2003;12:1413–1426. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 86.Liou Y.C., Ryo R., Huang H.K., Lu P.J., Bronson R., Fujimori F., et al. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc. Natl. Acad. Sci. USA. 2002;99:1335–1340. doi: 10.1073/pnas.032404099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suizu F., Ryo A., Wulf G., Lim J., Lu K.P. Pin1 regulates centrosome duplication, and its overexpression induces centrosome amplification, chromosome instability, and oncogenesis. Mol. Cell. Biol. 2006;26:1463–1479. doi: 10.1128/MCB.26.4.1463-1479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Min S.H., Lau A.W., Lee T.H., Inuzuka H., Wei S., Huang P., et al. Negative regulation of the stability and tumor suppressor function of Fbw7 by the Pin1 prolyl isomerase. Mol. Cell. 2012;46(6):771–783. doi: 10.1016/j.molcel.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee T.H., Tun-Kyi A., Shi R., Lim J., Soohoo C., Finn G., et al. Essential role of Pin1 in the regulation of TRF1 stability and telomere maintenance. Nat. Cell Biol. 2009;11(1):97–105. doi: 10.1038/ncb1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wulf G., Garg P., Liou Y.C., Iglehart D., Lu K.P. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004;23:3397–3407. doi: 10.1038/sj.emboj.7600323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wulf G.M., Liou Y.C., Ryo A., Lee S.W., Lu K.P. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J. Biol. Chem. 2002;277:47976–47979. doi: 10.1074/jbc.C200538200. [DOI] [PubMed] [Google Scholar]
- 92.Kozono S., Lin Y.M., Seo H.S., Pinch B., Lian X., Qiu C., et al. Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat. Commun. 2018;9(1):3069. doi: 10.1038/s41467-018-05402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang D., Luo W., Wang J., Zheng M., Liao X.H., Zhang N., et al. A novel controlled release formulation of the Pin1 inhibitor ATRA to improve liver cancer therapy by simultaneously blocking multiple cancer pathways. J. Control. Release. 2018;269:405–422. doi: 10.1016/j.jconrel.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zheng M., Xu H., Liao X.H., Chen C.P., Zhang A.L., Lu W., et al. Inhibition of the prolyl isomerase Pin1 enhances the ability of sorafenib to induce cell death and inhibit tumor growth in hepatocellular carcinoma. Oncotarget. 2017;8(18):29771–29784. doi: 10.18632/oncotarget.15967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liao X.H., Zhang A.L., Zheng M., Li M.Q., Chen C.P., Xu H., et al. Chemical or genetic Pin1 inhibition exerts potent anticancer activity against hepatocellular carcinoma by blocking multiple cancer-driving pathways. Sci. Rep. 2017;7:43639. doi: 10.1038/srep43639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wei S., Kozono S., Kats L., Nechama M., Li W., Guarnerio J., et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015;21(5):457–466. doi: 10.1038/nm.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Luo M.L., Gong C., Chen C.H., Lee D.Y., Hu H., Huang P., et al. Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer. Cancer Res. 2014;74(13):3603–3616. doi: 10.1158/0008-5472.CAN-13-2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen C.H., Chang C.C., Lee T.H., Luo M., Huang P., Liao P.H., et al. SENP1 deSUMOylates and regulates Pin1 protein activity and cellular function. Cancer Res. 2013;73(13):3951–3962. doi: 10.1158/0008-5472.CAN-12-4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu K.P., Hanes S.D., Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380(6574):544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 100.Ryo A., Liou Y.C., Wulf G., Nakamura N., Lee S.W., Lu K.P. Pin1 is an E2F target gene essential for the Neu/Ras-induced transformation of mammary epithelial cells. Mol. Cell. Biol. 2002;22:5281–5295. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rippmann J.F., Hobbie S., Daiber C., Guilliard B., Bauer M., Birk J., et al. Phosphorylation-dependent proline isomerization catalyzed by Pin1 is essential for tumor cell survival and entry into mitosis. Cell Growth Differ. 2000;11(7):409–416. [PubMed] [Google Scholar]
- 102.Takahashi K., Akiyama H., Shimazaki K., Uchida C., Akiyama-Okunuki H., Tomita M., et al. Ablation of a peptidyl prolyl isomerase Pin1 from p53-null mice accelerated thymic hyperplasia by increasing the level of the intracellular form of Notch1. Oncogene. 2007;26:3835–3845. doi: 10.1038/sj.onc.1210153. [DOI] [PubMed] [Google Scholar]
- 103.Kuo J.C., Lin J.R., Staddon J.M., Hosoya H., Chen R.H. Uncoordinated regulation of stress fibers and focal adhesions by DAP kinase. J. Cell Sci. 2003;116(Pt 23):4777–4790. doi: 10.1242/jcs.00794. [DOI] [PubMed] [Google Scholar]
- 104.Ittner L.M., Gotz J. Amyloid-beta and tau: A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011;12(2):65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 105.Jack C.R., Jr, Holtzman D.M. Biomarker modeling of Alzheimer’s disease. Neuron. 2013;80(6):1347–1358. doi: 10.1016/j.neuron.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ballatore C., Lee V.M., Trojanowski J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007;8(9):663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 107.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 108.Lee G., Cowan N., Kirschner M. The primary structure and heterogeneity of tau protein from mouse brain. Science. 1988;239(4837):285–288. doi: 10.1126/science.3122323. [DOI] [PubMed] [Google Scholar]
- 109.Lee G., Neve R.L., Kosik K.S. The microtubule binding domain of tau protein. Neuron. 1989;2(6):1615–1624. doi: 10.1016/0896-6273(89)90050-0. [DOI] [PubMed] [Google Scholar]
- 110.Esmaeli-Azad B., McCarty J.H., Feinstein S.C. Sense and antisense transfection analysis of tau function: tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J. Cell Sci. 1994;107(Pt 4):869–879. doi: 10.1242/jcs.107.4.869. [DOI] [PubMed] [Google Scholar]
- 111.Stoothoff W.H., Johnson G.V. Tau phosphorylation: physiological and pathological consequences. Biochim. Biophys. Acta. 2005;1739(2-3):280–297. doi: 10.1016/j.bbadis.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 112.Goedert M., Spillantini M.G., Cairns N.J., Crowther R.A. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8(1):159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 113.Matsuo E.S., Shin R.W., Billingsley M.L., Van deVoorde A., O’Connor M., Trojanowski J.Q., et al. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron. 1994;13(4):989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- 114.Lee V.M., Balin B.J., Otvos L., Jr, Trojanowski J.Q. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251(4994):675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 115.Selkoe D.J., Yamazaki T., Citron M., Podlisny M.B., Koo E.H., Teplow D.B., et al. The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann. N. Y. Acad. Sci. 1996;777:57–64. doi: 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- 116.Giasson B.I., Lee V.M., Trojanowski J.Q. Interactions of amyloidogenic proteins. Neuromolecular Med. 2003;4(1-2):49–58. doi: 10.1385/NMM:4:1-2:49. [DOI] [PubMed] [Google Scholar]
- 117.Lewis J., Dickson D.W., Lin W.L., Chisholm L., Corral A., Jones G., et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 118.Gotz J., Chen F., van Dorpe J., Nitsch R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 119.Roberson E.D., Scearce-Levie K., Palop J.J., Yan F., Cheng I.H., Wu T., et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 120.Bolmont T., Clavaguera F., Meyer-Luehmann M., Herzig M.C., Radde R., Staufenbiel M., et al. Induction of tau pathology by intracerebral infusion of amyloid-beta-containing brain extract and by amyloid-beta deposition in APPx Tau transgenic mice. Am. J. Pathol. 2007;171(6):2012–2020. doi: 10.2353/ajpath.2007.070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yukawa K., Tanaka T., Bai T., Li L., Tsubota Y., Owada-Makabe K., et al. Deletion of the kinase domain from death-associated protein kinase enhances spatial memory in mice. Int. J. Mol. Med. 2006;17(5):869–873. [PubMed] [Google Scholar]
- 122.Shu S., Zhu H., Tang N., Chen W., Li X., Li H., et al. Selective degeneration of entorhinal-CA1 Synapses in Alzheimer’s disease via activation of DAPK1. J. Neurosci. 2016;36(42):10843–10852. doi: 10.1523/JNEUROSCI.2258-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Luna-Munoz J., Chavez-Macias L., Garcia-Sierra F., Mena R. Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer’s disease. J. Alzheimers Dis. 2007;12(4):365–375. doi: 10.3233/jad-2007-12410. [DOI] [PubMed] [Google Scholar]
- 124.Lu P.J., Wulf G., Zhou X.Z., Davies P., Lu K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399(6738):784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 125.Liou Y.C., Sun A., Ryo A., Zhou X.Z., Yu Z.X., Huang H.K., et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424(6948):556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 126.Lim J., Balastik M., Lee T.H., Nakamura K., Liou Y.C., Sun A., et al. Pin1 has opposite effects on wild-type and P301L tau stability and tauopathy. J. Clin. Invest. 2008;118(5):1877–1889. doi: 10.1172/JCI34308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lu P.J., Zhou X.Z., Shen M., Lu K.P. A function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]
- 128.Pastorino L., Sun A., Lu P.J., Zhou X.Z., Balastik M., Finn G., et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 129.Kondo A., Albayram O., Zhou X.Z., Lu K.P. Pin1 knockout mice: A model for the study of tau pathology in Alzheimer’s disease. Methods Mol. Biol. 2017;1523:415–425. doi: 10.1007/978-1-4939-6598-4_28. [DOI] [PubMed] [Google Scholar]
- 130.Chen C.H., Li W., Sultana R., You M.H., Kondo A., Shahpasand K., et al. Pin1 cysteine-113 oxidation inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol. Dis. 2015;76:13–23. doi: 10.1016/j.nbd.2014.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kondo A., Shahpasand K., Mannix R., Qiu J., Moncaster J., Chen C.H., et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523(7561):431–436. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nakamura K., Greenwood A., Binder L., Bigio E.H., Denial S., Nicholson L., et al. Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer’s disease. Cell. 2012;149(1):232–244. doi: 10.1016/j.cell.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pei L., Wang S., Jin H., Bi L., Wei N., Yan H., et al. A Novel mechanism of spine damages in stroke via DAPK1 and tau. Cereb. Cortex. 2015;25(11):4559–4571. doi: 10.1093/cercor/bhv096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Duan D.X., Chai G.S., Ni Z.F., Hu Y., Luo Y., Cheng X.S., et al. Phosphorylation of tau by death-associated protein kinase 1 antagonizes the kinase-induced cell apoptosis. J. Alzheimers Dis. 2013;37(4):795–808. doi: 10.3233/JAD-130377. [DOI] [PubMed] [Google Scholar]
- 135.Pastorino L., Lu K.P. Phosphorylation of the amyloid precursor protein (APP): Is this a mechanism in favor or against Alzheimer’s disease? Neurosci. Res. Commun. 2005;35:213–231. [Google Scholar]
- 136.Suzuki T., Nakaya T. Regulation of amyloid beta-protein precursor by phosphorylation and protein interactions. J. Biol. Chem. 2008;283(44):29633–29637. doi: 10.1074/jbc.R800003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lee M.S., Kao S.C., Lemere C.A., Xia W., Tseng H.C., Zhou Y., et al. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003;163(1):83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ma S.L., Pastorino L., Zhou X.Z., Lu K.P. Prolyl isomerase Pin1 promotes amyloid precursor protein (APP) turnover by inhibiting glycogen synthase kinase-3beta (GSK3beta) activity: novel mechanism for Pin1 to protect against Alzheimer disease. J. Biol. Chem. 2012;287(10):6969–6973. doi: 10.1074/jbc.C111.298596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Craft J.M., Watterson D.M., Frautschy S.A., Van Eldik L.J. Aminopyridazines inhibit beta-amyloid-induced glial activation and neuronal damage in vivo. Neurobiol. Aging. 2004;25(10):1283–1292. doi: 10.1016/j.neurobiolaging.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 140.Farag A.K., Roh E.J. Death-associated protein kinase (DAPK) family modulators: Current and future therapeutic outcomes. Med. Res. Rev. 2018;4:120–129. doi: 10.1002/med.21518. [DOI] [PubMed] [Google Scholar]
- 141.Gandesiri M., Chakilam S., Ivanovska J., Benderska N., Ocker M., Di Fazio P., et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis. 2012;17(12):1300–1315. doi: 10.1007/s10495-012-0757-7. [DOI] [PubMed] [Google Scholar]
- 142.Zhang H., Chen G.G., Zhang Z., Chun S., Leung B.C., Lai P.B. Induction of autophagy in hepatocellular carcinoma cells by SB203580 requires activation of AMPK and DAPK but not p38 MAPK. Apoptosis. 2012;17(4):325–334. doi: 10.1007/s10495-011-0685-y. [DOI] [PubMed] [Google Scholar]
- 143.Wu J., Hu C.P., Gu Q.H., Li Y.P., Song M. Trichostatin A sensitizes cisplatin-resistant A549 cells to apoptosis by up-regulating death-associated protein kinase. Acta Pharmacol. Sin. 2010;31(1):93–101. doi: 10.1038/aps.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Luo X.J., Li L.L., Deng Q.P., Yu X.F., Yang L.F., Luo F.J., et al. Grifolin, a potent antitumour natural product upregulates death-associated protein kinase 1 DAPK1 via p53 in nasopharyngeal carcinoma cells. Eur. J. Cancer. 2011;47(2):316–325. doi: 10.1016/j.ejca.2010.09.021. [DOI] [PubMed] [Google Scholar]
- 145.Wu B., Yao H., Wang S., Xu R. DAPK1 modulates a curcumin-induced G2/M arrest and apoptosis by regulating STAT3, NF-kappaB, and caspase-3 activation. Biochem. Biophys. Res. Commun. 2013;434(1):75–80. doi: 10.1016/j.bbrc.2013.03.063. [DOI] [PubMed] [Google Scholar]
- 146.Puto L.A., Reed J.C. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 2008;22(8):998–1010. doi: 10.1101/gad.1632208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pulling L.C., Grimes M.J., Damiani L.A., Juri D.E., Do K., Tellez C.S., et al. Dual promoter regulation of death-associated protein kinase gene leads to differentially silenced transcripts by methylation in cancer. Carcinogenesis. 2009;30(12):2023–2030. doi: 10.1093/carcin/bgp276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Satoh A., Toyota M., Itoh F., Kikuchi T., Obata T., Sasaki Y., et al. DNA methylation and histone deacetylation associated with silencing DAP kinase gene expression in colorectal and gastric cancers. Br. J. Cancer. 2002;86(11):1817–1823. doi: 10.1038/sj.bjc.6600319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Toyooka S., Toyooka K.O., Miyajima K., Reddy J.L., Toyota M., Sathyanarayana U.G., et al. Epigenetic down-regulation of death-associated protein kinase in lung cancers. Clin. Cancer Res. 2003;9(8):3034–3041. [PubMed] [Google Scholar]
- 150.Chico L.K., Van Eldik L.J., Watterson D.M. Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discov. 2009;8(11):892–909. doi: 10.1038/nrd2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Velentza A.V., Wainwright M.S., Zasadzki M., Mirzoeva S., Schumacher A.M., Haiech J., et al. An aminopyridazine-based inhibitor of a pro-apoptotic protein kinase attenuates hypoxia-ischemia induced acute brain injury. Bioorg. Med. Chem. Lett. 2003;13(20):3465–3470. doi: 10.1016/s0960-894x(03)00733-9. [DOI] [PubMed] [Google Scholar]
- 152.Mirzoeva S., Sawkar A., Zasadzki M., Guo L., Velentza A.V., Dunlap V., et al. Discovery of a 3-amino-6-phenyl-pyridazine derivative as a new synthetic antineuroinflammatory compound. J. Med. Chem. 2002;45(3):563–566. doi: 10.1021/jm015573g. [DOI] [PubMed] [Google Scholar]
- 153.Okamoto M., Takayama K., Shimizu T., Ishida K., Takahashi O., Furuya T. Identification of death-associated protein kinases inhibitors using structure-based virtual screening. J. Med. Chem. 2009;52(22):7323–7327. doi: 10.1021/jm901191q. [DOI] [PubMed] [Google Scholar]
- 154.Okamoto M., Takayama K., Shimizu T., Muroya A., Furuya T. Structure-activity relationship of novel DAPK inhibitors identified by structure-based virtual screening. Bioorg. Med. Chem. 2010;18(7):2728–2734. doi: 10.1016/j.bmc.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 155.Fan X., Jin W.Y., Lu J., Wang J., Wang Y.T. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat. Neurosci. 2014;17(3):471–480. doi: 10.1038/nn.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shamloo M., Soriano L., Wieloch T., Nikolich K., Urfer R., Oksenberg D. Death-associated protein kinase is activated by dephosphorylation in response to cerebral ischemia. J. Biol. Chem. 2005;280(51):42290–42299. doi: 10.1074/jbc.M505804200. [DOI] [PubMed] [Google Scholar]
- 157.Carlson D.A., Franke A.S., Weitzel D.H., Speer B.L., Hughes P.F., Hagerty L., et al. Fluorescence linked enzyme chemoproteomic strategy for discovery of a potent and selective DAPK1 and ZIPK inhibitor. ACS Chem. Biol. 2013;8(12):2715–2723. doi: 10.1021/cb400407c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yokoyama T., Kosaka Y., Mizuguchi M. Structural insight into the interactions between death-associated protein kinase 1 and natural flavonoids. J. Med. Chem. 2015;58(18):7400–7408. doi: 10.1021/acs.jmedchem.5b00893. [DOI] [PubMed] [Google Scholar]
- 159.Albayram O., Kondo A., Mannix R., Smith C., Tsai C.Y., Li C., et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun. 2017;8(1):1000. doi: 10.1038/s41467-017-01068-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lu K.P., Kondo A., Albayram O., Herbert M.K., Liu H., Zhou X.Z. Potential of the antibody against cis-phosphorylated tau in the early diagnosis, treatment, and prevention of Alzheimer disease and brain injury. JAMA Neurol. 2016;73(11):1356–1362. doi: 10.1001/jamaneurol.2016.2027. [DOI] [PubMed] [Google Scholar]
- 161.Nakamura K., Zhen Zhou X., Ping Lu K. Cis phosphorylated tau as the earliest detectable pathogenic conformation in Alzheimer disease, offering novel diagnostic and therapeutic strategies. Prion. 2013;7(2):117–120. doi: 10.4161/pri.22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nakamura K., Zhou X.Z., Lu K.P. Distinct functions of cis and trans phosphorylated tau in Alzheimer’s disease and their therapeutic implications. Curr. Mol. Med. 2013;13(7):1098–1109. doi: 10.2174/1566524011313070001. [DOI] [PubMed] [Google Scholar]
- 163.Albayram O., Herbert M.K., Kondo A., Tsai C.Y., Baxley S., Lian X., et al. Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci. 2016;6:59. doi: 10.1186/s13578-016-0124-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Albayram O., Angeli P., Bernstein E., Baxley S., Gao Z., Lu K.P., et al. Targeting prion-like cis phosphorylated tau pathology in neurodegenerative diseases. J. Alzheimers Dis. Parkinsonism. 2018;8(3):25–32. doi: 10.4172/2161-0460.1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Califano J. Detection of head and neck cancer using hypermethylated gene detection. US20140323321. 2014 [Google Scholar]
- 166.Mansour H., Incitti R., Bajic V. Methylation biomarkers for ovarian cancer. US10041124. 2018 [Google Scholar]
- 167.Brock M.V., Baylin S.B., Heman J.G. DNA methylation markers and methods of use. US20150031022. 2015 [Google Scholar]
- 168.Corvalan A. Non-invasive method for the early detection of stomach cancer. US20130095477. 2013 [Google Scholar]
- 169.Cha T., Chang S., Sun G., Lin C., Tsai Y. Pharmaceutical compositions and methods for treating cancer and biomarkers for drug screening. US9782393. 2017 [Google Scholar]
- 170.Schneider-Stock R., Neufert C. Death-associated protein kinases, inhibitors and activators thereof for use in pharmaceutical compositions and in predictive medicine. EP2767273. 2014 [Google Scholar]
- 171.Jin Y., Blue E.K., Dixon S., Hou L., Wysolmerski R.B., Gallagher P.J. Identification of a new form of death-associated protein kinase that promotes cell survival. J. Biol. Chem. 2001;276:39667–39678. doi: 10.1074/jbc.M101886200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Jin Y., Blue E.K., Gallagher P.J. Control of death-associated protein kinase (DAPK) activity by phosphorylation and proteasomal degradation. J. Biol. Chem. 2006;281:39033–39040. doi: 10.1074/jbc.M605097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kimchi A., Carlessi R. Compositions and methods for treating cancer and neurodegenerative diseases. US9149523. 2015 [Google Scholar]
- 174.Watterson M., Van Eldik L., Haiech J., Hibert M., Bourguignon J.J., Velentza A., Hu W., Zasadzki M. Pyridazine compounds, compositions and methods. US9527819. 2016 [Google Scholar]
- 175.Hertz N.T., Shokat K.M., Devita R. Compositions and methods for treating neurodegenerative diseases and cardiomyopathy. US20180072731. 2018 [Google Scholar]
- 176.Shantha T.R. Alzheimer's disease treatment with multiple therapeutic agents delivered to the olfactory region through a special delivery catheter and iontophoresis. US20150080785. 2015 [Google Scholar]
- 177.Panitch A., Seal B., Ward B. Kinase inhibitors and uses thereof. US20140342993. 2014 [Google Scholar]
- 178.Yang J., Hsu K., Sung T., Lin S., Wang Y., Hsu K., Lin H., Liu W. Selective inhibitors for protein kinases and pharmaceutical composition and use thereof. US20160096848. 2016 [Google Scholar]
- 179.Wang Y.T., Lan S.X., Jin W.J. Peptide directed protein knockdown. US20150266935. 2015 [Google Scholar]
- 180.Bradner J., Ishoey M., Buckley D., Paulk J., Cohen M.A., Zeid R. Targeted protein degradation using a mutant E3 ubiquitin ligase. US20180327419. 2018 [Google Scholar]
- 181.Lin Y., Henderson P., Pettersson S., Satsangi J., Hupp T., Stevens C. Tuberous sclerosis-2 (TSC2) regulates the stability of death-associated protein kinase-1 (DAPK) through a lysosome-dependent degradation pathway. FEBS J. 2011;278(2):354–370. doi: 10.1111/j.1742-4658.2010.07959.x. [DOI] [PubMed] [Google Scholar]
- 182.Pardridge W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx. 2005;2(1):3–14. doi: 10.1602/neurorx.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Park M.H., Hyun H., Ashitate Y., Wada H., Park G., Lee J.H., et al. Prototype nerve-specific near-infrared fluorophores. Theranostics. 2014;4:823–833. doi: 10.7150/thno.8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.