

Abstract
Quantitative bottom-up shotgun lipidomics relies on molecular species-specific “signature” fragments consistently detectable in tandem mass spectra of analytes and standards. Molecular species of glycerophospholipids are typically quantified using carboxylate fragments of their fatty acid moieties produced by higher-energy collisional dissociation of their molecular anions. However, employing standards whose fatty acids moieties are similar, yet not identical, to the target lipids could severely compromise their quantification. We developed a generic and portable fragmentation model implemented in the open-source LipidXte software that harmonizes the abundances of carboxylate anion fragments originating from fatty acid moieties having different sn-1/2 positions at the glycerol backbone, length of the hydrocarbon chain, and number and location of double bonds. The postacquisition adjustment enables unbiased absolute (molar) quantification of glycerophospholipid species independent of instrument settings, collision energy, and employed internal standards.
The concept of omics quantification differs from a classical paradigm of small molecules analysis that relies upon authentic internal standards whose chemical structure is identical to target analytes (reviewed in refs 1 and 2). In a single analysis shotgun lipidomics (reviewed in refs (3−6)) typically detects several hundred species from more than 20 major lipid classes, whose molar abundance can differ by more than 1000-fold.7−9 Individual lipids are quantified using a few (or sometimes just one) internal standard(s) per each class by presuming the same molar response for standards and analytes. Because of the direct infusion of total lipid extracts into a high-resolution mass spectrometer both standards and analytes are ionized and detected together. This helps to equalize (but not alleviate) matrix suppression and reduce (but not eliminate) quantification bias. For example, more than 50 species of triacylglycerols (TAG) having fatty acid moieties with 10–24 atoms of carbon and 0–6 double bonds were consistently quantified in human plasma by top-down analysis using a single internal standard 50:0 d5 TAG.7,10
Top-down lipidomics detects and quantifies intact lipid molecules, yet it does not reveal their molecular structure or distinguish isomeric species. Higher molecular specificity of lipid quantification is achieved by bottom-up analyses based on tandem mass spectrometry.11−13 Here lipids are quantified by considering the abundance of species-specific “signature” fragments consistently detected in their MSn spectra.14 For example, glycerophospholipid (GPL) species are typically quantified using carboxylate anion (CA) fragments produced from their molecular anions by higher-energy collisional dissociation (HCD).13,15 While the bottom-up identification of lipid species has become routine,11 their quantification requires semiempirical adjustment of CA abundances for several structure-, instrument-, and experiment-dependent factors.12,13,16 The quantification bias is most dramatic for GPL comprising polyunsaturated fatty acid (PUFA) moieties.12 Upon HCD, CA of PUFA readily release CO213,17,18 and also yield a large number of poorly accountable low-abundant fragments. Therefore, the quantification of unsaturated lipids using available, yet nonauthentic (typically, saturated), lipid standards tends to underrepresent their true abundance. To balance the response toward saturated and unsaturated GPL species Ejsing et al. adjusted CA abundances for CO2-loss fragments.13 Yang et al. described a system of linear equations with several semiempirical coefficients for compensating position-specific biases of PUFA fragment intensities.16 However, these and similar approaches are only applicable to a limited scope of lipids analyzed under fixed experimental conditions. Lack of flexible and comprehensive analytical routines severely compromises the interlaboratory consistency of lipidomes quantification at the molecular species level.1,19,20
We developed, validated, and tested a computational model and software that harmonizes the abundance of CA fragments depending on their chemical structure and employed collision energy. It enabled consistent and accurate absolute (molar) quantification of structurally diverse GPL species using nonauthentic internal standards at different mass spectrometers and experimental conditions.
Experimental Section
Annotation of lipid species followed common conventions.21 FA stands for fatty acids, PE for 1,2-diacyl-sn-glycero-3-phosphoethanolamines, PC for 1,2-diacyl-sn-glycero-3-phosphocholines, and PC O- for 1-alkyl,2-acyl-glycero-3-phosphocholines. For presentation clarity, for unsaturated FA we only specified the position of the first double bond [e.g., FA 20:4 (5Z) for arachidonic acid]. In acronyms of GPL species, fatty acid moieties at undefined sn-1/-2 position are spaced by “–” (minus); spacing with “/” (slash) indicates that their position is defined. In this way, PC 16:0–18:1 represents a PC species having 16:0 and 18:1 moieties at an unspecified location; PC 16:0/18:1 stands for the individual species with 16:0 and 18:1 moieties located at the sn-1 and sn-2 positions, respectively.
Lipid Standards and Common Chemicals
Common lipid standards and total PC extracts were purchased from Avanti Polar Lipids (Alabaster, AL). Common chemicals and solvents were of LC–MS or Chromasolv/LiChrosolv grade purchased from Sigma-Aldrich (Darmstadt, Germany). Chloroform of HPLC grade was purchased from Rathburn Chemicals (Walkerburn, U.K.).
Synthesis of Lipids Standards
Standards that are not commercially available were synthesized in house. The series of PC O- standards were synthesized by acylating 1-O-hexadecyl-2-hydroxy-sn-glycero-3-phosphocholine [Avanti Polar Lipids (Alabaster, AL)] with free fatty acids [Larodan (Malmö, Sweden)] and fatty acid chlorides [Nu-Chek Prep (Elysian, CA)] in dry chloroform in the presence of polymer-bound DMAP (Sigma-Aldrich). Reaction mixtures were extracted with MTBE/methanol/water (10:3:3, v/v/v); the organic phase was collected, diluted down to the concentration of 0.5 μM, and subjected to HCD Fourier transform tandem mass spectrometry (FT MS/MS).
PC 20:4 (5Z)/16:0 and PC 22:6 (4Z)/16:0 were synthesized from glycerol-3-phosphocholine via 2-palmitoyl-sn-lysophosphatidylcholine (Figure S1). They were purified by high-performance liquid chromatography (HPLC) and analyzed by 1H, 13C, and 31P NMR. Ion mobility MS22 was employed to establish their isomeric purity (Figure S2).
Acquisition and Processing of HCD Fragment Intensity Curves
Synthetic standards were diluted with 2-propanol/methanol/chloroform (4:2:1 v/v/v) mixture containing 7.5 mM ammonium formate (further termed MSmix) and infused into LTQ Orbitrap Velos or Q Exactive mass spectrometers using a Triversa NanoMate nanoflow robotic ion source (Advion BioSciences) or, where specified, an electrospray ionization (ESI) probe (Thermo Fisher Scientific) such that total ion current (TIC) varied within the range of ±10%. HCD FT MS/MS was performed under the following settings (values for Q Exactive are in parentheses): mass isolation window of 1.6 (0.8) Th; normalized collision energy (NCE) ramped from 0% to 120% [10–70 with 2% (1%) increment]; acquisition started at m/z 100 (180); target mass resolution Rm/z=200, 200 000 (140 000); automated gain control (AGC) value, 2 × 104; S-lens rf value, 50%; capillary temperature, 200 °C (Figures S3–S5). Each precursor was fragmented four times; raw files were converted to mzXML and processed by LipidXte software (see below). Intensities of precursor and fragment peaks were normalized to the value of TIC at NCE of 0–5% (10–15%), under which precursor fragmentation was negligible. Details of the data preprocessing routine are in Figures S6 and S7.
Quantitative Bottom-Up Shotgun Lipidomics
Samples were diluted with MSmix to a total lipid concentration below 2 μM and analyzed by FT MS and HCD FT MS/MS in negative mode. FT MS spectra were acquired in reduced profile mode within the range of m/z 400–1200 under AGC 3 × 106; injection time of 1 s. HCD FT MS/MS were acquired with Rm/z=200 of 140 000; AGC of 2 × 104; injection time of 1 s; precursor isolation width of 1 Th; starting m/z of 180. Precalculated m/z of plausible PC precursors were compiled into an inclusion list, and for each precursor tandem mass spectra were acquired under NCE of 25%, 30%, 35%.
The acquired *.raw files were filtered using the PeakStrainer software.23 Lipids were identified, and abundances of correspondent precursor and fragment ions reported by LipidXplorer14,24 software. The output data files in *.csv format were further processed by LipidXte for automated correction of the intensity of CA fragments and quantification of individual molecular lipid species.
Stand-Alone and Web Versions of LipidXte Software
LipidXte creates a computational model by aligning fragment ions intensity curves and corrects the abundances of CA fragments for quantifying GPL molecular species. Stand-alone LipidXte is a Java-based program that uses ReAdW and mzxml-parser package in jmzReader25 for generating fragment intensity curves and computing correction factors for CA intensities. The web version of LipidXte available at https://doi.org/21.11101/0000-0007-D64D-2 is a JavaScript-based interface that processes *.raw files produced by data-independent acquisition and whose size does not exceed 30 MB. While processing spectra, it interacts with the LipidXplorer and PeakStrainer programs installed at the same web server.
Results and Discussion
Rationale and Experimental Basis of the Model
We aimed to develop a computational model and software for bottom-up shotgun quantification of molecular species of GPL that should lessen its dependence on internal standards and experiment settings. The model should adjust the abundance of CA fragments of fatty acid moieties detected in HCD FT MS/MS spectra of molecular anions of GPL and equalize the molar response independent of their chemical structure and collision energy. We also presumed that, if adjusted correctly, molar concentrations of lipids determined by top-down (FT MS) and, independently, bottom-up (HCD FT MS/MS) analyses should match.
Although we eventually aimed at quantifying a broad scope of lipid classes, we used phosphatidylcholines (PC) to develop and validate the computational model. PC show a “classical” fragmentation behavior, and many PC standards are commercially available or could be chemically synthesized from available precursors. As a resource for computational modeling, we used a collection of full CA intensity curves (Figures S8–S10) acquired on a Q Exactive mass spectrometer for, in total, 148 standards (including 26 PC and 39 PC O- species) by ramping NCE from 10% to 70%. To make curves comparable, their intensity was normalized to the total ion current observed in the same experiment under low NCE (Figure S7). Curves were remarkably reproducible over a long (more than 900 days) period of time (Figure S11).
Importantly, curves acquired for the same CA by fragmenting different PC precursors were very similar and unaffected by complementary FA moieties (Figure S12). At the same time, they strongly differed from the curves acquired from [M – H]− molecular anions of free fatty acids that (unfortunately) could not be used for lipid fragmentation modeling (Figures S13 and S14).
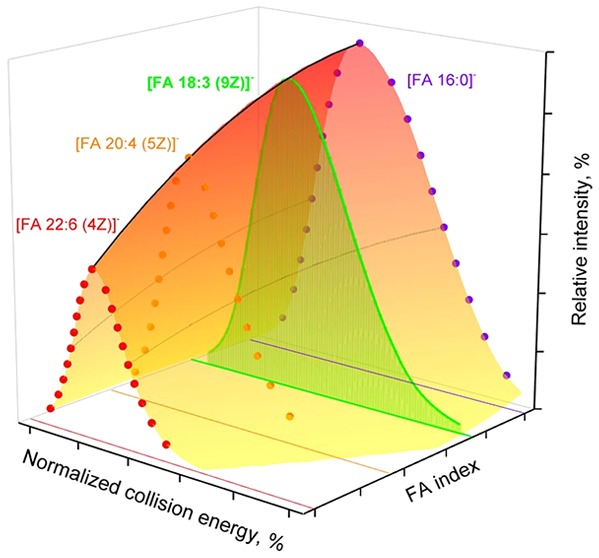
The alignment of normalized CA curves acquired from a series of structurally related PC O- species having 16:0 fatty alcohol at the sn-1 and varying FA moieties at the sn-2 position suggested that the length, unsaturation, and location of double bonds affected them in different ways (Figure 1). The relative intensity of CA fragments increased with the length and decreased with the number of double bonds in corresponding moieties. The curves of all saturated CA maxed at the same NCE (ca. 30%), while maxima of the curves of unsaturated CA were shifted toward lower NCE depending on the number (Figure 1B) and position of double bonds (Figure 1C). It is also known that in diester PC CA produced from sn-2 fatty acid moiety are more abundant than from sn-1.15,26
Figure 1.

Intensity curves of CA fragments acquired from molecular anions [M + HCOO]− of a series of PC O- species having different sn-2 fatty acids moieties (see insets). Curves grouping highlights the impact of (A) hydrocarbon chain length, (B) unsaturation, and (C) double bonds location.
We, therefore, concluded that, for harmonizing the abundances of CA fragments, four major corrections accounting for the hydrocarbon chain length, number and position of double bonds, and sn-1/2 location of the FA moiety should be applied successively and independently.
Building and Testing the Computational Model
First, we aligned the CA curves of three symmetric saturated PC 12:0/12:0; PC 18:0/18:0, and PC 22:0/22:0 as a 3D plot in coordinates [m/z; relative intensity (normalized to TIC); NCE] (Figure S15). Next, we used second-order polynomial regression to connect the data points at each curve having the same relative abundance (normalized to the curve maximum) by monotonous convex lines. In turn, this mathematical representation allowed us to project the relative intensity (normalized to TIC) for any CA at any NCE and to compute individual mass-dependent adjustment factors using CA 16:0 as an arbitrary reference (Figure S16). This adjustment was applied to curves of all CA and (if detected) their CO2-loss fragments before other corrections.
The relationship between the abundance and chemical structure of CA fragment(s) does not lend itself to a simple mathematical description using a few intuitive structure-related terms. Therefore, we set out to experimentally define a single numeric measure for CA fragmentation propensity. We termed it FI for fatty acid index and defined FI = 0 for the fatty acid with the lowest (22:6) and FI = 1.0 with the highest (e.g., 16:0 or 18:0) relative intensity at the maxima of their curves (Figure 1B). Next, we acquired CA curves from 39 PC O- species (Table S1) that differed by their sn-2 fatty acid moiety. Curves were preprocessed, subjected to m/z-dependent correction as described above, and plotted in coordinates [FI; relative intensity (normalized to TIC); NCE]. We positioned individual curves along the FI axes such that their maxima fit a monotonous convex line. We also requested that, upon scaling along FI axes, the experimental and projected curves had the best Pearson correlation. In this way, each FA moiety received its individual FI index (full list of FI is in Table S1). Finally, we interpolated intermediate values by second-order polynomial regression emulating a 3D shape.
Using CA curves acquired from PC O- species alleviated the impact of sn-1/2 positioning (all fatty acid moieties were at sn-2) and contributed to the alignments consistency. Next, we asked if FI determined by fragmenting PC O- species would be applicable to asymmetric and symmetric diacyl PC having, respectively, different or identical fatty acid moieties (Figure 2). We produced and aligned CA curves from four symmetric PC [PC 18:0/18:0, PC 22:0/22:0, PC 20:4 (5Z)/20:4 (5Z), PC 22:6 (4Z)/22:6 (4Z)] and five asymmetric PC [PC 12:0/13:0, PC 16:0/20:4 (5Z), PC 16:0/22:6 (4Z), PC 20:4 (5Z)/16:0, PC 22:6 (4Z)/16:0] (Figures S17 and S18). We underscore that, for building the model, it was critical to include a PC standard with the polyunsaturated moiety at the sn-1 position that was synthesized in house.
Figure 2.

Computational model and its validation using symmetric PC. (A) The 3D plot computed using CA curves of PC 16:0/16:0 (FI = 1), PC 20:4 (5Z)/20:4 (5Z) (FI = 0.35), and PC 22:6 (4Z)/22:6 (4Z) (FI = 0). The curves of PC 18:2/18:2 and PC 18:3 (9Z)/18:3 (9Z) were fitted to the model using FI = 0.91 (for 18:2) and FI = 0.73 [for 18:3 (9Z)] according to Table S1. Computed and experiment curves are shown as normal and dashed lines, respectively (panels B and C). Curves of CA 16:0 (FI = 1) and 20:4 (5Z) (FI = 0) are shown as thin lines for reference. Shading of the curves indicates that they are cross sections of the 3D-shape model at corresponding FI.
As expected, saturated CA were all having FI = 1.0, and there was no difference in FI of isomers of monounsaturated FA (Table S1). Importantly, FI (but not intensities at the maxima of the curve) showed no positional specificity and were less than 2.5% different between CA from the ether- and diacyl-PC (Table S2).
To test if this model correctly predicted CA profiles, we acquired CA curves from two symmetrical unsaturated PCs and compared the experimental curves with curves computed for FI from Table S1. Within the working range of NCE of 25–35%, they differed by less than 5% (Figure 2, parts B and C).
Altogether, we produced four 3D alignments suitable for subsequent adjustment of CA fragment intensities: one for PC O- (fatty acid at the sn-2 position) and three for diacyl PC: separately for the fatty acids at sn-1 and sn-2 positions and symmetric PC. The same FI (Table S1) were assigned to corresponding CA fragments in all models.
One remarkable observation was that source CA curves, FI, and the entire models were highly concordant between different mass spectrometers. FI determined by HCD FT MS/MS on LTQ Orbitrap Velos and Q Exactive instruments differed by less than 10% (Table S1). Taken together, our models harmonized the abundance of CA fragments of common fatty acid moieties produced within a broad range of NCE in the sn-position-specific manner.
Quantification of sn-1/sn-2 Position Isomers
We assumed that each asymmetric GPL species could occur as a mixture of sn-1/2 isomers of varying molar ratio. We further reasoned that, permitting the data quality, this ratio could be deduced from the actual intensities of complementary CA fragments adjusted according to their FI and applied NCE. The molar ratio of isomers was computed from the intensities of CA peaks (see Figures S19 and S20 for further details) according to the following equation:
where Ia and Ib are the intensities of the two complementary CA peaks; mol%a(sn-2) is the mol % of the isomer having the fatty acid moiety “a” at the sn-2 position; in this way
We tested the accuracy of isomers ratio determination by analyzing mixtures of synthetic PC standards whose isomeric purity was established by ion mobility mass spectrometry.22 We quantified the relative abundances in three pairs of synthetic isomers, including PC with saturated and polyunsaturated fatty acid moieties (Table S4).
Next, we used a commercial porcine brain PC extract, in which the isomeric composition of several PC species was previously determined by two independent experiments using ion mobility mass spectrometry.22,27 We analyzed it by HCD FT MS/MS and applied our model to quantify positional isomers now only using the adjusted relative abundances of CA fragments (Table S4). Altogether, we were able to dissect 39 positional isomers. Expectantly, PUFA moieties (fatty acids with four or more double bonds) were found almost exclusively at the sn-2 position, while the location of saturated and monounsaturated moieties was promiscuous (Table S4 only includes the species whose abundance exceeded 0.5 mol %). We note that it was not always possible to quantify the isomers because of interfering fragments or limited ion statistics, despite being able to detect both complementary fatty acid moieties.
Improved Consistency between Top-Down and Bottom-Up Quantification
We next tested if adjusting the abundance of CA fragments resulted in accurate and consistent quantification of lipid species. To this end, we quantified a mixture of 23 PC standards (Table S3) by top-down (FT MS) and bottom-up (HCD FT MS/MS) analysis using PC 12:0/13:0 as an internal standard. The abundance of precursor and fragment ions was reported by LipidXplorer, and then adjusted by LipidXte (Figure S25).
We observed poor consistency (R2 = 0.388) between FT MS and uncorrected HCD FT MS/MS quantification at NCE = 30% (Figure 3A). Concentrations of polyunsaturated species were strongly underrepresented in bottom-up profiles because of profound (up to 80%) losses of CA fragments, while adding up CO2-loss fragments did not restore the anticipated abundances. At the same time, saturated species were overestimated by more than 20%. Varying the collision energy did not improve the quantification concordance (Figures S21 and S23; Table S5). At the same time, adjusting CA abundances by LipidXte eliminated the quantification bias (R2 = 0.993) and reduced the spread of errors down to ±10% at all applied collision energies.
Figure 3.

Adjustment of abundances of CA fragments improved the concordance between top-down and bottom-up quantification. (A) Uncorrected intensities of CA fragments at different NCE (inset) in MS/MS spectra of four PC species (indicated below the bars). Lines above the bars indicate the sum of intensities of sn-1 and sn-2 fragments; shaded parts of the bars stand for intensities added by accounting for the products of CO2 loss. Gray shaded bars indicate the intensities of intact precursor peaks in the FT MS spectrum. (B) Correlation plot of lipid standards quantified by FT MS and by HCD FT MS/MS. (C) The intensities of the same CA peaks after LipidXte correction. (D) Correlation plot after LipidXte correction.
Therefore, we concluded that automated adjustment of CA abundances is required for consistent quantification of both saturated and polyunsaturated molecular species.
The Model is Portable between Orbitrap Instruments
The fragmentation model used for calculating the adjustment factors relied upon the alignment of CA curves acquired at the same Q Exactive mass spectrometer. However, we noticed that FI were similar even if CA curves were acquired by HCD FT MS/MS on an LTQ Orbitrap Velos (Tables S1 and S2). We wondered if the computational model could be also portable between several Q Exactive instruments installed in different laboratories and operating under their own optimized settings.
We found that CA curves acquired on five instruments in five laboratories were having a similar shape, yet we observed that their maxima were slightly shifted along the NCE axes. Therefore, LipidXte offers an automated routine for adjusting the basis model that only required a single analysis of a mixture of two PC standards (PC 12:0/13:0 and PC 22:6/22:6) by FT MS and HCD FT MS/MS (Figures S24 and S25).
By adjusting the model, we achieved excellent concordance of top-down and bottom-up quantification on four Q Exactive instruments (Figure 4). We analyzed a mixture of 23 synthetic PC standards at NCE of 25%, 30%, and 35% and computed FT MS versus FT MS/MS correlation plots (similar to Figure 3) for each instrument. With no LipidXte correction, the concordance between top-down and bottom-up estimates was poor with R2 values within the range of 0.1–0.8. Upon LipidXte correction, R2 values were better than 0.9 for all PC and NCE on all four mass spectrometers. For a yet unknown reason, we failed to achieve an equally good correlation on one (out of, in total, five) tested instruments. Therefore, we recommend performing the bottom-up/top-down correlation test with a mixture of standards prior to the analysis of real samples.
Figure 4.

LipidXte improves the concordance between top-down and bottom-up quantification at four Q Exactive instruments in three laboratories. (A) R2 of the correlation plots with (in blue) and without (in red) LipidXte adjustment. (B) Slope of the same plots after LipidXte correction. Slopes of uncorrected plots differed by more than 5-fold and, for presentation clarity, are not shown here (see Figure S22 for the quantification of sn-positional isomers). At the instrument no. 1 full data sets were independently acquired twice, and both replicates are presented here for the reference.
We concluded that our fragmentation model is portable and achieves concordant quantification if fine-tuned by LipidXte.
Bottom-Up Quantification of Polyunsaturated Phosphatidylcholines in Porcine Brain
We next applied LipidXplorer with LipidXte to quantify PC in porcine brain extract that contains a sizable fraction of polyunsaturated lipids (Figure 5). The extract was subjected to top-down analysis and to bottom-up analyses under several NCE. Then, the top-down profile was compared with the nonadjusted and with LipidXte-adjusted bottom-up profiles (Figure 5A). The adjustment almost doubled the number of quantifiable PC species (Figure 5B) (for presentation clarity, only lipids above 0.5 mol % are shown in Figure 5A) and practically eliminated significant (up to 60%) bias of the abundance of PUFA lipids (Figure 5C). Qualitative and quantitative underrepresentation of polyunsaturated lipids was the major source of profiles discordance also in the analyses of total extracts (Figure 5D). However, it was compensated by LipidXte software. We, therefore, concluded that consistent absolute bottom-up quantification of lipid species can be achieved by a combination of appropriate analytical and computational procedures.1,19,20
Figure 5.

LipidXte reduced quantification bias in bottom-up analyses of total extracts. (A) Profiles of PC (only species above 0.5 mol % shown) acquired by FT MS and uncorrected and corrected HCD FT MS/MS. (B) Number of quantifiable species before and after LipidXte correction. (C) Quantification discordance (in percent) for individual species before and after correction. (D) Quantification discordance and total unsaturation of species.
Conclusion and Perspectives
A common and persisting bottleneck for many omics sciences, including lipidomics, is the dramatic discrepancy between a large number of detectable molecules and a much lower number of authentic standards available for their quantification. Therefore, accurate and precise lipidome-wide quantification should rely upon a combination of expertly chosen experimental conditions and spectra processing software that, together, should be harmonizing instrument response toward similar, yet not identical, lipid molecules.
Here we demonstrate that the computational model based on the alignment of full CA fragment intensity curves enables the unbiased quantification of molecular species within a broad range of unsaturation, length, and also different sn-1/2 positioning of their fatty acid moieties. To our delight, the model was remarkably stable toward variable instrument-dependent settings and was portable between different mass spectrometers. Further work should extend the approach to other lipid classes, possibly including those that do not produce CA fragments. It also looks promising to combine our approach with complementary fragmentation methods, e.g., collision-induced dissociation (CID) in linear traps, chemical fragmentation, e.g., ozone-induced dissociation (OzID),27,28 and Paterno–Buechi photochemical cleavage,29 UV 193 nm laser excitation,30,31 and also with ion mobility separation (reviewed in ref (32)).
Eventually, the field should be able to close the gap between “one standard” and “one standard per analyte” quantification and, eventually, deliver consistent absolute lipidome-wide quantification that is particularly critical for molecular medicine and disease diagnostics.20
Acknowledgments
Work in A.S. and A.N.’s laboratories was supported by the Max Planck Gesellschaft, by the TRR83 collaborative research center (TP17 and TP27, respectively) from the Deutsche Forschungsgemeinschaft (DFG), and by the Indo-German Max-Planck Lipid Center. The A.S. laboratory was also supported by the Liver System Medicine (LiSyM) program and the Lipidomics and Informatics for Life Sciences (LIFS) Unit of the de.NBI Consortium funded by the Bundesministerium für Bildung und Forschung (BMBF). A.N. acknowledges funding by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement GA758334 ASYMMEM).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b03270.
Schemes of the chemical synthesis of polyunsaturated PCs and their characterization by 1H, 13C, and 31P NMR spectrometry and high-resolution mass spectrometry, characterization of isomeric purity of PC standards by ion mobility mass spectrometry, intensity curves of carboxylate anions and impact of acquisition and preprocessing settings, reproducibility of fragmentation curves, the impact of common lipid structure features, the impact of fatty acid moieties on CO2-loss fragments, fragment intensity curves of free fatty acids anions, tables of fatty acid indexes (FI) of individual fatty acids, table presenting internal standards used for the method validation, evidence on the accuracy and precision of the quantification of positional isomers of lipids, and detailed description of algorithms and procedures of fragment ion intensity curves alignment and isomers quantification (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Vvedenskaya O.; Wang Y.; Ackerman J. M.; Knittelfelder O.; Shevchenko A.. TrAC, Trends Anal. Chem. [Online early access]. 10.1016/j.trac.2018.10.013. Published Online: Oct 20, 2018. https://www.sciencedirect.com/science/article/abs/pii/S016599361830428X. [DOI] [Google Scholar]
- Wang M.; Wang C.; Han X. Selection of internal standards for accurate quantification of complex lipid species in biological extracts by electrospray ionization mass spectrometry-What, how and why?. Mass Spectrom. Rev. 2017, 36 (6), 693–714. 10.1002/mas.21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.; Han X. Lipidomics: Techniques, Applications, and Outcomes Related to Biomedical Sciences. Trends Biochem. Sci. 2016, 41 (11), 954–969. 10.1016/j.tibs.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wang C.; Han X. Tutorial on lipidomics. Anal. Chim. Acta 2019, 1061, 28–41. 10.1016/j.aca.2019.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Wang C.; Han R. H.; Han X. Novel advances in shotgun lipidomics for biology and medicine. Prog. Lipid Res. 2016, 61, 83–108. 10.1016/j.plipres.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustam Y. H.; Reid G. E. Analytical Challenges and Recent Advances in Mass Spectrometry Based Lipidomics. Anal. Chem. 2018, 90 (1), 374–397. 10.1021/acs.analchem.7b04836. [DOI] [PubMed] [Google Scholar]
- Sales S.; Graessler J.; Ciucci S.; Al-Atrib R.; Vihervaara T.; Schuhmann K.; Kauhanen D.; Sysi-Aho M.; Bornstein S. R.; Bickle M.; Cannistraci C. V.; Ekroos K.; Shevchenko A. Gender, contraceptives and individual metabolic predisposition shape a healthy plasma lipidome. Sci. Rep. 2016, 6, 27710. 10.1038/srep27710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho M.; Sampaio J. L.; Palm W.; Brankatschk M.; Eaton S.; Shevchenko A. Effects of diet and development on the Drosophila lipidome. Mol. Syst. Biol. 2012, 8, 600. 10.1038/msb.2012.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejsing C. S.; Sampaio J. L.; Surendranath V.; Duchoslav E.; Ekroos K.; Klemm R. W.; Simons K.; Shevchenko A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (7), 2136–41. 10.1073/pnas.0811700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmann K.; Almeida R.; Baumert M.; Herzog R.; Bornstein S. R.; Shevchenko A. Shotgun lipidomics on a LTQ Orbitrap mass spectrometer by successive switching between acquisition polarity modes. J. Mass Spectrom. 2012, 47 (1), 96–104. 10.1002/jms.2031. [DOI] [PubMed] [Google Scholar]
- Schwudke D.; Oegema J.; Burton L.; Entchev E.; Hannich J. T.; Ejsing C. S.; Kurzchalia T.; Shevchenko A. Lipid profiling by multiple precursor and neutral loss scanning driven by the data-dependent acquisition. Anal. Chem. 2006, 78 (2), 585–95. 10.1021/ac051605m. [DOI] [PubMed] [Google Scholar]
- Schuhmann K.; Herzog R.; Schwudke D.; Metelmann-Strupat W.; Bornstein S. R.; Shevchenko A. Bottom-up shotgun lipidomics by higher energy collisional dissociation on LTQ Orbitrap mass spectrometers. Anal. Chem. 2011, 83 (14), 5480–7. 10.1021/ac102505f. [DOI] [PubMed] [Google Scholar]
- Ejsing C. S.; Duchoslav E.; Sampaio J.; Simons K.; Bonner R.; Thiele C.; Ekroos K.; Shevchenko A. Automated identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning. Anal. Chem. 2006, 78 (17), 6202–14. 10.1021/ac060545x. [DOI] [PubMed] [Google Scholar]
- Herzog R.; Schwudke D.; Schuhmann K.; Sampaio J. L.; Bornstein S. R.; Schroeder M.; Shevchenko A. A novel informatics concept for high-throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biol. 2011, 12 (1), R8. 10.1186/gb-2011-12-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekroos K.; Chernushevich I. V.; Simons K.; Shevchenko A. Quantitative profiling of phospholipids by multiple precursor ion scanning on a hybrid quadrupole time-of-flight mass spectrometer. Anal. Chem. 2002, 74 (5), 941–9. 10.1021/ac015655c. [DOI] [PubMed] [Google Scholar]
- Yang K.; Zhao Z.; Gross R. W.; Han X. Identification and quantitation of unsaturated fatty acid isomers by electrospray ionization tandem mass spectrometry: A shotgun lipidomics approach. Anal. Chem. 2011, 83 (11), 4243–4250. 10.1021/ac2006119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F. F.; Turk J. Characterization of phosphatidylinositol, phosphatidylinositol-4- phosphate, and phosphatidylinositol-4,5-bisphosphate by electrospray ionization tandem mass spectrometry: a mechanistic study. J. Am. Soc. Mass Spectrom. 2000, 11 (11), 986–99. 10.1016/S1044-0305(00)00172-0. [DOI] [PubMed] [Google Scholar]
- Hsu F. F.; Turk J. Electrospray ionization with low-energy collisionally activated dissociation tandem mass spectrometry of glycerophospholipids: mechanisms of fragmentation and structural characterization. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2009, 877 (26), 2673–95. 10.1016/j.jchromb.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden J. A.; Heckert A.; Ulmer C. Z.; Jones C. M.; Koelmel J. P.; Abdullah L.; Ahonen L.; Alnouti Y.; Armando A. M.; Asara J. M.; Bamba T.; Barr J. R.; Bergquist J.; Borchers C. H.; Brandsma J.; Breitkopf S. B.; Cajka T.; Cazenave-Gassiot A.; Checa A.; Cinel M. A.; Colas R. A.; Cremers S.; Dennis E. A.; Evans J. E.; Fauland A.; Fiehn O.; Gardner M. S.; Garrett T. J.; Gotlinger K. H.; Han J.; Huang Y.; Neo A. H.; Hyotylainen T.; Izumi Y.; Jiang H.; Jiang H.; Jiang J.; Kachman M.; Kiyonami R.; Klavins K.; Klose C.; Kofeler H. C.; Kolmert J.; Koal T.; Koster G.; Kuklenyik Z.; Kurland I. J.; Leadley M.; Lin K.; Maddipati K. R.; McDougall D.; Meikle P. J.; Mellett N. A.; Monnin C.; Moseley M. A.; Nandakumar R.; Oresic M.; Patterson R.; Peake D.; Pierce J. S.; Post M.; Postle A. D.; Pugh R.; Qiu Y.; Quehenberger O.; Ramrup P.; Rees J.; Rembiesa B.; Reynaud D.; Roth M. R.; Sales S.; Schuhmann K.; Schwartzman M. L.; Serhan C. N.; Shevchenko A.; Somerville S. E.; St. John-Williams L.; Surma M. A.; Takeda H.; Thakare R.; Thompson J. W.; Torta F.; Triebl A.; Trotzmuller M.; Ubhayasekera S. J. K.; Vuckovic D.; Weir J. M.; Welti R.; Wenk M. R.; Wheelock C. E.; Yao L.; Yuan M.; Zhao X. H.; Zhou S. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58 (12), 2275–2288. 10.1194/jlr.M079012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burla B.; Arita M.; Arita M.; Bendt A. K.; Cazenave-Gassiot A.; Dennis E. A.; Ekroos K.; Han X.; Ikeda K.; Liebisch G.; Lin M. K.; Loh T. P.; Meikle P. J.; Oresic M.; Quehenberger O.; Shevchenko A.; Torta F.; Wakelam M. J. O.; Wheelock C. E.; Wenk M. R. MS-based lipidomics of human blood plasma: a community-initiated position paper to develop accepted guidelines. J. Lipid Res. 2018, 59 (10), 2001–2017. 10.1194/jlr.S087163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebisch G.; Vizcaino J. A.; Kofeler H.; Trotzmuller M.; Griffiths W. J.; Schmitz G.; Spener F.; Wakelam M. J. Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 2013, 54 (6), 1523–30. 10.1194/jlr.M033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groessl M.; Graf S.; Knochenmuss R. High resolution ion mobility-mass spectrometry for separation and identification of isomeric lipids. Analyst 2015, 140 (20), 6904–6911. 10.1039/C5AN00838G. [DOI] [PubMed] [Google Scholar]
- Schuhmann K.; Thomas H.; Ackerman J. M.; Nagornov K. O.; Tsybin Y. O.; Shevchenko A. Intensity-Independent Noise Filtering in FT MS and FT MS/MS Spectra for Shotgun Lipidomics. Anal. Chem. 2017, 89 (13), 7046–7052. 10.1021/acs.analchem.7b00794. [DOI] [PubMed] [Google Scholar]
- Herzog R.; Schwudke D.; Shevchenko A. LipidXplorer: software for quantitative shotgun lipidomics compatible with multiple mass spectrometry platforms. Curr. Protoc. Bioinf. 2013, 43, 14.12.1. 10.1002/0471250953.bi1412s43. [DOI] [PubMed] [Google Scholar]
- Griss J.; Reisinger F.; Hermjakob H.; Vizcaino J. A. jmzReader: A Java parser library to process and visualize multiple text and XML-based mass spectrometry data formats. Proteomics 2012, 12 (6), 795–8. 10.1002/pmic.201100578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekroos K.; Ejsing C. S.; Bahr U.; Karas M.; Simons K.; Shevchenko A. Charting molecular composition of phosphatidylcholines by fatty acid scanning and ion trap MS3 fragmentation. J. Lipid Res. 2003, 44 (11), 2181–2192. 10.1194/jlr.D300020-JLR200. [DOI] [PubMed] [Google Scholar]
- Maccarone A. T.; Duldig J.; Mitchell T. W.; Blanksby S. J.; Duchoslav E.; Campbell J. L. Characterization of acyl chain position in unsaturated phosphatidylcholines using differential mobility-mass spectrometry. J. Lipid Res. 2014, 55 (8), 1668–77. 10.1194/jlr.M046995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R. A.; May J. C.; Stinson C. A.; Xia Y.; McLean J. A. Determining Double Bond Position in Lipids Using Online Ozonolysis Coupled to Liquid Chromatography and Ion Mobility-Mass Spectrometry. Anal. Chem. 2018, 90 (3), 1915–1924. 10.1021/acs.analchem.7b04007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. P.; Zhang D. H.; Chen Q. H.; Wu J. H.; Ouyang Z.; Xia Y. Online photochemical derivatization enables comprehensive mass spectrometric analysis of unsaturated phospholipid isomers. Nat. Commun. 2019, 10, 79. 10.1038/s41467-018-07963-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D. R.; Brodbelt J. S. Structural Characterization of Phosphatidylcholines Using 193 nm Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem. 2017, 89 (3), 1516–1522. 10.1021/acs.analchem.6b03353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams P. E.; Klein D. R.; Greer S. M.; Brodbelt J. S. Pinpointing Double Bond and sn-Positions in Glycerophospholipids via Hybrid 193 nm Ultraviolet Photodissociation (UVPD) Mass Spectrometry. J. Am. Chem. Soc. 2017, 139 (44), 15681–15690. 10.1021/jacs.7b06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R. A.; Leaptrot K. L.; May J. C.; McLean J. A. New frontiers in lipidomics analyses using structurally selective ion mobility-mass spectrometry. TrAC, Trends Anal. Chem. 2019, 116, 316–323. 10.1016/j.trac.2019.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.