

Abstract
We report a series of ferrocene-based derivatives and their corresponding oxidized forms in which the introduction of simple electron donating groups like methyl or tert-butyl units on cyclopentadienyl-rings afford great tunability of Fe+III/Fe+II redox potentials from +0.403 V down to −0.096 V versus saturated calomel electrode. The spin forbidden d–d transitions of ferrocene derivatives shift slightly toward the blue region with an increasing number of electron-donating groups on the cyclopentadienyl-rings with very little change in absorptivity values, whereas the ligand-to-metal transitions of the corresponding ferricinium salts move significantly to the near-IR region. The electron-donating groups also contribute in the strengthening of electron density of Fe+III d-orbitals, which therefore improves the chemical stability against the oxygen reaction. Further, density functional theory calculations show a reducing trend in outer shell reorganization energy with an increasing number of the electron donating units.
Introduction
Ferrocene is a well-known organometallic redox mediator, which has a number of applications in almost all fields related to electrochemistry.1 The sandwich structure of ferrocene is based on two negatively charged aromatic cyclopentadienyl (Cp) rings which creates a stable 18-electron system for Fe(+II) ions.2 It confers to the complex a high temperature robustness up to 400 °C in air,1a,3 a reversible electrochemical characteristic at a redox potential of +0.403 V [vs saturated calomel electrode (SCE)] for which this latter is independent of the solvent properties, and good solubility in most of the common organic solvents. However, ferrocene is predisposed to various electrophilic attacks because of the electron-rich aromatic Cp-rings.1a,4 Taking this as an advantage, it can thus serve as a potential precursor for synthesizing a variety of derivatives to finely reach the targeted asset. Ferrocene is also being used in the design of novel ligand architectures for the synthesis of various metal catalysts,5 suitable redox couples in mixed valence states,6,7 redox shuttles in catalysis,8 and for the development of electrochemically active dendrons, dendrimers, and polymers.1a,9−11 Ferrocene can be used as part of a battery electrode material12 and has been proposed as a fast redox mediator in dye-sensitized solar cells to decrease energy losses related to the dye regeneration step.13
This paper reports a complete study on the characterization and density functional theory (DFT) calculations of four differently modified ferrocenes by means of the introduction of methyl and tertiary-butyl units to the Cp-rings. This study provides an insight, how and to which extent the characteristics of ferrocene can be controlled or tuned with the introduction of a simple electron donating unit to the Cp-rings. The increased electron density on the Cp-rings further influences the overall electrochemical and optical properties of the whole molecule. The consequences in terms of electrochemistry, stability, optical and magnetic properties are herein discussed.
Results and Discussion
The electrochemical properties of all the ferrocene derivatives were evaluated by cyclic voltammetry (CV). All the Fe+III/Fe+II redox potentials and their electrochemical reversibility were also investigated taking ferrocene as the standard.4b,4c The voltammograms recorded for the different ferrocene molecules in acetonitrile are shown in Figure 1. The Fe+III/Fe+II redox potential for Me10Fc+/Me10Fc was observed at −0.096 V (vs SCE) compared to standard Fc+/Fc at 0.403 V (vs SCE) without affecting electrochemical reversibility. The increasing number of methyl groups on the Cp-rings gradually decreases the redox potential by ca. 100 mV (one methyl group per one Cp ring), that is, +0.302 V (vs SCE) for Me2Fc+/Me2Fc and +0.003 V for Me8Fc+/Me8Fc. We found a linear relationship between the redox potential of the complex and the Hammett constant (σp) of the substituent and its total number according to the equation E1/2 (vs SCE) = 0.403 + 0.291σp (Figure S1). The synthesis of Me4Fc and Me6Fc still revealed to be challenging despite our efforts. Indeed, the corresponding Cp ring with two and three methyl groups (σp = 0.17) are found to be highly unstable and forms structural isomers during synthesis. These structural isomers revealed afterward to be rather difficult to separate also. Other synthetic routes involving the multistep methylation of a suitable ferrocene derivative (e.g., Fc or Me2Fc) also resulted in a mixture of isomers with very low yields. As an alternative to these two complexes, we proposed the introduction of two tert-butyl units which has a similar Hammett constant (σp = 0.16) on each Cp ring, which affords lowering of the redox potential to +0.170 V (vs SCE) while keeping excellent electrochemical reversibility. This complex has been proposed by Bläser et al. and then by Gleiter et al.18 This potential value lies exactly between Me2Fc+/Me2Fc and Me8Fc+/Me8Fc filling the gap with an identical synthetic procedure (Figures 1 and 2). The result is in good agreement with predicting the energetic destabilization of the iron d-orbitals within the crystal field with the increasing electron density on the Cp-rings by methyl or tert-butyl (tBu) groups. The greater effect of the tert-butyl group is due to its stronger inductive effect (σi = 0.07 vs 0.04 for CH3).19 The diffusion coefficients of these different ferrocene molecules have been determined by cyclic voltamperometry with different scan rates (Figures 2 and S2–S7). This parameter is particularly important for electrochemical applications in which the mass transport limitation can control the overall cell kinetic such as in dye-sensitized solar cells,13c molecular redox shuttle for batteries,20 or as an electroactive component in redox flow batteries.21 Randles–Sevcik equation has been utilized after verifying (i) the linear relationship between the peak currents (both anodic and cathodic) as a function of the square root of scan rate, and (ii) that the system remains rapid and reversible in the whole domain of scan rates. The obtained values of diffusion coefficients for the different complexes are reported in Table 1. Although the introduction of the methyl or tert-butyl group(s) onto the Cp ring will lead to a slightly more bulky complex, which can penalize its diffusion capacity into confined mesoporosity, it is however noteworthy to see a beneficial effect on the diffusion property in acetonitrile likely as a result of the increased electron density redistribution between two Cp-rings which may have improved the ion pairing interactions between ferrocene and PF6– (TBAPF6 used as a supporting electrolyte) in the electrolyte solution. Twofold enhancement in the experimental diffusion coefficient value (from DFc = 2.60 × 10–9 m2/s for ferrocene, a value in good agreement with the literature,22 to DMe2Fc = 5.73 × 10–9 m2/s for the dimethyl ferrocene counterpart) also suggests that there is a better ion pair interaction despite being heavier. More methyl groups result in a greater diffusion coefficient value, reaching as high as DMe10Fc+ = 7.87 × 10–9 m2/s and DtBuFc+ = 4.99 × 10–9 m2/s for the deca-methyl and tert-butyl counterparts, respectively. There are no noticeable differences on the diffusion coefficient between the ferrocene and oxidized ferricinium·TFSI derivatives except for the deca-methyl ferrocene complex regardless of the metal centers being at different oxidation states. The Fe+III/Fe+II redox potential values for both oxidized and reduced ferrocene molecules are almost identical. There is a slight downward shift of the whole voltamperogram compared to the parental reduced form without any significant difference in peak current or Faradaic current values (Figure 3). This downward shift was also noticed while CVs were being recorded after each successive addition of AgTFSI to the electrochemical cell in order to confirm that it is not because of any impurity (Figure S8). The reason for this downward shift of voltamperogram for oxidized forms is not very clear at the moment and needs further investigation.
Figure 1.
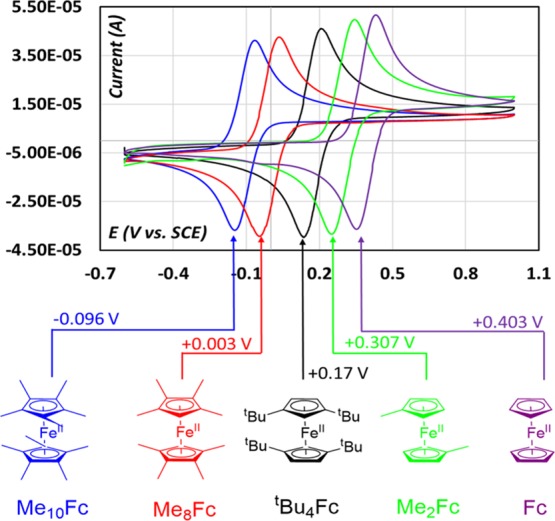
CV and redox potential for the Fe(III)/Fe(II) redox couple of different ferrocene derivatives. (Solvent = MeCN; supporting electrolyte = 0.1 (M) TBAPF6; working electrode = glassy carbon (3 mm dia); counter electrode = Pt wire, reference electrode = SCE; scan rate = 100 mV s–1).
Figure 2.

CV at different scan rates from 20 to 1000 mV s–1 of 2 mmol/L tBu4Fc in acetonitrile (supporting electrolyte: 0.1 mol/L TBAPF6, glassy carbon working electrode (3 mm dia), Pt wire counter electrode, SCE as a reference electrode).
Table 1. Electrochemical and Spectroscopic Data of All Ferrocene and Ferricinium Derivatives in Acetonitrilea.
| compounds | E1/2(Fe3+/Fe2+) (V) | ΔE1/2 (mV) | Dox × 10–9 (m2 s–1) | Dred × 10–9 (m2 s–1) | Dox/Dred | λ (nm)/ε (M–1 cm–1) |
|---|---|---|---|---|---|---|
| Fc21 | +0.403 | 43 | 2.6 | 2.6 | 1 | 322/61 |
| 442/95 | ||||||
| [Fc][TFSI] | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. |
| Me2Fc | +0.307 | 56 | 5.73 | 5.94 | 0.96 | 326/59 |
| 438/96 ± 2 | ||||||
| [Me2Fc][TFSI] | n.s. | n.s. | n.s. | n.s. | n.s. | 386(s) |
| 457(s) | ||||||
| 552(s) | ||||||
| 647/97 | ||||||
| tBu4Fc | +0.170 | 55 | 4.99 | 5.30 | 0.94 | 334/83 |
| 462/105 | ||||||
| [tBu4Fc][TFSI] | +0.170 | 38 | 4.67 | 4.81 | 0.97 | 390(s) |
| 484(s) | ||||||
| 597(s) | ||||||
| 680/370 | ||||||
| Me8Fc | +0.003 | 43 | 7.02 | 7.22 | 0.97 | 325(s) |
| 428/95 | ||||||
| [Me8Fc][TFSI] | –0.034 | 34 | 5.45 | 6.03 | 0.90 | 463(s) |
| 496(s) | ||||||
| 617(s) | ||||||
| 750/380 | ||||||
| Me10Fc | –0.096 | 46 | 6.47 | 6.71 | 0.96 | 325(s) |
| 423/97 | ||||||
| [Me10Fc][TFSI] | –0.120 | 44 | 7.37 | 7.87 | 0.94 | 470(s) |
| 518(s) | ||||||
| 637(s) | ||||||
| 698(s) | ||||||
| 778/488 |
n.s. stands for not stable, s for shoulder.
Figure 3.

Difference in CV between reduced and chemically oxidized forms. (a) tBu4Fc and tBu4Fc+, (b) Me8Fc and Me8Fc+, and (c) Me10Fc and Me10Fc+. (Solvent = MeCN; supporting electrolyte = 0.1 (M) TBAPF6; working electrode = glassy carbon (3 mm dia); counter electrode = Pt wire, reference electrode = SCE; scan rate = 100 mV s–1).
Chemical and electrochemical stabilities of these complexes are other aspects while considering them to be applicable. Pure ferricinium salt is stable in the solid-state when air is excluded. However, ferricinium salt in the solution becomes unstable in de-aerated aqueous solution when the pH is lower than 4 or in de-aerated aprotic solvents. The rapid decomposition of ferrocene occurs at a pH greater than 4 or in non-aqueous solvents which can act as proton acceptors.23 The ferricinium salt reacts with molecular oxygen in polar solvents such as acetonitrile, leading to a reactive μ-peroxy iron radical intermediate which again reacts with ferricinium salt further resulting in a μ-peroxy di-iron complex.24 This reactivity toward oxygen is also a function of the type of counter ions and atmosphere of cycling.25 To better assess the electrochemical stability of the different complexes in pure acetonitrile under ambient conditions, Figures 4 and 5 report the cyclic voltamperograms for ferrocene and the modified complexes including either different methyl units or tert-butyl groups over 100 cycles at a scan rate of 100 mV s–1. Figure 4a shows that a gradual loss in peak currents and deteriorating shape of original CVs for Fc and Me2Fc while carrying out successive oxidation/reduction cycles under ambient conditions, indicating instability of oxidized forms in solution due to a reaction with oxygen in a facile manner as described elsewhere. This is favorable to an irreversible steady-state current in oxidation and reduction, which is maintained over cycles with a large overpotential greater than 500 mV. This result highlights the rapid and continuous decomposition of electrochemically formed ferricinium after the very few first cycles. In the case of Me2Fc, a similar decomposition of the complex is experienced (Figure 4b). However, this decomposition leads to the occurrence of an irreversible anodic faradaic peak at 0.92 V (vs SCE) and a broad steady-state cathodic current starting at −0.35 V (vs SCE) with similar features as Fc+ except the onset, which is at a potential 200 mV greater compared to Fc+. Interestingly, the introduction of more than one methyl or tert-butyl unit per Cp ring affords to greatly enhance electrochemical stability of the corresponding oxidized forms as reported in Figure 5a–f, showing a perfectly reversible fingerprint without any fading of peak currents upon cycling. This result suggests that the formation of the μ-peroxo iron radicals could have been hampered by strengthening the electron density to Fe+III orbitals. The electrochemical signature and stability are similar regardless if we start by the ferrocene derivatives or the corresponding ferricinium salts obtained from chemical oxidation using AgTFSI.
Figure 4.

CV on glassy carbon electrode (3 mm dia.) in a three-electrode configuration of the 2 mM ferrocene derivative salt over 100 cycles in an acetonitrile/ambient atmosphere (supporting electrolyte: 0.1 M TBAPF6, counter electrode: Pt, reference electrode: SCE, scan rate: 100 mV s–1) for (a) Fc and (b) Me2Fc.
Figure 5.

CV on glassy carbon electrode (3 mm dia.) in a three-electrode configuration of the 2 mM ferrocene derivative salt over 100 cycles in an acetonitrile/ambient atmosphere (supporting electrolyte: 0.1 M TBAPF6, counter electrode: Pt, reference electrode: SCE, scan rate: 100 mV s–1) for (a) Me2Fc, (a) tBu4Fc, (b) Me8Fc, (c) Me10Fc, (d) tBu4Fc+, (e) Me8Fc+, and (f) Me10Fc+.
The inner shell reorganization energies of the different Fc derivatives
have been calculated from the fundamental definition, that is, the
energy required to bring the oxidized state at the equilibrium geometry
from the reduced state. This parameter is particularly important in
photocatalysis and in dye-sensitized solar cells for instance because
the energy requisite to undergo the “red-to-ox” reaction
results in an internal energy loss of the system. In this case, the
molecular geometry is first optimized for the cation (C) state and
the energy Ecat(C) is computed, then the
geometry is optimized for the neutral state, one electron is removed,
and the electronic energy Ecat(N) of the
cation state is computed. The reorganization energy is then defined
as λin = Ecat(N) – Ecat(C). In order to verify the accuracy and
reliability of the results, the reorganization energies of Fc, Me2Fc, and Me8Fc have also been computed using the
expression 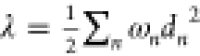 , valid
in the harmonic approximation of
the potential energy surfaces (values reported into bracket in Table 2). Here, ωn stands for the frequencies of the normal
modes of the molecule vibration and dn for their dimensionless displacements upon oxidation/reduction.
The results, reported in Table 2, show excellent agreement between the two approaches confirming
the reliability of the calculations. In all the cases, one interesting
feature of ferrocene is the niche in their very low reorganization
energies which are in the order of a few tenths of meV, in agreement
with previous studies on ferrocene.26 Such
a small reorganization energy is associated with minor variations
of the molecular geometry upon oxidation and reduction, in contrast
to polypyridyl cobalt complexes for instance used in dye-sensitized
solar cells which require more than 200 meV of reorganization energy.27 The introduction of methyl or tert-butyl units on Cp-rings induces only a slight increase of the reorganization
energy from 16 meV for Fc to 46 meV for tBu4Fc.
, valid
in the harmonic approximation of
the potential energy surfaces (values reported into bracket in Table 2). Here, ωn stands for the frequencies of the normal
modes of the molecule vibration and dn for their dimensionless displacements upon oxidation/reduction.
The results, reported in Table 2, show excellent agreement between the two approaches confirming
the reliability of the calculations. In all the cases, one interesting
feature of ferrocene is the niche in their very low reorganization
energies which are in the order of a few tenths of meV, in agreement
with previous studies on ferrocene.26 Such
a small reorganization energy is associated with minor variations
of the molecular geometry upon oxidation and reduction, in contrast
to polypyridyl cobalt complexes for instance used in dye-sensitized
solar cells which require more than 200 meV of reorganization energy.27 The introduction of methyl or tert-butyl units on Cp-rings induces only a slight increase of the reorganization
energy from 16 meV for Fc to 46 meV for tBu4Fc.
Table 2. Inner Shell and Outer Shell Reorganization Energies of Fc and Substituted Fca.
| molecule | λin (meV) | λout (meV) |
|---|---|---|
| Fc | 16(15) | 780 |
| Me2Fc | 28(22) | 713 |
| Me8Fc | 42(40) | 589 |
| Me10Fc | 42 | 563 |
| tBu4Fc | 46 | 498 |
Values in parenthesis are obtained within the harmonic approximation. The outer shell reorganization energy is computed in acetonitrile using a two-zone heterogeneous electron-transfer model.
In addition, outer shell reorganization energy λo for electrode reactions have been computed considering a two-zone heterogeneous electron-transfer model28
where ϵop and ϵst are the optical and static dielectric constants of the solvent, a is the cavity radius of the redox species, and 2d is the distance between the redox species and its image in the metal electrode, that is twice the distance d of the redox species from the surface of the electrode. The effective radius a of the cavity of the redox species is obtained by using an effective value of a sphere with the same volume of the cavity obtained from the tessellation of molecular surfaces. This distance d is usually assumed to coincide with the position of the outer Helmholtz plane. This latter can be estimated as the sum of the diameter of the solvent molecule (Dsolv) and the radius of the solvated ions (Rion). The parameters used in our calculations are tabulated in Table 3.29 In contrast to the inner reorganization energy, the introduction of methyl or tert-butyl groups on Cp-rings leads to a decrease of the outer-sphere reorganization energy from 780 meV for Fc to 498 meV for tBu4Fc suggesting that the modification of the pentadiene units with methyl or tert-butyl units affords reducing the energy of the first solvation shell.
Table 3. Parameters Used in the Calculation of the Outer-Shell Reorganization Energya.
| ϵst | ϵop | d | Dsolv | Rion |
|---|---|---|---|---|
| 35.688 | 1.806 | 8.31 | 4.31 | 4.00 |
Note that the radius of the TFSI– in the electrolyte has never been reported in the literature. We assumed its size to be half of its largest atom–atom distance.
Absorption Spectra and Kinetic Study
The UV–visible absorption spectra of all ferrocene and ferricinium derivatives in acetonitrile are shown in Figure 6. Ferrocene exhibits two distinct absorption bands, at 322 nm with a very low molar extinction coefficient (ε) of 61 L mol–1 cm–1 and at 442 nm giving the typical orange color of the complex with ε = 95 L mol–1 cm–1. The optical absorption spectrum of the ferricinium counterpart has not been determined because of fast decomposition in the solution under ambient conditions. A stronger and broad absorption band deeper in UV is also visible with a maximum at 205 nm (ε ≈ 50 000 L mol–1 cm–1). This latter corresponds to an allowed π–π* transition in the Cp-rings because of its high oscillator strength (f ≈ 0.77) in analogy with the absorption fingerprint of benzene molecules.30 It has also some ligand-to-metal charge transfer (LMCT) characters in the red part of this band, explaining its broadness and the molar extinction coefficient dropping in the range of 8000 L mol–1 cm–1.31 The origin of the electronic transitions at 332 nm is still not clear from the literature.32b−32d Indeed, it seems that the origin is not entirely a symmetry-forbidden d–d transition which occurred because of the vibrational distortion of the molecular symmetry as one could expect.32e Indeed, Becker et al. reported that the position of this band is independent of temperature, whereas sharpening and shifting of the band maximum with a decrease in temperature would be expected in this case.30b Our results show a red-shift and intensity increase of this band by the introduction of one methyl or two tert-butyl units in each cyclopentadienyl ring. The introduction of further methyl units leads even to a disappearance of this band or becoming curtained off by the broadening and red-shifting of the π–π* transition of Cp-rings and LMCT contribution. Consequently, this gives the credit that molecular orbitals of the rings are involved in this electronic transition at 322 nm, in agreement with Becker et al.30b This transition is thus assigned either to a n−π* type symmetry-allowed transition of the type 3d-MO* or alternatively to a symmetry-forbidden π–π* transition. The band situated in the visible range at 442 nm is assigned to a forbidden d–d transition localized on the iron metal center. It is only slightly affected by the substitution on the cyclopentadienyl rings. Indeed, all ferrocene derivatives have λmax at a very similar region between 442 and 423 nm, except for tBu4Fc which has λmax at 462 nm. The introduction of methyl groups in the Cp core shifts therefore this optical transition to blue, signifying symmetry modification by the substitution with more localized d–d transitions on iron. The molar extinction coefficients for all the ferrocene derivatives are comparatively identical (ε ≈ 96 L mol–1 cm–1) except for tBu4Fc which has a slightly higher molar extinction coefficient (ε ≈ 105 L mol–1 cm–1). Considering the low molar extinction coefficient which is due to the spin forbidden d–d transition, all these ferrocene derivatives absorb a very negligible amount of light in the visible region.
Figure 6.

UV–visible absorption spectra of the different (a) ferrocene and derivatives and (b) ferricinium derivatives in acetonitrile at a concentration of 2 mmol/L.
Regardless of the derivatives, the chemical oxidation by AgTFSI leads to a color modification of the solution turning green, which translates into a drastic modification in the absorption spectra as shown in Figure 6b. All the oxidized forms absorb part in blue up to ca. 550 nm and a stronger band for which the maximum varies more noticeably in energy from 647 nm for [Me2Fc][TFSI] to 778 nm for [Me10Fc][TFSI]. The molar extinction coefficient is also enhanced by the substitution in the Cp-rings, for example, from 97 L mol–1 cm–1 for [Me2Fc][TFSI] to 488 L mol–1 cm–1 [Me10Fc][TFSI]. This band is ascribed to an allowed LMCT transition (2E1u ← 2E2g) in agreement with the literature.32b−32f The reason this band is more sensitive to substitution when in the ferricinium oxidation state is explained on the basis of the d5-Fe(III) electron configuration which leads to a more compact metal-to-ligand bonding and extended electron delocalization between the metal center and Cp-rings. The substitution in the cyclopentadienyl ring with a number of electron-donating groups, enhances the electron density in the ligand.
The chemical oxidation process for all the ferrocene derivatives was also monitored by UV–vis spectroscopy (see Figures S9–S11) with successive addition of AgTFSI. The UV–vis absorption experiment suggested a very slow oxidation process occurred for Fc → Fc+ with AgTFSI and probably Fc being decomposed as soon as it is getting oxidized as stated elsewhere. While adding the successive amount of AgTFSI to the Me2Fc solution in acetonitrile, it shows slow oxidation of Me2Fc and gradual decomposition indicated by the color change from green to brown. For ferrocene, the oxidation reaction competes with the ferricinium decomposition. This is also the case for Me2Fc to a lower extent for which the absorption value at 653 nm decreases gradually over a few hours because of the possible decomposition of Me2Fc·TFSI in acetonitrile (Figure 7). The chemical oxidation for tBu4Fc, Me8Fc, and Me10Fc is almost instantaneous. The stability of the Fc+ form has been investigated by monitoring in situ the modification of the UV–visible absorption spectrum over a period of 10 h (Figure S12). For this, the absorption value at the highest absorption peak was monitored at intervals of 5 min. The oxidized forms of tBu4Fc, Me8Fc, and Me10Fc show no noticeable change over 10 h demonstrating the higher stability against the oxygen reaction in good agreement with the improved reversibility in electrochemistry upon cycling (Figure 7).
Figure 7.

In situ evolution of the UV–visible absorption spectrum of 2 mmol/L solution of Me2Fc, tBu4Fc, Me8Fc, and Me10Fc oxidation with AgTFSI in acetonitrile and stability over 10 h ageing in ambient conditions. Each spectrum was recorded at 5 min intervals. The evolution of the absorption band maximum as a function of time is reported in the inset.
Magnetic Properties
The room-temperature M(H) hysteresis loops of Fc, Me2Fc, tBu4Fc, Me8Fc, and Me10Fc ferrocene derivatives are shown in Figure 8. Figures S13–S15 show an example of the hysteresis loop measured at 20 K. A negative slope is measured for all ferrocene derivatives indicating a diamagnetic behavior and the absence of unpaired electrons. The absence of unpaired electrons is in agreement with the six electrons occupying the t2g levels of the d-orbitals in Fe+II ions. As expected, the ferricinium derivatives exhibit a different magnetic behavior within the whole range of temperature from 300 K down to 20 K (Figure 8b–d). The linear and slight hysteresis loop is the signature of a paramagnetic behavior induced by the one unpaired electron of the Fe+III metal center. In order to extract quantitative information, magnetic susceptibilities of the three oxidized compounds were estimated from the slope of the M(H) hysteresis loops. The susceptibilities were also corrected for the overall diamagnetism of the investigated oxidized substances with an approximation given by χD = −MW/2 × 10–6 emu g–1; MW denotes the molecular weight of the molecule.33 The temperature dependence of the susceptibilities is linear with a Curie–Weiss behavior (χ = C/(T – θ)) without any hint of a magnetic phase transition. Figure 7 shows χ–1 and the extrapolation to T = 0 K of χ–1 gives positive values to θ reminiscent of ferromagnetic interactions between the spins (see Figures S13–S15). Effective magnetic moments were also deduced for the three oxidized derivatives μeff = 2.823(χ × T)1/2; see Miller et al. and Gray et al. for a discussion on magnetic properties and ordering in ferrocene-based complexes.32 The expected moment value for a single spin is 1.73 μB. A deviation is observed for the obtained values in agreement with previous investigations on closely related molecules (Figure 8b).34 Indeed, the obtained values depend on temperature and are in the range of 2.15–2.25 μB for tBu4Fc·TFSI and 1.95–2.20 μB for Me8Fc·TFSI and Me10Fc·TFSI, respectively. According to Miller et al. and Reiners et al. deviation to the spin only value and temperature dependence of the effective magnetic moment is attributed to electronic excitations to higher levels, spin–orbit coupling and lower symmetry (Figure 9).32,34
Figure 8.

(a) Diamagnetic loops of the three ferrocene derivatives tBu4Fc, Me8Fc, and Me10Fc at 300 K and (b–d) paramagnetic loops as a function of temperature for tBu4FcTFSI, Me8FcTFSI, and Me10FcTFSI.
Figure 9.

(a) Inverse magnetic susceptibility and (b) effective magnetic moment for the oxidized ferrocene derivatives (T = [TFSI]).
Conclusions
A series of ferrocene-based compounds and their corresponding oxidized derivatives were synthesized and characterized by optical, electrochemical, and magnetic means. The introduction of electron-donating groups on the Cp-rings affords to lower the redox potential to a greater extent from +0.403 V for Fc+/Fc down to −0.096 V (vs SCE) for Me10Fc+/Me10Fc without drastically increasing the reorganization energy between -red and -ox forms, remaining well below 50 meV showing high structural robustness upon electron transfer. This modification offers a substantial enhancement in the chemical/electrochemical stability of the oxidized forms in acetonitrile under ambient conditions. The introduction of methyl or tert-butyl groups on Cp-rings hampers the complex decomposition against oxygen reaction. Finally, our results show that the chemical modification of the Cp-ring leads to a slight blue-shift of the main absorption band for the reduced forms related to the spin forbidden d–d transition. However, a significant red-shift and molar extinction coefficient enhancement occurred for the oxidized forms related to the LMCT band.
Experimental Section
Materials
All solvents were purchased from Sigma-Aldrich and were of reagent grade except for dried tetrahydrofuran (THF) which was at 99.9% purity. Ferrocene (Fc) and 1,1′-dimethylferrocene (Me2Fc) were purchased from Fisher Scientific. Bis(η5-pentamethylcyclopentadienyl)iron(II) (Me10Fc) was purchased from Acros Organics. AgTFSI, 2,3,4-tetramethyl-1,3-cyclopentadiene, 1,2,3,4-tetramethyl-1,3-cyclopentadiene,1,3-ditertiarybutylcyclopetadiene, 1.9 mol/L t-BuLi in pentane, and 3 mol/L n-BuLi solution in hexane were purchased from Sigma-Aldrich. 1H, and 13C NMR spectra were recorded on a Bruker 400 MHz ADVANCE III HD spectrometer at 400 and 100 MHz, respectively. When necessary, assignments of 1H and 13C signals were performed using correlation spectroscopy and heteronuclear single quantum correlation. High-resolution electrospray mass spectra (HRMS) in the positive ion mode were obtained on a Q-Tof Ultima Global hybrid quadrupole/time-of-flight instrument.
Synthesis of Me8Fc
2.47 mL (2 g, 16.35 mmol) of 2,3,4,5-tetra methylcyclopentadienyl was taken in 20 mL THF under Ar and at 0 °C. 10 mL (18 mmol) of n-BuLi (1.9 mol/L in pentane) was slowly added to the reaction mixture over a period of 30 min in agreement with the literature.14 A white slurry was obtained at the end of n-BuLi addition. The reaction was further allowed to stir for another 30 min. 1.28 g (7.89 mmol) of FeCl2 was then added to this slurry followed by gentle heating of the reaction mixture to room temperature (20 °C). The stirring was continued for another 1 h at room temperature under Ar. The reaction mixture was then quenched by the dropwise addition of ice-cold water. Once quenched, 50 mL of extra water was added. THF was evaporated by a rotary evaporator followed by the solvent extraction of the aqueous reaction mixture with hexane (100 mL × 3). The organic layer was dried over MgSO4 and a yellow-color product was collected by evaporating the solvent with a yield of 1.9 g (6.37 mmol, 81%).
Synthesis of tBu4Fc
tBu4Fc was synthesized following rigorously the same synthetic procedure as for Me8Fc in agreement with the literature.14 2 mL (1.67 g, 9.37 mmol) of bis(tertiarybutyl)cyclopentadienyl, 3.70 mL (9.37 mmol) of n-BuLi (2.5 M in hexane), and 593.19 mg of FeCl2 were taken for the reaction. Yield: 3 g (7.49 mmol, 80%).
Synthesis and Structural Characterization of Fc, Me2Fc, Me8Fc, Me10Fc, and tBu4Fc
Fc, Me2Fc and Me10Fc are commercially available. Me8Fc and tBu4Fc were synthesized. Each ferrocene derivative was fully characterized by their melting points, NMR and high-resolution mass spectrometry and results are gathered in ESI materials.
Oxidation Procedure for [Me2Fc][TFSI], [tBu4Fc][TFSI], [Me8Fc][TFSI], and [Me10Fc][TFSI]
200 mg of each ferrocene derivatives were reacted with one equivalent of AgTFSI (silver trifluorosulfone-imide) in 20 mL of acetone under ambient conditions.13b,15 The reaction mixture turned instantly from yellow to green. However, chemical oxidation was maintained under stirring for another 30 min at RT. The reaction mixture was then filtered through a 1 in. silica pad in order to remove the Ag particles. The crude product was collected by evaporating the solvent. The latter was further washed with hexane (10 mL × 3) to remove any unreacted ferrocene derivatives. Finally, the desired pure oxidized product was collected and dried.
Electrochemistry
All electrochemical experiments were performed using a potentiostat/galvanostat VMP3 (BioLogic Science Instruments). 100 mmol/L of TBAPF6 was used as a supporting electrolyte and acetonitrile as a solvent. A 3 mm diameter glassy carbon electrode (CHI instrument) was used as a working electrode, a platinum wire as a counter electrode and a SCE as a reference electrode. All electrochemical experiments were systematically carried out using 2 mM solution the desired compound.
Absorption Spectroscopy and Kinetic Study
The UV–visible absorption spectra were recorded using an Agilent Cary 5000 UV–vis–NIR spectrophotometer in acetonitrile. Spectra were recorded in the range of 300–850 nm for the kinetic study over a period of 10 h with intervals of 5 min.
Magnetic Properties
The magnetic properties were investigated using a Cryogen Free Quantum Design system equipped with a vibrating sample magnetometer. M(H) hysteresis loops were measured under a field of up to 1 T and from 300 K down to 20 K.
Computational Details
Equilibrium geometries of all molecules in their neutral and cationic states have been optimized using DFT with the B3PW91 hybrid functional and an Ahlrichs’ def2-TZVP full electron basis for all atoms including the iron center. This combination of the functional and basis set has already proven to give reliable estimates of geometry parameters for metallocenes.16 Normal modes of vibration of Fc, Me2Fc, and Me8Fc have been computed at the same level of theory. The displacement of normal modes dn have been obtained by projecting the Cartesian displacements onto the direction of normal modes by the relation17
where ω is the diagonal matrix of the vibrational wavenumbers in γ = 2πωc/ℏ, L is the rectangular matrix of normal modes of the neutral state expressed as linear combinations of Cartesian displacements, and xN/C0 are the equilibrium coordinates of the molecules in their neutral and cation states, respectively.
Acknowledgments
F.S. wishes to acknowledge the financial support of the ANR “VISION-NIR” under the grant agreement ANR-17-CE05-0037-01 and H2020 “IMPRESSIVE” LC-SC3-RES-2-2018 under the grant agreement number 826013. F.S. wishes also to thanks Dr. David Mathiron for HRMS measurements and Dr. Gwladys Pourceau for NMR analysis, reading, and discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01341.
Structural characterization of Fc, Me2Fc, Me8Fc, Me10Fc, and t-Bu4Fc, dependence of the redox potential as a function of the Hammett constant for the different ferrocene derivatives, CV at different scan rates from 20 to 1000 mV s–1 of 2 mmol/L Me2Fc, Me8Fc, Me10Fc, Bu4FcTFSI, Me8FcTFSI, and Me10FcTFSI in acetonitrile and corresponding linear dependence of the faradaic anodic and cathodic peak current as a function of square root of scan rate, cyclic voltamograms of different ferrocene molecules and change after successive addition of AgTFSI, monitoring chemical oxidation of Me2Fc, tBu4Fc, and Me10Fc with successive manual addition of AgTFSI using UV–visible spectroscopy, in situ evolution of the UV–visible absorption spectrum of 2 mmol/L solution of Fc, oxidation with AgTFSI in acetonitrile and stability over 10 h ageing in ambient conditions, hysteresis loops of the tBu4Fc, Me8Fc, and Me10Fc derivatives from 300 K down to 20 K (PDF)
Author Contributions
AP carried out all the synthesis, characterizations of ferrocenes, absorption properties, kinetic studies, electrochemistry, associated data processing, manuscript writing, results interpreting, and partly contributed in magnetic measurement. R.B. carried out all the calculations related to reorganization energies for ferrocene molecules. H.B. carried out the magnetic study, associated data processing, and manuscript writing. S.G. contributed in assistance for synthesis and manuscript correction. F.S. contributed in experiment design, planning, in result interpreting, manuscript writing, and correcting.
The authors declare no competing financial interest.
Supplementary Material
References
- a Astruc D. Why is Ferrocene so Exceptional?. Eur. J. Inorg. Chem. 2016, 6–29. 10.1002/ejic.201600983. [DOI] [Google Scholar]; b Werner H. At Least 60 Years of Ferrocene: The Discovery and Rediscovery of the Sandwich Complexes. Angew. Chem., Int. Ed. 2012, 51, 6052–6058. 10.1002/anie.201201598. [DOI] [PubMed] [Google Scholar]
- a Kealy T. J.; Pauson P. L. A New Type of Organo-Iron Compound. Nature 1951, 168, 1039. 10.1038/1681039b0. [DOI] [Google Scholar]; b Miller S. A.; Tebboth J. A.; Tremaine J. F. 114. Dicyclopentadienyliron. J. Chem. Soc. 1952, 632–635. 10.1039/jr9520000632. [DOI] [Google Scholar]
- Bhattacharjee A.; Rooj A.; Roy D.; Roy M. Thermal Decomposition Study of Ferrocene [(C5H5)2Fe]. J. Exp. Phys. 2014, 2014, 1. 10.1155/2014/513268. [DOI] [Google Scholar]
- a Pauson P. L.; Sandhu M. A.; Watts W. E.; Haley R. C.; Knox G. R. Ferrocene derivatives. Part XVIII. The aminomethylation of methyl- and dimethyl-ferrocene. J. Chem. Soc. C 1967, 1851–1853. 10.1039/j39670001851. [DOI] [Google Scholar]; b Batterjee S. M.; Marzouk M. I.; Aazab M. E.; El-Hashash M. A. The electrochemistry of some ferrocene derivatives: redox potential and substituent effects. Appl. Organomet. Chem. 2003, 17, 291–297. 10.1002/aoc.414. [DOI] [Google Scholar]; c Silva M. E. N. P. R. A.; Pombeiro A. J. L.; Herrmann R.; Deus N.; Bozak R.; Bozak R. Redox potential and substituent effects in ferrocene derivatives: II. J. Organomet. Chem. 1994, 480, 81–90. 10.1016/0022-328x(94)87105-1. [DOI] [Google Scholar]
- a Colacot T. J. A Concise Update on the Applications of Chiral Ferrocenyl Phosphines in Homogeneous Catalysis Leading to Organic Synthesis. Chem. Rev. 2003, 103, 3101–3118. 10.1021/cr000427o. [DOI] [PubMed] [Google Scholar]; b Barbaro P.; Bianchini C.; Giambastiani G.; Parisel S. L. Progress in stereoselective catalysis by metal complexes with chiral ferrocenyl phosphines. Coord. Chem. Rev. 2004, 248, 2131–2150. 10.1016/j.ccr.2004.03.022. [DOI] [Google Scholar]
- Connelly N. G.; Geiger W. E. Chemical Redox Agents for Organometallic Chemistry. Chem. Rev. 1996, 96, 877–910. 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- a Richardson D. E.; Taube H. Mixed-valence molecules: Electronic delocalization and stabilization. Coord. Chem. Rev. 1984, 60, 107–129. 10.1016/0010-8545(84)85063-8. [DOI] [Google Scholar]; b Launay J.-P. Electron transfer in molecular binuclear complexes and relation with electron transport through nanojunctions. Coord. Chem. Rev. 2013, 257, 1544–1554. 10.1016/j.ccr.2012.09.005. [DOI] [Google Scholar]; c Ceccon A.; Santi S.; Orian L.; Bisello A. Electronic communication in heterobinuclear organometallic complexes through unsaturated hydrocarbon bridges. Coord. Chem. Rev. 2004, 248, 683–724. 10.1016/j.ccr.2004.02.007. [DOI] [Google Scholar]; d Murata M.; Yamada M.; Fujita T.; Kojima K.; Kurihara M.; Kubo K.; Kobayashi Y.; Nishihara H. Structural Conversion and Spin Separation in Bis(ferrocenylethynyl)anthraquinones Triggered by Proton-Coupled Intramolecular Electron Transfer. J. Am. Chem. Soc. 2001, 123, 12903–12904. 10.1021/ja0113504. [DOI] [PubMed] [Google Scholar]
- a Chauvin Y. Olefin Metathesis: The Early Days (Nobel Lecture). Angew. Chem., Int. Ed. 2006, 45, 3740–3747. 10.1002/anie.200601234. [DOI] [PubMed] [Google Scholar]; b Studer A.; Curran D. P. The electron is a catalyst. Nat. Chem. 2014, 6, 765. 10.1038/nchem.2031. [DOI] [PubMed] [Google Scholar]; c Saveant J. M. Catalysis of chemical reactions by electrodes. Acc. Chem. Res. 1980, 13, 323–329. 10.1021/ar50153a005. [DOI] [Google Scholar]; d Bunnett J. F.; Zahler R. E. Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 1951, 49, 273–412. 10.1021/cr60153a002. [DOI] [Google Scholar]; e Foo K.; Sella E.; Thomé I.; Eastgate M. D.; Baran P. S. A Mild, Ferrocene-Catalyzed C–H Imidation of (Hetero)Arenes. J. Am. Chem. Soc. 2014, 136, 5279–5282. 10.1021/ja501879c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Foucher D. A.; Tang B. Z.; Manners I. Ring-opening polymerization of strained, ring-tilted ferrocenophanes: a route to high-molecular-weight poly(ferrocenylsilanes). J. Am. Chem. Soc. 1992, 114, 6246–6248. 10.1021/ja00041a053. [DOI] [Google Scholar]; b Ni Y.; Rulkens R.; Manners I. Transition Metal-Based Polymers with Controlled Architectures: Well-Defined Poly(ferrocenylsilane) Homopolymers and Multiblock Copolymers via the Living Anionic Ring-Opening Polymerization of Silicon-Bridged [1]Ferrocenophanes. J. Am. Chem. Soc. 1996, 118, 4102–4114. 10.1021/ja953805t. [DOI] [Google Scholar]; c Tanabe M.; Vandermeulen G. W. M.; Chan W. Y.; Cyr P. W.; Vanderark L.; Rider D. A.; Manners I. Photocontrolled living polymerizations. Nat. Mater. 2006, 5, 467. 10.1038/nmat1649. [DOI] [PubMed] [Google Scholar]; d Rulkens R.; Lough A. J.; Manners I.; Lovelace S. R.; Grant C.; Geiger W. E. Linear Oligo(ferrocenyldimethylsilanes) with between Two and Nine Ferrocene Units: Electrochemical and Structural Models for Poly(ferrocenylsilane) High Polymers. J. Am. Chem. Soc. 1996, 118, 12683–12695. 10.1021/ja962470s. [DOI] [Google Scholar]; e Boott C. E.; Lunn D. J.; Manners I. Versatile and controlled functionalization of polyferrocenylsilane-b-polyvinylsiloxane block copolymers using a N-hydroxysuccinimidyl ester strategy. J. Polym. Sci., Part A: Polym. Chem. 2015, 54, 245–252. 10.1002/pola.27727. [DOI] [Google Scholar]; f Heilmann J. B.; Scheibitz M.; Qin Y.; Sundararaman A.; Jäkle F.; Kretz T.; Bolte M.; Lerner H.-W.; Holthausen M. C.; Wagner M. A Synthetic Route to Borylene-Bridged Poly(ferrocenylene)s. Angew. Chem., Int. Ed. 2006, 45, 920–925. 10.1002/anie.200503515. [DOI] [PubMed] [Google Scholar]; g Alkan A.; Natalello A.; Wagner M.; Frey H.; Wurm F. R. Ferrocene-Containing Multifunctional Polyethers: Monomer Sequence Monitoring via Quantitative 13C NMR Spectroscopy in Bulk. Macromolecules 2014, 47, 2242–2249. 10.1021/ma500323m. [DOI] [Google Scholar]; h Buchmeiser M. R. Homogeneous Metathesis Polymerization by Well-Defined Group VI and Group VIII Transition-Metal Alkylidenes: Fundamentals and Applications in the Preparation of Advanced Materials. Chem. Rev. 2000, 100, 1565–1604. 10.1021/cr990248a. [DOI] [PubMed] [Google Scholar]
- a Casado C. M.; Cuadrado I.; Morán M.; Alonso B.; García B.; González B.; Losada J. Redox-active ferrocenyl dendrimers and polymers in solution and immobilised on electrode surfaces. Coord. Chem. Rev. 1999, 185–186, 53–80. 10.1016/s0010-8545(98)00252-5. [DOI] [Google Scholar]; b Astruc D. Electron-transfer processes in dendrimers and their implication in biology, catalysis, sensing and nanotechnology. Nat. Chem. 2012, 4, 255. 10.1038/nchem.1304. [DOI] [PubMed] [Google Scholar]; c Ornelas C.; Ruiz J.; Belin C.; Astruc D. Giant Dendritic Molecular Electrochrome Batteries with Ferrocenyl and Pentamethylferrocenyl Termini. J. Am. Chem. Soc. 2009, 131, 590–601. 10.1021/ja8062343. [DOI] [PubMed] [Google Scholar]
- a Long B.; Liang S.; Xin D.; Yang Y.; Xiang J. Synthesis, characterization and in vitro antiproliferative activities of new 13-cis-retinoyl ferrocene derivatives. Eur. J. Med. Chem. 2009, 44, 2572–2576. 10.1016/j.ejmech.2009.01.029. [DOI] [PubMed] [Google Scholar]; b Jaouen G.; Vessières A.; Top S. Ferrocifen type anti cancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. 10.1039/c5cs00486a. [DOI] [PubMed] [Google Scholar]
- a Ding Y.; Zhao Y.; Yu G. A Membrane-Free Ferrocene-Based High-Rate Semiliquid Battery. Nano Lett. 2015, 15, 4108–4113. 10.1021/acs.nanolett.5b01224. [DOI] [PubMed] [Google Scholar]; b Park K.-S.; Schougaard S. B.; Goodenough J. B. Conducting-Polymer/Iron-Redox- Couple Composite Cathodes for Lithium Secondary Batteries. Adv. Mater. 2007, 19, 848–851. 10.1002/adma.200600369. [DOI] [Google Scholar]; c Beladi-Mousavi S. M.; Sadaf S.; Walder L.; Gallei M.; Rüttiger C.; Eigler S.; Halbig C. E. Poly(vinylferrocene)-Reduced Graphene Oxide as a High Power/High Capacity Cathodic Battery Material. Adv. Energy Mater. 2016, 6, 1600108. 10.1002/aenm.201600108. [DOI] [Google Scholar]
- a Ponseca C. S.; Chábera P.; Uhlig J.; Persson P.; Sundström V. Ultrafast Electron Dynamics in Solar Energy Conversion. Chem. Rev. 2017, 117, 10940. 10.1021/acs.chemrev.6b00807. [DOI] [PubMed] [Google Scholar]; b Daeneke T.; Mozer A. J.; Kwon T.-H.; Duffy N. W.; Holmes A. B.; Bach U.; Spiccia L. Dye regeneration and charge recombination in dye-sensitized solar cells with ferrocene derivatives as redox mediators. Energy Environ. Sci. 2012, 5, 7090. 10.1039/c2ee21257a. [DOI] [Google Scholar]; c Daeneke T.; Kwon T.-H.; Holmes A. B.; Duffy N. W.; Bach U.; Spiccia L. High-efficiency dye-sensitized solar cells with ferrocene-based electrolytes. Nat. Chem. 2011, 3, 211. 10.1038/nchem.966. [DOI] [PubMed] [Google Scholar]
- a Petrov A. R.; Derheim A.; Oetzel J.; Leibold M.; Bruhn C.; Scheerer S.; Oßwald S.; Winter R. F.; Siemeling U. A Stable Planar-Chiral N-Heterocyclic Carbene with a 1,1′-Ferrocenediyl Backbone. Inorg. Chem. 2015, 54, 6657–6670. 10.1021/acs.inorgchem.5b01064. [DOI] [PubMed] [Google Scholar]; b Roemer M.; Skelton B. W.; Piggott M. J.; Koutsantonis G. A. 1,1′-Diacetyloctamethylferrocene: an overlooked and overdue synthon leading to the facile synthesis of an octamethylferrocenophane. Dalton Trans. 2016, 45, 18817–18821. 10.1039/c6dt03820d. [DOI] [PubMed] [Google Scholar]
- Mochida T.; Funasako Y.; Ishida M.; Saruta S.; Kosone T.; Kitazawa T. Crystal Structures and Phase Sequences of Metallocenium Salts with Fluorinated Anions: Effects of Molecular Size and Symmetry on Phase Transitions to Ionic Plastic Crystals. Chem.—Eur. J. 2016, 22, 15725–15732. 10.1002/chem.201603170. [DOI] [PubMed] [Google Scholar]
- Latouche C.; Palazzetti F.; Skouteris D.; Barone V. High-Accuracy Vibrational Computations for Transition-Metal Complexes Including Anharmonic Corrections: Ferrocene, Ruthenocene, and Osmocene as Test Cases. J. Chem. Theory Comput. 2014, 10, 4565–4573. 10.1021/ct5006246. [DOI] [PubMed] [Google Scholar]
- a Borrelli R.; Peluso A. Elementary electron transfer reactions: from basic concepts to recent computational advances. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2013, 3, 542–559. 10.1002/wcms.1147. [DOI] [Google Scholar]; b Borrelli R.; Peluso A. Quantum Dynamics of Radiationless Electronic Transitions Including Normal Modes Displacements and Duschinsky Rotations: A Second-Order Cumulant Approach. J. Chem. Theory Comput. 2015, 11, 415–422. 10.1021/ct500966c. [DOI] [PubMed] [Google Scholar]
- a Kuhn N.; Jendral K.; Boese R.; Bläser D. Heterocycles as Ligands, X1). — 2,2′,5,5′-Tetra-tert-butyl-1,1′-diazaferrocene – Stabilization of the Diheterometallocene Structure by Steric Shielding. Chem. Ber. 1991, 124, 89. 10.1002/cber.19911240115. [DOI] [Google Scholar]; b Janiak C.; Kuhn N.; Gleiter R. Effects of isolobal substitution in cyclopentadienyl ligands: the azacyclopentadienyl system C,R,N. Comparative photoelectron spectra of the tetra-tert-butyl ferrocene derivatives. J. Organomet. Chem. 1994, 475, 223–227. 10.1016/0022-328x(94)84025-3. [DOI] [Google Scholar]
- Leo A.Substituent Constants for Correlation Analysis in Chemistry and Biology; Wiley-Interscience: NY, 1979. [Google Scholar]
- Ates M. N.; Allen C. J.; Mukerjee S.; Abraham K. M. Electronic Effects of Substituents on Redox Shuttles for Overcharge Protection of Li-ion Batteries. J. Electrochem. Soc. 2012, 159, A1057–A1064. 10.1149/2.064207jes. [DOI] [Google Scholar]
- Hwang B.; Park M.-S.; Kim K. Ferrocene and Cobaltocene Derivatives for Non-Aqueous Redox Flow Batteries. ChemSusChem 2015, 8, 310–314. 10.1002/cssc.201403021. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Rogers E. I.; Compton R. G. The measurement of the diffusion coefficients of ferrocene and ferrocenium and their temperature dependence in acetonitrile using double potential step microdisk electrode chronoamperometry. J. Electroanal. Chem. 2010, 648, 15–19. 10.1016/j.jelechem.2010.07.006. [DOI] [Google Scholar]
- Horsfield A.; Wassermann A. Electron spin resonance spectra of ferricinium. J. Chem. Soc., Dalton Trans. 1972, 187–188. 10.1039/dt9720000187. [DOI] [Google Scholar]
- Hurvois J. P.; Moinet C. Reactivity of ferrocenium cations with molecular oxygen in polar organic solvents: Decomposition, redox reactions and stabilization. J. Organomet. Chem. 2005, 690, 1829–1839. 10.1016/j.jorganchem.2005.02.009. [DOI] [Google Scholar]
- a Buyukserin F.; Martin C. R. Investigation of Ferricinium Stability Inside the Constrained Geometry of Gold Nanotube Membranes via the Utilization of Argon Plasma. Electrochim. Acta 2016, 188, 619–624. 10.1016/j.electacta.2015.12.020. [DOI] [Google Scholar]; b Nesmeyanov A. N.; Materikova R. B.; Getnarski B. Izv. Akad. Nauk SSSR, Ser. Khim. 1965, 4, 731–733. [Google Scholar]; c Popenoe D. D.; Deinhammer R. S.; Porter M. D. Infrared spectroelectrochemical characterization of ferrocene-terminated alkanethiolate monolayers at gold. Langmuir 1992, 8, 2521–2530. 10.1021/la00046a028. [DOI] [Google Scholar]; d Abbott N. L.; Whitesides G. M. Potential-Dependent Wetting of Aqueous Solutions on Self-Assembled Monolayers Formed from 15-(Ferrocenylcarbonyl)pentadecanethiol on Gold. Langmuir 1994, 10, 1493–1497. 10.1021/la00017a029. [DOI] [Google Scholar]
- Chidsey C. E. D. Free Energy and Temperature Dependence of Electron Transfer at the Metal-Electrolyte Interface. Science 1991, 251, 919. 10.1126/science.251.4996.919. [DOI] [PubMed] [Google Scholar]
- Hagfeldt A.; Boschloo G.; Sun L.; Kloo L.; Pettersson H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. 10.1021/cr900356p. [DOI] [PubMed] [Google Scholar]
- Liu Y.-P.; Newton M. D. Reorganization Energy for Electron Transfer at Film-Modified Electrode Surfaces: A Dielectric Continuum Model. J. Phys. Chem. 1994, 98, 7162–7169. 10.1021/j100080a011. [DOI] [Google Scholar]
- Ghosh S.; Horvath S.; Soudackov A. V.; Hammes-Schiffer S. Electrochemical Solvent Reorganization Energies in the Framework of the Polarizable Continuum Model. J. Chem. Theory Comput. 2014, 10, 2091–2102. 10.1021/ct500051e. [DOI] [PubMed] [Google Scholar]
- a Platt J. R.; Klevens H. B. Absolute Absorption Intensities of Alkylbenzenes in the 2250-1700 A. Region. Chem. Rev. 1947, 41, 301–310. 10.1021/cr60129a009. [DOI] [PubMed] [Google Scholar]; b Scott D. R.; Becker R. S. Comprehensive Investigation of the Electronic Spectroscopy and Theoretical Treatments of Ferrocene and Nickelocene. J. Chem. Phys. 1961, 35, 516–531. 10.1063/1.1731962. [DOI] [Google Scholar]; c Maki G. Ligand Field Theory of Ni(II) Complexes. II. Electronic Spectra and Structure of Some Paramagnetic Chelates. J. Chem. Phys. 1958, 29, 162–172. 10.1063/1.1744417. [DOI] [Google Scholar]
- Fery-Forgues S.; Delavaux-Nicot B. Ferrocene and ferrocenyl derivatives in luminescent systems. J. Photochem. Photobiol., A 2000, 132, 137–159. 10.1016/s1010-6030(00)00213-6. [DOI] [Google Scholar]
- a Miller J. S.; Epstein A. J.; Reiff W. M. Ferromagnetic molecular charge-transfer complexes. Chem. Rev. 1988, 88, 201–220. 10.1021/cr00083a010. [DOI] [Google Scholar]; b Gray H. B.; Hendrickson D. N.; Sohn Y. S. Magnetic susceptibility study of various ferricenium and iron(III) dicarbollide compounds. Inorg. Chem. 1971, 10, 1559–1563. 10.1021/ic50102a001. [DOI] [Google Scholar]; c Rowe M. D.; Gale R.; McCaffery A. J. Distortion parameters of the ferricenium ion from low temperature absorption, MCD and ESR spectra. Chem. Phys. Lett. 1973, 21, 360–362. 10.1016/0009-2614(73)80157-5. [DOI] [Google Scholar]; d Sohn Y. S.; Hendrickson D. N.; Gray H. B. Electronic structure of ferricenium ion. J. Am. Chem. Soc. 1970, 92, 3233–3234. 10.1021/ja00713a079. [DOI] [Google Scholar]; e Hendrickson D. N.; Sohn Y. S.; Duggan D. M.; Gray H. B. Low-temperature (4.2°K) study of the 2E1u ←2E2g band system in the electronic spectra of various ferricenium compounds. J. Chem. Phys. 1973, 58, 4666–4675. 10.1063/1.1679029. [DOI] [Google Scholar]; f Duggan D. M.; Hendrickson D. N. Electronic structure of various ferricenium systems as inferred from Raman, Infrared, low-temperature electronic absorption, and electron paramagnetic resonance measurements. Inorg. Chem. 1975, 14, 955–970. 10.1021/ic50147a001. [DOI] [Google Scholar]
- Bain G. A.; Berry J. F. Diamagnetic Corrections and Pascal’s Constants. J. Chem. Educ. 2008, 85, 532–536. 10.1021/ed085p532. [DOI] [Google Scholar]
- Reiners M.; Baabe D.; Schweyen P.; Freytag M.; Jones P. G.; Walter M. D. Teaching Ferrocenium How to Relax: A Systematic Study on Spin–Lattice Relaxation Processes in tert-Butyl-Substituted Ferrocenium Derivatives. Eur. J. Inorg. Chem. 2017, 388–400. 10.1002/ejic.201600873. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.