

SUMMARY
Deficiency in DNA double-strand break (DSB) repair mechanisms has been widely exploited for the treatment of different malignances, including homologous recombination (HR)-deficient breast and ovarian cancers. Here we demonstrate that diffuse large B cell lymphomas (DLBCL) expressing LMO2 protein are functionally deficient in HR-mediated DSB repair. Mechanistically, LMO2 inhibits BRCA1 recruitment to DSBs by interacting with 53BP1 during repair. Similar to BRCA1-deficient cells, LMO2 positive DLBCL and T cell acute lymphoblastic leukemia (T-ALL) cells exhibit a high sensitivity to Poly (ADP-ribose) polymerase (PARP) inhibitors. Further, chemotherapy and PARP inhibitors synergize to inhibit the growth of LMO2 positive tumors. Together, our results reveal that LMO2 expression predicts HR-deficiency and the potential therapeutic utility of PARP inhibitors in DLBCL and T-ALL.
Keywords: Diffuse Large B Cell Lymphoma (DLBCL), LMO2, DNA damage, Homologous recombination, 53BP1, BRCA1, PARP, Olaparib, Synthetic lethality, RCHOP
Graphical Abstract:
INTRODUCTION
LIM-domain only 2 (LMO2) is a cysteine-rich protein containing two zinc-binding LIM domains. LMO2 is expressed in all tissues with the exception of mature T cells (Neale et al., 1995) and is implicated in angiogenesis, hematopoiesis, and hematopoietic stem cell maintenance (Chambers and Rabbitts, 2015; Nam and Rabbitts, 2006). In T cells, LMO2 is expressed up to the stage of immature CD4/CD8 double-negative thymocytes and is turned off thereafter (Chambers and Rabbitts, 2015; Nam and Rabbitts, 2006). Aberrant expression of LMO2 in mature T cells induces T cell leukemogenesis; indeed, LMO2 is one of the most frequently deregulated genes in T-ALL (Chambers and Rabbitts, 2015; Hirose et al., 2010; McCormack and Rabbitts, 2004; Van Vlierberghe et al., 2006).
We have reported that in normal B cells, the expression of LMO2 protein is specifically up-regulated in germinal centers (GC) (Lossos et al., 2004; Natkunam et al., 2007). GCs are morphologic and functional structures within secondary lymphoid organs in which B cell responses to antigens are amplified and refined in specificity. This is accomplished via repeated rounds of B cell proliferation, somatic hypermutation and rearrangement of the immunoglobulin genes to generate high affinity antigen-specific antibodies. To allow for somatic hypermutation and the recombination of the antibody genes in GC B cells, error-free DNA repair pathways such as HR need to be downregulated or inhibited at the IGH locus (Di Virgilio et al., 2013). Due to the cycles of DNA mutation and recombination, GC B cells are believed to be the cell of origin of many subtypes of non-Hodgkin lymphoma (NHL), including diffuse large B cell lymphomas (DLBCL), the most common subtype (Armitage and Weisenburger, 1998; Zelenetz et al., 2016).
DLBCL are genetically heterogeneous tumors. Marked advances in the understanding of DLBCL pathobiology have been made by the application of gene expression arrays, comparative genomic hybridization arrays, and next-generation sequencing (Alizadeh et al., 2000; Lenz et al., 2008a; Lenz et al., 2008b; Morin et al., 2010; Rosenwald et al., 2002). Gene expression array studies lead to the cell of origin (COO) classification identifying GC B cell type (GCB) and activated B cell type (ABC) DLBCLs and providing insights into pathogenesis, prognosis and potential treatment targets. Approximately 73% of GCB and 45% of ABC DLBCL tumors express levels of LMO2 protein similar to that of normal GCB cells (Alizadeh et al., 2000; Malumbres et al., 2008; Natkunam et al., 2008). Despite marked improvement in therapy, about half of DLBCL patients succumb to their disease (Coiffier et al., 2010; Habermann et al., 2006). Therefore, there is a strong need for better therapeutic approaches to improve DLBCL patient survival.
Although the function of LMO2 in B cells and DLBCL is unknown, expression of LMO2 serves as one of the best prognostic markers of longer survival following rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) immunochemotherapy (Alizadeh et al., 2009; Lossos et al., 2004; Malumbres et al., 2008). Additionally, LMO2 expression in DLBCL cells results in genomic instability (Cubedo et al., 2012). This observation, together with the longer survival of LMO2 expressing DLBCL patients treated with genotoxic agents compared to their non-expressing counterparts, suggest that LMO2 may affect DNA repair efficiency and could be therapeutically exploited. To address this question, we investigated the effects of LMO2 on DNA double-strand break (DSB) repair in DLBCL.
RESULTS
LMO2 expression induces the accumulation of DSBs in DLBCL
Using the S139 phosphorylated histone H2AX (γH2AX) as a DSB marker, we first noticed that patient-derived DLBCL expressing high levels of LMO2 protein (LMO2HIGH DLBCL) had more DSBs than tumors with low levels of LMO2 protein (LMO2LOW). This was observed using immunofluorescence (IF) analysis for γH2AX foci in tumor samples with different LMO2 levels (Figure 1A), as well as by Western blot (Figure 1B). This positive correlation between LMO2 expression and γH2AX accumulation was also observed in patient-derived DLBCL cell lines (Figures 1C and 1D).
Figure 1. Expression of LMO2 protein induces the accumulation of DSBs in DLBCL.

(A) Representative immunohistochemistry (IHC) and immunofluorescence (IF) micrographs showing LMO2 and γH2AX expression, respectively, in untreated patient-derived LMO2LOW or LMO2HIGH DLBCL samples. Green scale bars represent 100 μm, black scale bars 50 μm, and white scale bars 10 μm.
(B) Western blots showing LMO2 and γH2AX levels in untreated patient-derived DLBCL samples.
(C) As in (B), for the indicated DLBCL cell lines.
(D) Representative IF micrographs showing γH2AX foci in DLBCL cell lines with and without LMO2 expression (left) and quantification of γH2AX foci per cell in the indicated DLBCL cell lines (right). Example of γH2AX foci are indicated with red arrows. White scale bars represent 1 μm. At least 500 cells analyzed per cell line/experiment in IF experiments. p values from Student’s t tests. Box plots show all points, median and the lower and upper quantile (box). Data obtained from 3 independent experiments.
(E) LMO2 protein levels determined via Western blots (left) and quantification of γH2AX foci per cell (right) in the indicated DLBCL cell lines expressing GFP, GFP-LMO2, shGFP or shLMO2. At least 500 cells analyzed per cell line/experiment. Box plots show all points, median and the lower and upper quantile (box). Data obtained from 3 independent experiments. p values from Student’s t tests.
(F) Representative micrographs showing SYBR Green signal in neutral comet assays for OCI-LY1 expressing indicated proteins (left) and quantification of tail-moment in comet assays (right). White scale bars represent 10 μm. Box plots show all points, median and the lower and upper quantile (box). Data obtained from 3 independent experiments. p value from Student’s t test.
(G) Metaphase spreads showing chromatid breaks in OCI-LY1 cells expressing either GFP or GFP-LMO2 proteins. The percentage of metaphases showing more than one chromatid break in each cell line is shown below the micrographs. p values from Student’s t tests. Data obtained from 3 independent experiments. Scale bars, 1 μm.
We next asked whether high levels of LMO2 protein was sufficient to induce DSB accumulation in DLBCL. Indeed, 48 hr post-induction of a GFP-LMO2 fusion protein via a doxycycline-inducible system in the LMO2LOW DLBCL cell line OCI-LY1 (OCI-LY1 LMO2 cells) we observed a significant increase in DSBs when compared to OCI-LY1 cells expressing only GFP. This increase in DSBs was visualized as an accumulation of γH2AX in IF and Western assays (Figure 1E and not shown), as well as via neutral comet assays and metaphase spreads (Figures 1F and 1G). On the other hand, knockdown of LMO2 in the DLBCL cell line OCI-LY8 resulted in a significant decrease in γH2AX foci (Figure 1E). Together, these findings revealed that high LMO2 expression in DLBCL cells induces the accumulation of DSBs. Since the accumulation of DNA lesions is generally associated with a defect in the DNA repair capacity of the cell, these results suggested that LMO2 expression compromises the activity of one or more DSB repair pathways.
LMO2 inhibits HR activity
DSBs are the most toxic kind of DNA lesion. Although there are at least five DSB repair pathways, the choice of which mechanism will repair the lesion is of paramount importance. Mammalian cells mainly rely on the error-prone non-homologous end-joining (NHEJ) and error-free homologous recombination (HR) pathways to repair DSBs. To determine whether high expression of LMO2 caused a defect in DSB repair pathways in DLBCL cells, we initially evaluated the accumulation of core factors for HR such as BRCA1 and the RAD51 recombinase at ionizing radiation (IR)-induced foci (IRIF) via IF. All DLBCL cell lines studied exhibited a comparable degree of DNA damage after radiation as similar γH2AX IRIF levels were observed (Figures 2A and 2B). However, LMO2HIGH DLBCL cells displayed significantly less BRCA1 and RAD51 IRIF than LMO2LOW DLBCL cells (Figure 2B). Similarly, LMO2HIGH DLBCL cells showed a defect in forming BRCA1 damage foci after exposure to the topoisomerase II inhibitor doxorubicin, the main genotoxic agent used for the treatment of DLBCL (Habermann et al., 2006) (Figure S1A). Importantly, no differences in BRCA1 or RAD51 expression (Figure 2C), or significant differences in cell-cycle phases distribution before or after DNA damage (Figure 2D) were observed between the cell lines studied.
Figure 2. High expression levels of LMO2 protein inhibits BRCA1 and RAD51 damage foci after DNA damage.

(A) Representative immunofluorescence (IF) micrographs showing γH2AX, BRCA1, RAD51 and 53BP1 IRIF in the indicated DLBCL cell lines at 6 hr after ionizing radiation (10 Gy). Insets zoom factor 2X. Scale bar, 5 μm.
(B) Quantification of IRIF in the indicated cell lines 6 hr post-exposure to 10 Gy. *, p<0.001 Student’s t tests. At least 50 cells analyzed per cell line/experiment in IF experiments. Bars represent SD obtained from 3 independent experiments.
(C) Western blot assays for the indicated proteins in DLBCL cells.
(D) Cell cycle analysis via propidium iodide staining before and 6 hr after ionizing radiation (10 Gy).
(E) Quantification of RAD51 IRIF in OCI-LY1 cells expressing GFP or GFP-LMO2 protein and OCI-LY8 cells expressing shGFP or shLMO2. *, p<0.001 Student’s t test. At least 50 cells analyzed per cell line/experiment in IF experiments. Bars represent SD obtained from 3 independent experiments.
(F) Western blots with total or chromatin-enriched extracts from OCI-LY1 cells expressing GFP or GFP-LMO2 protein at 0 and 6 hr post-IR (10 Gy) using the indicated antibodies (left) and quantification of Western blot signal intensities in chromatin-enriched fractions (right). A.U., arbitrary units. SD obtained from 3 independent experiments.
(G) Schematic representation of the structure of the HR reporter substrate DR-GFP and the HR product expressing a functional GFP (left), representative FACS results (middle), and frequency of HR activity (right). The relative HR activity of HEK293T cells expressing LMO2 (LMO2) to that in HEK293T control cells is measured with the DR-GFP reporter after I-SceI transfection. Box plots show all cells, median and the lower and upper quantile (box). Data obtained from 3 independent experiments. p value from Student’s t test.
(H) DLBCL cells grown for 32 hr in the presence of BrdU and 10 nM olaparib (PARPi) were arrested with colcemid, fixed and metaphases processed to visualize individual sister chromatids. SCEs in metaphase chromosomes are indicated with red arrows. The bar graph shows the mean+SD SCEs in the indicated cells obtained from 3 independent experiments. Scale bars, 2 μm. p value from Student’s t tests. At least 50 metaphases analyzed per cell line/experiment.
See also Figure S1.
We next asked whether the deficiency in BRCA1 and RAD51 recruitment to IRIF observed in LMO2HIGH DLBCL cells was due to LMO2. Indeed, shRNA-mediated LMO2 knockdown in LMO2HIGH DLBCL cells led to an increase of RAD51 IRIF levels (Figure 2E). Concordantly, over-expression of LMO2 in LMO2LOW OCI-LY1 cells produced the opposite effect (Figure 2E). Similarly, analysis of chromatin-enriched fractions showed a defective accumulation of BRCA1 and RAD51 at the chromatin after DNA damage in OCI-LY1 LMO2 cells when compared to OCI-LY1 control cells (Figure 2F).
Since the recruitment of RAD51 to the break site depends on a DSB-end resection step to generate 3’ single-stranded DNA tails, we asked whether LMO2 expression affects DSB-end processing in DLBCL. In support of this, analysis of chromatin-enriched fractions revealed that over-expression of LMO2 in LMO2LOW cells decreases the accumulation of phosphorylated (S4 and S8) replication protein A (RPA32 subunit), a marker of single-stranded DNA generated by DSB-end resection (Bunting et al., 2010), in total and chromatin-enriched protein extracts after ionizing radiation (Figure 2F). Overall, these data suggested that high levels of LMO2 inhibit HR activity. To evaluate this, we used two different systems. First, we used the direct repeat-green fluorescent protein (DR-GFP) reporter system (Pierce et al., 1999) in human HEK293T cells. The DR-GFP reporter system allows to measure repair via HR of a DSB produced by the endonuclease I-SceI in a mutated GFP gene. Error-free repair of the lesion leads to restoration of a functional GFP gene, which can be scored by measuring the fraction of GFP-positive cells (Figure 2G). Knockdown of BRCA1 via siRNA was used as a positive control (Figure S1B). In agreement with our previous data, LMO2 expression in HEK293T cells decreased HR activity when compared to control cells (Figure 2G). Furthermore, this result suggested that the inhibitory effect of LMO2 on HR may not be limited to lymphoid cells. As a second measure of HR activity, we examined RAD51-dependent sister chromatid exchanges (SCEs) (Sonoda et al., 1998) in DLBCL cells exposed to the PARP1 and PARP2 inhibitor olaparib. PARP1/2 inhibition increases the frequency of replication-dependent DSBs and consequently the need for HR-dependent DSB repair. Although LMO2HIGH cells showed an increase in SCEs after exposure to olaparib, it was significantly lower than the SCEs observed in LMO2LOW cells (Figure 2H). Compared to LMO2LOW cells, LMO2HIGH cells also showed an increase in asymmetric radial chromosome structures, a type of chromatid exchange characteristic of HR deficiency (Figure S1C) (Venkitaraman, 2004). In addition, we used the GFP-based reporter system EJ5-GFP (Bennardo et al., 2008) to evaluate NHEJ activity in HEK293T cells expressing or not LMO2 protein. As expected for cells that relies on NHEJ when the HR pathway is defective, LMO2-expressing cells showed a significant increase in NHEJ activity (Figure S1D).
Noteworthy, although LMO2 is principally implicated in regulating expression of genes implicated in kinetochore function and chromosome assembly in DLBCL cells (Cubedo et al., 2012), the gene sets related to DNA repair significantly skewed towards a positive correlation with LMO2 indicating that these functions are slightly up-regulated when LMO2 is high (Figure S1E). However, LMO2 transcriptional effects do not lead to significant differences in BRCA1 or RAD51 protein expression (Figure 2C).
Compromised HR can contribute to genomic instability and increase the mutational burden. To determine whether primary LMO2HIGH DLBCL tumors show an increase in somatic mutations when compared to LMO2LOW tumors, we searched publicly available DLBCL datasets with information on LMO2 expression and somatic mutations in the same tumors (Chapuy et al., 2018). These analyses revealed a positive correlation between the number of somatic mutations and LMO2 mRNA expression in primary untreated DLBCL (Figure S1F). Similarly, LMO2HIGH DLBCL tumors (defined as second and third tertile based on the spectrum of LMO2 mRNA expression) showed a statistically significant increase in the number of somatic mutations in LMO2HIGH DLBCL tumors (Figure S1G). Finally, in an independent cohort of DLBCL patients, we also observed a statistically significant increase in the number of mutations per sequenced coding megabase (identified by FoundationOne Heme test) in LMO2HIGH DLBCL (using a previously defined 30% threshold via immunohistochemistry (IHC) for LMO2 protein expression, (Natkunam et al., 2008)) (Figure S1H). These observations suggest that LMO2 by inhibiting HR may lead to increased genomic instability not only in cell lines but also in primary DLBCL tumors.
Together, our studies revealed that high levels of LMO2 protein inhibit HR activity. Considering the essential role of HR in repairing DSBs during DNA replication, it is likely that the higher accumulation of DSBs observed in LMO2HIGH DLBCL (Figure 1) is due to this LMO2-dependent HR-dysfunction.
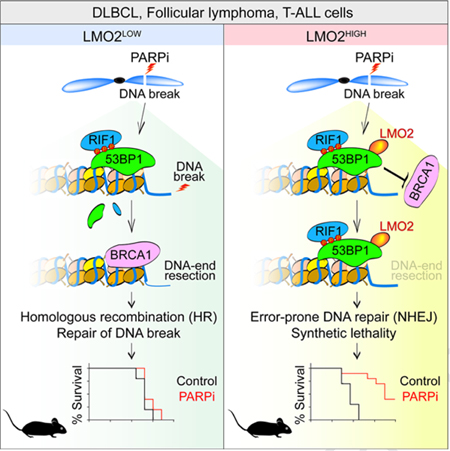
LMO2 interacts with and requires 53BP1 to inhibit HR
53BP1 is a NHEJ core factor that plays a critical role in defining DSBs repair pathway choice (Bothmer et al., 2011; Bothmer et al., 2010; Chapman et al., 2012). The association of 53BP1 with DSBs in the G1 phase of the cell cycle promotes NHEJ by suppressing the DSB-end resection necessary for repair via HR (Bothmer et al., 2011; Bothmer et al., 2010). However, when cells enter S phase, BRCA1 drives the removal of 53BP1 from DSBs and allows DSB-end resection for repair by HR (Bothmer et al., 2010; Bouwman et al., 2010; Cao et al., 2009; Chapman et al., 2012). Based on our data showing the inhibitory effect of LMO2 on BRCA1 and phospho-RPA32 accumulation at damaged chromosomes, we hypothesized that LMO2 functionally interacts with 53BP1. To investigate this, we studied 53BP1 behavior in DLBCL cells after DNA damage. Although no difference in 53BP1 expression was observed between the different cell lines (Figure 2C), we observed that LMO2HIGH DLBCL cells have higher levels of 53BP1 IRIF than LMO2LOW cells (Figure 2B). Similarly, over-expression of LMO2 in LMO2LOW DLBCL cells increased the number of IRIF with 53BP1 and the 53BP1-associated protein RIF1 (Di Virgilio et al., 2013; Escribano-Diaz et al., 2013; Zimmermann et al., 2013) (Figure 3A), as well as their accumulation in chromatin fractions after DNA damage (Figure 2F). Furthermore, knockdown of LMO2 decreased 53BP1 and RIF1 IRIF in LMO2HIGH DLBCL cells (Figure 3A). These data suggest that high levels of LMO2 favors the presence of the 53BP1 complex over BRCA1 at DSBs.
Figure 3. LMO2 forms a complex with 53BP1 during DNA damage HR.

(A) Quantification of 53BP1 and RIF1 ionizing radiation (IR)-induced foci (IRIF) in OCI-LY1 cells expressing GFP or GFP-LMO2 protein and OCI-LY8 cells expressing shGFP or shLMO2 4 hr post-exposure to 10 Gy. p values from Student’s t test are shown. At least 50 cells analyzed per cell line/experiment in IF experiments. Bars represent SD obtained from 3 independent experiments.
(B) Representative 3D confocal microscopy projections in OCI-LY8 cells showing co-localization of LMO2 and 53BP1 in IRIF (left) and quantification of LMO2 and 53BP1 foci colocalization in OCI-LY8 cells before and 4 hr post-exposure to 10 Gy (right). Inset zoom factor 3X. Scale bar, 1 μm. Bars represent SD obtained from 3 independent experiments.
(C) Representative fluorescence micrographs (left) and quantification (right) of LMO2 and KU70 co-localization in LMO2LOW U2932 cells transfected with plasmids encoding mCherry-LMO2 and GFP-KU70 after micro-irradiation with 405 nm laser (5 min). Inset zoom factor 3X. Scale bar, 1 μm. Error bars represent SD obtained from 3 independent experiments.
(D) LMO2 and γH2AX foci per cell in OCI-LY8 cells expressing shGFP or sh53BP1 4 hr post-exposure to 10 Gy. p value from Student’s t tests. Error bars represent SD obtained from 3 independent experiments. At least 50 cells analyzed per cell line/experiment in IF experiments.
(E) Immunoprecipitation (IP) assays with anti-GFP or anti-53BP1 antibodies and DNAseI-treated total extracts from OCI-LY1 cells expressing GFP or GFP-LMO2 fusion protein exposed or not to IR (10 Gy) for 4 hr.
(F) Immunoprecipitation (IP) assays with anti-LMO2 or anti-53BP1 antibodies and DNAseI-treated (nuclear for 53BP1 IP, total for LMO2 IP) extracts from OCI-LY19 cells expressing endogenous LMO2 exposed or not to IR (10 Gy) for 4 hr.
(G) Levels of BRCA1 and RAD51 IRIF in the indicated cells 6 hr post-10Gy. Error bars represent SD obtained from 3 independent experiments.
(H) Proposed model for LMO2-dependent inhibition of HR.
See also Figure S2.
We also observed that after DNA damage LMO2 spatially co-localized with 53BP1 IRIF (Figure 3B) and it was found in increased levels in chromatin-enriched extracts (Figure 2F), suggesting that LMO2 is present at the break site. To further test this, we performed laser-induced DNA break assays in LMO2LOW U2932 cells expressing mCherry-LMO2 and GFP-KU70 fusions proteins. In these assays we observed a high co-localization between LMO2 and the NHEJ factor KU70 (Figure 3C). Additionally, we observed that 53BP1 depletion in LMO2HIGH DLBCL decreased LMO2 and γH2AX IRIF co-localization but not LMO2 expression (Figure 3D and not shown), revealing that the presence of LMO2 at DSBs depends on 53BP1. These data suggested that LMO2 may associate with 53BP1 after DNA damage. Indeed, reciprocal immunoprecipitation assays using DNAse-treated protein extract from OCI-LY1 LMO2 cells revealed that LMO2, 53BP1 and RIF1 form a complex after DNA damage (Figure 3E). In these assays, both 53BP1 and RIF1 showed a weaker signal in the LMO2 precipitates from non-irradiated cells, probably due to the increased DNA damage observed in cells expressing LMO2 protein (Figures 1 and 2B). No signal for BRCA1 or RAD51 was observed in the LMO2 precipitates before or after DNA damage (Figure 3E). Similar conclusions were obtained when using extracts from LMO2HIGH OCI-LY19 cells exposed or not to IR (Figure 3F). Finally, GST pull-down assays confirmed that LMO2 forms a complex with 53BP1 but not BRCA1 after DNA damage (Figure S2A). Since 53BP1 inhibits HR (Daley and Sung, 2014), we wanted to determine whether 53BP1 knockdown rescued the BRCA1 and RAD51 defects observed in LMO2HIGH DLBCL cells. Indeed, silencing of 53BP1 increased the levels of BRCA1 and RAD51 IRIF to values similar to control cells without affecting LMO2 levels (Figure 3G and not shown). Furthermore, 53BP1 knockdown decreased the level of γH2AX in non-radiated LMO2HIGH cells (Figure S2B), supporting our previous hypothesis that the breaks observed in these cells are due to a deficient HR pathway. Finally, human GC B cells, which have high levels of LMO2 protein, also show expression of 53BP1 (Figure S2C), suggesting the possibility that LMO2 and 53BP1 could play a similar role in controlling homology-dependent DNA repair in normal B cells. Altogether, these results show that LMO2 depends on 53BP1 to inhibit HR, by in fact associating with the 53BP1 complex after DNA damage and possibly increasing its inhibitory effect over the BRCA1-dependent DSB-end resection step (Figure 3H).
LMO2 expression sensitizes tumor cells to PARP1/2 inhibition
Trapping of PARP1/2 on single-stranded DNA breaks via small molecules PARP inhibitors (PARPi) such as olaparib blocks the progression of the replication fork and induces a DNA damage response (Murai et al., 2012; Murai et al., 2014; Pommier et al., 2016). Although these stalled replication forks can be repaired via HR, they are toxic in BRCA1- or BRCA2-deficient cells (Bryant et al., 2005; Farmer et al., 2005; Jackson and Bartek, 2009). This synthetic lethality is used for treatment of breast and ovarian cancers, where BRCA1 or BRCA2 are mutated (Fong et al., 2009; Jackson and Bartek, 2009). Since our studies revealed that LMO2HIGH DLBCL cells have a defective HR-pathway (Figure 2), we wanted to explore the therapeutic potential of PARPi in DLBCL. Indeed, olaparib induced a significant decrease in cell proliferation in all LMO2HIGH DLBCL cell lines, compared to human primary lung fibroblasts (IMR90), EBV-transduced normal lymphoblasts (HCC1187) and LMO2LOW DLBCL cell lines (Figures 4A and 4B). LMO2 protein levels significantly correlated with the sensitivity to olaparib expressed as IC50 values (Figure 4C). Similarly, we observed a pronounced olaparib-mediated decrease in colony formation and increase in cell death via apoptosis in LMO2HIGH DLBCL cell lines, while there was no or minimal effect on LMO2LOW cell lines (Figures 4D and 4E). The proliferation defect in LMO2HIGH DLBCL cells is most likely due to increased DNA damage caused by exposure to PARPi, as we observed an increase in γH2AX foci in these cells (Figure 4F). The proliferation inhibition induced by olaparib was largely independent of p53 status as it affected both p53-proficient OCI-LY19 and p53-defective OCI-LY8 cell lines (Figure 4B). Furthermore, OCI-LY1 LMO2 cells showed a high sensitivity to olaparib when compared to OCI-LY1 GFP control cells, demonstrating that the proliferation defect induced by olaparib was dependent on LMO2 expression (Figure 4G). Accordingly, silencing of LMO2 via shRNA in OCI-LY8 and OCI-LY19 or LMO2 disruption via CRISPR/Cas9 in VAL cells, both LMO2HIGH cell lines (Figure 4A), rescues the proliferation defect caused by olaparib (Figure 4G). Furthermore, knockdown of 53BP1 reverses the sensitivity to olaparib in LMO2HIGH OCI-LY19 cells (Figure S3A), which is in agreement with our previous conclusions that the deficiency of HR induced by LMO2 expression is 53BP1 dependent. This last result also supports the conclusion of synthetic lethality induced by PARPi in these cells.
Figure 4. Olaparib induces apoptosis and decreases proliferation of lymphoma cells expressing LMO2.

(A) Western blots showing LMO2 level in the indicated human cell lines. O.E., over-exposure.
(B) MTS assay evaluating the effect of olaparib on proliferation of the indicated human cell lines. In black are control cell lines, in blue are LMO2LOW cell lines and in red are LMO2HIGH cell lines as determined in (A). p value from two-way ANOVA. Error bars represent SD obtained from 3 independent experiments.
(C) Correlation between olaparib IC50 as determined in (B) and LMO2 protein levels as determined in (A) for DLBCL cell lines.
(D) Quantification of colony formation assays in DLBCL cells exposed to increasing concentrations of olaparib. Error bars represent SD obtained from 3 independent experiments.
(E) Representative charts (left) and quantification (right) of flow cytometry results for propidium iodide and annexin V staining in the indicated cell lines after exposure to olaparib for 4 days. p value from two-way ANOVA. Error bars represent SD obtained from 3 independent experiments.
(F) Quantification of γH2AX foci in the indicated cell lines 48 hr after exposure to olaparib (1 μM). White scale bars represent 2 μm. p values from Student’s t tests. At least 50 cells analyzed per cell line/experiment in IF experiments. Error bars represent SD obtained from 3 independent experiments.
(G) MTS assay to evaluate the proliferation of DLBCL cell lines after exposure to olaparib for 72 hr. p value from two-way ANOVA. Error bars represent SD obtained from 3 independent experiments.
(H) Western blots showing LMO2 and γH2AX levels in the indicated human T-cell lymphoma (Jurkat), T-ALL and DLBCL cell lines.
(I) MTS assay evaluating proliferation of the indicated cell lines after exposure to olaparib for 4 days. Error bars represent SD obtained from 3 independent experiments.
(J) Representative FACS results (left) and quantification (right) of the indicated cell lines (right). p value from two-way ANOVA. Error bars represent SD obtained from 3 independent experiments.
See also Figure S3.
Although LMO2 is not normally expressed in mature T cells, high levels of LMO2 expression is found in a significant proportion of T-ALL (Chambers and Rabbitts, 2015; Hirose et al., 2010; McCormack and Rabbitts, 2004; Van Vlierberghe et al., 2006). Similar to LMO2HIGH DLBCL, we observed that LMO2HIGH T-ALL but not LMO2LOW T-ALL cells have a proliferation defect when exposed to olaparib (Figures 4H and 4I). The higher sensitivity to olaparib in LMO2HIGH T-ALL correlated with an increase in apoptosis and cell death (Figure 4J).
Given that LMO2 inhibits HR, we hypothesized that PARP1/2 inhibition in DLBCL would synergize with genotoxic chemotherapy. In support of this, the same LMO2HIGH DLBCL cell lines that are sensitive to olaparib, are also sensitive to doxorubicin, a standard-of-care drug used for upfront treatment of DLBCL patients (Coiffier et al., 2002; Habermann et al., 2006) (Figure S3B). Accordingly, olaparib induced a proliferation deficiency in LMO2HIGH DLBCL cells that was synergistic with doxorubicin (Figure 5A and 5B). These results support a model where high levels of LMO2 inhibits HR-dependent DNA repair and increases the cellular sensitivity to PARPi, inducing synthetic lethality that can be potentiated by standard chemotherapy. This conclusion was further supported by increased sensitivity to olaparib alone or in combination with doxorubicin in untreated patient-derived LMO2HIGH but not LMO2LOW DLBCL (Figure 5C). Similar results were obtained in untreated patient-derived follicular lymphoma and T-ALL expressing high levels of LMO2 protein (Figure 5D), but not in normal lymph nodes and small lymphocytic, peripheral T cell, and angioimmunoblastic lymphomas not expressing LMO2 protein (Figure 5D and not shown).
Figure 5. PARPi and doxorubicin show synergy in killing tumor cells expressing LMO2.

(A) Colony formation assays evaluating proliferation of DLBCL cell lines exposed for 2 days to increasing concentrations of olaparib and doxorubicin, as indicated. Error bars represent SD obtained from 3 independent experiments.
(B) Graphics showing the combination indexes (CI) calculated from colony formation assays shown in (A).
(C) Quantification of FACS results for annexin V staining of primary patient-derived DLBCL cells exposed for 4 days to increasing concentrations of olaparib and doxorubicin, as indicated. Combination index (CI) lower than 0.25 (*) or 0.1 (#) are shown. Error bars represent SD obtained from 3 independent experiments. p values from two-way ANOVA.
(D) IHC showing LMO2 protein expression in patient-derived samples (top) and quantification of FACS results for annexin V staining of tumor cells exposed for 4 days to increasing concentrations of olaparib and doxorubicin as indicated (bottom). Combination index (CI) lower than 0.25 (*) or 0.1 (#) are shown. Error bars represent SD obtained from 3 independent experiments. p values from two-way ANOVA. Green scale bars, 100 μm. White scale bars, 400 μm. Black scale bars 40 μm.
The pro-apoptotic and anti-proliferative activity of olaparib in LMO2HIGH cell lines and primary tumors suggest that this drug may be useful to treat DLBCL patients after stratification by LMO2 levels. Consequently, we examined the in vivo efficacy of olaparib in mice using human cell lines or patient-derived xenograft (PDX) DLBCL models with different levels of LMO2 expression: low LMO2 expression (OCI-LY1), intermediate LMO2 expression (PDX 75549) (Chapuy et al., 2016) or high LMO2 expression at levels similar to GC lymphocytes (OCI-LY19) (Figures 6A and S4). To this end, non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice were xenotransplanted with patient-derived OCI-LY1 and OCI-LY19 DLBCL cell lines and non-obese diabetic/severe combined immunodeficiency/gamma (NSG) mice with PDX 75549. Treatment with olaparib after the tumor reached a volume of 150 mm3 did not prolong survival of mice harboring OCI-LY1 tumors, while survival of mice harboring LMO2LOW DLBCL PDX 75549 tumors was slightly prolonged (Figure 6B). Statistically significant long-term survival prolongation was observed in the LMO2HIGH DLBCL xenografts (OCI-LY19) treated with olaparib (Figure 6B). Olaparib, as a single agent, delayed tumor growth and increased cell death via apoptosis in LMO2HIGH xenografts (Figures 6B and 6C). These studies demonstrated a positive correlation between the level of LMO2 protein expression and olaparib-induced prolongation of survival in mice harboring human DLBCL tumors, similar to what we observed in vitro (Figures 6A and 4C). Furthermore, similar to the in vitro results observed in patient-derived primary tumors (Figure 5), olaparib in combination with the current standard R-CHOP immuno-chemotherapy for DLBCL significantly prolonged murine survival compared to cohorts treated with either olaparib or R-CHOP alone (Figures 6D–6H). Importantly, while olaparib, alone did not prolong survival of mice harboring DLBCL expressing intermediate levels of LMO2 (OCI-LY8, Figure 6D), it still synergized with chemotherapy leading to statistically longer survival of animals treated with olaparib and R-CHOP in comparison to R-CHOP treatment alone (Figures 6E and 6F). Together, these data support the therapeutic potential of olaparib as a single agent and specifically in combination with immuno-chemotherapy in LMO2HIGH DLBCL.
Figure 6. Olaparib alone or in combination with R-CHOP slows tumor growth and prolongs survival of mice with LMO2HIGH DLBCL xenografts.

(A) Western blots for LMO2 levels in DLBCL GC naïve patient-derived xenograft (PDX) 75549 (after sorting CD45+ cells) and OCI-LY1 and OCI-LY19 DLBCL cell lines.
(B) Kaplan-Meier survival plots for OCI-LY1, OCI-LY19, and PDX 75549 xenografts treated daily with intraperitoneal (IP) olaparib or PBS (10 mice per cohort) after tumor reached a volume of 150 mm3. Mice were euthanized when tumor volume reached 1500 mm3 in OCI-LY1 and OCI-LY19 models or 1000 mm3 in PDX 75549 model. p value from Log-rank (Mantel-Cox) test are shown.
(C) Quantification of flow cytometry results for propidium iodide and annexin V staining of tumors excised from OCI-LY19 xenografts animals treated (IP) for 3 days with olaparib or PBS. p value from Student’s t test. Error bars represent SD obtained from 3 independent experiments.
(D) Western blots for LMO2 levels in DLBCL cell lines.
(E) Kaplan-Meier survival plots for OCI-LY8 xenografts. After tumors reached a volume of 150 mm3 mice were treated with intravenous PBS (control) or R-CHOP, and daily with olaparib (IP), or their combination (R-CHOP plus olaparib). Mice were euthanized when tumor volume reached 1500 mm3. The number of mice (n) per group is indicated. p values from Log-rank test are shown.
(F) Tumor volume versus time of individual mice treated as shown in (E).
(G) As in (E), for OCI-LY19 xenografts. The number of mice (n) per group is indicated. p value from Log-rank test is shown.
(H) Tumor volume versus time of individual OCI-LY19 mice treated as shown in (G).
See also Figure S4.
DISCUSSION
Elucidating the biological basis and pathogenesis of DLBCL should ideally lead to insights into tumor vulnerabilities. LMO2 is one of the best prognostic biomarkers for survival of DLBCL patients treated with RCHOP irrespective of the cell of origin of the tumors (Alizadeh et al., 2009; Lossos et al., 2004; Malumbres et al., 2008). Herein we propose a molecular mechanism underlying its prognostic significance by directly implicating LMO2 in DSB repair pathway choice, resulting in HR-dysfunction and tumor cell sensitization to genotoxic agents. This is a function that differs from the previously well-characterized roles of LMO2 in transcriptional regulation (Chambers and Rabbitts, 2015; Cubedo et al., 2012; Davenport et al., 2000; El Omari et al., 2011; Grutz et al., 1998).
The key step that determines the choice between error-free HR and error-prone NHEJ is DNA-end processing to generate 3’ tails, a process controlled by an interplay between 53BP1 and BRCA1 (Daley and Sung, 2014). Herein we show that in LMO2HIGH cells, 53BP1 displays an increased association with DSBs while BRCA1 and RAD51 show the opposite behavior. This BRCA1/RAD51 deficiency in LMO2HIGH cells correlates with a reduction in the generation of 3’ single-strand DNA tails at DSB breaks and consequently lower HR activity. Although further work will be needed to fully understand how LMO2 inhibits HR in LMO2HIGH cells, our results provide evidence that at high concentrations, LMO2 inhibits the recruitment of BRCA1 to the DSB site via its association with the 53BP1 complex after DNA damage. It is accepted that DNA-end resection inhibition by 53BP1 and RIF1 is more effective in G1 phase than in S and G2 phases since BRCA1 can induce the removal of 53BP1 from the break site and allow the end-resection step (Daley and Sung, 2014). Our results are consistent with a role of LMO2 in inhibiting HR and consequently affecting genomic stability. This is probably through its interaction with 53BP1 and RIF1 which could affect the DNA-end resection step. However, a direct role for LMO2 in DNA-end resection is yet to be demonstrated. Thus, LMO2HIGH cells have to rely on the mutagenic NHEJ for repair of DSBs. Interestingly, we have shown that LMO2 expression level in primary human normal germinal center B cells is similar to LMO2HIGH DLBCL cells (Malumbres et al., 2008; Natkunam et al., 2008). During the germinal center (GC) reaction the immunoglobulin genes are recombined to allow the diversification of the antibody response against antigens. This molecular process, known as antibody class-switch recombination (CSR), is an intra-chromosomal recombination that requires the production and joining of distant DSBs. These DSBs, located up to ~200 kb apart in the immunoglobulin locus, are joined by NHEJ. This process depends on 53BP1 and RIF1, as deletion of either abolishes CSR (Stavnezer et al., 2008); (Di Virgilio et al., 2013). On the other hand, repair of the DSBs at the immunoglobulin locus by the error-free HR pathway restores the genomic structure and inhibits CSR (Di Virgilio et al., 2013). Therefore, it is likely that LMO2 also plays a significant role in the choice of DNA repair pathway during CSR in normal B cells by inhibiting HR to allow CSR; and that this LMO2-dependent inhibition of HR in normal GC B cells is maintained in GC-derived tumors.
Our results show that high expression of LMO2 results in HR-dysfunction that phenocopies BRCA1 and BRCA2 mutations, or BRCAness, observed in other tumors (Lord and Ashworth, 2016). In agreement with this, and similarly to breast, ovarian and castration-resistant prostate cancers, this HR-dysfunction can be exploited for PARPi-mediated killing of LMO2 positive lymphoma cells. While the 5-year progression-free survival of patients with LMO2HIGH DLBCL tumors is 74%, the remaining quarter of these patients (about 3,500 patients per year) do not respond to standard upfront therapy (Natkunam et al., 2008). This underlies an urgent need for better therapeutic approaches in these patients. Herein we show that in DLBCL expressing LMO2, PARPi-induced killing is synergistic with chemotherapeutic agents such as doxorubicin that are routinely clinically used in DLBCL patients, thus providing a clear path for therapeutic development of PARPi in DLBCL.
The information about the clinical relevance of PARPi in lymphoid malignancies is limited. Preclinical studies with olaparib in chronic lymphocytic leukemia and mantle cell lymphoma, which are usually LMO2 negative (Natkunam et al., 2008; Natkunam et al., 2007), demonstrated greater dose-dependent reduction in the number of cells harboring ATM deletions and/or mutations compared with ATM-proficient cells (Weston et al., 2010; Williamson et al., 2012). However, no meaningful clinical responses were observed in a multi-center Phase-I trial of olaparib in patients with relapsed chronic lymphocytic leukemia and mantle cell lymphoma (Pratt et al., 2018). In contrast, the PARP inhibitor veliparib in combination with bendamustine and rituximab demonstrated preliminary evidence of activity in patients with NHL (Soumerai et al., 2017). Interestingly, all 6 evaluable patients with follicular lymphoma, in which LMO2 is expressed in majority of tumors, achieved complete responses (Soumerai et al., 2017).
Aberrations in DNA damage repair (DDR) mechanisms are common in cancer cells (Knijnenburg et al., 2018). Usually they are caused by loss of function mechanisms by gene mutations, deletions or silencing. However, most of the large-scale genetic studies in DLBCL demonstrated that mutations/deletions of genes implicated in DDR in general and specifically in HR dysfunction are uncommon. Indeed, a recent study found that BRCA1 mutations are present in less than 10% of ABC and absent in GCB DLBCL tumors (Schmitz et al., 2018). In other studies BRCA1 was not identified as DLBCL associated cancer genes (Chapuy et al., 2018; de Miranda et al., 2013; Reddy et al., 2017). Instead, we demonstrate a “gain of function” mechanism where high LMO2 expression at levels similar to GC, as found in ~50% DLBCL, impairs HR function. Further, we also demonstrate that this LMO2-dependent HR dysfunction can be exploited for synthetic lethality to PARPi in follicular lymphoma and T-ALL, suggesting that LMO2 expression can be used as a biomarker in different genetic backgrounds. This is relevant since LMO2 expression is not limited to lymphoid cells and has been reported in a subset of breast and prostate tumors (Agostinelli et al., 2012; Ma et al., 2007). Therefore, our findings may lead to therapeutic approaches in variety of LMO2 expressing tumors. While the precise molecular mechanisms by which LMO2 mediates HR-dysfunction need further investigations, our data provide a sound mechanistic basis for employing LMO2 expression as a biomarker for HR-dysfunction that will help to stratify DLBCL patients for therapy with PARPi in clinical trials.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Izidore S. Lossos (ilossos@med.miami.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
The human DLBCL cell lines OCI-LY1, OCI-LY8, SUDHL-6, G452, VAL, DOHH2, U2932 and OCI-LY19 and HCC1187 lymphoblasts were grown in Iscove modified Dulbecco medium (IMDM) (Mediatech Inc.) supplemented with 20% human plasma (Florida’s Blood Centers, Orlando, FL), 2 mM glutamine (Mediatech Inc.), 50 mM 2-mercaptoethanol (SIGMA) and supplemented with 1% penicillin/streptomycin (Mediatech Inc.) and non-essential amino acids (Mediatech Inc.). Jurkat, DND41, MOLT4 and MOLT16 were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. HEK293T cells were grown in DMEM medium (Mediatech Inc.) supplemented with 10% FBS (Hyclone), 2 mM glutamine, and penicillin/streptomycin.
Mice
The University of Miami Institutional Animal Care and Use Committee (IACUC) approved all mouse procedures. All mice were bred and housed on a 12 hr light/dark cycle. NOD/SCID mice were purchased from Envigo and NSG mice from the Jackson laboratory.
Human DLBCL samples
B cells were enriched from primary tissues and human tonsils that were obtained during routinely scheduled biopsies and tonsillectomies, respectively. The tissue collection protocol was approved by the Institutional Review Board (IRB) of the University of Miami and all the patients signed a written informed consent. Fresh tissues were rinsed in RPMI medium, cut into small sections and ground in gentleMACS Dissociator (Miltenyi) machine. Cell suspension was passed through several layers of sterile mesh to remove clumps. B cells were enriched using the B cells Isolation Kit (StemCell Technology) according to the manufacturers’ instructions.
METHODS DETAILS
Plasmids
pDRGFP was a gift from Maria Jasin (Addgene plasmids # 26475 and 26477) (Pierce et al., 1999; Richardson et al., 1998), pimEJ5GFP was a gift from Jeremy Stark (Addgene plasmid # 44026) (Bennardo et al., 2008), and pEGFP-C1-FLAG-Ku70 was a gift from Steve Jackson (Addgene plasmid # 46957) (Britton et al., 2013). The mcherry-LMO2 lentiviral vector was purchased from Genecopoeia (CS-A2951-Lv113).
For expression of GFP-LMO2 fusion protein in DLBCL cells a doxycycline-inducible lentiviral vector, a generous gift of Dr. Ari Melnick (Weill Cornell Medical College,NY), was used. We generated stable short hairpin RNA (shRNA)-mediated knockdowns in DLBCL cells as described previously (Cortizas et al., 2016). Transduced cells were selected with puromycin at 1 μg/ml final concentration. shLMO2–1, TRCN0000017136. shLMO2–2, TRCN0000017132. sh53BP1, TRCN0000018866. sh53BP1–2, TRCN0000018869.
Western Blots and Antibodies sources
Whole-cell extracts were prepared by lysing cells for 10 min on ice in RIPA lysis buffer (50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1.0% NP-40, 0.1% SDS, and 0.1% Na-deoxycholic acid) supplemented with protease and phosphatase cocktail inhibitors (Thermo Fisher). Cellular lysates were assayed for protein concentration using Coomassie Protein Assay Reagent (Pierce) in 96 well-plates using a Bio-Rad Benchmark Microplate Reader. Whole cell lysates were separated through SDS-polyacrylamide gels (4–12%) and transferred to nitrocellulose membrane (Bio-Rad Laboratories, Inc.). Membranes were blocked with 5% milk powder in 0.1% Tween20 in 1x PBS (PBS-T) for 1 hr at room temperature followed by incubation with primary antibodies diluted in 2.5% milk PBS-T. ImmunoCruz Western Blotting Luminol Reagent (Santa Cruz Biotechnology, Inc.) was used to visualize protein levels with light sensitive-films (Phenix Research). Immunoblots were quantified using ImageJ software. The following antibodies were used in this study: 53BP1 (Millipore), BRCA1 (D-9) (Santa Cruz), H2A (Abcam), H2AX antibody (Bethyl labs), HA clone HA-7 (SIGMA), γ-H2AX (2F3) [S139] (Novus), γ-H2AX (S139) clone JBW301 (Millipore), Phospho RPA32 (S4/S8) (Bethyl labs), RAD51 (H92) (Santa Cruz), RIF1 (Bethyl labs), RPA1 (SIGMA). Monoclonal antibody against LMO2 was generated in our laboratory as previously reported (Cubedo et al., 2012; Natkunam et al., 2007).
Immunoprecipitations and GST Pull-down assays
Proteins were immunoprecipitated from whole-cell extracts. Briefly, cells were collected, washed and lysed on ice for 20 min in a lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.8% NP-40, 5% glycerol, 1 mM MgCl2, 1 mM ZnCl2, DNAseI (20 μg/mg of protein), N-ethylmaleimide (NEM 10mM) and proteases and phosphatases cocktail inhibitors (Roche). Sonication was performed with eight 30 s ON/OFF cycles with a Bioruptor (Diagenode) at full power yielding soluble chromatin. Protein concentration from cleared supernatants was estimated by a Bradford assay (Bio-Rad). Two milligrams of whole-cell extracts were incubated for 30 min at 4 °C with anti-GFP micro-beads (Miltenyi Biotec), columns were washed ten times with 200 μl of lysis buffer before elution in 50 μl of heated up Laemmli buffer. Eluted proteins were analyzed by immunoblotting as described above.
GST pull-down assays were performed by incubating whole-cell lysates with 10 μg of recombinant GST-LMO2 protein for 2 hr at 4 °C and then with anti-GST micro-beads (Miltenyi Biotec) for 30 min. Micro-Beads were washed 10 times with 200 μl of lysis buffer before elution in 50 μl of heated up Laemmli buffer. Eluted proteins were analyzed by immunoblotting as described above.
Chromatin fractionation
Chromatin-enrichment was performed as previously described (Ochs et al., 2016). The cytosolic protein fraction was removed by incubation in hypotonic buffer (10 mM HEPES (pH 7), 50 mM NaCl, 0.3 M sucrose, and 0.5% NP-40), supplemented with NEM 10mM, protease and phosphatase cocktail inhibitors (Thermo Fisher) for 15 min on ice, and centrifuged at 1,500g for 5 min. The soluble nuclear fraction was removed by incubation with 10 mM HEPES (pH 7), 200 mM NaCl, 0.5% NP-40, and NEM 10mM, protease and phosphatase cocktail inhibitors (Thermo Fisher) during 20 min on ice and then centrifuged at 1,500g for 5 min. The pellets were resuspended in 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1.0% NP-40, 0.1% SDS, 0.1% Na-deoxycholic acid, 5% glycerol, DNAse 20 μg/mg, N-Ethylmaleimide 10 mM and protease and phosphatase cocktail inhibitors (Thermo Fisher), sonicated with eight 30 s ON/OFF cycles with a Bioruptor (Diagenode), and centrifuged for 2 min at 15,000g; the supernatant was then transferred to a new tube. Total chromatin protein was quantified using Bradford’s method, and a total of 30 μg protein was used for Western blots analyses as described above.
Analysis of mutations in DLBCL
Mutation data for DLBCL were obtained for each patient as previously described (Chapuy et al., 2018). We considered single nucleotide variants (SNVs) and small insertions and deletions. All such variants (synonymous as well as non-synonymous) were summed per patient. Expression data were obtained as raw Affymetrix CEL files from the Gene Expression Omnibus, accession GSE98588. Microarrays were processed using the mas5 function of the affy package v. 1.6 (Gautier et al., 2004) under R 3.5.2. Expression intensities were calculated using Custom CDF chip annotation files to map probes to Entrez gene identifiers.
Immunofluorescence assays
After washes with 1X PBS, cells were fixed with 2% formaldehyde in PBS for 10 min at room temperature (RT), washed again with PBS and bound to poly-L-lysine coated slides. After wash with PBS for 5 min, cells were incubated with Blocking Solution (1 mg/ml BSA, 3% goat serum, 0.1 % Triton X-100 in PBS) for 30 min at RT. Next, the cells were incubated with the primary antibodies in Blocking Solution for 1 hr at RT, washed three times with PBS, and incubated with Alexa 488 or 594 coupled antibodies (ThermoFisher Sc.) for another 30 min. Finally, cells were incubated with 0.5 μg/ml 4’, 6-diamino-2-phenylindole (DAPI) (Sigma) in PBS for 5 min, washed with PBS for 2 min, air dried, and mounted in SlowFade Diamond antifade mounting reagent (ThermoFisher Sc.). Samples were analyzed using a Leica DMI6000B microscope with LASX software (Leica). Antibodies to the following proteins were used in this study: 53BP1 (Millipore), RIF1 (Bethyl labs), BRCA1 (D-9) (Santa Cruz), γ-H2AX (Ser 139) clone JBW301 (Millipore), γ-H2AX (Cell Signalling), RAD51 (B-Bridge International). Monoclonal antibody against LMO2 was described before (Cubedo et al., 2012; Natkunam et al., 2007).
Comet assay
Agarose-precoated slides were prepared by dipping the slides into molten 1% agarose. Twenty thousand cells were resuspended in 250 μl of ice-cold PBS and mixed with 250 μl of 1% low-melting agarose (Lonza) and the mixture was poured onto the pre-coated agarose slides. Next, slides were immersed in lysis buffer (Trevigen) for 1 hr. Slides were placed into the Trevigen electrophoresis system and subjected to conduct electrophoresis at 16V for 25 min. After electrophoresis, the slides were dried at 37 °C overnight at RT. Cellular DNA was stained in a solution containing 40 μg/ml propidium iodide (Sigma) supplemented with 50 μg/ml RNAse and then slides were dried at 37 °C. The slides were visualized using a Leica DMI6000B microscope with LASX software (Leica) and analyzed using CometScore 2.0. A minimum of 100 cells were analyzed for each experimental point.
Mitotic Spreads and SCE
For quantification of chromosome breaks, metaphase spreads were prepared as described previously (Cortizas et al., 2016) and stained with Giemsa. Cells were grown in the presence of BrdU (10 μg/ml) for two rounds of cell division after which colcemid (Sigma) was added to the cell culture to enrich mitotic cells. Metaphase spreads were stained with Hoechst 33528 (200 μg/ml) for 30 min, rinsed with water and air-dried. Slides were covered with McIlvaines buffer (164 mM Na2HPO4, 16 mM citric acid, pH 7.0) and then irradiated with UV light (λ= 365 nm) for 30 min on slide warmer at 60 °C, then rinsed with MilliQ water and air-dried. The slides were stained in Giemsa for about 10 min, air-dried and fixed with xylene for 1 min and then again air-dried. After mounting with Permount (Electron Microscopy Sciences) metaphases spreads were visualized using a Leica DMI6000B microscope with LASX software (Leica).
Proliferation assays, cell apoptosis and colony-formation assays
Cell proliferation inhibition was assayed using the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (Promega) on an ELx800 Universal Microplate Reader (Bio-TEK Instruments). Cells (1×105/ml) were plated and treated for 4 days in triplicates for each experimental condition. Each well was incubated with 20 μl of MTS reagent in 100 μl of cell suspension, the plate was then incubated for 1–4 hr at 37 °C in a humidified, 5% CO2 atmosphere. The absorbance was corrected relative to blank wells containing MTS reagent and media only. The MTS readings at specified time points were normalized to vehicle controls readings.
For cell death and apoptosis assays, 5×105 cells were collected, washed twice with 4 °C Annexin binding buffer (ABB) (BD Pharming en), and then resuspended in 500 μl of ABB. Phycoerythrin (PE) or FITC-conjugated Annexin V (5 μl) and propidium iodide (5 μl) were added to the solution and mixed. After incubation for 15 min at RT in the dark, the cells were analyzed using flow cytometry.
Cell cycle phases profiles were captured by flow cytometry after ethanol fixation and permeabilization and propidium iodide staining and analyzed using FlowJo software (FlowJo, LLC).
Colony formation assays were performed as described elsewhere (McDonnell et al., 2012). Briefly, cells were pretreated with indicated olaparib concentration alone or in combination with Doxorubicin for 48 hr and 500 cells seeded in triplicates in MethoCult (Stem Cell Technologies). After 10–14 days, macroscopic colonies were stained with iodonitrotetrazolium chloride (Sigma) overnight and counted with ImageJ software. Data was normalized to vehicle controls.
Synergy was estimated with Isobologram analysis using the median-effect principle of Chou & Talalay using the CalcuSyn software (Biosoft).
In vivo tumor studies
Animal studies were conducted according to an approved Institutional Animal Care and Use Committee protocol. For xenograft assays, five million of OCI-LY19 or OCI-LY1 cells or seven million of OCI-LY8 cells were injected subcutaneously into the flanks of 4-week old NOD/SCID mice. For the PDX (DLBCL GC type, #75549, (Chapuy et al., 2016)) experiments, one million cells were injected subcutaneously into 6–8-week-old NSG mice. Once the tumors reached 150 mm3 of volume, mice were treated intraperitoneally daily with vehicle or olaparib (50 mg/kg). For R-CHOP and R-CHOP plus olaparib cohorts, Rituximab (20 mg/kg), Cyclophosphamide (40 mg/kg), Doxorubicin (3.3 mg/kg) and Vincristine (0.5 mg/kg) were administered intravenously on Day 1. Prednisone was given via gavage daily for 5 days at 0.2 mg/kg. Daily IP of olaparib or vehicle were injected into the respective animal cohorts. Mice were monitored for tumor growth and overall health. When the tumor size reached 1500 mm3 (xenograft) or 1000 mm3 (PDX) as measured by caliper or high-frequency ultrasound using Vevo 3100 from FUJIFILM Visualsonics (PDX), mice were euthanized. Survival was estimated with the Kaplan-Meier survival curve method and differences in survival were calculated by log-rank test (Graph Pad Prism 6.0).
Immunohistochemistry
Tumor sections were embedded in paraffin, and thin histologic sections were prepared and stained with hematoxylin and eosin following standard protocols. Briefly, tissue sections were deparaffinized in xylene and rehydrated in ethanol following treatment in pre-heated target retrieval solution. Following washes, serum-free blocking solution was applied for 40 min at RT. Expression of LMO2 and CD20 was determined using an anti-human antibody (L26; Agilent; 1:1000) and mouse monoclonal anti-LMO2 antibody (generated in our laboratory, as previously reported (Natkunam et al., 2007)) in formalin fixed, paraffin-embedded tissue sections. The slides were counterstained with hematoxylin, dried and mounted with Permount. Micrographs of the morphology and CD20 and LMO2 expression were captured using an Olympus BX43 dual-head light microscope equipped with an Olympus Q-Color 5 digital camera (Olympus America) (all at a magnification of x400).
STATISTICAL ANALYSIS
Statistical parameters including sample size and statistical significance are reported in the figures and corresponding figure legends. Two-sided Student’s t test was used to compare differences between two groups of cells in vitro. Data are presented as means ± SD and p≤0.05 was considered significant. Log rank tests ≤0.05 were standard for significance between subgroup of animals. Analysis of variance (ANOVA) was used to compare differences among multiple groups. Data were analyzed and plotted using GraphPad Prism 6.0.
DATA AND CODE AVAILABILITY
This study did not generate new dataset for analysis. Previously published datasets analyzed in this study are referenced in the manuscript.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 53BP1 | Millipore | Cat# MAB3804, RRID:AB_2256673 |
| Anti-53BP1 | Novus | Cat# NB100–304, RRID:AB_10003037 |
| BRCA1 (D-9) | Santa Cruz | Cat# sc-6954, RRID:AB_626761 |
| H2AX | Bethyl labs | Cat# A300–083A, RRID:AB_203289 |
| HA (clone HA-7) | Sigma-Aldrich | Cat# H9658, RRID:AB_260092 |
| γ-H2AX (Ser 139) clone JBW301 | Millipore | Cat# 05–636, RRID:AB_309864 |
| γ-H2AX | Cell Signaling Tech. | Cat# 2577, RRID:AB_2118010 |
| Phospho RPA32 (S4/S8) | Bethyl labs | Cat# A300–245A, RRID:AB_210547 |
| RAD51 (H92) | Santa Cruz | Cat# sc-8349, RRID:AB_2253533 |
| RAD51 | B-Bridge International | Cat# 70–001, RRID:AB_2177110 |
| RIF1 | Bethyl | Cat# A300–244A, RRID:AB_185548 |
| RPA2 | Bethyl | Cat# A300–245A, RRID:AB_210547 |
| LMO2 | Natkunam et al., 2008 | N/A |
| GAPDH | Santa Cruz | Cat# sc-25778, RRID:AB_10167668 |
| CD20 | Agilent | Cat# M0755, RRID:AB_2282030 |
| γ-Tubulin | Sigma-Aldrich | Cat# T5326, RRID:AB_532292 |
| Actin | Sigma-Aldrich | Cat# A2066, RRID:AB_476693 |
| 488 Fluor Donkey anti-Rabbit antibody | ThermoFisher Sc. | Cat# A-21207, RRID:AB_141637 |
| 488 Fluor Donkey anti-mouse antibody | ThermoFisher Sc. | Cat# A-21202, RRID:AB_141607 |
| 594 Fluor Goat anti-Mouse antibody | ThermoFisher Sc. | Cat# A-11020, RRID:AB_2534087 |
| 594 Fluor Goat anti-Rabbit antibody | ThermoFisher Sc. | Cat# A-11012, RRID:AB_2534079 |
| 647 Fluor Donkey anti-goat antibody | ThermoFisher Sc. | Cat# A-21447, RRID:AB_2535864 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, HRP | ThermoFisher Sc. | Cat# A16110, RRID:AB_2534782 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, HRP | ThermoFisher Sc. | Cat# A16078, RRID:AB_2534751 |
| Biological Samples | ||
| Patient Derived Xenografts (PDXs) | Chapuy et al., 2016 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Olaparib | LC Labs | Cat# O9201 |
| BrdU | Sigma-Aldrich | Cat# B5002 |
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| Demecolcine | Sigma-Aldrich | Cat# D7385 |
| Permount | Electron Microscope Sciences | Cat# 17986–01 |
| DAPI | Millipore-Sigma | Cat# 268298 |
| CometAssay®Lysis Solution | Trevigen | Cat# 4250–010-01 |
| Bacterial and Virus Strains | ||
| pLKO.1 shLMO2–1 | Dharmacon Inc. | TRCN0000017136 |
| pLKO.1 shLMO2–2 | Dharmacon Inc. | TRCN0000017132 |
| pLKO.1 sh53BP1–1 | Dharmacon Inc. | TRCN0000018866 |
| pLKO.1 sh53BP1–2 | Dharmacon Inc. | TRCN0000018869 |
| Critical Commercial Assays | ||
| CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (MTS) | Promega | Cat# G5421 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | ThermoFisher Sc. | Cat# 34095 |
| Halt™ Protease and Phosphatase Inhibitor Cocktails | ThermoFisher Sc. | Cat# 78441 |
| Slowfade Diamond antifade reagent | ThermoFisher Sc. | Cat# S36963 |
| MethoCult | Stem Cell Technologies | Cat# H4100 |
| JetPrime Transfection Reagent | Polyplus Transfection | Cat# 114–07 |
| Experimental Models: Cell Lines | ||
| OCI-LY19 | Ontario Cancer Institute (OCI) | N/A |
| OCI-LY1 | Ontario Cancer Institute (OCI) | N/A |
| Doxycycline induced LMO2-GFP OCILY-1 | Gift from Ari Melnick Weill Cornell MC, NY |
N/A |
| OCI-LY7 | Ontario Cancer Institute (OCI) | N/A |
| OCI-LY8 | Ontario Cancer Institute (OCI) | N/A |
| DOHH2 | Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) | Cat# ACC-47, RRID: CVCL_1179 |
| VAL | Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) | Cat# ACC-586, RRID: CVCL_1819 |
| SUDHL6 | American Type Culture Collection (ATCC) | Cat# CRL-2959, RRID: CVCL_2206 |
| U2932 | Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) | Cat# ACC-633, RRID: CVCL_1896 |
| Jurkat | American Type Culture Collection (ATCC) | Cat# TIB-152, RRID: CVCL_0367 |
| DND41 | Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) | Cat# ACC 525, RRID: CVCL_2022 |
| MOLT16 | Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) | Cat# ACC 29, RRID: CVCL_1424 |
| MOLT4 | Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) | Cat# ACC 362, RRID: CVCL_0013 |
| HCC1187-BL | American Type Culture Collection (ATCC) | Cat# CRL-2323, RRID: CVCL_1248 |
| IMR90 | American Type Culture Collection (ATCC) | Cat# CCL-186, RRID: CVCL_0347 |
| HEK-293T | American Type Culture Collection (ATCC) | Cat# CRL-3216, RRID: CVCL_0063 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.CB17-Prkdcscid/NCrHsd | Envigo | N/A |
| Mouse: NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ | The Jackson Laboratory | Cat# 005557 |
| Oligonucleotides | ||
| ON-TARGETplus Human BRCA1 (672) siRNA - SMARTpool | Dharmacon | L-003461–00- 0010 |
| ON-TARGETplus Non-targeting Control Pool | Dharmacon | D-001810–10- 20 |
| Recombinant DNA | ||
| pDRGFP | Addgene | Cat# 26475 |
| pCBASceI | Addgene | Cat# 26477 |
| mcherry-LMO2 lentiviral vector | Genecopoeia | Cat# CS-A2951-Lv113 |
| pEGFP-C1-FLAG-Ku70 | Addgene | Cat# 46957 |
| pimEJ5GFP | Addgene | Cat# 44026 |
| Software and Algorithms | ||
| LASX software | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| CometScore IV | Instem | https://www.instem.com/ |
| Prism 6.0 | Graph Pad | https://www.graphpad.com/scientific-software/prism/ |
| CalcuSyn software | Biosoft | http://www.biosoft.com/w/calcusyn.htm |
| FlowJo | FlowJo, LLC | https://www.flowjo.com/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
SIGNIFICANCE.
Effective therapies are needed for DLBCL, the most common subtype of non-Hodgkin lymphomas. LMO2 expression serves as one of the best prognostic markers of longer survival of DLBCL patients following immunochemotherapy; however, the reason for its prognostic significance remains poorly understood. Herein we demonstrate that LMO2 controls DSB pathway choice, resulting in HR-dysfunction and tumor cell sensitization to genotoxic agents that could be further enhanced with PARP1/2 inhibitors. This synthetic lethality provides the rationale for clinical trials testing PARP inhibitors in combination with immunochemotherapy in LMO2 expressing tumors such as DLBCL, follicular lymphoma and T-ALL.
HIGHLIGHTS.
DLBCL expressing LMO2 protein have deficient homologous recombination DNA repair
LMO2 interacts with and requires 53BP1 to inhibit homologous recombination
LMO2 expression sensitizes tumor cells to PARP1/2 inhibitors (PARPi)
PARPi and chemotherapy synergistically inhibit the growth of LMO2 positive tumors
Parvin et al. show that LMO2 inhibits BRCA1 recruitment to DNA double-strand breaks (DSB) by interacting with 53BP1. Thus diffuse large B cell lymphomas expressing LMO2 are functionally deficient in homologous recombination-mediated DSB repair and are sensitive to Poly (ADP-ribose) polymerase inhibitors.
ACKNOWLEDGMENTS
We are indebted to the University of Miami Sylvester Comprehensive Cancer Center (SCCC) Flow Cytometry Shared Resource (FCSR), Cancer Modeling Shared Resource (CMSR), and Radiation Control Center (RCC) for assistance.
This work was supported by grant 1R01CA233945 from the National Cancer Institute, Florida Health Bankhead-Coley Cancer Research Award AWD-005151 and the Intramural Funding Program from the University of Miami SCCC (to R.E.V. and I.S.L.). I.S.L. is supported by the Dwoskin and Anthony Rizzo Families Foundations and Jaime Erin Follicular Lymphoma Research Consortium. R.E.V. is supported R01GM121595 from the National Institute of General Medical Science to REV and funds from the University of Miami SCCC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Dr. Verdun and Dr. Lossos have a patent related to this work entitled “DIAGNOSIS AND TREATMENT OF LYMPHOMA AND LYMPHOPROLIFERATIVE DISORDERS WITH PARP INHIBITORS”. The authors have no additional financial interests to declare.
REFERENCES
- Agostinelli C, Paterson JC, Gupta R, Righi S, Sandri F, Piccaluga PP, Bacci F, Sabattini E, Pileri SA, and Marafioti T (2012). Detection of LIM domain only 2 (LMO2) in normal human tissues and haematopoietic and non-haematopoietic tumours using a newly developed rabbit monoclonal antibody. Histopathology 61, 33–46. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Gentles AJ, Lossos IS, and Levy R (2009). Molecular outcome prediction in diffuse large-B-cell lymphoma. The New England journal of medicine 360, 2794–2795. [DOI] [PubMed] [Google Scholar]
- Armitage JO, and Weisenburger DD (1998). New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 16, 2780–2795. [DOI] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, and Stark JM (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al. (2011). Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Molecular cell 42, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, and Nussenzweig MC (2010). 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med 207, 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 17, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton S, Coates J, and Jackson SP (2013). A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol 202, 579–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. (2009). A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Molecular cell 35, 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J, and Rabbitts TH (2015). LMO2 at 25 years: a paradigm of chromosomal translocation proteins. Open Biol 5, 150062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Taylor MR, and Boulton SJ (2012). Playing the end game: DNA double-strand break repair pathway choice. Molecular cell 47, 497–510. [DOI] [PubMed] [Google Scholar]
- Chapuy B, Cheng H, Watahiki A, Ducar MD, Tan Y, Chen L, Roemer MG, Ouyang J, Christie AL, Zhang L, et al. (2016). Diffuse large B-cell lymphoma patient-derived xenograft models capture the molecular and biological heterogeneity of the disease. Blood 127, 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et al. (2018). Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med 24, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, Morel P, Van Den Neste E, Salles G, Gaulard P, et al. (2002). CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. The New England journal of medicine 346, 235–242. [DOI] [PubMed] [Google Scholar]
- Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, Lefort S, Marit G, Macro M, Sebban C, et al. (2010). Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood 116, 2040–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortizas EM, Zahn A, Safavi S, Reed JA, Vega F, Di Noia JM, and Verdun RE (2016). UNG protects B cells from AID-induced telomere loss. J Exp Med 213, 2459–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubedo E, Gentles AJ, Huang C, Natkunam Y, Bhatt S, Lu X, Jiang X, Romero-Camarero I, Freud A, Zhao S, et al. (2012). Identification of LMO2 transcriptome and interactome in diffuse large B-cell lymphoma. Blood 119, 5478–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, and Sung P (2014). 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol 34, 1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport J, Neale GA, and Goorha R (2000). Identification of genes potentially involved in LMO2-induced leukemogenesis. Leukemia 14, 1986–1996. [DOI] [PubMed] [Google Scholar]
- de Miranda NF, Peng R, Georgiou K, Wu C, Falk Sorqvist E, Berglund M, Chen L, Gao Z, Lagerstedt K, Lisboa S, et al. (2013). DNA repair genes are selectively mutated in diffuse large B cell lymphomas. J Exp Med 210, 1729–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. (2013). Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 339, 711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Omari K, Hoosdally SJ, Tuladhar K, Karia D, Vyas P, Patient R, Porcher C, and Mancini EJ (2011). Structure of the leukemia oncogene LMO2: implications for the assembly of a hematopoietic transcription factor complex. Blood 117, 2146–2156. [DOI] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. (2013). A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular cell 49, 872–883. [DOI] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, et al. (2009). Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. The New England journal of medicine 361, 123–134. [DOI] [PubMed] [Google Scholar]
- Gautier L, Cope L, Bolstad BM, and Irizarry RA (2004). affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315. [DOI] [PubMed] [Google Scholar]
- Grutz GG, Bucher K, Lavenir I, Larson T, Larson R, and Rabbitts TH (1998). The oncogenic T cell LIM-protein Lmo2 forms part of a DNA-binding complex specifically in immature T cells. The EMBO journal 17, 4594–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habermann TM, Weller EA, Morrison VA, Gascoyne RD, Cassileth PA, Cohn JB, Dakhil SR, Woda B, Fisher RI, Peterson BA, and Horning SJ (2006). Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 24, 3121–3127. [DOI] [PubMed] [Google Scholar]
- Hirose K, Inukai T, Kikuchi J, Furukawa Y, Ikawa T, Kawamoto H, Oram SH, Gottgens B, Kiyokawa N, Miyagawa Y, et al. (2010). Aberrant induction of LMO2 by the E2A-HLF chimeric transcription factor and its implication in leukemogenesis of B-precursor ALL with t(17;19). Blood 116, 962–970. [DOI] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. (2018). Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell reports 23, 239–254 e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, Zhao H, Xu W, Rosenwald A, et al. (2008a). Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319, 1676–1679. [DOI] [PubMed] [Google Scholar]
- Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al. (2008b). Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A 105, 13520–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2016). BRCAness revisited. Nat Rev Cancer 16, 110–120. [DOI] [PubMed] [Google Scholar]
- Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D, and Levy R (2004). Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. The New England journal of medicine 350, 1828–1837. [DOI] [PubMed] [Google Scholar]
- Ma S, Guan XY, Beh PS, Wong KY, Chan YP, Yuen HF, Vielkind J, and Chan KW (2007). The significance of LMO2 expression in the progression of prostate cancer. J Pathol 211, 278–285. [DOI] [PubMed] [Google Scholar]
- Malumbres R, Chen J, Tibshirani R, Johnson NA, Sehn LH, Natkunam Y, Briones J, Advani R, Connors JM, Byrne GE, et al. (2008). Paraffin-based 6-gene model predicts outcome in diffuse large B-cell lymphoma patients treated with R-CHOP. Blood 111, 5509–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack MP, and Rabbitts TH (2004). Activation of the T-cell oncogene LMO2 after gene therapy for X-linked severe combined immunodeficiency. The New England journal of medicine 350, 913–922. [DOI] [PubMed] [Google Scholar]
- McDonnell SR, Hwang SR, Basrur V, Conlon KP, Fermin D, Wey E, Murga-Zamalloa C, Zeng Z, Zu Y, Elenitoba-Johnson KS, and Lim MS (2012). NPM-ALK signals through glycogen synthase kinase 3beta to promote oncogenesis. Oncogene 31, 3733–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. (2010). Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42, 181–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, and Pommier Y (2012). Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, and Pommier Y (2014). Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther 13, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam CH, and Rabbitts TH (2006). The role of LMO2 in development and in T cell leukemia after chromosomal translocation or retroviral insertion. Mol Ther 13, 15–25. [DOI] [PubMed] [Google Scholar]
- Natkunam Y, Farinha P, Hsi ED, Hans CP, Tibshirani R, Sehn LH, Connors JM, Gratzinger D, Rosado M, Zhao S, et al. (2008). LMO2 protein expression predicts survival in patients with diffuse large B-cell lymphoma treated with anthracycline-based chemotherapy with and without rituximab. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 26, 447–454. [DOI] [PubMed] [Google Scholar]
- Natkunam Y, Zhao S, Mason DY, Chen J, Taidi B, Jones M, Hammer AS, Hamilton Dutoit S, Lossos IS, and Levy R (2007). The oncoprotein LMO2 is expressed in normal germinal-center B cells and in human B-cell lymphomas. Blood 109, 1636–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale GA, Mao S, Parham DM, Murti KG, and Goorha RM (1995). Expression of the proto-oncogene rhombotin-2 is identical to the acute phase response protein metallothionein, suggesting multiple functions. Cell Growth Differ 6, 587–596. [PubMed] [Google Scholar]
- Ochs F, Somyajit K, Altmeyer M, Rask MB, Lukas J, and Lukas C (2016). 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol 23, 714–721. [DOI] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, and Jasin M (1999). XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13, 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, O’Connor MJ, and de Bono J (2016). Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 8, 362ps317. [DOI] [PubMed] [Google Scholar]
- Pratt G, Yap C, Oldreive C, Slade D, Bishop R, Griffiths M, Dyer MJS, Fegan C, Oscier D, Pettitt A, et al. (2018). A multi-centre phase I trial of the PARP inhibitor olaparib in patients with relapsed chronic lymphocytic leukaemia, T-prolymphocytic leukaemia or mantle cell lymphoma. Br J Haematol 182, 429–433. [DOI] [PubMed] [Google Scholar]
- Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L, Karjalainen-Lindsberg ML, et al. (2017). Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 171, 481–494 e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson C, Moynahan ME, and Jasin M (1998). Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev 12, 3831–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al. (2002). The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. The New England journal of medicine 346, 1937–1947. [DOI] [PubMed] [Google Scholar]
- Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. (2018). Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. The New England journal of medicine 378, 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, and Takeda S (1998). Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. The EMBO journal 17, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumerai JD, Zelenetz AD, Moskowitz CH, Palomba ML, Hamlin PA Jr., Noy A, Straus DJ, Moskowitz AJ, Younes A, Matasar MJ, et al. (2017). The PARP Inhibitor Veliparib Can Be Safely Added to Bendamustine and Rituximab and Has Preliminary Evidence of Activity in B-Cell Lymphoma. Clin Cancer Res 23, 4119–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JE, and Schrader CE (2008). Mechanism and regulation of class switch recombination. Annu Rev Immunol 26, 261–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe P, van Grotel M, Beverloo HB, Lee C, Helgason T, Buijs-Gladdines J, Passier M, van Wering ER, Veerman AJ, Kamps WA, et al. (2006). The cryptic chromosomal deletion del(11)(p12p13) as a new activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood 108, 3520–3529. [DOI] [PubMed] [Google Scholar]
- Venkitaraman AR (2004). Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat Rev Cancer 4, 266–276. [DOI] [PubMed] [Google Scholar]
- Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P, et al. (2010). The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 116, 4578–4587. [DOI] [PubMed] [Google Scholar]
- Williamson CT, Kubota E, Hamill JD, Klimowicz A, Ye R, Muzik H, Dean M, Tu L, Gilley D, Magliocco AM, et al. (2012). Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol Med 4, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenetz AD, Gordon LI, Wierda WG, Abramson JS, Advani RH, Andreadis CB, Bartlett N, Byrd JC, Fayad LE, Fisher RI, et al. (2016). Diffuse Large B-Cell Lymphoma Version 1.2016. J Natl Compr Canc Netw 14, 196–231. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, and de Lange T (2013). 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 339, 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new dataset for analysis. Previously published datasets analyzed in this study are referenced in the manuscript.