

Abstract
A new family of cyclic cell-penetrating peptides (CPPs) is discovered and differs from previously reported cyclic CPPs by containing only a single hydrophobic residue. The optimal CPP structure consists of four arginine residues and a hydrophobic residue of long alkyl chain (e.g., a decyl group) in a cyclohexapeptide ring. The most active member of this family, CPP 17, has an intrinsic cellular entry efficiency similar to that of cyclic CPP12, the most active CPP reported to date. However, CPP 17 is 2.8-fold more active than CPP12 under high serum protein concentrations, presumably because of less protein binding. CPP 17 enters the cell primarily by direct translocation at relatively low concentration (≥5 μM).
Keywords: cell-penetrating peptide, cyclic peptide, drug delivery, permeability
Graphical Abstract
A new family of cyclic cell-penetrating peptides with high cytosolic delivery efficiency and lower sensitivity to inhibition by serum proteins.
The cell membrane presents a major barrier in drug discovery, especially for biologics such as peptides, proteins and nucleic acids. Over the past three decades, cell-penetrating peptides (CPPs) have been intensely investigated as a potential solution to intracellular delivery of biologics.[1–3] However, conventional linear CPPs have low cytosolic delivery efficiency as well as poor metabolic stability, bioavailability, and biodistribution, hampering their clinical application. These limitations prompted us and other researchers to explore cyclic peptides as CPPs, since the latter should at least have improved proteolytic stability. Cyclization of CPPs (e.g., Tat and R9) substantially enhances their cellular uptake efficiency, in addition to improved metabolic stability.[4–6] In particular, we discovered a family of small, amphipathic cyclic peptides as exceptionally active CPPs, exhibiting up to 120% cytosolic delivery efficiency (which is defined as the ratio of cytosolic versus extracellular concentration), good bioavailability, and broad biodistribution.[7–9] These cyclic CPPs, as exemplified by cyclo(Phe-Nal-Arg-Arg-Arg-Arg-Gln) (CPP1, where Nal is L-2-naphthylalanine), cyclo(phe-Nal-Arg-arg-Arg-arg-Gln) (CPP9, where arg is D-arginine and phe is D-phenylalanine), and cyclo(Phe-phe-Nal-Arg-arg-Arg-arg-Gln) (CPP12), typically have four arginine residues, two (or occasionally three) hydrophobic residues, and a ring size of 7 or 8 residues. In this study, we explored cyclic CPPs containing a single hydrophobic group and different ring sizes, with the goal of understanding the structure-activity relationship (SAR) of cyclic CPPs and potentially discovering new CPPs of improved properties.
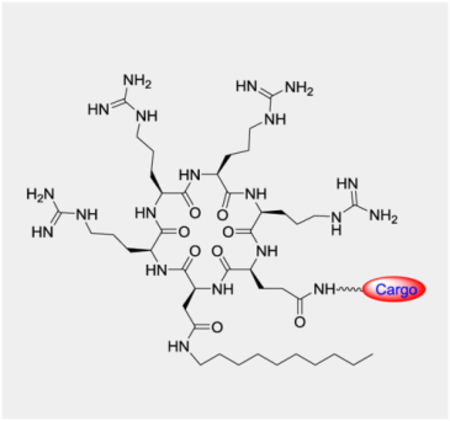
We previously showed that both hydrophobic and arginine residues are required for high cellular entry efficiency of cyclic CPPs.[7] To determine whether both (or all three) hydrophobic residues are required for high CPP activity, we synthesized a series of cyclohexapeptides in the form of cyclo(X-Arg-Arg-Arg-Arg-Gln) where X is an aromatic hydrophobic amino acid or L-2,3-diaminopropionic acid (Dap) derivatized with alkyl or aryl hydrophobic groups of increasing sizes (Figure 1A, peptides 1–8). Each peptide was labeled with naphthofluorescein (NF) at the Gln side chain through a long, flexible linker, miniPEG-Lys. HeLa cells were treated with 5 μM peptide for 2 h in the presence of 10% fetal bovine serum (FBS) and the cellular entry efficiency of the peptides was quantitated by flow cytometry analysis. NF is pH sensitive (pKa = 7.8) and fluorescent in the neutral cytosol and nucleus (pH 7.4) but nonfluorescent inside the acidic endosomal/lysosomal compartment (pH 4.5–6.5). The mean fluorescence intensity (MFI) values as determined by flow cytometry therefore reflect the cytosolic entry efficiencies of the CPPs.[9,10] It is clear that the CPP activity increases with the size of the hydrophobic group (Figure 1B). Cyclic peptide 4, which contains a decanoyl group is substantially more active than peptides 2 and 3, which have hexanoyl and octanoyl groups, respectively. Likewise, the CPP activity also increases with the size of the aryl groups for cyclic peptides 1, 5, 6, 7, and 8. Remarkably, the most active peptide of this series, 4, has a cellular entry efficiency that is 58% of CPP12, the most active cyclic CPP reported to date.[2] These results demonstrate that a single hydrophobic group of the proper structure is both necessary and sufficient for high CPP activity.
Figure 1.

Structures (A) and cytosolic entry efficiencies (B) of cyclic peptides 1–8 into HeLa cells in the presence of 10% FBS as determined by flow cytometry. MFI values reported represent the mean ± SD of at least three independent experiments and relative to that of CPP12 (100%).
We previously observed that variation of the stereochemistry of cyclic CPPs produced diastereomers of improved cellular entry efficiencies (e.g., CPP9 and CPP12).[9] We attempted to improve the activity of peptide 4 by changing the stereochemistry of its arginine residues from all-L configuration to LDLD and DLDL configurations (Figure 2, peptides 9 and 10). Interestingly, both stereochemical variations substantially decreased the CPP activity (by ~3-fold; Figure 3A). To determine whether all four arginine residues are required for CPP activity, we replaced the arginine distal or adjacent to the hydrophobic residue with alanine, producing peptides 11 and 12, respectively (Figure 2). Both substitutions reduced the CPP activity by ~2-fold (Figure 3b). Thus, in agreement with our earlier finding,[7,9] three arginine residues are sufficient to generate a functional CPP, but four arginine residues are optimal for CPP activity. To examine the effect of ring size on CPP activity, we deleted the alanine residue of 11 (or 12) to give cyclopentapeptide 13 and inserted a fifth arginine into peptide 4 to produce cycloheptapeptide 14 (Figure 2). Comparison of cyclic peptides 4 and 11-14 reveals that the cyclohexapeptide ring is optimal for CPP activity; either contraction (13) or expansion of the CPP ring (14) dramatically decreases the cellular entry efficiency (by ~10- and 5-fold, respectively) (Figure 3A). We have previously shown that expansion of the ring of cyclic CPP1 by inserting increasing numbers of alanine residues resulted in progressive loss of its CPP activity.[7] In the previous study, however, the smallest ring tested was a cycloheptapeptide, which is necessary to accommodate four arginine residues, two hydrophobic residues, and a Gln for cargo attachment. We hypothesize that a cyclohexapeptide ring may have the proper balance between conformational rigidity and spatial presentation of the arginine and hydrophobic groups for optimal interaction with the plasma and endosomal membranes and stabilization of the negative Gaussian curvature at the budding neck during endosomal escape.[9,11] Larger rings (e.g., 14) may be too flexible to bind tightly to the plasma and/or endosomal membranes, while smaller rings (e.g., 13) may be too rigid, locking the arginine and hydrophobic side chains out of the optimal conformation(s) for membrane binding.
Figure 2.

Structure of cyclic peptides 9–17
Figure 3.

Cytosolic entry efficiencies of cyclic peptides 4 and 9-17 into HeLa cells in the presence of 10% (A) or 1% FBS (B) as determined by flow cytometry. MFI values reported represent the mean ± SD of at least three independent experiments and relative to that of CPP12 (100%).
Next, we elongated the hydrophobic side chain of residue X by replacing the Dap residue of peptide 4 with ornithine or lysine, anticipating that the latter might allow the decanoyl group to insert more deeply into the lipid bilayer and increase the membrane-binding affinity (Figure 2, peptides 15 and 16). Indeed, substitution of ornithine increased the CPP activity by 2-fold, while lysine had a smaller effect (1.3-fold) (Figure 3A). Finally, we replaced the Dap residue of peptide 4 with an aspartic acid and the acid functionality was then amidated with decylamine to give cyclic peptide 17 (Figure 2). Gratifyingly, peptide 17 showed ~5-fold improvement in cellular entry efficiency relative to peptide 4 and is 2.8-fold more active than CPP12 when assayed in the presence of 10% FBS (Figure 3A). This increase in CPP activity relative to peptide 4 is likely due to elongation of the hydrocarbon length by one carbon atom (10 vs 9 atoms). We anticipate that still longer hydrocarbon chains (e.g., fatty acyl or alkylamino groups of ≥11 carbon atoms) may further increase the CPP activity, but may cause cytotoxicity and aqueous solubility problems.[12–14] Variation of the stereochemistry of cyclic peptide 16 and 17 greatly reduced their CPP activities (data not shown).
The “intrinsic” cytosolic entry efficiencies of peptides 4, 15-17, and CPP12 were tested again with HeLa cells, but in the presence of reduced serum concentration (1% FBS). The absolute MFI values of the treated cells were much greater than the corresponding values measured in the presence of 10% FBS. For CPP12, increase of the FBS concentration from 1% to 10% decreased the cellular entry efficiency by ~25-fold, primarily as the result of slowed uptake kinetics, whereas the corresponding reduction was ~7-fold for peptide 17. In the presence of 1% FBS, CPP12 is the most active CPP (100%), followed by peptides 17 (94%), 15 (58%), 4 (44%), and 16 (43%) (Figure 3B). Thus, the higher activity of peptides 15 and 17 (relative to CPP12) in the presence of 10% FBS is likely the result of less binding to serum proteins and therefore greater availability for membrane binding and cellular entry. The reduced sensitivity of peptides 15 and 17 to serum proteins may result in greater efficacy during in vivo applications, a notion that will be experimentally tested in due course.
Cellular entry by peptide 17 was also examined by live-cell confocal microscopy. Treatment of HeLa cells with 5 μM fluorescein isothiocyanate (FITC)-labeled peptide 17 for 2 h resulted in diffuse fluorescence throughout the cell volume (Figure 4A). Small intensely fluorescent spots are also visible, usually at the cell surface. These bright fluorescent spots are already formed at the cell surface 3 min after the addition of the peptide to the cells (Figure 4B). At ≥6 min, intense, diffuse fluorescence accumulated inside the cells, which became more intensely fluorescent than the surrounding environment. Similar cellular entry efficiency and intracellular fluorescence distribution were observed when peptide 17 was labeled with positively charged tetramethylrhodamine (TMR) or a negatively charged dye, Alexa488 (Figure S1 in Supporting Information). The rapid internalization of peptide 17 and the presence of bright fluorescent spots at the cell surface suggest that peptide 17 enters the cell primarily through direct translocation, a well-established route of cellular entry by some CPPs, usually ocurring at high CPP concentrations.[11] The bright fluorescent spots at the cell surface have been described as “nucleation zones“.[15,16] Under similar conditions, cyclic CPP1, CPP9, and CPP12 enter cells primarily through endocytosis, followed by endosomal escape.[7–9]
Figure 4.

Confocal microscopic images of HeLa cells after treatment with 5 μM FITC-labeled peptide 17. (A) Cells after 2 h treatment and washing. I, Green fluorescence of FITC-labeled peptide 17; II, DIC. (B) Time-lapse images of cells (no washing) at indicated time points following the addition of FITC-labeled peptide 17. All experiments were performed in the presence of 1% FBS. Scale bars = 20 μm.
Peptide 17 was tested for potential cytotoxicity against HeLa cells. It resulted in a small (but not concentration-dependent) reduction of cell viability (~30%) at ≤25 μM concentration as judged by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Figure S2). The origin of this apparent loss of viability is yet unclear. Higher concentrations of peptide 17 (≥50 μM) resulted in significant loss of HeLa cell viability.
In summary, we have discovered a new family of highly efficient cyclic CPPs consisting of three and preferably four arginine residues and a single hydrophobic residue. The most preferred hydrophobic group is a long-chain alkyl or fatty acyl group and the optimal ring size is a cyclohexapeptide. The most active member of this family, peptide 17, has an intrinsic cellular entry efficiency rivaling that of cyclic CPP12, but outperforms CPP12 in the presence of high concentrations of FBS. Peptide 17 appears to enter cells by direct translocation at relatively low concentration (≥5 μM).
While this paper was in preparation, Burlina and co-workers reported a family of structurally similar cyclic CPPs, which enter the mammalian cell by a combination of endocytosis and direct translocation mechanisms.[17]
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health (GM122459) and Entrada Therapeutics. J.S. was supported by a visiting professorship from China Scholarship Council.
Footnotes
Conflict of Interest
D.P. is a co-Founder and shareholder of Entrada Therapeutics.
References
- [1].Milletti F, Drug Discovery Today 2012, 17, 850–860. [DOI] [PubMed] [Google Scholar]
- [2].Gallo M, Defaus S, Andreu D, Arch. Biochem. Biophys 2019, 661, 74–86. [DOI] [PubMed] [Google Scholar]
- [3].Guidotti G, Brambilla L, Rossi D, Trends Pharmacol. Sci 2017, 38, 406–424. [DOI] [PubMed] [Google Scholar]
- [4].Lättig-Tünnemann G, Prinz M, Hoffmann D, Behlke J, Palm-Apergi C, Morano I, Herce HD, Cardoso MC, Nat. Commun 2011, 2, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mandal D, Nasrolahi Shirazi A, Parang K, Angew. Chem. Int. Ed 2011, 50, 9633–9637. [DOI] [PubMed] [Google Scholar]
- [6].Horn M, Reichart F, Natividad-Tietz S, Diaz D, Neundorf I, Chem. Commun 2016, 52, 2261–2264. [DOI] [PubMed] [Google Scholar]
- [7].Qian Z, Liu T, Liu Y-Y, Briesewitz R, Barrios AM, Jhiang SM, Pei D, ACS Chem. Biol 2013, 8, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Qian Z, LaRochelle JR, Jiang B, Lian W, Hard RL, Selner NG, Luechapanichkul R, Barrios AM, Pei D, Biochemistry 2014, 53, 4034–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, Phelps MA, Rossman JS, Pei D, Biochemistry 2016, 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Qian Z, Dougherty PG, Pei D, Chem. Commun 2015, 51, 2162–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dougherty PG, Sahni A, D. Pei. Chem. Rev 2019, 119, ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Katayama S, Hirose H, Takayama K, Nakase I, Futaki S, J. Controlled Release 2011, 149, 29–35. [DOI] [PubMed] [Google Scholar]
- [13].Bode SA, Thevenin M, Bechara C, Sagan S, Bregant S, Lavielle S, Chassaing G, Burlina F, Chem. Commun 2012, 48, 7179–7181. [DOI] [PubMed] [Google Scholar]
- [14].Lehto T, Vasconcelos L, Margus H, Figueroa R, Pooga M, Hällbrink M, Langel Ü, Bioconjugate Chem. 2017, 28, 782–795. [DOI] [PubMed] [Google Scholar]
- [15].Duchardt F, Fotin-Mleczek M, Schwarz H, Fischer R, Brock R, Traffic 2007, 8, 848–866. [DOI] [PubMed] [Google Scholar]
- [16].Hirose S, Takeuchi T, Osakada H, Pujals S, Katayama S, Nakase I, Kobayashi S, Haraguchi T, Futaki S, Mol. Ther 2012, 20, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Amoura M, Illien F, Joliot A, Guitot K, Offer J, Sagana S, Burlina F, Chem. Commun 2019, 55, 4566–4569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.