

ABSTRACT
Chronic kidney disease (CKD) is a common public health problem worldwide characterized by gradual decline of renal function over months/years accompanied by renal fibrosis and failure in tissue wound healing after sustained injury. Patients with CKD frequently present with profound signs/symptoms that require medical treatment, mostly culminating in hemodialysis and renal transplantation. To prevent CKD more efficiently, there is an urgent need for better understanding of the pathogenic mechanisms and molecular pathways of the disease pathogenesis and progression, and for developing novel therapeutic targets. Recently, several lines of evidence have shown that epigallocatechin-3-gallate (EGCG), an abundant phytochemical polyphenol derived from Camellia sinensis, might be a promising bioactive compound for prevention of CKD development/progression. This review summarizes current knowledge of molecular mechanisms underlying renoprotective roles of EGCG in CKD based on available preclinical evidence (from both in vitro and in vivo animal studies), particularly its antioxidant property through preservation of mitochondrial function and activation of Nrf2 (nuclear factor erythroid 2-related factor 2)/HO-1 (heme oxygenase-1) signaling, anti-inflammatory activity, and protective effect against epithelial mesenchymal transition. Finally, future perspectives, challenges, and concerns regarding its clinical use in CKD and renal fibrosis are discussed.
Keywords: Camellia sinensis, CKD, diabetic nephropathy, EGCG, glomerulonephritis, lupus nephritis, renoprotection, tea
Introduction
Chronic kidney disease (CKD) is one of the major noncommunicable diseases worldwide, especially in the aging population (1, 2). A deceptive feature of renal failure, it can remain unnoticed by patients until the disease progresses, making CKD one of the most challenging diseases to diagnose, and is considered a silent killer (3). Progression of CKD has been thought to be initiated by renal injury, followed by a defective repair process that subsequently causes impaired nephron function and compensatory response of the remaining nephrons to maintain renal function (4). However, a vicious cycle of these alterations continues, resulting in mechanical and metabolic stresses and ultimately loss of the nephron (4). Because of the increasing expenditure on CKD, much greater efforts have been made during the last decade toward its prevention (5).
Epigallocatechin-3-gallate (EGCG) is the most abundant catechin derived from the tea plant (Camelliasinensis), especially green tea, which contains ∼77.8 mg EGCG per gram of dried leaves (accounting for 60% of all catechins found in tea polyphenols) (6). Computational molecular docking and X-ray crystallographic studies have revealed that EGCG can anchor a galloyl moiety in a protein cleft and interact with the protein through its hydroxyl groups, giving it greater biological activity than nongalloylated catechins (7). EGCG is widely recognized as a potent antioxidant that effectively eliminates intracellular free radicals by its phenol ring structure, which can act as an electron trap (8). In addition, anti-inflammatory, antiaging, and
antifibrosis properties of EGCG appear to involve several molecular signaling pathways and cellular machineries that are responsible for specified cellular functions under physiological and pathological conditions (9, 10). Therefore, EGCG has received wide attention for its beneficial roles in chronic diseases (e.g., cardiovascular and neurodegenerative diseases and various types of cancers), which frequently involve sustained oxidative stress and inflammation (11). Recently, several lines of evidence from various in vitro and in vivo animal studies have suggested that EGCG exerts renoprotection against CKD (12, 13). This review summarizes the current knowledge and recent progress made in this research area. Also discussed are future perspectives, challenges, and concerns over the clinical use of EGCG for CKD and renal fibrosis.
Preclinical Evidence for Molecular Mechanisms of EGCG in CKD and Renal Fibrosis
Antioxidant property of EGCG
Preservation of mitochondrial function
Mitochondria play several important roles in cellular adaptive responses to a stressful environment. In general, mitochondria are important for biosynthesis of macromolecules, regulation of calcium homeostasis, maintenance of cellular redox status, and regulation of apoptotic cell death (14). In addition to the heart, the kidney is another mitochondria-rich organ per tissue mass (15). Therefore, renal functions are highly dependent on mitochondrial performance to maintain body homeostasis (16). Moreover, the kidney is highly sensitive to cellular redox imbalance, resulting in overproduction of reactive oxygen species (ROS) and subsequently oxidative stress, which is critical for development of several kidney diseases (17, 18).
It has been hypothesized that mitochondrial dysfunction contributes to the progression of CKD, particularly in diabetic nephropathy (DN) (17, 19). Renoprotective effects of EGCG against mitochondrial dysfunction have been extensively studied in CKD. In rats with DN induced by streptozotocin injection and nephrectomy, oral administration of EGCG for 50 d suppressed hyperglycemia and proteinuria (20). Decreases in lipid peroxidation and advanced glycation end products (AGEs) were observed, indicating an improvement in mitochondrial oxidative stress (20). An in vitro study using human embryonic kidney (HEK) cells treated with high glucose concentration revealed that EGCG in combination with lipoic acid attenuated suppression of superoxide dismutase expression and ROS overproduction caused by high glucose (21). Regarding the role of mitochondria in regulating apoptotic cell death, a recent study has revealed antiapoptotic activity of EGCG in DN rats (22). Supplementation with EGCG suppressed expression of the apoptotic protein Bax and active caspase 3, but maintained expression of the antiapoptotic protein B cell lymphoma-2 (Bcl-2) in the diabetic kidney (22). Similar findings were observed in models of renal ischemia-reperfusion injury in rats (23) and unilateral ureteral obstruction (UUO) in mice (24).
Cisplatin has been used for chemotherapy in many solid tumors but can induce toxic renal effects. It is thus worth mentioning a protective role of EGCG against mitochondrial oxidative stress and dysfunction in cisplatin-induced nephrotoxicity (25). Patients who survive acute kidney injury (AKI) can have a 2-fold greater risk of developing CKD in their lifetime (26). In a mouse model of cisplatin-induced nephrotoxicity, EGCG preserved renal mitochondrial function by maintaining electron transport and mitochondrial antioxidant enzyme activities, leading to protection against inflammation and apoptotic cell death (27). In addition, EGCG reduced mitochondrial ROS production in HK-2 cells (27). Consistent with the previous study, combined treatment of EGCG and coenzyme Q10 in a rat model of cisplatin-induced renal damage attenuated oxidative stress, inflammation, and renal cell death (28). Additionally, EGCG attenuated cisplatin-induced nephrotoxicity in animals with solid Ehrlich ascites carcinoma by increasing concentration of the antioxidant glutathione (10). Likewise, EGCG protected renal cells against cisplatin-induced apoptosis by inhibiting Bax, while promoting Bcl-2 expression (29). Moreover, beneficial effects of EGCG on aging and kidney function have been also reported (30). Supplementation of EGCG for 6 mo in Fischer 344 rats reduced oxidative damage (by lowering H2O2 and malondialdehyde), increased mitochondrial membrane potential of lymphocytes, and increased antioxidant enzyme activities (30).
Activation of Nrf2/HO-1 signaling
Kelch-like ECH-associated protein 1 (Keap1) coordinates with nuclear factor erythroid 2-related factor 2 (Nrf2) to govern the defense mechanism against oxidative and electrophilic stresses (31). Under quiescent conditions, Keap1 quenches Nrf2 activity by forming an E3 ubiquitin ligase complex with cullin-3 (Cul3) that links Nrf2 with E2 ubiquitin-conjugating enzyme, which transfers ubiquitin to Nrf2 and targets Nrf2 for proteasomal degradation (32). However, exposure to an electrophile can directly modify the reactive cysteine residues inside Keap1, and consequently reduce the E3 ubiquitin ligase activity of the Keap1-Cul3 complex and release Nrf2 from E2 ubiquitin-conjugating enzyme (33). Nascent Nrf2 then translocates to the nucleus, where it combines with small musculoaponeurotic fibrosarcoma (sMAF) protein, and subsequently binds to the antioxidant response elements of a myriad cytoprotective genes and activates their transcripts (34). Prolonged activation of Nrf2 can exert the dark side of Nrf2 in cancer progression and chemoresistance (35, 36). However, this is not the case for CKD, in which pharmacological activation of Nrf2 is nowadays considered as one of the strategies to reduce oxidative stress and to prevent the disease progression (37).
EGCG has been documented as a phytochemical that can modulate Nrf2 signaling, resulting in upregulation of antioxidant or detoxifying enzymes (38). In the context of CKD, antioxidative activity of EGCG has been evaluated in lupus nephritis (LN) (39). In such a study, daily supplementation of EGCG in a mouse model of LN improved renal function and prevented proteinuria. In addition, oxidative stress parameters in serum, urine, and kidney were significantly lowered in the EGCG-treated mice. Moreover, nuclear translocation of Nrf2 was observed in the EGCG-treated mice in concordance with the increased expression or activities of its downstream phase II enzymes [i.e., NAD(P)H:quinone oxidoreductase, heme oxygenase-1 (HO-1) and glutathione peroxidase (GPx)]), indicating Nrf2 activation by electrophilic EGCG (39).
Similarly, the therapeutic potential of EGCG has been shown in a mouse model of crescentic glomerulonephritis (GN) (40). In this study, EGCG-treated mice showed improved renal function and histopathology. EGCG treatment also restored expression of nuclear Nrf2 and its downstream targets, including glutamate–cysteine ligase catalytic subunit, glutamate–cysteine ligase modifier subunit, and GPx (40). In addition, the renal glutathione expression level was increased whereas the renal malondialdehyde expression level was decreased in the EGCG-treated mice, suggesting a reduction of oxidative stress by Nrf2 restoration (40). These findings imply that EGCG could reactivate the Nrf2 pathway, thereby reversing the progression of crescentic GN.
In a mouse model of high-fat-diet–induced obesity, the EGCG-treated animals showed significant reduction in fasting blood glucose and AGE formation, most likely via Nrf2 activation as shown by the increased expression levels of nuclear Nrf2 and HO-1 (41). Interestingly, EGCG prevented DN in streptozotocin-induced diabetic mice via Nrf2 activation (42). Nrf2-knockout mice showed a complete abolition of such protective effects of ECGC against DN, resulting in sustained oxidative stress, inflammation, and fibrosis (42). In addition, direct evidence by molecular docking assay revealed that EGCG directly interacted with the Keap1 interior by hydrogen bonding (42). This direct interaction of EGCG with Keap1 was confirmed by a silencing assay using small interfering RNA (siRNA) specific to Keap1 (42).
Activation of Nrf2 by EGCG has been also reported in a UUO model of obstructive nephropathy (43, 44). Intraperitoneal injection of EGCG after unilateral ureteral ligation improved renal ultrastructure, especially in renal tubules (43). The amelioration of the renal injury was inferred from the increased expression of Nrf2 and its nuclear translocation as well as the increased expression of γ-glutamylcysteine synthetase (γ-GCS) in the kidney (43). Similarly, another study demonstrated that 14-d administration of EGCG in UUO mice ameliorated renal damage and renal dysfunction through activation of Nrf2 as shown by the increase in Nrf2 and its nuclear accumulation, resulting in enhanced HO-1 production (44). Moreover, a recent in vitro study has also shown that EGCG protects renal cells from oxidative damage induced by H2O2 through Nrf2 activation, resulting in increased γ-GCS expression (45).
Anti-inflammatory activity of EGCG
Uncontrolled inflammation is an intermediate mechanism in the development of many chronic diseases including CKD. The NF-κB pathway is recognized as a classical pathway regulating expression of proinflammatory cytokines/chemokines in response to inflammation (46). Both canonical and noncanonical NF-κB pathways have been discovered in various inflammatory kidney diseases (47, 48). However, the most common form of activated NF-κB stimulated by a pathological condition is the heterodimeric complex of p65/p50 (46). During the resting state, heterodimeric p65/p50 is kept inactive by the IκΒ inhibitory protein. Shortly after stimulation, inhibitor of kappa B (IκB) is phosphorylated by the NF-κB essential modulator (NEMO)-containing IκB kinase complex and is marked for degradation via proteasomes, resulting in nuclear translocation of p65/p50 and binding of the complex to promoter regions of genes encoding proinflammatory mediators (49, 50). Therefore, targeting the NF-κB signaling pathway in order to alleviate the inflammatory response and to restore a proper balance of anti-inflammatory signaling pathways is a strategic approach in the context of chronic diseases (51).
The anti-inflammatory property of EGCG has been reported in many models of CKD, including DN, immune-mediated kidney diseases, and renal fibrosis. In general, EGCG exhibits anti-inflammatory effects by inhibiting the NF-κB pathway, resulting in reduction of proinflammatory mediators and decreased recruitment of inflammatory cells. An in vitro study of AGE-induced inflammation of HEK cells and human mesangial cells revealed that both of these renal cells had significantly increased concentration of the proinflammatory cytokine TNF-α upon AGE treatment, but pretreatment with EGCG could attenuate such an increase (52). Interestingly, the inhibitory property of EGCG was comparable to that of L-165,041, a selective ligand for peroxisome proliferator-activated receptor delta (PPARδ) (52). In addition, both EGCG and L-165,041 induced PPARδ while suppressing expression of receptor of AGE (RAGE). Accordingly, EGCG suppressed activation of NF-κB as indicated by the decreased expression of nuclear NF-κB and the remaining IκB in the cytoplasm (52). However, this effect was abolished by siRNA specific to PPARδ. These findings indicate that the inhibitory effect of EGCG on AGE-induced inflammation is mediated through PPARδ activation (52). Similarly, EGCG in combination with alpha-lipoic acid had an anti-inflammatory effect on high glucose-treated HEK cells by reducing the production of proinflammatory cytokines (i.e., TNF-α and IL-6) and RAGE expression (21).
The anti-inflammatory action of EGCG has been also demonstrated in vivo using a rat model of streptozotocin-induced DN (20). EGCG treatment significantly inhibited NF-κB signaling, resulting in decreased expression levels of NF-κB and p-IκB-α as well as downstream enzymes, namely, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (20). EGCG treatment also ameliorated renal pathological changes (including mesangial matrix expansion and glomerular enlargement) and markedly reduced expression of inflammatory markers, including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) in the diabetic mice (42). However, such protective effects of EGCG were not found in the Nrf2-knockout diabetic mice, suggesting that the anti-inflammatory effects of EGCG were mediated through Nrf2 signaling (42).
Furthermore, the anti-inflammatory effects of EGCG have been demonstrated in immune-mediated CKD, including GN and LN. EGCG inhibited inflammation induced by anti–glomerular basement membrane antibody in 129/svJ GN mice (53). Histopathology showed improvement of glomerular and tubulointerstitial injury and significant reduction of macrophage and lymphocyte infiltration together with downregulation of osteopontin, a known marker for monocyte/macrophage recruitment, in the EGCG-treated mice (53). Additionally, p65/NF-κB molecules were stained strongly positive in the GN renal tissues, indicating NF-κB activation in GN. EGCG pretreatment ameliorated such NF-κB activation markers and also reduced iNOS expression (53). The same group of investigators later demonstrated that the inhibitory activity of EGCG against inflammation observed in crescentic GN was Nrf2-dependent (40). Another study in mesangial cells isolated from the kidney of mice with GN induced by systemic lupus erythematosus–like disease demonstrated that EGCG suppressed nuclear translocation of NF-κB induced by LPS and IFN-γ, resulting in downregulation of iNOS and proinflammatory mediators (54). Likewise, a reduction of renal expression of p65/NF-κB has been found in EGCG-treated LN mice (39). This was consistent with the decreased number of infiltrating immune cells (i.e., T-cells and macrophages). In addition, EGCG markedly reduced expression of nucleotide-binding domain, leucine-rich repeat-containing family, pyrin domain-containing 3 (NLRP3) and production of IL-1β and IL-18. This effect was confirmed by decreased expression of mature caspase-1 (p20 subunit), suggesting an inhibitory effect of EGCG on NLRP3-dependent inflammasome activation (39).
In an obstructive nephropathy model, EGCG inhibited NF-κB signaling as shown by significantly decreased expression levels of NF-κB and p-IκB, which were related to a decrease in DNA binding activity of NF-κB determined by electrophoretic mobility shift assay (44). The decreased expression levels of proinflammatory cytokines (i.e., TNF-α, IL-6, and IL-1β) were also observed in the EGCG-treated group (44). In addition, the same group of investigators demonstrated by histological staining of renal sections that EGCG inhibited infiltrating macrophages (55).
Besides CKD, a beneficial effect of EGCG on age-associated kidney inflammation has been demonstrated in healthy rats (56). Long-term consumption of EGCG decreased renal expression of NF-κB and proinflammatory mediators IL-6 and TNF-α in Wistar rats. Interestingly, these findings were correlated with the increase in longevity factors, sirtuin (SIRT1) and forkhead box O3 (FOXO3a), in liver and kidney as well as the extended lifespan of EGCG-treated rats. These data suggest that the anti-inflammatory activity of EGCG might be mediated by increasing SIRT1 and FOXO3a expression (56).
EGCG and other signaling pathways
Apart from the 2 major signaling pathways (Nrf2 and NF-κB pathways), EGCG can also regulate other signaling pathways. Pharmacological inhibition of PPAR-γ (an anti-inflammatory transcription factor) by using GW9662 diminished the renoprotective effect of EGCG (53). This finding was concomitant with an inability of EGCG to mitigate activation of Akt and extracellular signal-regulated kinases 1 and 2 (ERK1/2), suggesting that the anti-inflammatory property of EGCG in GN could be mediated at least through PPAR-γ, Akt, and mitogen-activated protein kinase (MAPK) pathways (53). EGCG also inhibited inflammation induced by both NF-κB activation and the phosphoinositide 3-kinase (PI3K)/Akt/mTOR pathway in a mouse model of GN (54). In this regard, EGCG blocked the PI3K/Akt/mTOR pathway as indicated by decreasing phospho-Akt and phospho-p70s6K (54). These results were further confirmed by another study providing evidence that the anti-inflammatory activity of EGCG in a model of GN was mediated by inhibiting multiple signaling pathways, including PI3K, Akt, and MAPK pathways [i.e., c-Jun N-terminal kinase (JNK), ERK1/2, and p38] (40).
Additionally, an antiapoptotic effect of EGCG has been demonstrated in UUO mice (24). Decreased expression levels of Bax, active caspase 3, and poly ADP-ribose polymerase were observed in the EGCG-treated mice, indicating inhibition of the apoptotic pathway (24). EGCG also inhibited activation of the MAPK pathway observed in the obstructed kidney as shown by decreasing phosphorylation of ERK, JNK, and p38 (24). In concordance, this inhibitory effect was confirmed in the TGF-β1–treated NRK-52E cell line (24). Taken together, the data suggest that EGCG can inhibit apoptosis of renal tubular cells in UUO mice by inactivation of the TGF-β1–induced MAPK pathway (24).
Anti–epithelial mesenchymal transition property of EGCG
Epithelial mesenchymal transition (EMT) is a process that normally occurs during embryogenesis, allowing phenotypic change of epithelial cells to acquire mesenchymal features to perform specific organ functions (57). However, EMT can occur under pathological conditions, for example, cancer metastasis and organ fibrogenesis (57, 58). The fibrogenic milieu is primed by sustained renal injury followed by an inflammatory response and activation/expansion of fibrogenic effector cells, for example, extracellular matrix (ECM)–producing cells (59, 60). These ECM-producing cells are of mesenchymal origin, including fibroblasts, myofibroblasts, fibrocytes, pericytes, and cells derived from transdifferentiation of tubular epithelial cells through EMT (61). Fibrotic mediators such as TGF-β1 are well documented to induce epithelial cells to undergo EMT, in which they lose their epithelial phenotypes [markers include E-cadherin, zonula occludens-1 (ZO-1), and cytokeratins] while gaining mesenchymal phenotypes [markers include α-smooth muscle actin (α-SMA) and vimentin] and contributing to ECM production (62, 63). Although a recent study has elucidated that EMT-derived myofibroblasts contributed only 5% to ECM production in renal fibrosis, it is imperative to prevent or inhibit such pathological cellular events (61).
The protective effect of EGCG against EMT has been reported in TGF-β1–treated renal tubular epithelial cells (64). EGCG reduced mesenchymal-like features of the TGF-β1–treated cells by restoring cytokeratin-18 and α-SMA expression. In addition, EGCG suppressed activation of Smad3, ERK1/2, and β-catenin, suggesting that its anti-EMT might be mediated through these signaling molecules (64). In the UUO mouse model (55, 65), EGCG alleviated renal injury and renal interstitial fibrosis (55). These findings were accompanied by a reduction of collagen production and α-SMA expression, and increase in expression of E-cadherin (55). An in vitro study showed that EGCG inhibited TGF-β1–induced Smad2/3 phosphorylation, and promoted Nrf2 expression and its nuclear accumulation in renal tubular cells (65). A similar effect was observed when Nrf2 was overexpressed. By contrast, Nrf2 knockdown abolished these beneficial effects of EGCG, as indicated by the increased expression of fibrotic proteins such as collagen and α-SMA (65). Collectively, these findings indicated that the anti-EMT property of EGCG against TGF-β1/Smad signaling was mediated through the Nrf2 pathway (65).
The antifibrotic effect of EGCG has been also investigated in another model of EMT induced by high concentration of oxalate (66). Pretreatment with EGCG suppressed expression of EMT markers (vimentin and fibronectin) whereas it maintained epithelial markers (E-cadherin, occludin, cytokeratin, and ZO-1) in the oxalate-treated renal tubular cells. In addition, EGCG significantly reduced oxalate-induced ROS overproduction and enhanced expression of an antioxidant catalase enzyme (66). Moreover, these beneficial effects of EGCG were abrogated by Nrf2 silencing, indicating that the protective mechanism of EGCG against oxalate-induced EMT was most likely due to its antioxidative property to reduce ROS overproduction through Nrf2 activation (66). In addition, EGCG exhibited its anti-EMT property in a rat model of cadmium-induced chronic renal injury and fibrosis (67). Supplementation with EGCG attenuated the increased expression levels of TGF-β1, p-Smad3, α-SMA, and vimentin. Interestingly, EGCG affected expression of several microRNAs regulated by TGF-β1/Smad signaling [TGF-β1–induced miR-21 and miR-192 were suppressed, whereas the antifibrotic miR-29 family (miR29a/b/c) was enhanced] (67).
Summary of Preclinical Evidence
Molecular targets of EGCG are mostly proteins that can be classified as membrane receptors, transcription factors, epigenetic regulators, inflammatory molecules, intracellular enzymes, cell cycle–related proteins, apoptosis-related proteins, and others (68). Studies of molecular interactions of EGCG and proteins have revealed that EGCG modifies enzymatic activities and bioactivities of other proteins by occupying protein active sites (69). In addition, dynamic interactions between EGCG and proteins can affect many biological processes through regulation of transcription, posttranslational modifications, and epigenetic changes, which largely depend on cell types (70–72). In the context of CKD, previous and recent preclinical studies have indicated that EGCG could prevent CKD by several mechanisms, including preservation of mitochondrial function, activation of Nrf2/HO-1 signaling, anti-inflammation, and prevention of EMT (see the summary in Figure 1). Continuing efforts are underway to prevent and mitigate progression of CKD. Although the precise mechanisms of the disease progression remain to be clarified, aberrant oxidative stress and inflammation are recognized as the crucial players contributing to the renal damage in CKD (73). Therefore, maintaining redox balance in the kidney would provide a key strategy for defining new therapeutic interventions. Based on the molecular mechanisms of EGCG and its renoprotective effects, it might be possible to develop EGCG or its derivatives for treatment of CKD in 2 ways: 1) as an Nrf2 activator, and 2) as a mitochondria-targeting agent.
FIGURE 1.

Summary of molecular basis on which epigallocatechin-3-gallate (EGCG) prevents chronic kidney disease (CKD) and renal fibrosis based on preclinical evidence. The anti–oxidative stress properties of EGCG are exerted through its inhibition of mitochondrial dysfunction and its induction of antioxidant enzymes via activation of the Nrf2/HO-1 signaling pathway. For its anti-inflammation property, EGCG inhibits NF-κB signaling and inflammasome formation resulting in inhibition of the inflammatory cascade. Moreover, EGCG exhibits an anti-EMT property through various signaling pathways, including TGF-β1/Smad, β-catenin, and Nrf2 pathways, to prevent fibrotic transdifferentiation by the EMT process. Shaded boxes (in light orange) represent molecular events that occur in CKD, which can be inhibited or prevented by EGCG. ARE, antioxidant responsive element; Cul3, cullin-3; E2, E2 ubiquitin-conjugating enzyme; ECM, extracellular matrix; EMT, epithelial mesenchymal transition; GPx, glutathione peroxidase; HO-1, heme oxygenase 1; Keap1, Kelch-like ECH-associated protein 1; MDA, malondialdehyde; MPO, myeloperoxidase; NLRP3, nucleotide-binding domain, leucine-rich repeat-containing family, pyrin domain-containing 3; NQO1, NAD(P)H:quinone oxidoreductase 1; Nrf2, nuclear factor erythroid 2-related factor 2; ROS, reactive oxygen species; SOD, superoxide dismutase; TGF-β1, transforming growth factor-β1; Ub, ubiquitin; ZO-1, zonula occludens-1; α-SMA, α-smooth muscle actin; 8-OHdG, 8-hydroxy-2'-deoxyguanosine.
Challenges and Concerns for Clinical Use of EGCG
The health-promoting effects of EGCG involve many cellular and molecular mechanisms that remain poorly understood. Based on recent preclinical evidence, the Keap1-Nrf2 system has gained considerable attention as the therapeutic target to prevent progression of CKD because of its main function to protect cells from oxidative stress and inflammation (32, 37). Nrf2 activators or Keap1 inhibitors have been extensively investigated in many kidney disease models and some have already entered into clinical trials (74). A phase 2/3 clinical trial (CARDINAL study) of an Nrf2 activator, bardoxolone methyl (also known as CDDO-methyl ester), in CKD patients is in progress (74). Bardoxolone methyl is classified as an electrophilic Nrf2 activator, which can alter the Keap1–Cul3 interaction by direct complexing with Cys151 residing at the BTB (broad complex, tramtrack, and bric-a-brac) domain of Keap1 protein, resulting in an inability of Keap1–Cul3 to target Nrf2 for proteasomal degradation (33, 75). A covalent modification of thiol groups in Keap1 is thus crucial for regulation of Nrf2 signaling.
It has been demonstrated that EGCG can interact with specific residues of Keap1. In a rat model of fluoride-induced oxidative stress in lungs, EGCG has been shown to be docked in Keap1, forming interaction bonds with Gly343, Thr595, Leu578, and Asp579 (76). Another molecular docking analysis has revealed that EGCG can directly interact with Ser508, Ser555, Ser602, Tyr525, Tyr572, Gln530, and Arg483 inside Keap1 through hydrogen bonding (42). These interacting residues fall into P1, P3, and P4 subpockets within the kelch domain of Keap1, where interaction with ETGE and DLG motifs of Nrf2 takes place (77–79). Therefore, it is possible that EGCG can disrupt the Keap1–Nrf2 interaction and thereby activate Nrf2 signaling through this molecular docking. Alternatively, EGCG probably induces Nrf2 activation via Keap1-independent mechanisms (34, 80), for example, transcription regulation (via microRNAs), posttranslational modification (phosphorylation, sumoylation, acetylation, methylation, etc.), and epigenetic factors, that cannot be ruled out and deserve further investigations in CKD models. This sheds light onto the potential use of EGCG or its derivatives as Nrf2 activators for preventing renal damage and progression of CKD. Nonetheless, because a relation between Nrf2 signaling and disease pathology is sophisticated and context-dependent, it is imperative to interpret and evaluate the information cautiously.
Although the Nrf2-based therapy has proven to be beneficial, another interesting approach is the mitochondria-targeting therapy. It is conceived that mitochondrial bioenergetics stress, increasing mitochondrial fission, and ROS overproduction in adaptation to nephron loss contribute to the CKD progression (19). These alterations are well documented in the chronic stage of DN (81–83). Maintaining mitochondrial homeostasis in terms of its dynamics and bioenergetics could offer beneficial effects over downstream events like inflammation and fibrogenesis (14). A pharmacological strategy to improve mitochondrial dysfunction has been evaluated in animal models of both AKI and CKD (84). Based on their disparate modes of action, such compounds are classified as cardiolipin-targeting peptides, mitochondria-targeting antioxidants, mitochondria-homing agents, mitochondrial biogenesis-stimulating agents, and fatty acid oxidation-stimulating agents (84). Among these, Szeto–Schiller peptide (SS-31, also known as elamipretide) targeting cardiolipin has been tested in a phase 2a trial, with positive results showing that this compound could reduce postprocedural hypoxia, increase renal blood flow, and improve kidney function in patients with atherosclerotic renal artery stenosis (85). Although the mitochondria-targeting antioxidants might help to preserve or improve mitochondrial function in CKD, only MitoQ has been entered into a phase 4 clinical trial (NCT02364648) of stages 3–5 CKD patients.
Interestingly, EGCG is also known as a mitochondria-targeting agent because of its well-documented roles in regulating mitochondrial biogenesis, bioenergetics, and mitochondria-dependent apoptosis (86–91). For example, EGCG can increase mRNA expression level of PPAR-γ coactivator-1α (PGC-1α), a master regulator of mitochondrial biogenesis, in HepG2 cells and can differentiate 3T3-L1 adipocytes by enhancing the activity of PGC-1α promoter (87). In a model of Down syndrome (DS), EGCG has been demonstrated to rescue oxidative phosphorylation and promote mitochondrial biogenesis in DS-phenotypically characterized fetal skin fibroblasts via cAMP/protein kinase A (PKA)- and SIRT1-dependent pathways (88). Similar results were obtained when EGCG or resveratrol (another natural polyphenol) were tested in neural progenitor cells from a DS mouse model (90). However, there are few experimental studies to support the protective effect of EGCG on renal mitochondria. Interestingly, a recent study has unveiled that a structurally modified EGCG prodrug (peracetylated EGCG, or AcEGCG), also known as 4″-O-alkyl AcEGCG derivative, can increase mitochondrial mass and mitochondrial DNA content and markedly enhance mitochondrial biogenesis (92). As an enhancer of mitochondrial biogenesis, 4″-O-alkyl AcEGCG could enhance expression of many regulators for mitochondrial function (i.e., PGC-1α, phospho-5' adenosine monophosphate-activated protein kinase (p-AMPK), SIRT1, estrogen-related receptor-α, nuclear respiratory factor 1, nuclear respiratory factor 2, and mitochondrial transcription factor A) (92). Additionally, it could increase the NAD+/NADH ratio, cytochrome c concentration, ATP synthesis, and oxygen consumption in Hepa1-6 cells (92). These findings underscore that an in-depth knowledge of compound structures and mitochondrial activities in combination with further development of powerful tools that can directly assess mitochondrial function is necessary for mitochondria-targeting pharmacotherapy in CKD.
Nonetheless, there is a big gap before EGCG is implemented in clinical use. Because of the low stability, poor absorption, and low bioavailability of EGCG (93, 94), clinical trials are quite sophisticated and translation of preclinical findings to clinical practice is currently handicapped. Recently, nanotechnology-based delivery systems, including metal-based and organic-based nanoparticles, have been applied to enhance EGCG bioavailability to overcome these difficulties (95, 96). A recent study in rats with nephrotic syndrome has shown improved bioavailability of EGCG when using EGCG-polymeric nanoparticles compared with free EGCG powder, resulting in improved renal function, reduced renal damage, and reduced proteinuria (93). Although most of the preclinical studies using EGCG-loaded nanoparticles have provided promising results, there were some inconsistencies in terms of nanoparticle size, cytotoxicity effects at high doses, and pharmacokinetics. Furthermore, it is imperative to understand the pharmacokinetic interaction between EGCG and other drugs when EGCG is used as an adjuvant. However, such information is currently limited, especially at the level of the clinical trial (97, 98).
Unlike the progress made for clinical trials of EGCG in various cancers, there is currently no clinical study to demonstrate a renoprotective role of EGCG or green tea in CKD in humans. Besides the poor bioavailability of EGCG, the complexity of CKD in terms of extremely varied etiologies and natural history also contributes to this hurdle. Moreover, the pan-specific bioactivity of EGCG is also a concern (Figure 2). According to a substructure-based drug screening tool, EGCG is defined as one of the Pan-Assay INterference compoundS (PAINS) (99). EGCG contains a PAINS motif, catechol, which frequently triggers signals in various bioassays by different mechanisms (100). For example, catechol can undergo redox cycling, which causes formation of reactive chelators or orthoquinones that subsequently modify proteins, nucleic acids, or lipids (Figure 2). Such a property might also contribute to its cytotoxicity and affect its therapeutic efficacy (100). However, care must be taken in interpreting the PAINS alerts because the existence of a PAINS substructure does not always convey PAINS behavior in a compound, and some FDA-approved drugs and dark chemical matters (compounds that have been tested in 100 or more assays but have never shown any substantial biological activity) contain the PAINS motif in their structures (101–103).
FIGURE 2.
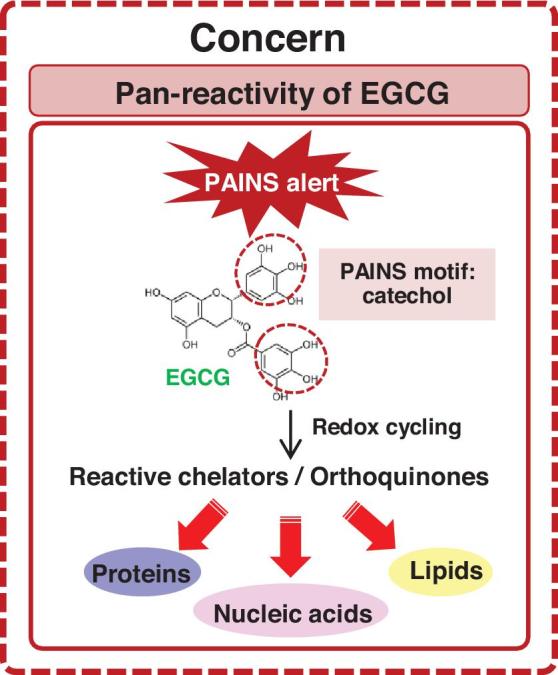
Potential pan-reactivity of epigallocatechin-3-gallate (EGCG). EGCG is one of the Pan-Assay INterference compoundS (PAINS) and contains a PAINS motif, catechol, that frequently triggers signals in various bioassays by different mechanisms. For example, catechol can undergo redox cycling that causes formation of reactive chelators or orthoquinones, which subsequently modify proteins, nucleic acids, or lipids.
In conclusion, although clinical research of EGCG in CKD is still immature, it is likely that EGCG might serve as a potential medication or adjuvant therapy for targeting many signaling pathways involved in CKD progression, including oxidative stress, inflammatory response, and energy sensing through mitochondria. Nonetheless, for safety, more efforts should be put into investigating specific targets of EGCG to minimize off-target effects and to better understand its mechanisms of action against CKD. More importantly, the clinical trials should be performed to precisely address whether EGCG is really beneficial for prevention of CKD and, if so, to what extent. Finally, connecting multiple pathways regulated by EGCG coupled with recent advances in drug delivery systems might accelerate the whole process to achieve the ultimate goal of effective prevention of CKD.
Acknowledgements
The authors’ responsibilities were as follows—both authors (RK and VT): drafted the manuscript, and read and approved the final manuscript.
Notes
This work was supported by a Mahidol University research grant and the Thailand Research Fund (IRN60W0004 and IRG5980006). Both authors are also supported by “Chalermphrakiat” and “Research Staff” grants from Faculty of Medicine Siriraj Hospital.
Author disclosures: RK and VT: no conflicts of interest.
Abbreviations used: AcEGCG, peracetylated epigallocatechin-3-gallate; AGE, advanced glycation end product; AKI, acute kidney injury; Bcl-2, B cell lymphoma-2; CKD, chronic kidney disease; Cul3, cullin-3; DN, diabetic nephropathy; DS, Down syndrome; ECM, extracellular matrix; EGCG, epigallocatechin-3-gallate; EMT, epithelial mesenchymal transition; ERK1/2, extracellular signal-regulated kinase 1 and 2; FOXO3, forkhead box O3; GN, glomerulonephritis; GPx, glutathione peroxidase; HEK, human embryonic kidney; HO-1, heme oxygenase-1; iNOS, inducible nitric oxide synthase; IκΒ, inhibitor of kappa B; JNK, c-Jun N-terminal kinase; Keap1, Kelch-like ECH-associated protein 1; LN, lupus nephritis; MAPK, mitogen-activated protein kinase; miR, microRNA; NLRP3, nucleotide-binding domain, leucine-rich repeat-containing family, pyrin-domain containing 3; Nrf2, nuclear factor erythroid 2-related factor 2; PI3K, phosphoinositide 3-kinase; PGC, PPAR-γ coactivator; PAINS, pan-assay interference compounds; PPAR, peroxisome proliferator-activated receptor; RAGE, receptor of AGE; ROS, reactive oxygen species; siRNA, small interfering RNA; SIRT1, sirtuin-1; TGF, transforming growth factor; UUO, unilateral ureteral obstruction; ZO-1, zonula occludens-1; α-SMA, alpha-smooth muscle actin; γ-GCS, γ-glutamylcysteine synthetase.
References
- 1. Ng JK, Li PK. Chronic kidney disease epidemic: how do we deal with it? Nephrology (Carlton) 2018;23(Suppl 4):116–20. [DOI] [PubMed] [Google Scholar]
- 2. Tonelli M, Riella M.. Chronic kidney disease and the ageing population. Nephron Clin Pract 2014;128:319–22. [DOI] [PubMed] [Google Scholar]
- 3. Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet 2017;389:1238–52. [DOI] [PubMed] [Google Scholar]
- 4. Schnaper HW. Remnant nephron physiology and the progression of chronic kidney disease. Pediatr Nephrol 2014;29:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Busink E, Canaud B, Schroder-Back P, Paulus ATG, Evers SMAA, Apel C, Bowry SK, Stopper A. Chronic kidney disease: exploring value-based healthcare as a potential viable solution. Blood Purif 2019;47:156–65. [DOI] [PubMed] [Google Scholar]
- 6. Hu J, Webster D, Cao J, Shao A. The safety of green tea and green tea extract consumption in adults—results of a systematic review. Regul Toxicol Pharmacol 2018;95:412–33. [DOI] [PubMed] [Google Scholar]
- 7. Nakano S, Megro SI, Hase T, Suzuki T, Isemura M, Nakamura Y, Ito S. Computational molecular docking and X-ray crystallographic studies of catechins in new drug design strategies. Molecules 2018;23(8):E2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu B, Yan W.. Lipophilization of EGCG and effects on antioxidant activities. Food Chem 2019;272:663–9. [DOI] [PubMed] [Google Scholar]
- 9. Chowdhury A, Sarkar J, Chakraborti T, Pramanik PK, Chakraborti S. Protective role of epigallocatechin-3-gallate in health and disease: a perspective. Biomed Pharmacother 2016;78:50–9. [DOI] [PubMed] [Google Scholar]
- 10. El Mowafy AM, Al Gayyar MM, Salem HA, El Mesery ME, Darweish MM. Novel chemotherapeutic and renal protective effects for the green tea (EGCG): role of oxidative stress and inflammatory-cytokine signaling. Phytomedicine 2010;17:1067–75. [DOI] [PubMed] [Google Scholar]
- 11. Shirakami Y, Sakai H, Kochi T, Seishima M, Shimizu M. Catechins and its role in chronic diseases. Adv Exp Med Biol 2016;929:67–90. [DOI] [PubMed] [Google Scholar]
- 12. Kanlaya R, Thongboonkerd V. Protective Effects of epigallocatechin-3-gallate from green tea in various kidney diseases. Adv Nutr 2019;10:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bao H, Peng A. The green tea polyphenol(−)-epigallocatechin-3-gallate and its beneficial roles in chronic kidney disease. J Transl Int Med 2016;4:99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bhargava P, Schnellmann RG.. Mitochondrial energetics in the kidney. Nat Rev Nephrol 2017;13:629–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Parikh SM, Yang Y, He L, Tang C, Zhan M, Dong Z. Mitochondrial function and disturbances in the septic kidney. Semin Nephrol 2015;35:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sharma K. Mitochondrial dysfunction in the diabetic kidney. Adv Exp Med Biol 2017;982:553–62. [DOI] [PubMed] [Google Scholar]
- 17. Pedraza-Chaverri J, Sanchez-Lozada LG, Osorio-Alonso H, Tapia E, Scholze A. New pathogenic concepts and therapeutic approaches to oxidative stress in chronic kidney disease. Oxid Med Cell Longev 2016;2016:6043601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gorin Y. The kidney: an organ in the front line of oxidative stress-associated pathologies. Antioxid Redox Signal 2016;25:639–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aparicio-Trejo OE, Tapia E, Sanchez-Lozada LG, Pedraza-Chaverri J. Mitochondrial bioenergetics, redox state, dynamics and turnover alterations in renal mass reduction models of chronic kidney diseases and their possible implications in the progression of this illness. Pharmacol Res 2018;135:1–11. [DOI] [PubMed] [Google Scholar]
- 20. Yamabe N, Yokozawa T, Oya T, Kim M. Therapeutic potential of (−)-epigallocatechin 3-O-gallate on renal damage in diabetic nephropathy model rats. J Pharmacol Exp Ther 2006;319:228–36. [DOI] [PubMed] [Google Scholar]
- 21. Leu JG, Lin CY, Jian JH, Shih CY, Liang YJ. Epigallocatechin-3-gallate combined with alpha lipoic acid attenuates high glucose-induced receptor for advanced glycation end products (RAGE) expression in human embryonic kidney cells. An Acad Bras Cienc 2013;85:745–52. [DOI] [PubMed] [Google Scholar]
- 22. Mohan T, Velusamy P, Chakrapani LN, Srinivasan AK, Singh A, Johnson T, Periandavan K. Impact of EGCG supplementation on the progression of diabetic nephropathy in rats: an insight into fibrosis and apoptosis. J Agric Food Chem 2017;65:8028–36. [DOI] [PubMed] [Google Scholar]
- 23. Lv J, Feng M, Zhang L, Wan X, Zeng YC, Liang PF, Xu AP. Protective effect of epigallocatechin gallate, a major constituent of green tea, against renal ischemia-reperfusion injury in rats. Int Urol Nephrol 2015;47:1429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y, Liu N, Bian X, Sun G, Du F, Wang B, Su X, Li D. Epigallocatechin-3-gallate reduces tubular cell apoptosis in mice with ureteral obstruction. J Surg Res 2015;197:145–54. [DOI] [PubMed] [Google Scholar]
- 25. Peres LA, da Cunha AD Jr. Acute nephrotoxicity of cisplatin: molecular mechanisms. J Bras Nefrol 2013;35:332–40. [DOI] [PubMed] [Google Scholar]
- 26. Bucaloiu ID, Kirchner HL, Norfolk ER, Hartle JE, Perkins RM. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int 2012;81:477–85. [DOI] [PubMed] [Google Scholar]
- 27. Pan H, Chen J, Shen K, Wang X, Wang P, Fu G, Meng H, Wang Y, Jin B. Mitochondrial modulation by epigallocatechin 3-gallate ameliorates cisplatin induced renal injury through decreasing oxidative/nitrative stress, inflammation and NF-kB in mice. PLoS One 2015;10:e0124775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fatima S, Al Mohaimeed N, Al Shaikh Y, Tyagi P, Banu N, Hasan S, Arjumand S. Combined treatment of epigallocatechin gallate and coenzyme Q10 attenuates cisplatin-induced nephrotoxicity via suppression of oxidative/nitrosative stress, inflammation and cellular damage. Food Chem Toxicol 2016;94:213–20. [DOI] [PubMed] [Google Scholar]
- 29. Zou P, Song J, Jiang B, Pei F, Chen B, Yang X, Liu G, Hu Z. Epigallocatechin-3-gallate protects against cisplatin nephrotoxicity by inhibiting the apoptosis in mouse. Int J Clin Exp Pathol 2014;7:4607–16. [PMC free article] [PubMed] [Google Scholar]
- 30. Meng Q, Velalar CN, Ruan R. Regulating the age-related oxidative damage, mitochondrial integrity, and antioxidative enzyme activity in Fischer 344 rats by supplementation of the antioxidant epigallocatechin-3-gallate. Rejuvenation Res 2008;11:649–60. [DOI] [PubMed] [Google Scholar]
- 31. Suzuki T, Yamamoto M.. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med 2015;88:93–100. [DOI] [PubMed] [Google Scholar]
- 32. Yamamoto M, Kensler TW, Motohashi H. The Keap1-Nrf2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 2018;98:1169–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eggler AL, Small E, Hannink M, Mesecar AD. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem J 2009;422:171–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bryan HK, Olayanju A, Goldring CE, Park BK. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem Pharmacol 2013;85:705–17. [DOI] [PubMed] [Google Scholar]
- 35. Furfaro AL, Piras S, Domenicotti C, Fenoglio D, De Luigi A, Salmona M, Moretta L, Marinari UM, Pronzato MA, Traverso N et al.. Role of Nrf2, HO-1 and GSH in neuroblastoma cell resistance to bortezomib. PLoS One 2016;11:e0152465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rojo de la Vega M, Dodson M, Chapman E, Zhang DD. NRF2-targeted therapeutics: new targets and modes of NRF2 regulation. Curr Opin Toxicol 2016;1:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nezu M, Suzuki N, Yamamoto M. Targeting the KEAP1-NRF2 system to prevent kidney disease progression. Am J Nephrol 2017;45:473–83. [DOI] [PubMed] [Google Scholar]
- 38. Na HK, Surh YJ. Modulation of Nrf2-mediated antioxidant and detoxifying enzyme induction by the green tea polyphenol EGCG. Food Chem Toxicol 2008;46:1271–8. [DOI] [PubMed] [Google Scholar]
- 39. Tsai PY, Ka SM, Chang JM, Chen HC, Shui HA, Li CY, Hua KF, Chang WL, Huang JJ, Yang SS et al.. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic Biol Med 2011;51:744–54. [DOI] [PubMed] [Google Scholar]
- 40. Ye T, Zhen J, Du Y, Zhou JK, Peng A, Vaziri ND, Mohan C, Xu Y, Zhou XJ. Green tea polyphenol (−)-epigallocatechin-3-gallate restores Nrf2 activity and ameliorates crescentic glomerulonephritis. PLoS One 2015;10:e0119543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sampath C, Rashid MR, Sang S, Ahmedna M. Green tea epigallocatechin 3-gallate alleviates hyperglycemia and reduces advanced glycation end products via nrf2 pathway in mice with high fat diet-induced obesity. Biomed Pharmacother 2017;87:73–81. [DOI] [PubMed] [Google Scholar]
- 42. Sun W, Liu X, Zhang H, Song Y, Li T, Liu X, Liu Y, Guo L, Wang F, Yang T et al.. Epigallocatechin gallate upregulates NRF2 to prevent diabetic nephropathy via disabling KEAP1. Free Radic Biol Med 2017;108:840–57. [DOI] [PubMed] [Google Scholar]
- 43. Zhou P, Yu JF, Zhao CG, Sui FX, Teng X, Wu YB. Therapeutic potential of EGCG on acute renal damage in a rat model of obstructive nephropathy. Mol Med Rep 2013;7:1096–102. [DOI] [PubMed] [Google Scholar]
- 44. Wang Y, Wang B, Du F, Su X, Sun G, Zhou G, Bian X, Liu N. Epigallocatechin-3-gallate attenuates oxidative stress and inflammation in obstructive nephropathy via NF-kappaB and Nrf2/HO-1 signalling pathway regulation. Basic Clin Pharmacol Toxicol 2015;117:164–72. [DOI] [PubMed] [Google Scholar]
- 45. Du X, Yu J, Sun X, Qu S, Zhang H, Hu M, Yang S, Zhou P. Impact of epigallocatechin-3-gallate on expression of nuclear factor erythroid 2-related factor 2 and gamma-glutamyl cysteine synthetase genes in oxidative stress-induced mouse renal tubular epithelial cells. Mol Med Rep 2018;17:7952–8. [DOI] [PubMed] [Google Scholar]
- 46. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 2009;1:a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lv W, Booz GW, Wang Y, Fan F, Roman RJ. Inflammation and renal fibrosis: recent developments on key signaling molecules as potential therapeutic targets. Eur J Pharmacol 2018;820:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Poveda J, Tabara LC, Fernandez-Fernandez B, Martin-Cleary C, Sanz AB, Selgas R, Ortiz A, Sanchez-Nino MD. TWEAK/Fn14 and non-canonical NF-kappaB signaling in kidney disease. Front Immunol 2013;4:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hayden MS, Ghosh S.. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 2012;26:203–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol 2010;2:a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Giridharan S, Srinivasan M.. Mechanisms of NF-kappaB p65 and strategies for therapeutic manipulation. J Inflamm Res 2018;11:407–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang YJ, Jian JH, Liu YC, Juang SJ, Shyu KG, Lai LP, Wang BW, Leu JG. Advanced glycation end products-induced apoptosis attenuated by PPARdelta activation and epigallocatechin gallate through NF-kappaB pathway in human embryonic kidney cells and human mesangial cells. Diabetes Metab Res Rev 2010;26:406–16. [DOI] [PubMed] [Google Scholar]
- 53. Peng A, Ye T, Rakheja D, Tu Y, Wang T, Du Y, Zhou JK, Vaziri ND, Hu Z, Mohan C et al.. The green tea polyphenol (−)-epigallocatechin-3-gallate ameliorates experimental immune-mediated glomerulonephritis. Kidney Int 2011;80:601–11. [DOI] [PubMed] [Google Scholar]
- 54. Peairs A, Dai R, Gan L, Shimp S, Rylander MN, Li L, Reilly CM. Epigallocatechin-3-gallate (EGCG) attenuates inflammation in MRL/lpr mouse mesangial cells. Cell Mol Immunol 2010;7:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Y, Wang B, Du F, Su X, Sun G, Zhou G, Bian X, Liu N. Epigallocatechin-3-gallate attenuates unilateral ureteral obstruction-induced renal interstitial fibrosis in mice. J Histochem Cytochem 2015;63:270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Niu Y, Na L, Feng R, Gong L, Zhao Y, Li Q, Li Y, Sun C. The phytochemical, EGCG, extends lifespan by reducing liver and kidney function damage and improving age-associated inflammation and oxidative stress in healthy rats. Aging Cell 2013;12:1041–9. [DOI] [PubMed] [Google Scholar]
- 57. Simeone P, Trerotola M, Franck J, Cardon T, Marchisio M, Fournier I, Salzet M, Maffia M, Vergara D. The multiverse nature of epithelial to mesenchymal transition. Semin Cancer Biol [Internet] 2018. doi: 10.1016/j.semcancer.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 58. Cruz-Solbes AS, Youker K. Epithelial to mesenchymal transition (EMT) and endothelial to mesenchymal transition (EndMT): role and implications in kidney fibrosis. Results Probl Cell Differ 2017;60:345–72. [DOI] [PubMed] [Google Scholar]
- 59. Rockey DC, Bell PD, Hill JA. Fibrosis—a common pathway to organ injury and failure. N Engl J Med 2015;372:1138–49. [DOI] [PubMed] [Google Scholar]
- 60. Djudjaj S, Boor P.. Cellular and molecular mechanisms of kidney fibrosis. Mol Aspects Med 2019;65:16–36. [DOI] [PubMed] [Google Scholar]
- 61. LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med 2013;19:1047–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sutariya B, Jhonsa D, Saraf MN. TGF-beta: the connecting link between nephropathy and fibrosis. Immunopharmacol Immunotoxicol 2016;38:39–49. [DOI] [PubMed] [Google Scholar]
- 63. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol 2016;12:325–38. [DOI] [PubMed] [Google Scholar]
- 64. Zhao CG, Zhou P, Wu YB. Impact and significance of EGCG on Smad, ERK, and beta-catenin pathways in transdifferentiation of renal tubular epithelial cells. Genet Mol Res 2015;14:2551–60. [DOI] [PubMed] [Google Scholar]
- 65. Wang Y, Liu N, Su X, Zhou G, Sun G, Du F, Bian X, Wang B. Epigallocatechin-3-gallate attenuates transforming growth factor-beta1 induced epithelial-mesenchymal transition via Nrf2 regulation in renal tubular epithelial cells. Biomed Pharmacother 2015;70:260–7. [DOI] [PubMed] [Google Scholar]
- 66. Kanlaya R, Khamchun S, Kapincharanon C, Thongboonkerd V. Protective effect of epigallocatechin-3-gallate (EGCG) via Nrf2 pathway against oxalate-induced epithelial mesenchymal transition (EMT) of renal tubular cells. Sci Rep 2016;6:30233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen J, Du L, Li J, Song H. Epigallocatechin-3-gallate attenuates cadmium-induced chronic renal injury and fibrosis. Food Chem Toxicol 2016;96:70–8. [DOI] [PubMed] [Google Scholar]
- 68. Gan RY, Li HB, Sui ZQ, Corke H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): an updated review. Crit Rev Food Sci Nutr 2018;58:924–41. [DOI] [PubMed] [Google Scholar]
- 69. Saeki K, Hayakawa S, Nakano S, Ito S, Oishi Y, Suzuki Y, Isemura M. In vitro and in silico studies of the molecular interactions of epigallocatechin-3-O-gallate (EGCG) with proteins that explain the health benefits of green tea. Molecules 2018;23(6):E1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lorenz M. Cellular targets for the beneficial actions of tea polyphenols. Am J Clin Nutr 2013;98:1642S–50S. [DOI] [PubMed] [Google Scholar]
- 71. Peter B, Bosze S, Horvath R. Biophysical characteristics of proteins and living cells exposed to the green tea polyphenol epigallocatechin-3-gallate (EGCg): review of recent advances from molecular mechanisms to nanomedicine and clinical trials. Eur Biophys J 2017;46:1–24. [DOI] [PubMed] [Google Scholar]
- 72. Negri A, Naponelli V, Rizzi F, Bettuzzi S. Molecular targets of epigallocatechin-gallate (EGCG): a special focus on signal transduction and cancer. Nutrients 2018;10(12):E1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Small DM, Coombes JS, Bennett N, Johnson DW, Gobe GC. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology (Carlton) 2012;17:311–21. [DOI] [PubMed] [Google Scholar]
- 74. Yamawaki K, Kanda H, Shimazaki R. Nrf2 activator for the treatment of kidney diseases. Toxicol Appl Pharmacol 2018;360:30–7. [DOI] [PubMed] [Google Scholar]
- 75. Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, Callahan JF, Carr R, Concha N, Kerns JK et al.. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS One 2014;9:e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Shanmugam T, Selvaraj M, Poomalai S. Epigallocatechin gallate potentially abrogates fluoride induced lung oxidative stress, inflammation via Nrf2/Keap1 signaling pathway in rats: an in-vivo and in-silico study. Int Immunopharmacol 2016;39:128–39. [DOI] [PubMed] [Google Scholar]
- 77. Jiang ZY, Lu MC, Xu LL, Yang TT, Xi MY, Xu XL, Guo XK, Zhang XJ, You QD, Sun HP. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J Med Chem 2014;57:2736–45. [DOI] [PubMed] [Google Scholar]
- 78. Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol 2006;26:2887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Suzuki T, Yamamoto M.. Stress-sensing mechanisms and the physiological roles of the Keap1-Nrf2 system during cellular stress. J Biol Chem 2017;292:16817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Na HK, Kim EH, Jung JH, Lee HH, Hyun JW, Surh YJ. (−)-Epigallocatechin gallate induces Nrf2-mediated antioxidant enzyme expression via activation of PI3K and ERK in human mammary epithelial cells. Arch Biochem Biophys 2008;476:171–7. [DOI] [PubMed] [Google Scholar]
- 81. Hallan S, Sharma K.. The role of mitochondria in diabetic kidney disease. Curr Diab Rep 2016;16:61. [DOI] [PubMed] [Google Scholar]
- 82. Guo K, Lu J, Huang Y, Wu M, Zhang L, Yu H, Zhang M, Bao Y, He JC, Chen H et al.. Protective role of PGC-1alpha in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PLoS One 2015;10:e0125176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Flemming NB, Gallo LA, Forbes JM Mitochondrial dysfunction and signaling in diabetic kidney disease: oxidative stress and beyond. Semin Nephrol 2018;38:101–10. [DOI] [PubMed] [Google Scholar]
- 84. Szeto HH. Pharmacologic approaches to improve mitochondrial function in AKI and CKD. J Am Soc Nephrol 2017;28:2856–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Saad A, Herrmann SMS, Eirin A, Ferguson CM, Glockner JF, Bjarnason H, McKusick MA, Misra S, Lerman LO, Textor SC. Phase 2a clinical trial of mitochondrial protection (elamipretide) during stent revascularization in patients with atherosclerotic renal artery stenosis. Circ Cardiovasc Interv 2017;10(9):e005487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shi W, Li L, Ding Y, Yang K, Chen Z, Fan X, Jiang S, Guan Y, Liu Z, Xu D et al.. The critical role of epigallocatechin gallate in regulating mitochondrial metabolism. Future Med Chem 2018;10:795–809. [DOI] [PubMed] [Google Scholar]
- 87. Lee MS, Lee S, Doo M, Kim Y. Green tea (−)-epigallotocatechin-3-gallate induces PGC-1alpha gene expression in HepG2 cells and 3T3-L1 adipocytes. Prev Nutr Food Sci 2016;21:62–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Valenti D, De Rasmo D, Signorile A, Rossi L, de Bari L, Scala I, Granese B, Papa S, Vacca RA. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down's syndrome. Biochim Biophys Acta 2013;1832:542–52. [DOI] [PubMed] [Google Scholar]
- 89. Wang L, Wang Z, Yang K, Shu G, Wang S, Gao P, Zhu X, Xi Q, Zhang Y, Jiang Q. Epigallocatechin gallate reduces slow-twitch muscle fiber formation and mitochondrial biosynthesis in C2C12 cells by repressing AMPK activity and PGC-1alpha expression. J Agric Food Chem 2016;64:6517–23. [DOI] [PubMed] [Google Scholar]
- 90. Valenti D, de Bari L, De Rasmo D, Signorile A, Henrion-Caude A, Contestabile A, Vacca RA. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim Biophys Acta 2016;1862:1093–104. [DOI] [PubMed] [Google Scholar]
- 91. Adikesavan G, Vinayagam MM, Abdulrahman LA, Chinnasamy T. (−)-Epigallocatechin-gallate (EGCG) stabilizes the mitochondrial enzymes and inhibits the apoptosis in cigarette smoke-induced myocardial dysfunction in rats. Mol Biol Rep 2013;40:6533–45. [DOI] [PubMed] [Google Scholar]
- 92. Ha T, Kim MK, Park KS, Jung W, Choo H, Chong Y. Structural modification of (−)-epigallocatechin gallate (EGCG) shows significant enhancement in mitochondrial biogenesis. J Agric Food Chem 2018;66:3850–9. [DOI] [PubMed] [Google Scholar]
- 93. Zhang G, Zhang J.. Enhanced oral bioavailability of EGCG using pH-sensitive polymeric nanoparticles: characterization and in vivo investigation on nephrotic syndrome rats. Drug Des Devel Ther 2018;12:2509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Krupkova O, Ferguson SJ, Wuertz-Kozak K. Stability of (−)-epigallocatechin gallate and its activity in liquid formulations and delivery systems. J Nutr Biochem 2016;37:1–12. [DOI] [PubMed] [Google Scholar]
- 95. Yang QQ, Wei XL, Fang YP, Gan RY, Wang M, Ge YY, Zhang D, Cheng LZ, Corke H. Nanochemoprevention with therapeutic benefits: an updated review focused on epigallocatechin gallate delivery. Crit Rev Food Sci Nutr [Internet] 2019. doi: 10.1080/10408398.2019.1565490. [DOI] [PubMed] [Google Scholar]
- 96. Cai ZY, Li XM, Liang JP, Xiang LP, Wang KR, Shi YL, Yang R, Shi M, Ye JH, Lu JL et al.. Bioavailability of tea catechins and its improvement. Molecules 2018;23(9):E2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Abe O, Ono T, Sato H, Muller F, Ogata H, Miura I, Shikama Y, Yabe H, Onoue S, Fromm MF et al.. Role of (−)-epigallocatechin gallate in the pharmacokinetic interaction between nadolol and green tea in healthy volunteers. Eur J Clin Pharmacol 2018;74:775–83. [DOI] [PubMed] [Google Scholar]
- 98. Albassam AA, Markowitz JS.. An appraisal of drug-drug interactions with green tea (Camellia sinensis). Planta Med 2017;83:496–508. [DOI] [PubMed] [Google Scholar]
- 99. Baell JB, Holloway GA.. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2010;53:2719–40. [DOI] [PubMed] [Google Scholar]
- 100. Baell JB. Feeling nature's PAINS: natural products, natural product drugs, and pan assay interference compounds (PAINS). J Nat Prod 2016;79:616–28. [DOI] [PubMed] [Google Scholar]
- 101. Capuzzi SJ, Muratov EN, Tropsha A. Phantom PAINS: problems with the utility of alerts for Pan-Assay INterference CompoundS. J Chem Inf Model 2017;57:417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Baell JB, Nissink JWM.. Seven year itch: pan-assay interference compounds (PAINS) in 2017—utility and limitations. ACS Chem Biol 2018;13:36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Siramshetty VB, Preissner R.. Drugs as habitable planets in the space of dark chemical matter. Drug Discov Today 2018;23:481–6. [DOI] [PubMed] [Google Scholar]